Genetic Syndromes Associated with Obesity
Inherited factors play a substantial role in determining adiposity across the full range of human body weight.1 In this chapter, we focus on the known Mendelian disorders that include obesity as a consistent clinical feature. Classically, patients affected by these obesity syndromes have been identified as a result of their association with mental retardation, dysmorphic features, and/or other developmental abnormalities. More recently, several monogenic disorders resulting from disruption of the leptin-melanocortin signaling pathway (see Chapter 1) have been identified. In these disorders, obesity itself is the predominant presenting feature, although it often is accompanied by characteristic patterns of neuroendocrine dysfunction. For the purposes of clinical assessment, it remains useful to categorize the genetic obesity syndromes as those with and without associated developmental delay.
Obesity Associated With Developmental Delay
Definition, Prevalence, Etiology, and Pathogenesis
Prader, Labhart, and Willi described the first patient with this syndrome in 1956.2 Prader-Willi syndrome (PWS) is the most common syndromal cause of human obesity, with an estimated prevalence of about 1 in 25,000 births and a population prevalence of 1 in 50,000.3 Prader-Willi syndrome is caused by deficiency of one or more paternally expressed imprinted transcripts within chromosome 15q11-q13, a region that includes SNURF-SNRPN and multiple small nucleolar RNAs (snoRNAs). The molecular pathophysiology of PWS remains unclear, although several candidate genes in this region have been studied and their expression has been shown to be absent in postmortem brains of PWS patients.4 Balanced translocations that leave the SNURF-SNRPN promoter and coding regions intact5 suggest that disruption of SNURF-SNRPN is less important, whereas a recently reported microdeletion of the HBII-85 snoRNAs in a child with PWS provides strong evidence that deficiency of HBII-85 snoRNAs plays a major role in the key characteristics of the PWS phenotype.6 However, some atypical features in the latter patient suggest that other genes in this region also may be important.
One suggested mediator of the obesity phenotype in PWS patients is the enteric hormone ghrelin, which is implicated in the regulation of meal-time hunger in rodents and humans and is also a stimulator of growth hormone (GH) secretion via the GH-secretagogue receptor (GHS-R).7 Fasting plasma ghrelin levels are 4.5-fold higher in PWS subjects than in equally obese controls and patients with other obesity syndromes, and thus they may be implicated in the pathogenesis of hyperphagia in these patients.8,9
Clinical Features
The Prader-Willi syndrome (PWS) is characterized by diminished fetal activity, hypotonia, mental retardation, short stature, hypogonadotropic hypogonadism, and obesity. The diagnostic criteria arrived at by a consensus group are based on a point system; 1 point each is allowed for each of five major criteria, and one-half point each for seven minor criteria.10 A minimum of 8.5 points is considered necessary for the clinical diagnosis of PWS (Table 3-1).
Table 3-1
Diagnostic Criteria for Prader-Willi Syndrome
Major Criteria
Neonatal and infantile hypotonia, with poor suck and subsequent improvement with age
Feeding problems with poor weight gain in infancy, needing gavage or other special feeding techniques
Weight gain (rapid onset 1 to 6 years old) that leads to central obesity
Characteristic facial features, including narrow bifrontal diameter, almond-shaped palpebral fissures, and turned-down mouth
Hypogonadism/hypogenitalism: genital hypoplasia (small labia minora and clitoris in females, and hypoplastic scrotum in males); incomplete and delayed puberty; and infertility
Developmental delay/mild to moderate mental retardation/multiple learning disabilities
Minor Criteria
Reduced fetal movement and infantile lethargy, which improves with age
Characteristic behavioral problems, including temper tantrums, obsessive-compulsive behavior, stubbornness, rigidity, stealing, and lying
Short stature for family by 15 years of age
Small hands and feet for height and age
Narrow hands with straight ulnar border
Eye abnormalities, including esotropia and myopia
Altered temperature sensitivity
Normal neuromuscular studies (e.g., muscle biopsy and electromyography)
Children with PWS display diminished growth, reduced muscle mass, and increased fat mass; body composition abnormalities resemble those seen in GH deficiency.11 Diminished GH responses to various provocative agents, low insulin-like growth factor-1 levels, and the presence of additional evidence of hypothalamic dysfunction support the presence of true GH deficiency (GHD) in many children with PWS. Boys with PWS usually have hypoplastic external genitalia, including micropenis, whereas girls have hypoplastic labia minora. Adrenarche can occur early, but gonadal maturation usually is delayed or incomplete as the result of hypogonadotropic hypogonadism.
Recent studies have demonstrated a particular pattern of fat distribution in adult patients with PWS, with large amounts of subcutaneous fat, in the presence of relatively normal intraabdominal fat stores. This is associated with relative protection from the insulin resistance and metabolic syndrome usually associated with morbid obesity.12
Diagnosis
Loss of the paternal chromosomal segment 15q11.2-q12 (usually de novo) is principally responsible for PWS. Such a loss can occur by either of two mechanisms: through deletion of the paternal “critical” segment (75%), or through loss of the entire paternal chromosome 15 with the presence of two maternal homologues (uniparental maternal disomy) in approximately 22% of patients.13 The opposite (i.e., maternal deletion or paternal uniparental disomy) causes another characteristic phenotype, the Angelman syndrome. In rare instances, imprinting errors due to a sporadic or inherited microdeletion in the imprinting center (3% of patients) or a paternal imprinted translocation (<1%) is observed.13
Deletions account for 70% to 80% of cases, many of which can be visualized by standard prometaphase banding examination. A minority consist of unbalanced translocations, which are detected easily by routine chromosome examination. Remaining cases are the result of maternal uniparental disomy wherein cytogenetic examinations yield normal results. However, distinct differences in DNA methylation are noted at the D15S9 locus on 15q11-q13, according to the parent of origin; thus DNA methylation can be used as a reliable postnatal diagnostic tool in PWS patients with a normal karyotype.14
Treatment
Traditionally, the mainstay of management has centered on early institution of a low-calorie diet with regular exercise, rigorous supervision, restriction of food and money, and appropriate psychological and behavioral counseling for the patient and family, often in the context of group homes for PWS adolescents and adults. Pharmacologic treatment, including anorexigenic agents that act through central monoamine and serotoninergic pathways, is not always beneficial in treating hyperphagia and obesity, although a few published controlled studies can be found in the literature. The choice and the use of specific antidiabetic, antihypertensive, and lipid-lowering agents will be guided by those in the general population with obesity, but possible differences in PWS have not been addressed systematically.15
In PWS children, therapy with GH significantly improves the rate of growth and final height. Long-term studies show that final height is in the average range for age, and GH is now licensed for use in PWS. GH treatment in PWS children also decreases body fat and increases muscle mass, fat oxidation, and energy expenditure.16 Physical strength and agility are also improved. These improvements are most dramatic during the first year of GH therapy, although prolonged treatment does not completely normalize these parameters.17 Although increases in fasting insulin and reduced glucose elimination rates have been seen during GH therapy, the development of glucose intolerance and diabetes mellitus does not appear to be a problem to date.
