[level-membership-for-neonatal-and-perinatal-medicine-category]
27. Genetic Disorders, Malformations, and Inborn Errors of Metabolism*
Anne Matthews and Nathaniel H. Robin
Aneonate born with a malformation, a genetic syndrome, or an acute metabolic disorder presents a management challenge for the neonatal intensive care unit (NICU) staff. If these conditions are not suspected and diagnosed in a critically ill neonate, an appropriate course of action might not be taken. Thus a specific diagnosis becomes imperative. An accurate diagnosis provides the staff with information about the cause of the condition, points the way toward appropriate treatment, and indicates the prognosis so that the most appropriate care of the infant can be initiated. Moreover, the broader issues of providing supportive care and counseling for the affected infant’s family can be addressed.
Genetic evaluation is a complex process that requires expertise in differentiating normal variations from abnormal findings and knowledge of the principles of embryology and dysmorphology to provide an accurate diagnosis. Skills in obtaining detailed information of prenatal and family histories may be equally important.
The field of genomics and genetic medicine has witnessed an explosion of new knowledge, much of which has been generated by the efforts of the Human Genome Project. 13 Advances in understanding of the genetic basis of development and function, as well as the interaction of genes and the environment, continue to provide new insights into human health.
This chapter presents a concise overview of the major categories of genetic disorders and the appropriate techniques to establish specific diagnoses. For an excellent review and detailed explanation of concepts, terminology, and specific genetic mechanisms, refer to Thompson and Thompson Genetics in Medicine.37 See Box 27-1 for a comprehensive list of terms.
BOX 27-1
Aerocentric chromosome A chromosome with the centromere near the end of the chromosome.
Allele One of a pair or series of alternate forms of a gene at the same locus.
Aneuploid Any chromosome number that is not an exact multiple of the haploid set.
Autosome A chromosome that is not a sex chromosome.
Centromere The primary constriction of a chromosome in which the long and the short arms meet.
Chromatid After replication of a chromosome, two subunits attached by the centromere can be seen; each is called a chromatid, and after separation, each becomes a chromosome of a daughter cell.
Chromosomes The microscopic structures in the cell nucleus composed of DNA and proteins that contain the genes.
Congenital Present at birth.
Dermatoglyphics The dermal ridge patterns on the digits, palms, and soles.
Diploid Two copies of all chromosomes; the number of chromosomes normally present in somatic cells. In humans, this is 46 and is sometimes symbolized as 2N.
Dominant A gene (allele) that is expressed clinically in the heterozygous state. In a dominant disorder, the mutant allele overshadows the normal allele.
Dysmorphic Morphologic abnormality, often a minor physical finding that may or may not have any cosmetic or functional significance and is present in less than 4% of the newborn population.
Fluorescence in situ hybridization (FISH) Molecular cytogenetic method for detection of microdeletions of chromosomes.
Gamete Mature reproductive cell, the egg or the sperm, containing the haploid number of chromosomes.
Gene The functional unit of heredity.
Genotype A person’s genetic constitution.
Haploid One copy of all chromosomes; the number of chromosomes present in the gamete; in humans this is 23 and can be symbolized as N.
Hemizygous The condition in which only one copy of a gene is normally present, so its effect is expressed because there is no counterpart gene present (e.g., the genes on the X or Y chromosome of the male).
Heterozygote An individual who has two different alleles at a given locus of two homologous chromosomes.
Homologous chromosomes Members of the same chromosome pair; normally they have the same number and arrangement of genes.
Homozygote An individual who has two identical alleles at a given locus of two homologous chromosomes.
Karyotype The standard pictorial arrangement of chromosome pairs, numbered according to centromere position and length.
Locus The position or place that a gene occupies on a chromosome.
Malformation A primary structural defect that results from a localized error of morphogenesis; abnormal development.
Metacentric chromosome Chromosome with the centromere in the center of the chromosome.
Monosomy Absence of one chromosome of one pair.
Mosaicism Presence in the same individual of two or more different chromosomal constitutions.
Mutation A heritable alteration in the genetic material.
Nondisjunction Failure of two homologous chromosomes to separate equally during cell division into two daughter cells, resulting in abnormal chromosome numbers in gametes or somatic cells.
Phenotype The observable expression of traits either physically or biochemically.
Recessive A gene (allele) that is expressed clinically in the homozygous state. In a recessive disorder, both genes at a given locus must be abnormal to manifest the disorder.
Sex chromosomes The X and Y chromosomes.
Syndrome Recognizable pattern of multiple malformations that occur together and have the same cause.
Transcription The process by which complementary messenger RNA is synthesized from a DNA template.
Translation The process whereby the amino acids in a given polypeptide are synthesized from the messenger RNA template.
Translocation Transfer of all or part of a chromosome to another location (i.e., on the same or another chromosome) after chromosome breakage.
Trisomy The presence of three homologous chromosomes rather than the normal two.
X-linked A gene located on an X chromosome.
Zygote A fertilized egg that develops into an embryo.
GENETIC PRINCIPLES
Genes
A gene is a segment of a deoxyribonucleic acid (DNA) molecule that codes for the synthesis of a single polypeptide and contains the hereditary information needed for development or function. DNA, which allows the storing, duplicating, and processing of hereditary information, consists of two long strands twisted around each other to form a double helix. Each strand of DNA is composed of four nucleotides: guanine (G), adenine (A), thymine (T), and cytosine (C). The specific order of the nucleotides determines the precise information that will be encoded at that site. Genes can (1) regulate other genes by turning them “on” or “off,” (2) specify the exact structure of proteins, which then control the activities of the cells, and (3) specify ribonucleic acid (RNA), which is necessary for protein synthesis.
Chromosomes
Genes are packed in linear order on chromosomes. Chromosomes are found in the nuclei of cells. In humans, normal somatic cells contain 46 chromosomes (diploid number), of which 44 are termed autosomes and 2 are sex chromosomes. Females have two X chromosomes (XX), and males have an X and a Y chromosome (XY). Gametes—eggs or sperm—contain 23 chromosomes (haploid number). In the zygote and somatic cells, chromosomes are paired (homologs). In each pair, one homolog is maternal and the other is paternal in origin. Each chromosomal pair has unique morphologic characteristics that allow it to be distinguished from other chromosomes, such as size, position of the centromere, and the unique banding pattern that is demonstrated by special staining techniques (Figure 27-1). 19 To pass on the genetic information to daughter cells, the chromosomes must replicate and then divide correctly. Somatic cells undergo mitosis, in which cells replicate and then divide chromosomal material into two genetically identical daughter cells with 46 chromosomes each. In gametes, the process is known as meiosis, which is different from mitotic division in that daughter cells contain the haploid number of chromosomes (23) and crossing over or recombination between two homologs occurs, thus facilitating genetic variation in offspring. 37
 |
| FIGURE 27-1
(Courtesy Dr. Loris McGavran, Ph.D., Cytogenics Laboratory at the University of Colorado at Denver and Health Sciences Center, Denver.)
|
An individual’s chromosome constitution can be determined by examining dividing body cells under certain laboratory conditions from any accessible tissue such as blood lymphocytes or skin fibroblasts. The resulting karyotype (see Figure 27-1), or pictorial arrangement, demonstrates the number and structure of that individual’s chromosomes.
ETIOLOGY
Malformations and genetic disorders caused wholly or partly by genetic factors can be categorized into four major areas: (1) chromosomal disorders caused by numeric or structural abnormalities of chromosomes; (2) single-gene or mendelian disorders, which are secondary to single-gene mutations; (3) complex or multifactorial disorders resulting from interaction of genes and environmental influences; and (4) abnormalities caused by environmental exposures of the fetus during development.
More recently, better understanding of molecular processes has allowed the identification of additional genetic mechanisms contributing to genetic disorders: germline mosaicism, genomic imprinting, and uniparental disomy.
Chromosomal Disorders
Chromosomal abnormalities are relatively common. Approximately 0.5% to 0.7% of all live newborns have a chromosomal abnormality, and 4% to 7% of perinatal deaths result from a chromosomal abnormality. Moreover, it is estimated that at least 50% of all recognized first-trimester miscarriages are caused by a chromosomal aberration. 18 Current cytogenetic techniques, such as high-resolution banding, fluorescence in situ hybridization (FISH), and microarray-based comparative genomic hybridization (array-CGH), have increased the detection rate of chromosomal aberrations. Submicroscopic deletions, duplications, or other abnormal rearrangements of chromosome material that may not have been identified a few years previously are now being detected in children with congenital malformations or mental retardation.
Chromosomal aberrations should be suspected in any of the following situations:
• Small for gestational age for weight, length, or head circumference
• Presence of one or more congenital malformations
• Presence of dysmorphic features
• Neurologic or neuromuscular dysfunction
• Family history of multiple miscarriages or siblings with mental retardation or birth defects along with one or more of the above
Chromosomal abnormalities can be classified into two major categories: (1) abnormalities of chromosome number (aneuploidy), in which there is an extra or missing chromosome; and (2) abnormalities of chromosome structure that result in the loss or duplication of part of the chromosomal material. Abnormalities of autosomes usually have more significant deleterious effects on the development of the infant than those seen with sex chromosome abnormalities.
ABNORMALITIES OF CHROMOSOME NUMBER
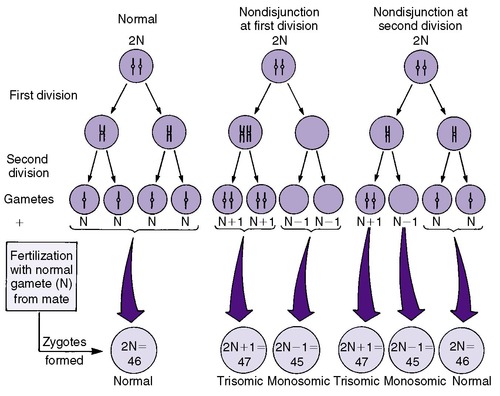
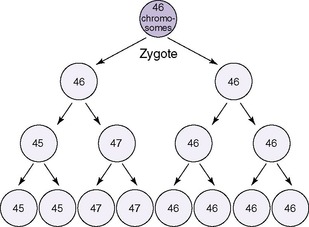
Numeric chromosomal abnormalities occur as a result of nondisjunction in which aberrant segregation leads to loss or gain of one or more chromosomes. Nondisjunction can occur during either meiosis or mitosis, resulting in an abnormal gamete (egg or sperm) or abnormal somatic cell, respectively (Figure 27-2). Fertilization of an aneuploid gamete by a normal gamete produces a zygote with an extra chromosome (trisomy) or missing chromosome (monosomy). Aneuploidy in somatic cells results in chromosomal mosaicism (i.e., the presence of some cells with the normal number of chromosomes and other cells with an abnormal number of chromosomes) (Figure 27-3). Although nondisjunction may affect any chromosomal pair, the most commonly recognized trisomies in liveborns are trisomy 21 (Down syndrome), trisomy 18 (Edward syndrome), and trisomy 13 (Patau syndrome). On the other hand, trisomy 16 has been found exclusively in spontaneous abortions. 18 The most common monosomy is 45,X, Turner syndrome. As a rule, numeric chromosomal abnormalities are associated with intrauterine growth restriction (IUGR), dysmorphic features, malformations, and mental retardation. Physical abnormalities may be milder or absent in the newborn with mosaicism.
ABNORMALITIES OF CHROMOSOME STRUCTURE
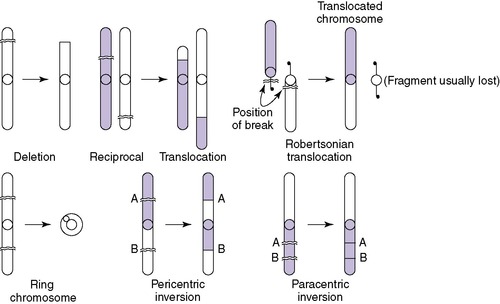
Structural abnormalities have been described in all chromosomes. These include deletions, translocations, duplications, and inversions (Figure 27-4). A deletion is a loss of chromosome material and results in partial monosomy for the chromosome involved. Loss of material from the end of a chromosome is known as a terminal deletion, as seen in 5p−, or cri du chat syndrome. An interstitial deletion involves a loss of chromosomal material that does not include the ends of the chromosome. A terminal deletion of both arms of a chromosome may result in reattachment of the remaining arms, leading to a formation of a ring chromosome. The presence of additional chromosome material results in duplication or partial trisomy of a chromosome. Translocation is the detachment of a chromosome segment from its normal location and its attachment to another chromosome. The translocation is balanced if the cell contains two complete copies of all chromosomal material, although in different order. In an unbalanced translocation, the rearrangement results in partial trisomy or monosomy.
 |
| FIGURE 27-4
(From Hathaway WE, Groothius J, Hay W, editors: Current pediatric diagnosis and treatment, ed 10, Norwalk, Conn, 1991, Appleton & Lange.)
|
Translocations can be reciprocal or robertsonian. A reciprocal translocation involves exchange of segments between two chromosomes (e.g., part of the short arm of chromosome 4 trades a place with a part of chromosome 10). Robertsonian translocations involve two acrocentric chromosomes fused at their centromeres. The most common robertsonian translocation is formed between chromosomes 14 and 21. 22
Inversions are the result of a double break in a single chromosome and reinsertion of the chromosomal material that has been inverted. Inversions are either pericentric (including the centromere) or paracentric (without the centromere). The most common inversion is a small pericentric inversion of chromosome 9, which is considered to be a normal variant, found in approximately 1% of the general population. 37 All other inversions may produce gametes that result in an individual with an unbalanced rearrangement (i.e., having both a duplication and a deletion of some chromosome material, such as that seen in recombinant 8 syndrome).
