Genetic Disorders Of Phosphate Homeostasis
Regulation of Phosphate Homeostasis
Phosphate in serum exists almost exclusively as the free ion or in association with cations. Unlike calcium, only 12% of phosphate is protein bound.1 Also in contrast to calcium, serum phosphate concentrations may vary substantially throughout the day. Carbohydrate ingestion may markedly reduce serum phosphate by moving serum phosphate from the extracellular to the intracellular space. Moreover, serum phosphate undergoes diurnal variation of as much as 1.5 mg/dL (0.5 mmol/L), with a nadir between 8 and 11 am.2 Interference with the measurement of phosphate in serum may occur during hypertriglyceridemia,3 hypergammaglobulinemia,4 or mannitol therapy,5 depending on the method of analysis.
Fasting serum phosphate remains stable throughout the menstrual cycle and during pregnancy.6–10 The placenta actively transports phosphate into the fetus, as reflected in the higher phosphate concentrations of newborn cord arterial and venous blood compared with maternal blood levels.8 Lactating women, who may lose 100 to 500 mg of phosphorus daily in milk, nevertheless maintain normal levels of serum phosphate.11,12 Serum phosphate concentrations are relatively high in the newborn (5 to 7 mg/dL),8,13 decrease gradually thereafter, and then increase again briefly at puberty before reaching adult levels by age 18 to 20 years. Serum phosphate typically increases in women after menopause but decreases in the elderly.14–16
Phosphate is a ubiquitous constituent of a vast array of biomolecules. Of particular importance is the fundamental role of inorganic phosphate as a substrate for intracellular enzymes involved in glycolysis and respiration that synthesize high-energy phosphate bonds for storage of chemical energy in organophosphate compounds such as ATP, creatine phosphate, diphosphoglycerate (DPG), phosphoenolpyruvate, and others. Severe phosphate depletion leads to a concentration-dependent inhibition of glycolysis, accumulation of “triose phosphates” immediately proximal to glyceraldehyde 3-phosphate dehydrogenase, and decreased production of ATP. Adequate extracellular phosphate is required for normal mineralization of bone and cartilage,17,18 and chronic hypophosphatemia of any cause may therefore lead to osteomalacia or, in children, rickets.
The average dietary intake of phosphate—derived largely from dairy products, cereals, and meats—is roughly twice the estimated minimum requirement of 400 mg/day.19 Absorptive efficiency is high, averaging about 70%, and may increase further (=90%) if the intake of dietary phosphate decreases to less than 2 mg/kg/day.20 Phosphate is avidly absorbed throughout the small intestine, but especially in the jejunum in animals and humans.21–23
In the jejunum, overall phosphate uptake consists of two components: a saturable, sodium-dependent process that is responsive to vitamin D (see later) and a nonsaturable, sodium-independent mechanism thought to represent paracellular diffusional transport.21,24 The saturable mechanism reflects active transport via the transcellular route, the energy for which is derived from the transmembrane sodium gradient. In animal tissues and in human jejunal biopsies, the sodium phosphate cotransporter exhibits a Km of approximately 0.05 mmol/L, half-maximal stimulation by 30 to 50 mmol/L sodium, and a ratio of two sodium molecules per molecule of phosphate transported.21,25,26 Sodium-dependent active phosphate absorption is mediated by the NPT2b cotransporters present in the luminal brush-border membranes of enterocytes.27,28 Sodium phosphate transporter NPT2b is the product of a different gene from that which encodes the predominant forms expressed in the renal proximal tubule (NPT2a and NPT2c).27,28 For example, mice lacking the renal cotransporter manifest striking hyperphosphaturia because of continued intestinal phosphate absorption.29 NPT2b is fully saturated at intraluminal phosphate concentrations of 1 to 2 mmol/L, which are easily achieved after most typical meals. Subsequent transport across the basolateral membrane does not require active transport and is thought to proceed via facilitated diffusion, although the transporter or channel involved has not been characterized.30
Regulation Of Phosphate Absorption
The central role of vitamin D in the regulation of intestinal phosphate transport has been recognized for many years. Absorption of phosphate, like that of calcium, is strikingly augmented by 1,25(OH)2D.21,22,31–34 Basal fractional phosphate absorption in the absence of 1,25(OH)2D is much higher than that of calcium, however. The action of 1,25(OH)2D on phosphate transport has been studied in vitro by using intact intestinal segments, isolated enterocytes, and brush-border membrane vesicles (BBMVs).24,35–39 In each case, stimulation by 1,25(OH)2D was shown to result from activation of the sodium-dependent active transport mechanism and not the passive diffusional component. Specifically, 1,25(OH)2D stimulates the maximal velocity of sodium-dependent phosphate cotransport by increasing expression of NPT2b, mainly or exclusively via a posttranscriptional mechanism.30,40–43 Other studies have pointed to an additional, very rapid (minutes), nongenomic mechanism of 1,25(OH)2D-dependent stimulation of intestinal phosphate transport, analogous to its nongenomic effect on duodenal calcium transport.44,45
Restriction of dietary phosphate increases intestinal NPT2b expression41,43 and thus, like 1,25(OH)2D, enhances the Vmax of the saturable component of intestinal phosphate absorption.46–48 This response also involves a posttranscriptional mechanism,41,43 results in part from augmented renal synthesis of 1,25(OH)2D,30 but can be seen also in vitamin D–deficient or vitamin D receptor–null animals.43,49–51
Phosphate Excretion
Eighty percent of filtered phosphate is reabsorbed by the proximal tubule, and the capacity for phosphate transport appears to diminish between the early convoluted and the straight (pars recta) portions of the proximal tubule.52–54 Additional phosphate may be reabsorbed in the distal tubule or cortical collecting tubule or both.55–60 Phosphate must be actively transported across the luminal brush-border membrane against a steep electrochemical gradient. Consistent with this, renal phosphate transport requires luminal sodium ions and is blocked by inhibitors of Na+/K+-ATPase.54,61–64 Dibasic phosphate is preferentially transported by the rat proximal tubule,65–67 and studies with isolated perfused tubules have shown that increased intraluminal pH and decreased intracellular pH accelerate phosphate reabsorption by the intact cell.68
The two primary transport proteins responsible for Pi reabsorption in the kidney are the type II sodium-phosphate cotransporters NPT2a and NPT2c, expressed in the apical membrane of the proximal tubule. In the mouse, NPT2a is a critical phosphate cotransporter in the renal proximal tubule. Ablation of the NPT2a gene in the mouse results in a decrease in proximal tubular sodium phosphate cotransport, hypophosphatemia, and loss of regulation of phosphate reabsorption by both parathyroid hormone (PTH) and dietary phosphate.29,69,70 Ablation of NPT2c in mice does not result in a phosphate phenotype,71 thereby indicating that it may play a less central role in phosphate transport in the mouse. However, mutations in the gene coding NPT2c, SLC34A3, result in hereditary hypophosphatemic rickets with hypercalciuria (HHRH, see later), indicating that this transporter plays an important role in maintenance of phosphate homeostasis in humans.
Measurement of Renal Phosphate Transport
For clinical purposes, a relatively convenient way of measuring a patient’s ability to reabsorb phosphate is to calculate the tubular maximum reabsorption of phosphate divided by the glomerular filtration rate (TMP/GFR). A simple nomogram has been developed by Walton and Bijvoet72 that works well in most situations (Fig. 6-1).
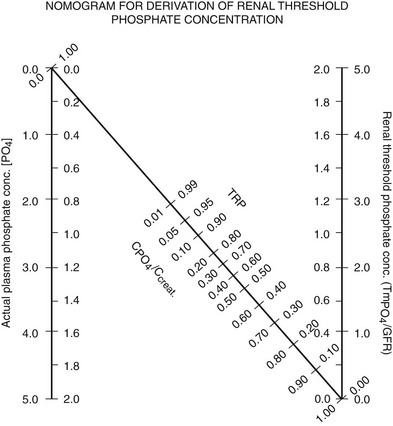
FIGURE 6-1 Nomogram for calculating TmP/GFR. Calculations are done from fasting samples obtained in the morning. The urine is collected for 1 to 2 hours (the time period is not critical). The CPO4/Ccreat = (UPO4 × [Creat])/(Ucreat × [Pi]). To use the nomogram to calculate TMP/GFR, a straight line is passed through the appropriate plasma phosphate concentration and the CPO4/Ccreat. The line will intersect with the corresponding TMP/GFR value. For SI units, use the inside scales. For metric (mg/dL), use the outside scales. (From Walton RJ, Bijvoet OL: Nomogram for derivation of renal threshold phosphate concentration. Lancet 2:309–310, 1975.)
Regulation of Phosphate Reabsorption
PTH has a major effect on serum phosphate. The clinical relevance of this effect is demonstrated in patients with hyperparathyroidism, who develop phosphaturia and hypophosphatemia, as well as those with hypoparathyroidism, who have increased phosphate reabsorption and hyperphosphatemia. PTH rapidly (15 to 60 minutes) reduces the number of NPT2a cotransporters on the apical surface of the cells in the renal proximal tubule. The effect appears to result from microtubule-dependent internalization into endocytic vesicles and subsequent destruction of the transporters.73,74 This acute down-regulation of NPT2a cotransporters does not involve reduction in NPT2a gene transcription,75 although transcriptional suppression is seen after more prolonged PTH exposure.76 After parathyroidectomy, rats manifest a two- to threefold increase in both protein and messenger RNA (mRNA) levels of NPT2a, which correlates with a striking increase in phosphate reabsorption.75 Evidence exists for involvement of both PKA and protein kinase C (PKC), as well as MAPK signaling via PTH/PTHrP receptors in bringing about these responses.77,78 Despite the fact that PTH clearly has a major effect on phosphate homeostasis, it should be kept in mind that the primary role of PTH is to regulate serum calcium level and not phosphate homeostasis. Recent studies of the role of FGF23 further illuminate this issue (see later).
Dietary Phosphate
Renal phosphate excretion is extremely sensitive to changes in dietary phosphate availability. Thus, dietary phosphate deprivation79–86 or supplementation80,82,87 rapidly evokes a compensatory increase or decrease, respectively, in renal phosphate reabsorption ([Pi]Th). Compelling clinical and experimental evidence has established that these adaptations to dietary phosphate occur quite independently of PTH.74,75,79–82,88
Increased sodium-dependent phosphate transport by isolated brush-border membrane vesicles occurs within a few hours of phosphate deprivation86,89 and reflects increased maximal velocity (rather than affinity) of the phosphate carrier,89–91 consistent with an increased number of membrane transporters. This has been corroborated by direct immunohistologic demonstration that institution of a low-phosphate diet causes rapid (within 2 hours) insertion of NPT2a cotransporters into the apical plasma membrane of rat proximal tubular cells by a microtubule-dependent mechanism.92–94 Up-regulation of NPT2a gene transcription occurs subsequently during more prolonged phosphate restriction (i.e., several days).74 Similarly, high dietary phosphate rapidly reduces apical membrane NPT2a protein levels, with no change in NPT2a gene transcription for at least several hours.74,95
Fibroblast Growth Factor-23
Fibroblast growth factor-23 (FGF23), the gene identified as causative for autosomal dominant hypophosphatemic rickets (see later) and some forms of familial hyperphosphatemic tumoral calcinosis, does not appear to play a role in the rapid changes in phosphate reabsorption due to acute changes in intestinal phosphate.96 However, there is substantial evidence that FGF23 plays a role in maintenance of normal phosphate homeostasis over the course of days.
Fibroblast Growth Factor-23 Activity
FGF23 has similar functions as PTH to reduce renal Pi reabsorption but has opposite effects on 1,25(OH)2D production. FGF23 delivery leads to renal Pi wasting through the down-regulation of both NPT2a and NPT2c.97 Under normal circumstances, hypophosphatemia is a strong positive stimulator for increasing serum 1,25(OH)2D production. However, patients with ADHR, TIO, XLH, and ARHR manifest hypophosphatemia with paradoxically low or inappropriately normal serum 1,25(OH)2D concentrations. In mice, the expression of the 1α(OH)ase enzyme and the catabolic 24(OH)ase are reduced and elevated, respectively, when the animals are exposed to FGF23 by injection or by transgenic approaches.98 Thus, the effects of FGF23 on the renal vitamin D metabolic enzymes is most likely responsible for the reductions in 1,25(OH)2D in the setting of often marked hypophosphatemia in ADHR, XLH, TIO, and ARHR patients.
Regulation of FGF23 Production
In humans, dietary Pi supplementation increased FGF23, whereas Pi restriction and the addition of Pi binders suppressed serum FGF23 (Fig. 6-2),99 indicating that FGF23 plays a role in maintenance of Pi homeostasis. In animal studies, the FGF23 response to serum Pi appears to be much more dramatic than in the human studies. Mice given high and low Pi diets have increased and decreased serum FGF23 levels, respectively.100
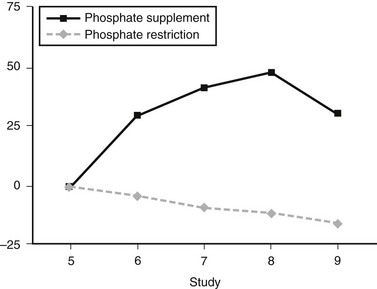
FIGURE 6-2 FGF23 levels in response to diet. Percent change in FGF23 during the intervention period ( , phosphate depletion intervention as measured with the intact FGF23 assay;
, phosphate depletion intervention as measured with the intact FGF23 assay;  , phosphate loading intervention as measured with the intact FGF23 assay). The intervention, dietary phosphate depletion, or loading was started after the day 5 samples were collected. Values are presented as mean ± SE. (Data from Burnett SM, Gunawardene SC, Bringhurst FR et al: Regulation of C-terminal and intact FGF-23 by dietary phosphate in men and women, J Bone Miner Res 21:1187–1196, 2006.)
, phosphate loading intervention as measured with the intact FGF23 assay). The intervention, dietary phosphate depletion, or loading was started after the day 5 samples were collected. Values are presented as mean ± SE. (Data from Burnett SM, Gunawardene SC, Bringhurst FR et al: Regulation of C-terminal and intact FGF-23 by dietary phosphate in men and women, J Bone Miner Res 21:1187–1196, 2006.)
