[level-membership-for-neurology-category]
Chapter 71 Genetic and Metabolic Disorders of the White Matter
Introduction
Over the past two decades, an increasing number of novel heritable disorders affecting the white matter of the brain, or leukodystrophies, have been described, often with identification of a causative gene (Table 71-1). Pathognomonic magnetic resonance imaging (MRI) patterns (see Figure 71-1) or clinical characteristics permit identification in a number of these disorders, and should guide the clinician’s molecular diagnosis for many conditions (see Figures 71-3, 71-5, and 71-6 for MRI features). Challenges remain, however, for the neurologist, geneticist, or primary-care provider evaluating patients with suspected genetic or metabolic disorders of the white matter. In many settings, over half of subjects with suspected heritable white-matter disease do not receive a diagnosis, and therefore the focus of this chapter is on assisting the treating neurologist in the diagnosis of inherited disorders of the white matter.
Table 71-1 Molecular Causes of Leukodystrophies and Leukoencephalopathies (a Nonexhaustive List)
| Disorder | Gene |
|---|---|
| Acyl-coenzyme A (acyl-CoA) oxidase deficiency | ACOX |
| Adenylosuccinate lyase deficiency | ADSL |
| Aicardi–Goutières syndrome (AGS) | TREX1, SMHD1, RNAseH2A, B, C |
| Alexander’s disease (AxD)* | GFAP |
| Autosomal-dominant leukodystrophy with autonomic dysfunction* | Duplication LaminB1 |
| Canavan’s disease | ASPA |
| Cerebrotendinous xanthomatosis (CTX) | CYP27A1 |
| D-bifunctional protein deficiency | HSD17B4 |
| eIF2B-related disorder/VWM disease* | EIF2B1-5 |
| Glutaric aciduria type II/multiple acylCoA dehydrogenase deficiency (MADD) | ETFA, ETFB, ETFDH |
| 3-hydroxy-3-methylglutaryl coenzyme A (HMG CoA) reductase deficiency | HMGCR |
| Hypomyelination with congenital cataracts (HCC) * | FAM126A |
| Infantile sialic acid storage disease (ISSD) | SLC17A5 |
| Krabbe’s disease | GALC |
| Leukoencephalopathy with brainstem and spinal cord involvement and elevated lactate (LBSL)* | DARS2 |
| Lowe’s syndrome (oculocerebrorenal syndrome of Lowe, OCRL) | OCRL |
| Megalencephalic leukoencephalopathy with subcortical cysts (MLC)* | MLC1 |
| Metachromatic leukodystrophy (MLD) | ARSA |
| Mitochondrial neurogastrointestinal encephalopathy (MNGIE) | TYMP |
| Mucolipidosis IV | MCOLN1 |
| Oculodental digital dysplasia (ODDD)* | GJA1 |
| Pelizaeus–Merzbacher disease (PMD)* | PLP1 |
| Pelizaeus–Merzbacher-like disease (PMLD)* | GJC2 |
| Peroxisomal thiolase deficiency | ACAA |
| Polymerase gamma 1 (POLG1)* | POLG1 |
| Polyglucosan body disease (PGBD) | GBE1 |
| RNAse T2-deficient leukoencephalopathy* | RNASET2 |
| Sjögren–Larsson syndrome | ALDH3A2 |
| X-linked adrenoleukodystrophy (XALD) | ABCD1 |
| 18q minus syndrome | Deletion of short arm chromosome 18 |
VWM, vanishing white matter
* No specific biochemical marker clinically available, and molecular testing is primary diagnostic tool.
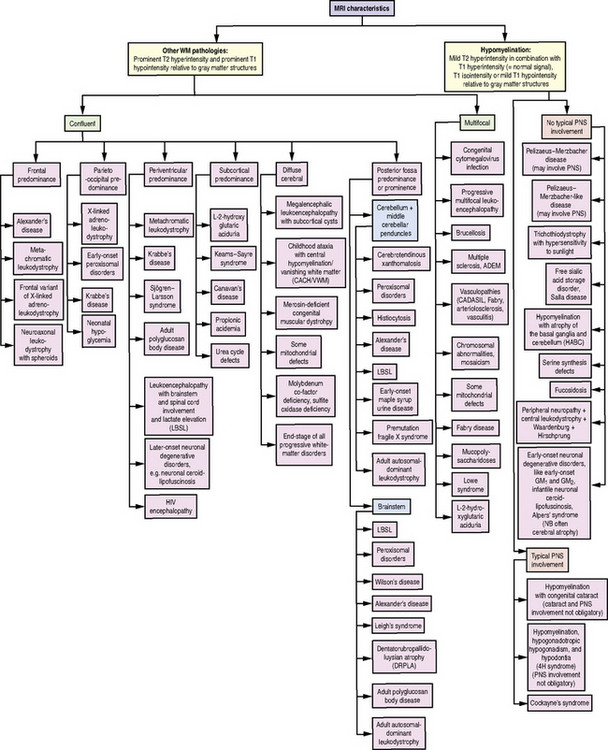
Fig. 71-1 Diagnostic algorithm for use in patients with abnormal myelination by MRI.
HIV, human immunodeficiency virus; PNS, peripheral nervous system; WM, white matter.
(Used with permission from Schiffmann R, van der Knaap MS. Invited article: an MRI-based approach to the diagnosis of white matter disorders. Neurology 2009;72:750–759.)
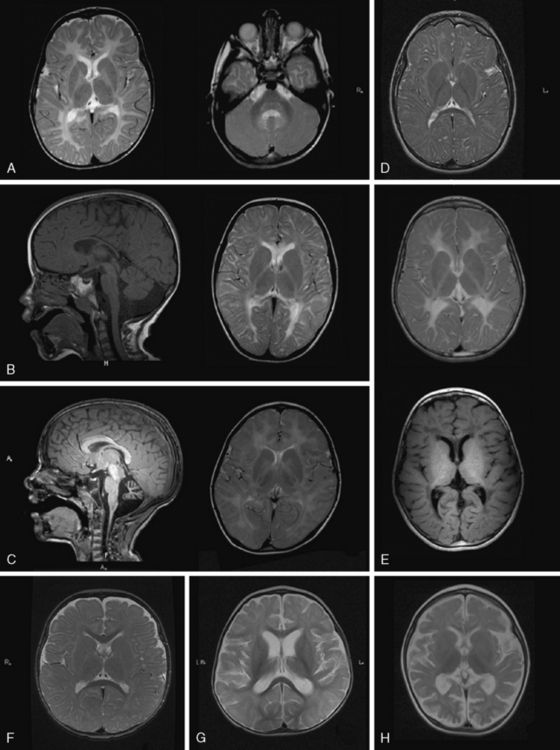
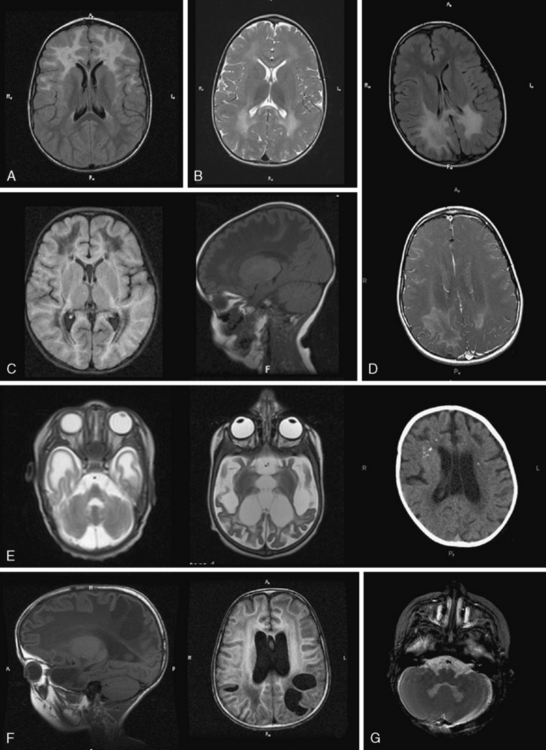
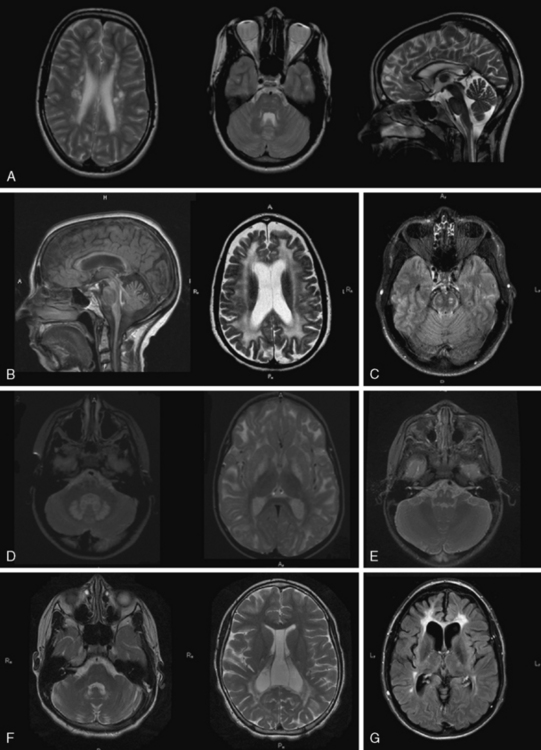
Fig. 71-6 Dysmyelinating and demyelinating leukodystrophies and pathognomonic MRI features, part II.
Given the heterogeneity and limited specificity in clinical findings, MRI pattern recognition is the most useful tool for evaluating suspected leukodystrophy patients [Schiffmann and van der Knaap, 2009] (Figure 71-1). MRIs should be reviewed comprehensively, with attention to changes over time, expected myelin development for the patient’s age, and characteristics of T1- and T2-weighted signal abnormalities. Broadly, there are two groups of leukodystrophies. Hypomyelinating leukodystrophies show increased white-matter signal on T2 relative to age, and often isointense or hyperintense white-matter signal on T1. Conversely, demyelinating leukodystrophies show increased white-matter signal on T2 relative to age, and hypointense white-matter signal on T1. Additional radiologic features should be examined, such as white-matter vacuolization or cysts, best seen on fluid-attenuated inversion recovery (FLAIR) or similar imaging paradigms; involvement of the basal ganglia, brainstem, cerebellum, or spinal cord; and abnormalities of cortical gray matter. Finally, findings consistent with calcifications should be noted, and if clinically indicated, a computed tomography (CT) scan or calcium-sensitive MRI sequences should be acquired to exclude the presence of calcifications that might be missed on standard MRI.
Hypomyelinating White-Matter Disorders
White-matter disorders with hypomyelination are relatively frequent; hypomyelinating leukodystrophies comprise 20 percent of leukodystrophies. They are characterized by a significant and permanent deficit of myelin [Schiffmann and van der Knaap, 2009]. Even with thorough genetic investigation, at least half of these hypomyelinating disorders lack definitive diagnosis, prognostic information, and potential for prenatal diagnosis. While most of these disorders show autosomal-recessive inheritance, both X-linked inheritance and de novo mutations are possible. Clinical manifestations common to hypomyelinating disorders are ataxia, spasticity, and nystagmus. Other non-neurological features can be valuable in diagnosis, such as hypodontia in 4H syndrome and cataracts in hypomyelination with congenital cataracts.
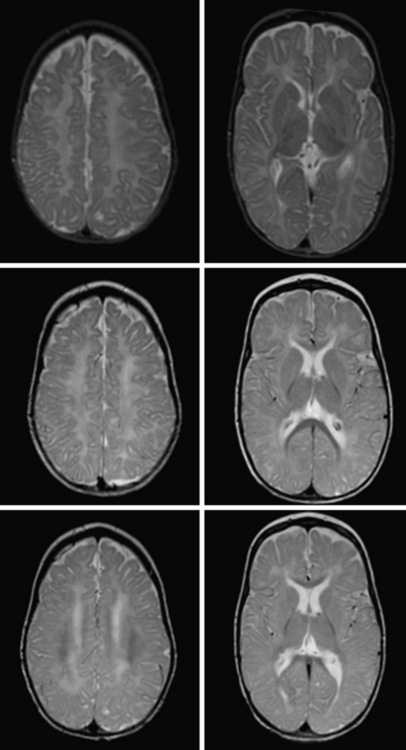
The diagnosis of hypomyelination may be made if two MRIs at least 6 months apart after the age of 12 months show little or no myelin development. Images in hypomyelinating leukodystrophies demonstrate diffusely hyperintense signal of the supratentorial white matter in T2-weighted images, and iso- or hyperintense white-matter signal on T1-weighted images. Certain areas, such as the posterior internal capsule, may have more normal-appearing myelin signal. Myelin deposition in infratentorial structures is usually higher. A definitive diagnosis of hypomyelination is not possible in young infants, as myelination is incomplete. If no myelin deposition is visible on the first MRI of a child older than 24 months, however, hypomyelination is highly probable [Schiffmann and van der Knaap, 2009].
Delayed myelination is sometimes misdiagnosed as hypomyelination. In contrast to hypomyelination, in delayed myelination, myelination progresses on serial MRI images to near-normal myelin development (Figure 71-2). Gray-matter disorders with early onset often show hypomyelination, presumably a result of defective axonal function. Features such as early atrophy and signal changes of cortex and basal ganglia may help distinguish between primary hypomyelination and hypomyelination secondary to neuronal dysfunction (see Figure 71-3F, G, and H below).
In cases of primary hypomyelination, the disorders described below should be considered.
Pelizaeus–Merzbacher Disease
Pelizaeus–Merzbacher disease (PMD, OMIM 312080) is the prototypic hypomyelinating disorder, and is caused by alterations in the proteolipid 1 (PLP1) gene [Hudson et al., 1989; Trofatter et al., 1989]. Located on Xq22.2, this gene encodes the PLP1 protein, which constitutes roughly half of all myelin protein. The most common abnormality is a duplication of the entire gene, found in 60–70 percent of PMD cases and associated with the classic form of the disease [Ellis and Malcolm, 1994; Sistermans et al., 1998]. The lack of common breakpoints in these duplications results in varying sizes of the duplicated area between patients. Missense mutations account for 10–15 percent of cases. Several missense mutations affecting splicing or putative regulation of expression have also been described. Deletions are also seen in a smaller number of cases. Triplications are present in 1–2 percent of cases; higher copy numbers have been described in one child. More complex chromosomal rearrangements involving PLP1 or its promoter region have been described in individual cases [Muncke et al., 2004]. Deletions or null mutations are rare. Roughly 15 percent of cases fulfilling diagnostic criteria for PMD do not possess identifiable PLP1 mutations, suggesting the possibility of mutations in regulatory regions beyond genetic assays.
PMD is allelic with a relatively mild disorder, X-linked spastic paraplegia type 2 (SPG2), also caused by PLP1 mutations [Saugier-Veber et al., 1994]. In its pure form, the sole symptom associated with SPG2 is slowly increasing spasticity, more of the legs than of the arms, which begins during childhood or adolescence. In the complicated forms, additional symptoms, such as nystagmus, ataxia, dysarthria, and mild cognitive impairment, are present. Life expectancy in this form is normal. Peripheral neuropathy is another possible finding with certain PLP1 mutations. Altogether, alterations of PLP1 mutations give rise to a spectrum of disorders, ranging from the very severe connatal phenotype to mild spastic paraplegia with adolescent onset.
Genotype–phenotype correlations have been established for most PLP1 alterations, although clinical heterogeneity among patients with common genetic alterations makes definitive characterization difficult. Symptoms do not correlate with duplication size, but high copy number appears to predict increased clinical severity. Severe epilepsy has been reported in children with triplications, an otherwise uncommon feature of PMD [Wolf et al., 2005]. Point mutations are associated with the full spectrum of PLP1-connected disorders [Cailloux et al., 2000]. Demyelinating neuropathy is associated with either null mutations or mutations in PLP-specific regions. Patients with functional null mutations show relatively mild clinical course; they usually achieve independent, albeit clumsy, ambulation, and show mild cognitive impairment and demyelinating neuropathy. As spasticity increases after the first decade, patients become wheelchair-bound and develop pseudobulbar palsy. Histopathologic investigations show evidence of length-dependent axonal degeneration in these patients and in corresponding mouse models [Garbern et al., 2002].
PLP1 is a highly conserved, hydrophobic membrane protein with four transmembrane domains and a large cytosolic loop between the second and third transmembrane domains. The N- and C-termini are located in the cytoplasm. Different splicing of PLP1 yields a second, smaller isoform, DM20. Both isoforms are primarily expressed within the CNS. While the function of PLP1 and DM20 remains poorly understood, mutations that leave DM20 intact are associated with relatively mild phenotype. Mutations associated with severe phenotype likely cause protein misfolding, which activates compensatory oligodendrocytic responses that ultimately cause oligodendrocyte apoptosis. The pathogenicity of PLP1 duplications is less well understood, although it has been speculated that overexpressed PLP1 in endosomal and lysosomal compartments depletes myelin rafts of lipids necessary for the production of functional myelin compounds [Garbern, 2007].
MRI in PMD shows hypomyelination, indicated by diffusely elevated white-matter signal on T2 (Figure 71-3A). Myelination is arrested and fails to progress on repeated scans. Some patients show a more mottled, “tigroid” signal, perhaps due to small myelinated areas surrounding blood vessels. In some patients, myelin deposition is seen in the posterior limb of the internal capsule or the optic radiations. Myelination is usually present in the brainstem and sometimes in the cerebellar hemispheres. Often – but not always – the pyramidal tracts in the brainstem show high T2 signal. At a young age, atrophy is not considerable, especially in the cerebellum, although white-matter volume may be decreased. The corpus callosum is thin, reflecting lack of myelinated axons. As children grow older, generalized atrophy develops. Proton MR spectroscopy reveals low choline, due to reduced membrane turnover in the absence of myelinating oligodendrocytes, and normal to elevated N-acetylaspartate (NAA), due to more densely packed axons. Patients with null mutations or deletions show more advanced supratentorial myelination. In SPG2 patients, MRI abnormalities are variable, ranging from diffuse mild hypomyelination to mild periventricular signal changes. Carrier females sometimes display small areas of elevated white-matter signal, but MRI is usually normal.
Diagnosis of PMD is based on typical clinical presentation, radiologic evidence of hypomyelination, and detection of PLP1 alterations. Routine laboratory and metabolic tests, including cerebrospinal fluid (CSF), are normal. As most PLP1 alterations are duplications, simple sequencing must also be accompanied by gene dose quantification by Southern blot, quantitative polymerase chain reaction (PCR), or, more recently, multiple ligation-dependent probe amplification (MLPA). Recently, elevation of the dipeptide N-acetylaspartylglutamate (NAAG) has been described in several PMD cases, with NAAG level appearing to predict clinical severity [Burlina et al., 2006]. The mechanism of NAAG elevation, however, remains unknown, and it is neither specific to PMD, nor a marker of hypomyelination.
Pelizaeus–Merzbacher-Like Disease
PMLD is a genetically heterogeneous disease. In most cases, no gene has been identified. In a small subset of PMLD patients (fewer than 10 percent), mutations in GJC2 (also called GJA12), coding for connexin 46.6 (Cx47), have been found (OMIM 608804) [Henneke et al., 2008]. Located on chromosome 1q41–q42, this gene was identified using a homozygosity mapping approach in a large consanguineous family [Uhlenberg et al., 2004]. Mutations were subsequently found in other, unrelated, patients, confirming GJC2 as the responsible gene. It remains the only gene identified in PMLD; other approaches to identify PMLD genes, such as sequencing candidate genes for structural myelin proteins, have so far been unsuccessful.
As in PMD, patients typically present with nystagmus apparent after the first few weeks of life. Motor development is delayed and children display ataxia during the first few years. Pyramidal signs and frank spasticity often develop later. Compared with boys with classic PMD, motor and cognitive performance in PMLD is better, especially during the first years of life, and children are often able to walk, some without support. However, patients show a more precipitous decline, with progressive spasticity, prominent pseudobulbar signs, and facial weakness reminiscent of myopathic face. Patients become wheelchair-bound in their teens. Epilepsy starting in late school age has been reported in a substantial proportion of cases, and may be more severe in isolated cases, a very unusual feature for boys with PMD. Mild demyelinating peripheral neuropathy revealed by nerve conduction studies has been described in some patients, although it does not influence the overall clinical manifestation. A recently described family carrying a homozygote missense mutation in GJC2 showed a phenotype of almost pure spastic paraplegia and minor cerebellar signs [Orthmann-Murphy et al., 2008]. There are no histopathological data on PMLD.
MRI shows hypomyelination, and prominent signal abnormalities of the pyramidal tract of the brainstem have been described (Figure 71-3B). Cerebellar atrophy is mild or absent, at least in the early stages; supratentorial atrophy with considerable white-matter loss is prominent in older patients [Wolf et al., 2007]. Brainstem atrophy is often seen in later stages. Routine metabolic investigations are normal. In CSF, NAAG is elevated, as it is in PMD [Sartori et al., 2008].
4H Syndrome
This recently described leukoencephalopathy (OMIM 612440) is also characterized by hypomyelination. Its name is derived from its three main clinical findings: hypomyelination, hypodontia, and hypogonadotropic hypogonadism [Wolf et al., 2005, 2007; Timmons et al., 2006]. The disorder is rare and a genetic locus has not yet been identified. As affected siblings of both sexes have been reported, inheritance is presumed to be autosomal-recessive.
Besides these neurological symptoms, hypodontia is the most prominent diagnostic sign (Figure 71-4). Eruption of deciduous teeth is delayed, and its order disturbed. Normally, the lower median deciduous incisors erupt first, followed by the upper median incisors. In 4H syndrome, the deciduous molars erupt first, followed by the incisors, and finally by the upper median incisors. Despite this delayed and disorganized eruption, the deciduous teeth are usually complete. In the permanent dentition, however, some teeth are missing. The incisors have an abnormal shape and often also a yellowish color. About 10 percent of patients show natal teeth, an otherwise very rare finding occurring only in 1 in 1000–3000 newborns.
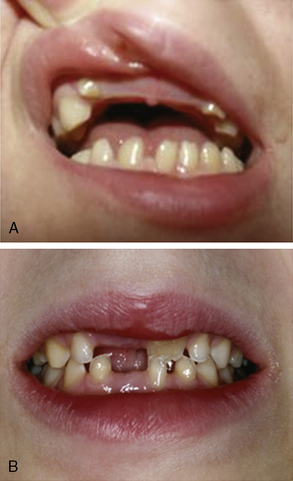
Fig. 71-4 Dental phenotype of children with 4H syndrome.
The upper median incisors erupt late. A, Child aged 5 years. The typical situation in a younger child with lacking upper median (and in this case also lateral) incisors. The left lower median incisor [Schiffmann et al., 1994] has not erupted yet. B, Child aged 8 years. No deciduous incisors have erupted. The right permanent upper median incisor [Paznekas et al., 2003] shows yellowish discoloration. The left median incisors have not erupted yet.
As the name of the disease suggests, MRI shows hypomyelinated white matter (see Figure 71-3C). T2 signal in the supratentorial white matter is diffusely elevated, with the exception of aspects of the posterior limb of the internal capsule and optic radiation. T1 white-matter signal varies from hypointense to hyperintense, depending on the amount of myelin deposited. Cerebellar white matter is usually myelinated on T2. Corpus callosum is thin. There is also early and considerable cerebellar atrophy, more of the vermis than of the hemispheres, in virtually all patients. Whether clinical severity correlates with amount of myelin deposited or degree of initial cerebellar atrophy has not yet been elucidated. Proton MR spectroscopy reveals low choline, as is usual in hypomyelination, and often also elevation of myo-inositol compatible with gliosis. In the later stages, considerable cortical atrophy and white-matter loss develop, indicating on-going myelin loss.
Metabolic investigations are all normal. In some children, a muscle biopsy, with assessment of respiratory chain enzymes, has been performed in the context of deterioration of symptoms with infections, but showed normal results. Analysis of the known genes involved in hypomyelination, including GJA1, in which mutations lead to oculodentodigital dysplasia, did not reveal abnormalities [Wolf et al., 2007].
Oculodentodigital Dysplasia
Oculodentodigital dysplasia (ODDD, OMIM 164200) is another hypomyelinating disorder characterized by dental abnormalities. Its inheritance is autosomal-dominant. Dominant mutations in another connexin gene on chromosome 6q21–23.2, GJA1, coding for connexin 43 (Cx43), cause ODDD. There is one family described with autosomal-recessive mutations leading to the same phenotype. Cx43 is expressed in the developing brain and teeth, and also in hands and feet [Paznekas et al., 2003].
Neurologic symptoms are common, although no large case series has been reported, so little is known about the exact time course and development of neurologic abnormalities. In childhood and adolescence, coordination problems and mild ataxia are common. In adulthood, slow neurologic deterioration is seen, with development of pyramidal tract lesions, ataxia, dysarthria, loss of bladder control, and finally, frank spasticity. Unsupported gait may become impossible in late stages. Optic atrophy and deafness are possible. Cognition is preserved in most patients, although learning disabilities have been reported. Whether and to what extent early cognitive deterioration occurs awaits further study. Peripheral nervous system appears unaffected [Loddenkemper et al., 2002].
Hypomyelination with Congenital Cataract
Affected patients present early with delayed motor development. Cognitive development is mildly to moderately delayed. Most HCC children learn to walk with support before their second birthday. Neurological examination reveals dysarthria, moderate to severe spasticity with elevated muscle tone, brisk reflexes, extensor plantar response, and cerebellar signs such as intention tremor and dysmetria. Nystagmus is rare. As in other hypomyelinating disorders, progression of secondary neurologic symptoms occurs, and children may be unable to walk, even with support, by the end of the first decade. An additional pathognomonic finding is congenital cataract [Zara et al., 2006]. Several HCC cases have been published, and one patient failed to develop cataract until age 9. A small subset of HCC patients suffers from occasional seizures. Peripheral neuropathy is seen in almost all patients, leading to loss of previously exaggerated tendon reflexes and distal muscle wasting. It is not yet known whether a broad phenotypic spectrum exists in HCC, and clinical presentation is heretofore remarkably homogeneous, with the exception of cataract and peripheral neuropathy, which are not present in all patients. There are no data yet about CNS pathology, nor experimental models to elucidate Hyccin function.