MICRODELETIONS AND SYNDROMES
At times, structural chromosomal abnormalities are submicroscopic and therefore cannot be detected by conventional cytogenetic techniques. FISH is a molecular cytogenetic method that facilitates the detection of microdeletions. FISH uses segments of fluorescently labeled DNA called probes, constructed so that each probe can attach only to a specific segment of a chromosome, which then will be fluorescent during a microscopic visualization. In the case of a deletion of that chromosome segment, the probe cannot attach to the chromosome; thus the fluorescent segment is missing from the deleted segment of that chromosome. 45
The most recent advance being used to detect very small submicroscopic deletions and duplications is comparative genomic hybridization (array-CGH). 15 This technology blends molecular techniques with cytogenetics and allows the genome to be scanned at a higher resolution than conventional techniques. DNA from a patient sample and DNA from a control sample are differentially labelled, mixed in equal proportions, and hybridized to DNA substrates fixed on an array platform (i.e., bacterial artificial chromosomes [BACs] or oligonucleotides [short segments of DNA usually 8-50 base pairs]). This technique can measure the difference between two different DNA samples in copy number (dosage) of a particular segment of DNA. Thus microscopic gains and losses from a patient sample can be quantified. 15
Microdeletions result in phenotypic abnormalities. A number of well-recognized microdeletion syndromes may be suspected in the NICU. Prader-Willi syndrome, caused by an interstitial deletion of chromosome 15 (q11q13), usually manifests in a newborn as severe hypotonia, feeding difficulties, and micropenis or hypoplastic labia. 9Williams syndrome is caused by an interstitial deletion or mutation of the elastin gene (ELN) on the long arm of chromosome 7 (7q11). 35 The condition is often first seen in an affected newborn in the postterm period; the infant is small for family size. There may be a congenital heart defect, in particular, supravalvular aortic stenosis or peripheral pulmonic stenosis; hypotonia; failure to thrive with gastroesophageal reflux; poor suck and swallow; and vomiting and irritability or colic. Infantile hypercalcemia is seen in approximately 20% of these infants. Subtle dysmorphic facial features may be noted in the newborn. 35
One of the most commonly seen microdeletion syndromes is velocardiofacial syndrome (VCFS), which is characterized by cleft palate or velopharyngeal insufficiency, hypernasal speech, learning disabilities, conotruncal heart defects, and characteristic facies. VCFS actually represents one of a spectrum of clinical disorders all known to be caused by a deletion in chromosome 22q11 (del22q11). These include DiGeorge syndrome (DGS) (conotruncal heart defect, hypocalcemia, and thymic hypoplasia) and conotruncal anomaly face syndrome (CTAF) (conotruncal heart defects and typical facies). In addition, del22q11 has been found in 11% to 16% of cases of nonsyndromic congenital conotruncal heart disease and has been reported to present as apparently isolated neonatal hypocalcemia or learning problems. 14 Overall, del22q11 has an estimated incidence of 1 in 2000 to 4000 newborns. The availability of molecular cytogenetic testing by FISH has led to appreciation of both the high incidence of the del22q11, as well as the increasing variety of clinical presentations that can be seen even within a single family. 32
In the newborn period, the characteristic facial features are seldom obvious. However, most affected individuals manifest some of these findings by early childhood. Most prominent is the nose, which is described as long, with a “built up nasal bridge, squared off nasal root, and bulbous nasal tip.”42 The eyes appear narrow and slitlike, the mala (cheeks) are flat, and the jaw is recessed. The ears usually are small and in some way abnormally formed. There may be an overt cleft of the secondary palate, a bifurcated uvula, a subtle submucosal cleft, or cleft lip with or without cleft palate. Other nonstructural palatal abnormalities can be seen, most commonly velopharyngeal insufficiency. In an older child or adult, this presents as hypernasal speech; in a newborn, one sees excessive nasal regurgitation. Congenital heart disease (CHD) is seen in 35% of del22q11 patients. The type of CHD is fairly specific and includes those lesions classified as “conotruncal heart defects” (truncus arteriosus, interrupted aortic arch, tetralogy of Fallot, left-sided aortic arch, vascular rings, and some types of ventricular septal defects [VSDs]). Perhaps the most consistent finding in patients of all age-groups is long, thin fingers and toes. Additional nonspecific findings include abundant scalp hair; hypospadias; renal abnormalities that can include renal agenesis; tortuous retinal vessels; ectopic/aberrant/unilateral absence of carotid and vertebral artery; microcephaly; and microdontia (there are 166 different findings to date). 42
Developmental delay, learning disabilities, or mental retardation is common and quite variable. Behavioral and psychiatric problems are common but underappreciated findings in VCFS. These individuals have a characteristic personality, marked by a flattened affect and abnormal social interaction, ranging from being intermittently withdrawn to socially precocious. A host of other psychiatric diagnoses have been seen in patients with VCFS.
Both DiGeorge syndrome and VCFS have been recognized as being caused by deletions in 22q11. More than 95% of cases of DGS and VCFS are deleted. In a small number of patients with DGS, point mutations in TBX1 have been found when no deletion was identified. 50 There are a few cases of VCFS in which no 22q11 deletion or TBX1 mutation has been found. Thus obtaining family histories and examining parents for subtle features of the syndrome are important. In many cases, the infant has inherited the abnormality from a parent.
CLINICAL EXAMPLES OF CHROMOSOMAL ABNORMALITIES
Down Syndrome
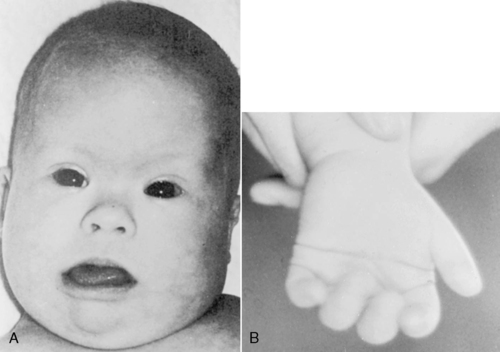
Down syndrome has an incidence of approximately 1 in 600 live births. Approximately 95% of cases are caused by nondisjunction involving chromosome 21, 4% are caused by a translocation, and 1% are mosaic. Down syndrome may manifest with marked hypotonia; a number of major malformations, most commonly congenital heart defects, duodenal atresia, and tracheoesophageal fistula; and a characteristic pattern of dysmorphic features. The classic phenotype seen in Down syndrome includes a flattened occiput, midfacial hypoplasia, depressed nasal bridge, upward-slanting palpebral fissures, epicanthic folds, grayish speckling of the iris (Brushfield spots), micrognathia, excess nuchal skin, single palmar creases (simian creases), single flexion creases and in-curving of the fifth fingers (clinodactyly), and increased distance between the first and second toes (Figure 27-5).
 |
| FIGURE 27-5
( A from Cohen MM: The child with multiple birth defects, ed 2, New York, 1997, Raven Press. B courtesy Dr. Eva Sujansky, Genetic Services at The Children’s Hospital, Denver, Colo.)
|
In full-term infants with the classic phenotype of Down syndrome, the clinical diagnosis is not difficult. However, it is imperative that cytogenetic studies be done to confirm the diagnosis and to differentiate a nondisjunctional trisomy from a translocation. This distinction has important implications for recurrence risks (see discussion in “Prevention” section). In premature infants, the classic facial phenotype is frequently missing, making clinical diagnosis more difficult. The presence of an atrioventricular (AV) canal or duodenal atresia with minor malformations, such as abnormal dermatoglyphics, should alert the clinician to the possibility of Down syndrome.
Trisomy 18
Trisomy 18 has an incidence of 1 in 6000 live births. The major phenotypic features include prenatal growth restriction, complex cardiac malformations, abnormal muscle tone, microcephaly, prominent occiput, short sternum, low-set and malformed ears, corneal opacities, micrognathia, peculiar hand posturing with the second and fifth digits overlapping the third and fourth, hypoplasia of fingernails, abnormal dermatoglyphics, prominent calcanei, and deep plantar furrows between the first and second toes (Figure 27-6). The prognosis is poor, and the majority of infants with trisomy 18 die within the first few months of life. Infants who have survived into childhood are profoundly retarded.
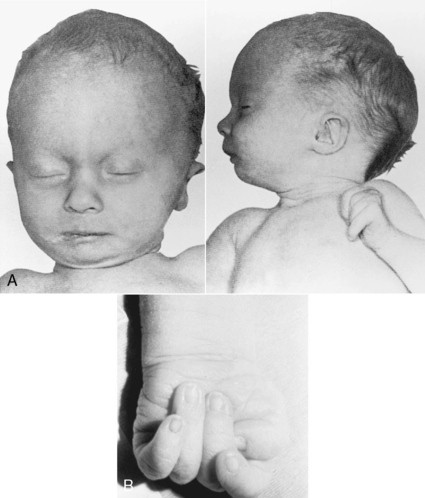 |
| FIGURE 27-6
(From Paerregaard P, Mikkelsen M, Froland A, et al: Trisomy no. 17-18: report of two cases, Acta Pathol Microbiol Scand 67:479, 1966.)
|
Trisomy 13
Trisomy 13 is seen in approximately 1 in 15,000 live births. Phenotypic features include prenatal and postnatal growth restriction, microcephaly, sloping forehead, coloboma of the iris, microphthalmia or anophthalmia, low-set or malformed ears, cleft lip and palate, postaxial polydactyly, and abnormal palmar creases and dermatoglyphics (Figure 27-7). Internal abnormalities may include a number of central nervous system (CNS) malformations, such as holoprosencephaly, cardiac malformations, omphalocele, renal malformations, and urogenital abnormalities such as cryptorchidism in males and uterine malformations in females. The prognosis is extremely poor for these infants, with most dying within the first few months of life.
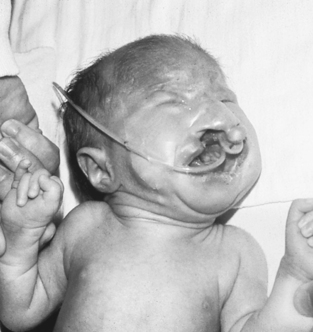 |
| FIGURE 27-7
(From Hathaway WE, Groothuis J, Hay W, editors: Current pediatric diagnoses and treatment, ed 10, Norwalk, Conn, 1991, Appleton & Lange.)
|
Turner Syndrome
The only monosomy to be seen in live births is that of Turner syndrome—females with a 45,X karyotype. In addition, it is the only numeric abnormality of the sex chromosome that may be identifiable at birth. Turner syndrome has an incidence of 1 in 5000 female births. 40 Clinical features that may be evident in the newborn period are a short, webbed neck or redundant skin on the back of the neck and marked lymphedema of the dorsum of the hands and feet (Figure 27-8). Congenital heart defects are seen in approximately half of the patients, with 30% having a coarctation of the aorta. Renal anomalies may also be present. 23 Prognosis is usually excellent but depends on the presence and severity of the congenital heart defect. Intelligence is normal; however, some females with Turner syndrome have been noted to have problems with spatial perception or fine motor abilities. 40
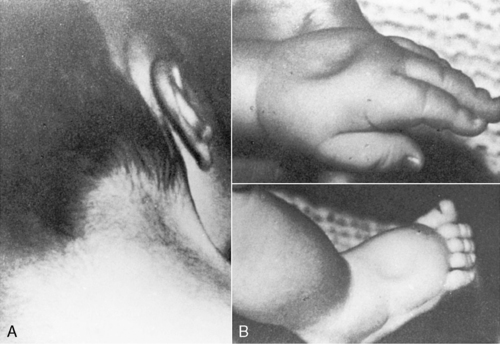 |
| FIGURE 27-8
(From Knuppel R, Drukker JD, editors: High risk pregnancy: a team approach, Philadelphia, 1988, Saunders.)
|
Cri du Chat
Cri du chat, or “cat cry” syndrome, is the result of loss of the terminal end of the short arm of chromosome 5 (5p−). The name of the syndrome reflects the unusual catlike, weak cry these infants have in the neonatal period. These infants are usually small for gestational age, hypotonic, and microcephalic and may have ocular hypertelorism, epicanthic folds, downward slant of the palpebral fissures, low-set ears, and micrognathia. They are significantly mentally retarded.
