[level-membership-for-opthalmology-category]
Chapter 34 Gene Therapy for Retinal Disease
 For additional online content visit http://www.expertconsult.com
For additional online content visit http://www.expertconsult.com
Background: preclinical gene therapy studies
Nonviral gene delivery
There are nonviral methods of delivering nucleic acids to cells, and those include use of physicochemical agents to compact the DNA and/or transport it across the membrane lipid bilayer.1,2 The transport across the bilayer can also be achieved through physical means, including propulsion with a “gene gun” or by electroporation or iontophoresis. Such methods can also be combined with reagents that enhance integration into the host chromosome.3 There are several potential advantages of nonviral approaches: First, they can be used to deliver DNA of unlimited size. Second, there is a smaller chance of detrimental immune response since the only antigen would be the nucleic acid itself plus any protein that is used as a condensation agent. Several studies have demonstrated proof of concept of retinal gene therapy using nonviral DNA delivery1,4,5 and additional studies will reveal the long-term safety, stability, and efficacy of this approach.
Viral vector-mediated gene delivery
Recombinant viruses are genetically neutered or crippled so that they cannot reproduce and cause an infectious disease once they infect a target cell. There is a large list of recombinant viruses that have been tested in the retina (Table 34.1). Different viruses have different attributes and challenges, including cargo capacity, ease of purification, cellular specificity, and immune response. However, a large portion of these have been used to demonstrate efficacy in animal models of retinal disease (Table 34.2).
Table 34.1 Vectors tested for transduction characteristics in animal models. Various retinal cell targets are listed; however, the exact targets depend on the route of administration, dose, species, and modifications to the vector
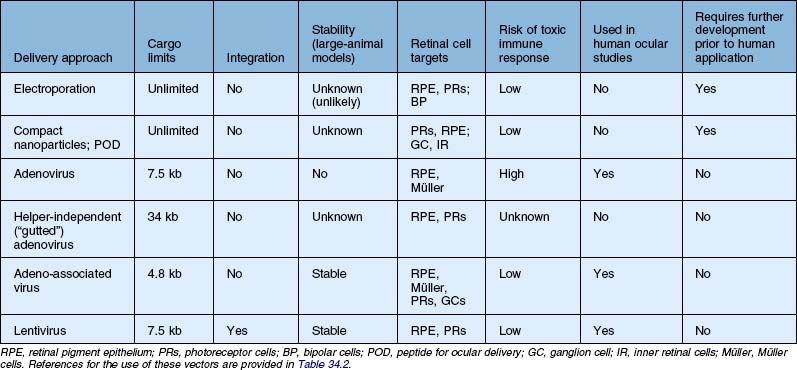
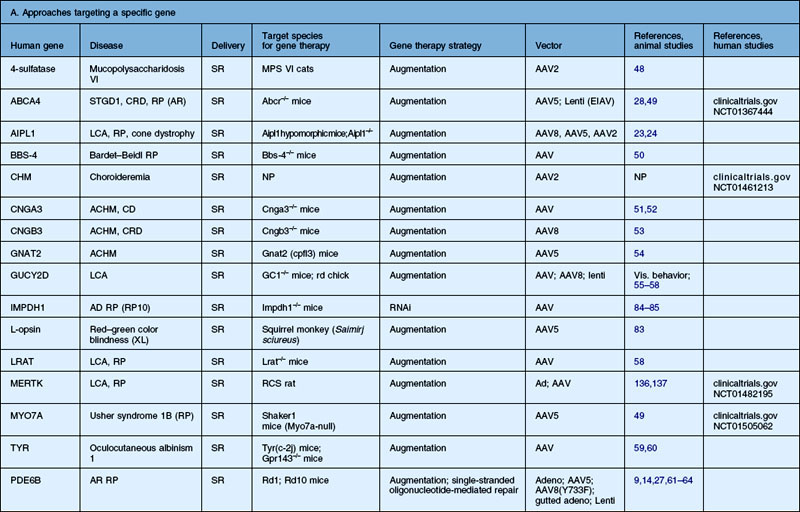
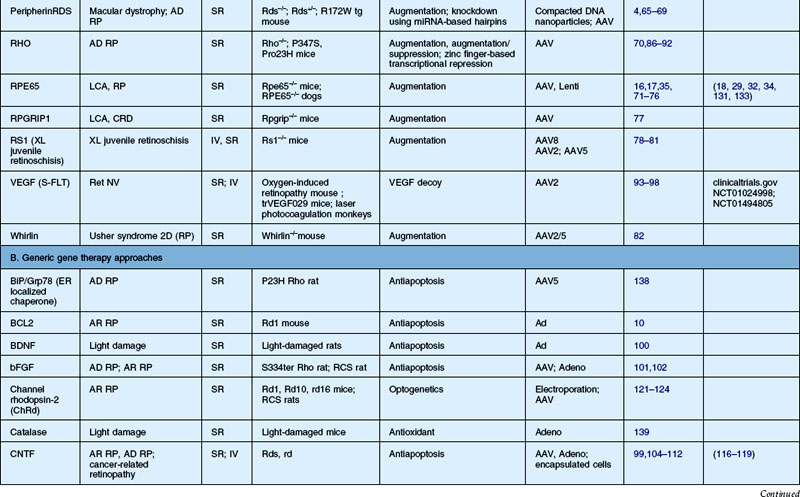
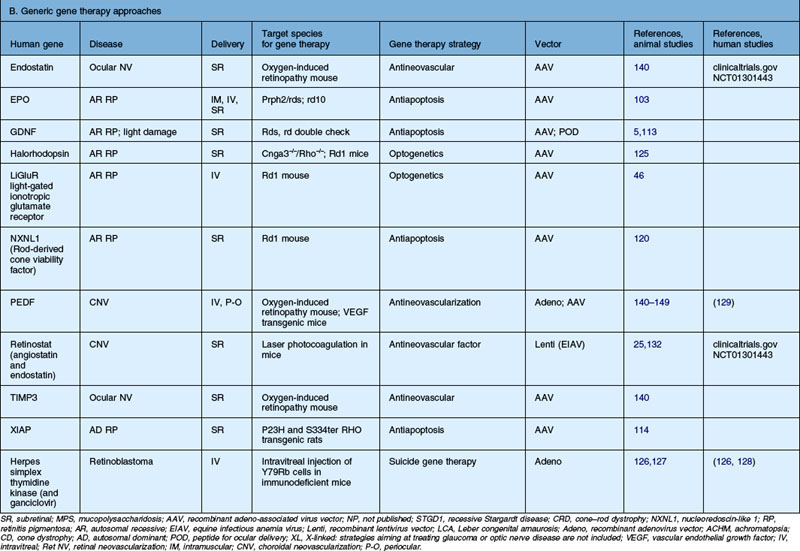
Table 34.2 Recent studies showing proof of concept of retinal gene therapy strategies using (A) selected approaches targeting a specific gene and (B) “generic” approaches that could potentially be used regardless of the disease-causing genetic defect. This list highlights many of the recent studies aiming to treat animal models of retinal degeneration, retinal development anomalies, and retinal neovascularization. Several of these studies have been carried forward to human clinical trial and those references are listed as well
The first recombinant adenovirus vectors, generated from the common respiratory virus, carried deletions in the adenoviral E1, E3 genes, and these Ad type 5 (Ad5) vectors were the first to be evaluated for retinal gene transfer in the differentiated retina.6,7 Adenovirus vectors result in high levels of gene expression within 24–48 hours. When injected subretinally, they target retinal pigment epithelial (RPE) cells efficiently in the adult eye and also Müller cells.6,7 When injected intravitreally, they target Müller cells and cells in the anterior segment, including corneal endothelium, lens and iris epithelium, and cells in the outflow tract (such as trabecular meshwork cells).8 Similar results are found when recombinant adenovirus is injected into the undifferentiated (early postnatal) retina; however, in addition to RPE cells, progenitor cells are targeted in the neonatal mouse eye.9,10 Additional manipulations of adenovirus vectors have yielded reagents that target photoreceptors more efficiently.11,12
A disadvantage of the early generations of E1, E3-deleted adenovirus is that it still carries viral open reading frames. These can enhance its immunogenicity, even in the immune-privileged environment of the eye. These vectors were thus also used to probe the nature of the intraocular immune response. When these vectors were injected subretinally, transgene expression persisted for several weeks to months. However, when injected intravitreally, transgene expression ceased within 2 weeks. Expression could be prolonged by incorporating immune-suppressant molecules, however.13 Efforts were made to generate adenovirus vectors lacking any viral open reading frames, the so-called “gutted” or helper independent vectors, thereby reducing immune clearance and allowing stable transgene expression.14 Such vectors do indeed result in more stable transgene expression than the first-generation vectors and, further, have a much greater cargo capacity than the original adenoviral vectors (Table 34.1).15 They are more difficult to generate, however, and preparations are likely to be contaminated by wild-type adenovirus.
Adeno-associated virus (AAV) vectors do not carry any virus open reading frames (and thus encode any virus-specific proteins) and therefore are generally more favorable from an immunologic standpoint than adenovirus vectors (Table 34.1). There is also an abundant amount of safety data related to AAV administration in animals and in humans, both systemically and intraocularly. Recombinant AAV (rAAV) vectors have an added benefit in that they target a more diverse set of cell types than adenoviral (or other) vectors. Unlike lentiviral vectors, AAV vectors do not integrate, or do so only rarely (Table 34.1). However, since the transgene persists in episomal fashion in the target retinal cells, rAAV vectors result in stable transgene expression. Expression persists for the life of small animals (mice and rats) and at least for many years in large animals and humans.16–19 rAAV vectors are useful for delivering genes efficiently to many types of retinal cells. A disadvantage of these vectors is their relatively limited cargo capacity (a maximum of 4.8 kb) (Table 34.1).
In AAV vectors, the transgene cassette is bordered by the inverted terminal repeats (ITRs) from an AAV2 genome. The original AAV vectors were generated by packaging the transgene cassette ITRs into an AAV serotype 2 capsid and this resulted in “AAV2/2” vectors, i.e., ITRs of an AAV serotype 2 genome packaged into an AAV2 serotype 2 capsid. (Often investigators skip the reference to the ITR serotype and refer to AAV2 vectors instead of AAV2/2 vectors.) More than a dozen AAVs of different serotypes have been described. Cross-packaged rAAVs (for example, rAAV2/5 vectors generated after packaging the AAV2 cis plasmid into AAV serotype 5 capsid) differ significantly from rAAV2/2 vectors with respect to cellular specificity, efficiency of transduction, and onset of transgene expression. While rAAV2/2 targets RPE cells efficiently (and photoreceptors less efficiently), it takes up to 6 weeks for transgene expression mediated by this vector to plateau.20–22 In comparison, rAAV2/5 and rAAV2/8 vectors transduce photoreceptors with much higher efficiency than rAAV2/2 and result in transgene expression within 5–10 days of delivery.20,21 This information is helpful in selecting vectors for particular applications. For example, in a relatively slowly progressive retinal degenerative disease (Leber congenital amaurosis (LCA) due to RPE65 mutations), rAAV2/2 performs well in delivering a therapeutic transgene to RPE cells. For an animal model with a much faster rate of degeneration (e.g., LCA due to AIPL1 mutations23,24), it is necessary to use a vector with a much faster onset of expression and which targets photoreceptors efficiently (such as a rAAV2/5 or 2/8 vector; Table 34.2).
Vectors originally based on the human immunodeficiency virus, lentiviral vectors, have been shown to be safe in animal models and their safety in humans is currently being tested (Table 34.2). A number of groups have since generated vectors based on viruses that were identified in nonhuman species, such as equine Lentivirus.25,26 Transgene expression is stable after lentiviral administration, because these vectors mediate integration into the host chromosome (Table 34.1). Lentiviral vectors target RPE cells efficiently after subretinal injection and, in undifferentiated retina, also target neural progenitor cells. They have thus been used to demonstrate efficacy in animal models of RPE disease, such as LCA-RPE65 or autosomal recessive (AR) retinitis pigmentosa (RP) due to PDE6B mutations27 (Table 34.2). Lentiviral vectors are also attractive in that they can carry a cargo of ~7.5 kb (Table 34.1). Specific modifications also allow lentiviral vectors to target mature photoreceptors.28 The lentiviral envelope can be swapped in order to alter cellular tropism.22 Since lentiviral vectors are integrating vectors, there is a concern about the potential for insertional mutagenesis. To date there has been no report of such an effect after retinal administration in animal models.