MRI shows hypomyelination, as evident from the diffusely elevated T2 white-matter signal. In contrast to those with other hypomyelinating disorders, HCC patients show additional areas of higher T2 white-matter signal and decreased signal intensity on corresponding T1-weighted images, indicating elevated water content in these areas, particularly in periventricular regions [Rossi et al., 2007] (Figure 71-3E). Cerebellar atrophy is not seen. In the early stages, normal myelin signal may be apparent in subcortical fibres and corpus callosum. In the late stages, white matter appears shrunken, with increased apparent diffusion quotients. Proton MR spectroscopy gives variable results, depending on the disease stage. Choline may even be slightly elevated in the early stages, which is unusual for a hypomyelinating disorder, and decreased in later stages.
Hypomyelination with Atrophy of the Basal Ganglia and Cerebellum
Hypomyelination with atrophy of the basal ganglia and cerebellum (HABC, OMIM 612438) is a rare disorder of unknown genetic origin [van der Knaap et al., 2002]. As no affected siblings of children with HABC have been reported, it is possible that this entity arises from a dominant de novo mutation, although autosomal-recessive inheritance remains possible. Disease severity ranges from severely affected infants presenting shortly after birth to more mildly affected children with initially normal development. In severe cases, children fail to achieve motor milestones and linguistic development, and show profound axial hypotonia. Some have nystagmus, and optic atrophy is possible. Spasticity is common. Extrapyramidal symptoms, comprising dystonia, rigidity, and choreoathetosis, are uniquely common to HABC relative to other white-matter disorders. Failure to thrive and microcephaly are common. In mildly affected children, unsupported walking is achieved within the first few years of life, sometimes on time, but is later lost. There is a combination of spasticity, extrapyramidal symptoms, and ataxia. cognitive impairment is mild, although patients tend to show cognitive decline in addition to deteriorating motor functions.
Recently published neuropathologic findings from one HABC patient detail slightly reduced white-matter volume, reduced oligodendrocytes, severe myelin deficiency, especially of deep white matter, and some white-matter loss, as indicated by the presence of macrophages in perivascular regions [van der Knaap et al., 2007]. Axons appeared relatively preserved. There was mild astrocytosis and a strong presence of microglia. The putamen was visible only as a small streak, and microscopy revealed substantial neuronal loss. Cerebral cortex was normal, both macroscopically and microscopically. In the atrophic cerebellum, there was loss of granule cells. Pyramidal tracts appeared degenerated in the brainstem and spinal cord. Metabolic investigations are normal in these children.
Sialic Acid Storage Disorders
Salla disease (OMIM 604369) and infantile sialic acid storage disease (ISSD, OMIM 269920) are both caused by autosomal-recessive mutations in SLC17A5, coding for Sialin, a lysosomal membrane protein transporting sialic acid from lysosomes [Verheijen et al., 1999]. The gene is located on chromosome 6q14/15. Recently, it was shown that Sialin also functions as a shuttle for aspartate and glutamate in synaptic vesicles [Miyaji et al., 2008]. Free sialic acid accumulates in lysosomes of many cell types, including liver and kidney cells and cultured fibroblasts. In leukocytes, this accumulation causes vacuoles visible at light microscopic examination. Electron microscopy reveals membrane-bound vacuoles filled with fibrillogranular amorphous material. The pathogenesis of free sialic acid storage disease and the role of sialic acid remain unclear.
Both diseases are characterized by elevated excretion of free sialic acid in urine. Sialic acid is also elevated in other fluids, such as CSF. Recently, two siblings have been described who lack the characteristic sialuria; sialic acid was elevated only in CSF [Mochel et al., 2009]. Prevalent in Finland, Salla disease is characterized by seemingly normal early development, followed by presentation with hypotonia and ataxia in the second half of the first year of life. Nystagmus is also common. It may be evident in the neonatal period and frequently disappears. Many children also show strabismus. Spasticity develops slowly, and mild extrapyramidal symptoms are common in later stages. Mean age at walking is 4 years, with roughly one-third of patients who do not develop independent ambulation. Language is severely affected and usually dysarthric; patients are, at best, able to produce short sentences. Epilepsy with short, complex focal seizures is relatively common. In some patients, there is evidence of peripheral hypomyelination with decreased nerve conduction velocities. The disease is stable over a long period of time, with ultimate late progression. Additional symptoms may include short stature or hypogonadotropic hypogonadism. Facial features become coarse in adulthood. Otherwise, there is no evidence for dysostosis multiplex or hepatosplenomegaly, despite the presence of free sialic acid storage in liver and spleen. Life expectancy is normal [Aula et al., 2000].
A phenotype with intermediate severity relative to Salla disease and ISSD has also been identified.
MRI in patients with Salla disease shows hypomyelination [Sonninen et al., 1999] (Figure 71-3D). There is white-matter volume loss. Corpus callosum may be stringlike, especially in severe cases. There is usually cerebellar atrophy, and supratentorial atrophy is found in older patients. Thickening of the calvarium is another common radiologic feature. Proton MR spectroscopy reveals a high NAA peak, likely due to elevated free sialic acid, whose N-acetyl peak co-resonates with the N-acetyl peak of NAA [Varho et al., 1999].
Fucosidosis
MRI shows hypomyelination. A characteristic feature of fucosidosis is high T1 and low T2 signal in globus pallidus, thalamus, and substantia nigra. Cerebral and cerebellar atrophy may be prominent in older patients [Prietsch et al., 2008].
Serine Synthesis Defects
Children with 3-phosphoglycerate dehydrogenase deficiency are born microcephalic, and their development is grossly delayed. Epilepsy develops in the second half of the first year of life; West’s syndrome is one possible manifestation. Supplementation with serine and glycine is effective in seizure management. If treatment is commenced prenatally, head circumference at birth and development are normal [de Koning et al., 2004]. In untreated children, MRI shows hypomyelination and white-matter volume loss [de Koning et al., 2000]. Corpus callosum is thin and short. Myelination improves under treatment.
The first documented patient with phosphoserine aminotransferase deficiency presented in the neonatal period with severe epilepsy resistant to medical treatment and rapidly developing microcephaly. MRI showed supratentorial and brainstem atrophy. Supplementation with serine and glycine did not attenuate the seizures. The same treatment, if started before the development of symptoms, was shown to prevent epilepsy and enable normal development in the sibling of the proband [Hart et al., 2007].
Cockayne’s Syndrome and Trichothiodystrophy
Cockayne’s syndrome (CS) is a rare disease combining neurologic and non-neurologic features. This disorder and related disorders of DNA repair, such as cerebro-oculo-facial syndrome (COFS) and trichothiodystrophy (TTD), are genetically heterogeneous and are caused by mutations in CSA (CKN1 or DNA excision repair protein ERCC-8, responsible for 20 percent of CS cases), CSB (CKN2 or ERCC-6, responsible for most of the remainder of CS cases), XPB (ERCC3, OMIM 610651), XPD (ERCC2), XPG (ERCC5), ERCC1-XPF, TTDA and TTDN1 genes, and possibly others [Weidenheim et al., 2009].
The classic form, Cockayne’s syndrome type I, presents in the first year of life with failure to thrive; weight is more affected than length (“cachectic dwarfism”), and there is loss of subcutaneous fat, leading to a “wizened,” bird-like, progeroid face. Microcephaly also develops, usually by the end of the second year. Children develop contractures of the large joints, giving them a typical posture. Hands and feet are disproportionally large. Dental caries is prominent. Psychomotor development is also delayed, resulting in mild to severe cognitive impairment. Predominant neurologic features include ataxia and spasticity, which show slow progression. In late stages, peripheral neuropathy leads to muscle wasting and loss of the initially increased tendon reflexes. Over half of individuals develop sensorineural hearing loss. Most suffer from pigmentary retinal degeneration and cataracts. Autonomic dysfunction (hypolacrimia, hypohydrosis, miosis, acrocyanosis) is possible. In Cockayne’s syndrome type II, the clinical picture is much more severe, with growth failure already evident at birth. Loss or even absence of subcutaneous fat is striking. Joint contractures and kyphosis develop rapidly. Hypotonia is prominent initially, followed by development of spastic tetraparesis. Psychomotor development is absent or extremely delayed, with subsequent deterioration and early death. Subcutaneous fat loss has been treated in both types by early hypercaloric tube feeding, which allows reasonable growth in some children. Type III describes patients with milder forms of disease. Cutaneous photosensitivity is also characteristic of CS, occurring in 75 percent of all patients [Rapin et al., 2000].
Both syndromes are caused by defective nucleotide excision repair. Over 30 proteins are involved in this process. It eliminates DNA lesions induced by ultraviolet light. There are two major subpathways of nuclear excision repair: transcription-coupled repair, dealing with reparation of transcribed genes, and global genome repair, removing lesions in the entire genome. Defective DNA repair can be demonstrated by irradiating cultured skin fibroblasts with ultraviolet light and subsequently measuring unscheduled DNA synthesis. This unscheduled DNA synthesis is diminished in TTD and xeroderma pigmentosum patients. In CS, this unscheduled DNA synthesis is not significantly attenuated, but the otherwise rapid recovery of RNA and DNA synthesis after ultraviolet irradiation is adversely affected, indicating that the global genome repair is still functional. Additionally, reduction in basal transcription is also seen, an important signal for apoptosis. These defects of transcription, perhaps combined with activation of apoptosis, are thought to be mainly responsible for the neurologic symptoms in CS. Complementation assays in cultured cells could distinguish two different complementation groups, CS type I (OMIM 216400), caused by mutations in the gene coding for group 8 excision-repair cross-complementing protein (ERCC8), and type II (OMIM 133540), due to mutations in ERCC6. TTD is also heterogeneous, genetic defects having been identified in at least three different genes [Cleaver et al., 2009].
MRI of patients with CS shows hypomyelination, its degree corresponding to clinical severity. In severe cases with CS type II, hypoplasia of cerebellum and brainstem is possible. Basal ganglia calcifications are common. Similar features are seen in TTD, although calcifications are less common than in CS [Rapin et al., 2000; Adachi et al., 2006].
18q Minus Syndrome
In this disorder (OMIM 601808), the distal region of the long arm of chromosome 18 is deleted. It occurs de novo most commonly. The contiguous gene deletion usually involves the bands 18q22.3→qter. The gene for myelin basic protein (MBP), a component of healthy myelin, is located within this region. It has been postulated that haploinsufficiency for MBP leads to the myelin abnormalities observed in 18q minus syndrome. Heterogeneity in severity of clinical symptoms and hypomyelination between patients, despite consistent loss of MBP, is a focus of on-going inquiry. There is a well-investigated mouse model, the shiverer mouse, with homozygous rearrangements in the MBP gene [Nave, 1994], which lead to CNS, but no peripheral, hypomyelination. It is unknown why peripheral myelin is spared, despite MBP expression in peripheral nerves.
MRI shows hypomyelination of variable, but usually mild, degree. The myelin signal in the cerebral hemispheres may be inhomogeneous, with patchy white matter abnormalities. Corpus callosum may be thin. There may be mild supratentorial atrophy [Linnankivi et al., 2006].
SOX10-Associated Disorders
These rare syndromes are caused by mutations in SOX10 on chromosome 22q13, which encodes a transcription factor for various genes, some involved in myelin formation and metabolism, such as GJB1 (connexin 32). These disorders are characterized by a white hair lock and hypomelanotic spots, sensorineural deafness, and Hirschsprung’s disease. Patients are affected with varying severity, ranging from antenatal onset with congenital arthrogryposis multiplex and severe neurologic abnormalities, to more mildly affected patients who lack neurologic manifestations (Waardenburg–Shah syndrome, WS4, OMIM 277580). The severe variant has been designated peripheral demyelinating neuropathy, central dysmyelinating leukodystrophy, Waardenburg’s syndrome, Hirschsprung’s disease, or PCWH (OMIM 609136). Consistent with a neurocristopathy, many features of this disease can be explained by defective differentiation or migration of neural crest cells. In the severe cases, MRI reveals hypomyelination [Touraine et al., 2000]. The milder phenotype of WS4 is explained by SOX10 haploinsufficiency, whereas the severe form, PCWH, is thought to be caused by a dominant negative mechanism. Mutations found in children with PCWH are all truncating and located in the last exon. These mutations, unlike SOX10 mutations leading to WS4, escape nonsense-mediated decay and can therefore exert their dominant negative effect on protein level [Inoue et al., 2004, 2007].
Neurologic symptoms of children with PCWH include delayed development, nystagmus, spasticity, and ataxia. In severe cases, neonates are already symptomatic, with profound hypotonia, seizures, or congenital arthrogryposis due to hypomyelinating neuropathy. Other possible symptoms are reduced tear and saliva production, anhidrosis, and severe failure to thrive, in addition to the classic syndromes of WS4. Neuropathologic investigations of a severely affected infant have shown absence of central and peripheral nervous system myelin at the age of 3 months [Inoue et al., 2002]. Detailed MRI features have not been reported for many patients, but preliminary cases suggest mild to severe hypomyelination, and possibly atrophic brainstem [Inoue et al., 1999].
White-Matter Disorders with Demyelination
If MRI is not consistent with hypomyelination, there is white-matter hypointensity on T1 instead of iso- or hyperintensity, and there is hyperintensity on T2, the imaging pattern fits the demyelinating leukodystrophies. They comprise the leukodystrophies with primary demyelination, leukodystrophies with white-matter vacuolization, calcifying leukoencephalopathies, cystic leukoencephalopathies, leukoencephalopathies with brainstem involvement, and most adult-onset leukoencephalopathies. In the assessment of patients with these leukodystrophies, careful attention should be paid to specific neuroimaging features, including basal ganglia or brainstem signal abnormalities, contrast enhancement, cysts, calcifications, contrast enhancement, or specific FLAIR imaging abnormalities (Figure 71-5 and Figure 71-6; see also Figure 71-3) to assist in the differential diagnosis.
Primary Demyelinating Leukodystrophies
Alexander’s Disease
Alexander’s disease (AxD, OMIM 203450)[Alexander, 1949] is associated with mutations in the gene encoding the glial fibrillary acidic protein (GFAP) [Brenner et al., 2001]. GFAP mutations are thought to confer gain-of-function mutations, and a mutation on a single allele is sufficient to cause disease. In most cases, mutations are sporadic, although familial cases are described in adult- or juvenile-onset cases. In familial cases, inheritance is autosomal-dominant. There is usually concordance in presentation within a family, such that a family with adult-onset presentation and subsequent infantile presentation has not been reported. There is no definite genotype–phenotype correlation, with the exception of the two most common mutations, R79 and R239. R239, in particular, is associated with earlier onset and poor prognosis.
GFAP mutations are thought to result in decreased solubility of the glial fibrillary acidic protein, and accumulation of GFAP, along with vimentin, αβ-crystallin, and heat shock protein 27, result in Rosenthal fiber (RF) aggregation. RFs, accumulating in astrocytes, are thought to obstruct normal glial cell function and result in myelin destruction. RF accumulation in the ventricular collecting system is believed to cause the hydrocephalus seen in the clinical setting. Studies to improve pathophysiologic understanding of this disease and possible treatment strategies are on-going in an existing murine model of AxD. AxD is typically suspected from clinical presentation and characteristic MRI features. Typical AxD imaging features include frontal predominance of white-matter abnormalities, basal ganglia and or brainstem involvement, a periventricular rim of altered signal on T1- and T2-weighted imaging, and contrast enhancement of specific intracranial structures (see Figure 71-5A). The presence of four of five of these features makes diagnosis of AxD very likely and should prompt mutation testing [van der Knaap et al., 2001]. In very young infants and in the juvenile- or adult-onset cases, MRI may be less typical and involve only restricted brain regions. Additional features described in AxD imaging include predominant or isolated involvement of posterior fossa structures, multifocal tumor-like brainstem lesions, brainstem atrophy, and garland-like abnormalities along the ventricular wall [van der Knaap et al., 2005, 2006]. MR spectroscopy can be helpful in the diagnosis if it shows a lactate peak in affected tissues, but may also lead to inappropriate evaluations for mitochondrial cytopathies. There have been reports of findings of elevations of GFAP protein in CSF [Kyllerman et al., 2005], but this test is not used as a clinical tool.
X-Linked Adrenoleukodystrophy
X-linked adrenoleukodystrophy (XALD, OMIM 300100) is associated with mutations in the ABCD1 gene [Mosser et al., 1993] encoding a peroxisomal membrane transporter. This disorder follows X-linked inheritance, and often, when male children are diagnosed with the childhood-onset cerebral form, disease manifestations are recognized in obligate female carriers or male relatives of the proband. Differences in disease manifestations are known to occur with identical genotype, and even within a sibship, underscoring the likely effect of other genetic factors on clinical presentation. Genotype does not predict very long chain fatty acid levels or specific clinical prognosis. Genetic changes reported include missense mutations, nonsense mutations, frameshift mutations, small deletions/insertions, and large deletions.
Of note, an allelic disorder, with a neonatal presentation of cholestasis, hypotonia, and developmental delay, is caused by a contiguous gene deletion syndrome involving the 5′ end of ABCD1. This disorder is called contiguous ABCD1 DXS1357E deletion syndrome (CADDS), and is clinically distinct from XALD [Corzo et al., 2002].
The estimated prevalence of XALD is estimated to be 1:20,000–1:50,000. The estimated prevalence of hemizygotes (affected males) and heterozygotes (carrier females) is estimated at 1:16,800 [Bezman et al., 2001].
The pathophysiology of XALD is believed to arise from accumulation of saturated very long chain fatty acids (SVLCFA) within the brain. This accumulation is thought to result from defective peroxisomal fatty acid oxidation of SVLCFA, caused by defective transport by the mutated ABCD1, possibly due to altered adenosine triphosphate (ATP) binding [Gartner et al., 2002; Roerig et al., 2001].
Diagnosis is based on characteristic clinical presentation and suggestive MRI features. MRI shows a predominance of occipital findings (see Figure 71-5D), although frontal and corpus callosum variants are recognized. The affected white matter appears hyperintense on T2 and hypointense on T1. Characteristically, there is a rim of enhancement around the abnormal tissue that can be very helpful in establishing the diagnosis, as few other leukodystrophies, with the exception of Alexander’s disease, show significant contrast enhancement. When XALD is suspected, appropriate clinical tests include fasting VLCFA testing on plasma, which shows an excess in SVLCFA with specific abnormalities in C26:0, C24:0/C22:0, and C26:0/C22:0 ratios [Moser et al., 1981]. Mutation testing of the ABCD1 gene provides molecular confirmation of the diagnosis, with attention to the 7 percent of cases with deletions or rearrangements.
Treatment with Lorenzo’s oil or diets rich in oleic and erucic acids, in addition to VLCFA restriction, can alter VLCFA levels. As treatment strategies for XALD evolve, interest in newborn screening is growing. This can be done using liquid chromatography-tandem mass spectrometry (LC-MS/MS) to test for the analyte 1-hexacosanoyl-2-lyso-sn-3-glycero-phosphorylcholine (26:0-lyso-PC)[Hubbard et al., 2009; Raymond et al., 2007]. As with many disorders now being considered for widespread newborn screening, the clinical benefit of early diagnosis remains to be established.
Treatment of XALD depends on the stage and type of disease manifestations, and is still research-based. Treatment with Lorenzo’s oil, which decreases hexacosanoic acid (C26:0), does not appear to stop or reverse cerebral disease once it has begun, although there are reports of improved long-term stability in presymptomatic patients in open-label studies with no placebo control [Moser et al., 2007]. The use of Lorenzo’s oil is investigational and should be performed in the context of a clinical research protocol. Bone marrow transplantation (BMT) is indicated in children with early-stage, cerebral-form XALD, as evidenced by active disease on MRI. Family members of an affected proband should be tested by VLCFA and monitored for early MRI evidence of cerebral involvement to identify candidates for BMT. These patients should be evaluated by clinicians with specific expertise in this disorder. Gene therapy is a potential future tool and research studies are under way. Supportive care can improve comfort and quality of life for XALD patients. Careful monitoring and treatment of adrenal insufficiency should be part of therapeutic management.
Metachromatic Leukodystrophy
Metachromatic leukodystrophy (MLD, OMIM 250100) [Greenfield, 1933; Von Hirsch and Peiffer, 1955; Suzuki and Chen, 1966] is caused by mutations in the ARSA gene encoding arylsulfatase A [Stein et al., 1989; Austin et al., 1964] on chromosome 22q13.31, and is inherited in an autosomal-recessive manner. Homozygous or compound heterozygous ARSA mutations impair arylsulfatase A degradation of sulfatides, causing sulfatide accumulation within the brain and peripheral nervous system. Complete loss of arylsulfatase A activity (“I” or “O” alleles) is typically associated with early infantile MLD. Partial loss of arylsulfatase A activity (“R” or “A” alleles) is typically associated with juvenile- or adult-onset MLD. Compound heterozygosity with an I and A allele usually results in juvenile-onset MLD. Rarely, subjects with microdeletions of 22q13 and deletion of the ARSA gene with an MLD-causing mutation on the other allele have been diagnosed with MLD.
MLD diagnosis is often suspected based on clinical manifestations of motor impairment with a peripheral neuropathy. Typical MRI features include sparing of arcuate fibers and a rim of subcortical white matter, with involvement of periventricular and deep white matter in the supratentorial CNS (see Figure 71-5B). Involved white matter takes on the appearance of radiating stripes that can be highly suggestive of the disorder and reflects accumulation of sulfatides in perivascular macrophages. These radiating stripes are also present in other disorders, however, including Krabbe’s disease. In early stages or in adult cases, incomplete neurologic findings can complicate diagnosis, and early involvement of the corpus callosum may provide a clue. Occasionally, isolated involvement of cranial or peripheral nerves has been seen in the early stages of disease.
Krabbe’s Disease or Globoid Cell Leukodystrophy
Krabbe’s disease (OMIM 245200) is caused by autosomal-recessive mutations in the gene encoding galactosylceramidase (GALC) on chromosome 14q31 [Austin et al., 1970; Suzuki and Suzuki, 1971; Wenger et al., 1974; Zlotogora et al., 1990; Chen et al., 1993]. Two recurring mutations are associated with specific phenotypes. One common 30-kb deletion results in the classic infantile form in the homozygous state or when compound heterozygous with another mutation associated with severe disease. The 809G>A mutation is associated with the late-onset form of Krabbe’s disease, even when connected to mutations associated with severe disease. There is limited genotype–phenotype correlation for other mutations, however, and different disease courses have been seen in the same family.
Prevalence has been reported to be about 1 in 100,000 births in the US and Europe.
Diagnosis is suspected based on combined motor and peripheral nerve involvement. Neuroimaging can also guide diagnosis, with CT demonstrating highly suggestive hyperdensity of the thalami, caudate, corona radiata, and, in some cases, cerebellum and brainstem. MRI is less pathognomonic than in other disorders; however, early involvement of the brainstem and cerebellar white matter, in particular the hilus of the dentate nucleus, can be helpful in establishing the diagnosis (see Figure 71-5G). Thickening of the optic nerves and chiasm has been reported. Over time, the most significant involvement occurs in deep and periventricular white matter, with relative sparing of U fibers early on. The corpus callosum is affected. Radiating stripes can be seen within the affected white matter. There is often prominent atrophy of the brain. Late-onset forms may present with similar MRI patterns, or less typical ones, such as changes limited to the corticospinal tracts in adult-onset cases. In some instances, enhancement of the gray–white matter junction, cranial nerves, or spinal nerve roots can be seen.
Saposin A Deficiency
An infant with abnormal myelination resembling Krabbe’s disease was found to have a mutation in the saposin A region of the prosaposin (PSAP) gene (OMIM 611722) [Spiegel et al., 2005]. Prosaposin is one of several known sphingolipid activator proteins, and interacts with the enzyme GALC to catalyze the hydrolysis of lipids.
White Matter Disorders with White-Matter Vacuolization
Among the specific radiologic features associated with white-matter disease, white-matter vacuolization is very helpful in the differential diagnosis of leukodystrophies. The disorders most likely to present with white-matter vacuolization are Canavan’s disease and eIF2B-related disorders, also known as vanishing white-matter disease and childhood-onset ataxia and central nervous system hypomyelination (CACH), although these disorders are clinically and radiologically distinct. One other disorder, not considered a leukoencephalopathy or leukodystrophy, but which may have vacuolization on MRI, is Lowe’s disorder. The oculocerebrorenal syndrome of Lowe (OCRL) is a multisystem disorder with major abnormalities in the eyes, the nervous system, and the kidneys. OCRL is an X-linked disorder caused by mutations in OCRL1, encoding a phosphatidylinositol-4,5-bisphosphate 5-phosphatase. The pathophysiology of this disorder remains unclear. Affected patients may have patchy white-matter abnormalities in the centrum semiovale on T1- and T2-weighted images, with vacuolization on FLAIR or proton density images [Yuksel et al., 2009]. Diagnosis is based on the constellation of cataracts, renal tubular acidosis, and neurologic features. Molecular testing is the mainstay of diagnosis. Symptomatic management of renal, ophthalmologic, and neurologic abnormalities is indicated.
Canavan’s Disease
Canavan’s disease (CD; OMIM 271900), previously known as spongy degeneration of the CNS of the Van Bogaert–Bertrand type, is caused by a deficiency of aspartoacylase, encoded by ASPA [Kaul et al., 1993; Matalon et al., 1988]. This disorder has been documented since at least the 1950s [De Vries et al., 1958], but a biochemical marker was not identified until nearly 30 years later. Inheritance is autosomal-recessive. Incidence is increased in patients of Ashkenazi Jewish descent, with two common mutations, p.Glu285Ala and p.Tyr231X, responsible for 98 percent of disease alleles, facilitating carrier screening in this population. In addition, p.Ala305Glu is responsible for 40–60 percent of the disease-causing alleles in non-Jewish populations, and for 1 percent of disease-causing alleles in the Ashkenazi Jewish population [Kaul et al., 1994]. A final splice-site mutation, c.433-2A>G, is responsible for 1 percent of disease-causing alleles in the Ashkenazi Jewish population. Beyond these, there are many pathogenic mutations with no clear genotype–phenotype correlation, and affected siblings may have a variable course, despite identical genotypes. Although less common, deletions and duplications can be disease-causing alleles.