San Luis Valley Syndrome
Recombinant 8 or the San Luis Valley syndrome, named for the area in which many of these individuals were first identified, is an example of an unbalanced pericentric inversion with both a duplication and a deletion of chromosome 8 material. The pericentric inversion of chromosome 8 found in a parent and other relatives of a child with recombinant 8 syndrome has no phenotypic consequence because it is a balanced rearrangement. However, a carrier is at risk for producing unbalanced gametes during meiosis. In recombinant 8 syndrome, there is a deletion of chromosomal material of the short arm of chromosome 8 and a duplication of chromosome material of the long arm of 8. The phenotype is characterized by unusual facial features, including a wide face, depressed nasal bridge, hypertelorism, down-slanting palpebral fissures, upturned nose, long philtrum, low-set and malformed ears, cleft lip or cleft palate, congenital heart disease, and renal abnormalities. 43
PREVENTION
The identification of chromosomal abnormalities in the newborn is important, not only for management issues about the infant but also because of the recurrence risks the abnormality carries for the family. In general, numeric chromosomal abnormalities carry low recurrence risks (approximately 1% to 2%). 18 In the presence of structural abnormalities, recurrence risks depend on whether one of the parents carries a balanced rearrangement. If parental chromosomes are normal, the recurrence risk is minimal. However, if a parent carries a balanced chromosomal rearrangement, the recurrence risk is significantly increased. The exact risk figure varies with the nature of the specific chromosomal rearrangement and, in some cases, the sex of the carrier parent. In either situation, prenatal diagnosis for chromosome analysis is available for parents and families concerned about recurrence risk.
Single-Gene Disorders
McKusick’s online catalog of mendelian inherited disorders currently lists more than 19,000 entries with approximately 6000 single-gene disorders with known patterns of inheritance. 38 Many of these disorders are singularly rare; however, collectively, they affect about 1% of the population. Single-gene disorders are the result of either a single or double dose of an abnormal gene. Single-gene disorders are classified as autosomal dominant, autosomal recessive, X-linked dominant, and X-linked recessive. Humans have two copies of each gene located at identical places (gene loci) on homologous chromosomes. In a single-gene disorder, an abnormal or mutated allele (an alternate form of a gene) is found on one or both members of a pair of chromosomes. 37 Individuals with identical alleles at a particular locus are homozygous for the gene. Individuals with different alleles are heterozygous for the gene. Because males have only one X chromosome and most genes located on the Y chromosome do not correspond to those located on the X, males are hemizygous for the genes on the X chromosome. Abnormal genes located on one of the 44 autosomes are the cause of autosomal disorders: disease-causing genes located on the X chromosome are the cause of X-linked disorders. Disorders are dominant when the phenotype is expressed in the presence of only one copy of the mutated gene. In recessive disorders, the phenotype is expressed only when both chromosomes carry the mutated gene.
AUTOSOMAL DOMINANT DISORDERS
Autosomal dominant disorders are ones in which the disorder is expressed in the heterozygous state. Major characteristics include the following: (1) multiple generations are affected (i.e., an infant would have an affected parent); (2) both males and females are affected, and both sexes can transmit the disorder to their offspring (i.e., male-to-male transmission can occur); (3) there is a 50% risk for each offspring to inherit the gene from an affected parent; and (4) individuals who do not have the gene cannot transmit the disorder to their offspring.
A negative family history does not rule out the presence of an autosomal dominant disorder. Possible explanations for a negative family history are the following: (1) the infant’s disorder is a result of a new mutation; (2) a parent has a very mild expression of the disorder and may not have been previously diagnosed; (3) nonpaternity; (4) decreased penetrance (i.e., not all individuals with the gene have phenotypic abnormalities [skipped generation]); and (5) germline mosaicism for the mutation (see the “Nontraditional Inheritance” section).
Dominant disorders that may be seen in the NICU include skeletal dysplasias, such as achondroplasia (abnormality in the FGFR3 gene), osteogenesis imperfecta (abnormality in COL1A1 or COL1A2), Apert and Crouzon syndromes (abnormality in the FGFR2 gene), Treacher Collins syndrome (abnormality in Treacle gene), and ectrodactyly (Figure 27-9). 1
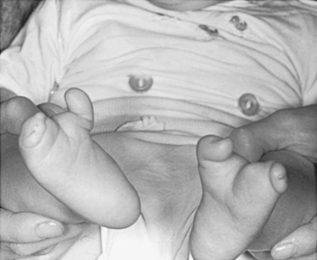 |
| FIGURE 27-9
(Courtesy Dr. Eva Sujansky, Genetic Services at The Children’s Hospital, Denver, Colo.)
|
AUTOSOMAL RECESSIVE DISORDERS
Autosomal recessive disorders are expressed only in the homozygous state. Thus to be affected, an individual usually inherits an abnormal gene from each parent. The parent who is heterozygous for a disease-causing gene is usually phenotypically normal and is called a carrier. Major characteristics of autosomal recessive inheritance include (1) phenotypically normal parents, (2) affected siblings, (3) both males and females affected, (4) offspring of two carrier parents (25% risk for being affected), (5) unaffected siblings have a two-thirds chance of being carriers, and (6) possibly an increased incidence of consanguinity (mating between blood relatives). Autosomal recessive disorders that may be identified in the neonatal period include many of the metabolic disorders (e.g., phenylketonuria [PKU], galactosemia, and isovaleric acidemia) and some of the multiple-malformation syndromes (e.g., Meckel-Gruber syndrome), cystic fibrosis presenting with meconium ileus, Zellweger (cerebrohepatorenal) syndrome (Figure 27-10), and skeletal dysplasias such as achondrogenesis. The specific gene or biochemical defect for many of these disorders is now known.
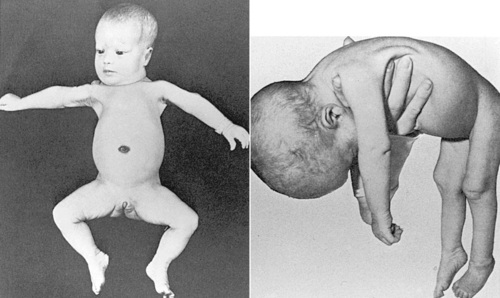 |
| FIGURE 27-10
( Left from Jan JE, Hardwick DF, Lowry RB, et al: Cerebro-hepato-renal syndrome of Zellweger, Am J Dis Child 119:274, 1970. Right from Passarge E, McAdams AJ: Cerebro-hepato-renal syndrome: a newly recognized hereditary disorder of multiple congenital defects, including sudanophilic leukodystrophy, cirrhosis of the liver, and polycystic kidneys, J Pediatr 71:691, 1967.)
|
X-LINKED DISORDERS
X-linked disorders are caused by an abnormal gene (or genes) located on the X chromosome. Most X-linked disorders are recessive. The X-linked recessive disorders are phenotypically expressed in hemizygous males; heterozygous females are generally phenotypically normal and are called carriers. Affected fathers do not have affected sons (no male-to-male transmission); however, all daughters of affected males are carriers. A carrier female has a 50% chance of having an affected male offspring.
Occasionally, heterozygous females may be phenotypically affected, although usually less severely than males. If females are severely affected, other mechanisms, including homozygosity for the X-linked gene, may be responsible for the phenotype. X-linked recessive disorders that may be recognizable in the newborn period include factor VIII and IX deficiency (classic hemophilia A and B), X-linked hydrocephalus, and Opitz syndrome.
X-linked dominant disorders occur when the abnormal gene located on the X chromosome is expressed in both the hemizygous and heterozygous states. As in X-linked recessive conditions, there is no male-to-male transmission, because the affected male passes his Y chromosome and not his X chromosome to his sons; however, all of the daughters of an affected male will inherit his X chromosome and thus be affected. Each son and daughter of an affected female has a 50% risk for being affected; males usually are more severely affected than females. Only a few disorders are known to be inherited as an X-linked dominant, such as incontinentia pigmenti, hypo-phosphatemia (vitamin D–resistant rickets), and ornithine transcarbamylase (OTC) deficiency. Early diagnosis of OTC deficiency is important because, if untreated, it leads to neonatal hyperammonemia and death in affected males. In affected females, the clinical picture can be variable, ranging from an asymptomatic infant to one who presents in the first week of life with lethargy, vomiting, and protein avoidance, ending in seizures and coma.
Complex and Multifactorial Disorders
Complex, common non-mendelian disorders, often called multifactorial disorders, are the result of both environmental and genetic factors. 21 Most isolated single malformations, including congenital heart defects, neural tube defects, cleft lip and palate, pyloric stenosis, and club feet, are inherited in this manner. In addition, the more complex, common familial disorders, such as diabetes mellitus, coronary artery disease, affective disorders, and mild mental retardation, are the result of multifactorial inheritance. In contrast to single-gene inheritance, multifactorial disorders recur within families without a characteristic pedigree pattern and recurrence risks are based on empiric data. 37
Multifactorial inheritance is explained as a liability model with a threshold effect. 21 The general population as a whole has an underlying genetic predisposition for multifactorial traits and disorders that follow a normal distribution curve; only in those individuals in whom the genetic predisposition exceeds the threshold will the malformation actually be expressed (Figure 27-11).
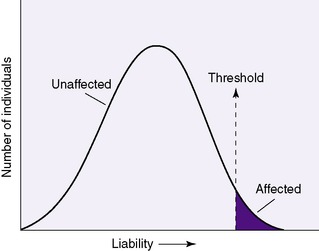 |
| FIGURE 27-11 |
Major characteristics of complex/multifactorial inheritance include the following: (1) no consistent pedigree pattern exists between families (i.e., there may be only an isolated occurrence, or the disorder may be seen among siblings, in multiple generations, or scattered throughout the family); and (2) recurrence risks are not constant, as in single-gene disorders, but are influenced by a number of factors. These factors include the following:
• The number of family members affected (i.e., the more family members affected, the higher the recurrence risk becomes)
• The degree of relatedness to those affected (i.e., first-degree relatives are at higher recurrence risk than second- or third-degree relatives)
• The severity of the defect (i.e., the more severely affected an individual is, the higher the recurrence risk)
• The frequency of the disorder, which may vary with ethnic background (e.g., neural tube defects have a higher incidence among English and Irish populations)
• The gender of the individual (for disorders in which one gender is more commonly affected than the other [e.g., pyloric stenosis is more common in males]; if the less commonly affected gender has the defect, then the recurrence risk is higher)
With the increasing sophistication of ultrasound diagnosis during pregnancy, a number of these isolated malformations can now be diagnosed before delivery. Moreover, in the case of neural tube defects, it has been shown that folic acid supplements may decrease the incidence of spina bifida by as much as 70% in women of reproductive age. 34
NONTRADITIONAL INHERITANCE
Germline Mosaicism
Spectacular growth in the field of molecular genetics and its technologies in only the past few years has enabled the clarification of the inheritance patterns of many genetic disorders and birth defects that were previously unknown or unclear. For example, the lethal form of osteogenesis imperfecta (OI) occurring in multiple offspring of unaffected parents was thought to be the result of autosomal recessive inheritance. However, improved molecular techniques have documented it to be the result of germline mosaicism; that is, the mutation occurs in the gonad of one of the parents, who then has some gametes with and others without the OI mutation. This distinction alters the recurrence risks and is very important for genetic counseling. 37
Genomic Imprinting
In the past, little notice was given to whether the sex of the parent who transmitted an abnormal gene to offspring had any effect on the expression of genes. It is now recognized that maternally and paternally derived genes may function differently, and this is called genomic imprinting.48 For example, offspring who inherit the gene for Huntington disease (autosomal dominant) from an affected father are more likely to have childhood onset of the disease than if they inherited the maternal gene.
Uniparental Disomy
Uniparental disomy is the result of inheriting both copies of a chromosome from one parent and none from the other. 48 It is assumed that for normal growth and development, a child must receive both maternal and paternal genes; if both copies of a gene originate only from one parent, the development is abnormal. Uniparental disomy has been seen in cystic fibrosis with short stature and in the Prader-Willi, Angelman, and Beckwith-Wiedemann syndromes. The parental origin of a child’s chromosomes can be identified only by molecular analysis; routine chromosome analysis usually is not helpful.
The possibility of a nontraditional pattern of inheritance makes genetic counseling more complex than previously thought. Thus it is imperative that health care providers be aware of such complexities and refer families to a geneticist or genetic counselor for a more detailed discussion when appropriate.
INBORN ERRORS OF METABOLISM
Genetic disorders in which defects of single genes cause clinically significant blocks in metabolic pathways are known as inborn errors of metabolism. Recognition of disorders caused by inborn errors of metabolism has increased rapidly in recent years, and they are now recognized as important causes of disease in the newborn and pediatric age-group. 44Inborn errors of metabolism include defects of carbohydrate, amino acid, organic acid, and purine metabolism; disorders of fatty acid oxidation; lysosomal storage diseases; and disorders of peroxisomes. Remember that inborn errors can present at any time and may affect almost any organ system. Specific disorders that should be considered in symptomatic newborns include galactosemia, OTC or carbamoyl-phosphate synthetase (CPS) deficiency, maple syrup urine disease, nonketotic hyperglycinemia, propionic and methylmalonic acidemias, isovaleric acidemia, and glutaric acidemia type II. 24
Although the majority of infants are not found to have an inborn error of metabolism as the etiology of their illness, early recognition is imperative and may be considered a medical emergency if appropriate treatment is to be initiated. Many of these disorders can be treated effectively; if untreated, they are lethal in the newborn period. Moreover, without the appropriate diagnosis, parents would not be aware of recurrence risks in future offspring.