Treatment with clomiphene citrate has been shown to raise plasma luteinizing hormone, testosterone, and urinary gonadotropin levels to normal and to result in normal spermatogenesis and physical signs of puberty.18 The prescription of testosterone therapy for PWS males has been complicated by anecdotal reports of increased aggressive behavior.
Fragile X Syndrome
Definition, Prevalence, Etiology, and Pathogenesis
The fragile X syndrome is the most common cause of inherited mental retardation. Recent epidemiologic studies indicate that it is responsible for moderate to severe mental retardation in 1 in 4000 to 6000 males of European descent and is responsible for mild to moderate mental retardation in 1 in 7000 to 10,000 females, with frequency of disease thought to be higher in some ethnic groups (e.g., Tunisian Jews, African Americans). In affected families, there are often clinically normal, transmitting males whose daughters, who are also clinically normal, have a high risk of having clinically affected children.19 In 1991, the molecular cloning of the fragile X locus revealed unstable expansions of a CGG trinucleotide repeat, located in the FMR1 (fragile X mental retardation 1) gene, which lead to transcriptional silencing. FMR1 encodes a specific RNA-binding protein, FMRP, that negatively regulates local protein synthesis in neuronal dendrites. In its absence, the transcripts normally regulated by FMRP are overtranslated. The resulting overabundance of certain proteins results in reduced synaptic strength and synaptic plasticity.20
Clinical Features
Fragile X syndrome is characterized by moderate to severe mental retardation, macroorchidism, large ears, macrocephaly, prominent jaw (mandibular prognathism), and high-pitched jocular speech. In affected boys, delay in language acquisition and/or behavioral problems are often the presenting symptoms. A Prader-Willi–like subphenotype of the fragile X syndrome has been described in a subset of patients with extreme obesity with a full, round face, small, broad hands and feet, and regional skin hyperpigmentation.21 Unlike with Prader-Willi syndrome, patients lack neonatal hypotonia and feeding problems during infancy followed by hyperphagia during toddlerhood.
Diagnosis and Treatment
The discovery of the fragile X expansion mutation has produced efficient and reliable tools for diagnosis, genetic counseling, and prenatal diagnosis.22 Approaches used in the management of the behavioral disturbance of these children include the use of clonidine and anticonvulsants, especially carbamazepine and valproate, which may have behavior-modifying effects in addition to their antiseizure actions, and some forms of behavioral therapy.23
Bardet-Biedl Syndrome
Definition, Prevalence, Etiology, and Pathogenesis
The earliest formal description of this syndrome was provided in 1920 by George Bardet, who described patients with polydactyly, retinitis pigmentosa, and obesity. In 1922, Artur Biedl, an Austrian professor of pathology and endocrinology, published a short independent account of two siblings with “congenital deformations (retinitis pigmentosa and polydactyly) and an intellectual torpidity.” Bardet-Biedl syndrome (BBS) is a rare (prevalence <1/100,000), genetically highly heterogeneous, autosomal recessive syndrome characterized by central obesity (in 75% of patients), mental retardation, dysphormic extremities (syndactyly, brachydactyly, or polydactyly), retinal dystrophy or pigmentary retinopathy, hypogonadism or hypogenitalism (limited to male patients), and structural abnormalities of the kidney or functional renal impairment. Some overlap has been noted with the syndrome described by John Laurence (an ophthalmic surgeon) and his house surgeon Robert Moon in the late 1800s, which was characterized by retinal pigmentary degeneration, mental retardation, and hypogonadism in conjunction with progressive spastic paraparesis and distal muscle weakness, but without polydactyly.24
Bardet-Biedl syndrome is a genetically heterogeneous disorder that now is known to map to at least twelve loci, many of which have now been identified at the molecular level.25–27 Although BBS is usually transmitted as a recessive disorder, some families have exhibited so called “tri-allelic” inheritance, where the clinical manifestation of the syndrome requires two mutations in one BBS gene plus an additional mutation in a second, unlinked BBS gene.28
Recent studies strongly indicate that most of the genes involved in the BBS are involved in the structure and/or function of the basal body, a modified centriole that is essential for the function of nonmotile cilia,29,30 subcellular organelles whose importance for intercellular communication is becoming increasingly evident. Some BBS proteins are involved in non-canonical Wnt and Sonic Hedgehog signaling within the cilium, suggesting that BBS proteins may contribute to disease pathogenesis through multiple molecular mechanisms.31
Borjeson-Forssman-Lehmann Syndrome
In 1962, Borjeson, Forssman, and Lehmann described a syndrome characterized by moderate to severe mental retardation, epilepsy, hypogonadism, obesity with marked gynecomastia, swelling of subcutaneous tissue of the face, narrow palpebral fissures, and large but not deformed ears.32 By linkage analysis, the gene associated with BFLS was localized to Xq26-q27, and recently, mutations in a novel, widely expressed zinc-finger gene (plant homeodomain finger gene 6) (PHF6) have been identified in affected families.33 Strongest PHF6 gene expression and nuclear localization of PHF6 protein are observed in the developing central nervous system, the anterior pituitary gland, the primordia of facial structures, and the limb buds, although as yet nothing is known about the cellular function of PHF6.34
Wilson-Turner Syndrome
In 1991, Wilson described a kindred in which males in five successive generations in an X-linked recessive pedigree pattern had a mental retardation syndrome.35 Clinical features include obesity, gynecomastia, speech difficulties, emotional lability, tapering fingers, and small feet. Some of the features resemble those of Borjeson-Forssman-Lehmann syndrome, but the patients of Wilson and Turner did not have hypermetropia or cataracts in later life and did not have elongated earlobes. Linkage studies have defined the physical location as Xp21.1-q22.