Surgical delivery
When a subretinal injection is performed through a small retinotomy, a retinal detachment or “bleb” is raised (Fig. 34.1). Most if not all of the volume of injected material is trapped between the outer retina and RPE as a localized retinal detachment. There is negligible escape of material back through the retinotomy site into the vitreous, as evidenced by the fact that, initially, the size of the bleb does not change once it is formed. There is apparently little pressure differential between the subretinal space and vitreous once the bleb is established and this is especially so when the scleral incisions are closed. In addition, the small-gauge cannulas used for the subretinal injection appear to be self-sealing, especially when a gas tamponade or formed vitreous is present. Since a bleb raised by subretinal injection tends not to expand beyond the border of the initial injection, the volume of distribution of the administered agent is limited, especially when compared to an intravitreal injection or systemic administration. The concentration of the compound contained in the bleb remains high and may even increase as the RPE cells extract free water from the vehicle used to dilute the agent. Limiting the volume of distribution in this manner may serve both to increase the efficiency of drug delivery and to decrease local and systemic toxicities by restricting diffusion of the drug. The location of the original subretinal detachment cannot be appreciated in most species (including humans) after it has flattened.18,29 (The location is often visible in dogs years after the injection, however, because of alterations in reflectivity of the underlying tapetum due to the procedure: Fig. 34.1.)
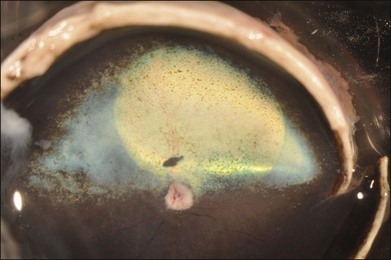
Since the RPE and Bruch’s membrane are typically not violated when subretinal injection is performed by the pars plana approach, there is further protection against systemic exposure of antigens via the highly vascular choroidal circulation. In addition, there is minimal disruption of retinal vasculature since the placement of the injection cannula is done under direct visualization. Therefore, the integrity of the blood–ocular barrier remains intact when subretinal delivery is performed in this manner. There is in addition an immunologic compartmentalization when antigenic material is delivered to the subretinal space. Limiting exposure to this area may result not only in characteristic immunoprivilege behavior evident with intraocular delivery but, when delivery is confined to the subretinal space, antigenic tolerance can be induced due to immune-deviant response.30,31 This unique property of the subretinal space is of great significance in the delivery of biologics and in gene therapy in particular as the development of antibody/immune response to both viral antigens and foreign transgene products may limit the effectiveness of treatment.
The major differences in surgical methods between various investigators who have carried out gene transfer in humans so far18,29,32–34 involve: (1) use of perioperative systemic corticosteroid therapy; (2) the removal of posterior cortical vitreous prior to subretinal injection; and (3) placement of a gas (air) bubble after the injection is performed. With regard to systemic corticosteroid use, there appears to be no important difference in efficacy. It should be noted that all studies employ the use of topical and periocular corticosteroid to suppress surgical inflammation.
The surgical protocol for the Children’s Hospital of Philadelphia (CHOP) gene therapy trial for LCA2 specifies removal of the posterior hyaloid.18,29 In many instances, a complete posterior vitreous detachment (PVD) is already present despite the young age of the subjects enrolled in this trial. This is not unexpected as vitreous abnormalities, including the presence of debris or posterior separation, are characteristic features of eyes with retinal degeneration. In cases without PVD, the posterior cortical vitreous is engaged with active suction and the hyaloid face is gently separated to create a complete PVD, as evidenced by the presence of a glial ring separating from the optic nerve head (Weiss ring). Once the presence of a PVD is confirmed, the mobilized vitreous is removed as completely as possible, with special attention to pare back any gel in the vicinity of the active sclerotomy sites. This is done both to avoid vitreoretinal traction induced by instruments passing into and out of the eye, and also to prevent vitreous traction which can bend the tip of the 39-gauge (and smaller) subretinal injection cannula. Most investigators recommend the removal of epiretinal membrane (ERM) if present in the macular area in order to prevent interference with the injection cannula and to avoid late-occurring complications such as macular hole resulting from membrane contraction.
When directing the injection into the posterior pole or macula, the cannula tip is usually placed in the vicinity of the papillomacular bundle. Even in eyes with advanced retinal degeneration, the retina in this area is usually thick enough to allow for successful placement of the cannula tip and the injection into the subretinal space. The cannula tip is positioned so as to avoid direct injury to retinal arterioles. The CHOP protocol specifies that the site of injection be a minimum of 3 mm from the foveal center in order to avoid development of a foveal dehiscence from fluid tracking directly to the central macula in a fistula-like manner. In addition, a small bubble of perfluoro-octane liquid is placed over the fovea to counteract the hydrodynamic force created during the subretinal injection, thereby buttressing this anatomically vulnerable area (Fig. 34.2; video 51 online).
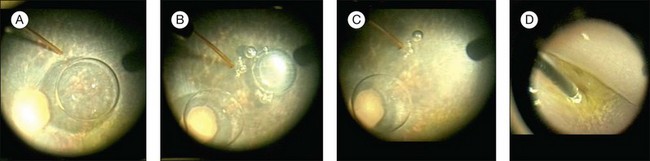
Fig. 34.2 Subretinal injection with protection to the fovea. (A) Subretinal injection cannula is apposed to the retina. Perfluoron had already been layered over the fovea. (B) The injection was initiated. A few small bubbles were expressed initially from the cannula (and are in the subretinal space). (C) The bleb has expanded. (D) The cannula has been removed and the Perfluoron is being removed. The inferior border of the bleb is visible. (See video 51 (online) showing subretinal injection of AAV2-hRPE65v2 in a human.29)
 VIDEO 51
VIDEO 51One unique feature of subretinal injection in retinal gene therapy when compared to other surgical indications is that the subretinal fluid, i.e., gene therapy agent, is not evacuated and the retina flattened at the time of fluid–air exchange. This is done in order to maximize the time of exposure to the subretinal injection, something that is not necessary, for example, after the extraction of a subretinal hemorrhage or translocation of the macula. Indeed, the creation of retinal detachment by subretinal injection is of itself a concern with respect to possible retinal toxicity. Fortunately, the extent of injury after acute retinal detachment created by injection of physiologic solutions appears to be small based on both laboratory and clinical data. Resorption of volumes less than 0.45 mL typically occurs in less than 24 hours.16–18,29,35–37 In the CHOP trial, resorption of 0.3 mL of fluid was observed within 6 hours of injection. Bainbridge et al.32 reported a longer period of detachment >24 hours after subretinal injection using a two-step injection technique where vector was administered into a bleb raised first with a physiologic solution. The longer period required for reattachment is likely due to the large volume of injection, i.e., 1.0 mL.32
Development of macular hole is a complication which appears to be unique to the subretinal injection procedure. Although creation of macular hole has been described in subretinal surgery for choroidal neovascularization (CNV), this typically occurs as a CNV is extracted in the presence of an adhesion to the overlying retina. In the CHOP trial, macular hole developed in one patient not during the surgical manipulation but rather several days postoperatively. Although ultimately the ophthalmoscopic appearance of the defect was typical for an idiopathic macular hole, several key differences were noted. First, prior to the development of a full-thickness defect, inner lamellar thinning was demonstrated on optical coherence tomography.18 In addition, intraretinal edema or cystoid macular edema was never present either during evolution of the hole or after hole formation. Finally, a subretinal fluid cuff was never apparent before or after hole formation. In the absence of inflammation, vasculopathy, or acute tissue injury, it was felt that the macular hole was unlikely to have been caused by drug toxicity. The appearance of the lamellar defect was reminiscent of pseudohole caused by ERM contracture. Since the ERM was recognized preoperatively and the posterior cortical vitreous was removed at surgery, it was thought that surgery-induced contraction of the pre-existing ERM was the most likely cause of macular hole formation. A second patient was observed to develop a foveal dehiscence at the time of subretinal injection as fluid channeled directly from the cannula through the fovea through a fistula-like tract.29 This phenomenon has been observed to occur infrequently during subretinal injection in nonhuman primates.38 Occurrence of a foveal dehiscence is presumably induced by intense hydrodynamic stress from injection in the vicinity of the fovea. No ERM was present in this case. Air tamponade with face-down positioning resulted in complete resolution of this induced hole within days. Since instituting a protocol modification specifying a minimum distance from the fovea for injection and use of perfluorocarbon liquid prior to injection, intraoperative foveal dehiscence has not occurred.29
History of retinal gene therapy
Preclinical studies: retinal transduction characteristics of different vectors
Initial gene transfer studies in animals generally evaluated the safety and stability of delivery of “reporter genes,” genes whose activity in terms of protein products can be evaluated noninvasively or in tissue samples through either histochemical/immunohistochemical measures or through a bioassay. In the early 1990s, these studies often utilized the LacZ (Escherichia coli β-galactosidase-encoding) gene. β-Galactosidase activity can be detected through either histochemical or spectophotometric activity. In the mid to late 1990s, many groups switched to use of green fluorescent protein (GFP), a bioluminescent, intracellular protein normally produced by the jellyfish, Aequorea victoria. A modified version of this protein, enhanced GFP (EGFP), has stronger fluorescence than the original protein, is excited by light in the blue range of the spectrum (498 nm), and has an emission peak at 509 nm, which is in the green portion of the visible spectrum. These absorption/fluorescence characteristics are very similar to those of fluorescein, which is used in the clinic to measure blood vessel integrity in the eye. Thus, EGFP can be measured using the same instruments/optics that are used in the clinic to measure fluorescein (Fig. 34.3). The only difference is that intracellular EGFP does not wash out with time.
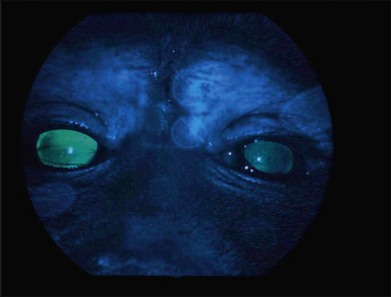
Fig. 34.3 Green fluorescent protein (GFP) is visible through illumination with blue light with an ophthalmoscope in this nonhuman primate that had received subretinal injection of 1E11 vector genomes (vg) AAV2/8.CMV.EGFP in its right eye. The left eye had received subretinal injection of the same material but with a dose that was 2 log units lower than the right eye.39
Evaluation of different vectors
Numerous studies in small and large animals have relied on vector-mediated delivery of reporter genes to elucidate the characteristics of one vector versus another.20–22,39 The transduction characteristics are affected by dose and age (or stage of development) of the animal. In the fetal mouse, one can deliver vector to retinal progenitor cells and carry out “birthday studies” by looking later in adulthood at which types of photoreceptor cells were being “born” at the time of injection.40 Such studies were initially elegantly performed using retroviral vectors, which only target dividing cells.41 Studies on fetal retina AAV-mediated transduction characteristics led to selection of an approach that could be used to reverse blindness in utero in mice.42
The majority of studies published to date involve postnatal delivery. Transduction characteristics are dependent on the surgical approach and the dose. Intravitreal injection of particular recombinant viruses can lead to transduction of ganglion cells and/or Müller glia. For example, intravitreal injection of AAV2 leads to ganglion cell transduction (and expression in the optic nerve, optic chiasms, and brain) in species ranging from mouse to dog to human, whereas intravitreal injection of AAV5 does not.43,44 Intravitreal injection of recombinant viral vectors does not usually result in transduction of the photoreceptors in the outer retina or the RPE (Table 34.1).8,16,17,21
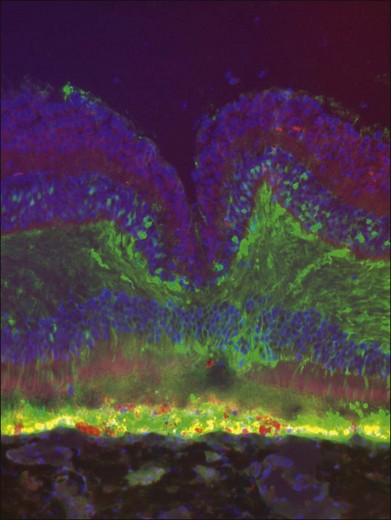
Most vectors target RPE cells efficiently even at low dose (Table 34.1). As the dose is increased and depending on the vector, photoreceptors and Müller cells can also be transduced (Fig. 34.4).39 Cellular transduction characteristics can also differ from species to species. A very active area at present involves engineering of AAV capsids in order to enhance their transduction properties.22,45,46 Efforts are underway to modify the capsid in order to expand its cargo capacity and to modify the capsid to target specific cell types. For example, Greenberg et al. generated lentiviral vectors that target Müller cells specifically after intravitreal injection.47 Recently, some groups have reported that modifications of the AAV capsid will allow it to penetrate from the vitreous through the various retinal cell layers in mice.45 Penetration of that sort has not yet been reported to occur in large-animal models. In smaller animals, it may be an artifact induced by localized retinal detachment produced by the need for a choroidal approach in order to deliver intravitreally or subretinally (see below).