Most patients with CD present with the infantile form. These children appear normal until approximately 3–6 months of life, when hypotonia with loss of head control, irritability, loss of milestones, and head circumference growth becomes notable. Over time, spasticity replaces hypotonia, and optic atrophy, extrapyramidal movement disorders, seizures, and autonomic disturbances develop. In time, a more chronic vegetative state develops, which can evolve over years. Tonic extensor spasms are often described. More rarely, a congenital variant is seen in which poor feeding, irritability, and hypotonia become evident within days after birth, followed by rapid deterioration and death. Some patients with prolonged development of early milestones have been characterized as having juvenile onset, but the course generally overlaps the infantile form and it is unclear whether this is truly a distinct variant [Traeger and Rapin, 1998].
Biochemical testing is diagnostic in CD, with NAA elevations detectable in urine, plasma, and CSF, and decreased aspartoacylase activity in cultured fibroblasts. Targeted mutation analysis is often used as a first-line diagnostic strategy. Panels testing the four most common disease-causing alleles will diagnose almost 100 percent of affected Ashkenazi Jewish patients, and 40–60 percent of affected non-Jewish patients (www.genereviews.org). Full sequencing and detection of deletions or duplications are used to identify the remainder of subjects with CD. Diagnosis may also be suggested by MRI features, such as confluent involvement of subcortical white matter, extending in severe cases to the central and periventricular white matter in a centripetal fashion, and signal abnormalities of the globus pallidus and thalamus, with sparing of the caudate and putamen. Signal abnormalities of the brainstem and cerebellar white matter can be seen. Over time, extensive white-matter atrophy leads to ventriculomegaly.
eIF2B-Related Disorder (Vanishing White Matter Disease)
eIF2B-related disorder and its various allelic clinical subtypes (vanishing white matter disease OMIM 603896 [van der Knaap et al., 1997] or CACH [Schiffmann et al., 1994], ovarioleukodystrophy [Fogli et al., 2003; Schiffmann et al., 1997], and Cree leukoencephalopathy [Fogli et al., 2002]) are caused by mutations in one of the five genes (EIF2B1–5) encoding a complex, eIF2B [Leegwater et al., 1999, 2001; van der Knaap et al., 2002]. eIF2B, or eukaryotic translation initiation protein 2B, is the guanine nucleotide exchange factor for eIF2, another critical protein in translation initiation. eIF2B-related disorders are inherited in an autosomal-recessive manner, with 65 percent of mutations found on the gene EIF2B5 [van der Knaap et al., 2002; Maletkovic et al., 2008]. Mutations across the five genes are numerous, and in most cases are private mutations, with a compound heterozygous presentation, although some patients are homozygous for the more common mutations. No phenotype is known to exist for heterozygous carriers. There is known genotype–phenotype correlation for certain more common genotypes, including homozygous R113H mutation-positive subjects [van der Knaap et al., 2004], who have an adult onset with milder phenotype, and the homozygous R195H mutation-positive subjects, who have a more severe early-onset presentation [Fogli et al., 2002].
At opposite ends of the phenotypic spectrum, eIF2B mutations can also present in a connatal form, where neurologic disability is evident from birth, with additional findings such as ovarian dysgenesis, cataract, or hepatomegaly, or may present in an adult form with progressive spastic paraparesis. Allelic disorders include Cree leukoencephalopathy, whose name derives from the Cree Indian population in Alaska; the R195H founder mutation results in aggressive early infantile presentation, with rapid progression and early death [Fogli et al., 2002]. Another allelic disorder is ovarioleukodystrophy, in which adult women present with primary ovarian failure and minimal, if any, neurologic features.
Incidence and prevalence of eIF2B-related disorders are not known. eIF2B-related disorder is thought to be caused by alterations in the cellular response to endoplasmic reticulum stress and alterations in protein translation [Kantor et al., 2008; van Kollenburg et al., 2006a,b; Kantor et al., 2005; Schiffmann and Elroy-Stein, 2006]. eIF2B plays a crucial role in the initiation of protein translation, acting as the guanine nucleotide exchange factor for eIF2alpha, another translation initiation factor. When a cell undergoes physiologic stress, protein translation is altered and misfolded proteins often accumulate within the endoplasmic reticulum. Cell-salvaging pathways include eIF2B as a critical modulatory element. It is unknown, however, how alterations in a ubiquitously expressed housekeeping gene result in a primarily glial cell phenotype.
Diagnosis of eIF2B-related disorder is based on appropriate clinical history, neuroimaging, and molecular genetics. Common MRI features include diffuse signal abnormality of the supratentorial white matter, with less constant signal abnormalities in the cerebellar white matter, brainstem, thalamus, and globus pallidus. In supratentorial white matter, T2 FLAIR shows low signal intensity, isointense with CSF, and suggestive of white-matter rarefaction and cystic degeneration. On sagittal T1-weighted images, a pattern of radiating tissue strands may be seen, compatible on pathology with preserved tissue (see Figure 71-5C). MRI findings of eIF2B-related disorder are nearly pathognomonic, except in cases with very early or late onset. In appropriate clinical conditions, molecular studies of the EIF2B genes is appropriate, usually beginning with EIF2B5, which harbors 65 percent of identified pathogenic mutations. If molecular testing does not clarify the diagnosis, biomarkers such as CSF glycine [van der Knaap et al., 1999] and decreased CSF asialotransferrin [Vanderver et al., 2008] can help further investigate the likelihood of eIF2B-related disorder.
Calcifying Leukoencephalopathies
Aicardi–Goutières Syndrome
Aicardi–Goutières syndrome (AGS; OMIM 225750 [AGS1], 610181 [AGS2], 610329 [AGS3], and 610333 [AGS4]) is a genetically heterogeneous disorder associated with mutations in a series of genes encoding nucleases or putative nucleases. These include TREX1 [Crow et al., 2006], SAMHD1 [Rice et al., 2009], and RNAseH2 A, B, and C [Crow et al., 2006]. In most cases, AGS appears to be autosomal-recessive, although rare cases of heterozygotes with AGS phenotypes are reported [Rice et al., 2007]. Additionally, rare patients with clinical diagnoses of systemic lupus erythematosus have been found to harbor heterozygous mutations in TREX1 [Lee-Kirsch et al., 2007]. In addition, a disorder now referred to as autosomal-dominant retinal vasculopathy with cerebral leukodystrophy (RVCL) is allelic with AGS. This disorder has previously been described as cerebroretinal vasculopathy/hereditary retinal vasculopathy/hereditary endotheliopathy with retinopathy, nephropathy, and stroke (CRV/HRV/HERNS) [Grand et al., 1988; Gutmann et al., 1989; Jen et al., 1997], associated with linkage to 3p21 [Ophoff et al., 2001] and with heterozygous mutations in TREX1 [Richards et al., 2007]. RVCL is a rare disorder with retinal vasculopathy, migraine, Raynaud’s phenomenon, stroke, and dementia with onset in middle age. Allelic disorders with AGS with an autosomal-recessive inheritance include familial chilblain lupus [Rice et al., 2007], Cree leukoencephalitis, and toxoplasmosis, rubella, cytomegalovirus, and herpes simplex virus (TORCH)-like disorders. Approximately 83 percent of patients with characteristic AGS-related clinical findings have mutations in TREX1, RNAseH2A, RNAseH2B, or RNAseH2C [Rice et al., 2007]. In those individuals with identified molecular changes, 65 percent had disease caused by TREX1 or RNAseH2B mutations. Moreover, almost all individuals with RNAseH2B mutations had at least one mutation in exons 2, 6, or 7. These observations may allow for a targeted initial screening strategy when performing diagnostic molecular testing.
AGS is characterized by leukoencephalopathy, basal-ganglia or white-matter calcifications, and elevated CSF interferon-alpha [Rice et al., 2007; Lebon et al., 1988; Bonnemann and Meinecke, 1992; Tolmie et al., 1995; Goutieres et al., 1998; Kuijpers, 2002], with no detectable infectious etiology. Patients present most often in the neonatal period or in infancy. Early-onset patients may present in the neonatal period with a syndrome that mimics in utero viral infections, including Coombs-positive hemolytic anemia and autoimmune thrombocytopenia, elevated transaminases, microcephaly, seizures, vasculitic skin lesions, and cerebral calcifications. Often, these patients are initially suspected of having a congenital CMV, rubella, or HIV infection [Crow and Livingston, 2008]. A genetic cause may be suspected only after the birth of a second affected child. This condition may also present in older infants with progressive microcephaly, dystonia, seizures, and developmental delay, as well as sterile pyrexias, lupus-like skin and joint manifestations [Aicardi and Goutieres, 2000], progressive intracranial calcifications, chronically elevated CSF lymphocytes [Giroud et al., 1986], autoantibodies[Ramantani et al., 2010], and elevated CSF pterins.
Suggested minimal criteria for diagnosis are intracranial calcifications with abnormal CNS white matter and no infectious explanation, characteristic clinical findings, such as chilblains, and/or CSF findings of leukocytes, pterins [Wassmer et al., 2009], or interferon-alpha. Molecular confirmation of mutations in TREX1 [Crow et al., 2006], SAMHD1 [Rice et al., 2009], RNAseH2 A, B and C [Crow et al., 2006] is helpful, and may obviate the need for CSF testing in the future. However, it is important to consider the genetic heterogeneity of this disorder, and the fact that many AGS patients do not have mutations in these genes. Neuroimaging characteristics can be very helpful in the diagnosis of AGS. The pattern of calcifications is usually that of punctate calcifications seen in the periventricular white matter and the basal ganglia, most often the globus pallidus and the putamen. In some cases, larger calcifications can be seen. Calcifications may also be present in the brainstem or the cerebellum. In some cases, in particular early in the course, calcifications may be absent altogether. MRI images show high T2 signal and low T1 signal abnormalities in the white matter, most prominently in the frontal and temporal lobes, and atrophy may be significant.
Cerebroretinal Microangiopathy with Calcifications and Cysts
Cerebroretinal microangiopathy with calcifications and cysts (CRMCC, OMIM 612199) [Briggs et al., 2008] is thought to be on a continuum with leukoencephalopathy with calcifications and cysts (LCC) [Labrune et al., 1996; Nagae-Poetscher et al., 2004] and Coats’ plus syndrome [Crow et al., 2004; Tolmie et al., 1988]. An underlying genetic etiology has not been identified, although reports of affected siblings, male and female probands, and children born to consanguineous families suggest an autosomal-recessive inheritance pattern.
A number of pathologic reports appear to suggest that CRMCC is an obliterative microangiopathy [Briggs et al., 2008]. This may explain the neurologic, ocular, intestinal, and hepatic findings, although it does not explain other pathogenic features.
Bandlike Intracranial Calcification with Simplified Gyration and Polymicrogyria
Bandlike intracranial calcification with simplified gyration and polymicrogyria is a recently described phenotypic group [Briggs et al., 2008; Abdel-Salam and Zaki, 2009]. Two series of patients have been characterized by severe postnatal microcephaly, dysmorphic facial features, developmental arrest, and seizures. Neuroimaging demonstrates simplified sulcation, and a large calcified band most often in the frontal white matter and basal ganglia, as well as brainstem calcifications. Inheritance pattern is autosomal-recessive, although a specific molecular etiology has yet to be identified. Reports of other patients with cerebral dysgenesis, intracranial calcification, and no evidence of TORCH infection exist in the literature [Reardon et al., 1994; Burn et al., 1986; Kalyanasundaram et al., 2003], but none exhibits all the symptoms described in these subjects. It remains to be seen whether this initial description represents a more widely distributed phenotype.
Cockayne’s Syndrome
Cockayne’s syndrome, as discussed above, is a disorder characterized by cachectic dwarfism, neurologic disability, cutaneous photosensitivity, pigmentary retinopathy, leukodystrophy, and occasionally, intracranial calcifications. Disorders allelic with Cockayne’s syndrome, including COFS and TTD, are less likely to have significant intracranial calcifications, although these have been described in a restricted number of cases [Linna et al., 1982].
Cerebral Autosomal-Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy
Cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL, OMIM 125310) is a primarily cerebral microangiopathy, presenting in adults with a history of migraine headaches, progressive early-onset cerebrovascular disease, and early-onset dementia; leukoencephalopathy and subcortical infarcts are revealed on neuroimaging. Over 90 percent of individuals have mutations in NOTCH3, the only gene known to be associated with CADASIL. CADASIL is inherited in an autosomal-dominant manner. De novo mutations appear rare, so family history may support the diagnosis. The pathologic diagnosis of CADASIL is based on the finding of electron-dense granules in the media of arterioles that can often be identified by electron microscopic (EM) evaluation of skin biopsies. MRI findings can also guide diagnosis, including early white-matter T2 hyperintensities in the temporal lobe and extreme capsule. Another common radiologic feature is subcortical lacunar lesions, typically linearly grouped at the gray–white matter junction. Microbleeds can result in calcifications in addition to the above-mentioned leukoencephalopathy. Several other hereditary disorders affecting cerebral vasculature, including cerebral autosomal-recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL) [Yanagawa et al., 2002], recently associated with mutations in the HtrA serine protease 1 (HTRA1) gene [Hara et al., 2009], and hereditary systemic angiopathy (HSA) [Winkler et al., 2008], can all be associated with leukoencephalopathy and intracranial calcifications.
Intracranial Calcification
Intracranial calcification associated with leukoencephalopathy is also seen in mitochondrial disease, and is sporadically reported for various disorders, including mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes (MELAS) [Pronicki et al., 2002].
Dihydropterine Reductase Deficiency
Dihydropterine reductase deficiency can be associated with leukoencephalopathy and calcifications [Miladi et al., 1998; Coskun et al., 1990]. This disorder is further discussed below.
Bilateral Occipital Calcifications
Bilateral occipital calcifications with leukoencephalopathy, seizures, and clinical or subclinical celiac disease have also been reported as a distinct entity [Iglesias et al., 2000; Lea et al., 1995; Crosato and Senter, 1992].
Familial Hemophagocytic Lymphohistiocytosis
Familial hemophagocytic lymphohistiocytosis (FHLH) is characterized by proliferation and infiltration of hyperactivated macrophages and T lymphocytes. It can be caused by mutations in a number of genes, including PRF1 (FHL2, OMIM 603553) [Stepp et al., 1999], UNC13D (FHL3, OMIM 608898) [Feldmann et al., 2003], STX11 (FHL4, OMIM 603552) [zur Stadt et al., 2005], and Munc 18-2 (FHL5, OMIM 613101) [zur Stadt et al., 2009], although a number of patients have no identified mutation. Inheritance is autosomal-recessive.
FHLH is characterized by acute illness with prolonged fever (lasting more than 7 days), and features associated with macrophage activating syndrome, including hepatosplenomegaly, cytopenias (anemia, thrombocytopenia, and neutropenia), hypertriglyceridemia and/or hypofibrinogenemia, and bone marrow hemophagocytosis. Neurologic symptoms are highly variable and may include motor deficits and seizures. Onset is usually within the first few months of life, but sometimes in the neonatal period or in older childhood. MRI may show hypomyelination or demyelination. MRI may also show multifocal abnormalities at the gray–white matter junction, hemorrhage, atrophy, focal necrosis, and edema. Calcifications can be seen in areas of abnormal brain tissue [Takano and Becker, 1995]. Allogeneic hematopoietic stem cell transplantation is the only curative therapy and should be undertaken as early as possible for children with confirmed FHLH.
Cystic Leukoencephalopathies
There are several disorders with cystic findings in the white matter not likely to be thought of classic leukodystrophies, and these must be considered in evaluations of white-matter cystic abnormalities. One such finding is greatly dilated Virchow–Robin or perivascular spaces. Many children, often with nonspecific developmental difficulties, including autistic spectrum difficulties [Boddaert et al., 2009; Taber et al., 2004], are referred for evaluation of cystic lesions of the white matter on MRI that are most consistent with dilated perivascular spaces. These are most often described in the centrum semiovale and posterior parietal white-matter structures, although they can be seen in other regions where Virchow–Robin spaces are typically seen. Macrocephaly is sometimes noted on clinical examination [Groeschel et al., 2006; Hartel et al., 2005]. Occasionally, a primary metabolic disorder, such as mucopolysaccharidosis (Hurler’s/Scheie’s syndrome), or structural chromosomal abnormalities will be identified, but no specific underlying disorder is found in the vast majority of cases. It is currently unknown whether identified MRI abnormalities have functional effects or are pathognomonic for specific disorders. Indeed, various sources dispute whether significantly dilated perivascular spaces can be seen in a small percentage of normal children [Boddaert et al., 2009; Groeschel et al., 2006; Artigas et al., 1999], and with higher frequency in children with migraines [Lewis and Dorbad, 2000]. A subset of cases shows frontonasal dysplasia and is referred to as Sener’s syndrome [Lynch et al., 2000], although no causative genetic change has been identified.
Other disorders beyond the classically defined leukodystrophies include mitochondrial leukoencephalopathies, which can present with cystic leukoencephalopathies in a number of molecularly defined mitochondrial disorders [Zafeiriou et al., 2008], or COL4A1 associated disorder. Mutations in the gene encoding type IV collagen α-chain 1, COL4A1, are thought to disrupt the vascular basement membrane. In patients with a history of familial intracranial hemorrhage and stroke, as well as familial intraventricular hemorrhage and porencephalic cysts in neonates, this disorder should be considered as a possible cause of cystic leukoencephalopathy [Gould et al., 2005].
Megalencephalic Leukoencephalopathy with Subcortical Cysts
Megalencephalic leukoencephalopathy with subcortical cysts (MLC, OMIM 604004) [van der Knaap et al., 1995] is an autosomal-recessive disease associated with mutations in the gene encoding MLC1 protein [Leegwater et al., 2001; Topcu et al., 2000], thus named because at least 15 percent of MLC subjects have no mutation at MLC1 and locus heterogeneity is anticipated. Synonyms include leukoencephalopathy with swelling and discrepantly mild course, cysts, infantile leukoencephalopathy and megalencephaly, van der Knaap’s disease, and vacuolating leukoencephalopathy. There is a founder effect in the Agarwal community in India, with a common genotype (p.Cys46LeufsX34), as well as some more common mutations in individual populations, including persons of Libyan Jewish descent, Turkish Jewish descent, and Japanese descent [Leegwater et al., 2001; Topcu et al., 2000; Ben-Zeev et al., 2002; Saijo et al., 2003; Singhal et al., 2003; Tsujino et al., 2003; Gorospe et al., 2004; Leegwater et al., 2002].
Postnatal macrocephaly is the predominant presenting symptom, noted in the first year, although macrocephaly may be present at birth. No molecularly confirmed cases of MLC without megalencephaly have been identified. This is of diagnostic importance, since a number of other disorders can present with temporal lobe subcortical cysts, including AGS, CMV infection, and RNAse T2-deficient leukoencephalopathy, among others [Henneke et al., 2005]. Stabilization of head growth rate often occurs after age 1, up to 4–6 standard deviations above normal. Patients may have normal early development, or mild delays in motor and cognitive milestones. Later in life, patients develop a spastic ataxia of variable severity. Epilepsy, often easily controlled, is common. Cognitive deterioration is usually late and mild. Dysarthric speech can be particularly debilitating in MLC.
Pathophysiology in MLC1-related MLC is thought to involve altered cellular trafficking in mutated MLC1, a cellular membrane protein expressed in astrocytes. It is localized with the dystrophin glycoprotein complex in astrocytic endfeet in perivascular, subpial, and subependymal regions [Boor et al., 2007]. MLC1 mutations are believed to reduce membrane expression of this protein [Duarri et al., 2008]. Downstream effects on transport across the blood– and CSF–brain barriers have been hypothesized [Boor et al., 2005], but have yet to be empirically established.
Diagnosis in MLC is based on clinical manifestations and typical MRI features. These include a swollen appearance of subcortical white matter; diffuse white-matter signal abnormality with relative preservation of central structures such as the corpus callosum, internal capsule, brainstem, and cerebellum; and development of subcortical cystic structures, most prominently in the anterior temporal and frontoparietal regions (see Figure 71-5F). Over time, severe white-matter structural atrophy can ensue. In typical cases, genetic testing for splice-site mutations and deletions, in addition to missense or nonsense mutations, can identify alterations in many cases, although in at least 15 percent of typical MLC presentations, no mutations in MLC1 can be found.
RNAse T2-Deficient Leukoencephalopathy
A subset of subjects with subcortical cysts and leukoencephalopathy without megalencephaly [Henneke et al., 2005] appear to have mutations in RNase T2, encoded by RNAseT2 [Henneke et al., 2009]. This is a recently described disorder and shares clinical and neuroradiological features with congenital CMV infection. In RNAse T2-deficient leukoencephalopathy there is no evidence of CMV infection, and familial cases suggest autosomal-recessive inheritance. Affected subjects have normo- or microcephaly, developmental delay, and epilepsy, in some cases. MRI reveals striking abnormalities, with multifocal white-matter abnormalities, and temporal lobe cystic structures similar to those seen in CMV. CT reveals scattered calcifications in some patients. The pathophysiologic mechanism eliciting a clinical and radiologic phenotype so reminiscent of congenital CMV remains unknown, and no known treatment for this disorder currently exists.
Congenital Cytomegalovirus Infection
Diagnosis of congenital CMV infection may be made in cases of known maternal infection, and confirmed by neonatal viremia established by CMV PCR in urine or blood. In these cases, identified white-matter abnormalities, with or without gyrational abnormalities involving the brain and cerebellum, should not necessarily raise concern for an additional underlying disorder, but are associated with the identified congenital infection. In many cases, however, infants become symptomatic after the neonatal period, when a definitive PCR diagnosis of infection can no longer easily be established. These patients are often characterized as having unsolved leukoencephalopathies. MRI criteria suggestive of the diagnosis of congenital CMV include multifocal cerebral white-matter lesions located in the deep white matter, predominantly in the parietal region, associated with dilated temporal horns and subcortical cysts in the temporal lobe [van der Knaap et al., 2004]. In cases in which abnormal gyration is suspected, such as polymicrogyria, white-matter abnormalities may be diffuse or multifocal. These MRI criteria are not specific, however, and cannot establish a definitive diagnosis. If saved, newborn blood spots can permit retrospective CMV PCR [van der Knaap et al., 2004; O’Rourke et al., 2008; Lopez-Pison et al., 2005].
Cerebroretinal Microangiopathy with Calcifications and Cysts
As discussed above, CRMCC [Briggs et al., 2008] can present with a significant progressive cystic leukoencephalomalacia, although the cerebral calcifications and multisystem involvement differentiate it from other cystic leukoencephalomalacias.
Leukoencephalopathies with Brainstem, Cerebellum and Spinal Cord Involvement
Certain leukoencephalopathies present with significant brainstem, cerebellum, and spinal cord involvement. This MRI feature should always be carefully noted and is often useful in differential diagnosis. In addition to the disorders discussed in detail below, other disorders, including Alexander’s disease, peroxisomal disorders, mitochondrial cytopathies (see Figure 71-6E), Krabbe’s disease, cerebrotendinous xanthomatosis, and familial histiocytosis, may have brainstem or cerebellar involvement. In addition, X-linked Charcot–Marie–Tooth disease, fragile X permutation, and dentatorubral-pallidoluysian atrophy (DRPLA), while not considered leukoencephalopathies, may have significant brainstem and cerebellar involvement, occasionally mischaracterized as primary leukoencephalopathies.
Leukoencephalopathy with Brainstem and Spinal Cord Involvement and Lactate Elevation
Leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation (LBSL, OMIM 611105) [van der Knaap et al., 2003] is caused by autosomal-recessive mutations in DARS2, encoding mitochondrial aspartyl-tRNA synthetase [Scheper et al., 2007]. Patients are compound heterozygous for pathogenic mutations [Scheper et al., 2007].
LBSL is characterized by slowly progressive cerebellar and sensory ataxia, spasticity, and dorsal column dysfunction. Onset ranges from late childhood to adulthood. In some patients, gait is never normal, but most patients have initial normal milestones. The predominant presenting feature is spastic ataxia, accompanied by distal abnormalities in vibration and proprioceptive sensation. Loss of motor function is slowly progressive and may result in loss of independent ambulation. Rarely, seizures are seen. There may be mild cognitive difficulties. A few patients have a paroxysmal and partially reversible neurologic decline associated with physiologic stressors, including febrile infection or minor head trauma [Marjo and van der Knaap, 2005].
Mitochondrial dysfunction results in a wide range of myelin abnormalities, suspected to arise from disturbance in energy production and use by glial cells. DARS2 encodes mitochondrial aspartyl-tRNA synthetase, which incorporates aspartic acid into mitochondrial DNA-encoded proteins. LBSL could be classified as a “mitochondrial” leukoencephalopathy, but although mutant proteins were found to have decreased ability to aminoacylate tRNAASP in experimental conditions, no abnormalities in mitochondrial complex activity have been identified [Scheper et al., 2007]. This finding leaves the role of DARS2 mutations in the clinical and radiologic abnormalities found in LBSL unresolved.
Before linkage studies identified DARS2 as the etiologic locus [Scheper et al., 2007], diagnosis of LBSL was based exclusively on MRI pattern recognition, as no biomarker exists for this disorder. Classic MRI features include patchy cortical and cerebellar white-matter abnormalities, with striking tract involvement over the entire length of the CNS, including the spinal cord (see Figure 71-6A). Initial reports note involvement of the pyramidal tracts from the internal capsule to the spinal cord corticospinal tracts; the sensory tracts, including the dorsal column, medial lemniscus, thalamus, and corona radiata; cerebellar peduncles; and the trigeminal nerve and mesenchymal trigeminal tracts [van der Knaap et al., 2003; Serkov et al., 2004; Linnankivi et al., 2004]. The corpus callosum is involved, usually more posteriorly than anteriorly. Elevated lactate in affected white matter on MR spectroscopy is often seen, but not always, DARS2 sequencing of this gene has become the diagnostic standard.
Alexander’s Disease
As described above, AxD patients may have significant and sometimes isolated brainstem signal abnormalities, particularly in late-onset cases, in which clinical and radiologic features are primarily related to brainstem structures [van der Knaap et al., 2005, 2006]. Of note, posterior fossa abnormalities are sometimes misdiagnosed as gliomas, and only correctly identified by histopathologic evidence of excessive Rosenthal fiber aggregation.