Cohen Syndrome
In 1973, Cohen and his collaborators observed three patients with a previously unrecognized pattern of abnormalities in association with truncal obesity.36 Cohen syndrome is one of the rare autosomal recessive disorders that are overrepresented in the Finnish population; it is characterized by nonprogressive mild to severe psychomotor retardation, motor clumsiness, microcephaly, characteristic facial features, childhood hypotonia and joint laxity, progressive retinochoroidal dystrophy, myopia, intermittent isolated neutropenia, and a cheerful disposition.37 Specific facial features include high-arched or wave-shaped eyelids, long, thick eyelashes, thick eyebrows, prominent root of nose, short philtrum (which is unable to cover the prominent upper central incisors), small or absent lobuli of the ears, thick hair and low hairline, narrow hands and feet, and mild syndactylies (in 50% to 60%). Progressive, often high-grade myopia and retinochoroidal dystrophy resembling retinitis pigmentosa are essential features in Cohen syndrome.38 Vision starts to deteriorate early but generally is preserved until adulthood, and by the age of 40, many patients are severely visually handicapped.
Linkage analysis based on a number of extended Finnish pedigrees led to the identification of a locus on 8q22, and homozygosity mapping in a consanguineous Lebanese kindred led to localization of the gene responsible for this condition to a region on chromosome 8q21.3-q22.1. A novel gene, COH1, which is mutated in some Finnish patients with Cohen syndrome, has been identified.39 Although the functional properties of this protein are unclear, comparison with structurally homologous proteins suggests a role in intracellular vesicle–mediated sorting and transport of proteins.
Albright’s Hereditary Osteodystrophy (AHO)
In 1942, Fuller Albright reported a syndrome of end-organ hormone resistance often accompanied by specific phenotype, including short stature, obesity, round facies, brachydactyly, ectopic soft tissue ossification (osteoma cutis), and mild developmental delay that was found in approximately 75% of patients in some series. Albright’s hereditary osteodystrophy is an autosomal dominant disorder caused by germline mutations in GNAS1 that decrease expression or function of the Gsα protein. Maternal transmission of GNAS1 mutations leads to AHO plus resistance to several hormones (e.g., parathyroid hormone) that activate Gs in their target tissues (pseudohypoparathyroidism type IA); paternal transmission leads only to the AHO phenotype (pseudopseudohypoparathyroidism). Thus GNAS1 is imprinted in a tissue-specific manner, being expressed primarily from the maternal allele in some tissues and biallelically expressed in most other tissues; thus multi-hormone resistance occurs only when Gsα mutations are inherited maternally.40
Bdnf and Trkb Deficiency
A single patient has been reported with a de novo missense mutation in the neurotropin receptor TrkB associated with severe hyperphagia and obesity, delayed speech and language development, impaired short-term memory, and loss of nociception.41 A de novo chromosomal inversion on the short arm of chromosome 11 that disrupts the expression of brain-derived neurotropic factor (BDNF), which signals through TrkB, also gives a comparable obesity and neurobehavioral phenotype.42 The neurotropins are involved in the development and maintenance of neurons and in mediating synaptic plasticity in the brain and peripheral nervous system.