Proof-of-concept studies
With progress in delineating the molecular genetic bases of inherited retinal degenerations in humans and in animals and the development of recombinant viral vectors with which to deliver transgenes to different retinal cell types, the logical next step is to determine how this information can be used to correct the diseases. Gene augmentation strategies, whereby a wild-type copy of a gene is delivered, have been tested successfully now in animal models of more than a dozen different conditions (Table 34.2). The animal model conditions have included AR RP, autosomal dominant (AD) RP, LCA, cone–rod dystrophy, macular dystrophy, oculocutaneous albinism, achromatopsia, mucopolysaccaridosis VI, AR Stargardt disease, and RP found in syndromes such as Bardet–Biedl and Usher syndrome (Table 34.2).4,9,14,16,17,23,24,27,28,35,48–82 Gene augmentation therapy has also been used to restore function to a nonhuman primate model of red–green color blindness (Table 34.2).83 There are a number of details that affect the success of retinal gene augmentation, including selection of the appropriate vector (see above), and when and where to deliver the vectors. The outcome measures used in the various studies include physiologic assays such as electroretinograms (ERGs), evaluations of pupillary light reflexes and optokinetic responses, visual behavior (ability to swim through a water maze or to select light or dark areas) and, in the monkey model of color blindness, to identify specific colors,83 and histology (including immunohistochemical demonstration of expression of the appropriate transgenic protein).
There has also been success with strategies aimed at rescuing disease due to toxic gain-of-function mutations.84–92 Such strategies are necessarily more complex than gene augmentation strategies. The best-studied examples of intervention with gain-of-function gene defects include rhodopsin mutations found in AD RP. Such defects result in abnormal cellular trafficking as well as altered functional properties. Deleterious effects of the endogenous mutant genes can be minimized by a knock-down or knock-down/gene augmentation strategy. The mutant messenger ribonucleic acid (mRNA) can be specifically targeted, leaving the wild-type mRNA (either endogenous or delivered via gene augmentation) intact. Knock-down has been achieved successfully by using ribozymes, RNA interference (RNAi), delivery of micro-RNAs, and use of zinc finger nucleases (Table 34.2 part A).
Gene therapy strategies have also been used successfully to target specific genes/proteins which may not be the primary cause of disease, but which are known to be involved in downstream pathways. An example involves manipulation of the vascular endothelial growth factor (VEGF) pathway implicated in ocular neovascularization (Table 34.2 part A).
Several groups have evaluated the possibility of ameliorating neovascularization by delivery of a gene that encodes a soluble decoy VEGF receptor, s-FLT.93–98 Such an approach has been used successfully in animal models of both retinal and CNV. These studies led to initiation of phase I human clinical trials to test the safety and efficacy of intravitreal delivery of an AAV2-sFLT vector in humans with CNV (Table 34.2 part A).
There are gene therapy strategies that can be used that are not specific to the disease-causing gene and could potentially be applied to a diverse set of conditions. One approach that has been used to evaluate development of a “generic” strategy with which to treat RP has been to use growth or neurotrophic factors or hormones to maintain the health of the diseased photoreceptors (Table 34.2 part B). Because growth factor proteins have very short half-lives (some as short as seconds), generation of a constant supply through a gene therapy approach is attractive. A growth factor could potentially be used to maintain the health of these cells until a disease-specific vector can be developed. Alternatively, such factors could be delivered as part of a cocktail.99 A number of different factors have been tested through direct viral gene transfer in animal models, including brain-derived neurotrophic factor, basic fibroblast growth factor, ciliary neurotrophic factor (CNTF), glial cell-derived neurotrophic factor, pigment epithelium-derived factor (PEDF), and X-linked inhibitor of apoptosis (Table 34.2 part B).5,99–114 The majority of these have ameliorated structure and function, although CNTF, while ameliorating structure, has been shown to diminish retinal function at high doses in some animal models.106,109,115 Sustained delivery of CNTF through use of a device that encapsulates CNTF-transfected cells (encapsulated cell therapy) has resulted in structural improvement without adverse functional effects (and in some cases, improvement in function), both in animals and in humans (Table 34.2 part B).104,116–119 The encapsulated cell therapy approach entails placement of a capsule containing a grid occupied by cells, transfected with a CNTF-containing plasmid, into the vitreous. There will likely be many other factors tested for rescue in gene therapy paradigms. One such factor which has attracted great interest due to data showing that it rescues cone photoreceptors in animal models is rod-derived neurotrophic factor (whose gene is known as NXNL1).120 Evaluation of the results in all of these growth/neurotrophic factor therapies reveals that, with the majority of these factors, efficacy is dependent on them being expressed in/by the retina. Interestingly, there is a hormone, erythropoietin, which can also delay retinal degeneration in certain animal models through gene therapy-mediated delivery, but this is only effective if it is delivered outside the eye (intramuscularly: Table 34.2 part B).103
Approaches using a generic gene therapy strategy to bring vision to retinas in which photoreceptor cells have been lost or severely damaged were also recently described. These involve delivering light-sensitive channels, originally isolated from single-celled organisms, to either inner retinal or remaining diseased cone photoreceptors. The gene encoding channel rhodopsin-2 (ChRd), originally identified in the algae Chlamydomonas reinhardii, has been delivered to either bipolar cells or ganglion cells; Halorhodopsin (NpHR), originally identified in halobacteria, has been delivered to diseased cone photoreceptors (Table 34.2 part B). Unlike mammalian opsins, however, these light-activated proteins directly form ion channels that polarize (NpHR) or depolarize (ChRd) upon photostimulation in a single molecule. Other groups have developed synthetic optogenetic molecules. With all of these molecules, however, after optogenetic gene therapy, animals that were previously insensitive to light showed development of retinal/visual behavior.46,121–125
Current status of retinal gene therapy trials: retinal diseases that have been/ARE currently being evaluated in human clinical trials
Studies that were completed, but discontinued
Retinoblastoma
Gene therapy has been used for treating tumor cells in the eye with the ultimate goal of avoiding the need to perform enucleation (and then chemotherapy and radiation therapy) in young children. If gene therapy were used to reduce the tumor burden, this would allow more control. Such an approach could potentially spare the patient disfigurement and loss of vision. Hurwitz and colleagues transduced murine retinoblastomas in vivo with an adenoviral vector containing the herpes simplex thymidine kinase gene followed by treatment with the prodrug ganciclovir (Table 34.2 part B).126,127 This resulted in a complete ablation of detectable tumors in 70% of animals and a significant prolongation of progression-free survival compared with untreated controls. The study was moved to clinical trial: 7 patients had resolution of their vitreous tumor and 1 patient remained free of active vitreous tumor 38 months after therapy.126,128
CNV using Ad.PEDF
Campochiaro and colleagues129 carried out a phase I clinical trial in individuals with advanced neovascular age-related macular degeneration (AMD). In this dose escalation study, a serotype 5 (Ad5), E1, partial E3-, E4-deleted adenoviral vector carrying human PEDF (AdPEDF.11) was injected intravitreally into one eye of each of 28 individuals (Table 34.2 part B). PEDF is an endogenous protein with potent antiangiogenic activity. It also has antiapoptotic activity. The vector was delivered in a dose escalation study and there were no serious adverse events or dose-limiting toxicities through the highest dose. There was evidence that injection of doses greater than 10E8 particle units of AdPEDF.11 resulted in antiangiogenic activity persisting for several months.
Studies in progress
Encapsulated cell therapy
Results of a phase I study of delivery of CNTF through encapsulated cell therapy (see above) were reported by Sieving et al.119 in 10 subjects with retinal degeneration. The implants were removed after 6 months. The delivery was safe save for one surgically related choroidal detachment and three of the individuals showed improved visual acuity. A phase II study followed. Two patients were followed with noninvasive imaging: one with RP and one with Usher syndrome type 2. While there were no changes in visual acuity, visual fields, or ERG responses, the outer retinal layers were thicker in CNTF-treated eye and cone spacing and density increased as judged with adaptive optics scanning laser ophthalmoscopy.118 The results of a multicenter, dose-ranging phase II study followed selected subjects with geographic atrophy and the results indicated that the implant slowed the progression of vision loss.117
Gene augmentation therapy for Leber congenital amaurosis
Three different clinical trials involving gene augmentation therapy for LCA due to RPE65 mutations were initiated near simultaneously in 2007. The disease in LCA-RPE65 is due to lack of a function of RPE 65 kDa (RPE65) protein in the RPE. RPE65 is an isomerohydrolase and breaks down retinyl ester, thereby allowing production of 11-cis retinal, the chromophore which contributes to the visual pigment rhodopsin.130 Each of the three studies used an AAV serotype 2 vector delivering the wild-type human RPE65 cDNA subretinally to the RPE in one eye but the studies differed in terms of dose, inclusion criteria, type of promoter, location of injection, and outcome measures. The early reports from all three trials revealed a high degree of safety and demonstrated efficacy as judged by increase in light sensitivity, improved visual acuity and visual fields, improved pupillary light reflex, and improved mobility.18,32,131
The entire set of results of the phase I/II study were reported from the group at CHOP and indicated that not only was the AAV delivery safe, but also each one of the 12 clinical trial subjects, ages 8–45 years, showed evidence of improved retinal and visual function as judged by any of several different test paradigms.29 The children in the study showed particularly large improvements, now being able to read books and play sports, although the older individuals also showed evidence of gain in function. The set of results from a second study were recently reported and were complementary yet did not describe any age-related effects of treatment.131a Two other clinical trials for LCA-RPE65 have recently been initiated (www.clinicaltrials.gov). One is designed to provide compassionate care and reported encouraging results for one patient.34
CNV and AAV-sFLT
Wet AMD is currently under consideration as a target for development of gene therapy. In this disease, people can go blind overnight due to leakage in blood vessels. Several therapies have been shown to be effective for the treatment of neovascular AMD, particularly anti-VEGF therapies. The downside of anti-VEGF therapies is that they must be readministered frequently, with the result that doctors are overburdened and patients must return frequently to receive intravitreal injections. There is a clear need for long-lasting production of a VEGF decoy in the eye – a stable product which could be delivered through a one-time injection. A strategy with which to interfere with proangiogenic activities of VEGF was employed and demonstrated to be effective by several groups in animal models using intravitreal delivery of an AAV-carrying soluble VEGF receptor (sFLT). This strategy is currently being tested in two different dose escalation studies in individuals with AMD (Table 34.2 part A).