Vitamin D has important regulatory effects on FGF23 in vivo. In mice, injections of 20 to 200 ng 1,25(OH)2D led to dose-dependent increases in serum FGF23 concentrations.98 These changes in FGF23 occurred before detectable changes in serum Pi, indicating that FGF23 may be directly regulated by vitamin D. Physiologically, this would be consistent with results examining the role of FGF23 in vitamin D metabolism, in that FGF23 has been shown to down-regulate the 1α(OH)ase mRNA.97,98 Thus as 1,25(OH)2D is elevated in the blood as a product of 1α(OH)ase activity, vitamin D would then increase FGF23 production, which would complete the negative-feedback loop and down-regulate 1α(OH)ase.
Serum Assays
FGF23 can be measured in the bloodstream via several assays. One widely used assay is a “C-terminal” FGF23 enzyme-linked immunosorbent assay (ELISA), with both the capture and detection antibodies binding C-terminal to the FGF23 176RXXR179/S cleavage site.101 This assay thus recognizes full-length FGF23 as well as C-terminal fragments that could arise through proteolytic processing. The C-terminal assay is quantified relative to standards composed of FGF23-conditioned media produced from stable cell lines expressing the human protein, and it only recognizes the human FGF23 isoform. The normal mean for this assay is 55 ± 50 reference units (RU)/mL, and the upper limit of normal is 150 RU/mL. In a study with a large number of controls and TIO patients, this ELISA was used to examine FGF23 concentrations in TIO and XLH101 and showed that serum FGF23 is detectable in normal individuals. The mean FGF23 was greater than 10-fold elevated in TIO patients, which rapidly fell after surgical removal of the tumor. Importantly, most XLH patients (13 out of 21) had elevated FGF23 compared to controls,101 and in those with “normal” FGF23, these levels may be “inappropriately normal” in the setting of hypophosphatemia.
An “intact” FGF23 assay has been developed that uses conformation-specific monoclonal antibodies that span the 176RXXR179/S180 SPC cleavage site (see later) and thus recognize N-terminal and C-terminal regions of the FGF23 polypeptide.102 In normal individuals, this assay has a mean circulating concentration of 29 pg/mL. The published upper limit of normal is 54 pg/mL.102 The results of these two assays generally agree with regard to the relative ranges of FGF23 concentrations in XLH and in TIO patients, and that FGF23 is elevated in most XLH patients. Based upon limited data from two TIO patients undergoing resection, the intact assay was used to determine that the half-life of FGF23 is between 20 and 50 minutes.103,104
FGF23-Associated Syndromes
FGF23-associated syndromes, summarized in Table 6-1, can be divided into three groups:
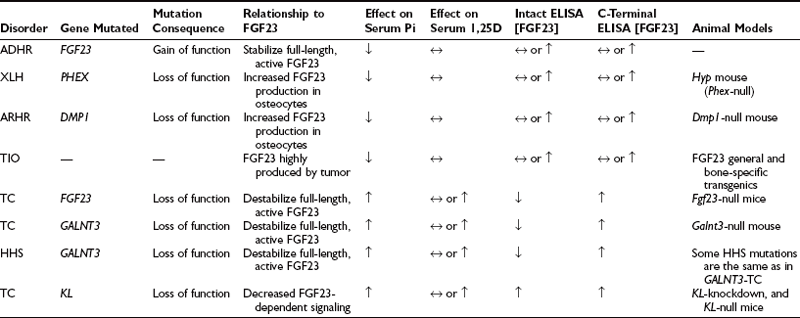
1. Disorders associated with increased FGF23 bioactivity
2. Disorders associated with reduced FGF23 bioactivity
3. Genetic hypophosphatemia not associated with elevated FGF23
Disorders Associated With Increased FGF23 Bioactivity
Autosomal-Dominant Hypophosphatemic Rickets (OMIM No. 193100)
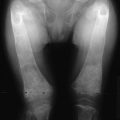
ADHR is a rare disorder characterized by low serum Pi concentrations due to decreased TmP/GFR and inappropriately low or normal circulating vitamin D concentrations.105 ADHR was first described in a small family,106 and subsequently, a large ADHR kindred with many affected individuals was evaluated.105 This kindred provided an opportunity to test the phenotypic variability of ADHR in a large number of individuals with the same mutation. There was no evidence of genetic anticipation or imprinting. In contrast to the other genetic renal phosphate-wasting disorders, ADHR displays incomplete penetrance and variable age of onset. Important to the diagnosis and clinical management of ADHR, it was observed that this expanded ADHR family contains two subgroups of affected individuals. One subgroup consists of patients who presented during childhood with Pi wasting, rickets, and lower-extremity deformity in a pattern similar to the classic presentation of XLH. The second group consists of individuals who presented clinically during adolescence or adulthood. These individuals had bone pain, weakness, and insufficiency fractures but did not have lower extremity deformities.105 The patients with adult-onset ADHR had clinical presentations essentially identical to patients who present with TIO (none of the ADHR patients developed tumors). The molecular mechanisms for early-onset ADHR resembling XLH and late-onset ADHR resembling TIO are currently unknown. To date, all patients that have been described with delayed onset of clinically evident disease are female. In addition to these two groups, we found unaffected individuals who are carriers for the ADHR mutation and two patients who were treated for hypophosphatemia and rickets but later lost the Pi wasting defect.105 Thus, the clinical manifestations of ADHR are more variable than those observed in XLH.
To identify the gene for ADHR, the ADHR Consortium undertook a family-based positional cloning approach. A genomewide linkage scan in the large ADHR kindred described earlier demonstrated linkage to chromosome 12p13.3 (homologous to mouse chromosome 6).107 FGF23 was identified using exon prediction programs on genomic DNA sequence from the Human Genome Project.108 The FGF23 gene is composed of three coding exons and contains an open reading frame of 251 residues.108 The tissue with the highest FGF23 expression is bone, where FGF23 mRNA is observed in osteoblasts, osteocytes, flattened bone-lining cells, and osteoprogenitor cells.109 Quantitative RT-PCR showed that FGF23 mRNA was most highly expressed in long bone, followed by thymus, brain, and heart.110
Western blot analysis has demonstrated that wild-type FGF23 is secreted as a full-length 32-kD protein, as well as cleavage products of 20 kD (N-terminal) and 12 kD (C-terminal).110–112 Cleavage of FGF23 occurs within a subtilisin-like proprotein convertase (SPC) proteolytic site (176RXXR179/S180) that separates the conserved FGF-like N-terminal domain from the variable C-terminal tail (Fig. 6-3).
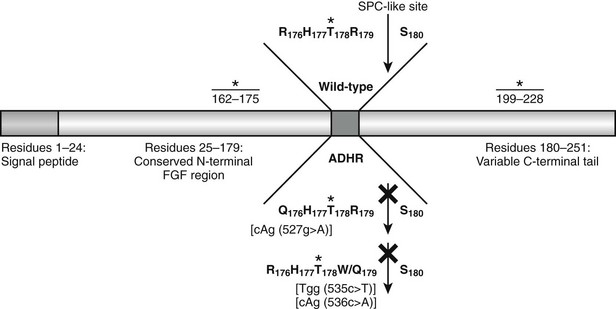
FIGURE 6-3 Model of FGF23 protein domains and effect of the ADHR mutations. FGF23 has a 24-residue signal peptide followed by residues 25 to 179 that comprise the conserved N-terminal FGF-like domain. The SPC-like site is interrupted by the ADHR mutations at R176 and R179 and divides the FGF-like domain from the variable C-terminal tail region. FGF23 undergoes glycosylation at three regions (denoted *), with the T178 most likely being the glycosylated residue that protects the mature protein from SPC degradation between R179 and S180 and is therefore critical for maintaining intact, active FGF23.
The ADHR mutations replace arginine (R) residues at FGF23 positions 176 or 179 with glutamine (Q) or tryptophan (W) within the SPC cleavage site, 176RXXR179/S180 (Table 6-2 and see Fig. 6-3).108,111,112 The SPCs are a family of serine proteases that process a wide variety of proteins including neuropeptides, peptide hormones, growth factors, membrane-bound receptors, blood coagulation factors, and plasma proteins.113 SPC substrates are cleaved C-terminal to the basic motif K/R-Xn-K/R, where Xn = 2, 4, or 6 residues of any amino acid.114,115 The SPCs, such as the furin protease, are largely expressed in the trans-Golgi network and possess similar but not exact substrate specificities. Following insertion of these mutations into wild-type FGF23, FGF23 secreted from mammalian cells was primarily produced as the full-length protein (32 kD) active polypeptide, as opposed to the 32-kD cleavage products typically observed for wild-type FGF23 expression.111 Peptide sequencing demonstrated that the 32-kD FGF23 form corresponded to full-length FGF23 after cleavage of the signal peptide (residues 25 to 251) and that the 12-kD isoform was the C-terminal portion of FGF23 downstream from the SPC cleavage site after R179 (residues 180 to 251) (see Fig. 6-3).112 As further evidence that FGF23 is processed intracellularly, the cleavage of wild-type FGF23 between R179/S180 is inhibited by a nonspecific SPC competitive inhibitor, Dec-RVKR-CMK, at concentrations between 25 and 50 µM.110,116 These studies show that the RXXR motif in FGF23 is central to its intracellular processing.
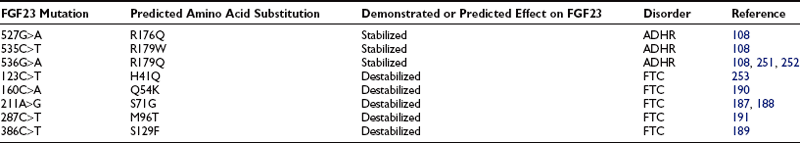
The SPC family is usually associated with the production of the active form of their substrate polypeptides. However, cleavage of FGF23 at the RXXR motif appears to be inactivating. In this regard, when full-length FGF23 or N-terminal (residues 25 to 179) and C-terminal (residues 180 to 251) fragments were injected into rodents, only the full-length protein lowered circulating phosphate concentrations.112 Since the full-length form of FGF23 induces hypophosphatemia, it is likely that the ADHR mutations increase the biological activity of FGF23 by stabilizing the full-length form and increasing its concentrations in the serum. Indeed, severely affected ADHR patients have increased circulating levels of FGF23.117
Tumor-Induced Osteomalacia
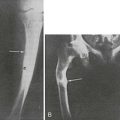
TIO is an acquired disorder of renal Pi wasting that is associated with tumors. Patients with TIO present with hypophosphatemia with inappropriately suppressed 1,25(OH)2D concentrations and elevated alkaline phosphatase levels.118 Osteomalacia is observed in bone biopsies. Clinical symptoms include gradual onset of muscle weakness, fatigue, and bone pain, especially from ankles, legs, hips, and back.118,119 Insufficiency fractures are common, and proximal muscle weakness can become severe enough for patients to require a wheelchair or become bed bound.118
The study of TIO introduced the idea for the existence of possible tumor-produced circulating factors, referred to as phosphatonins, that act upon the kidney to decrease Pi reabsorption.120,121 Support for these factors primarily comes from the knowledge that if the responsible tumor is surgically removed, the abnormalities in Pi wasting and vitamin D metabolism are rapidly corrected, as well as the fact that PTH, which decreases renal Pi reabsorption, is usually within normal ranges in TIO patients. Other studies have supported this hypothesis by showing that implantation of tumor tissue into nude mice resulted in increased urinary Pi excretion.122 To determine whether FGF23 could be involved in TIO as phosphatonin, five TIO tumors and several control tissues were tested by Northern blot for the presence of FGF23 transcripts, and it was determined that FGF23 mRNA was highly expressed in all of these tumors.123 Furthermore, FGF23 was present in a tumor lysate by Western blot analysis, with an anti–human FGF23 antibody.123
FGF23 was also independently identified in RNA from TIO tumors. Transcripts from tumors were isolated by differential selection using comparisons to mRNAs present in normal bone.124 The highly expressed transcripts were then subcloned, and the individual mRNAs were stably expressed in Chinese hamster ovary (CHO) cells, then injected into nude mice to form tumor masses.124 The cells that produced FGF23 recapitulated the TIO phenotype in vivo by causing hypophosphatemia, elevated alkaline phosphatase, and inappropriately low 1,25(OH)2D concentrations.124 In addition, the mice that received implanted cells also showed growth retardation, kyphosis, and osteomalacia. Further, there was marked decrease in the renal 1α(OH)ase. Both the biochemical and metabolic bone profiles were remarkably similar to those observed in TIO and ADHR patients. These experiments provided evidence that FGF23 was a phosphaturic substance and had dramatic effects on enzymes involved in vitamin D metabolism, and increased circulating FGF23 concentrations were consistent with the idea that FGF23 was at least in part responsible for the TIO phenotype. Serum FGF23 is elevated in patients with TIO,101,102 and tumors that cause TIO have a dramatic overexpression of FGF23 mRNA.123 Surgical resection of the tumor results in rapid decreases in serum FGF23.101
X-Linked Hypophosphatemic Rickets (OMIM No. 307800)
XLH is an X-linked dominant disorder and the most common form of heritable rickets.125 XLH is fully penetrant with variable severity. XLH patients present with laboratory findings that include hypophosphatemia with normocalcemia and inappropriately normal or low 1,25(OH)2D concentrations.125 Skeletal defects include lower-extremity deformities from rickets, bone pain, osteomalacia, fracture, and enthesopathy (calcification of the tendons and ligaments).125 It was determined by the Hyp Consortium that XLH is caused by inactivating mutations in PHEX (phosphate-regulating gene with homologies to endopeptidases on the X chromosome).126 Based upon sequence homology, PHEX encodes a protein that is a member of the M13 family of membrane-bound metalloproteases. Other members of this enzyme class include neutral endopeptidase (NEP) and endothelin-converting enzymes 1 and 2 (ECE-1 and ECE-2).126,127 This protease family is known to cleave small peptide hormones, therefore it is likely that PHEX has similar activity. Over 160 inactivating PHEX mutations have been described in XLH patients, including genomic variations that cause missense, nonsense, frameshift, and splicing changes (see PHEX Locus database: www.phexdb.mcgill.ca). Of note, although XLH is a renal Pi wasting disorder, PHEX shows the highest expression in bone cells such as osteoblasts, osteocytes, and odontoblasts in teeth, as well as lower expression in the parathyroid glands, lung, brain, and skeletal muscle but no expression in kidney.128 Taken together with the biochemical phenotype of XLH, PHEX protein homology and tissue expression are consistent with the hypothesis that PHEX interacts with small circulating factors outside of the kidney.