Additional Syndromes
Biemond syndrome type 2 (BS2) is a recessively inherited condition that comprises mental retardation, coloboma of the retina, early-onset obesity, polydactyly, and hypogonadism.43 MEHMO syndrome is characterized by Mental retardation, Epileptic seizures, Hypogonadism and hypogenitalism, Microcephaly, and Obesity. The life expectancy of affected patients is less than 2 years. Haplotype and linkage analyses in a large three-generation family assigned the disease locus to Xp22.13-p21.1.44
Obesity Without Developmental Delay
In 1959, Alstrom and colleagues45 reported patients with a disorder that bears some similarities to the Bardet-Biedl syndrome (retinitis pigmentosa, deafness, obesity, diabetes mellitus with recessive inheritance); however, classically, mental retardation, polydactyly, and hypogonadism are not features. In a large series of 22 UK Alstrom patients, all were suspected of having a severe visual defect in infancy, and severe photophobia and nystagmus were often reported by 4 months of age.46 Early loss of central vision was seen (usually by 1 year) in contrast to loss of peripheral vision in other pigmentary retinopathies. Electroretinograms (ERGs) can be used to classify the severity and pattern of cone and rod involvement. It is now recognized that subsets of affected individuals present with additional features such as dilated cardiomyopathy (often diagnosed in infancy), hepatic dysfunction, renal dysfunction, hypothyroidism, male hypogonadism, short stature, and mild to moderate developmental delay.47
Although obesity is common in this syndrome, it is rarely severe. In contrast, insulin resistance is frequently severe, and once diabetes develops, it may be very difficult to control. Hypertriglyceridemia may be severe and may result in acute pancreatitis. Mutations in a single gene (ALMS1) have been found to be responsible for all cases of Alstrom syndrome thus far characterized.48 ALMS1 is localized to the centrosome and the ciliary basal bodies in vitro,49 consistent with a role in the structure of the basal body or in the transport of proteins between the cytoplasm and the ciliary axoneme. ALMS1 mutant mice exhibit a lack of sperm flagella, a modified ciliary structure, and defective rhodopsin transport through the connecting cilia of photoreceptor cells.50 Thus, like Bardet-Biedl syndrome (BBS), Alstrom syndrome can be considered a “ciliopathy.”27
Ulnar-Mammary Syndrome
In 1973, Schinzel studied a Swiss kindred in which the proband, brother, father, and nephew had ulnar ray defects, small penis, delayed puberty, and obesity; the proband and his father also had anal atresia. Schinzel subsequently suggested that this syndrome is identical to the ulnar-mammary syndrome of Pallister reported in 1976, and major features are defined as ulnar finger and fibular toe ray defects, delayed growth, obesity, hypogenitalism, and hypoplasia of nipples and apocrine glands with subsequent diminished ability to perspire.51 Additional findings in single cases included pyloric, anal, and subglottic stenosis. In 1995, Bamshad and colleagues identified 33 affected individuals in a large Utah family with ulnar-mammary syndrome (UMS), a number greater than the sum of all previously reported cases at that time.52 By linkage analysis, the UMS gene was mapped to 12q23-q24.1, and in 1997, these investigators demonstrated that mutations in the TBX3 gene are responsible for UMS.53 Mutations in the closely linked and structurally related TBX5 gene cause anterior limb abnormalities in Holt-Oram syndrome in association with cardiac anomalies, compared with those in TBX3, which cause posterior (ulnar or postaxial) limb changes. Recently, TBX3 has been demonstrated to be a downstream target of the Wnt/beta-catenin pathway, playing a role in cell proliferation and survival.54
Simpson-Golabi-Behmel, Type 2
In 1975, Simpson reported patients with a collection of features (broad stocky appearance, large protruding jaw, widened nasal bridge, upturned nasal tip) and referred to the appearance as “bulldog”-like.55 Behmel observed 11 male newborns with a similar syndrome, and in 1984, Golabi and Rosen reported similar features in a family in which four males in three generations exhibited features consistent with X-linked recessive inheritance. Some patients with Simpson-Golabi-Behmel syndrome (SGBS) harbor mutations in glypican 3 (GPC3), a putative extracellular proteoglycan.56 GPC3 acts as a negative regulator of hedgehog signaling during mammalian development, and recent studies have suggested that the overgrowth observed in SGBS patients is, at least in part, the consequence of hyperactivation of the hedgehog signaling pathway.57
Congenital Leptin Deficiency
Homozygous frameshift, nonsense, and missense mutations in the gene encoding leptin (LEP) have been identified in 12 patients to date58–61 (and own observations). These mutations result in an inability to produce the protein product, leptin, with undetectable leptin levels in the serum of affected patients. All affected subjects show normal neurobehavioral development and have no dysmorphic features. Leptin-deficient subjects are of normal weight at birth but exhibit rapid weight gain in the first few months of life, resulting in severe obesity.62 Body composition measurements show that leptin deficiency is characterized by the preferential deposition of fat mass, giving a distinct clinical appearance with excessive amounts of subcutaneous fat over the trunk and limbs. Hyperinsulinemia consistent with the severity of obesity is observed, and some adults have developed type 2 diabetes in the third and fourth decades. All subjects in these families are characterized by intense hyperphagia with food-seeking behavior and aggressive behavior when food is denied,60 as well as an inability to discriminate between appetizing and bland foods.63 Although no changes in resting metabolic rate or total energy expenditure are detectable,60 abnormalities in sympathetic nerve function have been reported.64
Leptin deficiency is associated with hypothalamic hypothyroidism and hypogonadotropic hypogonadism.60,64 However, some evidence for the delayed but spontaneous onset of menses has been found in leptin-deficient adults.64 Children with leptin deficiency have impaired T cell number and function,60 consistent with high rates of childhood infection and a high reported rate of childhood mortality.64
Congenital leptin deficiency can be treated with daily subcutaneous injections of recombinant human leptin, leading to correction of all phenotypic abnormalities seen in these patients.60,65 Leptin administration reduces food intake60 and modulates the reward value of food, a response that is mediated by activation of striatal brain regions as measured by functional MRI.63 Administration of leptin permits the progression of appropriately timed pubertal development in children of appropriate age60 and induces the development of secondary sexual characteristics and pulsatile gonadotropin secretion in adults with leptin deficiency.65
Diagnosis and Treatment
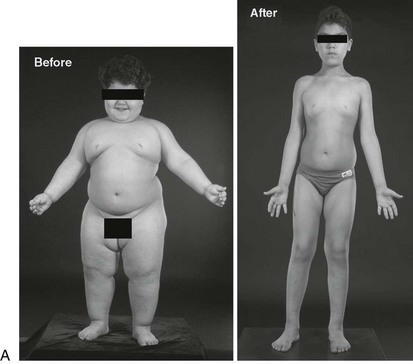