CNV and lentivirus-mediated delivery of angiostatin and endostatin
Another strategy that is being tested in clinical trial for wet AMD is subretinal delivery of RetinoStat, a Lentivirus vector delivering angiostatin and endostatin.25,132 Both angiostatin and endostatin have strong and well-documented antineovascularization effects. The approach is similar to what was used in a previous phase I clinical trial using adenovirus-mediated delivery of PEDF (see above).
Promises and challenges of bringing retinal gene transfer from bench to bedside
Safety/efficacy/stability Issues
Several studies have been carried out showing that retinal gene transfer can be both safe and effective. The successes of the first human gene augmentation therapy studies involving retinal degeneration, the LCA-RPE65 studies,18,29,32,34,131,133 provide the foundation for gene therapy approaches for the treatment of other forms of inherited retinal degenerative diseases. LCA-RPE65 may well be the easiest target for study as it is slowly progressive, is caused by a gene whose cDNA fits within the limited AAV cargo capacity, and is caused by a defect in RPE cells. The latter are easy to target using AAV vectors. In predicting what other disease(s) could be targeted using a similar approach, one confronts the following challenges:
1. Not all transgene cassettes fit within the cargo confines of the AAV vector (Table 34.1). Studies in progress aim to modify AAV capsids so that they can carry larger cargo or, alternatively, deliver different portions of large transgenes in multiple AAVs so that, after infection has taken place, the cargo recombines in the target cell and leads to the production of a full-length therapeutic protein. Alternative strategies to packaging large transgene cassettes into rAAVs include delivery of a cDNA encoding a truncated but functional protein, or delivering the cDNA in segments through a “trans-splicing” approach. For the latter approach, the cDNA is split into two separate rAAV vectors using an engineered intron to mediate splicing of the two cDNA segments within the cell. Feasibility of this approach has been demonstrated in vivo in the mouse retina.134
2. Photoreceptors – not RPE cells – are the primarily diseased cells in many retinal degenerative diseases and those cells are not targeted as efficiently by AAV2 or by most forms of Lentivirus (Table 34.1). Fortunately, a number of hybrid vectors are now available that can target these cells efficiently and this work continues (Fig. 34.4; see above). Further, some of them result in transgene expression using 10-fold less vector than AAV2.39 Additional modifications of capsids, envelopes, and surface proteins will continue to provide improved toolkits with which to deliver these large genes efficiently to photoreceptors and other retinal cell types.
3. Some diseases progress very quickly and there will be challenges making sure that the gene is delivered before the cells have died. In some cases (such as in diseases which involve mutation in a gene expressed early in development), the optimal result will likely require delivery early in infancy, or even prenatally. In those cases, besides the need to identify the disease early in life, there will be difficult ethical issues to consider, in addition to risk-to-benefit ratios.
4. There are formidable economic challenges. For many of the potential disease targets, very few patients have been identified. Even though the cost of carrying out genotyping has decreased considerably over the years, it is still expensive. It would be possible to establish widespread genetic screening programs to identify the disease-causing gene in every individual with retinal degeneration, but who will pay for this? Another challenge involves unavailability or lack of an appropriate animal model. It is possible to engineer rodent models but, again, this is costly and time-consuming. Also, rodent models do not mirror some of the unique issues of the primate (human retina), for example, presence of a macula. Finally, the costs of establishing and running clinical trials will limit the number of therapies that can be tested. It is possible, however, that once the first gene therapy reagent is designated as a Food and Drug Administration-approved drug in the USA or in Europe, the regulatory (and thus financial) burden for developing additional drugs will decrease. For example, data from development of one particular vector could be used to support the development of another.
5. Gene therapy is not going to be effective for every retinal disease. There are some diseases in which the efficiency of transduction and/or levels of gene expression are going to be critical. Delivery of either too much or too little of the reagent will be either ineffective or possibly even toxic. There may also be immune responses to particular vectors/gene products that could limit efficacy. The recent successes in retinal gene therapy are very exciting but we have much to learn about the safety limitations not only with respect to type of gene, route of delivery, and dose, but also with respect to readministration.
Outcome measures in human clinical trials
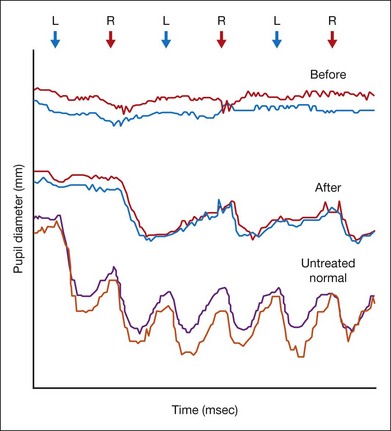
A number of outcome measures (visual acuity, visual field, color vision, and area of nonseeing retina) have been used or accepted in the development of drugs for ophthalmologic indications. Except for visual acuity, however, there is no generally accepted level of improvement considered statistically significant. (In visual acuity, this level is 15 letters, or three lines on an ETDRS eye chart.) For some of the diseases that are being considered for treatment with gene therapy, individuals may only have light perception vision and so an ETDRS chart is meaningless. In addition, endpoints that are used in adults may not be useful for small children, who do not have the attention span or cognitive abilities to participate in the study procedures. Some endpoints may not have enough sensitivity to detect a change in clinical status. Improvement on visual field testing can be considered clinically meaningful when results of multiple points in the visual field meet specific criteria. This test cannot be completed accurately, however, by an individual who cannot fixate due to nystagmus, such as individuals with LCA or cone–rod dystrophy. Thus, it is going to be very important to expand and validate additional efficacy outcomes such as pupillometry (Fig. 34.5) for particular target populations.
Plasticity of the visual system
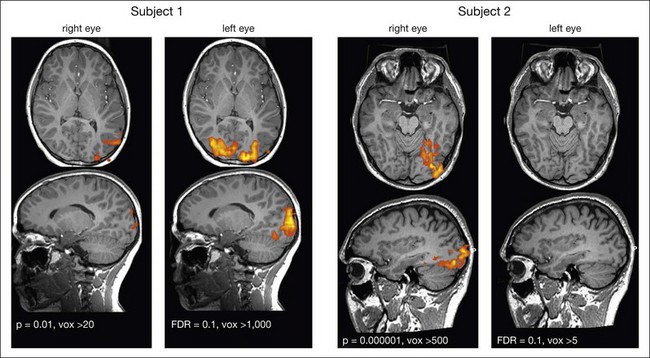
Of all sensory systems, vision provides the most information to the brain. Both laboratory and clinical studies have demonstrated that early onset of blindness can lead to structural and functional brain changes. Severe impairment of the visual pathway early in life due to developmental or retinal degenerative diseases is likely to limit the responsiveness of neurons in the visual cortex. Thus, while gene therapy for some early-onset retinal degenerative diseases may be able to rescue retinal function, it may not necessarily allow vision. The limits to restoration of the retinal-cortical pathways are not well defined, however, as shown by recent results from a functional magnetic resonance imaging (fMRI) study in children and young adults who had been treated at CHOP with gene therapy for LCA due to RPE65 mutations. Ashtari and colleagues135 carried out an fMRI study in human LCA-RPE65 patients to investigate how the cortex responds to the recovery of function after gene therapy in specific areas of the retina after prolonged visual deprivation. That study employed only dim light stimuli, since it is known that young LCA-RPE65 patients are able to see and function (albeit poorly) when their environment is brightly lit. Functional analyses were carried out separately for each individual patient to account for disease stage and treatment area in each of the subjects. The untreated eyes served as an internal control for each subject. The results showed that the visual cortex could become responsive to visual input, even after prolonged (up to 35 years in the oldest patient) visual deprivation (Fig. 34.6).135 This provides hope for being able to use gene therapy to resuscitate not just the retina but also the visual cortex even after chronic visual deprivation.
Genotyping issues
Over the past two decades, more than 220 different genes have been identified which, when mutated, cause retinal disease (http://www.sph.uth.tmc.edu/RetNet). Molecular diagnostics of inherited retinal diseases is performed in order to provide a definitive disease diagnosis and to be able to provide prognostic information to patients as well as genetic counseling. With the progress in developing gene therapy for retinal diseases, genotyping has become increasingly more important as patients want to know whether they are eligible for certain trials. In addition, genotype/phenotype data have become very important in planning ahead for other clinical trials. This information is invaluable for determining the optimal age for intervention, selecting appropriate outcome measures, and predicting how long it would take to document an improvement in the phenotype given the appropriate gene therapy intervention.
Generic strategies for reversing blindness in “dead” retinas
It is for that reason that generic strategies to treat retinal disease are appealing. As described above, there has long been an interest in using growth factors or neurotrophic factors to maintain the health of the retina for longer (Table 34.2 part B). In many diseases, a stabilization of the disease progression would be clinically meaningful. In “dead” retinas, it may be feasible to harness the remaining circuitry to provide some useful vision using “optogenetic therapy”. There are many technical challenges associated with this endeavor, including the desirability of engineering the molecules so that they would be useful with typical indoor lighting parameters. There are also potential biologic challenges with this approach. Will the mammalian retina recognize optogenetic (algal or bacterial) proteins produced in inner retinal cells as foreign? Nevertheless, the ability to deliver clinically meaningful vision to individuals who have none is very exciting and could supply hope to millions of patients with end-stage retinal or macular degeneration.
 http://www.expertconsult.com
http://www.expertconsult.com1 Farjo R, Skaggs J, Quiambao AB, et al. Efficient non-viral ocular gene transfer with compacted DNA nanoparticles. PLoS ONE. 2006;1:e38.
2 Johnson LN, Cashman SM, Kumar-Singh R. Cell-penetrating peptide for enhanced delivery of nucleic acids and drugs to ocular tissues including retina and cornea. Mol Ther. 2008;16:107–114.
3 Ortiz-Urda S, Thyagarajan B, Keene DR, et al. Stable nonviral genetic correction of inherited human skin disease. Nat Med. 2002;8:1166–1170.
4 Cai X, Nash Z, Conley SM, et al. A partial structural and functional rescue of a retinitis pigmentosa model with compacted DNA nanoparticles. PLoS ONE. 2009;4:e5290.
5 Read SP, Cashman SM, Kumar-Singh R. POD nanoparticles expressing GDNF provide structural and functional rescue of light-induced retinal degeneration in an adult mouse. Mol Ther. 2010;18:1917–1926.
6 Bennett J, Wilson J, Sun D, et al. Adenovirus vector-mediated in vivo gene transfer into adult murine retina. Invest Ophthalmol Vis Sci. 1994;35:2535–2542.
7 Li T, Adamian M, Roof DJ, et al. In vivo transfer of a reporter gene to the retina mediated by an adenoviral vector. Invest Ophthalmol Vis Sci. 1994;35:2543–2549.
8 Budenz D, Bennett J, Alonso L, et al. In vivo gene transfer into murine trabecular meshwork and corneal endothelial cells. Invest Ophthalmol Vis Sci. 1995;36:2211–2215.
9 Bennett J, Tanabe T, Sun D, et al. Photoreceptor cell rescue in retinal degeneration (rd) mice by in vivo gene therapy. Nat Med. 1996;2:649–654.
10 Bennett J, Zeng Y, Bajwa R, et al. Adenovirus-mediated delivery of rhodopsin-promoted bcl-2 results in a delay in photoreceptor cell death in the rd/rd mouse. Gene Ther. 1998;5:1156–1164.
11 Von Seggern D, Aguilar E, Kinder K. al. In vivo transduction of photoreceptors or ciliary body by intravitreal injection of pseudotyped adenoviral vectors. Mol Ther. 2003;7:27–34.
12 Cashman SM, McCullough L, Kumar-Singh R. Improved retinal transduction in vivo and photoreceptor-specific transgene expression using adenovirus vectors with modified penton base. Mol Ther. 2007;15:1640–1646.