A valuable tool for the study of the pathophysiology of XLH has been the Hyp mouse, which has 3′ deletion in the Phex gene from intron 15 through the 3′ UTR129 and does not make a stable Phex transcript.128 This rodent model parallels the XLH phenotype, characterized by hypophosphatemia with inappropriately normal 1,25(OH)2D levels and normal serum calcium, as well as growth retardation and bone mineralization defects.130 Parabiosis studies between the Hyp mouse and a normal mouse pointed to the presence of a humoral factor, a phosphatonin, being transferred through the circulation of the Hyp mouse to the normal mouse to cause isolated renal Pi wasting.131 After the identification of PHEX/Phex, it was logically postulated that the enzyme may directly degrade a phosphaturic substance; however, recent studies suggest a more complex pathophysiology.
X-Linked Hypophosphatemic Rickets and Fibroblast Growth Factor-23: As described earlier, patients with XLH have overlapping phenotypes with ADHR patients. Because XLH results from a mutation in PHEX, which shares homology to a family of extracellular proteases, and ADHR arises from mutations in a protease cleavage site, it was logically hypothesized that circulating FGF23 would be cleaved and inactivated by PHEX. Thus, by mutational inactivation of PHEX in XLH, serum FGF23 concentrations would then elevate and cause renal Pi wasting. As described earlier, lending further support to this hypothesis were parabiosis studies between Hyp and normal mice, which pointed to the presence of a humoral phosphaturic factor in the Hyp mouse being transferred to the normal mouse. However, evidence to date has not supported a direct enzyme-substrate relationship between FGF23 and PHEX. In this regard, it was shown that recombinant PHEX did not cleave FGF23 but did cleave a positive control substrate.110 Furthermore, another report provided evidence that recombinant FGF23 was not cleaved by PHEX in cultured HEK293 cells coexpressing the proteins.116 This latter study expressed native FGF23 that was not epitope tagged to ensure that the additional residues did not cause conformational changes within FGF23 and interfere with potential PHEX activity.116
Several reports have established that FGF23 is elevated in many XLH patients.101,102,132 To understand the possible relationship between PHEX and FGF23, quantitative real-time RT-PCR was used to test Hyp bone for FGF23 mRNA concentrations versus wild-type bone. Interestingly, FGF23 mRNA in bone tissue from Hyp mice was elevated compared to levels present in control mice,110 and serum concentrations of FGF23 have been reported to be 10-fold higher in Hyp mice when compared to normal mice (our unpublished results and Ref. 133). This finding provides support for the idea that there is a cellular connection between inactive PHEX mutants (or lack of Phex expression in Hyp mice) and the up-regulation of FGF23 mRNA in bone cells. The elevated FGF23 mRNA levels may indicate that the increase in serum FGF23 in XLH patients is due to overproduction and secretion of FGF23 by skeletal cells, as opposed to the alternative hypothesis of a decreased rate of FGF23 degradation by cell surface proteases after secretion into the circulation. Although the interactions between FGF23 and PHEX are most likely indirect, the encoded proteins are coexpressed in osteoblasts and osteocytes.109,110,128 At present, the PHEX substrate and the mechanisms for phosphate sensing are unknown.
The current therapy for XLH, ADHR, and TIO includes oral replacement of phosphorus in combination with high-dose 1,25(OH)2D. This regimen “treats” XLH by increasing serum Pi concentrations and ameliorates much of the metabolic bone disease, but it does not directly “cure” the disorder by reversing the underlying molecular defects in kidney and in bone. In this regard, several studies have attempted to reverse the XLH phenotype. Transgenic expression of wild-type PHEX under the control of the bone-specific mouse pro-alpha(I) collagen gene134 and the osteocalcin (OG2)135 promoters on the Hyp background was undertaken. Interestingly, the defective mineralization of bone and teeth in the Hyp mice was partially resolved with PHEX under the regulation of the collagen promoter, and dry ash weight increased with the OG2 PHEX, indicating improved mineralization. However, the hypophosphatemia was not normalized in either study, indicating that expression of PHEX under the temporal regulation of an osteoblast-specific promoter is not sufficient to rescue the Hyp phenotype. Furthermore, expression of PHEX to levels observed in wild-type animals was not obtained in all studies. Importantly, a recent report of a transgenic model overexpressing PHEX in the Hyp mouse—using the human beta-actin promoter for directing expression in a wider tissue distribution (bone, skin, lung, muscle, heart)—resulted in similar results as the bone-specific promoter studies,136 further demonstrating that proper spatial-temporal expression of Phex is critical for normal mineral metabolism.
Treatment of X-Linked Hypophosphatemic Rickets: The current standard of care is combination therapy with phosphate and 1,25(OH)2D (calcitriol) or 1-hydroxyvitamin D3.137–139 Therapy is labor intensive, and patients and caregivers should understand this before initiating treatment. As in any medical encounter, patients should be fully informed about potential side effects of therapy.
The indications for treatment of adult patients are more controversial. There are no data to suggest that current treatment regimens prevent enthesopathy (calcifications of tendons and ligaments). Pseudofractures are common in moderately to severely affected adult XLH patients. Since pseudofractures are often painful, may lead to fracture, and generally respond well to treatment, we generally recommend medical therapy for patients with pseudofractures. XLH patients frequently complain of bone pain,121 presumably due to osteomalacia. Treatment lessens osteomalacia and bone pain, and it is therefore reasonable to treat patients with this complaint. Additionally, it is advisable to treat patients who have nonunion after fractures or osteotomies, because treatment may improve fracture healing. In light of the complexity of therapy, potential side effects (see later), and lack of increased risk of fracture in patients without pseudofractures,121 therapy is not recommended in asymptomatic patients who do not have pseudofractures.
Once the decision is made to initiate therapy, it is best to start at a low dose of calcitriol and phosphate (to avoid diarrhea from phosphate) and gradually increase therapy over several months. Some clinicians maintain a “high dose” phase for up to 1 year. During this phase of therapy, the calcitriol dose may be as high as 50 ng/kg per day in two divided doses, but not more than 3.0 mcg/day. However, not all experienced clinicians use this high-dose phase and some will use a maximum of 25 ng/kg. Also, 20 to 40 mg/kg of phosphate is administered in four divided doses (up to a maximum of 2 g per day). Serum calcium, phosphorus, and creatinine levels, as well as urine calcium and creatinine concentrations, are routinely monitored on a monthly basis during the high-dose phase. The doses of calcitriol and phosphate are adjusted based on the laboratory results. Hypercalciuria may be the first sign of healing of osteomalacia (theoretically, osteomalacic bone is capable of taking up more calcium and phosphate than bone that is histologically normal). Although some clinicians recommend administering phosphate over five doses per day initially, we do not ask patients to wake up to take medication at night for the following reasons: (1) serum phosphate concentrations tend to rise at night in XLH patients,140 (2) there is an exaggerated nocturnal rise in PTH concentrations in XLH patients, and (3) it is very difficult for patients to wake up to take medication every night for a chronic condition. After approximately 1 year on high-dose therapy, patients are switched to a long-term “maintenance” phase with approximately 10 to 20 ng/kg per day of calcitriol and no change in the dose of phosphate. While patients are on maintenance therapy, we continue to see them and monitor serum and urine biochemistries at least every 3 to 4 months.
Preliminary data indicate that standard therapy may have the undesirable affect of increasing FGF23 concentrations. It is possible that calcitriol, phosphate, or both stimulate the production of FGF23 in XLH patients despite the fact that they are still hypophosphatemic.141 This is currently an active area of research; however, physicians may want to consider not using a high-dose phase.
The most common and potentially serious complication of therapy is the development of nephrocalcinosis.142–147 With the widespread use of renal ultrasound, nephrocalcinosis has been noted with increasing frequency. In most cases, the nephrocalcinosis is observed without evident changes in glomerular filtration rate (GFR). Its occurrence may be related to phosphate dosage, and some authors assert that phosphate administration leads to hyperabsorption of oxalate, which results in nephrocalcinosis.148 However, this assertion remains unproven, and patients who received vitamin D2 but no phosphate can develop nephrocalcinosis.149 Thus hypercalciuria and hypercalcemia may contribute to (or at least aggravate) this problem. Kidney biopsy specimens from three treated patients with nephrocalcinosis showed that renal calcifications are located mainly intratubularly and are composed exclusively of calcium phosphate.150 Of note, a report of two patients with nephrocalcinosis for 2 decades indicates that it was not associated with impaired renal function.149 Although these data are somewhat reassuring, the available data are not strong enough to completely allay concerns about renal complications of even carefully monitored therapy, and we are aware of at least one XLH patient with end-stage renal failure after long-term therapy.
A second, less common complication is the development of tertiary hyperparathyroidism.151–154 Presumably, tertiary hyperparathyroidism results from chronic stimulation of the parathyroid glands by phosphate therapy. Although this complication is more likely in patients who are getting too much phosphate, it also can occur in optimally treated individuals. In tertiary hyperparathyroidism, renal function may decline, necessitating a reduction in the dose of phosphate and calcitriol or cessation of therapy. This will lead to the redevelopment or worsening of the bone disease. A small number of such patients have undergone total parathyroidectomy with autotransplant to the forearm. In a number of these patients, the transplanted tissue has shown a propensity to proliferate and lead to the redevelopment of tertiary hyperparathyroidism. Removal of this hyperplastic tissue from the forearm site leads to a reduction in serum PTH levels. Theoretically, total parathyroidectomy followed by treatment with 1,25(OH)2D and phosphate is a potential option. However, this approach is not recommended, because it results in lifelong hypoparathyroidism.
In light of the difficulty with combined therapy and the complications attributable at least in part to therapy, additional therapeutic options are being explored. These include the use of human GH,155 24,25(OH)2D,156 and diuretics.157 None of these are alternatives to combined therapy, but each has been employed as adjuvant therapy.
A continued search for better therapeutic agents is needed because it is increasingly clear that the present-day combined therapy is not curative and is accompanied by a high complication rate. However, if therapy with combined phosphate and 1,25(OH)2D is monitored carefully, growth rates increase, rickets is corrected, and bowing of the lower extremities is significantly reduced.137,139,147,158–161
Autosomal Recessive Hypophosphatemic Rickets (OMIM No. 241520)
Dentin matrix protein-1 (DMP1) is a member of the small integrin-binding ligand, N-linked glycoprotein (SIBLING) family and is highly and specifically expressed in osteocytes. Both Dmp1-null mice and patients with ARHR manifest rickets and osteomalacia with isolated renal Pi wasting associated with elevated FGF23. Mutational analyses revealed that one ARHR family carried a mutation that ablated the DMP1 start codon (Met1Val), and a second family exhibited a deletion in the DMP1 C-terminus (1484-1490del).162 Mutations have also been identified in DMP1 splicing sites, which likely result in nonfunctional protein.163
Mechanistic studies using the Dmp1-null mouse have demonstrated that loss of Dmp1 impairs osteocyte maturation, leading to markedly elevated FGF23 mRNA and protein expression. The hypophosphatemia results in pathological changes in bone mineralization,162 which can be largely but not completely abrogated by high-phosphate diet in Dmp1-null mice. Importantly, Dmp1-null mice are biochemical phenocopies of the Hyp mouse, and patients with ARHR and XLH (as well as the Dmp1-null and Hyp mice) share a distinctive bone histology characterized by periosteocytic lesions of nonmineralized bone.162 Thus, these findings suggest that PHEX (mutated in XLH) may also have a role in osteocyte maturation in a parallel pathway to DMP1 that leads to overexpression of FGF23.
Other Disorders Involving FGF23
Fibrous Dysplasia: Fibrous dysplasia (FD) is a disorder caused by activating somatic mutations in the GNAS1 gene, encoding the α subunit of the stimulatory G protein, Gs.164,165 The skeletal defects in FD are characterized by fibrous lesions and co-localized mineralization defects, which contribute to the morbidity in these patients.166 A significant proportion of FD patients also manifest various degrees of renal Pi wasting and subsequent hypophosphatemia,167 which can lead to hypophosphatemic rickets and osteomalacia. Extraskeletal clinical manifestations of FD can occur, such as abnormal skin pigmentation, premature puberty, and hyperthyroidism; the disease is then referred to as McCune-Albright syndrome (MAS). In one study of FD patients, increased FGF23 serum levels correlated negatively to serum Pi and 1,25(OH)2D3 but positively to skeletal disease burden.109 FGF23 mRNA and protein were localized to fibrous cells in the fibrous bone lesions of FD, as well as osteogenic and endothelial cells associated with microvascular walls in bone.109 Therefore, it is likely that FGF23 plays an important role in the pathogenesis of the phosphate wasting that is often seen in FD/MAS.
FD patients are often treated with bisphosphonates, which can lead to improvement of symptoms. It was reported that several MAS patients significantly reduced their elevated FGF23 levels after pamidronate therapy, further supporting a central role of FGF23 in FD/MAS.168 The mechanisms of action for this reduction of serum FGF23 levels is unclear, but one plausible explanation is that osteogenic cells overproducing FGF23 in FD/MAS may undergo apoptosis, thus leading to decreased production of FGF23.