Congenital leptin deficiency can be diagnosed on the basis of an undetectable serum leptin measurement, followed by genotyping of the LEP gene. Although this syndrome is rare, it is unique in being amenable to a specific form of hormone replacement therapy. Eleven patients are currently receiving recombinant human leptin by subcutaneous injection, and all have shown dramatic improvement in their clinical and biochemical status (Figure 3-1).60,65 Leptin therapy results in a dramatic improvement in hyperphagia and correction of thyroid and T cell dysfunction. Leptin also permits the onset of puberty at an appropriate developmental age.62
Leptin Receptor Deficiency
Up to 3% of patients with severe obesity have been found to harbor mutations in the leptin receptor gene that are associated with loss of function in vitro as measured by an inability to phosphorylate STAT3.66 Although heterozygosity for LEP or LEPR mutations is associated with an increase in body weight,66,67 severe obesity requires the loss of two alleles as the result of homozygous or compound heterozygous mutations. Serum leptin levels are not elevated disproportionately in LEPR deficiency,66 although particular mutations located near the transmembrane domain can result in a truncated extracellular domain that may act as a false binding protein, resulting in abnormally elevated leptin levels.68,69 The clinical phenotype of congenital leptin receptor deficiency is similar to that of leptin deficiency, with hyperphagia, severe early-onset obesity, hypogonadism, and frequent infections.66
POMC Deficiency
Leptin activates hypothalamic neurons expressing pro-opiomelanocortin (POMC), and this action is functionally important for leptin’s action on appetite and body weight.70 In 1998, Krude and colleagues reported two unrelated obese German children who were homozygous or compound heterozygous for mutations in POMC (pro-opiomelanocortin)71; subsequently, several other children have been reported.72,73 Initial presentation occurs in neonatal life with adrenal crisis due to adrenocorticotropic hormone (ACTH) deficiency (POMC is a precursor of ACTH in the pituitary), and children require lifelong glucocorticoid replacement. Children have pale skin and red hair caused by the lack of melanocyte-stimulating hormone (MSH) action at melanocortin 1 receptors in the skin and hair follicles,72 although hypopigmentation may be less obvious in children from different ethnic backgrounds.73 POMC deficiency leads to hyperphagia and early-onset obesity, caused by loss of melanocortin signaling at the melanocortin 4 receptor (MC4R). Although, as yet, no specific treatment is available, selective small molecule MC4R agonists are being developed, and it is likely that these children would be highly responsive to such agents. It is notable that a high prevalence of overweight/obesity has been observed in the heterozygous relatives of children with complete POMC deficiency,72,73 suggesting that heterozygous point mutations in POMC might contribute to inherited obesity.74–76
Prohormone Convertase 1 Deficiency
Three unrelated subjects with compound heterozygous or homozygous mutations in the prohormone convertase (PC1/3) gene have been described.77–79 PC1/3 encodes an enzyme involved in the posttranslational processing of multiple prohormones. A prominent clinical feature is severe small intestinal absorptive dysfunction in neonatal life, which is likely to be attributable to impaired prohormone processing within the enteroendocrine cells and nerves that express PC1 throughout the gut.78 Postprandial hypoglycemia due to impaired processing of proinsulin to mature insulin is a key clinical feature and serves as the basis for a convenient diagnostic test, as elevated plasma levels of proinsulin and 32/33 split proinsulin in the context of low levels of mature insulin are found in PC1-deficient patients.78 Hypogonadotropic hypogonadism due to impaired processing of preprogonadotropin-releasing hormone contributes to infertility, and impaired processing of POMC contributes to hyperphagic severe early-onset obesity.
Melanocortin 4 Receptor Deficiency
Mutations in MC4R have been reported in up to ≈6% of patients with severe early-onset obesity80 and are found at a frequency of approximately 1 in 1000 in the general UK population,81 making this one of the most common human monogenic diseases. Although we found a 100% penetrance of early-onset obesity in heterozygous probands, others have described obligate carriers who were not obese.82 Given the large number of potential influences on body weight, perhaps it is not surprising that both genetic and environmental modifiers will have important effects in some pedigrees. When all of these observations are considered, codominance, with modulation of expressivity and penetrance of the phenotype, is the most appropriate descriptor for the mode of inheritance.83
Affected subjects exhibit hyperphagia; this is not as severe as that seen in leptin deficiency, although it often starts in the first year of life. Of particular note is the finding that the severity of receptor dysfunction seen in in vitro assays can predict the amount of food ingested at a test meal by the subject who is harboring that particular mutation.80 An elevated respiratory quotient (ratio of carbohydrate to fat oxidation) in MC4R deficiency is consistent with an impaired ability to mobilize fat, as has been seen in MC4R knockout mice.84 Alongside the increase in fat mass, MC4R-deficient subjects also have an increase in lean mass that is not seen in leptin deficiency and a marked increase in bone mineral density. Linear growth of these subjects is striking, with affected children having a height standard deviation score (SDS) of +2 compared with population standards, and adults having an increased final height when compared with equally obese adults with a normal MC4R genotype. MC4R-deficient subjects also have higher levels of fasting insulin than age- and BMI SDS–matched children.
Other Syndromes
Holder and colleagues studied a girl with hyperphagia and early-onset obesity and a balanced translocation between 1p22.1 and 6q16.2.85 The translocation disrupted the SIM1 gene on 6q. The Drosophila single-minded (Sim) gene is a regulator of neurogenesis, and in the mouse, Sim1 is expressed in the developing central nervous system and is essential for formation of the supraoptic and paraventricular (PVN) nuclei, which express the melanocortin 4 receptor. Mice heterozygous for loss of function mutations in Sim1 are obese, making it very likely that disruption of this gene in the child was the cause of her obesity.86 A number of patients with obesity, hypotonia, and developmental delay in association with interstitial chromosome 6q deletions have been described, although whether this syndrome can be attributed to SIM1 remains unclear.87
The WAGR syndrome (Wilms tumor, anorexia, ambiguous genitalia, and mental retardation) is one of the best studied contiguous gene syndromes associated with chromosomal deletions at 11p13, the location of the WT1 gene.88 A few patients with WAGR syndrome and obesity have been reported with deletions of chromosome 11p14-p12, a region that encompasses BDNF, the disruption of which is known to cause obesity.42
References
1. Barsh, GS, Farooqi, IS, O’Rahilly, S. Genetics of body-weight regulation. Nature. 2000;404:644–651.
2. Prader, A, Labhart, A, Willi, H. Ein Syndrom von Adipositas, Kleinwuchs, Kryptorchismus und Oligophrenie nach Myatonieartigem Zustand im Neugeborenenalter. Schweiz Med Wschr. 1956;86:1260–1261.