13 Hoffman LM, Maguire AM, Bennett J. Cell-mediated immune response and stability of intraocular transgene expression after adenovirus-mediated delivery. Invest Ophthalmol Vis Sci. 1997;38:2224–2233.
14 Kumar-Singh R, Chamberlain JS. Encapsidated adenovirus minichromosomes allow delivery and expression of a 14 kb dystrophin cDNA to muscle cells. Hum Mol Genet. 1996;5:913–921.
15 Kumar-Singh R, Yamashita C, Tran K, et al. Construction of encapsidated (gutted) adenovirus minichromosomes and their application to rescue of photoreceptor degeneration. Methods Enzymol. 2000;316:724–743.
16 Acland GM, Aguirre GD, Bennett J, et al. Long-term restoration of rod and cone vision by single dose rAAV-mediated gene transfer to the retina in a canine model of childhood blindness. Mol Ther. 2005;12:1072–1082.
17 Acland GM, Aguirre GD, Ray J, et al. Gene therapy restores vision in a canine model of childhood blindness. Nat Genet. 2001;28:92–95.
18 Maguire AM, Simonelli F, Pierce EA, et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med. 2008;358:2240–2248.
19 Lebherz C, Auricchio A, Maguire AM, et al. Long-term inducible gene expression in the eye via adeno-associated virus gene transfer in nonhuman primates. Hum Gene Ther. 2005;16:178–186.
20 Allocca M, Mussolino C, Garcia-Hoyos M, et al. Novel adeno-associated virus serotypes efficiently transduce murine photoreceptors. J Virol. 2007;81:11372–11380.
21 Lebherz C, Maguire A, Tang W, et al. Novel AAV serotypes for improved ocular gene transfer. J Gene Med. 2008;10:375–382.
22 Auricchio A, Kobinger G, Anand V, et al. Exchange of surface proteins impacts on viral vector cellular specificity and transduction characteristics: the retina as a model. Hum Mol Genet. 2001;10:3075–3081.
23 Sun X, Pawlyk B, Xu X, et al. Gene therapy with a promoter targeting both rods and cones rescues retinal degeneration caused by AIPL1 mutations. Gene Ther. 2011;17:117–131.
24 Tan M, Smith AJ, Pawlyk B, et al. Gene therapy for retinitis pigmentosa and Leber congenital amaurosis caused by defects in AIPL1: effective rescue of mouse models of partial and complete Aipl1 deficiency. Hum Mol Genet. 2009;18:2099–2114.
25 Kachi S, Binley K, Yokoi K, et al. EIAV vector-mediated co-delivery of endostatin and angiostatin driven by the RPE-specific VMD2 promoter inhibits choroidal neovascularization. Hum Gene Ther. 2008;20:31–39.
26 Balaggan KS, Binley K, Esapa M, et al. Stable and efficient intraocular gene transfer using pseudotyped EIAV lentiviral vectors. J Gene Med. 2006;8:275–285.
27 Takahashi M, Miyoshi H, Verma IM, et al. Rescue from photoreceptor degeneration in the rd mouse by human immunodeficiency virus vector-mediated gene transfer. J Virol. 1999;73:7812–7816.
28 Kong J, Kim SR, Binley K, et al. Correction of the disease phenotype in the mouse model of Stargardt disease by lentiviral gene therapy. Gene Ther. 2008;15:1311–1320.
29 Maguire AM, High KA, Auricchio A, et al. Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: a phase I dose-escalation trial. Lancet. 2009;374:1597–1605.
30 Jiang LG, Jorquera M, Streilein JW. Subretinal space and vitreous cavity as immunologically privileged sites for retinal allografts. Invest Ophthalmol Vis Sci. 1993;34:3347–3354.
31 Anand V, Chirmule N, Maguire AM, et al. Induction of immune deviant response in the subretinal space is dependent on the nature of antigen introduced. Invest Ophthalmol Vis Sci. 2000;41:S117.
32 Bainbridge JW, Smith AJ, Barker SS, et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med. 2008;358:2231–2239.
33 Hauswirth WW, Aleman TS, Kaushal S, et al. Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum Gene Ther. 2008;19:979–990.
34 Banin E, Bandah-Rozenfeld D, Obolensky A, et al. Molecular anthropology meets genetic medicine to treat blindness in the north African Jewish population: Hum Gene Therapy initiated in Israel. Hum Gene Ther. 2010;21:1749–1757.
35 Bennicelli J, Wright JF, Komaromy A, et al. Reversal of blindness in animal models of Leber congenital amaurosis using optimized AAV2-mediated gene transfer. Mol Ther. 2008;16:458–465.
36 Bennett J, Maguire AM, Cideciyan AV, et al. Recombinant adeno-associated virus-mediated gene transfer to the monkey retina. Proc Natl Acad Sci U S A. 1999;96:9920–9925.
37 Amado D, Mingozzi F, Hui D, et al. Safety and efficacy of subretinal re-administration of an AAV2 vector in large animal models: implications for studies in humans. Sci Transl Med. 2010;2:21ra16.
38 Vandenberghe L, Bell P, Maguire A, et al. AAV9 targets cone photoreceptors in non-human primates. submitted.
39 Vandenberghe L, Bell P, Maguire A, et al. Dosage thresholds for AAV2 and AAV8 photoreceptor gene therapy in monkey. Sci Transl Med. 2011;3:88ra54.
40 Surace EM, Auricchio A, Reich SJ, et al. Delivery of adeno-associated virus vectors to the fetal retina: impact of viral capsid proteins on retinal neuronal progenitor transduction. J Virol. 2003;77:7957–7963.
41 Cepko C, Ryder E, Austin C, et al. Lineage analysis using retroviral vectors. Methods. 1998;14:393–406.
42 Dejneka NS, Surace EM, Aleman TS, et al. In utero gene therapy rescues vision in a murine model of congenital blindness. Mol Ther. 2004;9:182–188.
43 Dudus L, Anand V, Acland G, et al. Persistent transgene product in retina, optic nerve and brain after intraocular injection of rAAV. Vis Res. 1999;39:2545–2553.
44 Bennett J, Anand V, Acland GM, et al. Cross-species comparison of in vivo reporter gene expression after recombinant adeno-associated virus-mediated retinal transduction. Methods Enzymol. 2000;316:777–789.
45 Petrs-Silva H, Dinculescu A, Li Q, et al. Novel properties of tyrosine-mutant AAV2 vectors in the mouse retina. Mol Ther. 2011;19:293–301.
46 Caporale N, Kolstad KD, Lee T, et al. LiGluR restores visual responses in rodent models of inherited blindness. Mol Ther. 2011;19:1212–1219.
47 Greenberg KP, Geller SF, Schaffer DV, et al. Targeted transgene expression in Müller glia of normal and diseased retinas using lentiviral vectors. Invest Ophthalmol Vis Sci. 2007;48:1844–1852.
48 Ho TT, Maguire AM, Aguirre GD, et al. Phenotypic rescue after adeno-associated virus-mediated delivery of 4-sulfatase to the retinal pigment epithelium of feline mucopolysaccharidosis VI. J Gene Med. 2002;4:613–621.
49 Allocca M, Doria M, Petrillo M, et al. Serotype-dependent packaging of large genes in adeno-associated viral vectors results in effective gene delivery in mice. J Clin Invest. 2008;118:1955–1964.
50 Simons DL, Boye SL, Hauswirth WW, et al. Gene therapy prevents photoreceptor death and preserves retinal function in a Bardet–Biedl syndrome mouse model. Proc Natl Acad Sci U S A. 2011;108:6276–6281.
51 Michalakis S, Muhlfriedel R, Tanimoto N, et al. Restoration of cone vision in the CNGA3–/– mouse model of congenital complete lack of cone photoreceptor function. Mol Ther. 2010;18:2057–2063.
52 Pang JJ, Alexander J, Lei B, et al. Achromatopsia as a potential candidate for gene therapy. Adv Exp Med Biol. 2010;664:639–646.
53 Carvalho LS, Xu J, Pearson RA, et al. Long-term and age-dependent restoration of visual function in a mouse model of CNGB3-associated achromatopsia following gene therapy. Hum Mol Genet. 2011;20:3161–3175.
54 Alexander JJ, Umino Y, Everhart D, et al. Restoration of cone vision in a mouse model of achromatopsia. Nat Med. 2007;13:685–687.
55 Mihelec M, Pearson RA, Robbie SJ, et al. Long-term preservation of cones and improvement in visual function following gene therapy in a mouse model of Leber congenital amaurosis (LCA) caused by GC1 deficiency. Hum Gene Ther. 2011;22:1179–1190.
56 Boye SE, Boye SL, Pang J, et al. Functional and behavioral restoration of vision by gene therapy in the guanylate cyclase-1 (GC1) knockout mouse. PLoS ONE. 2010;5:e11306.
57 Williams ML, Coleman JE, Haire SE, et al. Lentiviral expression of retinal guanylate cyclase-1 (RetGC1) restores vision in an avian model of childhood blindness. PLoS Med. 2006;3:e201.
58 Batten ML, Imanishi Y, Tu DC, et al. Pharmacological and rAAV gene therapy rescue of visual functions in a blind mouse model of Leber congenital amaurosis. PLoS Med. 2005;2:e333.
59 Gargiulo A, Bonetti C, Montefusco S, et al. AAV-mediated tyrosinase gene transfer restores melanogenesis and retinal function in a model of oculo-cutaneous albinism type I (OCA1). Mol Ther. 2009;17:1347–1354.
60 Surace EM, Domenici L, Cortese K, et al. Amelioration of both functional and morphological abnormalities in the retina of a mouse model of ocular albinism following AAV-mediated gene transfer. Mol Ther. 2005;12:652–658.
61 Pang JJ, Boye SL, Kumar A, et al. AAV-mediated gene therapy for retinal degeneration in the rd10 mouse containing a recessive PDEbeta mutation. Invest Ophthalmol Vis Sci. 2008;49:4278–4283.
62 Pang JJ, Dai X, Boye SE, et al. Long-term retinal function and structure rescue using capsid mutant AAV8 vector in the rd10 mouse, a model of recessive retinitis pigmentosa. Mol Ther. 2011;19:234–242.
63 Andrieu-Soler C, Halhal M, Boatright JH, et al. Single-stranded oligonucleotide-mediated in vivo gene repair in the rd1 retina. Mol Vis. 2007;13:682–706.
64 Allocca M, Manfredi A, Iodice C, et al. AAV-mediated gene replacement either alone or in combination with physical and pharmacological agents results in partial and transient protection from photoreceptor degeneration associated with β-PDE deficiency. Invest Ophthalmol Vis Sci. 2011;52(8):5713–5719.
65 Cai X, Conley SM, Naash MI. Gene therapy in the retinal degeneration slow model of retinitis pigmentosa. Adv Exp Med Biol. 2010;664:611–619.
66 Georgiadis A, Tschernutter M, Bainbridge JW, et al. AAV-mediated knockdown of peripherin-2 in vivo using miRNA-based hairpins. Gene Ther. 2010;17:486–493.
67 Conley SM, Naash MI. Nanoparticles for retinal gene therapy. Prog Retin Eye Res. 2010;29:376–397.
68 Ali R, Sarra G-M, Stephens C, et al. Restoration of photoreceptor ultrastructure and function in retinal degeneration slow mice by gene therapy. Nature Genet. 2000;25:306–310.
69 Sarra G-M, Stephens C, de Alwis M, et al. Gene replacement therapy in the retinal degeneration slow (rds) mouse: the effect on retinal degeneration following partial transduction of the retina. Human Mol Genet. 2001;10:2353–2361.
70 Mao H, James T, Jr., Schwein A, et al. AAV delivery of wild-type rhodopsin preserves retinal function in a mouse model of autosomal dominant retinitis pigmentosa. Hum Gene Ther. 2011;22:567–575.
71 Pang J, Boye SE, Lei B, et al. Self-complementary AAV-mediated gene therapy restores cone function and prevents cone degeneration in two models of Rpe65 deficiency. Gene Ther. 2010;17:815–826.