The reason why the bone cells associated with lesions in FD overexpress FGF23 is not understood. Normally, FGF23 is expressed in osteogenic cells,109,110 but it is possible that improperly differentiated cells of FD lesions may have lost regulatory mechanisms required to suppress FGF23 production. Speculatively, this is analogous to FGF23 overexpression in TIO tumors, which are not subject to normal regulatory mechanisms.169
Osteoglophonic Dysplasia (OMIM No. 166250): Activating mutations in fibroblast growth factor receptors 1-3 (FGFR1-3) are responsible for a diverse group of skeletal disorders. In general, mutations in FGFR1-2 cause syndromes involving craniosynostosis, whereas the dwarfing syndromes are associated with FGFR3 mutations. Osteoglophonic dysplasia (OGD) is a “crossover” disorder that has skeletal phenotypes associated with FGFR1-2 mutations as well as with FGFR3 mutations. In this regard, OGD patients present with craniosynostosis, prominent supraorbital ridge, and depressed nasal bridge, as well as with rhizomelic dwarfism and nonossifying bone lesions that show some similarity on x-ray to FD lesions.170 OGD is an autosomal dominant disorder that is caused by missense mutations in highly conserved residues comprising the ligand-binding and transmembrane domain of FGFR1, thus defining novel roles for this receptor as a negative regulator of long-bone growth. Of significance, three out of the four OGD patients studied had isolated renal Pi wasting with inappropriately low 1,25(OH)2D concentrations.171 In one of these patients, a sample was available for analysis of plasma FGF23 concentrations, which were significantly elevated above control levels.171 It was hypothesized that the associated metaphyseal lesions, which may be similar to FD lesions, produce FGF23 which causes renal Pi wasting. Although only a few patients have been assessed, owing to the rarity of the disorder, OGD may have parallels with FD whereby the lesional burden of a patient is proportional to the FGF23 production and the extent of Pi wasting.108
Linear Nevus Sebaceous Syndrome: Linear nevus sebaceous syndrome (LNSS) is a rare congenital disorder involving cutaneous lesions characterized by papillomatous epidermal hyperplasia and excess sebaceous glands.172,173 Additional aberrations are often present in LNSS patients, including developmental defects of the brain which are associated with seizures, as well as eye complications.174
Another rare association with LNSS is hypophosphatemic rickets, which usually presents within the first years of life, often as skeletal pain and insufficiency fractures.175 The primary cause of LNSS in currently unknown, but a report described elevated serum FGF23 in a patient with simultaneous therapy-resistant hypophosphatemic rickets.176 Treatment with octreotide and excision of the nevus were followed by normalization of serum FGF23 and regression of symptoms, implying a role of FGF23 in the development of hypophosphatemic rickets in this disorder.176
Disorders Associated With Reduced FGF23 Bioactivity
Tumoral Calcinosis (OMIM No. 211900)
Familial TC is an heritable autosomal recessive disorder characterized by ectopic calcified tumoral masses, dental abnormalities, and soft-tissue ectopic and vascular calcification.177–179 Biochemical abnormalities include hyperphosphatemia, increased TmP/GFR, and inappropriately normal or elevated 1,25(OH)2D concentrations. Calcium and PTH are typically within the normal reference range, although suppressed PTH levels also occur.178 Biochemically, TC is the mirror image of the Pi-wasting disorder ADHR.105,106 Hyperostosis-hyperphosphatemia syndrome (HHS) is a rare disorder in which patients present with a similar biochemical profile to TC and have localized hyperostosis.180,181
Tumoral Calcinosis/HHS Due to GALNT3 Mutations: The gene first identified for the heritable form of TC was UDP-N-acetyl-alpha-d-galactosamine: polypeptide N-acetylgalactosaminyl transferase-3 (GALNT3), in which potentially inactivating mutations in GALNT3 were detected.182 GALNT3 is expressed in the trans-Golgi network and initiates O-linked glycosylation of nascent proteins. In another family with a reported autosomal dominant TC, the clinical symptoms in this family were in fact caused by two different bi-allelic GALNT3 mutations, further supporting an autosomal recessive inheritance of this disorder.183 The patients with TC due to GALNT3 mutations were originally reported to have serum FGF23 levels approximately 30-fold above the normal mean when assessed with the C-terminal FGF23 ELISA.182 Importantly, it was demonstrated that the TC patients did indeed have elevated C-terminal FGF23, but the same individuals had low circulating FGF23 when measured with the intact FGF23 ELISA.184,185 These findings were then confirmed by demonstrating that loss of GALNT3 resulted in the production of nonfunctional FGF23 protein due to intracellular degradation.186 FGF23 is O-glycosylated on residues within the 176RHTR179/S180 site (specifically at threonine 178), thus the lack of glycosylation at this residue is thought to expose the SPC site and lead to degradation and destabilization of intact, active FGF23.186
HHS is due to inactivating mutations in GALNT3,180 and these patients also manifest inappropriate ELISA ratios of C-terminal to intact FGF23.180,181 It has been shown that some of the HHS mutations are the same as those that result in TC,181 indicating that genetic background may influence disease phenotype and/or that TC and HHS may represent ranges of severity within the same disease.
Tumoral Calcinosis Due to FGF23 Mutations: It was also discovered that the TC syndrome is caused by recessive mutations in the FGF23 gene. Groups reported mutations giving rise to an amino acid change from a serine to a glycine at residue 71 (S71G),187,188 or from a serine to a phenylalanine at residue 129 (S129F).189 Other missense mutations have also been identified, including Q54K190 and M96T.191 These FGF23 TC mutations (summarized in Table 6-2) occur within the FGF23 N-terminal FGF-like domain (residues 25 to 180).190 The TC alterations apparently destabilize intact FGF23, as supported by the findings that the TC patients with FGF23 mutations have the same FGF23 ELISA pattern as GALNT3-TC patients, that is, markedly elevated C-terminal concentrations in concert with low intact values187,188 and the fact that these mutants are proteolytically cleaved prior to cellular secretion.187–189 This cleavage is in part mediated by SPC proteases, inasmuch as addition of the ADHR mutations in tandem with the S71G TC mutations in the FGF23 cDNA resulted in rescue of full-length FGF23 production.192 Thus the common element in GALNT3-TC and FGF23-TC is the lack of secretion of intact FGF23. This lack of active FGF23 then results in elevation of serum Pi through increased renal reabsorption, which in turn most likely results in elevated secretion of nonfunctional FGF23 fragments through a positive-feedback cycle.
Tumoral Calcinosis Due to Klotho Mutations: FGF23 is a member of class of FGFs including FGF19 and FGF21 that are endocrine, as opposed to paracrine/autocrine, factors. FGF23 requires the coreceptor alpha-Klotho (KL) for bioactivity, whereas FGF19 and FGF21 require beta-Klotho.193–195 KL-null mice have severe calcifications as well as markedly elevated serum Pi,196 which parallels Fgf23-null mice197 and that of TC patients. Both the KL-null and Fgf23-null mice, however, have more severe phenotypes than that observed in TC patients. Importantly, the defects in the KL-null and Fgf23-null mice can largely be ameliorated with a low Pi diet to reduce serum Pi.198,199 In parallel with Fgf23-null mice, KL-null mice have increased NPT2a in the proximal tubule,200 indicating that the hyperphosphatemia in these animals is secondary to increased renal reabsorption of Pi.
KL is produced as two isoforms due to alternative splicing of the same gene. Membrane-bound KL (mKL) is a 130-kD single-pass transmembrane protein characterized by a large extracellular domain and a very short (10-residue) intracellular domain that does not possess signaling capabilities.201 The secreted form of KL (sKL) is approximately 80 kD and is spliced within exon 3 to result in a KL protein that does not contain the transmembrane domain and is thus secreted into the circulation.201
The most likely mechanism for FGF23 signaling through KL is the recruitment of canonical FGF receptors (FGFRs) to form active signaling complexes. One group has identified a specific complex between FGFR1c and KL.193 In contrast, others demonstrated that multiple FGFRs (FGFR1c, FGFR3c, and FGFR4) can interact with KL and FGF23 and signal through mitogen-activated protein kinase (MAPK) cascades.202 Importantly, within the kidney, KL localizes to the distal tubule,200 but FGF23 is known to mediate its effects on NPT2a, NPT2c, and the vitamin D metabolizing enzymes within the proximal tubule.97,124 It has been shown that p-ERK1/2 signaling occurs in the DCT within minutes of FGF23 delivery, indicating that a potential intrarenal signaling axis occurs for FGF23.203 However, the mechanisms underlying this local DCT-PT axis in the kidney following FGF23 delivery remain largely unclear.
Since Klotho (KL) is a coreceptor for FGF23, this gene was tested as a candidate for TC in a 13-year-old female with hypothesized end-organ resistance to renal FGF23 activity. This patient presented with hyperphosphatemia, hypercalcemia, elevated PTH, elevated intact and C-terminal FGF23 over 100- to 550-fold elevation of the normal means204 (in contrast to the differential C-terminal to intact ELISA ratios in GALNT3- and FGF23-TC), as well as ectopic calcifications in the heel and brain. She had normal pubertal development, and her disease paralleled KL-null mice with regard to ectopic calcifications and dramatic elevation of circulating FGF23.193 This patient was shown to have a recessive mutation in a highly conserved residue (Histidine193Arginine, or H193R) in the extracellular domain of KL (the “KL1” domain). Mutant KL expression was markedly reduced in vitro compared to that of wild-type KL, which resulted in a striking reduction in the ability of KL to mediate FGF23-dependent signaling.204 Thus, an inactivating H193R KL mutation results in a TC phenotype and demonstrates that KL is required for FGF23 bioactivity. Interestingly, recent findings in a rickets patient indicated that overexpression of KL (through a genomic rearrangement) resulted in hypophosphatemia and hyperparathyroidism.205
In sum, it is clear that in human disorders of phosphate handling and the corresponding mouse models, increased FGF23 bioactivity is associated with renal phosphate wasting and inappropriately normal 1,25(OH)2 vitamin D concentrations, and that the converse biochemical relationships exist with loss of FGF23 bioactivity in the hormone itself or in its receptor complex (summarized in Fig. 6-4).
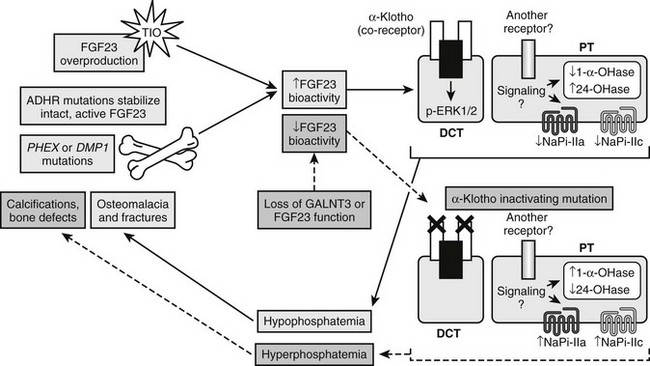
FIGURE 6-4 Genes involved in FGF23 activity. Summary of genes involved in gain of FGF23 bioactivity through intrinsic stabilization (FGF23 gene [ADHR]) or overproduction (TIO; PHEX gene [XLH]; DMP1 gene [ARHR]) (solid pathways) and involved in loss of FGF23 bioactivity through mutations that compromise intact FGF23 production (FGF23 and GALNT3 genes) or its signaling complex (KL gene) (dashed pathways).
Genetic Hypophosphatemia Not Associated With Elevated FGF23
Hereditary Hypophosphatemic Rickets with Hypercalciuria
HHRH was first described in a consanguineous Bedouin tribe.206 Similar to the disease entities described earlier, HHRH is characterized by hypophosphatemia; however, HHRH is distinguished by elevated 1,25(OH)D concentrations and suppressed PTH. The distinguishing biochemical characteristic of this disorder when compared to the FGF23-related diseases such as ADHR and XLH is the notable increase in serum 1,25(OH)D as a compensatory response to the prevailing hypophosphatemia.206 Affected individuals also have markedly elevated urine calcium excretion, with increased intestinal absorption of phosphorus and calcium that can lead to nephrolithiasis. Clinically, patients with HHRH have rickets and short stature. Additional analysis of this kindred revealed 21 members with “intermediate” phenotypes, which led to an initial hypothesis that HHRH was inherited through an autosomal codominant mode. This group of patients manifested hypercalciuria, mild hypophosphatemia, and elevated calcitriol levels but no apparent bony abnormalities.207
To identify the genetic defect responsible for HHRH, a genome wide linkage scan was performed in combination with homozygosity mapping in the large Bedouin kindred in which HHRH was first described. Using this approach, the disorder was mapped to a 1.6-Mb region on chromosome 9q34, which contains the SLC34A3 gene that encodes NPT2c.208,209 A recessive, single-nucleotide deletion was then identified in this gene (228delC). Subsequently, in three unrelated HHRH families, a compound heterozygous deletion and missense mutations were found,208 and additional mutations have more recently been identified.210–212 These are loss-of-function changes and most likely result in impaired Pi reabsorption in the kidney nephron through reduced plasma membrane expression of NPT2c or the uncoupling of Na-Pi cotransport in the proximal tubule.211,212 One of the key physiologic implications of identifying disease-causing mutations in NPT2c was the fact that this transporter now appears to have a significant role in renal Pi reabsorption throughout life and is not limited to perinatal Pi homeostasis as previously hypothesized. The fact that NPT2c mutations lead to disease in humans but not in mice71 may indicate that this transporter plays a more important role in humans than in mice.
Clinical Consequences and Treatment of Hypophosphatemia
Chronic hypophosphatemia reflects ongoing renal phosphate wasting, either from one of the genetic disorders described or from diseases associated with primary or secondary hyperparathyroidism, vitamin D deficiency or resistance, elevated PTHrP, or more generalized proximal tubular dysfunction (i.e., Fanconi’s syndrome). Sustained phosphate wasting may occur also in Dent’s disease213; in neurofibromatosis214; with various metabolic, hormonal, or electrolyte disturbances (poorly controlled diabetes,215 hypokalemia,216 or hypomagnesemia217); or following exposure to certain drugs or toxins (ethanol,218 toluene,219 heavy metals, cisplatin, or foscarnet220). The major clinical manifestations of sustained hypophosphatemia relate to the skeletal complications of rickets and osteomalacia noted earlier. Chronic phosphate depletion also may predispose to the development of acute, severe hypophosphatemia in certain clinical settings associated with rapid phosphate translocation into cells, such as intravenous glucose administration, insulin therapy of diabetic ketoacidosis, acute respiratory alkalosis, treatment of pernicious anemia, uncontrolled hematologic malignancy, or use of hematopoietic cytokines.221,222 Management of chronic hypophosphatemia involves correction of the underlying disorder (where possible) or measures directed at improving the skeletal disease, as described above for XLH.