Polyglucosan Body Disease and Adult Polyglucosan Body Disease
Polyglucosan body disease (PGBD, OMIM 263570) [Robitaille et al., 1980] is an adult-onset leukoencephalopathy with upper and lower neuron motor impairment, distal sensory neuropathy, and neurogenic bladder, characterized by polyglucosan body accumulation in nervous structures. GBE1, encoding 1,4-alpha-glucan-branching enzyme [Lossos et al., 1991], is the only gene known to be associated with adult polyglucosan body disease (APBD) [Lossos et al., 1998]. Inherited in an autosomal-recessive manner, this disorder is allelic with glycogen storage disorder type IV, although these usually much younger patients often have little to no residual enzyme activity. Further, a distinct clinical picture dominated by hepatic, cardiac, and muscle symptoms, with rare clinically important neurologic features, is seen [McMaster et al., 1979; Schroder et al., 1993]. Rare patients with PGBD have been reported with a childhood onset of neurologic disability [Felice et al., 1997]. Increased prevalence of APBD is thought to exist in the Ashkenazi Jewish population, with a common homozygous GBE1 mutation, p.Tyr329Ser, although other ethnic groups are known to be involved as well. Of importance in genetic counseling, manifesting carriers have been noted, including those with the common p.Tyr329Ser mutation [Ubogu et al., 2005; Massa et al., 2008].
The hallmark of APBD is accumulation of polyglucosan bodies in the brain and nerves. Polyglucosan bodies (PBs) are diastase-negative glucose polymers that stain with periodic acid–Schiff (PAS) on histological samples. When examined ultrastructurally, PBs are characterized by granular and filamentous material within the axonal structure and may be associated with loss of myelination. PB accumulation is believed secondary to glycogen branching enzyme deficiency. Within central and peripheral nervous structures, PB accumulation with resultant axonal demyelination [Massa et al., 2008] is thought to result in the clinical phenotype. Importantly, PBs are not specific to APBD, and their presence should be interpreted in the context of the clinical picture.
Diagnosis is usually suggested by the clinical features and the pattern of white-matter abnormalities on MRI. Patients with APBD have confluent but poorly demarcated periventricular white-matter abnormalities, with early sparing of the U fibers and corpus callosum. Cerebellar and brainstem signal abnormalities, most extensive around the fourth ventricle and the long tracts of the brainstem, are also seen. Most pathognomonic of APBD are sagittal FLAIR signal abnormalities outlining the pons (see Figure 71-6B). In the early stages, MRI abnormalities are sufficiently nonspecific that misdiagnosis of multiple sclerosis is common. Definitive diagnosis requires additional supportive evidence. GBE1 sequencing is abnormal in over 90 percent of patients with clinically suggested APBD (www.genereviews.org). It is unknown whether the remainder of subjects have mutations in another unidentified gene, or whether other occult genetic changes result in an altered glycogen branching enzyme. Sural nerve biopsy or skin biopsy can demonstrate evidence of excess PBs. Deficiency of glycogen branching enzyme can also be demonstrated in cultured skin fibroblasts if GBE1 sequencing is equivocal.
Autosomal-Dominant Leukodystrophy with Autonomic Disease (Lamin B1)
Autosomal-dominant leukodystrophy with autonomic disease (ADLD, OMIM 169500) [Marklund et al., 2006; Melberg et al., 2006; Eldridge et al., 1984; Schwankhaus et al., 1994; Coffeen et al., 2000; Leombruni et al., 1998; Quattrocolo et al., 1997] is an adult-onset leukoencephalopathy caused by duplications in LMNB1 on 5q23, encoding lamin B1 [Padiath et al., 2006; Meijer et al., 2008]. Apparent de novo cases are reported in addition to cases of autosomal inheritance. Recently, linkage to the 5q23 region was established in a family with ADLD-like features, but without identifiable LMNB1 alterations. These patients did, however, show increased expression of lamin B1 in lymphoblastoid cells, suggesting a degree of locus heterogeneity [Brussino et al., 2009].
Before the genetic locus was known, clinical history and MRI features were the mainstay of diagnosis. Notable radiologic features include confluent white-matter changes in supratentorial regions, predominantly affecting the deep white matter, with relative sparing of the periventricular white matter and the subcortical fibers. In some cases, predominance in the rolandic regions and pyramidal tracts is seen at onset, evolving over time into diffuse white-matter abnormalities. The corpus callosum is relatively spared, but can show posterior abnormalities. The posterior limb of the internal capsule is often involved. Abnormal signal is seen in brainstem tracts, and the upper and middle cerebellar peduncles are often involved (see Figure 71-6C) [Bergui et al., 1997; Schwankhaus et al., 1988]. Testing for LMNB1 duplications can provide important diagnostic and genetic counseling information.
Adult-Onset Leukoencephalopathies
Neuroaxonal Leukodystrophy with Spheroids
Neuroaxonal leukodystrophy with spheroids is an important leukoencephalopathy with primarily adult presentation. Neuroaxonal leukodystrophy with spheroids, also known as hereditary diffuse leukoencephalopathy with spheroids (HDLS, OMIM 221820) [Axelsson et al., 1984], has no known genetic cause. It is possibly the same entity as pigmentary orthochromatic leukodystrophy (POLD) [Marotti et al., 2004], with overlap in clinical symptoms, histopathology, and neuroimaging [Wider et al., 2009]. Until a biomarker or genetic etiology is identified, however, it will be difficult to clarify the spectrum of this disorder. Inheritance appears to be autosomal-dominant, although autosomal-recessive inheritance has been reported in POLD, and reports of sporadic mutations exist for both disorders.
The clinical manifestations of HDLS are fairly protean, and it is likely underdiagnosed. Onset occurs most commonly in the fifth decade, although a broader range of age of presentation has been reported, including in adolescence [Wider et al., 2009]. Behavioral and cognitive changes tend to predominate in the early stages, and are most often initially classified as depression and anxiety [Wider et al., 2009]. These neuropsychiatric symptoms may be severely debilitating, and often result in diagnosis of frontal dementia. Gait impairment, with pyramidal and extrapyramidal features, is often seen, sometimes with ataxia. Epilepsy is not uncommon. Peripheral nerve involvement is not reported. Profound dementia leads to death, usually within a decade of onset.
Pathophysiology remains poorly understood in this disorder. Pathologic specimens show myelin loss and axonal destruction, alongside axonal spheroids and lipid-laden, autofluorescent sudanophilic macrophages. The primary pathophysiologic event in HDLS is unknown, although mechanisms such as oxidative damage [Ali et al., 2007] and post-translational modification of myelin-associated proteins [Giordana et al., 2005] have been hypothesized.
Diagnosis is usually suspected based on familial inheritance, clinical features, and neuroimaging. Neuroimaging is nonspecific, and may show predominance of frontoparietal lobe involvement, with periventricular, corpus callosum, and central white matter. White-matter abnormalities may be nonconfluent and patchy, particularly in early-onset cases. White-matter abnormalities extend through the internal capsule, involving the pyramidal tracts in the brainstem [van der Knaap et al., 2000; Freeman et al., 2009]. Severe frontal atrophy and ventricular enlargement often develops (see Figure 71-6G). Definitive diagnosis can only be established by autopsy or brain biopsy. Identification of axonal spheroids and autofluorescent sudanophilic macrophages on pathologic specimens is particularly helpful for establishing this diagnosis. There is no specific treatment for HDLS at this time.
Secondary Leukoencephalopathies Associated with Inborn Errors of Metabolism, Excluding the Classical Lysosomal and Peroxisomal Disorders
Inborn errors of metabolism result in significant disturbances of cellular function, and the resultant disruption in energy metabolism, or accumulation of toxic metabolites, interferes with a normal function of a number of organs. The brain, specifically glial cells, is sensitive to such disturbances and, as such, secondary abnormalities of cerebral white matter can be seen in a broad range of inborn errors of metabolism. In some cases, such as the metachromatic leukodystrophies and Krabbe’s disease, the phenotype almost exclusively affects the white matter of the brain, and these disorders have long been classified as leukodystrophies. In a number of other disorders, however, multisystem abnormalities are seen, and white-matter involvement is one of many disease symptoms. These disorders are grouped below into disorders of small molecule metabolism and dysfunction of specific organelles. In the majority of cases, straightforward biochemical testing will exclude these disorders, and comprehensive testing should be considered in unsolved leukodystrophies since many of these disorders show nonspecific white-matter abnormalities (Table 71-2).
Table 71-2 Metabolic Blood, Urine, and Cerebrospinal Fluid Testing to Consider in the Evaluation of a Patient with Leukoencephalopathy of Unknown Cause
| Biochemical Test | Disorders |
|---|---|
| Plasma lactate and pyruvate | Pyruvate dehydrogenase, pyruvate carboxylase, other mitochondrial leukoencephalopathies |
| Ammonia | Urea cycle disorders |
| Plasma amino acids | Urea cycle disorders, tyrosinemia, NKH, disorders of transulfuration (cobalamin C), PKU, MSUD |
| Urine amino acids | Aspartylglucosaminuria |
| Urine organic acids | Canavan’s disease, glutaric aciduria type 1, glutaric aciduria type 2, L2-hydroxyglutaric aciduria, HMG CoA deficiency, multiple carboxylase deficiency, SSADH |
| Lysosomal enzymes on leukocytes | Krabbe’s disease, MLD, sialidoses, fucosidosis, α-mannosidase, β-mannosidase, GM1, GM2, Fabry’s disease |
| Plasma very long chain fatty acids, plasmalogens | XALD and other peroxisomal leukodystrophies |
| Copper/ceruloplasmin | Menkes’ and Wilson’s disease |
| Cholestanol | Cerebrotendinous xanthomatosis |
| 7-dehydrocholesterol/plasma sterols | Smith–Lemli–Opitz syndrome |
| Serum gastrin | Mucolipidosis type IV |
| Isoelectric focusing of transferrin | N-linked glycosylation defects |
| Electron micrography of buffy coat, palmitoyl protein thioesterase, tripeptidylpeptidase 1, cathepsin D | CLN1 and CLN2 |
| Plasma thymidine and deoxyuridine levels | MNGIE |
| GALT enzyme activity and gal-1-P | Galactosemia |
| Biotinidase | Biotinidase deficiency |
| Vitamin B12, folic acid | Nutritional deficiencies |
| Cortisol, TSH, FSH, LH | Primary endocrine abnormality, or secondary to primary leukodystrophy (4H syndrome or XALD) |
| Urinary sulfatides | MLD, MSD and saposin B deficiency |
| Urine mucopolysaccharides/oligosaccharides | Mucopolysaccharidoses, disorders of glycoprotein degradation |
| Urine S sulfocysteine | Molybdenum co-factor deficiency and isolated sulfite oxidase deficiency |
| Urine free and total sialic acid (or occasionally CSF) | Salla disease |
| Urine and CSF S-Ado and SAICAR | Adenylsuccinate lyase deficiency |
| Urinary ribitol and D-arabitol | Ribose 5 phosphate isomerase deficiency |
| CSF amino acids | NKH, serine synthesis defects |
| CSF neurochemistry (GABA, neopterin, HVA, 5HIAA) | SSADH, Aicardi–Goutières syndrome |
| CSF alpha interferon | Aicardi–Goutières syndrome |
CLN, neuronal ceroid-lipofuscinosis; CSF, cerebrospinal fluid; 5HIAA, 5-hydroxyindoleacetic acid; FSH, follicle stimulating hormone; GABA, gamma-aminobutyric acid; GALT, galactose-1-phosphate uridylyltransferase; HMG CoA, 3-hydroxy-3-methylglutaryl coenzyme A; HVA, homovanillic acid; LH, luteinizing hormone; MLD, metachromatic leukodystrophy; MNGIE, mitochondrial neurointestinal gastrointestinal encephalopathy; MSD, multiple sulfatase deficiency; MSUD, maple syrup urine disease; NKH, nonketotic hyperglycinemia; PKU, phenylketonuria; S-Ado, succinyladenosine; SAICAR, succinylaminoimidazole carboxamide ribotide; SSADH, succinic semialdehyde dehydrogenase deficiency; TSH, thyroid stimulating hormone; XALD, X-linked adrenoleukodystrophy.
Metabolic Disturbance in Pathways Involving Small Molecules
Defects in Glucose/Carbohydrate Metabolism
Defects of N-glycan synthesis
Many proteins require post-translational modifications for normal cellular function. One such modification is N-linked glycosylation, so named for linkage of glycans to the amide group of asparagines via an N-acetylglucosamine residue. Congenital disorders of glycosylation (CDG) are the result of defects of N-glycan synthesis in the cytosol or endoplasmic reticulum, or N-glycan processing in the Golgi apparatus. These disorders present with a range of symptoms, depending on the specific defect, and can include facial anomalies, liver dysfunction, hematologic disturbances, abnormal distribution of subcutaneous fat, and neurologic abnormalities, including cerebellar hypoplasia. Delayed myelination has been reported in CDG-IL [Weinstein et al., 2005], and regions of demyelination were reported in CDG-1x [de Lonlay et al., 2001]. Studies of blood protein glycosylation, such as that performed using isoelectric focusing of transferrin, can aid diagnosis. Molecular testing is available in a number of CDG.
Defects of O-glycan synthesis or dystroglycanopathies
A number of congenital muscular dystrophies are caused by deficiency in the genes responsible for the modification of alpha dystroglycan by O-mannosyl-linked glycans. These include POMT1 and POMT2 (Walker–Warburg syndrome [WWS] [Beltran-Valero et al., 2002; van Reeuwijk et al., 2005]), POMGnT1 (muscle-eye-brain disease [MEB] [Yoshida et al., 2001]), Fukutin (Fukuyama congenital muscular dystrophy [FCMD] [Kobayashi et al., 1998]), Fukutin related protein (FKRP) (congenital muscular dystrophy 1C [MDC1C] [Brockington et al., 2001], limb girdle muscular dystrophy 2I [LGMD2I] [Brockington et al., 2001], MEB and WWS [Beltran-Valero de Bernabe et al., 2004]), and LARGE (congenital muscular dystrophy 1D [MDC1D] [Longman et al., 2003]), although the complexity of this pathway and the number of alpha dystroglycanopathies without mutations in one of these genes suggests that more will be identified in the future. A number of these disorders, particularly WWS and MEB, show significant malformations of cerebral development of the brainstem in association with prominent muscular dystrophy, and altered myelination is often present. There is no peripheral biomarker for this series of disorders, and diagnosis is based on clinical manifestations, staining for dystroglycans on muscle biopsy, and molecular testing.
Ribose-5-phosphate isomerase deficiency
Deficiency of ribose-5-phosphate isomerase (OMIM 608611) has been identified in a single patient with a slowly progressive leukoencephalopathy and peripheral neuropathy [Huck et al., 2004]. This subject had elevations of the polyols ribitol and D-arabitol in body fluids and also on cerebral 1H-MR spectroscopy. No further patients have been identified to date.
Galactosemia
Galactosemia (OMIM 230350) is a defect in galactose metabolism. Several steps occur in the conversion of galactose, and enzymes galactokinase (GALK), galactose-1-phosphate uridylyltransferase (GALT), and uridine diphosphate galactose 4′-epimerase (GALE) have all been implicated in human disease. Classic galactosemia is caused by deficiencies in GALT. These subjects typically present on exposure to galactose with poor feeding, weight loss, vomiting, encephalopathy, and hypotonia. Medical evaluation may document liver failure, bleeding diathesis, septicemia, and cataracts. Treatment is stringent dietary restriction of galactose. Diagnosis is almost always made on newborn screening. These subjects may show moderate to mild hypomyelination, and in some cases cerebellar atrophy, despite appropriate dietary treatment [Hughes et al., 2009].
Disorders Involving Amino Acid Metabolism
Phenylketonuria/dihydropteridine reductase deficiency
Phenylketonuria (PKU, OMIM 261600) is caused by mutations in the phenylalanine hydroxylase (PAH) gene. This autosomal-recessively inherited disorder results in accumulation of phenylalanine (Phe) in tissues and body fluids. Phe is believed to be directly neurotoxic, and untreated individuals suffer permanent neurologic damage. Tetrahydrobiopterin (BH4) is a co-factor for Phe hydroxylation, and dihydropteridine reductase deficiency (OMIM 261640), with its resultant BH4 deficiency, causes hyperphenylalaninemia without PAH deficiency. Diagnosis of PKU is based on detection of elevated Phe on newborn screening or plasma amino acids. Exclusion of dihydropteridine reductase deficiency is based on urinary pterins and neurotransmitter derivatives in urine, plasma, and cerebrospinal fluid. Treatment of PKU is based on strict dietary Phe restriction. Dihydropteridine reductase deficiency may be treated with exogenous BH4. Untreated PKU can result in significant white-matter abnormalities, with diffuse white-matter volume loss. With the advent of newborn screening, these situations are rare, but patients can still manifest abnormal neuroimaging, with patchy T2-weighted and FLAIR signal abnormalities more prominent in the frontal and parietal periventricular regions [Phillips et al., 2001; Leuzzi et al., 1995; Thompson et al., 1993]. This appears to be more prominent in late or poorly treated cases. Dihydropteridine reductase deficiency is notable for a leukoencephalopathy that may be diffuse in nature, and is associated with globus pallidus, central white-matter, and subcortical calcifications [Gudinchet et al., 1992].
Maple syrup urine disease
Maple syrup urine disease (MSUD, OMIM 248600), or branched-chain ketoaciduria, is caused by deficiency in the branched-chain α-ketoacid dehydrogenase (BCKD) complex, resulting in accumulation of the branched-chain amino acids (BCAAs) leucine, isoleucine, and valine, and the corresponding branched-chain α-ketoacids (BCKAs). Diagnosis is based on increased BCAAs in plasma, as well as the finding of allo-isoleucine on plasma amino acid screening. Inheritance is autosomal-recessive. Treatment includes both dietary management and aggressive metabolic support during acute decompensation. Certain sources suggest that adequate metabolic control can minimize white-matter abnormalities [Muller et al., 1993], but others describe chronic changes in the mesencephalon, brainstem, thalamus, and globus pallidus [Schonberger et al., 2004]. These subjects can have acute changes in cerebral white matter during episodes of acute metabolic decompensation, and, if left untreated, develop delayed myelin maturation and symmetrical signal abnormality within the globi pallidi and brainstem [Ben-Omran et al., 2006]. It is possible that these discrepant views represent different levels of disease severity.
Disorders of transsulfuration
Methionine adenosyltransferase deficiency (MAT I and MAT III), S-adenosylhomocysteine hydrolase deficiency, methylenetetrahydrofolate reductase deficiency, and disorders affecting methylcobalamin metabolism (cobalamin C, D, E, and G disorders) may all result in hypomyelinating or demyelinating CNS lesions. These subjects are often detected when newborn screening demonstrates hypermethioninemia. Some authors postulate that disorders of transsulfuration interfere with appropriate myelin basic protein methylation [Horster et al., 2005].
Classic homocystinuria may also result in a similar pattern of demyelination in rare cases [Vatanavicharn et al., 2008], although the pathophysiology is less well understood. Homocystinuria is most often caused by cystathionine β-synthase (CBS) deficiency. Patients typically present with dislocation of the optic lens, osteoporosis, thinning and lengthening of the long bones, cognitive impairment, and thromboembolism. Some patients are pyridoxine-dependent. Most patients are managed with low-methionine, cystine-supplemented diets and betaine.
Disorders of GABA metabolism
Succinic semialdehyde dehydrogenase deficiency (4-hydroxybutyric aciduria, OMIM 271980) impairs the oxidative conversion of succinic semialdehyde (SSA) to succinic acid, resulting in its conversion to 4-hydroxybutyric acid (4-HBA), detectable in body fluids. An array of neurologic abnormalities is seen on presentation, including hypotonia, ataxia, neurocognitive disturbances, and seizures. Neuroimaging most commonly reveals signal abnormalities of the globus pallidus, dentate nucleus, and brainstem, but has also been documented to show T2 signal abnormalities in subcortical white matter [Pearl et al., 2003; Pearl and Gibson, 2004].
Nonketotic hyperglycinemia
Nonketotic hyperglycinemia (NKH, OMIM 605899) is an inborn error of glycine degradation, with symptoms caused by accumulation of glycine related to deficient glycine cleavage. Neonatal presentation is predominant, with apnea, hypotonia, and intractable seizures, although some patients present later in life with progressive symptoms. Diagnosis is made based on the presence of plasma and CSF glycine elevations on amino acid profiles. Demonstrating a CSF/plasma glycine concentration ratio greater than 0.08 is diagnostic. No effective treatment exists. MRI demonstrates delayed myelin development [Shah et al., 2005; Mourmans et al., 2006], as well as disturbances in myelin on diffusion-weighted imaging. Callosal abnormalities have been described in some cases.
Tyrosinemia
Tyrosinemia type I (OMIM 276700) is an autosomal-recessive disease caused by fumarylacetoacetate hydrolase (FAH) deficiency. Clinical presentation can include renal Fanconi’s syndrome, liver dysfunction, bleeding diathesis, peripheral neuropathy, and central neurologic dysfunction. Hypertyrosinemia can be found on plasma amino acids. Elevated succinylacetone in plasma or urine is diagnostic. Treatment includes dietary restriction of phenylalanine and tyrosine, and hepatic transplantation. MRI white-matter abnormalities, with diffuse delay in myelination and posterior centrum semiovale T2 signal abnormalities, have been described in tyrosinemia [Sener, 2005].
Urea cycle disorders
The urea cycle removes waste nitrogen and provides de novo synthesis of arginine. Most urea cycle disorders are characterized by hyperammonemia, encephalopathy, and respiratory alkalosis (carbamyl phosphate synthetase [CPS], ornithine transcarbamylase [OTC], argininosuccinic acid synthetase [AS], and argininosuccinate lyase [AL]). A fifth disorder, arginase deficiency, is characterized by progressive spastic quadriplegia and cognitive impairment. Diagnosis is considered based on plasma amino acid and ammonia measurements. Final confirmation is usually molecular. Treatment requires restriction of dietary protein intake and activation of other pathways for waste nitrogen synthesis and excretion, such as sodium phenylbutyrate or arginine. Patients usually have significant diffuse white-matter abnormalities related to generalized edema during acute episodes. Osmotic changes during the acute episodes can result in permanent white-matter damage [Cardenas and Bodensteiner, 2009], and patchy multifocal white-matter changes can be seen chronically in OTC [Kurihara et al., 2003], and CPS [Call et al., 1984].
Purines and Pyrimidines
Adenylosuccinate lyase deficiency
Adenylosuccinate lyase deficiency (OMIM 608222) causes cognitive impairment, epilepsy, and autistic features. Measurements in urine and CSF of succinyladenosine (S-Ado) and succinylaminoimidazole carboxamide ribotide (SAICAR) can aid diagnosis. No effective therapeutic approach has been established. MRI shows atrophy and, in some patients, elevated signal of the dentate nucleus; in very young children, white-matter areas show elevated T2 signal [Mierzewska et al., 2009].
Disorders Involving Urine Organic Acid Excretion
Multiple carboxylase deficiency
Multiple carboxylase deficiency (OMIM 253270) leads to impaired activity in four biotin-dependent enzymes: acetyl CoA carboxylase, propionyl CoA carboxylase, 3-methylcrotonyl CoA carboxylase, and pyruvate carboxylase. The clinical signs of this disorder include episodes of ketoacidosis, alopecia, diffuse erythematous rash, hypotonia, seizures, and developmental delay. These patients may have significant white-matter abnormalities. A single patient with isolated 3-methylcrotonyl CoA carboxylase deficiency had neuroimaging remarkable for multifocal patchy white-matter abnormalities in deep white matter [de Kremer et al., 2002].
L2-hydroxyglutaric aciduria
L2-hydroxyglutaric aciduria (L2HGA, OMIM 236792) is a deficiency of L-2-hydroxyglutarate dehydrogenase, which catalyzes the conversion of L-2-hydroxyglutarate to 2-ketoglutarate. It is characterized by the accumulation of L2 hydroxyglutaric acid in body fluids. Clinically, it presents with nonspecific neurodevelopmental difficulties, ataxia, and seizures. L2-hydroxyglutaric aciduria has a distinct MRI pattern, combining predominantly subcortical cerebral white-matter abnormalities, preservation of periventricular white-matter tissue, and abnormalities of the dentate nucleus, globus pallidus, putamen, and caudate nucleus [Steenweg et al., 2009] (see Figure 71-6D).
Glutaric academia type I
Glutaric academia type I (OMIM 231670) is caused by deficiency of glutaryl-CoA dehydrogenase. Clinically, this disorder is characterized by episodic decompensations, sometimes after illness or surgery, with resulting dystonia or dyskinesia. Diagnosis is often made on newborn screening, or by glutaric and 3-hydroxyglutaric acids in urine. MRI features include frontotemporal atrophy creating the appearance of “open” opercula and degeneration of the caudate and the putamen. However, periventricular white-matter abnormalities are also seen [Twomey et al., 2003].
Sjögren–larsson syndrome
Sjögren–Larsson syndrome (OMIM 270200) is an autosomal-recessive disorder characterized by ichthyosis, spastic diplegia or tetraplegia, and cognitive impairment. Inheritance is autosomal-recessive. The disorder is caused by mutations in the fatty aldehyde dehydrogenase (FALDH) gene, which result in impaired oxidation of long-chain aliphatic aldehydes derived from fatty alcohol metabolism. Neuroimaging reveals white-matter disease in most patients, with mild periventricular signal abnormalities on T2-weighted images. 1H MR spectroscopy reveals characteristic elevation of lipids to approximately 1.3 ppm [Willemsen et al., 2004].
Vitamin/Co-Factor Deficiencies
Biotinidase deficiency
Biotin is a co-factor for four important carboxylases: pyruvate carboxylase, propionyl CoA carboxylase, β-methylcrotonyl CoA carboxylase, and acetyl CoA carboxylase, and its deficiency results in significant alterations in gluconeogenesis, fatty acid synthesis, and amino acid metabolism. Patients can exhibit ketolactic acidosis with elevated concentrations of β-hydroxyisovalerate, lactate, β-methylcrotonylglycine, β-hydroxypropionate, and methylcitrate. Treatment with oral biotin is often successful. Untreated biotinidase deficiency (OMIM 253260) can be associated with significant white-matter abnormalities, most often delayed myelination [Grunewald et al., 2004].