3. Butler, M. Prader-Willi syndrome: current understanding of cause and diagnosis. Am J Med Genet. 1990;35:319–332.
4. Swaab, DF, Purba, JS, Hofman, MA. Alterations in the hypothalamic paraventricular nucleus and its oxytocin neurons (putative satiety cells) in Prader-Willi syndrome: a study of five cases. J Clin Endocrinol Metab. 1995;80:573–579.
5. Schulze, A, Hansen, C, Skakkebaek, NE, et al. Exclusion of SNRPN as a major determinant of Prader-Willi syndrome by a translocation breakpoint. Nat Genet. 1996;12:452–454.
6. Sahoo, T, del Gaudio, D, German, JR, et al. Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster. Nat Genet. 2008;40:719–721.
7. Cummings, DE WD, Frayo, RS, Breen, PA, et al. Plasma ghrelin levels after diet-induced weight loss or gastric bypass surgery. N Engl J Med. 2002;346:1623–1630.
8. Cummings, DE CK, Purnell, JQ, Vaisse, C, et al. Elevated plasma ghrelin levels in Prader Willi syndrome. Nat Med. 2002;8:643–644.
9. Haqq, AM, Farooqi, IS, O’Rahilly, S, et al. Serum ghrelin levels are inversely correlated with body mass index, age, and insulin concentrations in normal children and are markedly increased in Prader-Willi syndrome. J Clin Endocrinol Metab. 2003;88:174–178.
10. Holm, VA CS, Butler, MG, Hanchett, JM, et al. Prader-Willi syndrome: consensus diagnostic criteria. Pediatrics. 1993;91:398–402.
11. van Mil, EG WK, Gerver, WJ, Van Marken Lichtenbelt, WD, et al. Body composition in Prader-Willi syndrome compared with nonsyndromal obesity: Relationship to physical activity and growth hormone function. J Pediatr. 2001;139:708–714.
12. Hoybye, C, Hilding, A, Jacobsson, H, et al. Metabolic profile and body composition in adults with Prader-Willi syndrome and severe obesity. J Clin Endocrinol Metab. 2002;87:3590–3597.
13. Ohta, T, Gray, TA, Rogan, PK, et al. Imprinting-mutation mechanisms in Prader-Willi syndrome. Am J Hum Genet. 1999;64:397–413.
14. Driscoll, DJ WM, Williams, CA, Zori, RT, et al. A DNA methylation imprint, determined by the sex of the parent, distinguishes the Angelman and Prader-Willi syndromes. Genomics. 1992;13:917–924.
15. Goldstone, AP. Prader-Willi syndrome: advances in genetics, pathophysiology and treatment. Trends Endocrinol Metab. 2004;15:12–20.
16. Carrel, AL MS, Whitman, BY, Allen, DB. Growth hormone improves body composition, fat utilization, physical strength and agility, and growth in Prader-Willi syndrome: A controlled study. J Pediatr. 1999;134:215–221.
17. Lindgren, AC, Lindberg, A. Growth Hormone Treatment Completely Normalizes Adult Height and Improves Body Composition in Prader-Willi Syndrome: Experience from KIGS (Pfizer International Growth Database). Horm Res. 2008;70:182–187.
18. Hamilton, CR, Jr SR., Kliman, B. Hypogonadotropinism in Prader-Willi syndrome. Induction of puberty and sperm altogenesis by clomiphene citrate. Am J Med. 1972;52:322–329.
19. Hagerman, PJ, Hagerman, RJ. The fragile-X premutation: a maturing perspective. Am J Hum Genet. 2004;74:805–816.
20. Zalfa, F, Achsel, T, Bagni, C. mRNPs, polysomes or granules: FMRP in neuronal protein synthesis. Curr Opin Neurobiol. 2006;16:265–269.
21. de Vries, BB FJ, Butler, MG, Canziani, F, et al. Clinical and molecular studies in fragile X patients with a Prader-Willi-like phenotype. J Med Genet. 1993;30:761–766.
22. Kaplan, G, Kung, M, McClure, M, et al. Direct mutation analysis of 495 patients for fragile X carrier status/proband diagnosis. Am J Med Genet. 1994;51:501–502.
23. Jin, P, Warren, ST. New insights into fragile X syndrome: from molecules to neurobehaviors. Trends Biochem Sci. 2003;28:152–158.
24. Laurence, JZ MR. Four cases of “retinitis pigmentosa” occurring in the same family, and accompanied by general imperfections of development. Ophthalmol Rev. 1866;2:32–41.
25. Katsanis, N, Lupski, JR, Beales, PL. Exploring the molecular basis of Bardet-Biedl syndrome. Hum Mol Genet. 2001;10:2293–2299.
26. Katsanis, N. The oligogenic properties of Bardet-Biedl syndrome. Hum Mol Genet. 2004;13(Spec No 1):R65–71.
27. Badano, JL, Mitsuma, N, Beales, PL, et al. The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet. 2006;7:125–148.
28. Katsanis, N AS, Badano, JL, Eichers, ER, et al. Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science. 2001;293:2256–2259.
29. Mykytyn, K, Sheffield, VC. Establishing a connection between cilia and Bardet-Biedl Syndrome. Trends Mol Med. 2004;10:106–109.
30. Ainsworth, C. Cilia: tails of the unexpected. Nature. 2007;448:638–641.
31. Tobin, JL, Beales, PL. Bardet-Biedl syndrome: beyond the cilium. Pediatr Nephrol. 2007;22:926–936.
32. Borjeson, M, Forssman, H, Lehmann, O. An X-linked, recessively inherited syndrome characterized by grave mental deficiency, epilepsy, and endocrine disorder. Acta Med Scand. 1962;171:13–21.