Clinical Features of Acute Hypophosphatemia
The clinical manifestations of acute severe hypophosphatemia reflect a generalized impairment of cellular energy metabolism, reduced generation of ATP and other intracellular high-energy organophosphates, and associated global tissue or organ dysfunction. Neuromuscular signs and symptoms are particularly common, with muscle weakness (with or without rhabdomyolysis223) and any of a number of other findings, including lethargy, confusion, disorientation, hallucinations, dysarthria, dysphagia, oculomotor palsies, anisocoria, nystagmus, ataxia, cerebellar tremor, ballismus, hyporeflexia, impaired sphincter control, distal sensory deficits, paresthesia, hyperesthesia, generalized or Guillain Barré-like ascending paralysis, central pontine myelinolysis, seizures, coma, and death.224–228 Confusion, flaccid paralysis, areflexia, seizures, and other major sequelae generally are observed only when serum phosphate falls below 0.8 mg/dL, although abnormalities in muscle electrolyte content and membrane potential were demonstrable in animals with experimental phosphate depletion and much less severe hypophosphatemia (1.5 to 2 mg/dL).229
Reversible respiratory failure due to respiratory muscle weakness may occur at serum phosphate levels below 2 mg/dL,230–232 and reversible left ventricular dysfunction has been described with both acute and chronic severe hypophosphatemia.227,233 In patients with septic shock and severe hypophosphatemia (less than 2 mg/dL, average of 1 mg/dL), correction of hypophosphatemia acutely improved left ventricular stroke work index and systolic blood pressure.234 Various abnormalities in renal tubular function have been detected in phosphate-depleted animals, including tubular acidosis and impaired reabsorption of glucose, sodium, and calcium.82,235–237 Erythrocyte concentrations of ATP and 2,3-DPG are directly linked to that of extracellular inorganic phosphate, and severe hypophosphatemia may cause increased erythrocyte fragility, abnormal membrane composition, and excessive oxyhemoglobin affinity. Hemolysis, with membrane rigidity and microspherocytosis, may occur when serum phosphate is below 0.5 mg/dL,238–240 whereas oxyhemoglobin dissociation may be sufficiently impaired when serum phosphate is less than 1 mg/dL to provoke a substantial increase in the cardiac output required for maintenance of adequate oxygen delivery to peripheral tissues.239,241–243 Studies in animals have disclosed significant impairment of critical leukocyte functions (chemotaxis, phagocytosis, bacterial killing), platelet and hemostatic dysfunction, and spontaneous gastrointestinal bleeding during severe hypophosphatemia (less than 1 mg/dL). The appearance of these abnormalities correlates with reductions in leukocyte and platelet ATP content.244
Treatment of Acute Hypophosphatemia
The accumulated evidence suggests that acute, severe hypophosphatemia (<1.5 mg/dL), particularly in the setting of underlying phosphate depletion or when associated with infection or neurologic, cardiopulmonary, or hematologic dysfunction, constitutes a dangerous electrolyte abnormality that should be corrected promptly with intravenous sodium or potassium phosphate, as appropriate. Although the cumulative deficit in body phosphate in such patients cannot be accurately predicted from knowledge of the serum phosphate alone,245 available data indicate that phosphate may be safely administered intravenously at initial doses of 0.2 to 0.8 mmol/kg in elemental phosphorous equivalents over 6 hours (i.e., 10 to 50 mmol over 6 hours), with doses greater than 4 mmol/hr reserved for those with serum phosphate less than 1.5 mg/dL and normal renal function.231,232,234,246,247 Higher doses (1.5 to 3 mmol/kg/12 hr) can cause significant hyperphosphatemia, particularly when renal function is diminished, are not necessary for prevention of severe hypophosphatemia, and thus should be avoided.248,249 One large study conducted in a surgical intensive care unit employed a weight-based algorithm (approximately 0.5 mmol phosphate/kg for severe hypophosphatemia (<1.5 mg/dL) and 0.25 mmol/kg for moderate hypophosphatemia (up to 2.2 mg/dL)) to empirically treat all patients with hypophosphatemia and successfully restored normal serum phosphate in 76% of cases.250 Patients were appropriately excluded for GFR < 25 mL/min, oliguria (<30 mL/hr), hypocalcemia or hypercalcemia, concurrent phosphate-containing parenteral nutrition, or extremes of body weight. The argument for empirical treatment of moderate or severe hypophosphatemia may be more compelling in an intensive care unit setting than for patients less acutely ill on general care units. In the latter case, oral repletion often may suffice, and the threshold for intravenous phosphate therapy and the dose administered should reflect consideration of the risk of hypophosphatemic sequelae, the likely severity and duration of the underlying phosphate depletion, and the presence and severity of symptoms consistent with those of hypophosphatemia. Renal function must be considered and serum phosphate and calcium monitored closely. Recommended protocols (as noted earlier) rarely engender hyperphosphatemia and more often are insufficient, requiring additional infusions on successive days for normalization of serum phosphate.
Possible Future Therapeutic Avenues
As evidence accumulates supporting the role of FGF23 in rare as well as more common disorders of phosphate homeostasis, this molecule is becoming an attractive therapeutic target. Using a novel approach to understanding the mechanisms underlying XLH, neutralizing antibodies targeting FGF23 were administered to Hyp mice.133 After 4 weeks of treatment, injection of the monoclonal antibodies resulted in complete normalization of the serum Pi and 1,25(OH)2D concentrations. Additionally, the Hyp rachitic lesions were ameliorated, and bone and tail length increased.133 Exploring the mechanisms for these physiologic changes indicated that the inactivation of FGF23 in the mice led to increases in NPT2a protein and in 1α(OH)ase mRNA in the renal proximal tubule.133 These studies reinforce the concept that FGF23 has a central role in XLH.
The most direct potential application for recombinant FGF23 could be in familial tumoral calcinosis. Several groups have demonstrated that inactivating mutations in FGF23 lead to FTC,187–189 thus delivering recombinant FGF23 may completely resolve the disorder by directly treating the molecular defect through replacement of missing or mutant FGF23. Additional data are required to determine if FGF23 would be a potential treatment for GALNT3-mediated TC. Whether ADHR-mutant FGF23 (mutant at positions 176 and/or 179) would provide a “longer-acting” form of therapy as a result of stabilization of the full-length polypeptide compared to the labile wild-type form remains to be determined.
References
1. Walser, M. Ion association. VI. Interactions between calcium, magnesium, inorganic phosphate, citrate and protein in normal human plasma. J Clin Invest. 1961;40:723–730.
2. Somell, A, Alveryd, A. Diurnal variations in the urinary excretion of calcium and phosphate in hyperparathyroidism. Acta Chir Scand. 1976;142:357–359.
3. Adam, A, Boulanger, J, Azzouzi, M, et al. Colorimetric vs enzymatic determination of serum phosphorus. Clin Chem. 1984;30:1724–1725.
4. Landowne, RA. Immunoglobulin interference with phosphorus and chloride determinations with the Coulter chemistry. Clin Chem. 1979;25:1189–1190.
5. Donhowe, JM, Freier, EF, Wong, ET, et al. Factitious hypophosphatemia related to mannitol therapy. Clin Chem. 1981;27:1765–1769.
6. Pitkin, RM, Reynolds, WA, Williams, GA, et al. Calcium metabolism in normal pregnancy: a longitudinal study. Am J Obstet Gynecol. 1979;133:781–790.
7. MacDonald, RG, MacDonald, HN. Erythrocyte 2,3-diphosphoglycerate and associated haematological parameters during the menstrual cycle and pregnancy. Br J Obstet Gynaecol. 1977;84:427–433.
8. Reitz, RE, Daane, TA, Woods, JR, et al. Calcium, magnesium, phosphorus, and parathyroid hormone interrelationships in pregnancy and newborn infants. Obstet Gynecol. 1977;50:701–705.
9. Cruikshank, DP, Pitkin, RM, Reynolds, WA, et al. Altered maternal calcium homeostasis in diabetic pregnancy. J Clin Endocrinol Metab. 1980;50:264–267.
10. Baran, DT, Whyte, MP, Haussler, MR, et al. Effect of the menstrual cycle on calcium-regulating hormones in the normal young woman. J Clin Endocrinol Metab. 1980;50:377–379.
11. Hillman, L, Sateesha, S, Haussler, M, et al. Control of mineral homeostasis during lactation: interrelationships of 25-hydroxyvitamin D, 24,25-dihydroxyvitamin D, 1,25-dihydroxyvitamin D, parathyroid hormone, calcitonin, prolactin, and estradiol. Am J Obstet Gynecol. 1981;139:471–476.
12. Greer, FR, Tsang, RC, Searcy, JE, et al. Mineral homeostasis during lactation: relationship to serum 1,25-dihydroxyvitamin D, 25-hydroxyvitamin D, parathyroid hormone and calcitonin. Am J Clin Nutr. 1982;36:431–437.
13. Pitkin, RM, Cruikshank, DP, Schauberger, CW, et al. Fetal calcitropic hormones and neonatal calcium homeostasis. Pediatrics. 1980;66:77–82.
14. Aitken, JM, Gallagher, MJ, Hart, DM, et al. Plasma growth hormone and serum phosphorus concentrations in relation to the menopause and to oestrogen therapy. J Endocrinol. 1973;59:593–598.
15. Halloran, BP, Lonergan, ET, Portale, AA. Aging and renal responsiveness to parathyroid hormone in healthy men. J Clin Endocrinol Metab. 1996;81:2192–2197.
16. Cirillo, M, Ciacci, C, De Santo, NG. Age, renal tubular phosphate reabsorption, and serum phosphate levels in adults. N Engl J Med. 2008;359:864–866.
17. Boskey, AL, Posner, AS. The role of synthetic and bone extracted Ca-phospholipid-PO4 complexes in hydroxyapatite formation. Calcif Tissue Res. 1977;23:251–258.
18. Lian, JB, Cohen-Solal, L, Kossiva, D, et al. Changes in phosphoproteins of chicken bone matrix in vitamin D-deficient rickets. FEBS Lett. 1982;149:123–125.
19. Marxhall, DH, Nordin, BE, Speed, R. Calcium, phosphorus and magnesium requirement. Proc Nutr Soc. 1976;35:163–173.
20. Wilz, DR, Gray, RW, Dominguez, JH, et al. Plasma 1,25-(OH)2-vitamin D concentrations and net intestinal calcium, phosphate, and magnesium absorption in humans. Am J Clin Nutr. 1979;32:2052–2060.
21. Wasserman, RH, Taylor, AN. Intestinal absorption of phosphate in the chick: effect of vitamin D and other parameters. J Nutr. 1973;103:586–599.
22. Kowarski, S, Schachter, D. Effects of vitamin D on phosphate transport and incorporation into mucosal constituents of rat intestinal mucosa. J Biol Chem. 1969;244:211–217.
23. Hurwitz, S, Bar, A. Absorption of calcium and phosphorus along the gastrointestinal tract of the laying fowl as influenced by dietary calcium and egg shell formation. J Nutr. 1965;86:433–438.
24. Lee, DB, Walling, MW, Corry, DB, et al. 1,25-Dihydroxyvitamin D3 stimulates calcium and phosphate absorption by different mechanisms: contrasting requirements for sodium. Adv Exp Med Biol. 1984;178:189–193.
25. Danisi, G, Murer, H, Straub, RW. Effects of pH and sodium on phosphate transport across brush border membrane vesicles of small intestine. Adv Exp Med Biol. 1984;178:173–180.
26. Borowitz, SM, Ghishan, FK. Phosphate transport in human jejunal brush-border membrane vesicles. Gastroenterology. 1989;96:4–10.
27. Hilfiker, H, Hattenhauer, O, Traebert, M, et al. Characterization of a murine type II sodium-phosphate cotransporter expressed in mammalian small intestine. Proc Natl Acad Sci U S A. 1998;95:14564–14569.
28. Feild, JA, Zhang, L, Brun, KA, et al. Cloning and functional characterization of a sodium-dependent phosphate transporter expressed in human lung and small intestine. Biochem Biophys Res Commun. 1999;258:578–582.
29. Beck, L, Karaplis, AC, Amizuka, N, et al. Targeted inactivation of Npt2 in mice leads to severe renal phosphate wasting, hypercalciuria, and skeletal abnormalities. Proc Natl Acad Sci U S A. 1998;95:5372–5377.
30. Peterlik, M, Wasserman, RH. Effect of vitamin D on transepithelial phosphate transport in chick intestine. Am J Physiol. 1978;234:E379–E388.
31. Murer, H, Hildmann, B. Transcellular transport of calcium and inorganic phosphate in the small intestinal epithelium. Am J Physiol. 1981;240:G409–G416.
32. Chen, TC, Castillo, L, Korycka-Dahl, M, et al. Role of vitamin D metabolites in phosphate transport of rat intestine. J Nutr. 1974;104:1056–1060.
33. Corradino, RA. Embryonic chick intestine in organ culture. A unique system for the study of the intestinal calcium absorptive mechanism. J Cell Biol. 1973;58:64–78.
34. Brickman, AS, Hartenbower, DL, Norman, AW, et al. Actions of 1 alpha-hydroxyvitamin D3 and 1,25-dihydroxyvitamin D3 on mineral metabolism in man. I. Effects on net absorption of phosphorus. Am J Clin Nutr. 1977;30:1064–1069.
35. Walling, MW. Intestinal Ca and phosphate transport: differential responses to vitamin D3 metabolites. Am J Physiol. 1977;233:E488–E494.
36. Birge, SJ, Miller, R. The role of phosphate in the action of vitamin D on the intestine. J Clin Invest. 1977;60:980–988.
37. Danisi, G, Straub, RW. Unidirectional influx of phosphate across the mucosal membrane of rabbit small intestine. Pflugers Arch. 1980;385:117–122.
38. Cross, HS, Peterlik, M. Vitamin D activates (Na+-K+)ATPase: a possible regulation of phosphate and calcium uptake by cultured embryonic chick small intestine. Adv Exp Med Biol. 1984;178:163–171.
39. Karsenty, G, Lacour, B, Ulmann, A, et al. Early effects of vitamin D metabolites on phosphate fluxes in isolated rat enterocytes. Am J Physiol. 1985;248:G40–G45.
40. Fuchs, R, Peterlik, M. Vitamin D-induced transepithelial phosphate and calcium transport by chick jejunum. Effect of microfilamentous and microtubular inhibitors. FEBS Lett. 1979;100:357–359.
41. Hattenhauer, O, Traebert, M, Murer, H, et al. Regulation of small intestinal Na-P(i) type IIb cotransporter by dietary phosphate intake. Am J Physiol. 1999;277:G756–G762.
42. Xu, H, Bai, L, Collins, JF, et al. Age-dependent regulation of rat intestinal type IIb sodium-phosphate cotransporter by 1,25-(OH)(2) vitamin D(3). Am J Physiol Cell Physiol. 2002;282:C487–493.