Disorders of Metal Metabolism
Wilson’s disease
Wilson’s disease (OMIM 277900) is a disorder of impaired biliary copper excretion. The dysfunction of copper-transporting adenosine triphosphatases (ATPases) impairs transport of copper into the hepatocyte secretory pathway for incorporation into ceruloplasmin and excretion into the bile. This disorder is autosomal-recessive. Copper accumulates in the liver, cornea (Kayser–Fleischer rings), and brain, resulting in the hepatic and neuropsychiatric manifestations of this disease. Patients have decreased serum ceruloplasmin, increased urinary copper, and elevated hepatic copper concentration. Copper chelators are the mainstay of treatment. Neuroimaging reveals basal ganglia lesions, as well as nonspecific white-matter changes, including cystic degeneration [Sinha et al., 2006; Sankhyan et al., 2008].
Menkes’ disease
Menkes’ disease (OMIM 309400) is an X-linked disorder of copper deficiency, resulting from impaired transport of copper across the placenta, gastrointestinal tract, and blood–brain barrier due to an abnormal ATPase transporter. Clinical characteristics include skin laxity, abnormal hair, light skin pigmentation, and failure to thrive. Patients have decreased serum copper and ceruloplasmin. There is no effective treatment in severe cases, though early treatment with copper histidine is given in mild cases. Neuroimaging in Menkes’ disease may show severe atrophy and white-matter abnormalities, with prominent temporal lobe changes and reports of temporal pole cystic degeneration [Ichihashi et al., 1990; Blaser et al., 1989]. Clinical features and prominent tortuous vessels apparent on neuroimaging should facilitate this diagnosis.
Fatty Acid Metabolism
27-Hydroxylase deficiency (cerebrotendinous xanthomatosis)
27-hydroxylase deficiency or cerebrotendinous xanthomatosis (CTX, OMIM 213700) is an inherited disorder of sterol storage, with accumulation of cholestanol and cholesterol in xanthomas, gallbladder, and brain. Affected patients have encephalopathy, myelopathy, cerebellar ataxia, early atherosclerosis, tendon xanthomas, and cataracts. Diagnosis is based on clinical manifestations and elevations in cholestanol in the serum. Neuroimaging is remarkable for deep white-matter changes, and hyperintense lesions seen on T2-weighted images in the dentate nucleus, globus pallidus, substantia nigra, and inferior olive [Barkhof et al., 2000]. Affected white matter may become calcified. Patients may be treated successfully using chenodeoxycholic acid.
Smith–Lemli–Opitz syndrome
Smith–Lemli–Opitz syndrome (OMIM 270400) is caused by a deficiency of 7-dehydrocholesterol reductase (DHCR7), resulting in markedly increased levels of 7-dehydrocholesterol (7DHC) and decreased levels of cholesterol. Affected patients show craniofacial abnormalities, extremity dysmorphisms (2–3 toe syndactyly), structural brain abnormalities, behavioral disturbances, and variable levels of neurologic disability. Neuroimaging is remarkable for delayed myelination [Korade et al., 2009].
Organelle Dysfunction Resulting in Leukoencephalopathy
Lysosomal Disorders
Disorders of sphingolipid metabolism
Fabry’s Disease
Fabry’s disease (OMIM 301500) is an X-linked disorder caused by deficiency of lysosomal hydrolase alpha-galactosidase A, with subsequent accumulation of glycosphingolipids (globotriaosylceramide) in body fluids and cells in the walls of blood vessels, as well as most other tissues, including the brain. Affected males have angiokeratoma, renal impairment, paresthesias, and cerebrovascular disease. MRIs are remarkable for patchy white-matter lesions that may become confluent and cystic over time, and are thought to represent lacunar infarctions [Ginsberg et al., 2006].
GM1 Gangliosidosis
GM1 gangliosidosis is caused by deficiency of lysosomal acid β-galactosidase (β-galactosidosis). GM1 gangliosidosis–affected patients show macular cherry-red spots, hepatosplenomegaly, generalized skeletal dysplasia, early developmental arrest, and progressive neurologic deterioration. Rarer, late-onset forms exist. Diagnosis can be made by measuring oligosaccharides and gangliosides in urine of affected individuals. In early-onset cases, severe hypomyelination with complete arrest in myelination can be seen (see Figure 71-3F), although in late-onset cases, myelination may develop on MRI and abnormal signal within the globus pallidus and cortical atrophy may be the only diagnostic indicator [De Grandis et al., 2009].
GM2 Gangliosidoses
The GM2 gangliosidoses are characterized by excessive accumulation of ganglioside GM2 in the lysosomes, mainly of neuronal cells. Three disorders – Tay–Sachs disease (mutations in HEXA and deficient activity of hexosaminidase A), Sandhoff’s disease (mutations in HEXB and deficient activity of hexosaminidase A and hexosaminidase B), and GM2 activator deficiency (mutations in GM2A and inability to form GM2/GM2 activator complex) – all have deficient hydrolysis of ganglioside GM2. Tay–Sachs disease and GM2 activator deficiency are clinically very similar, while Sandhoff’s disease is distinguished by its visceral involvement. In all cases, disease presentation may vary widely with age at disease onset. All three disorders are marked by atrophy, hypomyelination, and basal ganglia signal abnormality in early-onset cases, and milder abnormalities in later-onset cases, with reports of isolated cerebellar atrophy [Grosso et al., 2003].
Niemann–Pick C Disease
Niemann–Pick C disease (NP-C, OMIM 257220) is caused by an error of cellular trafficking of cholesterol resulting in lysosomal accumulation of unesterified cholesterol (NPC1 mutations in most, but not all, subjects). Symptoms vary with age of onset, and include childhood neurologic presentations with vertical ophthalmoplegia, ataxia, dystonia, dementia, hepatosplenomegaly, and fatal neonatal liver disease, as well as adult-onset neuropsychiatric presentations. Diagnosis is based on clinical presentation, filipin cholesterol staining of tissues, and functional cell studies. Neuroimaging of affected patients can show delayed myelination or parietal white-matter changes and thinning of the corpus callosum [Palmeri et al., 1994] in addition to supra- and infratentorial atrophy.
Mucopolysaccharidoses, mucolipidoses, and oligosaccharidoses
Mucopolysaccharidoses
The mucopolysaccharidoses (MPS) are lysosomal storage disorders caused by deficient degradation of glycosaminoglycans (mucopolysaccharides). Seven distinct MPS exist, with multisystem clinical involvement, including organomegaly, dysostosis multiplex, abnormal facies, and variable cognitive deficits. Diagnosis is based on glycosaminoglycan urinary excretion and analysis of specific enzyme activity. Neuroimaging abnormalities involving the white matter with dilated perivascular spaces, periventricular confluent signal abnormalities, and multifocal subcortical white-matter lesions are seen [Wang et al., 2009; Vedolin et al., 2007]. Over time, these may progress to confluency. Sanfilippo’s syndrome may present with hypomyelination. Some of these disorders are currently treated with enzyme replacement therapy. In other disorders, bone marrow transplant treatment studies are on-going.
Mucolipidosis Type IV
Mucolipidosis type IV (MLIV, OMIM 252650) is caused by alteration of a transmembrane channel (MCOLN1 encoding mucolipin-1), whose function is poorly understood. It results in lysosomal inclusions, and a clinical phenotype characterized by severe developmental delay, retinal degeneration, corneal clouding, and achlorhydria. Elevated blood gastrin can further suggest the diagnosis. Neuroimaging shows thinning of the corpus callosum, diffuse nonhomogeneous white-matter signal abnormality throughout the cortical white matter, T2 hypointensity of the basal ganglia and thalamus attributed to ferritin deposition, and cerebellar atrophy [Frei et al., 1998]. No specific treatment exists.
Disorders of Glycoprotein Degradation: α-Mannosidosis, β-Mannosidosis, Fucosidosis, and Sialidosis
These disorders are deficiencies in glycoprotein or oligosaccharide degradation. α-mannosidosis (deficiency of α-mannosidase) is characterized by variable symptoms depending on age at onset; symptoms include neurologic, bony, and liver/spleen substrate accumulation. β-mannosidosis (deficiency of β-mannosidase) is characterized by neurologic, hearing, skin (angiokeratoma), and respiratory manifestations. Fucosidosis (deficiency of the lysosomal hydrolase α-fucosidase) is characterized by variable age-dependent symptoms, including neurologic, skin, and bony substrate accumulation. Patients may have an increase in sweat sodium chloride. Sialidosis (deficiency of α2 → 3 and α2 → 6 neuraminidase) has a variable phenotype, with older-onset patients (type I) developing cherry-red spots and myoclonus in adulthood, and type II patients presenting in infancy with an MPS-like phenotype. In animal models and humans, both α-mannosidosis, and β-mannosidosis are known to cause myelin lesions, seen on MRI as focal signal abnormalities in the parieto-occipital white matter [Dietemann et al., 1990]. Hypomyelination tends to be the predominant neuroradiologic sign in fucosidosis [Prietsch et al., 2008], although involvement of the basal ganglia, especially the pallidum, and thalami is seen over time, with low signal intensity on T2-weighted images.
Aspartylglucosaminuria
Aspartylglucosaminuria (OMIM 208400) is caused by deficiency of the lysosomal enzyme aspartylglucosaminidase, glycoasparaginase (AGA), resulting in accumulation of glycoasparagines in lysosomes. Developmental delay presents in toddlers, with slowly progressive deterioration into adulthood. Patients may develop a coarse facies, thick calvarium, osteoporosis, and seizures. Glycoasparagines are excreted in urine and AGA activity can be assayed in leukocytes. Imaging is characterized by delayed myelination, and decreased distinction between gray and white matter [Autti et al., 1997].
Neuronal ceroid-lipofuscinoses
The neuronal ceroid-lipofuscinoses (NCL) are a group of etiologically heterogeneous neurodegenerative disorders. They are remarkable for accumulation of autofluorescent material in brain and other tissues, and for clinical presentations with severe refractory epilepsy, blindness, and dementia. Several of the genes causing NCL encode lysosomal proteins, including CLN1 (palmitoyl-protein thioesterase) and CLN2 (tripeptidyl peptidase 1), both soluble lysosomal enzymes, and CLN3 (battenin), a lysosomal membrane protein. Affected subjects have high-intensity, hazy, periventricular white-matter signal abnormality in the corona radiata, associated with low signal in the basal ganglia on T2-weighted images (see Figure 71-3H) [Vanhanen et al., 2004; D’Incerti, 2000; Autti et al., 1996]. Significant cerebral and cerebellar atrophy can be seen over time.
Peroxisomal Disorders
Peroxisome biogenesis disorders
Peroxisome biogenesis disorders (PBD; see also Chapter 38) are abnormalities of peroxisome biosynthesis caused by mutations in a series of PEX genes (at least 15 in humans), encoding various peroxins. A variety of clinical presentations exists. Rhizomelic chondrodysplasia punctata, characterized by skeletal deformities, facial dysmorphisms, and developmental abnormalities, is associated with hypomyelination or focal white-matter signal abnormalities [Viola et al., 2002]. A second clinical phenotype is characterized by the Zellweger spectrum, in which patients can present at variable ages with facial dysmorphisms, hypotonia, seizures, liver dysfunction, and developmental delay. Neuroimaging can show cerebral malformations and both hypo- and demyelination in cerebral and cerebellar white matter (see Figure 71-6F) [Barth et al., 2004]. Metabolic testing reveals abnormalities of plasmalogens and very long chain fatty acids.
Mitochondrial Leukoencephalopathies
A broad range of mitochondrial disorders (see also Chapter 37) can present with significant white-matter abnormalities. Beyond the representative disorders discussed here, white-matter involvement can also be seen in MELAS, Leber’s hereditary optic neuropathy, Kearns–Sayre syndrome, Leigh’s syndrome, and a growing list of other nuclear mutations causing mitochondrial disease.
DNA polymerase gamma 1
DNA polymerase gamma 1 (OMIM 203700), encoded by POLG1, is involved in human mitochondrial DNA replication, and POLG1 mutations can cause mitochondrial depletion and a variety of mitochondrial phenotypes, including MELAS-like presentations and progressive external ophthalmoplegia. Patients with less specific presentations may show significantly delayed myelination (see Figure 71-3G) or demyelination [Bao et al., 2008].
Mitochondrial neurogastrointestinal encephalopathy
Mitochondrial neurogastrointestinal encephalopathy (MNGIE, OMIM 603041) is caused by deficiency of thymidine phosphorylase (encoded by ECGF1). MNGIE is characterized by progressive gastrointestinal dysmotility, ophthalmoplegia, hearing loss, and demyelinating peripheral neuropathy. Patients show increased plasma thymidine and plasma deoxyuridine concentrations. Significant diffuse leukoencephalopathy is seen on neuroimaging [Schupbach et al., 2007].
Pyruvate carboxylase and pyruvate dehydrogenase
Deficiency of pyruvate carboxylase (anabolic utilization of pyruvate, OMIM 266150) or pyruvate dehydrogenase (oxidative metabolism of pyruvate, OMIM 312170) causes pyruvate and lactate accumulation. Pyruvate carboxylase-deficient patients present in infancy with lactic acidosis. MRI may show Leigh’s syndrome or predominant leukoencephalopathy involving central white matter and the brainstem [Higgins et al., 1994]. Pyruvate dehydrogenase-deficient patients present in infancy with lactic acidosis or early developmental arrest and seizures. Inheritance is autosomal-recessive or X-linked, depending on the defective subunit. Patients can present with a Leigh’s syndrome-like encephalopathy, or with a leukoencephalopathy associated with structural abnormalities of the corpus callosum and posterior fossa, and cystic degeneration of affected white matter.
Inborn errors of ketone body metabolism
3-Hydroxy-3-Methylglutaryl Coenzyme A Lyase (HMG-CoA) Deficiency
3-Hydroxy-3-methylglutaryl coenzyme A lyase (HMG-CoA) deficiency (OMIM 246450) is a disorder of ketogenesis from fatty acids and ketogenic amino acids. Patients may have episodes of hypoketotic hypoglycemia and high levels of 3-hydroxy-3-methylglutaric acid (HMG), with related leucine metabolites in urine. Patients may have cognitive impairment and seizures. MRI findings are characterized by patchy white-matter changes in central and subcortical white matter [Bischof et al., 2004], which may or may not improve with metabolic support to prevent hypoglycemic episodes.
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Abdel-Salam G.M., Zaki M.S. Band-like intracranial calcification (BIC), microcephaly and malformation of brain development: a distinctive form of congenital infection like syndromes. Am J Med Genet A. 2009;149A:1565-1568.
Adachi M., Kawanami T., Ohshima F., et al. MR findings of cerebral white matter in Cockayne syndrome. Magn Reson Med Sci. 2006;5:41-45.
Aicardi J., Goutieres F. Systemic lupus erythematosus or Aicardi-Goutieres syndrome? Neuropediatrics. 2000;31:113.
Alexander W.S. Progressive fibrinoid degeneration of fibrillary astrocytes associated with mental retardation in a hydrocephalic infant. Brain. 1949;72:373-381. 373 pl,
Ali Z.S., Van Der Voorn J.P., Powers J.M. A comparative morphologic analysis of adult onset leukodystrophy with neuroaxonal spheroids and pigmented glia – a role for oxidative damage. J Neuropathol Exp Neurol. 2007;66:660-672.
Artigas J., Poo P., Rovira A., et al. Macrocephaly and dilated Virchow-Robin spaces in childhood. Pediatr Radiol. 1999;29:188-190.
Aula N., Salomaki P., Timonen R., et al. The spectrum of SLC17A5-gene mutations resulting in free sialic acid-storage diseases indicates some genotype-phenotype correlation. Am J Hum Genet. 2000;67:832-840.
Austin J., McAfee D., Armstrong D., et al. Abnormal sulphatase activities in two human diseases (metachromatic leucodystrophy and gargoylism). Biochem J. 1964;93:15C-17C.
Austin J., Suzuki K., Armstrong D., et al. Studies in globoid (Krabbe) leukodystrophy (GLD). V. Controlled enzymic studies in ten human cases. Arch Neurol. 1970;23:502-512.
Autti T., Raininko R., Haltia M., et al. Aspartylglucosaminuria: radiologic course of the disease with histopathologic correlation. J Child Neurol. 1997;12:369-375.
Autti T., Raininko R., Vanhanen S.L., et al. MRI of neuronal ceroid lipofuscinosis. I. Cranial MRI of 30 patients with juvenile neuronal ceroid lipofuscinosis. Neuroradiology. 1996;38:476-482.
Axelsson R., Roytta M., Sourander P., et al. Hereditary diffuse leucoencephalopathy with spheroids. Acta Psychiatr Scand Suppl. 1984;314:1-65.
Bao X., Wu Y., Wong L.J., et al. Alpers syndrome with prominent white matter changes. Brain Dev. 2008;30:295-300.
Barkhof F., Verrips A., Wesseling P., et al. Cerebrotendinous xanthomatosis: the spectrum of imaging findings and the correlation with neuropathologic findings. Radiology. 2000;217:869-876.
Barth P.G., Majoie C.B., Gootjes J., et al. Neuroimaging of peroxisome biogenesis disorders (Zellweger spectrum) with prolonged survival. Neurology. 2004;62:439-444.
Beltran-Valero de Bernabe D., Currier S., Steinbrecher A., et al. Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am J Hum Genet. 2002;71:1033-1043.
Beltran-Valero de Bernabe D., Voit T., Longman C., et al. Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker-Warburg syndrome. J Med Genet. 2004;41:e61.
Ben-Omran T.I., Blaser S., Phillips H., et al. Atypical phenotype in a boy with a maple syrup urine disease. J Inherit Metab Dis. 2006;29:195-200.
Ben-Zeev B., Levy-Nissenbaum E., Lahat H., et al. Megalencephalic leukoencephalopathy with subcortical cysts; a founder effect in Israeli patients and a higher than expected carrier rate among Libyan Jews. Hum Genet. 2002;111:214-218.
Bergui M., Bradac G.B., Leombruni S., et al. MRI and CT in an autosomal-dominant, adult-onset leukodystrophy. Neuroradiology. 1997;39:423-426.
Bezman L., Moser A.B., Raymond G.V., et al. Adrenoleukodystrophy: incidence, new mutation rate, and results of extended family screening. Ann Neurol. 2001;49:512-517.
Bischof F., Nagele T., Wanders R.J., et al. 3-hydroxy-3-methylglutaryl-CoA lyase deficiency in an adult with leukoencephalopathy. Ann Neurol. 2004;56:727-730.
Blaser S.I., Berns D.H., Ross J.S., et al. Serial MR studies in Menkes disease. J Comput Assist Tomogr. 1989;13:113-115.
Boddaert N., Zilbovicius M., Philipe A., et al. MRI findings in 77 children with non-syndromic autistic disorder. PLoS ONE. 2009;4:e4415.
Bonnemann C.G., Meinecke P. Encephalopathy of infancy with intracerebral calcification and chronic spinal fluid lymphocytosis – another case of the Aicardi-Goutieres syndrome. Neuropediatrics. 1992;23:157-161.
Boor I., Nagtegaal M., Kamphorst W., et al. MLC1 is associated with the dystrophin-glycoprotein complex at astrocytic endfeet. Acta Neuropathol. 2007;114:403-410.
Boor P.K., de Groot K., Waisfisz Q., et al. MLC1: a novel protein in distal astroglial processes. J Neuropathol Exp Neurol. 2005;64:412-419.
Brenner M., Johnson A.B., Boespflug-Tanguy O., et al. Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander disease. Nat Genet. 2001;27:117-120.
Briggs T.A., Abdel-Salam G.M., Balicki M., et al. Cerebroretinal microangiopathy with calcifications and cysts (CRMCC). Am J Med Genet A. 2008;146A:182-190.
Briggs T.A., Wolf N.I., D’Arrigo S., et al. Band-like intracranial calcification with simplified gyration and polymicrogyria: a distinct “pseudo-TORCH” phenotype. Am J Med Genet A. 2008;146A:3173-3180.
Brockington M., Blake D.J., Prandini P., et al. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha2 deficiency and abnormal glycosylation of alpha-dystroglycan. Am J Hum Genet. 2001;69:1198-1209.
Brockington M., Yuva Y., Prandini P., et al. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum Mol Genet. 2001;10:2851-2859.
Brussino A., Vaula G., Cagnoli C., et al. A family with autosomal dominant leukodystrophy linked to 5q23.2–q23.3 without lamin B1 mutations. Eur J Neurol. 2009.
Burlina A.P., Ferrari V., Burlina A.B., et al. N-acetylaspartylglutamate (NAAG) in Pelizaeus-Merzbacher disease. Adv Exp Med Biol. 2006;576:353-359. discussion 361–353
Burn J., Wickramasinghe H.T., Harding B., et al. A syndrome with intracranial calcification and microcephaly in two sibs, resembling intrauterine infection. Clin Genet. 1986;30:112-116.
Cailloux F., Gauthier-Barichard F., Mimault C., et al. Genotype-phenotype correlation in inherited brain myelination defects due to proteolipid protein gene mutations. Clinical European Network on Brain Dysmyelinating Disease. Eur J Hum Genet. 2000;8:837-845.
Call G., Seay A.R., Sherry R., et al. Clinical features of carbamyl phosphate synthetase-I deficiency in an adult. Ann Neurol. 1984;16:90-93.
Cardenas J.F., Bodensteiner J.B. Osmotic demyelination syndrome as a consequence of treating hyperammonemia in a patient with ornithine transcarbamylase deficiency. J Child Neurol. 2009;24:884-886.
Chen Y.Q., Rafi M.A., de Gala G., et al. Cloning and expression of CDNA encoding human galactocerebrosidase, the enzyme deficient in globoid cell leukodystrophy. Hum Mol Genet. 1993;2:1841-1845.
Cleaver J.E., Lam E.T., Revet I. Disorders of nucleotide excision repair: the genetic and molecular basis of heterogeneity. Nat Rev Genet. 2009;10:756-768.
Coffeen C.M., McKenna C.E., Koeppen A.H., et al. Genetic localization of an autosomal dominant leukodystrophy mimicking chronic progressive multiple sclerosis to chromosome 5q31. Hum Mol Genet. 2000;9:787-793.
Corzo D., Gibson W., Johnson K., et al. Contiguous deletion of the X-linked adrenoleukodystrophy gene (ABCD1) and DXS1357E: a novel neonatal phenotype similar to peroxisomal biogenesis disorders. Am J Hum Genet. 2002;70:1520-1531.
Coskun T., Besim A., Ozalp I., et al. Intracranial calcification in dihydropteridine reductase deficiency. Turk J Pediatr. 1990;32:259-264.
Crosato F., Senter S. Cerebral occipital calcifications in celiac disease. Neuropediatrics. 1992;23:214-217.
Crow Y.J., Hayward B.E., Parmar R., et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet. 2006;38:917-920.
Crow Y.J., Leitch A., Hayward B.E., et al. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection. Nat Genet. 2006;38:910-916.
Crow Y.J., Livingston J.H. Aicardi-Goutieres syndrome: an important Mendelian mimic of congenital infection. Dev Med Child Neurol. 2008;50:410-416.
Crow Y.J., McMenamin J., Haenggeli C.A., et al. Coats’ plus: a progressive familial syndrome of bilateral Coats’ disease, characteristic cerebral calcification, leukoencephalopathy, slow pre- and post-natal linear growth and defects of bone marrow and integument. Neuropediatrics. 2004;35:10-19.
De Grandis E., Di Rocco M., Pessagno A., et al. MR imaging findings in 2 cases of late infantile GM1 gangliosidosis. Am J Neuroradiol. 2009;30:1325-1327.
de Koning T.J., Jaeken J., Pineda M., et al. Hypomyelination and reversible white matter attenuation in 3-phosphoglycerate dehydrogenase deficiency. Neuropediatrics. 2000;31:287-292.
de Koning T.J., Klomp L.W., van Oppen A.C., et al. Prenatal and early postnatal treatment in 3-phosphoglycerate-dehydrogenase deficiency. Lancet. 2004;364:2221-2222.
de Kremer R.D., Latini A., Suormala T., et al. Leukodystrophy and CSF purine abnormalities associated with isolated 3-methylcrotonyl-CoA carboxylase deficiency. Metab Brain Dis. 2002;17:13-18.
de Lonlay P., Seta N., Barrot S., et al. A broad spectrum of clinical presentations in congenital disorders of glycosylation I: a series of 26 cases. J Med Genet. 2001;38:14-19.
De Vries E., Van Bogaert L., Edgar G.W. [New observations on familial idiocy with spongy degeneration of nerve centers (progressive edematous cerebral disease of infants)]. Rev Neurol (Paris). 1958;98:271-295.
Dietemann J.L., Filippi de la Palavesa M.M., Tranchant C., et al. MR findings in mannosidosis. Neuroradiology. 1990;32:485-487.
D’Incerti L. MRI in neuronal ceroid lipofuscinosis. Neurol Sci. 2000;21:S71-S73.
Duarri A., Teijido O., Lopez-Hernandez T., et al. Molecular pathogenesis of megalencephalic leukoencephalopathy with subcortical cysts: mutations in MLC1 cause folding defects. Hum Mol Genet. 2008;17:3728-3739.
Eldridge R., Anayiotos C.P., Schlesinger S., et al. Hereditary adult-onset leukodystrophy simulating chronic progressive multiple sclerosis. N Engl J Med. 1984;311:948-953.
Ellis D., Malcolm S. Proteolipid protein gene dosage effect in Pelizaeus-Merzbacher disease. Nat Genet. 1994;6:333-334.
Feldmann J., Callebaut I., Raposo G., et al. Munc13–4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell. 2003;115:461-473.
Felice K.J., Grunnet M.L., Rao K.R., et al. Childhood-onset spinocerebellar syndrome associated with massive polyglucosan body deposition. Acta Neurol Scand. 1997;95:60-64.
Fogli A., Rodriguez D., Eymard-Pierre E., et al. Ovarian failure related to eukaryotic initiation factor 2B mutations. Am J Hum Genet. 2003;72:1544-1550.
Fogli A., Wong K., Eymard-Pierre E., et al. Cree leukoencephalopathy and CACH/VWM disease are allelic at the EIF2B5 locus. Ann Neurol. 2002;52:506-510.