Inborn errors of metabolism should be included in the differential diagnosis for any critically ill newborn in the following instances: (1) suspicion of neonatal sepsis; (2) recurrent vomiting or altered consciousness; (3) clinical findings of hypoglycemia, seizures, parenchymal liver disease, unusual odor, hyperammonemia, or unexplained acidosis; or (4) a family history of a sibling affected with similar symptoms, mental retardation, or sudden infant death syndrome. 24
In general, laboratory analysis depends on the presenting symptoms seen in the newborn. Laboratory studies that should be obtained before any treatment is begun are electrolytes, ammonia, glucose, urine pH, urine-reducing substances, and urine ketones. Clues to a possible inborn error are (1) hypoglycemia and ketonuria in the newborn, (2) acidosis with recurrent vomiting and hyperammonemia, and (3) acidosis that is difficult to correct and is out of proportion to the clinical state. If other underlying disorders are not readily apparent, additional laboratory tests that may be appropriate are serum and urine amino acids and urine organic acids. 44 Moreover, molecular (DNA) testing is available for several disorders. 27
NEWBORN SCREENING
Inborn errors of metabolism, when unrecognized and untreated, may lead to severe consequences, including mental retardation and death in some instances. Thus the goal is to identify, treat, and prevent major sequelae whenever possible. Newborn screening accomplishes this goal for a growing number of disorders (Box 27-2). Screening criteria that should be met are relatively high frequency of the disorder, severity of symptomatology in untreated individuals, availability of treatment, simplicity of obtaining tissue for testing, and availability of a simple screening test with high sensitivity and specificity and reasonable cost. Recently, there have been major changes in the laboratory technologies available for newborn screening—specifically, the introduction of tandem mass spectrometry (MS/MS). 10 Using MS/MS technology, a single blood spot from a newborn can detect more than 50 inborn errors of metabolism. 36 Although many of the disorders detectable by MS/MS are rare, screening has been instituted in most states because screening remains inexpensive and a variety of disorders can be identified in a single assay. In most instances, additional testing adds approximately $25 to $60 to the cost of the newborn screen. 31
BOX 27-2
Defects Identifiable by Tandem Mass Spectrometry (MS/MS)
Amino Acid Disorders and Urea Cycle Disorders
Phenylketonuria
Homocystinuria
Hypermethioninemia
Argininosuccinic acidemia
Citrullinemia
Argininemia
Tyrosinemia types I and II
Organic Acid Disorders
Maple syrup urine disease
Isovaleric acidemia
Methylmalonic acidemia
Propionic acidemia
Glutaric acidemia type I
Isobutyryl-CoA dehydrogenase deficiency
3-Hydroxy-3-methylglutaryl-CoA lyase deficiency
2-Methylbutyryl-CoA dehydrogenase deficiency
3-Methylcrotonyl-CoA carboxylase deficiency
Fatty Acid Oxidation Disorders
Medium-chain acyl-CoA dehydrogenase deficiency
Short-chain acyl-CoA dehydrogenase deficiency
Very-long–chain acyl-CoA dehydrogenase deficiency
Long-chain hydroxyacyl-CoA dehydrogenase deficiency
Glutaric acidemia type II
Carnitine palmityl transferase deficiency type II
Carnitine/acylcarnitine translocase deficiency
Multiple CoA carboxylase deficiency
Trifunctional protein deficiency
Disorders Screened by Other Methodologies
Congenital adrenal hyperplasia
Galactosemia
Sickle cell disease and hemoglobinopathies
Hypothyroidism (congenital)
Data from the National Newborn Screening and Genetics Resource Center; website: http://genes-r-us.uthscsa.edu.
MS/MS has been used for many years to measure metabolites in blood and urine. However, the technology has been applied recently to newborn screening programs. A mass spectrometer is an instrument that separates and quantifies ions based on their mass/charge ratios (m/z). In MS/MS, there are two spectrometers in a series. After sample preparation from the dried blood spot, the process of tandem mass is automated and the analysis is computerized. The process takes only a few seconds, and an entire screen takes less than 2 minutes. 31 The MS/MS system is capable of handling a high volume of samples; thus it is an excellent technology for use in newborn screening. Currently the false-positive rate is 0.3% for all disorders. 52
Screening for metabolic disorders is mandated by individual states, and all states require screening for phenylketonuria (PKU), hypothyroidism, congenital adrenal hyperplasia, sickle cell disease, s-beta thalassemia, and galactosemia. 36 However, the American Academy of Pediatrics (AAP) and the American College of Medical Genetics have recommended that all states screen for a core panel of 29 treatable disorders and an additional 25 conditions that may be detected by screening. 3 The additional disorders most often screened for include amnio acid disorders (homocystinuria, maple syrup urine disease), organic acids (glutaric, methylmalonic, and propionic acidemias), disorders of fatty acid metabolism (medium-chain and very-long–chain acyl-CoA dehydrogenase deficiency), hemoglobinopathies, and others such as biotinidase deficiency, congenital adrenal hyperplasia, and cystic fibrosis. 3
Each state decides individually what will be included in its newborn screening program. Although some states require all of the tests on their list to be mandatory, other states offer a supplemental program in addition to the mandated program, often called expanded newborn screening. Testing for disorders listed on the mandated program is necessary, and only parents objecting on religious grounds may decline the mandated screen (e.g., Ohio Revised Code 3701.501). In those states offering a supplemental program, parents can choose whether or not to screen their child for the disorders listed. The program is optional, and no extra blood or additional tissue is necessary. 26 In most situations, parents are asked to sign a participation form to opt-in to the supplemental program. With more than 30 states now using MS/MS in their newborn screening process, 36 state legislators are constantly changing and updating their screening practices. It behooves the clinician to periodically check with the state newborn screening program for updates. The National Newborn Screening and Genetics Resource Center (NNSGRC) provides up-to-date information.
Any screening test may give both false-positive and false-negative results; this includes those identified by MS/MS. Thus a positive screen result must be followed by a confirmatory diagnostic test.3 Moreover, if there is clinical suspicion of a particular disorder in spite of a negative screening result, further diagnostic testing is warranted. In addition to confirming positive screens, clinicians may have to deal with the concern for increased parental anxiety based on false-positive screens. Waisbren et al found in a prospective interview study of 254 mothers and 153 fathers that stress levels of parents whose infants had false-positive screening results were significantly higher than those with normal results. 46 Thus for clinicians in the NICU setting, being cognizant of the potential for an increased number of positive newborn screening results is important for the care of the neonate, as well as for parent teaching. (For an in-depth review of those disorders that may be part of the newborn screening program, refer to the AAP’s newborn screening recommendations.) 3
Newborn Hearing Screening
Newborn hearing screening tests are now available that have high sensitivity when administered properly. 4 Hearing loss is present in approximately 1 to 2 per 1000 infants. Research has demonstrated that there are both genetic and nongenetic causes of deafness. Sixty percent of prelingual deafness has a recognizable genetic etiology, and of these, the most common cause of autosomal recessively inherited nonsyndromic hearing loss results from mutations in the connexin 26 (Cx26) gene, a member of the connexin family of gap junction proteins. 51 Commercial DNA–based screening tests are now available to detect common genetic forms of deafness including Cx26 and mitochondrial deafness. 5 The AAP has recommended that all newborns be screened for hearing loss before the age of 3 months. 2 The American College of Medical Genetics has recommended, in addition to screening and subsequent confirmation of hearing loss by diagnostic tests, that protocols be developed to ensure that appropriate genetic counseling be provided if diagnostic testing includes genetic testing. 5 This would provide families with accurate information about causes and recurrence risks for parents, siblings, and other family members.
ABNORMALITIES RESULTING FROM ENVIRONMENTAL EXPOSURES
Environmental exposures may have adverse (teratogenic) effects on fetal development, resulting in malformations and functional neurodevelopmental abnormalities in infants and children. The four major prerequisites needed for teratogenic action are as follows20:
1. The agent must have the potential to be teratogenic. Few conclusive data are available about the teratogenicity of most chemicals and drugs in humans. Animal studies provide most of the currently available data on the teratogenicity of agents; however, not all are always applicable to human situations. To prove that an agent is teratogenic, a causal relationship between the exposure and presence of a malformation must be documented; just the history of an exposure to an agent is not sufficient. Although very few agents have been documented to be teratogenic in humans, a few stand out, such as alcohol, cocaine, anticonvulsants, and isotretinoin (Accutane) (Figure 27-12).
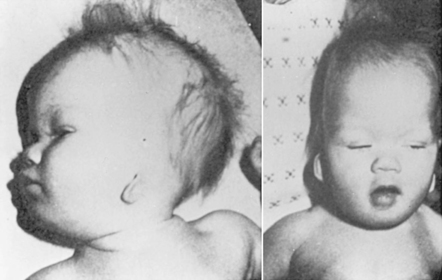 |
| FIGURE 27-12
(Courtesy Dr. Eva Sujansky, Genetic Services at The Children’s Hospital, Denver, Colo.)
|
2. The timing of the exposure during pregnancy is of major importance. For the agent to adversely affect the fetus, it must be present during organogenesis or histogenesis. Exposures occurring within the first 2 weeks after conception, before cell differentiation, will cause no damage or result in fetal wastage. Exposures occurring from 2 to 12 weeks of gestation (period of organogenesis) may result in major malformations. After completion of development of the major organ systems, harmful exposures usually do not result in malformations but can be responsible for organ dysfunction. However, some agents may morphologically disrupt previously intact organs. 11 On the other hand, it has been shown that if the harmful exposure to an agent such as alcohol has been discontinued, the damage is less severe than if the exposure continues throughout the pregnancy.
3. Dosage of the teratogen is related to the severity of the teratogenic effect; the higher the dose, the more severe the effect and the higher the frequency of affected fetuses.
4. Finally, genetic makeup or genetic susceptibility of the mother and fetus may affect the metabolism, as well as tissue sensitivity to the teratogen.
Teratogenic agents may be divided into four categories: (1) infectious agents, (2) chemical agents (drugs and environmental agents), (3) radiation, and (4) maternal factors. Chapter 2 provides excellent overviews of most of these exposures.
Traditionally, only maternal exposures to teratogens have been implicated in malformations. There has been a concern that some paternal exposures also may be teratogenic. Theoretically, a teratogen excreted in the semen could be introduced into the fetal environment and potentially be teratogenic to the developing fetus. 12
Teratogenic exposures should be considered in the differential diagnosis of congenital malformations and CNS dysfunction if one can document fetal exposure and the phenotype is compatible with the known effects of the suspected teratogen. The recognition of exposures is important for genetic counseling; if they can be avoided during subsequent pregnancies, recurrence risk is not increased. Frequently there is phenotypic overlap between the fetal abnormalities caused by specific teratogens and other syndromes. The family should be referred to a genetics clinic to rule out chromosomal, single-gene, and sporadic syndromes with overlapping phenotypes.
DATA COLLECTION
A genetic evaluation consists of the same components found in any medical evaluation; however, the emphasis may be different. Moreover, to make an accurate diagnosis and assessment, medical information about extended family members may have to be obtained. An excellent overview of the evaluation of the neonate with single or multiple congenital anomalies is published by the American College of Medical Genetics and available at their website. 5
History
Prenatal and perinatal histories, from a genetic standpoint, should elicit information about potential teratogenic exposures including maternal disease and acute illness. Fetal growth and behavior (e.g., fetal movement, swallowing) provide important clues for the assessment of fetal neuromuscular function. Thus information about fetal position, movement, and amount of amniotic fluid should be obtained. Perinatal history should include the duration of gestation, anthropometric birth measurements including head circumference, and information about perinatal adaptation. In a newborn with abnormal CNS functioning, it may be difficult to differentiate between primary maldevelopment and dysfunction caused by perinatal complications. An abnormal newborn with a genetic disorder may present with signs suggestive of birth asphyxia (i.e., hypoxia, acidosis, hypotonia, seizures). Moreover, because many a priori abnormal newborns have an increased frequency of perinatal complications, a documented birth injury does not rule out the presence of a genetic cause.
Family history may be extremely helpful in clarifying the causes and risk for recurrence. The information obtained from the parents may have to be complemented by physical examination of the parents and other family members and review of the medical records. This may be necessary because parents may not be aware that different defects in family members may be an expression of the same disorder. For example, an autosomal dominant gene may cause mild hypoplasia of thumbs in one family member and complete absence of thumb and radii in another.
A three-generation pedigree also should be obtained that includes health information about parents, siblings, grandparents, aunts, uncles, and cousins. Specifically, information about miscarriages, stillbirths, childhood deaths, relatives born with congenital malformations and birth defects, mental retardation, and other disorders that “run in the family” should be obtained. 6 Information about ethnic background and consanguinity also should be collected.