43. Segawa, H, Kaneko, I, Yamanaka, S, et al. Intestinal Na-P(i) cotransporter adaptation to dietary P(i) content in vitamin D receptor null mice. Am J Physiol Renal Physiol. 2004;287:F39–F47.
44. Nemere, I, Yoshimoto, Y, Norman, AW. Calcium transport in perfused duodena from normal chicks: enhancement within fourteen minutes of exposure to 1,25-dihydroxyvitamin D3. Endocrinology. 1984;115:1476–1483.
45. Nemere, I. Apparent nonnuclear regulation of intestinal phosphate transport: effects of 1,25-dihydroxyvitamin D3,24,25-dihydroxyvitamin D3, and 25-hydroxyvitamin D3. Endocrinology. 1996;137:2254–2261.
46. Quamme, GA. Phosphate transport in intestinal brush-border membrane vesicles: effect of pH and dietary phosphate. Am J Physiol. 1985;249:G168–G176.
47. Caverzasio, J, Danisi, G, Straub, RW, et al. Adaptation of phosphate transport to low phosphate diet in renal and intestinal brush border membrane vesicles: influence of sodium and pH. Pflugers Arch. 1987;409:333–336.
48. Danisi, G, Caverzasio, J, Trechsel, U, et al. Phosphate transport adaptation in rat jejunum and plasma level of 1,25-dihydroxyvitamin D3. Scand J Gastroenterol. 1990;25:210–215.
49. Armbrecht, HJ. Age-related changes in calcium and phosphorus uptake by rat small intestine. Biochim Biophys Acta. 1986;882:281–286.
50. Lee, DB, Walling, MW, Brautbar, N. Intestinal phosphate absorption: influence of vitamin D and non-vitamin D factors. Am J Physiol. 1986;250:G369–G373.
51. Cramer, CF, McMillan, J. Phosphorus adaptation in rats in absence of vitamin D or parathyroid glands. Am J Physiol. 1980;239:G261–G265.
52. Knox, FG, Osswald, H, Marchand, GR, et al. Phosphate transport along the nephron. Am J Physiol. 1977;233:F261–F268.
53. Dennis, VW, Stead, WW, Myers, JL. Renal handling of phosphate and calcium. Annu Rev Physiol. 1979;41:257–271.
54. Dennis, VW, Brazy, PC. Divalent anion transport in isolated renal tubules. Kidney Int. 1982;22:498–506.
55. Amiel, C, Kuntziger, H, Richet, G. Micropuncture study of handling of phosphate by proximal and distal nephron in normal and parathyroidectomized rat. Evidence for distal reabsorption. Pflugers Arch. 1970;317:93–109.
56. Haas, JA, Berndt, T, Knox, FG. Nephron heterogeneity of phosphate reabsorption. Am J Physiol. 1978;234:F287–F290.
57. Le Grimellec, C, Roinel, N, Morel, F. Simultaneous Mg, Ca, P, K and Cl analysis in rat tubular fluid. IV. During acute phosphate plasma loading. Pflugers Arch. 1974;346:189–204.
58. Pastoriza-Munoz, E, Colindres, RE, Lassiter, WE, et al. Effect of parathyroid hormone on phosphate reabsorption in rat distal convolution. Am J Physiol. 1978;235:F321–F330.
59. Peraino, RA, Suki, WN. Phosphate transport by isolated rabbit cortical collecting tubule. Am J Physiol. 1980;238:F358–F362.
60. Shareghi, GR, Agus, ZS. Phosphate transport in the light segment of the rabbit cortical collecting tubule. Am J Physiol. 1982;242:F379–F384.
61. Schneider, EG, McLane, LA. Evidence for a peritubular-to-luminal flux phosphate in the dog kidney. Am J Physiol. 1977;232:F159–F166.
62. Ullrich, KJ. Mechanisms of cellular phosphate transport in rat kidney proximal tubule. Adv Exp Med Biol. 1978;103:21–35.
63. Ullrich, KJ, Capasso, G, Rumrich, G, et al. Coupling between proximal tubular transport processes. Studies with ouabain, SITS and HCO3-free solutions. Pflugers Arch. 1977;368:245–252.
64. Ullrich, KJ, Murer, H. Sulphate and phosphate transport in the renal proximal tubule. Philos Trans R Soc Lond B Biol Sci. 1982;299:549–558.
65. Hoffmann, N, Thees, M, Kinne, R. Phosphate transport by isolated renal brush border vesicles. Pflugers Arch. 1976;362:147–156.
66. Burckhardt, G, Stern, H, Murer, H. The influence of pH on phosphate transport into rat renal brush border membrane vesicles. Pflugers Arch. 1981;390:191–197.
67. Cheng, L, Sacktor, B. Sodium gradient-dependent phosphate transport in renal brush border membrane vesicles. J Biol Chem. 1981;256:1556–1564.
68. Ullrich, KJ, Rumrich, G, Kloss, S. Phosphate transport in the proximal convolution of the rat kidney. III. Effect of extracellular and intracellular pH. Pflugers Arch. 1978;377:33–42.
69. Hoag, HM, Martel, J, Gauthier, C, et al. Effects of Npt2 gene ablation and low-phosphate diet on renal Na(+)/phosphate cotransport and cotransporter gene expression. J Clin Invest. 1999;104:679–686.
70. Zhao, N, Tenenhouse, HS. Npt2 gene disruption confers resistance to the inhibitory action of parathyroid hormone on renal sodium-phosphate cotransport. Endocrinology. 2000;141:2159–2165.
71. Segawa, H, Onitsuka, A, Kuwahata, M, et al. Type IIc sodium-dependent phosphate transporter regulates calcium metabolism. J Am Soc Nephrol. 2008;20:104–113. [–].
72. Walton, RJ, Bijvoet, OL. Nomogram for derivation of renal threshold phosphate concentration. Lancet. 1975;2:309–310.
73. Pfister, MF, Lederer, E, Forgo, J, et al. Parathyroid hormone-dependent degradation of type II Na+/Pi cotransporters. J Biol Chem. 1997;272:20125–20130.
74. Lotscher, M, Wilson, P, Nguyen, S, et al. New aspects of adaptation of rat renal Na-Pi cotransporter to alterations in dietary phosphate. Kidney Int. 1996;49:1012–1018.
75. Takahashi, F, Morita, K, Katai, K, et al. Effects of dietary Pi on the renal Na+-dependent Pi transporter NaPi-2 in thyroparathyroidectomized rats. Biochem J. 1998;333(Pt 1):175–181.
76. Kempson, SA, Lotscher, M, Kaissling, B, et al. Parathyroid hormone action on phosphate transporter mRNA and protein in rat renal proximal tubules. Am J Physiol. 1995;268:F784–F791.
77. Traebert, M, Volkl, H, Biber, J, et al. Luminal and contraluminal action of 1-34 and 3-34 PTH peptides on renal type IIa Na-P(i) cotransporter. Am J Physiol Renal Physiol. 2000;278:F792–F798.
78. Bacic, D, Schulz, N, Biber, J, et al. Involvement of the MAPK-kinase pathway in the PTH-mediated regulation of the proximal tubule type IIa Na+/Pi cotransporter in mouse kidney. Pflugers Arch. 2003;446:52–60.
79. Wen, SF. Micropuncture studies of phosphate transport in the proximal tubule of the dog. The relationship to sodium reabsorption. J Clin Invest. 1974;53:143–153.
80. Trohler, U, Bonjour, JP, Fleisch, H. Inorganic phosphate homeostasis. Renal adaptation to the dietary intake in intact and thyroparathyroidectomized rats. J Clin Invest. 1976;57:264–273.
81. Goldfarb, S, Westby, GR, Goldberg, M, et al. Renal tubular effects of chronic phosphate depletion. J Clin Invest. 1977;59:770–779.
82. Steele, TH, DeLuca, HF. Influence of dietary phosphorus on renal phosphate reabsorption in the parathyroidectomized rat. J Clin Invest. 1976;57:867–874.
83. Crawford, JD, Osborne, MM, Jr., Talbot, NB, et al. The parathyroid glands and phosphorus homeostasis. J Clin Invest. 1950;29:1448–1461.
84. Chambers, EL, Jr., Goldman, L, Gordan, GS, et al. Tests for hyperparathyroidism: tubular reabsorption of phosphate, phosphate deprivation, and calcium infusion. J Clin Endocrinol Metab. 1956;16:1507–1521.
85. McCrory, WW, Forman, CW, Mc, NH, et al. Renal excretion of inorganic phosphate in newborn infants. J Clin Invest. 1952;31:357–366.
86. Caverzasio, J, Bonjour, JP. Mechanism of rapid phosphate (Pi) transport adaptation to a single low Pi meal in rat renal brush border membrane. Pflugers Arch. 1985;404:227–231.
87. Thompson, DD, Hiatt, HH. Effects of phosphate loading and depletion on the renal excretion and reabsorption of inorganic phosphate. J Clin Invest. 1957;36:566–572.
88. Hruska, KA, Klahr, S, Hammerman, MR. Decreased luminal membrane transport of phosphate in chronic renal failure. Am J Physiol. 1982;242:F17–F22.
89. Levine, BS, Ho, K, Hodsman, A, et al. Early renal brush border membrane adaptation to dietary phosphorus. Miner Electrolyte Metab. 1984;10:222–227.
90. Brunette, MG, Chan, M, Maag, U, et al. Phosphate uptake by superficial and deep nephron brush border membranes. Effect of the dietary phosphate and parathyroid hormone. Pflugers Arch. 1984;400:356–362.
91. Murer, H, Stern, H, Burckhardt, G, et al. Sodium-dependent transport of inorganic phosphate across the renal brush border membrane. Adv Exp Med Biol. 1980;128:11–23.
92. Boyer, CJ, Xiao, Y, Dugre, A, et al. Phosphate deprivation induces overexpression of two proteins related to the rat renal phosphate cotransporter NaPi-2. Biochim Biophys Acta. 1996;1281:117–123.
93. Lotscher, M, Kaissling, B, Biber, J, et al. Role of microtubules in the rapid regulation of renal phosphate transport in response to acute alterations in dietary phosphate content. J Clin Invest. 1997;99:1302–1312.
94. Ritthaler, T, Traebert, M, Lotscher, M, et al. Effects of phosphate intake on distribution of type II Na/Pi cotransporter mRNA in rat kidney. Kidney Int. 1999;55:976–983.
95. Levi, M, Lotscher, M, Sorribas, V, et al. Cellular mechanisms of acute and chronic adaptation of rat renal P(i) transporter to alterations in dietary P(i). Am J Physiol. 1994;267:F900–F908.
96. Berndt, T, Thomas, LF, Craig, TA, et al. Evidence for a signaling axis by which intestinal phosphate rapidly modulates renal phosphate reabsorption. Proc Natl Acad Sci U S A. 2007;104:11085–11090.
97. Larsson, T, Marsell, R, Schipani, E, et al. Transgenic mice expressing fibroblast growth factor 23 under the control of the alpha1(I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology. 2004;145:3087–3094.
98. Shimada, T, Hasegawa, H, Yamazaki, Y, et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19:429–435.
99. Burnett, SM, Gunawardene, SC, Bringhurst, FR, et al. Regulation of C-terminal and intact FGF-23 by dietary phosphate in men and women. J Bone Miner Res. 2006;21:1187–1196.
100. Perwad, F, Azam, N, Zhang, MY, et al. Dietary and serum phosphorus regulate fibroblast growth factor 23 expression and 1,25-dihydroxyvitamin D metabolism in mice. Endocrinology. 2005;146:5358–5364.
101. Jonsson, KB, Zahradnik, R, Larsson, T, et al. Fibroblast growth factor 23 in oncogenic osteomalacia and X-linked hypophosphatemia. N Engl J Med. 2003;348:1656–1663.
102. Yamazaki, Y, Okazaki, R, Shibata, M, et al. Increased circulatory level of biologically active full-length FGF-23 in patients with hypophosphatemic rickets/osteomalacia. J Clin Endocrinol Metab. 2002;87:4957–4960.
103. Khosravi, A, Cutler, CM, Kelly, MH, et al. Determination of the elimination half-life of fibroblast growth factor-23. J Clin Endocrinol Metab. 2007;92:2374–2377.
104. Takeuchi, Y, Suzuki, H, Ogura, S, et al. Venous sampling for fibroblast growth factor-23 confirms preoperative diagnosis of tumor-induced osteomalacia. J Clin Endocrinol Metab. 2004;89:3979–3982.
105. Econs, MJ, McEnery, PT. Autosomal dominant hypophosphatemic rickets/osteomalacia: clinical characterization of a novel renal phosphate-wasting disorder. J Clin Endocrinol Metab. 1997;82:674–681.
106. Bianchine, JW, Stambler, AA, Harrison, HE. Familial hypophosphatemic rickets showing autosomal dominant inheritance. Birth Defects Orig Artic Ser. 1971;7:287–295.
107. Econs, MJ, McEnery, PT, Lennon, F, et al. Autosomal dominant hypophosphatemic rickets is linked to chromosome 12p13. J Clin Invest. 1997;100:2653–2657.
108. ADHR Consortium. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet. 2000;26:345–348.
109. Riminucci, M, Collins, MT, Fedarko, NS, et al. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest. 2003;112:683–692.
110. Liu, S, Guo, R, Simpson, LG, et al. Regulation of fibroblastic growth factor 23 expression but not degradation by PHEX. J Biol Chem. 2003;278:37419–37426.
111. White, KE, Carn, G, Lorenz-Depiereux, B, et al. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int. 2001;60:2079–2086.
112. Shimada, T, Muto, T, Urakawa, I, et al. Mutant FGF-23 responsible for autosomal dominant hypophosphatemic rickets is resistant to proteolytic cleavage and causes hypophosphatemia in vivo. Endocrinology. 2002;143:3179–3182.
113. Seidah, NG, Chretien, M. Proprotein and prohormone convertases: a family of subtilases generating diverse bioactive polypeptides. Brain Res. 1999;848:45–62.
114. Molloy, SS, Bresnahan, PA, Leppla, SH, et al. Human furin is a calcium-dependent serine endoprotease that recognizes the sequence Arg-X-X-Arg and efficiently cleaves anthrax toxin protective antigen. J Biol Chem. 1992;267:16396–16402.