Freeman S.H., Hyman B.T., Sims K.B., et al. Adult onset leukodystrophy with neuroaxonal spheroids: clinical, neuroimaging and neuropathologic observations. Brain Pathol. 2009;19:39-47.
Frei K.P., Patronas N.J., Crutchfield K.E., et al. Mucolipidosis type IV: characteristic MRI findings. Neurology. 1998;51:565-569.
Garbern J.Y. Pelizaeus-Merzbacher disease: Genetic and cellular pathogenesis. Cell Mol Life Sci. 2007;64:50-65.
Garbern J.Y., Yool D.A., Moore G.J., et al. Patients lacking the major CNS myelin protein, proteolipid protein 1, develop length-dependent axonal degeneration in the absence of demyelination and inflammation. Brain. 2002;125:551-561.
Gartner J., Dehmel T., Klusmann A., et al. Functional characterization of the adrenoleukodystrophy protein (ALDP) and disease pathogenesis. Endocr Res. 2002;28:741-748.
Ginsberg L., Manara R., Valentine A.R., et al. Magnetic resonance imaging changes in Fabry disease. Acta Paediatr Suppl. 2006;95:57-62.
Giordana M.T., Piccinini M., Palmucci L., et al. Myelin-associated glycoprotein is altered in a familial late-onset orthochromatic leukodystrophy. Brain Pathol. 2005;15:116-123.
Giroud M., Gouyon J.B., Chaumet F., et al. A case of progressive familial encephalopathy in infancy with calcification of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Childs Nerv Syst. 1986;2:47-48.
Gorospe J.R., Singhal B.S., Kainu T., et al. Indian Agarwal megalencephalic leukodystrophy with cysts is caused by a common MLC1 mutation. Neurology. 2004;62:878-882.
Gould D.B., Phalan F.C., Breedveld G.J., et al. Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science. 2005;308:1167-1171.
Goutieres F., Aicardi J., Barth P.G., et al. Aicardi-Goutieres syndrome: an update and results of interferon-alpha studies. Ann Neurol. 1998;44:900-907.
Grand M.G., Kaine J., Fulling K., et al. Cerebroretinal vasculopathy. A new hereditary syndrome. Ophthalmology. 1988;95:649-659.
Greenfield J.G. A Form of Progressive Cerebral Sclerosis in Infants associated with Primary Degeneration of the Interfascicular Glia. Proc R Soc Med. 1933;26:690-697.
Groeschel S., Brockmann K., Dechent P., et al. Magnetic resonance imaging and proton magnetic resonance spectroscopy of megalencephaly and dilated Virchow-Robin spaces. Pediatr Neurol. 2006;34:35-40.
Groeschel S., Chong W.K., Surtees R., et al. Virchow-Robin spaces on magnetic resonance images: normative data, their dilatation, and a review of the literature. Neuroradiology. 2006;48:745-754.
Grosso S., Farnetani M.A., Berardi R., et al. GM2 gangliosidosis variant B1 neuroradiological findings. J Neurol. 2003;250:17-21.
Grunewald S., Champion M.P., Leonard J.V., et al. Biotinidase deficiency: a treatable leukoencephalopathy. Neuropediatrics. 2004;35:211-216.
Gudinchet F., Maeder P., Meuli R.A., et al. Cranial CT and MRI in malignant phenylketonuria. Pediatr Radiol. 1992;22:223-224.
Gutmann D.H., Fischbeck K.H., Sergott R.C. Hereditary retinal vasculopathy with cerebral white matter lesions. Am J Med Genet. 1989;34:217-220.
Hara K., Shiga A., Fukutake T., et al. Association of HTRA1 mutations and familial ischemic cerebral small-vessel disease. N Engl J Med. 2009;360:1729-1739.
Hart C.E., Race V., Achouri Y., et al. Phosphoserine aminotransferase deficiency: a novel disorder of the serine biosynthesis pathway. Am J Hum Genet. 2007;80:931-937.
Hartel C., Bachmann S., Bonnemann C., et al. Familial megalencephaly with dilated Virchow-Robin spaces in magnetic resonance imaging: an autosomal recessive trait? Clin Dysmorphol. 2005;14:31-34.
Henneke M., Combes P., Diekmann S., et al. GJA12 mutations are a rare cause of Pelizaeus-Merzbacher-like disease. Neurology. 2008;70:748-754.
Henneke M., Diekmann S., Ohlenbusch A., et al. RNASET2-deficient cystic leukoencephalopathy resembles congenital cytomegalovirus brain infection. Nat Genet. 2009;41:773-775.
Henneke M., Preuss N., Engelbrecht V., et al. Cystic leukoencephalopathy without megalencephaly: a distinct disease entity in 15 children. Neurology. 2005;64:1411-1416.
Higgins J.J., Glasgow A.M., Lusk M., et al. MRI, clinical, and biochemical features of partial pyruvate carboxylase deficiency. J Child Neurol. 1994;9:436-439.
Horster F., Surtees R., Hoffmann G.F. Disorders of intermediary metabolism: toxic leukoencephalopathies. J Inherit Metab Dis. 2005;28:345-356.
Hubbard W.C., Moser A.B., Liu A.C., et al. Newborn screening for X-linked adrenoleukodystrophy (X-ALD): validation of a combined liquid chromatography-tandem mass spectrometric (LC-MS/MS) method. Mol Genet Metab. 2009;97:212-220.
Huck J.H., Verhoeven N.M., Struys E.A., et al. Ribose-5-phosphate isomerase deficiency: new inborn error in the pentose phosphate pathway associated with a slowly progressive leukoencephalopathy. Am J Hum Genet. 2004;74:745-751.
Hudson L.D., Puckett C., Berndt J., et al. Mutation of the proteolipid protein gene PLP in a human X chromosome-linked myelin disorder. Proc Natl Acad Sci USA. 1989;86:8128-8131.
Hughes J., Ryan S., Lambert D., et al. Outcomes of siblings with classical galactosemia. J Pediatr. 2009;154:721-726.
Ichihashi K., Yano S., Kobayashi S., et al. Serial imaging of Menkes disease. Neuroradiology. 1990;32:56-59.
Iglesias S., Chapon F., Baron J.C., et al. Familial occipital calcifications, hemorrhagic strokes, leukoencephalopathy, dementia, and external carotid dysplasia. Neurology. 2000;55:1661-1667.
Inoue K., Khajavi M., Ohyama T., et al. Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat Genet. 2004;36:361-369.
Inoue K., Ohyama T., Sakuragi Y., et al. Translation of SOX10 3′ untranslated region causes a complex severe neurocristopathy by generation of a deleterious functional domain. Hum Mol Genet. 2007;16:3037-3046.
Inoue K., Shilo K., Boerkoel C.F., et al. Congenital hypomyelinating neuropathy, central dysmyelination, and Waardenburg-Hirschsprung disease: phenotypes linked by SOX10 mutation. Ann Neurol. 2002;52:836-842.
Inoue K., Tanabe Y., Lupski J.R. Myelin deficiencies in both the central and the peripheral nervous systems associated with a SOX10 mutation. Ann Neurol. 1999;46:313-318.
Jen J., Cohen A.H., Yue Q., et al. Hereditary endotheliopathy with retinopathy, nephropathy, and stroke (HERNS). Neurology. 1997;49:1322-1330.
Kalyanasundaram S., Dutta S., Narang A., et al. Microcephaly with plate-like cortical calcification. Brain Dev. 2003;25:130-132.
Kantor L., Harding H.P., Ron D., et al. Heightened stress response in primary fibroblasts expressing mutant eif2b genes from CACH/VWM leukodystrophy patients. Hum Genet. 2005;118:99-106.
Kantor L., Pinchasi D., Mintz M., et al. A point mutation in translation initiation factor 2B leads to a continuous hyper stress state in oligodendroglial-derived cells. PLoS ONE. 2008;3:e3783.
Kaul R., Gao G.P., Aloya M., et al. Canavan disease: mutations among Jewish and non-Jewish patients. Am J Hum Genet. 1994;55:34-41.
Kaul R., Gao G.P., Balamurugan K., et al. Cloning of the human aspartoacylase CDNA and a common missense mutation in Canavan disease. Nat Genet. 1993;5:118-123.
Kobayashi K., Nakahori Y., Miyake M., et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature. 1998;394:388-392.
Korade Z., Kenworthy A.K., Mirnics K. Molecular consequences of altered neuronal cholesterol biosynthesis. J Neurosci Res. 2009;87:866-875.
Kuijpers T.W. Aicardi-Goutieres syndrome: immunophenotyping in relation to interferon-alpha. Eur J Paediatr Neurol. 2002;6(Suppl A):A59-A64. discussion A65–56, A77–86
Kurihara A., Takanashi J., Tomita M., et al. Magnetic resonance imaging in late-onset ornithine transcarbamylase deficiency. Brain Dev. 2003;25:40-44.
Kyllerman M., Rosengren L., Wiklund L.M., et al. Increased levels of GFAP in the cerebrospinal fluid in three subtypes of genetically confirmed Alexander disease. Neuropediatrics. 2005;36:319-323.
Labrune P., Lacroix C., Goutieres F., et al. Extensive brain calcifications, leukodystrophy, and formation of parenchymal cysts: a new progressive disorder due to diffuse cerebral microangiopathy. Neurology. 1996;46:1297-1301.
Lea M.E., Harbord M., Sage M.R. Bilateral occipital calcification associated with celiac disease, folate deficiency, and epilepsy. Am J Neuroradiol. 1995;16:1498-1500.
Lebon P., Badoual J., Ponsot G., et al. Intrathecal synthesis of interferon-alpha in infants with progressive familial encephalopathy. J Neurol Sci. 1988;84:201-208.
Leegwater P.A., Boor P.K., Yuan B.Q., et al. Identification of novel mutations in MLC1 responsible for megalencephalic leukoencephalopathy with subcortical cysts. Hum Genet. 2002;110:279-283.
Leegwater P.A., Konst A.A., Kuyt B., et al. The gene for leukoencephalopathy with vanishing white matter is located on chromosome 3q27. Am J Hum Genet. 1999;65:728-734.
Leegwater P.A., Vermeulen G., Konst A.A., et al. Subunits of the translation initiation factor eif2b are mutant in leukoencephalopathy with vanishing white matter. Nat Genet. 2001;29:383-388.
Leegwater P.A., Yuan B.Q., van der Steen J., et al. Mutations of MLC1 (KIAA0027), encoding a putative membrane protein, cause megalencephalic leukoencephalopathy with subcortical cysts. Am J Hum Genet. 2001;68:831-838.
Lee-Kirsch M.A., Gong M., Chowdhury D., et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet. 2007;39:1065-1067.
Leombruni S., Vaula G., Coletti Moja M., et al. Neurophysiological study in an Italian family with autosomal dominant late-onset leukodystrophy. Electromyogr Clin Neurophysiol. 1998;38:131-135.
Leuzzi V., Trasimeni G., Gualdi G.F., et al. Biochemical, clinical and neuroradiological (MRI) correlations in late-detected PKU patients. J Inherit Metab Dis. 1995;18:624-634.
Lewis D.W., Dorbad D. The utility of neuroimaging in the evaluation of children with migraine or chronic daily headache who have normal neurological examinations. Headache. 2000;40:629-632.
Linnankivi T., Lundbom N., Autti T., et al. Five new cases of a recently described leukoencephalopathy with high brain lactate. Neurology. 2004;63:688-692.
Linnankivi T., Tienari P., Somer M., et al. 18q deletions: clinical, molecular, and brain MRI findings of 14 individuals. Am J Med Genet A. 2006;140:331-339.
Linna S.L., Finni K., Simila S., et al. Intracranial calcifications in cerebro-oculo-facio-skeletal (COFS) syndrome. Pediatr Radiol. 1982;12:28-30.
Loddenkemper T., Grote K., Evers S., et al. Neurological manifestations of the oculodentodigital dysplasia syndrome. J Neurol. 2002;249:584-595.
Longman C., Brockington M., Torelli S., et al. Mutations in the human LARGE gene cause MDC1D, a novel form of congenital muscular dystrophy with severe mental retardation and abnormal glycosylation of alpha-dystroglycan. Hum Mol Genet. 2003;12:2853-2861.
Lopez-Pison J., Rubio-Rubio R., Urena-Hornos T., et al. [Retrospective diagnosis of congenital infection by cytomegalovirus in the case of one infant]. Rev Neurol. 2005;40:733-736.
Lossos A., Barash V., Soffer D., et al. Hereditary branching enzyme dysfunction in adult polyglucosan body disease: a possible metabolic cause in two patients. Ann Neurol. 1991;30:655-662.
Lossos A., Meiner Z., Barash V., et al. Adult polyglucosan body disease in Ashkenazi Jewish patients carrying the Tyr329Ser mutation in the glycogen-branching enzyme gene. Ann Neurol. 1998;44:867-872.
Lynch S.A., Hall K., Precious S., et al. Two further cases of Sener syndrome: frontonasal dysplasia and dilated Virchow-Robin spaces. J Med Genet. 2000;37:466-470.
Maletkovic J., Schiffmann R., Gorospe J.R., et al. Genetic and clinical heterogeneity in eif2b-related disorder. J Child Neurol. 2008;23:205-215.
Marjo S., van der Knaap J.V. Magnetic Resonance of Myelination and Myelin Disorders, ed 3. Berlin: Springer; 2005.
Marklund L., Melin M., Melberg A., et al. Adult-onset autosomal dominant leukodystrophy with autonomic symptoms restricted to 1.5 Mbp on chromosome 5q23. Am J Med Genet B Neuropsychiatr Genet. 2006;141B:608-614.
Marotti J.D., Tobias S., Fratkin J.D., et al. Adult onset leukodystrophy with neuroaxonal spheroids and pigmented glia: report of a family, historical perspective, and review of the literature. Acta Neuropathol. 2004;107:481-488.
Massa R., Bruno C., Martorana A., et al. Adult polyglucosan body disease: proton magnetic resonance spectroscopy of the brain and novel mutation in the GBE1 gene. Muscle Nerve. 2008;37:530-536.
Matalon R., Michals K., Sebesta D., et al. Aspartoacylase deficiency and N-acetylaspartic aciduria in patients with Canavan disease. Am J Med Genet. 1988;29:463-471.
McMaster K.R., Powers J.M., Hennigar G.R.Jr, et al. Nervous system involvement in type IV glycogenosis. Arch Pathol Lab Med. 1979;103:105-111.
Meijer I.A., Simoes-Lopes A.A., Laurent S., et al. A novel duplication confirms the involvement of 5q23.2 in autosomal dominant leukodystrophy. Arch Neurol. 2008;65:1496-1501.
Melberg A., Hallberg L., Kalimo H., et al. MR characteristics and neuropathology in adult-onset autosomal dominant leukodystrophy with autonomic symptoms. Am J Neuroradiol. 2006;27:904-911.
Mierzewska H., Schmidt-Sidor B., Jurkiewicz E., et al. Severe encephalopathy with brain atrophy and hypomyelination due to adenylosuccinate lyase deficiency – MRI, clinical, biochemical and neuropathological findings of Polish patients. Folia Neuropathol. 2009;47:314-320.
Miladi N., Larnaout A., Dhondt J.L., et al. Dihydropteridine reductase deficiency in a large consanguineous Tunisian family: clinical, biochemical, and neuropathologic findings. J Child Neurol. 1998;13:475-480.
Miyaji T., Echigo N., Hiasa M., et al. Identification of a vesicular aspartate transporter. Proc Natl Acad Sci USA. 2008;105:11720-11724.
Mochel F., Yang B., Barritault J., et al. Free sialic acid storage disease without sialuria. Ann Neurol. 2009;65:753-757.
Moser H.W., Moser A.B., Frayer K.K., et al. Adrenoleukodystrophy: increased plasma content of saturated very long chain fatty acids. Neurology. 1981;31:1241-1249.
Moser H.W., Moser A.B., Hollandsworth K., et al. “Lorenzo’s oil” therapy for X-linked adrenoleukodystrophy: rationale and current assessment of efficacy. J Mol Neurosci. 2007;33:105-113.
Mosser J., Douar A.M., Sarde C.O., et al. Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature. 1993;361:726-730.
Mourmans J., Majoie C.B., Barth P.G., et al. Sequential MR imaging changes in nonketotic hyperglycinemia. Am J Neuroradiol. 2006;27:208-211.
Muller K., Kahn T., Wendel U. Is demyelination a feature of maple syrup urine disease? Pediatr Neurol. 1993;9:375-382.
Muncke N., Wogatzky B.S., Breuning M., et al. Position effect on PLP1 may cause a subset of Pelizaeus-Merzbacher disease symptoms. J Med Genet. 2004;41:e121.
Nagae-Poetscher L.M., Bibat G., Philippart M., et al. Leukoencephalopathy, cerebral calcifications, and cysts: new observations. Neurology. 2004;62:1206-1209.
Nave K.A. Neurological mouse mutants and the genes of myelin. J Neurosci Res. 1994;38:607-612.
Ophoff R.A., DeYoung J., Service S.K., et al. Hereditary vascular retinopathy, cerebroretinal vasculopathy, and hereditary endotheliopathy with retinopathy, nephropathy, and stroke map to a single locus on chromosome 3p21.1-p21.3. Am J Hum Genet. 2001;69:447-453.
O’Rourke D., Bradley L., King M.D., et al. Leukoencephalopathy with Anterior Temporal Cysts Due to Congenital CMV Infection Diagnosed Retrospectively. J Neuroimaging. 2008.
Orthmann-Murphy J.L., Salsano E., Abrams C.K., et al. Hereditary spastic paraplegia is a novel phenotype for GJA12/GJC2 mutations. Brain. 2008.
Padiath Q.S., Saigoh K., Schiffmann R., et al. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat Genet. 2006;38:1114-1123.
Palmeri S., Battisti C., Federico A., et al. Hypoplasia of the corpus callosum in Niemann-Pick type C disease. Neuroradiology. 1994;36:20-22.
Paznekas W.A., Boyadjiev S.A., Shapiro R.E., et al. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am J Hum Genet. 2003;72:408-418.
Pearl P.L., Gibson K.M. Clinical aspects of the disorders of GABA metabolism in children. Curr Opin Neurol. 2004;17:107-113.
Pearl P.L., Novotny E.J., Acosta M.T., et al. Succinic semialdehyde dehydrogenase deficiency in children and adults. Ann Neurol. 2003;54(Suppl 6):S73-S80.
Phillips M.D., McGraw P., Lowe M.J., et al. Diffusion-weighted imaging of white matter abnormalities in patients with phenylketonuria. Am J Neuroradiol. 2001;22:1583-1586.
Prietsch V., Arnold S., Kraegeloh-Mann I., et al. Severe hypomyelination as the leading neuroradiological sign in a patient with fucosidosis. Neuropediatrics. 2008;39:51-54.
Pronicki M., Sykut-Cegielska J., Mierzewska H., et al. Diversity of clinical symptoms in A3243G mitochondrial DNA mutation (MELAS syndrome mutation). Med Sci Monit. 2002;8:CR767-CR773.
Quattrocolo G., Leombruni S., Vaula G., et al. Autosomal dominant late-onset leukoencephalopathy. Clinical report of a new Italian family. Eur Neurol. 1997;37:53-61.
Ramantani G., Kohlhase J., Hertzberg C., et al. Expanding the phenotypic spectrum of lupus erythematosus in Aicardi-Goutieres syndrome. Arthritis Rheum. 2010;62(5):1469-1477.
Rapin I., Lindenbaum Y., Dickson D.W., et al. Cockayne syndrome and xeroderma pigmentosum. Neurology. 2000;55:1442-1449.
Raymond G.V., Jones R.O., Moser A.B. Newborn screening for adrenoleukodystrophy: implications for therapy. Mol Diagn Ther. 2007;11:381-384.
Reardon W., Hockey A., Silberstein P., et al. Autosomal recessive congenital intrauterine infection-like syndrome of microcephaly, intracranial calcification, and CNS disease. Am J Med Genet. 1994;52:58-65.
Rice G., Newman W.G., Dean J., et al. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. Am J Hum Genet. 2007;80:811-815.
Rice G., Patrick T., Parmar R., et al. Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am J Hum Genet. 2007;81:713-725.
Rice G.I., Bond J., Asipu A., et al. Mutations involved in Aicardi-Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet. 2009;41:829-832.
Richards A., van den Maagdenberg A.M., Jen J.C., et al. C-terminal truncations in human 3′-5′ DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nat Genet. 2007;39:1068-1070.
Robitaille Y., Carpenter S., Karpati G., et al. A distinct form of adult polyglucosan body disease with massive involvement of central and peripheral neuronal processes and astrocytes: a report of four cases and a review of the occurrence of polyglucosan bodies in other conditions such as Lafora’s disease and normal ageing. Brain. 1980;103:315-336.
Roerig P., Mayerhofer P., Holzinger A., et al. Characterization and functional analysis of the nucleotide binding fold in human peroxisomal ATP binding cassette transporters. FEBS Lett. 2001;492:66-72.
Rossi A., Biancheri R., Zara F., et al. Hypomyelination and Congenital Cataract: Neuroimaging Features of a Novel Inherited White Matter Disorder. Am J Neuroradiol. 2007.
Saijo H., Nakayama H., Ezoe T., et al. A case of megalencephalic leukoencephalopathy with subcortical cysts (van der Knaap disease): molecular genetic study. Brain Dev. 2003;25:362-366.
Sankhyan N., Sharma S., Kalra V., et al. Cystic white-matter changes in childhood Wilson’s disease. Pediatr Neurol. 2008;39:281-282.
Sartori S., Burlina A.B., Salviati L., et al. Increased level of N-acetylaspartylglutamate (NAAG) in the CSF of a patient with Pelizaeus-Merzbacher-like disease due to mutation in the GJA12 gene. Eur J Paediatr Neurol. 2008;12:348-350.
Saugier-Veber P., Munnich A., Bonneau D., et al. X-linked spastic paraplegia and Pelizaeus-Merzbacher disease are allelic disorders at the proteolipid protein locus. Nat Genet. 1994;6:257-262.
Scheper G.C., van der Klok T., van Andel R.J., et al. Mitochondrial aspartyl-TRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat Genet. 2007;39:534-539.
Schiffmann R., Elroy-Stein O. Childhood ataxia with CNS hypomyelination/vanishing white matter disease – a common leukodystrophy caused by abnormal control of protein synthesis. Mol Genet Metab. 2006;88:7-15.
Schiffmann R., Moller J.R., Trapp B.D., et al. Childhood ataxia with diffuse central nervous system hypomyelination. Ann Neurol. 1994;35:331-340.
Schiffmann R., Tedeschi G., Kinkel R.P., et al. Leukodystrophy in patients with ovarian dysgenesis. Ann Neurol. 1997;41:654-661.
Schiffmann R., van der Knaap M.S. Invited article: an MRI-based approach to the diagnosis of white matter disorders. Neurology. 2009;72:750-759.
Schonberger S., Schweiger B., Schwahn B., et al. Dysmyelination in the brain of adolescents and young adults with maple syrup urine disease. Mol Genet Metab. 2004;82:69-75.
Schroder J.M., May R., Shin Y.S., et al. Juvenile hereditary polyglucosan body disease with complete branching enzyme deficiency (type IV glycogenosis). Acta Neuropathol. 1993;85:419-430.
Schupbach W.M., Vadday K.M., Schaller A., et al. Mitochondrial neurogastrointestinal encephalomyopathy in three siblings: clinical, genetic and neuroradiological features. J Neurol. 2007;254:146-153.
Schwankhaus J.D., Katz D.A., Eldridge R., et al. Clinical and pathological features of an autosomal dominant, adult-onset leukodystrophy simulating chronic progressive multiple sclerosis. Arch Neurol. 1994;51:757-766.
Schwankhaus J.D., Patronas N., Dorwart R., et al. Computed tomography and magnetic resonance imaging in adult-onset leukodystrophy. Arch Neurol. 1988;45:1004-1008.
Sener R.N. Tyrosinemia: computed tomography, magnetic resonance imaging, diffusion magnetic resonance imaging, and proton spectroscopy findings in the brain. J Comput Assist Tomogr. 2005;29:323-325.
Serkov S.V., Pronin I.N., Bykova O.V., et al. Five patients with a recently described novel leukoencephalopathy with brainstem and spinal cord involvement and elevated lactate. Neuropediatrics. 2004;35:1-5.
Shah D.K., Tingay D.G., Fink A.M., et al. Magnetic resonance imaging in neonatal nonketotic hyperglycinemia. Pediatr Neurol. 2005;33:50-52.
Singhal B.S., Gorospe J.R., Naidu S. Megalencephalic leukoencephalopathy with subcortical cysts. J Child Neurol. 2003;18:646-652.
Sinha S., Taly A.B., Ravishankar S., et al. Wilson’s disease: cranial MRI observations and clinical correlation. Neuroradiology. 2006;48:613-621.
Sistermans E.A., de Coo R.F., De Wijs I.J., et al. Duplication of the proteolipid protein gene is the major cause of Pelizaeus-Merzbacher disease. Neurology. 1998;50:1749-1754.
Sonninen P., Autti T., Varho T., et al. Brain involvement in Salla disease. Am J Neuroradiol. 1999;20:433-443.
Spiegel R., Bach G., Sury V., et al. A mutation in the saposin A coding region of the prosaposin gene in an infant presenting as Krabbe disease: first report of saposin A deficiency in humans. Mol Genet Metab. 2005;84:160-166.
Steenweg M.E., Salomons G.S., Yapici Z., et al. L-2-Hydroxyglutaric aciduria: pattern of MR imaging abnormalities in 56 patients. Radiology. 2009;251:856-865.
Stein C., Gieselmann V., Kreysing J., et al. Cloning and expression of human arylsulfatase A. J Biol Chem. 1989;264:1252-1259.
Stepp S.E., Dufourcq-Lagelouse R., Le Deist F., et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 1999;286:1957-1959.
Suzuki K., Chen G. Metachromatic leucodystrophy: isolation and chemical analysis of metachromatic granules. Science. 1966;151:1231-1233.
Suzuki K., Suzuki Y. Krabbe’s globoid cell leukodystrophy: deficiency of galactocerebroside beta-galactosidase activity. J Neuropathol Exp Neurol. 1971;30:145.