33. Turner, G, Gedeon, A, Mulley, J, et al. Borjeson-Forssman-Lehmann syndrome: clinical manifestations and gene localization to Xq26–27. Am J Med Genet. 1989;34:463–469.
34. Voss, AK, Gamble, R, Collin, C, et al. Protein and gene expression analysis of Phf6, the gene mutated in the Borjeson-Forssman-Lehmann Syndrome of intellectual disability and obesity. Gene Expr Patterns. 2007;7:858–871.
35. Wilson, M, Mulley, J, Gedeon, A, et al. New X-linked syndrome of mental retardation, gynecomastia, and obesity is linked to DXS255. Am J Med Genet. 1991;40:406–413.
36. Cohen, MM, Jr., Hall, BD, Smith, DW, et al. A new syndrome with hypotonia, obesity, mental deficiency, and facial, oral, ocular, and limb anomalies. J Pediatr. 1973;83:280–284.
37. Chandler, KE, Kidd, A, Al-Gazali, L, et al. Diagnostic criteria, clinical characteristics, and natural history of Cohen syndrome. J Med Genet. 2003;40:233–241.
38. Seifert, W, Holder-Espinasse, M, Spranger, S, et al. Mutational spectrum of COH1 and clinical heterogeneity in Cohen syndrome. J Med Genet. 2006;43:e22.
39. Tahvanainen, E, Norio, R, Karila, E, et al. Cohen syndrome gene assigned to the long arm of chromosome 8 by linkage analysis. Nat Genet. 1994;7:201–204.
40. Weinstein, LS, Chen, M, Liu, J. Gs(alpha) mutations and imprinting defects in human disease. Ann N Y Acad Sci. 2002;968:173–197.
41. Yeo, GS, Connie Hung, CC, Rochford, J, et al. A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat Neurosci. 2004;7:1187–1189.
42. Gray, J, Yeo, GS, Cox, JJ, et al. Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain-derived neurotrophic factor (BDNF) gene. Diabetes. 2006;55:3366–3371.
43. Verloes, A, Temple, IK, Bonnet, S, et al. Coloboma, mental retardation, hypogonadism, and obesity: critical review of the so-called Biemond syndrome type 2, updated nosology, and delineation of three “new” syndromes. Am J Med Genet. 1997;69:370–379.
44. Steinmuller, R, Steinberger, D, Muller, U. MEHMO (mental retardation, epileptic seizures, hypogonadism and -genitalism, microcephaly, obesity), a novel syndrome: assignment of disease locus to xp21.1-p22.13. Eur J Hum Genet. 1998;6:201–206.
45. Alstrom, CH, Hallgren, B, Nilsson, LB, et al. Retinal degeneration combined with obesity, diabetes mellitus and neurogenous deafness: a specific syndrome (not hitherto described) distinct from the Laurence-Moon-Bardet-Biedl syndrome: a clinical, endocrinological and genetic examination based on a large pedigree. Acta Psychiatr Neurol Scand. 1959;34:1–35.
46. Russell-Eggitt, IM, Clayton, PT, Coffey, R, et al. Alstrom syndrome, Report of 22 cases and literature review. Ophthalmology. 1998;105:1274–1280.
47. Michaud, JL, Heon, E, Guilbert, F, et al. Natural history of Alstrom syndrome in early childhood: onset with dilated cardiomyopathy. J Pediatr. 1996;128:225–229.
48. Collin, GB, Marshall, JD, Ikeda, A, et al. Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alstrom syndrome. Nat Genet. 2002;31:74–78.
49. Hearn, T, Spalluto, C, Phillips, VJ, et al. Subcellular localization of ALMS1 supports involvement of centrosome and basal body dysfunction in the pathogenesis of obesity, insulin resistance, and type 2 diabetes. Diabetes. 2005;54:1581–1587.
50. Arsov, T, Silva, DG, O’Bryan, MK, et al. Fat aussie–a new Alstrom syndrome mouse showing a critical role for ALMS1 in obesity, diabetes, and spermatogenesis. Mol Endocrinol. 2006;20:1610–1622.
51. Schinzel, A. Ulnar-mammary syndrome. J Med Genet. 1987;24:778–781.
52. Bamshad, M, Krakowiak, PA, Watkins, WS, et al. A gene for ulnar-mammary syndrome maps to 12q23-q24.1. Hum Mol Genet. 1995;4:1973–1977.
53. Bamshad, M, Lin, RC, Law, DJ, et al. Mutations in human TBX3 alter limb, apocrine and genital development in ulnar-mammary syndrome. Nat Genet. 1997;16:311–315.
54. Renard, CA, Labalette, C, Armengol, C, et al. Tbx3 is a downstream target of the Wnt/beta-catenin pathway and a critical mediator of beta-catenin survival functions in liver cancer. Cancer Res. 2007;67:901–910.
55. Simpson, JL, Landey, S, New, M, et al. A previously unrecognized X-linked syndrome of dysmorphia. Birth Defects Orig Artic Ser. 1975;11:18–24.
56. Brzustowicz, LM FS, Khan, MB, Weksberg, R. Mapping of a new SGBS locus to chromosome Xp22 in a family with a severe form of Simpson-Golabi-Behmel syndrome. Am J Hum Genet. 1999;65:779–783.
57. Capurro, MI, Xu, P, Shi, W, et al. Glypican-3 inhibits Hedgehog signaling during development by competing with patched for Hedgehog binding. Dev Cell. 2008;14:700–711.
58. Montague, CT, Farooqi, IS, Whitehead, JP, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387:903–908.
59. Strobel, A, Issad, T, Camoin, L, et al. A leptin missense mutation associated with hypogonadism and morbid obesity. Nat Genet. 1998;18:213–215.