Physical Examination
A physical examination should enable the examiner to detect major and minor malformations (dysmorphic features). Minor malformations are defined as structural variations found in less than 4% of the general population and that have no significant medical or cosmetic effect. This is in contrast to structural variations that are found in more than 4% of the newborn population and represent a normal variation, such as a mongolian spot and capillary hemangioma on the forehead. Minor malformations may provide important clues to the identification of a specific syndrome. None of the minor malformations as an isolated finding are clinically significant; however, a combination or pattern of minor malformations may indicate a specific disorder. For example, dysmorphic features such as up-slanted eyes, epicanthic folds, hypertelorism (Figure 27-13), and abnormal dermatoglyphic pattern (Figure 27-14) in an infant with a congenital heart defect are suggestive of Down syndrome. Minor malformations also may alert the clinician to the presence of major malformations. For example, preauricular ear tags are associated with an increased frequency of inner ear malformations and hearing impairment. In addition, the greater the number of minor malformations an infant has, the higher the chance of finding one or more major malformations.28
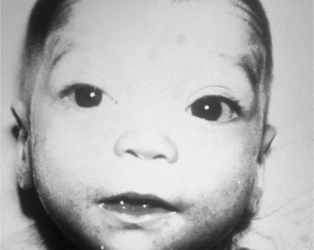 |
| FIGURE 27-13
(From Gilbert-Barness E, Kapur RP, Oligny LL et al: Potter’s pathology of the fetus, infant, and child, ed 2, St Louis, 2007, Mosby.)
|
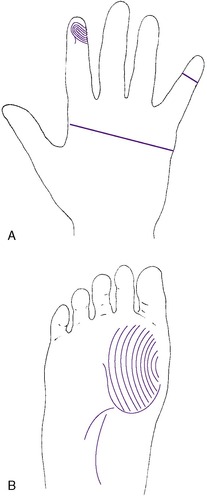
If minor malformations are identified, the parents of the infant should be examined. Presence of the same minor malformation in one of the parents may indicate a benign familial feature. Alternatively, finding the same dysmorphic features in other family members may represent an inherited genetic disorder. A mild syndactyly between the second and third toes is frequently an isolated, inherited finding without clinical significance. However, syndactyly associated with craniosynostosis may represent an autosomal dominant disorder with variable expression and significant clinical sequelae.
If an infant looks dysmorphic, documentation of specific features should be recorded. Actual measurements compared with age-related norms should be used to measure body proportions, length of extremities, and such facial features as distance between eyes, length of eye fissures, size of ears, and length of philtrum. Description of the other features, such as the shape of the neck (webbed) or the chest size (widely spaced nipples), or a specific description of any skin lesions including size, shape, location, and color (hyperpigmented or hypopigmented) may provide important clues for a specific diagnosis. Dermatoglyphic analysis—the analysis of the dermal ridges on the digits, palms, and soles—may prove useful for determining the timing of a fetal insult. 49 Development of ridges begins during the thirteenth week of gestation and is complete by the nineteenth week. Thus many chromosomal and genetic disorders have disruptions of the dermal ridge patterns. For example, an infant with Down syndrome may have a single palmar crease (simian crease), a single flexion crease of the fifth digit, and an open field pattern (tibial arch) on the hallucal area of the foot. 49 Moreover, specific descriptors or, even more useful, photographs, should be used to describe dysmorphic findings.
Photographs are particularly important if the infant is critically ill and the constellation of findings does not immediately suggest a specific syndrome. Thus the patient’s findings may be more accurately shared with other clinicians in the future.
Smith’s Recognizable Patterns of Human Malformation23 and other texts document a large number of syndromes, some of which are quite rare and may not be immediately recognized by neonatal staff. It may be helpful and important to consult a clinician who is familiar with dysmorphology and syndromology to help establish a diagnosis.
Laboratory Data
When a genetic disorder is suspected, a number of diagnostic studies may be useful in delineating a diagnosis, including chromosome analysis, CGH array, molecular DNA testing, biochemical studies to rule out inborn errors of metabolism, radiographs, organ imaging, and when appropriate, autopsy. Not all of these studies are routinely used in all patients, but selection of studies is based on clinical suspicion of a particular disorder.
CHROMOSOME ANALYSIS
Routinely, results of chromosome analysis may not be available for several weeks. However, if results of chromosome analysis are urgently needed for clinical management, chromosome analysis from bone marrow can be available within a few hours and preliminary results from blood lymphocyte culture can be obtained in 48 hours. Indications for chromosome analysis have been listed previously. However, chromosome analysis should be obtained in all critically ill infants for whom there is no plausible explanation for their grave clinical course, before death occurs. For postmortem examination, chromosomes also may be obtained from any tissue—in particular, intracardiac blood, thymus, skin, and gonad. Under sterile conditions, these tissues should be obtained as soon as possible after the infant’s demise. Tissue should be transported to the laboratory in a tissue culture medium or sterile saline solution, not in formalin.
MOLECULAR DNA ANALYSIS
There is a growing list of disorders for which the gene has been identified, and thus molecular diagnostic tests are commercially available. Although the Genetic Alliance, an international coalition comprising more than 600 advocacy, research, and health care organizations, lists 830 genetic conditions at their website (www.geneticalliance.org), not all those disorders are amenable to DNA testing. Moreover, often a clinical diagnosis of a recognized syndrome does not require additional diagnostic testing. 5 However, if the diagnosis is not clear, if there is a need to confirm a diagnosis for management issues, or if additional information about a clinically recognized disorder is needed for family planning and genetic counseling, then DNA testing may be extremely helpful. 5
Whenever the health care professional has a reasonable suspicion of a specific diagnosis based on clinical phenotype, it is reasonable to suggest molecular testing if it is available to confirm the diagnosis and provide appropriate genetic counseling. However, the clinician must remember that mutational analysis is complex and, although it can confirm a diagnosis when the test result is positive, a negative result may not determine conclusively that the neonate is not affected. 1 Some disorders (listed with their gene symbol or chromosomal locus) that might be seen in the neonate for which molecular gene testing is available are listed in Table 27-1.
| Syndrome | Gene | Chromosomal Location |
|---|---|---|
| Apert and Crouzon | FGFR2 | 10q26 |
| Achondroplasia and thanatophoric dysplasia | FGFR3 | 4p16 |
| Cystic fibrosis | CFTR | 7q31 |
| Congenital myotonic dystrophy | DMPK | 19q13 |
| Miller-Dieker | Microdeletion | 17p13.3 |
| Osteogenesis imperfecta | COL1A1 or COL1A2 | 17q21.3-q22 |
| Pfeiffer | FGFR1 | 10q26 |
| Prader-Willi and Angelman | Uniparental disomy, imprinting, deletion | 15q11.3-q13 |
| Smith-Lemli-Opitz | DHCR7 | 11q12-q13 |
| Treacher Collins | Treacle (TCOF1) | 5q32-q33 |
| Velocardiofacial/DiGeorge | Microdeletion | 22q11 |
| Waardenburg type 1 | PAX3 | 2q35 |
| Waardenburg type 2 | MITF | 3p14.1-p12.3 |
| Williams | ELN (elastin) | 7q11.2 |
| Wolf-Hirschhorn | Microdeletion | 4p16.3 |
BIOCHEMICAL STUDIES
A critically ill neonate who has a condition suggestive of an inborn error of metabolism or who has no specific diagnosis should have blood and urine sent for appropriate biochemical studies (specified in the “Inborn Errors of Metabolism” section). If such studies have not been obtained, postmortem tissue such as liver should be obtained and frozen for later biochemical analysis. Care providers should request detailed instructions from a laboratory specializing in testing for inherited metabolic disorders about which tissue is appropriate and how it should be obtained, stored, and shipped to the laboratory.
RADIOGRAPHS
X-ray examination should be obtained if a skeletal dysplasia or other skeletal abnormality is suspected or if the differential diagnosis includes a genetic syndrome that has skeletal defects as part of the phenotype. Moreover, if a localized skeletal defect is found, a skeletal survey should be obtained to identify other possible skeletal defects.
ORGAN IMAGING
Organ imaging by ultrasonography, magnetic resonance imaging (MRI), and computed tomography (CT) scan should be used to rule out structural abnormalities of major organs such as the brain, heart, and kidneys. Malformations may be suspected on the basis of clinical symptoms such as anuria or on the basis of known nonrandom associations of certain birth defects, such as the vertebral/anal/tracheoesopageal fistula/renal/radial (VATER) association. In addition, some dysmorphic features are associated with major malformations, and one must rule out these features. For example, there is an increased incidence of underlying midline brain defects associated with some facial dysmorphic features.
AUTOPSY
In the event of a neonate’s death, an autopsy may provide crucial information for the establishment of a correct diagnosis. As outlined, chromosome analysis, biochemical studies, x-ray examination, and photographs all should be included. In the absence of a specific, confirmed diagnosis, the family should be strongly encouraged to consent to an autopsy, and a tissue sample should be frozen for further testing. Without this valuable information, subsequent genetic counseling of the parents, including clarification of the causes and recurrence risks, becomes impossible.
TREATMENT AND INTERVENTION
For most genetic disorders and malformations, there are no “cures” and only symptomatic treatment is available; that is, conventional medical and surgical interventions are instituted, although the basic genetic defect is not corrected. Surgical intervention for specific malformations will depend on the malformation, its cause, and prognosis. For example, surgical repair of a cleft lip and palate usually has an excellent outcome, although the underlying genetic cause has not been altered. In other instances, the diagnosis may provide direction and guidance to the health care professionals and family as to the appropriate course of action. An infant born with a hypoplastic left side of the heart may be considered a candidate for a heart transplant. However, if the cardiac malformation is the result of a chromosomal abnormality with an extremely poor prognosis, such as trisomy 13, the management of that infant may be palliative rather than corrective.
With the diagnosis of a metabolic disorder, treatment may be one of a nutritional or pharmacologic approach, such as the restriction of phenylalanine in an infant with PKU or the replacement of a deficient hormone, such as thyroid supplements in hypothyroidism. For some conditions, such as OI, the best approach may be educating the parents on specific techniques of holding and caring for an infant to prevent further fractures. In some disorders, organ or tissue transplantation may be appropriate. Bone marrow transplantation has been found to be effective in treating select genetic disorders, including lysosomal storage disorders39 and beta thalassemia. 33 Because specific treatment leading to a cure is not available for most genetic disorders, the use of genetic counseling and available reproductive alternatives, such as prenatal diagnosis, in vitro fertilization, preimplantation diagnosis, and artificial insemination by donor, are acceptable alternatives for some families.
THE HUMAN GENOME PROJECT
The year 2000 marked the announcement that the vast majority of the human genome had been sequenced. 13 This international effort, funded in part by the National Institutes of Health (NIH), began in 1990 with the development of genetic and physical maps of the human genome and completed its initial goals in 2000 with a draft of the sequencing of the 3 billion base pairs of the human genome. Defining the sequence of the human genome is only the beginning of the application of that knowledge to current and future research opportunities that will provide avenues for innovative therapies. New powerful technologies for understanding gene expression are being applied to designing drugs that will moderate disease pathways. Collins et al have noted that “microarray technologies have catapulted many laboratories from studying the expression of one or two genes in a month to studying the expression of tens of thousands of genes in a day.”13 The field of genomics is also providing opportunities to predict responsiveness to drug therapies, because reactions to drugs often are based on individual genetic variations. With the identification of common gene variants involved in drug action or metabolism, health care professionals might be able to predict an infant’s response—good or bad—to a particular drug regimen. 13
With the numerous advances in molecular genetics, it is predicted that many genetic disorders and malformations may be amenable to treatment in utero or after birth. In utero correction of such birth defects as urinary tract malformations, myelomeningocele, and diaphragmatic hernia has been successful. 7,16,17 In 1990, the first human trial of gene therapy was undertaken at the NIH. The treatment was somatic gene replacement in a 4-year-old child with adenosine deaminase deficiency, a rare inherited disorder that destroys the immune system. 29 Successful efforts using improved gene therapy techniques continue with this disorder. 8 Since that time, a number of gene therapy trials for other genetic disorders have been done. Although the arena of gene therapy in general has been somewhat disappointing, the results have led to new areas of research and experimentation with new promising techniques. 41 The development of safer and more effective vectors based on technologies spearheaded by the Human Genome Project should provide significant improvements in gene therapy, such as that seen in an application of gene therapy with hemophilia B. 25
PARENT TEACHING
The birth of any infant with a malformation or genetic disorder is a devastating event for any family. The neonatal staff find themselves on the front lines helping families deal with the infant’s problems and providing the best environment for both the critically ill newborn and his or her family. In general, the most difficult factor for most parents and families to deal with is the unknown. Thus once again, the need for an accurate diagnosis becomes paramount (see the Parent Teaching box on p. 809). Moreover, even when the diagnosis carries a very poor prognosis, parents would prefer having the information so they can realistically anticipate and prepare for what is to come. 47 Currently, many parents obtain information about their infant’s malformation or genetic disorder through prenatal diagnosis and have already begun the process of anticipatory grief by the time the infant is admitted to the NICU. Parental feelings of disbelief, shock, anger, or despair may have already been replaced with a “sense of relief” about confirmation of the abnormalities and a need to deal with the situation at hand. 30
• Genetic diagnosis is essential not only for management of the neonate’s condition but also for counseling.