Taber K.H., Shaw J.B., Loveland K.A., et al. Accentuated Virchow-Robin spaces in the centrum semiovale in children with autistic disorder. J Comput Assist Tomogr. 2004;28:263-268.
Takano T., Becker L.E. Intracerebral vascular occlusion in familial erythrophagocytic lymphohistiocytosis: a case report of two siblings. Acta Neuropathol. 1995;90:532-538.
Thompson A.J., Tillotson S., Smith I., et al. changes in phenylketonuria. Associations with dietary status. Brain. 1993;116(Pt 4):811-821.
Timmons M., Tsokos M., Asab M.A., et al. Peripheral and central hypomyelination with hypogonadotropic hypogonadism and hypodontia. Neurology. 2006;67:2066-2069.
Tolmie J.L., Browne B.H., McGettrick P.M., et al. A familial syndrome with coats’ reaction retinal angiomas, hair and nail defects and intracranial calcification. Eye (Lond). 1988;2(Pt 3):297-303.
Tolmie J.L., Shillito P., Hughes-Benzie R., et al. The Aicardi-Goutieres syndrome (familial, early onset encephalopathy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis). J Med Genet. 1995;32:881-884.
Topcu M., Gartioux C., Ribierre F., et al. Vacuoliting megalencephalic leukoencephalopathy with subcortical cysts, mapped to chromosome 22qtel. Am J Hum Genet. 2000;66:733-739.
Touraine R.L., Attie-Bitach T., Manceau E., et al. Neurological phenotype in Waardenburg syndrome type 4 correlates with novel SOX10 truncating mutations and expression in developing brain. Am J Hum Genet. 2000;66:1496-1503.
Traeger E.C., Rapin I. The clinical course of Canavan disease. Pediatr Neurol. 1998;18:207-212.
Trofatter J.A., Dlouhy S.R., DeMyer W., et al. Pelizaeus-Merzbacher disease: tight linkage to proteolipid protein gene exon variant. Proc Natl Acad Sci USA. 1989;86:9427-9430.
Tsujino S., Kanazawa N., Yoneyama H., et al. A common mutation and a novel mutation in Japanese patients with van der Knaap disease. J Hum Genet. 2003;48:605-608.
Twomey E.L., Naughten E.R., Donoghue V.B., et al. Neuroimaging findings in glutaric aciduria type 1. Pediatr Radiol. 2003;33:823-830.
Ubogu E.E., Hong S.T., Akman H.O., et al. Adult polyglucosan body disease: a case report of a manifesting heterozygote. Muscle Nerve. 2005;32:675-681.
Uhlenberg B., Schuelke M., Ruschendorf F., et al. Mutations in the gene encoding gap junction protein alpha 12 (connexin 46.6) cause Pelizaeus-Merzbacher-like disease. Am J Hum Genet. 2004;75:251-260.
van der Knaap M.S., Barth P.G., Gabreels F.J., et al. A new leukoencephalopathy with vanishing white matter. Neurology. 1997;48:845-855.
van der Knaap M.S., Barth P.G., Stroink H., et al. Leukoencephalopathy with swelling and a discrepantly mild clinical course in eight children. Ann Neurol. 1995;37:324-334.
van der Knaap M.S., Leegwater P.A., Konst A.A., et al. Mutations in each of the five subunits of translation initiation factor eIF2B can cause leukoencephalopathy with vanishing white matter. Ann Neurol. 2002;51:264-270.
van der Knaap M.S., Leegwater P.A., van Berkel C.G., et al. Arg113His mutation in eIF2Bepsilon as cause of leukoencephalopathy in adults. Neurology. 2004;62:1598-1600.
van der Knaap M.S., Linnankivi T., Paetau A., et al. Hypomyelination with atrophy of the basal ganglia and cerebellum: follow-up and pathology. Neurology. 2007;69:166-171.
van der Knaap M.S., Naidu S., Breiter S.N., et al. Alexander disease: diagnosis with MR imaging. Am J Neuroradiol. 2001;22:541-552.
van der Knaap M.S., Naidu S., Kleinschmidt-Demasters B.K., et al. Autosomal dominant diffuse leukoencephalopathy with neuroaxonal spheroids. Neurology. 2000;54:463-468.
van der Knaap M.S., Naidu S., Pouwels P.J., et al. New syndrome characterized by hypomyelination with atrophy of the basal ganglia and cerebellum. Am J Neuroradiol. 2002;23:1466-1474.
van der Knaap M.S., Ramesh V., Schiffmann R., et al. Alexander disease: ventricular garlands and abnormalities of the medulla and spinal cord. Neurology. 2006;66:494-498.
van der Knaap M.S., Salomons G.S., Li R., et al. Unusual variants of Alexander’s disease. Ann Neurol. 2005;57:327-338.
van der Knaap M.S., van der Voorn P., Barkhof F., et al. A new leukoencephalopathy with brainstem and spinal cord involvement and high lactate. Ann Neurol. 2003;53:252-258.
van der Knaap M.S., Vermeulen G., Barkhof F., et al. Pattern of white matter abnormalities at MR imaging: use of polymerase chain reaction testing of Guthrie cards to link pattern with congenital cytomegalovirus infection. Radiology. 2004;230:529-536.
van der Knaap M.S., Wevers R.A., Kure S., et al. Increased cerebrospinal fluid glycine: a biochemical marker for a leukoencephalopathy with vanishing white matter. J Child Neurol. 1999;14:728-731.
Vanderver A., Hathout Y., Maletkovic J., et al. Sensitivity and specificity of decreased CSF asialotransferrin for eIF2B-related disorder. Neurology. 2008;70:2226-2232.
Vanhanen S.L., Puranen J., Autti T., et al. Neuroradiological findings (MRS, MRI, SPECT) in infantile neuronal ceroid-lipofuscinosis (infantile CLN1) at different stages of the disease. Neuropediatrics. 2004;35:27-35.
van Kollenburg B., Thomas A.A., Vermeulen G., et al. Regulation of protein synthesis in lymphoblasts from vanishing white matter patients. Neurobiol Dis. 2006;21:496-504.
van Kollenburg B., van Dijk J., Garbern J., et al. Glia-specific activation of all pathways of the unfolded protein response in vanishing white matter disease. J Neuropathol Exp Neurol. 2006;65:707-715.
van Reeuwijk J., Janssen M., van den Elzen C., et al. POMT2 mutations cause alpha-dystroglycan hypoglycosylation and Walker-Warburg syndrome. J Med Genet. 2005;42:907-912.
Varho T., Komu M., Sonninen P., et al. A new metabolite contributing to N-acetyl signal in 1H MRS of the brain in Salla disease. Neurology. 1999;52:1668-1672.
Vatanavicharn N., Pressman B.D., Wilcox W.R. Reversible leukoencephalopathy with acute neurological deterioration and permanent residua in classical homocystinuria: A case report. J Inherit Metab Dis. 2008.
Vedolin L., Schwartz I.V., Komlos M., et al. Brain MRI in mucopolysaccharidosis: effect of aging and correlation with biochemical findings. Neurology. 2007;69:917-924.
Verheijen F.W., Verbeek E., Aula N., et al. A new gene, encoding an anion transporter, is mutated in sialic acid storage diseases. Nat Genet. 1999;23:462-465.
Viola A., Confort-Gouny S., Ranjeva J.P., et al. MR imaging and MR spectroscopy in rhizomelic chondrodysplasia punctata. Am J Neuroradiol. 2002;23:480-483.
Von Hirsch T., Peiffer J. [Histological methods in differential diagnosis of leukodystrophy from lipoidosis]. Arch Psychiatr Nervenkr Z Gesamte Neurol Psychiatr. 1955;194:88-104.
Wang R.Y., Cambray-Forker E.J., Ohanian K., et al. Treatment reduces or stabilizes brain imaging abnormalities in patients with MPS I and II. Mol Genet Metab. 2009;98:406-411.
Wassmer E., Singh J., Agrawal S., et al. Elevated pterins in cerebral spinal fluid–biochemical marker of Aicardi-Goutieres syndrome. Dev Med Child Neurol. 2009;51:841-842.
Weidenheim K.M., Dickson D.W., Rapin I. Neuropathology of Cockayne syndrome: Evidence for impaired development, premature aging, and neurodegeneration. Mech Ageing Dev. 2009;130:619-636.
Weinstein M., Schollen E., Matthijs G., et al. CDG-IL: an infant with a novel mutation in the ALG9 gene and additional phenotypic features. Am J Med Genet A. 2005;136:194-197.
Wenger D.A., Sattler M., Clark C., et al. An improved method for the identification of patients and carriers of Krabbe’s disease. Clin Chim Acta. 1974;56:199-206.
Wider C., Van Gerpen J.A., DeArmond S., et al. Leukoencephalopathy with spheroids (HDLS) and pigmentary leukodystrophy (POLD): a single entity? Neurology. 2009;72:1953-1959.
Willemsen M.A., Van Der Graaf M., Van Der Knaap M.S., et al. MR imaging and proton MR spectroscopic studies in Sjogren-Larsson syndrome: characterization of the leukoencephalopathy. Am J Neuroradiol. 2004;25:649-657.
Winkler D.T., Lyrer P., Probst A., et al. Hereditary systemic angiopathy (HSA) with cerebral calcifications, retinopathy, progressive nephropathy, and hepatopathy. J Neurol. 2008;255:77-88.
Wolf N.I., Cundall M., Rutland P., et al. Frameshift mutation in GJA12 leading to nystagmus, spastic ataxia and CNS dys-/demyelination. Neurogenetics. 2007;8:39-44.
Wolf N.I., Harting I., Boltshauser E., et al. Leukoencephalopathy with ataxia, hypodontia, and hypomyelination. Neurology. 2005;64:1461-1464.
Wolf N.I., Harting I., Innes A.M., et al. Ataxia, delayed dentition and hypomyelination: a novel leukoencephalopathy. Neuropediatrics. 2007;38:64-70.
Wolf N.I., Sistermans E.A., Cundall M., et al. Three or more copies of the proteolipid protein gene PLP1 cause severe Pelizaeus-Merzbacher disease. Brain. 2005;128:743-751.
Yanagawa S., Ito N., Arima K., et al. Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy. Neurology. 2002;58:817-820.
Yoshida A., Kobayashi K., Manya H., et al. Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev Cell. 2001;1:717-724.
Yuksel A., Karaca E., Albayram M.S. Magnetic resonance imaging, magnetic resonance spectroscopy, and facial dysmorphism in a case of Lowe syndrome with novel OCRL1 gene mutation. J Child Neurol. 2009;24:93-96.
Zafeiriou D.I., Rodenburg R.J., Scheffer H., et al. MR spectroscopy and serial magnetic resonance imaging in a patient with mitochondrial cystic leukoencephalopathy due to complex I deficiency and NDUFV1 mutations and mild clinical course. Neuropediatrics. 2008;39:172-175.
Zara F., Biancheri R., Bruno C., et al. Deficiency of hyccin, a newly identified membrane protein, causes hypomyelination and congenital cataract. Nat Genet. 2006;38:1111-1113.
Zlotogora J., Chakraborty S., Knowlton R.G., et al. Krabbe disease locus mapped to chromosome 14 by genetic linkage. Am J Hum Genet. 1990;47:37-44.
zur Stadt U., Rohr J., Seifert W., et al. Familial hemophagocytic lymphohistiocytosis type 5 (FHL-5) is caused by mutations in Munc18–2 and impaired binding to syntaxin 11. Am J Hum Genet. 2009;85:482-492.
zur Stadt U., Schmidt S., Kasper B., et al. Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type-4 to chromosome 6q24 and identification of mutations in syntaxin 11. Hum Mol Genet. 2005;14:827-834.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 71 Genetic and Metabolic Disorders of the White Matter
Introduction
Over the past two decades, an increasing number of novel heritable disorders affecting the white matter of the brain, or leukodystrophies, have been described, often with identification of a causative gene (Table 71-1). Pathognomonic magnetic resonance imaging (MRI) patterns (see Figure 71-1) or clinical characteristics permit identification in a number of these disorders, and should guide the clinician’s molecular diagnosis for many conditions (see Figures 71-3, 71-5, and 71-6 for MRI features). Challenges remain, however, for the neurologist, geneticist, or primary-care provider evaluating patients with suspected genetic or metabolic disorders of the white matter. In many settings, over half of subjects with suspected heritable white-matter disease do not receive a diagnosis, and therefore the focus of this chapter is on assisting the treating neurologist in the diagnosis of inherited disorders of the white matter.
Table 71-1 Molecular Causes of Leukodystrophies and Leukoencephalopathies (a Nonexhaustive List)
| Disorder | Gene |
|---|---|
| Acyl-coenzyme A (acyl-CoA) oxidase deficiency | ACOX |
| Adenylosuccinate lyase deficiency | ADSL |
| Aicardi–Goutières syndrome (AGS) | TREX1, SMHD1, RNAseH2A, B, C |
| Alexander’s disease (AxD)* | GFAP |
| Autosomal-dominant leukodystrophy with autonomic dysfunction* | Duplication LaminB1 |
| Canavan’s disease | ASPA |
| Cerebrotendinous xanthomatosis (CTX) | CYP27A1 |
| D-bifunctional protein deficiency | HSD17B4 |
| eIF2B-related disorder/VWM disease* | EIF2B1-5 |
| Glutaric aciduria type II/multiple acylCoA dehydrogenase deficiency (MADD) | ETFA, ETFB, ETFDH |
| 3-hydroxy-3-methylglutaryl coenzyme A (HMG CoA) reductase deficiency | HMGCR |
| Hypomyelination with congenital cataracts (HCC) * | FAM126A |
| Infantile sialic acid storage disease (ISSD) | SLC17A5 |
| Krabbe’s disease | GALC |
| Leukoencephalopathy with brainstem and spinal cord involvement and elevated lactate (LBSL)* | DARS2 |
| Lowe’s syndrome (oculocerebrorenal syndrome of Lowe, OCRL) | OCRL |
| Megalencephalic leukoencephalopathy with subcortical cysts (MLC)* | MLC1 |
| Metachromatic leukodystrophy (MLD) | ARSA |
| Mitochondrial neurogastrointestinal encephalopathy (MNGIE) | TYMP |
| Mucolipidosis IV | MCOLN1 |
| Oculodental digital dysplasia (ODDD)* | GJA1 |
| Pelizaeus–Merzbacher disease (PMD)* | PLP1 |
| Pelizaeus–Merzbacher-like disease (PMLD)* | GJC2 |
| Peroxisomal thiolase deficiency | ACAA |
| Polymerase gamma 1 (POLG1)* | POLG1 |
| Polyglucosan body disease (PGBD) | GBE1 |
| RNAse T2-deficient leukoencephalopathy* | RNASET2 |
| Sjögren–Larsson syndrome | ALDH3A2 |
| X-linked adrenoleukodystrophy (XALD) | ABCD1 |
| 18q minus syndrome | Deletion of short arm chromosome 18 |
VWM, vanishing white matter
* No specific biochemical marker clinically available, and molecular testing is primary diagnostic tool.
Fig. 71-1 Diagnostic algorithm for use in patients with abnormal myelination by MRI.
HIV, human immunodeficiency virus; PNS, peripheral nervous system; WM, white matter.
(Used with permission from Schiffmann R, van der Knaap MS. Invited article: an MRI-based approach to the diagnosis of white matter disorders. Neurology 2009;72:750–759.)
Fig. 71-6 Dysmyelinating and demyelinating leukodystrophies and pathognomonic MRI features, part II.
Given the heterogeneity and limited specificity in clinical findings, MRI pattern recognition is the most useful tool for evaluating suspected leukodystrophy patients [Schiffmann and van der Knaap, 2009] (Figure 71-1). MRIs should be reviewed comprehensively, with attention to changes over time, expected myelin development for the patient’s age, and characteristics of T1- and T2-weighted signal abnormalities. Broadly, there are two groups of leukodystrophies. Hypomyelinating leukodystrophies show increased white-matter signal on T2 relative to age, and often isointense or hyperintense white-matter signal on T1. Conversely, demyelinating leukodystrophies show increased white-matter signal on T2 relative to age, and hypointense white-matter signal on T1. Additional radiologic features should be examined, such as white-matter vacuolization or cysts, best seen on fluid-attenuated inversion recovery (FLAIR) or similar imaging paradigms; involvement of the basal ganglia, brainstem, cerebellum, or spinal cord; and abnormalities of cortical gray matter. Finally, findings consistent with calcifications should be noted, and if clinically indicated, a computed tomography (CT) scan or calcium-sensitive MRI sequences should be acquired to exclude the presence of calcifications that might be missed on standard MRI.
Hypomyelinating White-Matter Disorders
White-matter disorders with hypomyelination are relatively frequent; hypomyelinating leukodystrophies comprise 20 percent of leukodystrophies. They are characterized by a significant and permanent deficit of myelin [Schiffmann and van der Knaap, 2009]. Even with thorough genetic investigation, at least half of these hypomyelinating disorders lack definitive diagnosis, prognostic information, and potential for prenatal diagnosis. While most of these disorders show autosomal-recessive inheritance, both X-linked inheritance and de novo mutations are possible. Clinical manifestations common to hypomyelinating disorders are ataxia, spasticity, and nystagmus. Other non-neurological features can be valuable in diagnosis, such as hypodontia in 4H syndrome and cataracts in hypomyelination with congenital cataracts.
The diagnosis of hypomyelination may be made if two MRIs at least 6 months apart after the age of 12 months show little or no myelin development. Images in hypomyelinating leukodystrophies demonstrate diffusely hyperintense signal of the supratentorial white matter in T2-weighted images, and iso- or hyperintense white-matter signal on T1-weighted images. Certain areas, such as the posterior internal capsule, may have more normal-appearing myelin signal. Myelin deposition in infratentorial structures is usually higher. A definitive diagnosis of hypomyelination is not possible in young infants, as myelination is incomplete. If no myelin deposition is visible on the first MRI of a child older than 24 months, however, hypomyelination is highly probable [Schiffmann and van der Knaap, 2009].
Delayed myelination is sometimes misdiagnosed as hypomyelination. In contrast to hypomyelination, in delayed myelination, myelination progresses on serial MRI images to near-normal myelin development (Figure 71-2). Gray-matter disorders with early onset often show hypomyelination, presumably a result of defective axonal function. Features such as early atrophy and signal changes of cortex and basal ganglia may help distinguish between primary hypomyelination and hypomyelination secondary to neuronal dysfunction (see Figure 71-3F, G, and H below).
In cases of primary hypomyelination, the disorders described below should be considered.
Pelizaeus–Merzbacher Disease
Pelizaeus–Merzbacher disease (PMD, OMIM 312080) is the prototypic hypomyelinating disorder, and is caused by alterations in the proteolipid 1 (PLP1) gene [Hudson et al., 1989; Trofatter et al., 1989]. Located on Xq22.2, this gene encodes the PLP1 protein, which constitutes roughly half of all myelin protein. The most common abnormality is a duplication of the entire gene, found in 60–70 percent of PMD cases and associated with the classic form of the disease [Ellis and Malcolm, 1994; Sistermans et al., 1998]. The lack of common breakpoints in these duplications results in varying sizes of the duplicated area between patients. Missense mutations account for 10–15 percent of cases. Several missense mutations affecting splicing or putative regulation of expression have also been described. Deletions are also seen in a smaller number of cases. Triplications are present in 1–2 percent of cases; higher copy numbers have been described in one child. More complex chromosomal rearrangements involving PLP1 or its promoter region have been described in individual cases [Muncke et al., 2004]. Deletions or null mutations are rare. Roughly 15 percent of cases fulfilling diagnostic criteria for PMD do not possess identifiable PLP1 mutations, suggesting the possibility of mutations in regulatory regions beyond genetic assays.
PMD is allelic with a relatively mild disorder, X-linked spastic paraplegia type 2 (SPG2), also caused by PLP1 mutations [Saugier-Veber et al., 1994]. In its pure form, the sole symptom associated with SPG2 is slowly increasing spasticity, more of the legs than of the arms, which begins during childhood or adolescence. In the complicated forms, additional symptoms, such as nystagmus, ataxia, dysarthria, and mild cognitive impairment, are present. Life expectancy in this form is normal. Peripheral neuropathy is another possible finding with certain PLP1 mutations. Altogether, alterations of PLP1 mutations give rise to a spectrum of disorders, ranging from the very severe connatal phenotype to mild spastic paraplegia with adolescent onset.
Genotype–phenotype correlations have been established for most PLP1 alterations, although clinical heterogeneity among patients with common genetic alterations makes definitive characterization difficult. Symptoms do not correlate with duplication size, but high copy number appears to predict increased clinical severity. Severe epilepsy has been reported in children with triplications, an otherwise uncommon feature of PMD [Wolf et al., 2005]. Point mutations are associated with the full spectrum of PLP1-connected disorders [Cailloux et al., 2000]. Demyelinating neuropathy is associated with either null mutations or mutations in PLP-specific regions. Patients with functional null mutations show relatively mild clinical course; they usually achieve independent, albeit clumsy, ambulation, and show mild cognitive impairment and demyelinating neuropathy. As spasticity increases after the first decade, patients become wheelchair-bound and develop pseudobulbar palsy. Histopathologic investigations show evidence of length-dependent axonal degeneration in these patients and in corresponding mouse models [Garbern et al., 2002].
PLP1 is a highly conserved, hydrophobic membrane protein with four transmembrane domains and a large cytosolic loop between the second and third transmembrane domains. The N- and C-termini are located in the cytoplasm. Different splicing of PLP1 yields a second, smaller isoform, DM20. Both isoforms are primarily expressed within the CNS. While the function of PLP1 and DM20 remains poorly understood, mutations that leave DM20 intact are associated with relatively mild phenotype. Mutations associated with severe phenotype likely cause protein misfolding, which activates compensatory oligodendrocytic responses that ultimately cause oligodendrocyte apoptosis. The pathogenicity of PLP1 duplications is less well understood, although it has been speculated that overexpressed PLP1 in endosomal and lysosomal compartments depletes myelin rafts of lipids necessary for the production of functional myelin compounds [Garbern, 2007].
MRI in PMD shows hypomyelination, indicated by diffusely elevated white-matter signal on T2 (Figure 71-3A). Myelination is arrested and fails to progress on repeated scans. Some patients show a more mottled, “tigroid” signal, perhaps due to small myelinated areas surrounding blood vessels. In some patients, myelin deposition is seen in the posterior limb of the internal capsule or the optic radiations. Myelination is usually present in the brainstem and sometimes in the cerebellar hemispheres. Often – but not always – the pyramidal tracts in the brainstem show high T2 signal. At a young age, atrophy is not considerable, especially in the cerebellum, although white-matter volume may be decreased. The corpus callosum is thin, reflecting lack of myelinated axons. As children grow older, generalized atrophy develops. Proton MR spectroscopy reveals low choline, due to reduced membrane turnover in the absence of myelinating oligodendrocytes, and normal to elevated N-acetylaspartate (NAA), due to more densely packed axons. Patients with null mutations or deletions show more advanced supratentorial myelination. In SPG2 patients, MRI abnormalities are variable, ranging from diffuse mild hypomyelination to mild periventricular signal changes. Carrier females sometimes display small areas of elevated white-matter signal, but MRI is usually normal.
Diagnosis of PMD is based on typical clinical presentation, radiologic evidence of hypomyelination, and detection of PLP1 alterations. Routine laboratory and metabolic tests, including cerebrospinal fluid (CSF), are normal. As most PLP1 alterations are duplications, simple sequencing must also be accompanied by gene dose quantification by Southern blot, quantitative polymerase chain reaction (PCR), or, more recently, multiple ligation-dependent probe amplification (MLPA). Recently, elevation of the dipeptide N-acetylaspartylglutamate (NAAG) has been described in several PMD cases, with NAAG level appearing to predict clinical severity [Burlina et al., 2006]. The mechanism of NAAG elevation, however, remains unknown, and it is neither specific to PMD, nor a marker of hypomyelination.
Pelizaeus–Merzbacher-Like Disease
PMLD is a genetically heterogeneous disease. In most cases, no gene has been identified. In a small subset of PMLD patients (fewer than 10 percent), mutations in GJC2 (also called GJA12), coding for connexin 46.6 (Cx47), have been found (OMIM 608804) [Henneke et al., 2008]. Located on chromosome 1q41–q42, this gene was identified using a homozygosity mapping approach in a large consanguineous family [Uhlenberg et al., 2004]. Mutations were subsequently found in other, unrelated, patients, confirming GJC2 as the responsible gene. It remains the only gene identified in PMLD; other approaches to identify PMLD genes, such as sequencing candidate genes for structural myelin proteins, have so far been unsuccessful.
As in PMD, patients typically present with nystagmus apparent after the first few weeks of life. Motor development is delayed and children display ataxia during the first few years. Pyramidal signs and frank spasticity often develop later. Compared with boys with classic PMD, motor and cognitive performance in PMLD is better, especially during the first years of life, and children are often able to walk, some without support. However, patients show a more precipitous decline, with progressive spasticity, prominent pseudobulbar signs, and facial weakness reminiscent of myopathic face. Patients become wheelchair-bound in their teens. Epilepsy starting in late school age has been reported in a substantial proportion of cases, and may be more severe in isolated cases, a very unusual feature for boys with PMD. Mild demyelinating peripheral neuropathy revealed by nerve conduction studies has been described in some patients, although it does not influence the overall clinical manifestation. A recently described family carrying a homozygote missense mutation in GJC2 showed a phenotype of almost pure spastic paraplegia and minor cerebellar signs [Orthmann-Murphy et al., 2008]. There are no histopathological data on PMLD.
MRI shows hypomyelination, and prominent signal abnormalities of the pyramidal tract of the brainstem have been described (Figure 71-3B). Cerebellar atrophy is mild or absent, at least in the early stages; supratentorial atrophy with considerable white-matter loss is prominent in older patients [Wolf et al., 2007]. Brainstem atrophy is often seen in later stages. Routine metabolic investigations are normal. In CSF, NAAG is elevated, as it is in PMD [Sartori et al., 2008].
4H Syndrome
This recently described leukoencephalopathy (OMIM 612440) is also characterized by hypomyelination. Its name is derived from its three main clinical findings: hypomyelination, hypodontia, and hypogonadotropic hypogonadism [Wolf et al., 2005, 2007; Timmons et al., 2006]. The disorder is rare and a genetic locus has not yet been identified. As affected siblings of both sexes have been reported, inheritance is presumed to be autosomal-recessive.