72 Pang JJ, Chang B, Kumar A, et al. Gene therapy restores vision-dependent behavior as well as retinal structure and function in a mouse model of RPE65 Leber congenital amaurosis. Mol Ther. 2006;13:565–572.
73 Dejneka N, Surace E, Aleman T, et al. Fetal virus-mediated delivery of the human RPE65 gene rescues vision in a murine model of congenital retinal blindness. Mol Ther. 2004;9:182–188.
74 Narfstrom K, Bragadottir R, Redmond TM, et al. Functional and structural evaluation after AAV.RPE65 gene transfer in the canine model of Leber’s congenital amaurosis. Adv Exp Med Biol. 2003;533:423–430.
75 Narfstrom K, Katz ML, Bragadottir R, et al. Functional and structural recovery of the retina after gene therapy in the RPE65 null mutation dog. Invest Ophthalmol Vis Sci. 2003;44:1663–1672.
76 Narfstrom K, Katz ML, Ford M, et al. In vivo gene therapy in young and adult RPE65–/– dogs produces long-term visual improvement. J Hered. 2003;94:31–37.
77 Pawlyk BS, Smith AJ, Buch PK, et al. Gene replacement therapy rescues photoreceptor degeneration in a murine model of Leber congenital amaurosis lacking RPGRIP. Invest Ophthalmol Vis Sci. 2005;46:3039–3045.
78 Kjellstrom S, Bush RA, Zeng Y, et al. Retinoschisin gene therapy and natural history in the Rs1h-KO mouse: long-term rescue from retinal degeneration. Invest Ophthalmol Vis Sci. 2007;48:3837–3845.
79 Zeng Y, Takada Y, Kjellstrom S, et al. RS-1 gene delivery to an adult Rs1h knockout mouse model restores ERG b-wave with reversal of the electronegative waveform of X-linked retinoschisis. Invest Ophthalmol Vis Sci. 2004;45:3279–3285.
80 Janssen A, Min SH, Molday LL, et al. Effect of late-stage therapy on disease progression in AAV-mediated rescue of photoreceptor cells in the retinoschisin-deficient mouse. Mol Ther. 2008;16:1010–1017.
81 Min SH, Molday LL, Seeliger MW, et al. Prolonged recovery of retinal structure/function after gene therapy in an Rs1h-deficient mouse model of X-linked juvenile retinoschisis. Mol Ther. 2005;12:644–651.
82 Zou J, Luo L, Shen Z, et al. Whirlin replacement restores the formation of the USH2 protein complex in whirlin knockout photoreceptors. Invest Ophthalmol Vis Sci. 2011;52:2343–2351.
83 Mancuso K, Hauswirth WW, Li Q, et al. Gene therapy for red-green colour blindness in adult primates. Nature. 2009;461:784–787.
84 Tam LC, Kiang AS, Chadderton N, et al. Protection of photoreceptors in a mouse model of RP10. Adv Exp Med Biol. 2010;664:559–565.
85 Tam LC, Kiang AS, Kennan A, et al. Therapeutic benefit derived from RNAi-mediated ablation of IMPDH1 transcripts in a murine model of autosomal dominant retinitis pigmentosa (RP10). Hum Mol Genet. 2008;17:2084–2100.
86 Millington-Ward S, Chadderton N, O’Reilly M, et al. Suppression and replacement gene therapy for autosomal dominant disease in a murine model of dominant retinitis pigmentosa. Mol Ther. 2011;19:642–649.
87 Palfi A, Ader M, Kiang AS, et al. RNAi-based suppression and replacement of rds-peripherin in retinal organotypic culture. Hum Mutat. 2006;27:260–268.
88 Chadderton N, Millington-Ward S, Palfi A, et al. Improved retinal function in a mouse model of dominant retinitis pigmentosa following AAV-delivered gene therapy. Mol Ther. 2009;17:593–599.
89 O’Reilly M, Palfi A, Chadderton N, et al. RNA interference-mediated suppression and replacement of human rhodopsin in vivo. Am J Hum Genet. 2007;81:127–135.
90 Mussolino C, Sanges D, Marrocco E, et al. Zinc-finger-based transcriptional repression of rhodopsin in a model of dominant retinitis pigmentosa. EMBO. Mol Med. 2011;3:118–128.
91 LaVail MM, Yasumura D, Matthes MT, et al. Ribozyme rescue of photoreceptor cells in P23H transgenic rats: Long-term survival and late stage therapy. Proc Natl Acad Sci U S A. 2000;97:11488–11493.
92 Lewin AS, Drenser KA, Hauswirth WW, et al. Ribozyme rescue of photoreceptor cells in a transgenic rat model of autosomal dominant retinitis pigmentosa. Nat Med. 1998;4:967–971.
93 Lai CM, Estcourt MJ, Wikstrom M, et al. rAAV.sFlt-1 gene therapy achieves lasting reversal of retinal neovascularization in the absence of a strong immune response to the viral vector. Invest Ophthalmol Vis Sci. 2009;50:4279–4287.
94 Lai CM, Shen WY, Brankov M, et al. Long-term evaluation of AAV-mediated sFlt-1 gene therapy for ocular neovascularization in mice and monkeys. Mol Ther. 2005;12:659–668.
95 Maclachlan TK, Lukason M, Collins M, et al. Preclinical safety evaluation of AAV2-sFLT01 – a gene therapy for age-related macular degeneration. Mol Ther. 2011;19:326–334.
96 Pechan P, Rubin H, Lukason M, et al. Novel anti-VEGF chimeric molecules delivered by AAV vectors for inhibition of retinal neovascularization. Gene Ther. 2009;16:10–16.
97 Tuo J, Pang JJ, Cao X, Shen D, Zhang J, Scaria A, et al. AAV5-mediated sFLT01 gene therapy arrests retinal lesions in Ccl2(–/–)/Cx3cr1(–/–) mice. Neurobiol Aging. 2012;33:433.e1–433.e10.
98 Lukason M, DuFresne E, Rubin H, et al. Inhibition of choroidal neovascularization in a nonhuman primate model by intravitreal administration of an AAV2 vector expressing a novel anti-VEGF molecule. Mol Ther. 2011;19:260–265.
99 Buch PK, MacLaren RE, Duran Y, et al. In contrast to AAV-mediated CNTF expression, AAV-mediated GDNF expression enhances gene replacement therapy in rodent models of retinal degeneration. Mol Ther. 2006;14:700–709.
100 Gauthier R, Joly S, Pernet V, et al. Brain-derived neurotrophic factor gene delivery to Müller glia preserves structure and function of light-damaged photoreceptors. Invest Ophthalmol Vis Sci. 2005;46:3383–3392.
101 McGee LH, Lau D, Zhou S, et al. Rescue of photoreceptor degeneration in S334ter(4) mutant rhodopsin transgenic rats by adeno-associated virus (AAV)-mediated delivery of basic fibroblast growth factor. Invest Ophthalmol Vis Sci. 1999;40:S936.
102 Akimoto M, Miyatake S-I, Kogishi J-I, et al. Adenovirally expressed basic fibroblast growth factor rescues photoreceptor cells in RCS rats. Invest Ophthalmol Vis Sci. 1999;40:273–279.
103 Rex TS, Allocca M, Domenici L, et al. Systemic but not intraocular Epo gene transfer protects the retina from light-and genetic-induced degeneration. Mol Ther. 2004;10:855–861.
104 Bush RA, Lei B, Tao W, et al. Encapsulated cell-based intraocular delivery of ciliary neurotrophic factor in normal rabbit: dose-dependent effects on ERG and retinal histology. Invest Ophthalmol Vis Sci. 2004;45:2420–2430.
105 Adamus G, Sugden B, Shiraga S, et al. Anti-apoptotic effects of CNTF gene transfer on photoreceptor degeneration in experimental antibody-induced retinopathy. J Autoimmun. 2003;21:121–129.
106 Bok D, Yasumura D, Matthes MT, et al. Effects of adeno-associated virus-vectored ciliary neurotrophic factor on retinal structure and function in mice with a P216L rds/peripherin mutation. Exp Eye Res. 2002;74:719–735.
107 Cayouette M, Gravel C. Adenovirus-mediated gene transfer of ciliary neurotrophic factor can prevent photoreceptor degeneration in the retinal degeneration (rd) mouse. Hum Gene Ther. 1997;8:423–430.
108 Huang SP, Lin PK, Liu JH, et al. Intraocular gene transfer of ciliary neurotrophic factor rescues photoreceptor degeneration in RCS rats. J Biomed Sci. 2004;11:37–48.
109 Liang F-Q, Aleman TS, Dejneka NS, et al. Long-term protection of retinal structure but not function using rAAV.CNTF in animal models of retinitis pigmentosa. Mol Ther. 2001;4:461–472.
110 Pearce-Kelling SE, Rein D, Acland GM, et al. Encapsulated cell-based intraocular delivery of CNTF slows inherited retinal degeneration in the rcd1 dog model. Invest Ophthalmol Vis Sci. 2000;41:S542.
111 Peterson WM, Flannery JG, Hauswirth WW, et al. Enhanced survival of photoreceptors in P23H mutant rhodopsin transgenic rats by adeno-associated virus (AAV)-mediated delivery of neurotrophic genes. Invest Ophthalmol Vis Sci. 1998;39:S1117.
112 Liang F-Q, Dejneka NS, Cohen DR, et al. AAV-mediated delivery of ciliary neurotrophic factor prolongs photoreceptor survival in the rhodopsin knockout mouse. Mol Ther. 2001;3:241–248.
113 Dalkara D, Kolstad KD, Guerin KI, et al. AAV mediated GDNF secretion from retinal glia slows down retinal degeneration in a rat model of retinitis pigmentosa. Mol Ther. 2011;19:1602–1608.
114 Leonard KC, Petrin D, Coupland SG, et al. XIAP protection of photoreceptors in animal models of retinitis pigmentosa. PLoS ONE. 2007;2:e314.
115 Schlichtenbrede FC, MacNeil A, Bainbridge JW, et al. Intraocular gene delivery of ciliary neurotrophic factor results in significant loss of retinal function in normal mice and in the Prph2Rd2/Rd2 model of retinal degeneration. Gene Ther. 2003;10:523–527.
116 MacDonald IM, Sauve Y, Sieving PA. Preventing blindness in retinal disease: ciliary neurotrophic factor intraocular implants. Can J Ophthalmol. 2007;42:399–402.
117 Zhang K, Hopkins JJ, Heier JS, et al. Ciliary neurotrophic factor delivered by encapsulated cell intraocular implants for treatment of geographic atrophy in age-related macular degeneration. Proc Natl Acad Sci U S A. 2011;108:6241–6245.
118 Talcott KE, Ratnam K, Sundquist SM, et al. Longitudinal study of cone photoreceptors during retinal degeneration and in response to ciliary neurotrophic factor treatment. Invest Ophthalmol Vis Sci. 2011;52:2219–2226.
119 Sieving PA, Caruso RC, Tao W, et al. Ciliary neurotrophic factor (CNTF) for human retinal degeneration: phase I trial of CNTF delivered by encapsulated cell intraocular implants. Proc Natl Acad Sci U S A. 2006;103:3896–3901.
120 Leveillard T, Mohand-Said S, Lorentz O, et al. Identification and characterization of rod-derived cone viability factor. Nat Genet. 2004;36:755–759.
121 Lagali PS, Balya D, Awatramani GB, et al. Light-activated channels targeted to ON bipolar cells restore visual function in retinal degeneration. Nat Neurosci. 2008;11:667–675.
122 Tomita H, Sugano E, Yawo H, et al. Restoration of visual response in aged dystrophic RCS rats using AAV mediated channelopsin-2 gene transfer. Invest Ophthalmol Vis Sci. 2007;48:3821–3826.
123 Bi A, Cui J, Ma YP, et al. Ectopic expression of a microbial-type rhodopsin restores visual responses in mice with photoreceptor degeneration. Neuron. 2006;50:23–33.