115. Nakayama, K. Furin: a mammalian subtilisin/Kex2p-like endoprotease involved in processing of a wide variety of precursor proteins. Biochem J. 1997;327(Pt 3):625–635.
116. Benet-Pages, A, Lorenz-Depiereux, B, Zischka, H, et al. FGF23 is processed by proprotein convertases but not by PHEX. Bone. 2004;35:455–462.
117. Imel, EA, Hui, SL, Econs, MJ. FGF23 concentrations vary with disease status in autosomal dominant hypophosphatemic rickets. J Bone Miner Res. 2007;22:520–526.
118. Ryan, EA, Reiss, E. Oncogenous osteomalacia. Review of the world literature of 42 cases and report of two new cases. Am J Med. 1984;77:501–512.
119. Schapira, D, Ben Izhak, O, Nachtigal, A, et al. Tumor-induced osteomalacia. Semin Arthritis Rheum. 1995;25:35–46.
120. Cai, Q, Hodgson, SF, Kao, PC, et al. Brief report: inhibition of renal phosphate transport by a tumor product in a patient with oncogenic osteomalacia. N Engl J Med. 1994;330:1645–1649.
121. Econs, MJ, Samsa, GP, Monger, M, et al. X-Linked hypophosphatemic rickets: a disease often unknown to affected patients. Bone Miner. 1994;24:17–24.
122. Chalew, SA, Lovchik, JC, Brown, CM, et al. Hypophosphatemia induced in mice by transplantation of a tumor-derived cell line from a patient with oncogenic rickets. J Pediatr Endocrinol Metab. 1996;9:593–597.
123. White, KE, Jonsson, KB, Carn, G, et al. The autosomal dominant hypophosphatemic rickets (ADHR) gene is a secreted polypeptide overexpressed by tumors that cause phosphate wasting. J Clin Endocrinol Metab. 2001;86:497–500.
124. Shimada, T, Mizutani, S, Muto, T, et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A. 2001;98:6500–6505.
125. Tenenhouse HS, Econs MJ: Mendelian hypophosphatemias. In Scriver CR, editor: The metabolic and molecular basis of inherited disease, 1998, pp 5039–5068.
126. PEXConsortium. A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. The HYP Consortium. Nat Genet. 1995;11:130–136.
127. Francis, F, Strom, TM, Hennig, S, et al. Genomic organization of the human PEX gene mutated in X-linked dominant hypophosphatemic rickets. Genome Res. 1997;7:573–585.
128. Beck, L, Soumounou, Y, Martel, J, et al. Pex/PEX tissue distribution and evidence for a deletion in the 3′ region of the Phex gene in X-linked hypophosphatemic mice. J Clin Invest. 1997;99:1200–1209.
129. Wang, Y, Spatz, MK, Kannan, K, et al. A mouse model for achondroplasia produced by targeting fibroblast growth factor receptor 3. Proc Natl Acad Sci U S A. 1999;96:4455–4460.
130. Eicher, EM, Southard, JL, Scriver, CR, et al. Hypophosphatemia: mouse model for human familial hypophosphatemic (vitamin D-resistant) rickets. Proc Natl Acad Sci U S A. 1976;73:4667–4671.
131. Meyer, RA, Jr., Meyer, MH, Gray, RW. Parabiosis suggests a humoral factor is involved in X-linked hypophosphatemia in mice. J Bone Miner Res. 1989;4:493–500.
132. Weber, TJ, Liu, S, Indridason, OS, et al. Serum FGF23 levels in normal and disordered phosphorus homeostasis. J Bone Miner Res. 2003;18:1227–1234.
133. Aono, Y, Yamazaki, Y, Yasutake, J, et al. Therapeutic effects of anti-FGF23 antibodies in hypophosphatemic rickets/ssteomalacia. J Bone Miner Res. 2009 May 6.
134. Bai, X, Miao, D, Panda, D, et al. Partial rescue of the Hyp phenotype by osteoblast-targeted PHEX (phosphate-regulating gene with homologies to endopeptidases on the X chromosome) expression. Mol Endocrinol. 2002;16:2913–2925.
135. Liu, S, Guo, R, Tu, Q, et al. Overexpression of Phex in osteoblasts fails to rescue the Hyp mouse phenotype. J Biol Chem. 2002;277:3686–3697.
136. Erben, RG, Mayer, D, Weber, K, et al. Overexpression of human PHEX under the human beta-actin promoter does not fully rescue the Hyp mouse phenotype. J Bone Miner Res. 2005;20:1149–1160.
137. Glorieux, FH, Marie, PJ, Pettifor, JM, et al. Bone response to phosphate salts, ergocalciferol, and calcitriol in hypophosphatemic vitamin D-resistant rickets. N Engl J Med. 1980;303:1023–1031.
138. Glorieux, FH, Scriver, CR, Reade, TM, et al. Use of phosphate and vitamin D to prevent dwarfism and rickets in X-linked hypophosphatemia. N Engl J Med. 1972;287:481–487.
139. Costa, T, Marie, PJ, Scriver, CR, et al. X-linked hypophosphatemia: effect of calcitriol on renal handling of phosphate, serum phosphate, and bone mineralization. J Clin Endocrinol Metab. 1981;52:463–472.
140. Carpenter, TO, Mitnick, MA, Ellison, A, et al. Nocturnal hyperparathyroidism: a frequent feature of X-linked hypophosphatemia. J Clin Endocrinol Metab. 1994;78:1378–1383.
141. Imel, EA, DiMeglio, LA, Hui, SL, et al. Treatment of XLH with calcitriol and phosphate increases FGF23 concentration. J Bone Miner Res. 2007;22(Suppl 1):W123.S123.
142. Reid, IR, Hardy, DC, Murphy, WA, et al. X-linked hypophosphatemia: a clinical, biochemical, and histopathologic assessment of morbidity in adults. Medicine (Baltimore). 1989;68:336–352.
143. Goodyer, PR, Kronick, JB, Jequier, S, et al. Nephrocalcinosis and its relationship to treatment of hereditary rickets. J Pediatr. 1987;111:700–704.
144. Stickler, GB, Morgenstern, BZ. Hypophosphataemic rickets: final height and clinical symptoms in adults. Lancet. 1989;2:902–905.
145. Balsan, S, Tieder, M. Linear growth in patients with hypophosphatemic vitamin D-resistant rickets: influence of treatment regimen and parental height. J Pediatr. 1990;116:365–371.
146. Bettinelli, A, Bianchi, ML, Mazzucchi, E, et al. Acute effects of calcitriol and phosphate salts on mineral metabolism in children with hypophosphatemic rickets. J Pediatr. 1991;118:372–376.
147. Verge, CF, Lam, A, Simpson, JM, et al. Effects of therapy in X-linked hypophosphatemic rickets. N Engl J Med. 1991;325:1843–1848.
148. Reusz, GS, Latta, K, Hoyer, PF, et al. Evidence suggesting hyperoxaluria as a cause of nephrocalcinosis in phosphate-treated hypophosphataemic rickets. Lancet. 1990;335:1240–1243.
149. Eddy, MC, McAlister, WH, Whyte, MP. X-linked hypophosphatemia: normal renal function despite medullary nephrocalcinosis 25 years after transient vitamin D2-induced renal azotemia. Bone. 1997;21:515–520.
150. Alon, U, Donaldson, DL, Hellerstein, S, et al. Metabolic and histologic investigation of the nature of nephrocalcinosis in children with hypophosphatemic rickets and in the Hyp mouse. J Pediatr. 1992;120:899–905.
151. Rasmussen, H, Anast, C. Familial hypophosphatemic rickets and vitamin D-dependent rickets. In: Stanbury JB, Wyngaarden JB, Fredrickson DS, et al, eds. The metabolic basis of inherited disease. ed 5. New York: McGraw-Hill; 1983:1743.
152. Rasmussen, H, Tenenhouse, HS. Hypophosphatemias. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic basis of inherited disease. ed 6. New York: McGraw Hill; 1989:2581.
153. Blydt-Hansen, TD, Tenenhouse, HS, Goodyer, P. PHEX expression in parathyroid gland and parathyroid hormone dysregulation in X-linked hypophosphatemia. Pediatr Nephrol. 1999;13:607–611.
154. Firth, RG, Grant, CS, Riggs, BL. Development of hypercalcemic hyperparathyroidism after long-term phosphate supplementation in hypophosphatemic osteomalacia. Report of two cases. Am J Med. 1985;78:669–673.
155. Wilson, DM, Lee, PD, Morris, AH, et al. Growth hormone therapy in hypophosphatemic rickets. Am J Dis Child. 1991;145:1165–1170.
156. Carpenter, TO, Keller, M, Schwartz, D, et al. 24,25 Dihydroxyvitamin D supplementation corrects hyperparathyroidism and improves skeletal abnormalities in X-linked hypophosphatemic rickets: a clinical research center study. J Clin Endocrinol Metab. 1996;81:2381–2388.
157. Hanna, JD, Niimi, K, Chan, JC. X-linked hypophosphatemia. Genetic and clinical correlates. Am J Dis Child. 1991;145:865–870.
158. Drezner, MK, Lyles, KW, Haussler, MR, et al. Evaluation of a role for 1,25-dihydroxyvitamin D3 in the pathogenesis and treatment of X-linked hypophosphatemic rickets and osteomalacia. J Clin Invest. 1980;66:1020–1032.
159. Rasmussen, H, Pechet, M, Anast, C, et al. Long-term treatment of familial hypophosphatemic rickets with oral phosphate and 1 alpha-hydroxyvitamin D3. J Pediatr. 1981;99:16–25.
160. Harrell, RM, Lyles, KW, Harrelson, JM, et al. Healing of bone disease in X-linked hypophosphatemic rickets/osteomalacia. Induction and maintenance with phosphorus and calcitriol. J Clin Invest. 1985;75:1858–1868.
161. Chesney, RW, Mazess, RB, Rose, P, et al. Long-term influence of calcitriol (1,25-dihydroxyvitamin D) and supplemental phosphate in X-linked hypophosphatemic rickets. Pediatrics. 1983;71:559–567.
162. Feng, JQ, Ward, LM, Liu, S, et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet. 2006;38:1310–1315.
163. Lorenz-Depiereux, B, Bastepe, M, Benet-Pages, A, et al. DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nat Genet. 2006;38:1248–1250.
164. Schwindinger, WF, Francomano, CA, Levine, MA. Identification of a mutation in the gene encoding the alpha subunit of the stimulatory G protein of adenylyl cyclase in McCune-Albright syndrome. Proc Natl Acad Sci U S A. 1992;89:5152–5156.
165. Weinstein, LS, Shenker, A, Gejman, PV, et al. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med. 1991;325:1688–1695.
166. Albright, F, Butler, A, Bloomberg, E. Rickets resistant to vitamin D therapy. Am J Dis Child. 1937;54:529–547.
167. Collins, MT, Chebli, C, Jones, J, et al. Renal phosphate wasting in fibrous dysplasia of bone is part of a generalized renal tubular dysfunction similar to that seen in tumor-induced osteomalacia. J Bone Miner Res. 2001;16:806–813.
168. Yamamoto, T, Imanishi, Y, Kinoshita, E, et al. The role of fibroblast growth factor 23 for hypophosphatemia and abnormal regulation of vitamin D metabolism in patients with McCune-Albright syndrome. J Bone Miner Metab. 2005;23:231–237.
169. Folpe, AL, Fanburg-Smith, JC, Billings, SD, et al. Most osteomalacia-associated mesenchymal tumors are a single histopathologic entity: an analysis of 32 cases and a comprehensive review of the literature. Am J Surg Pathol. 2004;28:1–30.
170. Beighton, P, Cremin, BJ, Kozlowski, K. Osteoglophonic dwarfism. Pediatr Radiol. 1980;10:46–50.
171. White, KE, Cabral, JM, Davis, SI, et al. Mutations that cause osteoglophonic dysplasia define novel roles for FGFR1 in bone elongation. Am J Hum Genet. 2005;76:361–367.
172. Lovejoy, FH, Jr., Boyle, WE, Jr. Linear nevus sebaceous syndrome: report of two cases and a review of the literature. Pediatrics. 1973;52:382–387.
173. Mehregan, AH, Hardin, I. Generalized follicular hamartoma. Complicated by multiple proliferating trichilemmal cysts and palmar pits. Arch Dermatol. 1973;107:435–438.
174. Brodsky, MC, Kincannon, JM, Nelson-Adesokan, P, et al. Oculocerebral dysgenesis in the linear nevus sebaceous syndrome. Ophthalmology. 1997;104:497–503.
175. Carey, DE, Drezner, MK, Hamdan, JA, et al. Hypophosphatemic rickets/osteomalacia in linear sebaceous nevus syndrome: a variant of tumor-induced osteomalacia. J Pediatr. 1986;109:994–1000.
176. Hoffman, WH, Jueppner, HW, Deyoung, BR, et al. Elevated fibroblast growth factor-23 in hypophosphatemic linear nevus sebaceous syndrome. Am J Med Genet A. 2005;134:233–236.
177. Inclan, A, Leon, P, Camjeo, M. Tumoral calcinosis. JAMA. 1943;121:490–495.
178. Mitnick, PD, Goldfarb, S, Slatopolsky, E, et al. Calcium and phosphate metabolism in tumoral calcinosis. Ann Intern Med. 1980;92:482–487.
179. Prince, MJ, Schaeffer, PC, Goldsmith, RS, et al. Hyperphosphatemic tumoral calcinosis: association with elevation of serum 1,25-dihydroxycholecalciferol concentrations. Ann Intern Med. 1982;96:586–591.
180. Frishberg, Y, Topaz, O, Bergman, R, et al. Identification of a recurrent mutation in GALNT3 demonstrates that hyperostosis-hyperphosphatemia syndrome and familial tumoral calcinosis are allelic disorders. J Mol Med. 2005;83:33–38.
181. Ichikawa, S, Guigonis, V, Imel, EA, et al. Novel GALNT3 mutations causing hyperostosis-hyperphosphatemia syndrome result in low intact fibroblast growth factor 23 concentrations. J Clin Endocrinol Metab. 2007;92:1943–1947.
182. Topaz, O, Shurman, DL, Bergman, R, et al. Mutations in GALNT3, encoding a protein involved in O-linked glycosylation, cause familial tumoral calcinosis. Nat Genet. 2004;36:579–581.