• Genetic counseling assists parents and care providers in addressing management issues such as cause and diagnosis, prognosis, treatment and interventions, short-term and long-term care, and follow-up.
• Genetic counseling assists parents in decisions about future pregnancies or the use of assistive reproductive technologies.
Chapter 30 provides an excellent review of the grief and mourning process that parents will experience when their anticipated “perfect baby” is born with a malformation or genetic disorder.
From a genetic counseling standpoint, a number of principles should be incorporated into the plan of care for the neonate and his or her family. First—and it cannot be overstated—an accurate diagnosis is essential if genetic counseling is to be provided. Even with what appears to be an isolated malformation, a genetics consultation may be appropriate to rule out other causes, such as single-gene disorders or chromosomal abnormalities. After establishing the diagnosis, one can realistically address the prognosis, treatment, and other management issues with the family. Finally, at the appropriate time for the family, recurrence risks and options for future pregnancies can be addressed.
Certainly, the busy and stressful environment of the NICU is not the most conducive atmosphere for obtaining and providing detailed information. However, it is very appropriate for the geneticist to make an initial contact with the family in the NICU, where basic information about the pregnancy and perinatal and family histories can be obtained that will aid in diagnosis and defining the cause. Diagnosis and management then can be addressed by the neonatal staff and geneticist. Later, at a time appropriate for the family, such issues as recurrence risks can be addressed. Eventually the family should receive a written summary of all the issues discussed for their own documentation.
The genetic evaluation is a complex and multifaceted process that cannot be done in isolation; it requires a team approach. The geneticist can assist the neonatal intensive care staff in determining the diagnosis, cause, and prognosis so that appropriate management of the infant can be implemented and aid in future counseling of the families they serve. The neonatal staff should use the genetics team as a resource for consultation and assistance in providing infants and families with the most appropriate and complete health care available.
REFERENCES
1. American Academy of Pediatrics, Molecular genetic testing in pediatric practice: a subject review, Pediatrics 106 (2000) 1494.
2. American Academy of Pediatrics, Newborn and infant hearing loss: principles and guidelines for early detection and intervention programs, Pediatrics 120 (2006) 898.
3. American Academy of Pediatrics, Newborn screening expands: recommendations for pediatricians and medical homes—implications for the system, Pediatrics 121 (1) ( 2008) 192.
4. American College of Medical Genetics, Newborn screening: toward a uniform screening panel and system, Genet Med 8 (2006) 15.
5. American College of Medical Genetics, Evaluation of the newborn with single or multiple congenital anomalies: a clinical guideline,, Accessed October 26, 2009, fromwww.acmg.net.
6. Bennett, R.L., The family medical history, Prim Care Clin Office Pract 31 (2004) 479.
7. Bruner, J.P.; Tulipan, N.; Paschall, R.L.; et al., Fetal surgery for myelomeningocele and the incidence of shunt-dependent hydrocephalus, JAMA 282 (1999) 1819.
8. Calazzana-Calvo, M.; Hacein-Bey, S.; de Saint Basile, G., Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease, Science 288 (2000) 669.
9. Cassidy, S.B.; McCandless, S., Prader Willi syndrome, In: (Editors: Cassidy, S.B.; Allanson, J.E.) Management of genetic syndromes,ed 2 ( 2004)Wiley-Liss, Hoboken, NJ.
10. Chace, D.H.; Kalas, T.A.; Naylor, E.W., Use of tandem mass spectrometry for multianalyte screening of dried blood specimens from newborns, Clin Chem 49 (2003) 1797.
11. Clayton-Smith, J.; Donnai, D., Human malformations, In: (Editors: Rimoin, D.L.; Connor, J.M.; Pyeritz, R.E.; et al.) Emery and Rimoin’s principles and practice of medical geneticsed 5 ( 2006)Churchill Livingstone, New York.
12. Colie, C.F., Male mediated teratogenesis, Reprod Toxicol 7 (1993) 3.
13. Collins, F.S.; Green, E.D.; Guttmarcher, A.E.; et al., A vision for the future of genomics research, Nature 422 (6934) ( 2003) 835.
14. Driscoll, D.A.; Salvin, J.; Sellinger, B.; et al., Prevalence of 22q11 microdeletions in DiGeorge and velocardiofacial syndromes: implications for genetic counseling and prenatal diagnosis, J Med Genet 30 (1993) 813.
15. Edelmann, L.; Hirschhorn, K., Clinical utility of array CGH for the detection of chromosomal imbalances associated with mental retardation and multiple congenital anomalies, Ann NY Acad Sci 1151 (2009) 157.
16. Flake, A.W., Fetal therapy: medical and surgical approaches, In: (Editors: Creasy, R.K.; Resnick, R.) Maternal and fetal medicine,ed 4 ( 1998)Saunders, Philadelphia.
17. Flake, A.W.; Crombleholme, T.M.; Johnson, M.P.; et al., Treatment of severe congenital diaphragmatic hernia by fetal tracheal occlusion: clinical experience with fifteen cases, Am J Obstet Gynecol 183 (2000) 1059.
18. Gardner, R.JM.; Sutherland, G.R., Chromosome abnormalities and genetic counseling,. ed 3 ( 2003)Oxford University Press, New York.
19. Gersen, S.L.; Keagle, M.B., Principles of clinical cytogenetics. ed 2 ( 2005)Humana Press, Totowa, NJ.
20. Hanson, J.W., Human teratogens, In: (Editors: Rimoin, D.L.; Connor, J.M.; Pyeritz, R.E.; et al.) Emery and Rimoin’s principles and practice of medical genetics,ed 5 ( 2006)Churchill Livingstone, New York.
21. Harper, P.S., Practical genetic counseling,. ed 6 ( 2004)Hodder Arnold, Oxford, England.
22. Hay, W.W.; Levin, M.J.; Deterding, R.R.; et al., Current pediatric diagnosis and treatment,. ed 19 ( 2008)McGraw Hill, Norwalk, Conn.
23. In: (Editor: Jones, K.L.) Smith’s recognizable patterns of human malformationed 6 ( 2005)Saunders, Philadelphia.
24. Kamboj, M., Clinical approach to the diagnosis of inborn errors of metabolism, Pediatr Clin North Am 55 (5) ( 2008) 1113; viii.
25. Kay, M.A.; Manno, C.S.; Ragni, M.V., Evidence for gene transfer and expression of factor IX in haemophilia B patients treated with an AAV vector, Nat Genet 24 (2000) 257.
26. Lamb, D.L., Current parent experiences with the Ohio Newborn Screening program since the implementation of the supplemental newborn screening program, dissertation. ( 2004)Case Western Reserve University, Cleveland, Ohio.
27. Levy, H.L.; Albers, S., Genetic screening of newborns, Annu Rev Genom Hum Genet 1 (2000) 139.
28. Marden, A.M.; Smith, D.W.; McDonald, M.N., Congenital anomalies in the newborn infant, including minor variations, J Pediatr 64 (1964) 357.
29. Marwick, C., Two more cell infusions on schedule for gene replacement therapy patient, JAMA 265 (1991) 2311.
30. Matthews, A.L., Known fetal malformations during pregnancy: a human experience of loss, Birth Defects Orig Artic Ser 26 (1990) 168.
31. McCandless, S.E., A primer on expanded newborn screening by tandem mass spectrometry, Prim Care Clin Office Pract 31 (2004) 583.
32. McDonald-McGinn, D.M.; Tonnesen, M.K.; Laufer-Cahana, A.; et al., Phenotype of the 22q11.2 deletion in individuals identified through an affected relative: cast a wide FISHing net!, Genet Med 3 (2001) 23.
33. Michlitsch, J.G.; Walters, M.C., Recent advances in bone marrow transplantation in hemoglobinopathies, Curr Mol Med 8 (7) ( 2008) 675.
34. Mitchell, L.E., Epidemiology of neural tube defects, Am J Med Genet C Semin Med Genet 135 (2005) 88.
35. Morris, C.A., Williams syndrome,, University of Washington. Accessed October 26, 2009, fromwww.geneclinics.org.
36. National Newborn Screening and Genetics Resource Center, website. Accessed December 20, 2008, fromhttp://genes-r-us.uthscsa.edu/resources/newborn/msmstests.htm.
37. Nussbaum, R.L.; McInnes, R.P.; Willard, H.F., Thompson and Thompson genetics in medicine,. ed 7 ( 2007)Saunders, Philadelphia.
38. Online Mendelian Inheritance in Man, OMIM (TM), McKusick-Nathans Institute for Genetic Medicine. Johns Hopkins University (Baltimore, Md) and National Center for Biotechnology Information. ( 2009)National Library of Medicine (Bethesda, Md); Accessed October 26, 2009, fromwww.ncbi.nlm.nih.gov/omim/.
39. Pastores, G.M.; Barnett, N.L., Current and emerging therapies for the lysosomal storage disorders, Expert Opin Emerg Drugs 10 (4) ( 2005) 891.
40. Robinson, A.; Bender, B.; Linden, M.; et al., Sex chromosome aneuploidy: the Denver prospective study, Birth Defects Orig Art Ser 26 (1990) 59.
41. Schuchman, E.H.; Desnick, R.J., Strategies for the treatment of genetic disease, In: (Editors: Rimoin, D.L.; Connor, J.M.; Pyeritz, R.E.) Emery and Rimoin’s principles and practice of medical genetics,ed 5 ( 2006)Churchill Livingstone, New York.
42. Sprintzen, R.J., Velo-cardio-facial syndrome, In: (Editors: Cassidy, S.B.; Allanson, J.E.) Management of genetic syndromes,ed 2 ( 2004)Wiley-Liss, Hoboken, NJ.
43. Sujansky, E.; Smith, A.C.; Prescott, K.E.; et al., Natural history of recombinant syndrome, Am J Med Genet 47 (1993) 512.
44. Thomas, J.A.; Van Hove, J.LK., Inborn errors of metabolism, In: (Editors: Hay, W.W.; Levin, M.J.; Deterding, R.R.; et al.) Current pediatric diagnosis and treatment,ed 19 ( 2008)McGraw Hill, Norwalk, Conn.
45. Tkachuk, D.C.; Pinkel, D.; Kuo, W.L.; et al., Clinical applications of fluorescence in situ hybridization, Genet Anal Tech Appl 7 (1991) 49.
46. Waisbren, S.E.; Albers, S.; Amato, S.; et al., Effect of expanded newborn screening for biochemical genetic disorders on child outcomes and parental stress,, JAMA 290 (2003) 2564.
47. Walker, A.P., Genetic counseling, In: (Editors: Rimoin, D.L.; Connor, J.M.; Pyeritz, R.E.; et al.) Emery and Rimoin’s principles and practice of medical genetics,ed 5 ( 2006)Churchill Livingstone, New York.
48. Walter, J.; Paulsen, M., Imprinting and disease, Semin Cell Dev Biol 14 (2003) 101.
49. Wertelecy, W., Dermatoglyphics, In: (Editors: Stevenson, R.E.; Hall, J.G.) Human malformations and related anomaliesed 3 ( 2006)Oxford University Press, New York.
50. Yagi, H.; Furutani, Y.; Hamada, H.; et al., Role of TBX1 in human del 22q.11 syndrome, Lancet 362 (2003) 1366.
51. Zelante, L.; Gasparini, P.; Estivill, X.; et al., Connexin 26 mutations associated with the most common form of non-syndromic neurosensory autosomal recessive deafness (DFNB1) in Mediterraneans, Hum Mol Genet 6 (1997) 1605.
52. Zythovicz, T.H.; Fitzgerald, E.F.; Marsden, D.; et al., Tandem mass spectrometric analysis for amino, organic and fatty acid disorders in newborn dried blood spots: a two-year summary from the New England Newborn Screening Program, Clin Chem 47 (2001) 1945.
RESOURCE MATERIALS
OMIM:(www.ncbi.nlm.nih.gov/) OMIM, www.ncbi.nlm.nih.gov/;
Online Mendelian Inheritance of Man database is a catalog of genetic disorders and human genes. It is overseen by its originator, Dr. Victor A. McKusick, and maintained by the National Center for Biotechnology Information (NCBI). The database contains information about known inherited disorders, including information about the genes responsible for the disorder, a clinical synopsis, and references.
Gene Reviews and Gene Tests, www.genereviews.org;
Gene Reviews and Gene Tests is an online database developed and maintained by the University of Washington. Gene Reviews lists over 466 genetic disorders that have information authored by experts for a particular disorder. A wealth of information is presented including (1) major clinical features of the disorder, (2) what is known about the genetics and inheritance, and (3) suggestions about a differential diagnosis for the findings. Each entry has an extensive reference list and information about support groups and is updated on a regular basis. The site also includes an international directory of laboratories for genetic testing (both clinical and research) and a listing of international genetics and prenatal diagnosis clinics. Educational materials are available about genetics, genetic disorders, and genetic testing including an illustrated glossary.
Genetic Alliance, www.geneticalliance.org;
The Genetic Alliance is a coalition of hundreds of genetic advocacy organizations health professionals, clinics, hospitals, and companies. Type in a disorder and be transported to that support group’s website for information about the disorder and resources for people with the disorder.