124 Doroudchi MM, Greenberg KP, Liu J, et al. Virally delivered Channelrhodopsin-2 safely and effectively restores visual function in multiple mouse models of blindness. Mol Ther. 2011;19:1220–1229.
125 Busskamp V, Duebel J, Balya D, et al. Genetic reactivation of cone photoreceptors restores visual responses in retinitis pigmentosa. Science. 2010;329:413–417.
126 Hurwitz M, Marcus K, Chevez-Barrios P, et al. Suicide gene therapy for treatment of retinoblastoma in a murine model. Hum Gene Ther. 1999;10:441–448.
127 Hurwitz RL, Marcus KT, Chevez-Barrios P, et al. Suicide gene therapy of retinoblastoma in a murine model. Invest Ophthalmol Vis Sci. 1998;39:S1118.
128 Chevez-Barrios P, Chintagumpala M, Mieler W, et al. Response of retinoblastoma with vitreous tumor seeding to adenovirus-mediated delivery of thymidine kinase followed by ganciclovir. J Clin Oncol. 2005;23:7927–7935.
129 Campochiaro PA, Nguyen QD, Shah SM, et al. Adenoviral vector-delivered pigment epithelium-derived factor for neovascular age-related macular degeneration: results of a phase I clinical trial. Hum Gene Ther. 2006;17:167–176.
130 Redmond TM, Poliakov E, Yu S, et al. Mutation of key residues of RPE65 abolishes its enzymatic role as isomerohydrolase in the visual cycle. Proc Natl Acad Sci U S A. 2005;102:13658–13663.
131 Hauswirth W, Aleman T, Kaushal S, et al. Treatment of Leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum Gene Ther. 2008;19:979–990.
131a Jacobson SG, Cideciyan AV, Ratnakaram R, et al. Gene therapy for Leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch Ophthalmol. 2012;130:9–24.
132 Kachi S, Binley K, Yokoi K, et al. Equine infectious anemia viral vector-mediated codelivery of endostatin and angiostatin driven by retinal pigmented epithelium-specific VMD2 promoter inhibits choroidal neovascularization. Hum Gene Ther. 2009;20:31–39.
133 Simonelli F, Maguire AM, Testa F, et al. Gene therapy for Leber’s congenital amaurosis is safe and effective through 1.5 years after vector administration. Mol Ther. 2010;18:643–650.
134 Reich SJ, Auricchio A, Hildinger M, et al. Efficient trans-splicing in the retina expands the utility of adeno-associated virus as a vector for gene therapy. Hum Gene Ther. 2003;14:37–44.
135 Ashtari M, Cyckowski LL, Monroe JF, et al. The human visual cortex responds to gene therapy-mediated recovery of retinal function. J Clin Invest. 2011;121:2160–2168.
136 Vollrath D, Feng W, Duncan J, et al. Correction of the retinal dystrophy phenotype of the RCS rat by viral gene transfer of Mertk. Proc Natl Acad Sci U S A. 2001;98:12584–12589.
137 Smith AJ, Schlichtenbrede FC, Tschernutter M, et al. AAV-mediated gene transfer slows photoreceptor loss in the RCS rat model of retinitis pigmentosa. Mol Ther. 2003;8:188–195.
138 Gorbatyuk MS, Knox T, LaVail MM, et al. Restoration of visual function in P23H rhodopsin transgenic rats by gene delivery of BiP/Grp78. Proc Natl Acad Sci U S A. 2010;107:5961–5966.
139 Rex T, Bennett J, Maguire A, et al. Adeno-catalase protects the Balb/C mouse retina from light damage. Ft. Lauderdale, FL: ARVO, 2003.
140 Auricchio A, Behling KC, Maguire AM, et al. Inhibition of retinal neovascularization by intraocular viral-mediated delivery of anti-angiogenic agents. Mol Ther. 2002;6:490–494.
141 Gehlbach P, Demetriades AM, Yamamoto S, et al. Periocular injection of an adenoviral vector encoding pigment epithelium-derived factor inhibits choroidal neovascularization. Gene Ther. 2003;10:637–646.
142 Miyazaki M, Ikeda Y, Yonemitsu Y, et al. Simian lentiviral vector-mediated retinal gene transfer of pigment epithelium-derived factor protects retinal degeneration and electrical defect in Royal College of Surgeons rats. Gene Ther. 2003;10:1503–1511.
143 Gehlbach P, Demetriades AM, Yamamoto S, et al. Periocular gene transfer of sFlt-1 suppresses ocular neovascularization and vascular endothelial growth factor-induced breakdown of the blood–retinal barrier. Hum Gene Ther. 2003;14:129–141.
144 Saishin Y, Silva RL, Kachi S, et al. Periocular gene transfer of pigment epithelium-derived factor inhibits choroidal neovascularization in a human-sized eye. Hum Gene Ther. 2005;16:473–478.
145 Behling K, Auricchio A, O’Connor E, et al. AAV-mediated retinal transfer of anti-angiogenic genes in a murine model of retinopathy of prematurity (ROP). Invest Ophthalmol Vis Sci. 2002;6:490–494.
146 Mori K, Gehlbach P, Ando A, et al. Regression of ocular neovascularization in response to increased expression of pigment epithelium-derived factor. Invest Ophthalmol Vis Sci. 2002;43:2428–2434.
147 Mori K, Gehlbach P, Yamamoto S, et al. AAV-mediated gene transfer of pigment epithelium-derived factor inhibits choroidal neovascularization. Invest Ophthalmol Vis Sci. 2002;43:1994–2000.
148 Li Q, Miller R, Han PY, et al. Intraocular route of AAV2 vector administration defines humoral immune response and therapeutic potential. Mol Vis. 2008;14:1760–1769.
149 Mori K, Duh E, Gehlbach P, et al. Pigment epithelium-derived factor inhibits retinal and choroidal neovascularization. J Cell Physiol. 2001;188:253–263.
[/level-membership-for-opthalmology-category][not-level-membership-for-opthalmology-category]
Chapter 34 Gene Therapy for Retinal Disease
For additional online content visit http://www.expertconsult.com
Background: preclinical gene therapy studies
Nonviral gene delivery
There are nonviral methods of delivering nucleic acids to cells, and those include use of physicochemical agents to compact the DNA and/or transport it across the membrane lipid bilayer.1,2 The transport across the bilayer can also be achieved through physical means, including propulsion with a “gene gun” or by electroporation or iontophoresis. Such methods can also be combined with reagents that enhance integration into the host chromosome.3 There are several potential advantages of nonviral approaches: First, they can be used to deliver DNA of unlimited size. Second, there is a smaller chance of detrimental immune response since the only antigen would be the nucleic acid itself plus any protein that is used as a condensation agent. Several studies have demonstrated proof of concept of retinal gene therapy using nonviral DNA delivery1,4,5 and additional studies will reveal the long-term safety, stability, and efficacy of this approach.
Viral vector-mediated gene delivery
Recombinant viruses are genetically neutered or crippled so that they cannot reproduce and cause an infectious disease once they infect a target cell. There is a large list of recombinant viruses that have been tested in the retina (Table 34.1). Different viruses have different attributes and challenges, including cargo capacity, ease of purification, cellular specificity, and immune response. However, a large portion of these have been used to demonstrate efficacy in animal models of retinal disease (Table 34.2).
Table 34.1 Vectors tested for transduction characteristics in animal models. Various retinal cell targets are listed; however, the exact targets depend on the route of administration, dose, species, and modifications to the vector
Table 34.2 Recent studies showing proof of concept of retinal gene therapy strategies using (A) selected approaches targeting a specific gene and (B) “generic” approaches that could potentially be used regardless of the disease-causing genetic defect. This list highlights many of the recent studies aiming to treat animal models of retinal degeneration, retinal development anomalies, and retinal neovascularization. Several of these studies have been carried forward to human clinical trial and those references are listed as well
The first recombinant adenovirus vectors, generated from the common respiratory virus, carried deletions in the adenoviral E1, E3 genes, and these Ad type 5 (Ad5) vectors were the first to be evaluated for retinal gene transfer in the differentiated retina.6,7 Adenovirus vectors result in high levels of gene expression within 24–48 hours. When injected subretinally, they target retinal pigment epithelial (RPE) cells efficiently in the adult eye and also Müller cells.6,7 When injected intravitreally, they target Müller cells and cells in the anterior segment, including corneal endothelium, lens and iris epithelium, and cells in the outflow tract (such as trabecular meshwork cells).8 Similar results are found when recombinant adenovirus is injected into the undifferentiated (early postnatal) retina; however, in addition to RPE cells, progenitor cells are targeted in the neonatal mouse eye.9,10 Additional manipulations of adenovirus vectors have yielded reagents that target photoreceptors more efficiently.11,12
A disadvantage of the early generations of E1, E3-deleted adenovirus is that it still carries viral open reading frames. These can enhance its immunogenicity, even in the immune-privileged environment of the eye. These vectors were thus also used to probe the nature of the intraocular immune response. When these vectors were injected subretinally, transgene expression persisted for several weeks to months. However, when injected intravitreally, transgene expression ceased within 2 weeks. Expression could be prolonged by incorporating immune-suppressant molecules, however.13 Efforts were made to generate adenovirus vectors lacking any viral open reading frames, the so-called “gutted” or helper independent vectors, thereby reducing immune clearance and allowing stable transgene expression.14 Such vectors do indeed result in more stable transgene expression than the first-generation vectors and, further, have a much greater cargo capacity than the original adenoviral vectors (Table 34.1).15 They are more difficult to generate, however, and preparations are likely to be contaminated by wild-type adenovirus.
Adeno-associated virus (AAV) vectors do not carry any virus open reading frames (and thus encode any virus-specific proteins) and therefore are generally more favorable from an immunologic standpoint than adenovirus vectors (Table 34.1). There is also an abundant amount of safety data related to AAV administration in animals and in humans, both systemically and intraocularly. Recombinant AAV (rAAV) vectors have an added benefit in that they target a more diverse set of cell types than adenoviral (or other) vectors. Unlike lentiviral vectors, AAV vectors do not integrate, or do so only rarely (Table 34.1). However, since the transgene persists in episomal fashion in the target retinal cells, rAAV vectors result in stable transgene expression. Expression persists for the life of small animals (mice and rats) and at least for many years in large animals and humans.16–19 rAAV vectors are useful for delivering genes efficiently to many types of retinal cells. A disadvantage of these vectors is their relatively limited cargo capacity (a maximum of 4.8 kb) (Table 34.1).
In AAV vectors, the transgene cassette is bordered by the inverted terminal repeats (ITRs) from an AAV2 genome. The original AAV vectors were generated by packaging the transgene cassette ITRs into an AAV serotype 2 capsid and this resulted in “AAV2/2” vectors, i.e., ITRs of an AAV serotype 2 genome packaged into an AAV2 serotype 2 capsid. (Often investigators skip the reference to the ITR serotype and refer to AAV2 vectors instead of AAV2/2 vectors.) More than a dozen AAVs of different serotypes have been described. Cross-packaged rAAVs (for example, rAAV2/5 vectors generated after packaging the AAV2 cis plasmid into AAV serotype 5 capsid) differ significantly from rAAV2/2 vectors with respect to cellular specificity, efficiency of transduction, and onset of transgene expression. While rAAV2/2 targets RPE cells efficiently (and photoreceptors less efficiently), it takes up to 6 weeks for transgene expression mediated by this vector to plateau.20–22 In comparison, rAAV2/5 and rAAV2/8 vectors transduce photoreceptors with much higher efficiency than rAAV2/2 and result in transgene expression within 5–10 days of delivery.20,21 This information is helpful in selecting vectors for particular applications. For example, in a relatively slowly progressive retinal degenerative disease (Leber congenital amaurosis (LCA) due to RPE65 mutations), rAAV2/2 performs well in delivering a therapeutic transgene to RPE cells. For an animal model with a much faster rate of degeneration (e.g., LCA due to AIPL1 mutations23,24), it is necessary to use a vector with a much faster onset of expression and which targets photoreceptors efficiently (such as a rAAV2/5 or 2/8 vector; Table 34.2).