183. Ichikawa, S, Lyles, KW, Econs, MJ. A novel GALNT3 mutation in a pseudoautosomal dominant form of tumoral calcinosis: evidence that the disorder is autosomal recessive. J Clin Endocrinol Metab. 2005;90:2420–2423.
184. Garringer, HJ, Fisher, C, Larsson, TE, et al. The role of mutant UDP-N-acetyl-alpha-D-galactosamine-polypeptide N-acetylgalactosaminyltransferase 3 in regulating serum intact fibroblast growth factor 23 and matrix extracellular phosphoglycoprotein in heritable tumoral calcinosis. J Clin Endocrinol Metab. 2006;91:4037–4042.
185. Ichikawa, S, Imel, EA, Sorenson, AH, et al. Tumoral calcinosis presenting with eyelid calcifications due to novel missense mutations in the glycosyl transferase domain of the GALNT3 gene. J Clin Endocrinol Metab. 2006;91:4472–4475.
186. Frishberg, Y, Ito, N, Rinat, C, et al. Hyperostosis-hyperphosphatemia syndrome: a congenital disorder of O-glycosylation associated with augmented processing of fibroblast growth factor 23. J Bone Miner Res. 2007;22:235–242.
187. Benet-Pages, A, Orlik, P, Strom, TM, et al. An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum Mol Genet. 2005;14:385–390.
188. Larsson, T, Yu, X, Davis, SI, et al. A novel recessive mutation in fibroblast growth factor-23 causes familial tumoral calcinosis. J Clin Endocrinol Metab. 2005;90:2424–2427.
189. Araya, K, Fukumoto, S, Backenroth, R, et al. A novel mutation in fibroblast growth factor 23 gene as a cause of tumoral calcinosis. J Clin Endocrinol Metab. 2005;90:5523–5527.
190. Garringer, HJ, Malekpour, M, Esteghamat, F, et al. Molecular genetic and biochemical analyses of FGF23 mutations in familial tumoral calcinosis. Am J Physiol Endocrinol Metab. 2008;295:E929–937.
191. Chefetz, I, Heller, R, Galli-Tsinopoulou, A, et al. A novel homozygous missense mutation in FGF23 causes familial tumoral calcinosis associated with disseminated visceral calcification. Hum Genet. 2005;118:261–266.
192. Larsson, T, Davis, SI, Garringer, HJ, et al. Fibroblast growth factor-23 mutants causing familial tumoral calcinosis are differentially processed. Endocrinology. 2005;146:3883–3891.
193. Urakawa, I, Yamazaki, Y, Shimada, T, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444:770–774.
194. Lin, BC, Wang, M, Blackmore, C, et al. Liver-specific activities of FGF19 require Klotho beta. J Biol Chem. 2007;282:27277–27284.
195. Kurosu, H, Choi, M, Ogawa, Y, et al. Tissue-specific expression of betaKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. J Biol Chem. 2007;282:26687–26695.
196. Tsujikawa, H, Kurotaki, Y, Fujimori, T, et al. Klotho, a gene related to a syndrome resembling human premature aging, functions in a negative regulatory circuit of vitamin D endocrine system. Mol Endocrinol. 2003;17:2393–2403.
197. Shimada, T, Kakitani, M, Yamazaki, Y, et al. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J Clin Invest. 2004;113:561–568.
198. Segawa, H, Yamanaka, S, Ohno, Y, et al. Correlation between hyperphosphatemia and type II Na-Pi cotransporter activity in klotho mice. Am J Physiol Renal Physiol. 2007;292:F769–F779.
199. Sitara, D, Razzaque, MS, Hesse, M, et al. Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol. 2004;23:421–432.
200. Li, SA, Watanabe, M, Yamada, H, et al. Immunohistochemical localization of Klotho protein in brain, kidney, and reproductive organs of mice. Cell Struct Funct. 2004;29:91–99.
201. Matsumura, Y, Aizawa, H, Shiraki-Iida, T, et al. Identification of the human klotho gene and its two transcripts encoding membrane and secreted klotho protein. Biochem Biophys Res Commun. 1998;242:626–630.
202. Kurosu, H, Ogawa, Y, Miyoshi, M, et al. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem. 2006;281:6120–6123.
203. Farrow, EG, Davis, SI, Summers, LJ, et al. Initial FGF23-mediated signaling occurs in the distal convoluted tubule. J Am Soc Nephrol. 2009;20:955–960.
204. Ichikawa, S, Imel, EA, Kreiter, ML, et al. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Clin Invest. 2007;117:2684–2691.
205. Brownstein, CA, Adler, F, Nelson-Williams, C, et al. A translocation causing increased alpha-klotho level results in hypophosphatemic rickets and hyperparathyroidism. Proc Natl Acad Sci U S A. 2008;105:3455–3460.
206. Tieder, M, Modai, D, Samuel, R, et al. Hereditary hypophosphatemic rickets with hypercalciuria. N Engl J Med. 1985;312:611–617.
207. Tieder, M, Modai, D, Shaked, U, et al. “Idiopathic” hypercalciuria and hereditary hypophosphatemic rickets. Two phenotypical expressions of a common genetic defect. N Engl J Med. 1987;316:125–129.
208. Bergwitz, C, Roslin, NM, Tieder, M, et al. SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodium-phosphate cotransporter NaPi-IIc in maintaining phosphate homeostasis. Am J Hum Genet. 2006;78:179–192.
209. Lorenz-Depiereux, B, Benet-Pages, A, Eckstein, G, et al. Hereditary hypophosphatemic rickets with hypercalciuria is caused by mutations in the sodium-phosphate cotransporter gene SLC34A3. Am J Hum Genet. 2006;78:193–201.
210. Ichikawa, S, Sorenson, AH, Imel, EA, et al. Intronic deletions in the SLC34A3 gene cause hereditary hypophosphatemic rickets with hypercalciuria. J Clin Endocrinol Metab. 2006;91:4022–4027.
211. Levi, M. Novel NaPi-2c mutations that cause mistargeting of NaPi-2c protein and uncoupling of Na-Pi cotransport cause HHRH. Am J Physiol Renal Physiol. 2008;295:F369–F370.
212. Jaureguiberry, G, Carpenter, TO, Forman, S, et al. A novel missense mutation in SLC34A3 that causes hereditary hypophosphatemic rickets with hypercalciuria in humans identifies threonine 137 as an important determinant of sodium-phosphate cotransport in NaPi-IIc. Am J Physiol Renal Physiol. 2008;295:F371–F379.
213. Lloyd, SE, Gunther, W, Pearce, SH, et al. Characterisation of renal chloride channel, CLCN5, mutations in hypercalciuric nephrolithiasis (kidney stones) disorders. Hum Mol Genet. 1997;6:1233–1239.
214. Konishi, K, Nakamura, M, Yamakawa, H, et al. Case report: hypophosphatemic osteomalacia in von Recklinghausen neurofibromatosis. Am J Med Sci. 1991;301:322–328.
215. Raskin, P, Pak, CYC. The effect of chronic insulin therapy on phosphate metabolism in diabetes mellitus. Diabetologia. 1981;21:50–53.
216. Dillon, MJ, Shah, V, Mitchell, MD. Bartter’s syndrome: 10 cases in childhood. Q J Med. 1979;48:429–446.
217. Jubiz, W, Canterbury, JM, Reiss, E, et al. Circadian rhythm in serum parathyroid hormone concentration in human subjects: correlation with serum calcium, phosphate, albumin and growth hormone levels. J Clin Invest. 1972;51:2040–2046.
218. Larsson, L, Rebel, K, Sorbo, B. Severe hypophosphatemia: a hospital survey. Acta Med Scand. 1983;214:221–223.
219. Weinstein, S, Scottolini, AG, Bhagavan, NV. Low neutrophl alkaline phosphatase in renal tubular acidosis with hypophosphatemia after toluene sniffing. Clin Chem. 1985;31:330–331.
220. Aschan, J, Ringden, O, Ljungman, P, et al. Foscarnet for treatment of cytomegalovirus infections in bone marrow transplant recipients. Scand J Infect Dis. 1992;24:143–150.
221. Perek, J, Mettelman, M, Gafter, U, et al. Hypophosphatemia accompanying blastic crisis in a patient with malignant lymphoma. J Cancer Res Clin Oncol. 1984;108:351–353.
222. Clark, R, Lee, E. Severe hypophosphatemia during stem cell harvesting in chronic myeloid leukaemia. Br J Haematol. 1995;90:450–452.
223. Knochel, JP. Neuromuscular manifestations of electrolyte disorders. Am J Med. 1982;72:521–535.
224. Silvis, SE, DiBartolomeo, AG, Aaker, HM. Hypophosphatemia and neurological changes secondary to oral caloric intake. Am J Gastroenterol. 1980;73:215–222.
225. Silvis, SE, Paragas, PU, Jr. Paresthesias, weakness, seizures and hypophosphatemia in patients receiving hyperalimentation. Gastroenterology. 1972;62:513.
226. Furlan, AJ, Hanson, M, Cooperman, A, et al. Acute areflexic paralysis. Association with hyperalimentation and hypophosphatemia. Arch Neurol. 1975;32:706–707.
227. O’Connor, LR, Klein, KL, Bethune, JE. Hyperphosphatemia in lactic acidosis. N Engl J Med. 1977;297:707–709.
228. Michell, AW, Burn, DJ, Reading, PJ. Central pontine myelinolysis temporally related to hypophosphataemia. J Neurol Neurosurg Psychiatry. 2003;74:820.
229. Fuller, TJ, Carter, NW, Barcenas, C, et al. Reversible changes of the muscle cell in experimental phosphorus deficiency. J Clin Invest. 1976;57:1019–1024.
230. Newman, JH, Neff, TA, Ziporin, P. Acute respiratory failure associated with hypophosphatemia. N Engl J Med. 1977;296:1101.
231. Aubier, M, Murciano, D, Lecocguic, Y, et al. Effect of hypophosphatemia on diaphragmatic contractility in patients with acute respiratory failure. N Engl J Med. 1985;313:420–424.
232. Gravelyn, T, Brophy, N, Siegert, C, et al. Hypophosphatemia-associated respiratory muscle weakness in a general inpatient population. Am J Med. 1988;84:870–876.
233. Rasmussen, A, Buus, S, Hessov, I. Postoperative myocardial performance during glucose-induced hypophosphatemia. Acta Chir Scand. 1985;151:13–15.
234. Bollaert, P-E, Levy, B, Nace, L, et al. Hemodynamic and metabolic effects of rapid correction of hypophosphatemia in patients with septic shock. Chest. 1995;107:1698–1701.
235. Coburn, JW, Massry, SG. Changes in serum and urinary calcium during phosphate depletion: studies on mechanisms. J Clin Invest. 1970;49:1073–1087.
236. Agus, ZS, Gardner, LB, Beck, LH, et al. Effects of parathyroid hornone on renal tubular reabsorption of calcium, sodium, and phosphate. Am J Physiol. 1973;224:1143–1148.
237. Emmett, M, Goldfarb, S, Agus, ZS, et al. The pathophysiology of acid-base changes in chronically phosphate-depleted rats. J Clin Invest. 1977;59:291.
238. Lichtman, MA, Miller, DR, Cohen, J, et al. Reduced red cell glycolysis, 2,3-diphosphoglycerate and adenosine triphosphate concentration and increased hemoglobin oxygen affinity caused by hypophosphatemia. Ann Intern Med. 1971;74:562–568.
239. Klock, JC, Williams, HE, Mentzer, WC. Hemolytic anemia and somatic cell dysfunction in severe hypophosphatemia. Arch Intern. 1974;134:360–364.
240. Jacob, JS, Amsden, P. Acute hemolytic anemia with rigid red cells in hypophosphatemia. N Engl J Med. 1971;285:1446.
241. Marshall, DH, Nordin, BEC, Speed, R. Calcium, phosphorus and magnesium requirements. Proc Nutr Soc. 1976;35:163–173.
242. Garner, GB, et al. Dietary phosphorus and salmonellosis in guinea pigs. Fed Proc. 1967;26:799.
243. Rajan, K. Hepatic hypoxia secondary to hypophosphatemia. Clin Res. 1973;23:521.
244. Craddock, PR, Yawata, Y, VanSanten, L, et al. Acquired phagocyte dysfunction. A complication of the hypophosphatemia of parenteral hyperalimentation. N Engl J Med. 1974;290:1403–1407.
245. Lentz, RD, Brown, DM, Kjellstrand, CM. Treatment of severe hypophosphatemia. Ann Intern Med. 1978;89:941–944.
246. Rosen, G, Boullata, J, O’Rangers, E, et al. Intravenous phosphate repletion regimen for critically ill patients with moderate hypophosphatemia. Crit Care Med. 1995;23:1204–1210.
247. Clark, C, Sacks, G, Dickerson, R, et al. Treatment of hypophosphatemia in patients receiving specialized nutrition support using a graduated dosing scheme: results from a prospective clinical trial. Crit Care Med. 1995;23:1504–1511.
248. Winter, RJ, Harris, CJ, Phillips, LS, et al. Diabetic ketoacidosis. Induction of hypocalcemia and hypomagnesemia by phosphate therapy. Am J Med. 1979;67:897–900.
249. Zipp, NB, Bacon, GE, Spencer, ML, et al. Hypocalcemia, hypomagnesemia and transient hypoparathyroidism during therapy with potassium phosphate in diabetic ketoacidosis. Diabetes Care. 1979;2:265.
250. Taylor, BE, Huey, WY, Buchman, TG, et al. Treatment of hypophosphatemia using a protocol based on patient weight and serum phosphorus level in a surgical intensive care unit. J Am Coll Surg. 2004;198:198–204.
251. Kruse, K, Woelfel, D, Strom, TM. Loss of renal phosphate wasting in a child with autosomal dominant hypophosphatemic rickets caused by a FGF23 mutation. Horm Res. 2001;55:305–308.
252. Negri, AL, Negrotti, T, Alonso, G, et al. [Different forms of clinical presentation of an autosomal dominant hypophosphatemic rickets caused by a FGF23 mutation in one family]. Medicina (B Aires). 2004;64:103–106.
253. Masi, L, Gozzini, A, Franchi, A, et al. A novel recessive mutation of fibroblast growth factor-23 in tumoral calcinosis. J Bone Joint Surg Am. 2009;91:1190–1198.