Besides these neurological symptoms, hypodontia is the most prominent diagnostic sign (Figure 71-4). Eruption of deciduous teeth is delayed, and its order disturbed. Normally, the lower median deciduous incisors erupt first, followed by the upper median incisors. In 4H syndrome, the deciduous molars erupt first, followed by the incisors, and finally by the upper median incisors. Despite this delayed and disorganized eruption, the deciduous teeth are usually complete. In the permanent dentition, however, some teeth are missing. The incisors have an abnormal shape and often also a yellowish color. About 10 percent of patients show natal teeth, an otherwise very rare finding occurring only in 1 in 1000–3000 newborns.
Fig. 71-4 Dental phenotype of children with 4H syndrome.
The upper median incisors erupt late. A, Child aged 5 years. The typical situation in a younger child with lacking upper median (and in this case also lateral) incisors. The left lower median incisor [Schiffmann et al., 1994] has not erupted yet. B, Child aged 8 years. No deciduous incisors have erupted. The right permanent upper median incisor [Paznekas et al., 2003] shows yellowish discoloration. The left median incisors have not erupted yet.
As the name of the disease suggests, MRI shows hypomyelinated white matter (see Figure 71-3C). T2 signal in the supratentorial white matter is diffusely elevated, with the exception of aspects of the posterior limb of the internal capsule and optic radiation. T1 white-matter signal varies from hypointense to hyperintense, depending on the amount of myelin deposited. Cerebellar white matter is usually myelinated on T2. Corpus callosum is thin. There is also early and considerable cerebellar atrophy, more of the vermis than of the hemispheres, in virtually all patients. Whether clinical severity correlates with amount of myelin deposited or degree of initial cerebellar atrophy has not yet been elucidated. Proton MR spectroscopy reveals low choline, as is usual in hypomyelination, and often also elevation of myo-inositol compatible with gliosis. In the later stages, considerable cortical atrophy and white-matter loss develop, indicating on-going myelin loss.
Metabolic investigations are all normal. In some children, a muscle biopsy, with assessment of respiratory chain enzymes, has been performed in the context of deterioration of symptoms with infections, but showed normal results. Analysis of the known genes involved in hypomyelination, including GJA1, in which mutations lead to oculodentodigital dysplasia, did not reveal abnormalities [Wolf et al., 2007].
Oculodentodigital Dysplasia
Oculodentodigital dysplasia (ODDD, OMIM 164200) is another hypomyelinating disorder characterized by dental abnormalities. Its inheritance is autosomal-dominant. Dominant mutations in another connexin gene on chromosome 6q21–23.2, GJA1, coding for connexin 43 (Cx43), cause ODDD. There is one family described with autosomal-recessive mutations leading to the same phenotype. Cx43 is expressed in the developing brain and teeth, and also in hands and feet [Paznekas et al., 2003].
Neurologic symptoms are common, although no large case series has been reported, so little is known about the exact time course and development of neurologic abnormalities. In childhood and adolescence, coordination problems and mild ataxia are common. In adulthood, slow neurologic deterioration is seen, with development of pyramidal tract lesions, ataxia, dysarthria, loss of bladder control, and finally, frank spasticity. Unsupported gait may become impossible in late stages. Optic atrophy and deafness are possible. Cognition is preserved in most patients, although learning disabilities have been reported. Whether and to what extent early cognitive deterioration occurs awaits further study. Peripheral nervous system appears unaffected [Loddenkemper et al., 2002].
Hypomyelination with Congenital Cataract
Affected patients present early with delayed motor development. Cognitive development is mildly to moderately delayed. Most HCC children learn to walk with support before their second birthday. Neurological examination reveals dysarthria, moderate to severe spasticity with elevated muscle tone, brisk reflexes, extensor plantar response, and cerebellar signs such as intention tremor and dysmetria. Nystagmus is rare. As in other hypomyelinating disorders, progression of secondary neurologic symptoms occurs, and children may be unable to walk, even with support, by the end of the first decade. An additional pathognomonic finding is congenital cataract [Zara et al., 2006]. Several HCC cases have been published, and one patient failed to develop cataract until age 9. A small subset of HCC patients suffers from occasional seizures. Peripheral neuropathy is seen in almost all patients, leading to loss of previously exaggerated tendon reflexes and distal muscle wasting. It is not yet known whether a broad phenotypic spectrum exists in HCC, and clinical presentation is heretofore remarkably homogeneous, with the exception of cataract and peripheral neuropathy, which are not present in all patients. There are no data yet about CNS pathology, nor experimental models to elucidate Hyccin function.
MRI shows hypomyelination, as evident from the diffusely elevated T2 white-matter signal. In contrast to those with other hypomyelinating disorders, HCC patients show additional areas of higher T2 white-matter signal and decreased signal intensity on corresponding T1-weighted images, indicating elevated water content in these areas, particularly in periventricular regions [Rossi et al., 2007] (Figure 71-3E). Cerebellar atrophy is not seen. In the early stages, normal myelin signal may be apparent in subcortical fibres and corpus callosum. In the late stages, white matter appears shrunken, with increased apparent diffusion quotients. Proton MR spectroscopy gives variable results, depending on the disease stage. Choline may even be slightly elevated in the early stages, which is unusual for a hypomyelinating disorder, and decreased in later stages.
Hypomyelination with Atrophy of the Basal Ganglia and Cerebellum
Hypomyelination with atrophy of the basal ganglia and cerebellum (HABC, OMIM 612438) is a rare disorder of unknown genetic origin [van der Knaap et al., 2002]. As no affected siblings of children with HABC have been reported, it is possible that this entity arises from a dominant de novo mutation, although autosomal-recessive inheritance remains possible. Disease severity ranges from severely affected infants presenting shortly after birth to more mildly affected children with initially normal development. In severe cases, children fail to achieve motor milestones and linguistic development, and show profound axial hypotonia. Some have nystagmus, and optic atrophy is possible. Spasticity is common. Extrapyramidal symptoms, comprising dystonia, rigidity, and choreoathetosis, are uniquely common to HABC relative to other white-matter disorders. Failure to thrive and microcephaly are common. In mildly affected children, unsupported walking is achieved within the first few years of life, sometimes on time, but is later lost. There is a combination of spasticity, extrapyramidal symptoms, and ataxia. cognitive impairment is mild, although patients tend to show cognitive decline in addition to deteriorating motor functions.
Recently published neuropathologic findings from one HABC patient detail slightly reduced white-matter volume, reduced oligodendrocytes, severe myelin deficiency, especially of deep white matter, and some white-matter loss, as indicated by the presence of macrophages in perivascular regions [van der Knaap et al., 2007]. Axons appeared relatively preserved. There was mild astrocytosis and a strong presence of microglia. The putamen was visible only as a small streak, and microscopy revealed substantial neuronal loss. Cerebral cortex was normal, both macroscopically and microscopically. In the atrophic cerebellum, there was loss of granule cells. Pyramidal tracts appeared degenerated in the brainstem and spinal cord. Metabolic investigations are normal in these children.
Sialic Acid Storage Disorders
Salla disease (OMIM 604369) and infantile sialic acid storage disease (ISSD, OMIM 269920) are both caused by autosomal-recessive mutations in SLC17A5, coding for Sialin, a lysosomal membrane protein transporting sialic acid from lysosomes [Verheijen et al., 1999]. The gene is located on chromosome 6q14/15. Recently, it was shown that Sialin also functions as a shuttle for aspartate and glutamate in synaptic vesicles [Miyaji et al., 2008]. Free sialic acid accumulates in lysosomes of many cell types, including liver and kidney cells and cultured fibroblasts. In leukocytes, this accumulation causes vacuoles visible at light microscopic examination. Electron microscopy reveals membrane-bound vacuoles filled with fibrillogranular amorphous material. The pathogenesis of free sialic acid storage disease and the role of sialic acid remain unclear.
Both diseases are characterized by elevated excretion of free sialic acid in urine. Sialic acid is also elevated in other fluids, such as CSF. Recently, two siblings have been described who lack the characteristic sialuria; sialic acid was elevated only in CSF [Mochel et al., 2009]. Prevalent in Finland, Salla disease is characterized by seemingly normal early development, followed by presentation with hypotonia and ataxia in the second half of the first year of life. Nystagmus is also common. It may be evident in the neonatal period and frequently disappears. Many children also show strabismus. Spasticity develops slowly, and mild extrapyramidal symptoms are common in later stages. Mean age at walking is 4 years, with roughly one-third of patients who do not develop independent ambulation. Language is severely affected and usually dysarthric; patients are, at best, able to produce short sentences. Epilepsy with short, complex focal seizures is relatively common. In some patients, there is evidence of peripheral hypomyelination with decreased nerve conduction velocities. The disease is stable over a long period of time, with ultimate late progression. Additional symptoms may include short stature or hypogonadotropic hypogonadism. Facial features become coarse in adulthood. Otherwise, there is no evidence for dysostosis multiplex or hepatosplenomegaly, despite the presence of free sialic acid storage in liver and spleen. Life expectancy is normal [Aula et al., 2000].
A phenotype with intermediate severity relative to Salla disease and ISSD has also been identified.
MRI in patients with Salla disease shows hypomyelination [Sonninen et al., 1999] (Figure 71-3D). There is white-matter volume loss. Corpus callosum may be stringlike, especially in severe cases. There is usually cerebellar atrophy, and supratentorial atrophy is found in older patients. Thickening of the calvarium is another common radiologic feature. Proton MR spectroscopy reveals a high NAA peak, likely due to elevated free sialic acid, whose N-acetyl peak co-resonates with the N-acetyl peak of NAA [Varho et al., 1999].
Fucosidosis
MRI shows hypomyelination. A characteristic feature of fucosidosis is high T1 and low T2 signal in globus pallidus, thalamus, and substantia nigra. Cerebral and cerebellar atrophy may be prominent in older patients [Prietsch et al., 2008].
Serine Synthesis Defects
Children with 3-phosphoglycerate dehydrogenase deficiency are born microcephalic, and their development is grossly delayed. Epilepsy develops in the second half of the first year of life; West’s syndrome is one possible manifestation. Supplementation with serine and glycine is effective in seizure management. If treatment is commenced prenatally, head circumference at birth and development are normal [de Koning et al., 2004]. In untreated children, MRI shows hypomyelination and white-matter volume loss [de Koning et al., 2000]. Corpus callosum is thin and short. Myelination improves under treatment.
The first documented patient with phosphoserine aminotransferase deficiency presented in the neonatal period with severe epilepsy resistant to medical treatment and rapidly developing microcephaly. MRI showed supratentorial and brainstem atrophy. Supplementation with serine and glycine did not attenuate the seizures. The same treatment, if started before the development of symptoms, was shown to prevent epilepsy and enable normal development in the sibling of the proband [Hart et al., 2007].
Cockayne’s Syndrome and Trichothiodystrophy
Cockayne’s syndrome (CS) is a rare disease combining neurologic and non-neurologic features. This disorder and related disorders of DNA repair, such as cerebro-oculo-facial syndrome (COFS) and trichothiodystrophy (TTD), are genetically heterogeneous and are caused by mutations in CSA (CKN1 or DNA excision repair protein ERCC-8, responsible for 20 percent of CS cases), CSB (CKN2 or ERCC-6, responsible for most of the remainder of CS cases), XPB (ERCC3, OMIM 610651), XPD (ERCC2), XPG (ERCC5), ERCC1-XPF, TTDA and TTDN1 genes, and possibly others [Weidenheim et al., 2009].
The classic form, Cockayne’s syndrome type I, presents in the first year of life with failure to thrive; weight is more affected than length (“cachectic dwarfism”), and there is loss of subcutaneous fat, leading to a “wizened,” bird-like, progeroid face. Microcephaly also develops, usually by the end of the second year. Children develop contractures of the large joints, giving them a typical posture. Hands and feet are disproportionally large. Dental caries is prominent. Psychomotor development is also delayed, resulting in mild to severe cognitive impairment. Predominant neurologic features include ataxia and spasticity, which show slow progression. In late stages, peripheral neuropathy leads to muscle wasting and loss of the initially increased tendon reflexes. Over half of individuals develop sensorineural hearing loss. Most suffer from pigmentary retinal degeneration and cataracts. Autonomic dysfunction (hypolacrimia, hypohydrosis, miosis, acrocyanosis) is possible. In Cockayne’s syndrome type II, the clinical picture is much more severe, with growth failure already evident at birth. Loss or even absence of subcutaneous fat is striking. Joint contractures and kyphosis develop rapidly. Hypotonia is prominent initially, followed by development of spastic tetraparesis. Psychomotor development is absent or extremely delayed, with subsequent deterioration and early death. Subcutaneous fat loss has been treated in both types by early hypercaloric tube feeding, which allows reasonable growth in some children. Type III describes patients with milder forms of disease. Cutaneous photosensitivity is also characteristic of CS, occurring in 75 percent of all patients [Rapin et al., 2000].
Both syndromes are caused by defective nucleotide excision repair. Over 30 proteins are involved in this process. It eliminates DNA lesions induced by ultraviolet light. There are two major subpathways of nuclear excision repair: transcription-coupled repair, dealing with reparation of transcribed genes, and global genome repair, removing lesions in the entire genome. Defective DNA repair can be demonstrated by irradiating cultured skin fibroblasts with ultraviolet light and subsequently measuring unscheduled DNA synthesis. This unscheduled DNA synthesis is diminished in TTD and xeroderma pigmentosum patients. In CS, this unscheduled DNA synthesis is not significantly attenuated, but the otherwise rapid recovery of RNA and DNA synthesis after ultraviolet irradiation is adversely affected, indicating that the global genome repair is still functional. Additionally, reduction in basal transcription is also seen, an important signal for apoptosis. These defects of transcription, perhaps combined with activation of apoptosis, are thought to be mainly responsible for the neurologic symptoms in CS. Complementation assays in cultured cells could distinguish two different complementation groups, CS type I (OMIM 216400), caused by mutations in the gene coding for group 8 excision-repair cross-complementing protein (ERCC8), and type II (OMIM 133540), due to mutations in ERCC6. TTD is also heterogeneous, genetic defects having been identified in at least three different genes [Cleaver et al., 2009].
MRI of patients with CS shows hypomyelination, its degree corresponding to clinical severity. In severe cases with CS type II, hypoplasia of cerebellum and brainstem is possible. Basal ganglia calcifications are common. Similar features are seen in TTD, although calcifications are less common than in CS [Rapin et al., 2000; Adachi et al., 2006].
18q Minus Syndrome
In this disorder (OMIM 601808), the distal region of the long arm of chromosome 18 is deleted. It occurs de novo most commonly. The contiguous gene deletion usually involves the bands 18q22.3→qter. The gene for myelin basic protein (MBP), a component of healthy myelin, is located within this region. It has been postulated that haploinsufficiency for MBP leads to the myelin abnormalities observed in 18q minus syndrome. Heterogeneity in severity of clinical symptoms and hypomyelination between patients, despite consistent loss of MBP, is a focus of on-going inquiry. There is a well-investigated mouse model, the shiverer mouse, with homozygous rearrangements in the MBP gene [Nave, 1994], which lead to CNS, but no peripheral, hypomyelination. It is unknown why peripheral myelin is spared, despite MBP expression in peripheral nerves.
MRI shows hypomyelination of variable, but usually mild, degree. The myelin signal in the cerebral hemispheres may be inhomogeneous, with patchy white matter abnormalities. Corpus callosum may be thin. There may be mild supratentorial atrophy [Linnankivi et al., 2006].
SOX10-Associated Disorders
These rare syndromes are caused by mutations in SOX10 on chromosome 22q13, which encodes a transcription factor for various genes, some involved in myelin formation and metabolism, such as GJB1 (connexin 32). These disorders are characterized by a white hair lock and hypomelanotic spots, sensorineural deafness, and Hirschsprung’s disease. Patients are affected with varying severity, ranging from antenatal onset with congenital arthrogryposis multiplex and severe neurologic abnormalities, to more mildly affected patients who lack neurologic manifestations (Waardenburg–Shah syndrome, WS4, OMIM 277580). The severe variant has been designated peripheral demyelinating neuropathy, central dysmyelinating leukodystrophy, Waardenburg’s syndrome, Hirschsprung’s disease, or PCWH (OMIM 609136). Consistent with a neurocristopathy, many features of this disease can be explained by defective differentiation or migration of neural crest cells. In the severe cases, MRI reveals hypomyelination [Touraine et al., 2000]. The milder phenotype of WS4 is explained by SOX10 haploinsufficiency, whereas the severe form, PCWH, is thought to be caused by a dominant negative mechanism. Mutations found in children with PCWH are all truncating and located in the last exon. These mutations, unlike SOX10 mutations leading to WS4, escape nonsense-mediated decay and can therefore exert their dominant negative effect on protein level [Inoue et al., 2004, 2007].
Neurologic symptoms of children with PCWH include delayed development, nystagmus, spasticity, and ataxia. In severe cases, neonates are already symptomatic, with profound hypotonia, seizures, or congenital arthrogryposis due to hypomyelinating neuropathy. Other possible symptoms are reduced tear and saliva production, anhidrosis, and severe failure to thrive, in addition to the classic syndromes of WS4. Neuropathologic investigations of a severely affected infant have shown absence of central and peripheral nervous system myelin at the age of 3 months [Inoue et al., 2002]. Detailed MRI features have not been reported for many patients, but preliminary cases suggest mild to severe hypomyelination, and possibly atrophic brainstem [Inoue et al., 1999].
White-Matter Disorders with Demyelination
If MRI is not consistent with hypomyelination, there is white-matter hypointensity on T1 instead of iso- or hyperintensity, and there is hyperintensity on T2, the imaging pattern fits the demyelinating leukodystrophies. They comprise the leukodystrophies with primary demyelination, leukodystrophies with white-matter vacuolization, calcifying leukoencephalopathies, cystic leukoencephalopathies, leukoencephalopathies with brainstem involvement, and most adult-onset leukoencephalopathies. In the assessment of patients with these leukodystrophies, careful attention should be paid to specific neuroimaging features, including basal ganglia or brainstem signal abnormalities, contrast enhancement, cysts, calcifications, contrast enhancement, or specific FLAIR imaging abnormalities (Figure 71-5 and Figure 71-6; see also Figure 71-3) to assist in the differential diagnosis.
Primary Demyelinating Leukodystrophies
Alexander’s Disease
Alexander’s disease (AxD, OMIM 203450)[Alexander, 1949] is associated with mutations in the gene encoding the glial fibrillary acidic protein (GFAP) [Brenner et al., 2001]. GFAP mutations are thought to confer gain-of-function mutations, and a mutation on a single allele is sufficient to cause disease. In most cases, mutations are sporadic, although familial cases are described in adult- or juvenile-onset cases. In familial cases, inheritance is autosomal-dominant. There is usually concordance in presentation within a family, such that a family with adult-onset presentation and subsequent infantile presentation has not been reported. There is no definite genotype–phenotype correlation, with the exception of the two most common mutations, R79 and R239. R239, in particular, is associated with earlier onset and poor prognosis.
GFAP mutations are thought to result in decreased solubility of the glial fibrillary acidic protein, and accumulation of GFAP, along with vimentin, αβ-crystallin, and heat shock protein 27, result in Rosenthal fiber (RF) aggregation. RFs, accumulating in astrocytes, are thought to obstruct normal glial cell function and result in myelin destruction. RF accumulation in the ventricular collecting system is believed to cause the hydrocephalus seen in the clinical setting. Studies to improve pathophysiologic understanding of this disease and possible treatment strategies are on-going in an existing murine model of AxD. AxD is typically suspected from clinical presentation and characteristic MRI features. Typical AxD imaging features include frontal predominance of white-matter abnormalities, basal ganglia and or brainstem involvement, a periventricular rim of altered signal on T1- and T2-weighted imaging, and contrast enhancement of specific intracranial structures (see Figure 71-5A). The presence of four of five of these features makes diagnosis of AxD very likely and should prompt mutation testing [van der Knaap et al., 2001]. In very young infants and in the juvenile- or adult-onset cases, MRI may be less typical and involve only restricted brain regions. Additional features described in AxD imaging include predominant or isolated involvement of posterior fossa structures, multifocal tumor-like brainstem lesions, brainstem atrophy, and garland-like abnormalities along the ventricular wall [van der Knaap et al., 2005, 2006]. MR spectroscopy can be helpful in the diagnosis if it shows a lactate peak in affected tissues, but may also lead to inappropriate evaluations for mitochondrial cytopathies. There have been reports of findings of elevations of GFAP protein in CSF [Kyllerman et al., 2005], but this test is not used as a clinical tool.
X-Linked Adrenoleukodystrophy
X-linked adrenoleukodystrophy (XALD, OMIM 300100) is associated with mutations in the ABCD1 gene [Mosser et al., 1993] encoding a peroxisomal membrane transporter. This disorder follows X-linked inheritance, and often, when male children are diagnosed with the childhood-onset cerebral form, disease manifestations are recognized in obligate female carriers or male relatives of the proband. Differences in disease manifestations are known to occur with identical genotype, and even within a sibship, underscoring the likely effect of other genetic factors on clinical presentation. Genotype does not predict very long chain fatty acid levels or specific clinical prognosis. Genetic changes reported include missense mutations, nonsense mutations, frameshift mutations, small deletions/insertions, and large deletions.
Of note, an allelic disorder, with a neonatal presentation of cholestasis, hypotonia, and developmental delay, is caused by a contiguous gene deletion syndrome involving the 5′ end of ABCD1. This disorder is called contiguous ABCD1 DXS1357E deletion syndrome (CADDS), and is clinically distinct from XALD [Corzo et al., 2002].
The estimated prevalence of XALD is estimated to be 1:20,000–1:50,000. The estimated prevalence of hemizygotes (affected males) and heterozygotes (carrier females) is estimated at 1:16,800 [Bezman et al., 2001].
The pathophysiology of XALD is believed to arise from accumulation of saturated very long chain fatty acids (SVLCFA) within the brain. This accumulation is thought to result from defective peroxisomal fatty acid oxidation of SVLCFA, caused by defective transport by the mutated ABCD1, possibly due to altered adenosine triphosphate (ATP) binding [Gartner et al., 2002; Roerig et al., 2001].
Diagnosis is based on characteristic clinical presentation and suggestive MRI features. MRI shows a predominance of occipital findings (see Figure 71-5D), although frontal and corpus callosum variants are recognized. The affected white matter appears hyperintense on T2 and hypointense on T1. Characteristically, there is a rim of enhancement around the abnormal tissue that can be very helpful in establishing the diagnosis, as few other leukodystrophies, with the exception of Alexander’s disease, show significant contrast enhancement. When XALD is suspected, appropriate clinical tests include fasting VLCFA testing on plasma, which shows an excess in SVLCFA with specific abnormalities in C26:0, C24:0/C22:0, and C26:0/C22:0 ratios [Moser et al., 1981]. Mutation testing of the ABCD1 gene provides molecular confirmation of the diagnosis, with attention to the 7 percent of cases with deletions or rearrangements.
Treatment with Lorenzo’s oil or diets rich in oleic and erucic acids, in addition to VLCFA restriction, can alter VLCFA levels. As treatment strategies for XALD evolve, interest in newborn screening is growing. This can be done using liquid chromatography-tandem mass spectrometry (LC-MS/MS) to test for the analyte 1-hexacosanoyl-2-lyso-sn-3-glycero-phosphorylcholine (26:0-lyso-PC)[Hubbard et al., 2009; Raymond et al., 2007]. As with many disorders now being considered for widespread newborn screening, the clinical benefit of early diagnosis remains to be established.
Treatment of XALD depends on the stage and type of disease manifestations, and is still research-based. Treatment with Lorenzo’s oil, which decreases hexacosanoic acid (C26:0), does not appear to stop or reverse cerebral disease once it has begun, although there are reports of improved long-term stability in presymptomatic patients in open-label studies with no placebo control [Moser et al., 2007]. The use of Lorenzo’s oil is investigational and should be performed in the context of a clinical research protocol. Bone marrow transplantation (BMT) is indicated in children with early-stage, cerebral-form XALD, as evidenced by active disease on MRI. Family members of an affected proband should be tested by VLCFA and monitored for early MRI evidence of cerebral involvement to identify candidates for BMT. These patients should be evaluated by clinicians with specific expertise in this disorder. Gene therapy is a potential future tool and research studies are under way. Supportive care can improve comfort and quality of life for XALD patients. Careful monitoring and treatment of adrenal insufficiency should be part of therapeutic management.
Metachromatic Leukodystrophy
Metachromatic leukodystrophy (MLD, OMIM 250100) [Greenfield, 1933; Von Hirsch and Peiffer, 1955; Suzuki and Chen, 1966] is caused by mutations in the ARSA gene encoding arylsulfatase A [Stein et al., 1989; Austin et al., 1964] on chromosome 22q13.31, and is inherited in an autosomal-recessive manner. Homozygous or compound heterozygous ARSA mutations impair arylsulfatase A degradation of sulfatides, causing sulfatide accumulation within the brain and peripheral nervous system. Complete loss of arylsulfatase A activity (“I” or “O” alleles) is typically associated with early infantile MLD. Partial loss of arylsulfatase A activity (“R” or “A” alleles) is typically associated with juvenile- or adult-onset MLD. Compound heterozygosity with an I and A allele usually results in juvenile-onset MLD. Rarely, subjects with microdeletions of 22q13 and deletion of the ARSA gene with an MLD-causing mutation on the other allele have been diagnosed with MLD.
MLD diagnosis is often suspected based on clinical manifestations of motor impairment with a peripheral neuropathy. Typical MRI features include sparing of arcuate fibers and a rim of subcortical white matter, with involvement of periventricular and deep white matter in the supratentorial CNS (see Figure 71-5B). Involved white matter takes on the appearance of radiating stripes that can be highly suggestive of the disorder and reflects accumulation of sulfatides in perivascular macrophages. These radiating stripes are also present in other disorders, however, including Krabbe’s disease. In early stages or in adult cases, incomplete neurologic findings can complicate diagnosis, and early involvement of the corpus callosum may provide a clue. Occasionally, isolated involvement of cranial or peripheral nerves has been seen in the early stages of disease.
[/not-level-membership-for-neurology-category]