Vectors originally based on the human immunodeficiency virus, lentiviral vectors, have been shown to be safe in animal models and their safety in humans is currently being tested (Table 34.2). A number of groups have since generated vectors based on viruses that were identified in nonhuman species, such as equine Lentivirus.25,26 Transgene expression is stable after lentiviral administration, because these vectors mediate integration into the host chromosome (Table 34.1). Lentiviral vectors target RPE cells efficiently after subretinal injection and, in undifferentiated retina, also target neural progenitor cells. They have thus been used to demonstrate efficacy in animal models of RPE disease, such as LCA-RPE65 or autosomal recessive (AR) retinitis pigmentosa (RP) due to PDE6B mutations27 (Table 34.2). Lentiviral vectors are also attractive in that they can carry a cargo of ~7.5 kb (Table 34.1). Specific modifications also allow lentiviral vectors to target mature photoreceptors.28 The lentiviral envelope can be swapped in order to alter cellular tropism.22 Since lentiviral vectors are integrating vectors, there is a concern about the potential for insertional mutagenesis. To date there has been no report of such an effect after retinal administration in animal models.
Surgical delivery
When a subretinal injection is performed through a small retinotomy, a retinal detachment or “bleb” is raised (Fig. 34.1). Most if not all of the volume of injected material is trapped between the outer retina and RPE as a localized retinal detachment. There is negligible escape of material back through the retinotomy site into the vitreous, as evidenced by the fact that, initially, the size of the bleb does not change once it is formed. There is apparently little pressure differential between the subretinal space and vitreous once the bleb is established and this is especially so when the scleral incisions are closed. In addition, the small-gauge cannulas used for the subretinal injection appear to be self-sealing, especially when a gas tamponade or formed vitreous is present. Since a bleb raised by subretinal injection tends not to expand beyond the border of the initial injection, the volume of distribution of the administered agent is limited, especially when compared to an intravitreal injection or systemic administration. The concentration of the compound contained in the bleb remains high and may even increase as the RPE cells extract free water from the vehicle used to dilute the agent. Limiting the volume of distribution in this manner may serve both to increase the efficiency of drug delivery and to decrease local and systemic toxicities by restricting diffusion of the drug. The location of the original subretinal detachment cannot be appreciated in most species (including humans) after it has flattened.18,29 (The location is often visible in dogs years after the injection, however, because of alterations in reflectivity of the underlying tapetum due to the procedure: Fig. 34.1.)
Since the RPE and Bruch’s membrane are typically not violated when subretinal injection is performed by the pars plana approach, there is further protection against systemic exposure of antigens via the highly vascular choroidal circulation. In addition, there is minimal disruption of retinal vasculature since the placement of the injection cannula is done under direct visualization. Therefore, the integrity of the blood–ocular barrier remains intact when subretinal delivery is performed in this manner. There is in addition an immunologic compartmentalization when antigenic material is delivered to the subretinal space. Limiting exposure to this area may result not only in characteristic immunoprivilege behavior evident with intraocular delivery but, when delivery is confined to the subretinal space, antigenic tolerance can be induced due to immune-deviant response.30,31 This unique property of the subretinal space is of great significance in the delivery of biologics and in gene therapy in particular as the development of antibody/immune response to both viral antigens and foreign transgene products may limit the effectiveness of treatment.
The major differences in surgical methods between various investigators who have carried out gene transfer in humans so far18,29,32–34 involve: (1) use of perioperative systemic corticosteroid therapy; (2) the removal of posterior cortical vitreous prior to subretinal injection; and (3) placement of a gas (air) bubble after the injection is performed. With regard to systemic corticosteroid use, there appears to be no important difference in efficacy. It should be noted that all studies employ the use of topical and periocular corticosteroid to suppress surgical inflammation.
The surgical protocol for the Children’s Hospital of Philadelphia (CHOP) gene therapy trial for LCA2 specifies removal of the posterior hyaloid.18,29 In many instances, a complete posterior vitreous detachment (PVD) is already present despite the young age of the subjects enrolled in this trial. This is not unexpected as vitreous abnormalities, including the presence of debris or posterior separation, are characteristic features of eyes with retinal degeneration. In cases without PVD, the posterior cortical vitreous is engaged with active suction and the hyaloid face is gently separated to create a complete PVD, as evidenced by the presence of a glial ring separating from the optic nerve head (Weiss ring). Once the presence of a PVD is confirmed, the mobilized vitreous is removed as completely as possible, with special attention to pare back any gel in the vicinity of the active sclerotomy sites. This is done both to avoid vitreoretinal traction induced by instruments passing into and out of the eye, and also to prevent vitreous traction which can bend the tip of the 39-gauge (and smaller) subretinal injection cannula. Most investigators recommend the removal of epiretinal membrane (ERM) if present in the macular area in order to prevent interference with the injection cannula and to avoid late-occurring complications such as macular hole resulting from membrane contraction.
When directing the injection into the posterior pole or macula, the cannula tip is usually placed in the vicinity of the papillomacular bundle. Even in eyes with advanced retinal degeneration, the retina in this area is usually thick enough to allow for successful placement of the cannula tip and the injection into the subretinal space. The cannula tip is positioned so as to avoid direct injury to retinal arterioles. The CHOP protocol specifies that the site of injection be a minimum of 3 mm from the foveal center in order to avoid development of a foveal dehiscence from fluid tracking directly to the central macula in a fistula-like manner. In addition, a small bubble of perfluoro-octane liquid is placed over the fovea to counteract the hydrodynamic force created during the subretinal injection, thereby buttressing this anatomically vulnerable area (Fig. 34.2; video 51 online).
Fig. 34.2 Subretinal injection with protection to the fovea. (A) Subretinal injection cannula is apposed to the retina. Perfluoron had already been layered over the fovea. (B) The injection was initiated. A few small bubbles were expressed initially from the cannula (and are in the subretinal space). (C) The bleb has expanded. (D) The cannula has been removed and the Perfluoron is being removed. The inferior border of the bleb is visible. (See video 51 (online) showing subretinal injection of AAV2-hRPE65v2 in a human.29)
One unique feature of subretinal injection in retinal gene therapy when compared to other surgical indications is that the subretinal fluid, i.e., gene therapy agent, is not evacuated and the retina flattened at the time of fluid–air exchange. This is done in order to maximize the time of exposure to the subretinal injection, something that is not necessary, for example, after the extraction of a subretinal hemorrhage or translocation of the macula. Indeed, the creation of retinal detachment by subretinal injection is of itself a concern with respect to possible retinal toxicity. Fortunately, the extent of injury after acute retinal detachment created by injection of physiologic solutions appears to be small based on both laboratory and clinical data. Resorption of volumes less than 0.45 mL typically occurs in less than 24 hours.16–18,29,35–37 In the CHOP trial, resorption of 0.3 mL of fluid was observed within 6 hours of injection. Bainbridge et al.32 reported a longer period of detachment >24 hours after subretinal injection using a two-step injection technique where vector was administered into a bleb raised first with a physiologic solution. The longer period required for reattachment is likely due to the large volume of injection, i.e., 1.0 mL.32
Development of macular hole is a complication which appears to be unique to the subretinal injection procedure. Although creation of macular hole has been described in subretinal surgery for choroidal neovascularization (CNV), this typically occurs as a CNV is extracted in the presence of an adhesion to the overlying retina. In the CHOP trial, macular hole developed in one patient not during the surgical manipulation but rather several days postoperatively. Although ultimately the ophthalmoscopic appearance of the defect was typical for an idiopathic macular hole, several key differences were noted. First, prior to the development of a full-thickness defect, inner lamellar thinning was demonstrated on optical coherence tomography.18 In addition, intraretinal edema or cystoid macular edema was never present either during evolution of the hole or after hole formation. Finally, a subretinal fluid cuff was never apparent before or after hole formation. In the absence of inflammation, vasculopathy, or acute tissue injury, it was felt that the macular hole was unlikely to have been caused by drug toxicity. The appearance of the lamellar defect was reminiscent of pseudohole caused by ERM contracture. Since the ERM was recognized preoperatively and the posterior cortical vitreous was removed at surgery, it was thought that surgery-induced contraction of the pre-existing ERM was the most likely cause of macular hole formation. A second patient was observed to develop a foveal dehiscence at the time of subretinal injection as fluid channeled directly from the cannula through the fovea through a fistula-like tract.29 This phenomenon has been observed to occur infrequently during subretinal injection in nonhuman primates.38 Occurrence of a foveal dehiscence is presumably induced by intense hydrodynamic stress from injection in the vicinity of the fovea. No ERM was present in this case. Air tamponade with face-down positioning resulted in complete resolution of this induced hole within days. Since instituting a protocol modification specifying a minimum distance from the fovea for injection and use of perfluorocarbon liquid prior to injection, intraoperative foveal dehiscence has not occurred.29
History of retinal gene therapy
Preclinical studies: retinal transduction characteristics of different vectors
Initial gene transfer studies in animals generally evaluated the safety and stability of delivery of “reporter genes,” genes whose activity in terms of protein products can be evaluated noninvasively or in tissue samples through either histochemical/immunohistochemical measures or through a bioassay. In the early 1990s, these studies often utilized the LacZ (Escherichia coli β-galactosidase-encoding) gene. β-Galactosidase activity can be detected through either histochemical or spectophotometric activity. In the mid to late 1990s, many groups switched to use of green fluorescent protein (GFP), a bioluminescent, intracellular protein normally produced by the jellyfish, Aequorea victoria. A modified version of this protein, enhanced GFP (EGFP), has stronger fluorescence than the original protein, is excited by light in the blue range of the spectrum (498 nm), and has an emission peak at 509 nm, which is in the green portion of the visible spectrum. These absorption/fluorescence characteristics are very similar to those of fluorescein, which is used in the clinic to measure blood vessel integrity in the eye. Thus, EGFP can be measured using the same instruments/optics that are used in the clinic to measure fluorescein (Fig. 34.3). The only difference is that intracellular EGFP does not wash out with time.
Fig. 34.3 Green fluorescent protein (GFP) is visible through illumination with blue light with an ophthalmoscope in this nonhuman primate that had received subretinal injection of 1E11 vector genomes (vg) AAV2/8.CMV.EGFP in its right eye. The left eye had received subretinal injection of the same material but with a dose that was 2 log units lower than the right eye.39
Evaluation of different vectors
Numerous studies in small and large animals have relied on vector-mediated delivery of reporter genes to elucidate the characteristics of one vector versus another.20–22,39 The transduction characteristics are affected by dose and age (or stage of development) of the animal. In the fetal mouse, one can deliver vector to retinal progenitor cells and carry out “birthday studies” by looking later in adulthood at which types of photoreceptor cells were being “born” at the time of injection.40 Such studies were initially elegantly performed using retroviral vectors, which only target dividing cells.41 Studies on fetal retina AAV-mediated transduction characteristics led to selection of an approach that could be used to reverse blindness in utero in mice.42
The majority of studies published to date involve postnatal delivery. Transduction characteristics are dependent on the surgical approach and the dose. Intravitreal injection of particular recombinant viruses can lead to transduction of ganglion cells and/or Müller glia. For example, intravitreal injection of AAV2 leads to ganglion cell transduction (and expression in the optic nerve, optic chiasms, and brain) in species ranging from mouse to dog to human, whereas intravitreal injection of AAV5 does not.43,44 Intravitreal injection of recombinant viral vectors does not usually result in transduction of the photoreceptors in the outer retina or the RPE (Table 34.1).8,16,17,21
Most vectors target RPE cells efficiently even at low dose (Table 34.1). As the dose is increased and depending on the vector, photoreceptors and Müller cells can also be transduced (Fig. 34.4).39
[/not-level-membership-for-opthalmology-category]