Genetics Home Reference, http://www.ghr.nlm.nih.gov/;
The Genetics Home Reference is the National Library of Medicine’s website for consumer information about genetic conditions and the genes or chromosomes responsible for those conditions. Summaries are provided about hundreds of genetic disorders. Each summary also contains information and web links to other resource materials.
[/level-membership-for-neonatal-and-perinatal-medicine-category][not-level-membership-for-neonatal-and-perinatal-medicine-category]
27. Genetic Disorders, Malformations, and Inborn Errors of Metabolism*
Anne Matthews and Nathaniel H. Robin
Aneonate born with a malformation, a genetic syndrome, or an acute metabolic disorder presents a management challenge for the neonatal intensive care unit (NICU) staff. If these conditions are not suspected and diagnosed in a critically ill neonate, an appropriate course of action might not be taken. Thus a specific diagnosis becomes imperative. An accurate diagnosis provides the staff with information about the cause of the condition, points the way toward appropriate treatment, and indicates the prognosis so that the most appropriate care of the infant can be initiated. Moreover, the broader issues of providing supportive care and counseling for the affected infant’s family can be addressed.
Genetic evaluation is a complex process that requires expertise in differentiating normal variations from abnormal findings and knowledge of the principles of embryology and dysmorphology to provide an accurate diagnosis. Skills in obtaining detailed information of prenatal and family histories may be equally important.
The field of genomics and genetic medicine has witnessed an explosion of new knowledge, much of which has been generated by the efforts of the Human Genome Project. 13 Advances in understanding of the genetic basis of development and function, as well as the interaction of genes and the environment, continue to provide new insights into human health.
This chapter presents a concise overview of the major categories of genetic disorders and the appropriate techniques to establish specific diagnoses. For an excellent review and detailed explanation of concepts, terminology, and specific genetic mechanisms, refer to Thompson and Thompson Genetics in Medicine.37 See Box 27-1 for a comprehensive list of terms.
BOX 27-1
Aerocentric chromosome A chromosome with the centromere near the end of the chromosome.
Allele One of a pair or series of alternate forms of a gene at the same locus.
Aneuploid Any chromosome number that is not an exact multiple of the haploid set.
Autosome A chromosome that is not a sex chromosome.
Centromere The primary constriction of a chromosome in which the long and the short arms meet.
Chromatid After replication of a chromosome, two subunits attached by the centromere can be seen; each is called a chromatid, and after separation, each becomes a chromosome of a daughter cell.
Chromosomes The microscopic structures in the cell nucleus composed of DNA and proteins that contain the genes.
Congenital Present at birth.
Dermatoglyphics The dermal ridge patterns on the digits, palms, and soles.
Diploid Two copies of all chromosomes; the number of chromosomes normally present in somatic cells. In humans, this is 46 and is sometimes symbolized as 2N.
Dominant A gene (allele) that is expressed clinically in the heterozygous state. In a dominant disorder, the mutant allele overshadows the normal allele.
Dysmorphic Morphologic abnormality, often a minor physical finding that may or may not have any cosmetic or functional significance and is present in less than 4% of the newborn population.
Fluorescence in situ hybridization (FISH) Molecular cytogenetic method for detection of microdeletions of chromosomes.
Gamete Mature reproductive cell, the egg or the sperm, containing the haploid number of chromosomes.
Gene The functional unit of heredity.
Genotype A person’s genetic constitution.
Haploid One copy of all chromosomes; the number of chromosomes present in the gamete; in humans this is 23 and can be symbolized as N.
Hemizygous The condition in which only one copy of a gene is normally present, so its effect is expressed because there is no counterpart gene present (e.g., the genes on the X or Y chromosome of the male).
Heterozygote An individual who has two different alleles at a given locus of two homologous chromosomes.
Homologous chromosomes Members of the same chromosome pair; normally they have the same number and arrangement of genes.
Homozygote An individual who has two identical alleles at a given locus of two homologous chromosomes.
Karyotype The standard pictorial arrangement of chromosome pairs, numbered according to centromere position and length.
Locus The position or place that a gene occupies on a chromosome.
Malformation A primary structural defect that results from a localized error of morphogenesis; abnormal development.
Metacentric chromosome Chromosome with the centromere in the center of the chromosome.
Monosomy Absence of one chromosome of one pair.
Mosaicism Presence in the same individual of two or more different chromosomal constitutions.
Mutation A heritable alteration in the genetic material.
Nondisjunction Failure of two homologous chromosomes to separate equally during cell division into two daughter cells, resulting in abnormal chromosome numbers in gametes or somatic cells.
Phenotype The observable expression of traits either physically or biochemically.
Recessive A gene (allele) that is expressed clinically in the homozygous state. In a recessive disorder, both genes at a given locus must be abnormal to manifest the disorder.
Sex chromosomes The X and Y chromosomes.
Syndrome Recognizable pattern of multiple malformations that occur together and have the same cause.
Transcription The process by which complementary messenger RNA is synthesized from a DNA template.
Translation The process whereby the amino acids in a given polypeptide are synthesized from the messenger RNA template.
Translocation Transfer of all or part of a chromosome to another location (i.e., on the same or another chromosome) after chromosome breakage.
Trisomy The presence of three homologous chromosomes rather than the normal two.
X-linked A gene located on an X chromosome.
Zygote A fertilized egg that develops into an embryo.
GENETIC PRINCIPLES
Genes
A gene is a segment of a deoxyribonucleic acid (DNA) molecule that codes for the synthesis of a single polypeptide and contains the hereditary information needed for development or function. DNA, which allows the storing, duplicating, and processing of hereditary information, consists of two long strands twisted around each other to form a double helix. Each strand of DNA is composed of four nucleotides: guanine (G), adenine (A), thymine (T), and cytosine (C). The specific order of the nucleotides determines the precise information that will be encoded at that site. Genes can (1) regulate other genes by turning them “on” or “off,” (2) specify the exact structure of proteins, which then control the activities of the cells, and (3) specify ribonucleic acid (RNA), which is necessary for protein synthesis.
Chromosomes
Genes are packed in linear order on chromosomes. Chromosomes are found in the nuclei of cells. In humans, normal somatic cells contain 46 chromosomes (diploid number), of which 44 are termed autosomes and 2 are sex chromosomes. Females have two X chromosomes (XX), and males have an X and a Y chromosome (XY). Gametes—eggs or sperm—contain 23 chromosomes (haploid number). In the zygote and somatic cells, chromosomes are paired (homologs). In each pair, one homolog is maternal and the other is paternal in origin. Each chromosomal pair has unique morphologic characteristics that allow it to be distinguished from other chromosomes, such as size, position of the centromere, and the unique banding pattern that is demonstrated by special staining techniques (Figure 27-1). 19 To pass on the genetic information to daughter cells, the chromosomes must replicate and then divide correctly. Somatic cells undergo mitosis, in which cells replicate and then divide chromosomal material into two genetically identical daughter cells with 46 chromosomes each. In gametes, the process is known as meiosis, which is different from mitotic division in that daughter cells contain the haploid number of chromosomes (23) and crossing over or recombination between two homologs occurs, thus facilitating genetic variation in offspring. 37
|
|
| FIGURE 27-1
(Courtesy Dr. Loris McGavran, Ph.D., Cytogenics Laboratory at the University of Colorado at Denver and Health Sciences Center, Denver.)
|
An individual’s chromosome constitution can be determined by examining dividing body cells under certain laboratory conditions from any accessible tissue such as blood lymphocytes or skin fibroblasts. The resulting karyotype (see Figure 27-1), or pictorial arrangement, demonstrates the number and structure of that individual’s chromosomes.
ETIOLOGY
Malformations and genetic disorders caused wholly or partly by genetic factors can be categorized into four major areas: (1) chromosomal disorders caused by numeric or structural abnormalities of chromosomes; (2) single-gene or mendelian disorders, which are secondary to single-gene mutations; (3) complex or multifactorial disorders resulting from interaction of genes and environmental influences; and (4) abnormalities caused by environmental exposures of the fetus during development.
More recently, better understanding of molecular processes has allowed the identification of additional genetic mechanisms contributing to genetic disorders: germline mosaicism, genomic imprinting, and uniparental disomy.
Chromosomal Disorders
Chromosomal abnormalities are relatively common. Approximately 0.5% to 0.7% of all live newborns have a chromosomal abnormality, and 4% to 7% of perinatal deaths result from a chromosomal abnormality. Moreover, it is estimated that at least 50% of all recognized first-trimester miscarriages are caused by a chromosomal aberration. 18 Current cytogenetic techniques, such as high-resolution banding, fluorescence in situ hybridization (FISH), and microarray-based comparative genomic hybridization (array-CGH), have increased the detection rate of chromosomal aberrations. Submicroscopic deletions, duplications, or other abnormal rearrangements of chromosome material that may not have been identified a few years previously are now being detected in children with congenital malformations or mental retardation.
Chromosomal aberrations should be suspected in any of the following situations:
• Small for gestational age for weight, length, or head circumference
• Presence of one or more congenital malformations
• Presence of dysmorphic features
• Neurologic or neuromuscular dysfunction
• Family history of multiple miscarriages or siblings with mental retardation or birth defects along with one or more of the above
Chromosomal abnormalities can be classified into two major categories: (1) abnormalities of chromosome number (aneuploidy), in which there is an extra or missing chromosome; and (2) abnormalities of chromosome structure that result in the loss or duplication of part of the chromosomal material. Abnormalities of autosomes usually have more significant deleterious effects on the development of the infant than those seen with sex chromosome abnormalities.
ABNORMALITIES OF CHROMOSOME NUMBER
Numeric chromosomal abnormalities occur as a result of nondisjunction in which aberrant segregation leads to loss or gain of one or more chromosomes. Nondisjunction can occur during either meiosis or mitosis, resulting in an abnormal gamete (egg or sperm) or abnormal somatic cell, respectively (Figure 27-2). Fertilization of an aneuploid gamete by a normal gamete produces a zygote with an extra chromosome (trisomy) or missing chromosome (monosomy). Aneuploidy in somatic cells results in chromosomal mosaicism (i.e., the presence of some cells with the normal number of chromosomes and other cells with an abnormal number of chromosomes) (Figure 27-3). Although nondisjunction may affect any chromosomal pair, the most commonly recognized trisomies in liveborns are trisomy 21 (Down syndrome), trisomy 18 (Edward syndrome), and trisomy 13 (Patau syndrome). On the other hand, trisomy 16 has been found exclusively in spontaneous abortions. 18 The most common monosomy is 45,X, Turner syndrome. As a rule, numeric chromosomal abnormalities are associated with intrauterine growth restriction (IUGR), dysmorphic features, malformations, and mental retardation. Physical abnormalities may be milder or absent in the newborn with mosaicism.
ABNORMALITIES OF CHROMOSOME STRUCTURE
Structural abnormalities have been described in all chromosomes. These include deletions, translocations, duplications, and inversions (Figure 27-4). A deletion is a loss of chromosome material and results in partial monosomy for the chromosome involved. Loss of material from the end of a chromosome is known as a terminal deletion, as seen in 5p−, or cri du chat syndrome. An interstitial deletion involves a loss of chromosomal material that does not include the ends of the chromosome. A terminal deletion of both arms of a chromosome may result in reattachment of the remaining arms, leading to a formation of a ring chromosome. The presence of additional chromosome material results in duplication or partial trisomy of a chromosome. Translocation is the detachment of a chromosome segment from its normal location and its attachment to another chromosome. The translocation is balanced if the cell contains two complete copies of all chromosomal material, although in different order. In an unbalanced translocation, the rearrangement results in partial trisomy or monosomy.
|
|
| FIGURE 27-4
(From Hathaway WE, Groothius J, Hay W, editors: Current pediatric diagnosis and treatment, ed 10, Norwalk, Conn, 1991, Appleton & Lange.)
|
Translocations can be reciprocal or robertsonian. A reciprocal translocation involves exchange of segments between two chromosomes (e.g., part of the short arm of chromosome 4 trades a place with a part of chromosome 10). Robertsonian translocations involve two acrocentric chromosomes fused at their centromeres. The most common robertsonian translocation is formed between chromosomes 14 and 21. 22
Inversions are the result of a double break in a single chromosome and reinsertion of the chromosomal material that has been inverted. Inversions are either pericentric (including the centromere) or paracentric (without the centromere). The most common inversion is a small pericentric inversion of chromosome 9, which is considered to be a normal variant, found in approximately 1% of the general population. 37 All other inversions may produce gametes that result in an individual with an unbalanced rearrangement (i.e., having both a duplication and a deletion of some chromosome material, such as that seen in recombinant 8 syndrome).