60. Farooqi, IS, Matarese, G, Lord, GM, et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest. 2002;110:1093–1103.
61. Gibson, WT, Farooqi, IS, Moreau, M, et al. Congenital leptin deficiency due to homozygosity for the Delta133G mutation: report of another case and evaluation of response to four years of leptin therapy. J Clin Endocrinol Metab. 2004;89:4821–4826.
62. Farooqi, IS, Jebb, SA, Langmack, G, et al. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med. 1999;341:879–884.
63. Farooqi, IS, Bullmore, E, Keogh, J, et al. Leptin regulates striatal regions and human eating behavior. Science. 2007;317:1355.
64. Ozata, M, Ozdemir, IC, Licinio, J. Human leptin deficiency caused by a missense mutation: multiple endocrine defects, decreased sympathetic tone, and immune system dysfunction indicate new targets for leptin action, greater central than peripheral resistance to the effects of leptin, and spontaneous correction of leptin-mediated defects. J Clin Endocrinol Metab. 1999;84:3686–3695.
65. Licinio, J, Caglayan, S, Ozata, M, et al. Phenotypic effects of leptin replacement on morbid obesity, diabetes mellitus, hypogonadism, and behavior in leptin-deficient adults. Proc Natl Acad Sci U S A. 2004;101:4531–4536.
66. Farooqi, IS, Wangensteen, T, Collins, S, et al. Clinical and molecular genetic spectrum of congenital deficiency of the leptin receptor. N Engl J Med. 2007;356:237–247.
67. Farooqi, IS, Keogh, JM, Kamath, S, et al. Partial leptin deficiency and human adiposity. Nature. 2001;414:34–35.
68. Clement, K, Vaisse, C, Lahlou, N, et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. 1998;392:398–401.
69. Lahlou, N, Landais, P, De Boissieu, D, et al. Circulating leptin in normal children and during the dynamic phase of juvenile obesity: relation to body fatness, energy metabolism, caloric intake, and sexual dimorphism. Diabetes. 1997;46:989–993.
70. Schwartz, MW, Woods, SC, Porte, D, Jr., et al. Central nervous system control of food intake. Nature. 2000;404:661–671.
71. Krude, H, Biebermann, H, Luck, W, et al. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet. 1998;19:155–157.
72. Krude, H, Biebermann, H, Schnabel, D, et al. Obesity due to proopiomelanocortin deficiency: three new cases and treatment trials with thyroid hormone and ACTH4–10. J Clin Endocrinol Metab. 2003;88:4633–4640.
73. Farooqi, IS, Drop, S, Clements, A, et al. Heterozygosity for a POMC-null mutation and increased obesity risk in humans. Diabetes. 2006;55:2549–2553.
74. Biebermann, H, Castaneda, TR, van Landeghem, F, et al. A role for beta-melanocyte-stimulating hormone in human body-weight regulation. Cell Metab. 2006;3:141–146.
75. Lee, YS, Challis, BG, Thompson, DA, et al. A POMC variant implicates beta-melanocyte-stimulating hormone in the control of human energy balance. Cell Metab. 2006;3:135–140.
76. Challis, BG, Pritchard, LE, Creemers, JW, et al. A missense mutation disrupting a dibasic prohormone processing site in pro-opiomelanocortin (POMC) increases susceptibility to early-onset obesity through a novel molecular mechanism. Hum Mol Genet. 2002;11:1997–2004.
77. Jackson, RS, Creemers, JW, Ohagi, S, et al. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene [see comments]. Nat Genet. 1997;16:303–306.
78. Jackson, RS, Creemers, JW, Farooqi, IS, et al. Small-intestinal dysfunction accompanies the complex endocrinopathy of human proprotein convertase 1 deficiency. J Clin Invest. 2003;112:1550–1560.
79. Farooqi, IS, Volders, K, Stanhope, R, et al. Hyperphagia and early-onset obesity due to a novel homozygous missense mutation in prohormone convertase 1/3. J Clin Endocrinol Metab. 2007;92:3369–3373.
80. Farooqi, IS, Keogh, JM, Yeo, GS, et al. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med. 2003;348:1085–1095.
81. Alharbi, KK, Spanakis, E, Tan, K, et al. Prevalence and functionality of paucimorphic and private MC4R mutations in a large, unselected European British population, scanned by meltMADGE. Hum Mutat. 2007;28:294–302.
82. Vaisse, C, Clement, K, Durand, E, et al. Melanocortin-4 receptor mutations are a frequent and heterogeneous cause of morbid obesity. J Clin Invest. 2000;106:253–262.
83. Stutzmann, F, Tan, K, Vatin, V, et al. Prevalence of MC4R deficiency in European population and their age-dependant penetrance in multi-generational pedigrees. Diabetes. 2008.
84. Nogueiras, R, Wiedmer, P, Perez-Tilve, D, et al. The central melanocortin system directly controls peripheral lipid metabolism. J Clin Invest. 2007;117:3475–3488.
85. Holder, JL, Jr., Butte, NF, Zinn, AR. Profound obesity associated with a balanced translocation that disrupts the SIM1 gene. Hum Mol Genet. 2000;9:101–108.
86. Michaud, JL, Boucher, F, Melnyk, A, et al. Sim1 haploinsufficiency causes hyperphagia, obesity and reduction of the paraventricular nucleus of the hypothalamus. Hum Mol Genet. 2001;10:1465–1473.
87. Faivre, L, Cormier-Daire, V, Lapierre, JM, et al. Deletion of the SIM1 gene (6q16.2) in a patient with a Prader-Willi-like phenotype. J Med Genet. 2002;39:594–596.
88. Rose, EA, Glaser, T, Jones, C, et al. Complete physical map of the WAGR region of 11p13 localizes a candidate Wilms’ tumor gene. Cell. 1990;60:495–508.