MICRODELETIONS AND SYNDROMES
At times, structural chromosomal abnormalities are submicroscopic and therefore cannot be detected by conventional cytogenetic techniques. FISH is a molecular cytogenetic method that facilitates the detection of microdeletions. FISH uses segments of fluorescently labeled DNA called probes, constructed so that each probe can attach only to a specific segment of a chromosome, which then will be fluorescent during a microscopic visualization. In the case of a deletion of that chromosome segment, the probe cannot attach to the chromosome; thus the fluorescent segment is missing from the deleted segment of that chromosome. 45
The most recent advance being used to detect very small submicroscopic deletions and duplications is comparative genomic hybridization (array-CGH). 15 This technology blends molecular techniques with cytogenetics and allows the genome to be scanned at a higher resolution than conventional techniques. DNA from a patient sample and DNA from a control sample are differentially labelled, mixed in equal proportions, and hybridized to DNA substrates fixed on an array platform (i.e., bacterial artificial chromosomes [BACs] or oligonucleotides [short segments of DNA usually 8-50 base pairs]). This technique can measure the difference between two different DNA samples in copy number (dosage) of a particular segment of DNA. Thus microscopic gains and losses from a patient sample can be quantified. 15
Microdeletions result in phenotypic abnormalities. A number of well-recognized microdeletion syndromes may be suspected in the NICU. Prader-Willi syndrome, caused by an interstitial deletion of chromosome 15 (q11q13), usually manifests in a newborn as severe hypotonia, feeding difficulties, and micropenis or hypoplastic labia. 9Williams syndrome is caused by an interstitial deletion or mutation of the elastin gene (ELN) on the long arm of chromosome 7 (7q11). 35 The condition is often first seen in an affected newborn in the postterm period; the infant is small for family size. There may be a congenital heart defect, in particular, supravalvular aortic stenosis or peripheral pulmonic stenosis; hypotonia; failure to thrive with gastroesophageal reflux; poor suck and swallow; and vomiting and irritability or colic. Infantile hypercalcemia is seen in approximately 20% of these infants. Subtle dysmorphic facial features may be noted in the newborn. 35
One of the most commonly seen microdeletion syndromes is velocardiofacial syndrome (VCFS), which is characterized by cleft palate or velopharyngeal insufficiency, hypernasal speech, learning disabilities, conotruncal heart defects, and characteristic facies. VCFS actually represents one of a spectrum of clinical disorders all known to be caused by a deletion in chromosome 22q11 (del22q11). These include DiGeorge syndrome (DGS) (conotruncal heart defect, hypocalcemia, and thymic hypoplasia) and conotruncal anomaly face syndrome (CTAF) (conotruncal heart defects and typical facies). In addition, del22q11 has been found in 11% to 16% of cases of nonsyndromic congenital conotruncal heart disease and has been reported to present as apparently isolated neonatal hypocalcemia or learning problems. 14 Overall, del22q11 has an estimated incidence of 1 in 2000 to 4000 newborns. The availability of molecular cytogenetic testing by FISH has led to appreciation of both the high incidence of the del22q11, as well as the increasing variety of clinical presentations that can be seen even within a single family. 32
In the newborn period, the characteristic facial features are seldom obvious. However, most affected individuals manifest some of these findings by early childhood. Most prominent is the nose, which is described as long, with a “built up nasal bridge, squared off nasal root, and bulbous nasal tip.”42 The eyes appear narrow and slitlike, the mala (cheeks) are flat, and the jaw is recessed. The ears usually are small and in some way abnormally formed. There may be an overt cleft of the secondary palate, a bifurcated uvula, a subtle submucosal cleft, or cleft lip with or without cleft palate. Other nonstructural palatal abnormalities can be seen, most commonly velopharyngeal insufficiency. In an older child or adult, this presents as hypernasal speech; in a newborn, one sees excessive nasal regurgitation. Congenital heart disease (CHD) is seen in 35% of del22q11 patients. The type of CHD is fairly specific and includes those lesions classified as “conotruncal heart defects” (truncus arteriosus, interrupted aortic arch, tetralogy of Fallot, left-sided aortic arch, vascular rings, and some types of ventricular septal defects [VSDs]). Perhaps the most consistent finding in patients of all age-groups is long, thin fingers and toes. Additional nonspecific findings include abundant scalp hair; hypospadias; renal abnormalities that can include renal agenesis; tortuous retinal vessels; ectopic/aberrant/unilateral absence of carotid and vertebral artery; microcephaly; and microdontia (there are 166 different findings to date). 42
Developmental delay, learning disabilities, or mental retardation is common and quite variable. Behavioral and psychiatric problems are common but underappreciated findings in VCFS. These individuals have a characteristic personality, marked by a flattened affect and abnormal social interaction, ranging from being intermittently withdrawn to socially precocious. A host of other psychiatric diagnoses have been seen in patients with VCFS.
Both DiGeorge syndrome and VCFS have been recognized as being caused by deletions in 22q11. More than 95% of cases of DGS and VCFS are deleted. In a small number of patients with DGS, point mutations in TBX1 have been found when no deletion was identified. 50 There are a few cases of VCFS in which no 22q11 deletion or TBX1 mutation has been found. Thus obtaining family histories and examining parents for subtle features of the syndrome are important. In many cases, the infant has inherited the abnormality from a parent.
CLINICAL EXAMPLES OF CHROMOSOMAL ABNORMALITIES
Down Syndrome
Down syndrome has an incidence of approximately 1 in 600 live births. Approximately 95% of cases are caused by nondisjunction involving chromosome 21, 4% are caused by a translocation, and 1% are mosaic. Down syndrome may manifest with marked hypotonia; a number of major malformations, most commonly congenital heart defects, duodenal atresia, and tracheoesophageal fistula; and a characteristic pattern of dysmorphic features. The classic phenotype seen in Down syndrome includes a flattened occiput, midfacial hypoplasia, depressed nasal bridge, upward-slanting palpebral fissures, epicanthic folds, grayish speckling of the iris (Brushfield spots), micrognathia, excess nuchal skin, single palmar creases (simian creases), single flexion creases and in-curving of the fifth fingers (clinodactyly), and increased distance between the first and second toes (Figure 27-5).
|
|
| FIGURE 27-5
( A from Cohen MM: The child with multiple birth defects, ed 2, New York, 1997, Raven Press. B courtesy Dr. Eva Sujansky, Genetic Services at The Children’s Hospital, Denver, Colo.)
|
In full-term infants with the classic phenotype of Down syndrome, the clinical diagnosis is not difficult. However, it is imperative that cytogenetic studies be done to confirm the diagnosis and to differentiate a nondisjunctional trisomy from a translocation. This distinction has important implications for recurrence risks (see discussion in “Prevention” section). In premature infants, the classic facial phenotype is frequently missing, making clinical diagnosis more difficult. The presence of an atrioventricular (AV) canal or duodenal atresia with minor malformations, such as abnormal dermatoglyphics, should alert the clinician to the possibility of Down syndrome.
Trisomy 18
Trisomy 18 has an incidence of 1 in 6000 live births. The major phenotypic features include prenatal growth restriction, complex cardiac malformations, abnormal muscle tone, microcephaly, prominent occiput, short sternum, low-set and malformed ears, corneal opacities, micrognathia, peculiar hand posturing with the second and fifth digits overlapping the third and fourth, hypoplasia of fingernails, abnormal dermatoglyphics, prominent calcanei, and deep plantar furrows between the first and second toes (Figure 27-6). The prognosis is poor, and the majority of infants with trisomy 18 die within the first few months of life. Infants who have survived into childhood are profoundly retarded.
|
|
| FIGURE 27-6
(From Paerregaard P, Mikkelsen M, Froland A, et al: Trisomy no. 17-18: report of two cases, Acta Pathol Microbiol Scand 67:479, 1966.)
|
Trisomy 13
Trisomy 13 is seen in approximately 1 in 15,000 live births. Phenotypic features include prenatal and postnatal growth restriction, microcephaly, sloping forehead, coloboma of the iris, microphthalmia or anophthalmia, low-set or malformed ears, cleft lip and palate, postaxial polydactyly, and abnormal palmar creases and dermatoglyphics (Figure 27-7). Internal abnormalities may include a number of central nervous system (CNS) malformations, such as holoprosencephaly, cardiac malformations, omphalocele, renal malformations, and urogenital abnormalities such as cryptorchidism in males and uterine malformations in females. The prognosis is extremely poor for these infants, with most dying within the first few months of life.
|
|
| FIGURE 27-7
(From Hathaway WE, Groothuis J, Hay W, editors: Current pediatric diagnoses and treatment, ed 10, Norwalk, Conn, 1991, Appleton & Lange.)
|
Turner Syndrome
The only monosomy to be seen in live births is that of Turner syndrome—females with a 45,X karyotype. In addition, it is the only numeric abnormality of the sex chromosome that may be identifiable at birth. Turner syndrome has an incidence of 1 in 5000 female births. 40 Clinical features that may be evident in the newborn period are a short, webbed neck or redundant skin on the back of the neck and marked lymphedema of the dorsum of the hands and feet (Figure 27-8). Congenital heart defects are seen in approximately half of the patients, with 30% having a coarctation of the aorta. Renal anomalies may also be present. 23 Prognosis is usually excellent but depends on the presence and severity of the congenital heart defect. Intelligence is normal; however, some females with Turner syndrome have been noted to have problems with spatial perception or fine motor abilities. 40
|
|
| FIGURE 27-8
(From Knuppel R, Drukker JD, editors: High risk pregnancy: a team approach, Philadelphia, 1988, Saunders.)
|
Cri du Chat
Cri du chat, or “cat cry” syndrome, is the result of loss of the terminal end of the short arm of chromosome 5 (5p−). The name of the syndrome reflects the unusual catlike, weak cry these infants have in the neonatal period. These infants are usually small for gestational age, hypotonic, and microcephalic and may have ocular hypertelorism, epicanthic folds, downward slant of the palpebral fissures, low-set ears, and micrognathia. They are significantly mentally retarded.
San Luis Valley Syndrome
Recombinant 8 or the San Luis Valley syndrome, named for the area in which many of these individuals were first identified, is an example of an unbalanced pericentric inversion with both a duplication and a deletion of chromosome 8 material. The pericentric inversion of chromosome 8 found in a parent and other relatives of a child with recombinant 8 syndrome has no phenotypic consequence because it is a balanced rearrangement. However, a carrier is at risk for producing unbalanced gametes during meiosis. In recombinant 8 syndrome, there is a deletion of chromosomal material of the short arm of chromosome 8 and a duplication of chromosome material of the long arm of 8. The phenotype is characterized by unusual facial features, including a wide face, depressed nasal bridge, hypertelorism, down-slanting palpebral fissures, upturned nose, long philtrum, low-set and malformed ears, cleft lip or cleft palate, congenital heart disease, and renal abnormalities. 43
PREVENTION
The identification of chromosomal abnormalities in the newborn is important, not only for management issues about the infant but also because of the recurrence risks the abnormality carries for the family. In general, numeric chromosomal abnormalities carry low recurrence risks (approximately 1% to 2%). 18 In the presence of structural abnormalities, recurrence risks depend on whether one of the parents carries a balanced rearrangement. If parental chromosomes are normal, the recurrence risk is minimal. However, if a parent carries a balanced chromosomal rearrangement, the recurrence risk is significantly increased. The exact risk figure varies with the nature of the specific chromosomal rearrangement and, in some cases, the sex of the carrier parent. In either situation, prenatal diagnosis for chromosome analysis is available for parents and families concerned about recurrence risk.
Single-Gene Disorders
McKusick’s online catalog of mendelian inherited disorders currently lists more than 19,000 entries with approximately 6000 single-gene disorders with known patterns of inheritance. 38 Many of these disorders are singularly rare; however, collectively, they affect about 1% of the population. Single-gene disorders are the result of either a single or double dose of an abnormal gene. Single-gene disorders are classified as autosomal dominant, autosomal recessive, X-linked dominant, and X-linked recessive. Humans have two copies of each gene located at identical places (gene loci) on homologous chromosomes. In a single-gene disorder, an abnormal or mutated allele (an alternate form of a gene) is found on one or both members of a pair of chromosomes. 37 Individuals with identical alleles at a particular locus are homozygous for the gene. Individuals with different alleles are heterozygous for the gene. Because males have only one X chromosome and most genes located on the Y chromosome do not correspond to those located on the X, males are hemizygous for the genes on the X chromosome. Abnormal genes located on one of the 44 autosomes are the cause of autosomal disorders: disease-causing genes located on the X chromosome are the cause of X-linked disorders. Disorders are dominant when the phenotype is expressed in the presence of only one copy of the mutated gene. In recessive disorders, the phenotype is expressed only when both chromosomes carry the mutated gene.
AUTOSOMAL DOMINANT DISORDERS
Autosomal dominant disorders are ones in which the disorder is expressed in the heterozygous state. Major characteristics include the following: (1) multiple generations are affected (i.e., an infant would have an affected parent); (2) both males and females are affected, and both sexes can transmit the disorder to their offspring (i.e., male-to-male transmission can occur); (3) there is a 50% risk for each offspring to inherit the gene from an affected parent; and (4) individuals who do not have the gene cannot transmit the disorder to their offspring.
A negative family history does not rule out the presence of an autosomal dominant disorder.
Buy Membership for Neonatal and Perinatal Medicine Category to continue reading. Learn more here
[/not-level-membership-for-neonatal-and-perinatal-medicine-category]
