CHAPTER 15 Gene Expression*
Each organism, whether it has 600 genes (Mycoplasma), 6000 genes (budding yeast), or 25,000 genes (humans), depends on reliable mechanisms to turn these genes on and off. This is called regulation of gene expression. In simple organisms, such as bacteria and yeast, environmental signals, such as temperature or nutrient levels, control much of gene expression. In multicellular organisms, genetically programmed gene expression controls development from a fertilized egg. Within these organisms, cells send each other signals that control gene expression either through direct contact or via secreted molecules, such as growth factors and hormones.
The Transcription Cycle
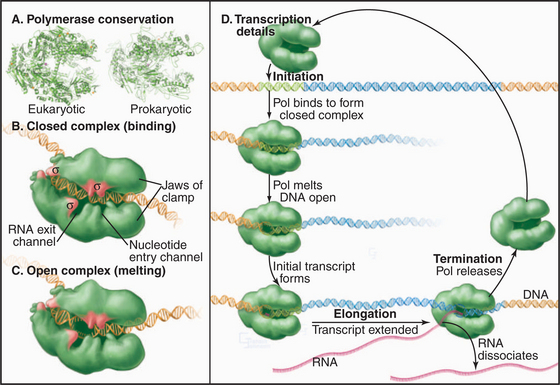
Synthesis of RNA by RNA polymerases is a cyclic process that can be broken down into three sets of events: initiation, elongation, and termination (Fig. 15-1). Each of these events consists of multiple individual steps. In the first step of the initiation process, RNA polymerase locates and binds to the chromosome near the beginning of the gene, forming a preinitiation complex at a sequence termed a promoter. This binding must be highly specific to distinguish promoter from nonpromoter DNA. Next, a conformational change in the polymerase-promoter complex results in formation of an open complex in which the DNA duplex is unpaired, allowing RNA polymerase access to nucleotide bases that are complementary to the start of the message. After formation of a phosphodiester bond between the first two complementary ribonucleotides, the polymerase translocates one base and repeats the process of phosphodiester bond formation, resulting in elongation of the nascent RNA. The elongation reaction cycle continues at an average rate of about 20 to 30 nucleotides per second until the complete gene has been transcribed. Elongation is not a uniform reaction, however, as RNA polymerase pauses at certain sequences. These pauses are important for regulation of transcription. The final step in the transcription cycle, termination, occurs when the polymerase reaches a signal on DNA that causes an extended pause in elongation. Given enough time and the appropriate sequence context, the nascent transcript dissociates from the elongating RNA polymerase, and the DNA template returns to a base-paired duplex conformation. Ultimately, RNA polymerase dissociates from the template and is free to begin a new search for a promoter.
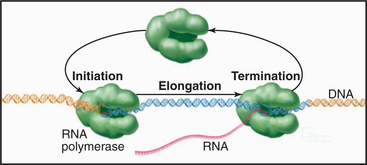
Each of the steps in the transcription cycle can potentially serve as the target of regulatory molecules. The frequency of initiation varies among different promoters as dictated by the need for the gene product. The initiation reaction is most often regulated, presumably because this prevents synthesis of messages that encode unneeded products. Elongation and termination can also be regulated, as can splicing and further processing of mRNAs (see Chapter 16). In eukaryotes, the sum of these nuclear regulatory steps, together with cytoplasmic regulation of mRNA stability and translation efficiency, contributes to the wide variation seen in the abundance of different mRNAs and proteins in particular types of cells.
The Transcription Unit
Coding information in genomes is transcribed in increments corresponding to one or a few genes. Gene-coding and regulatory (cis-acting) DNA sequences that direct transcription initiation, elongation, and termination are collectively called a transcription unit. Prokaryotic transcription units, called operons, contain more than one gene, often encoding physiologically related proteins (Fig. 15-2A). Operons are flanked by sequences that direct the initiation and termination of transcription. Figure 15-2B shows a simple eukaryotic transcription unit encoding the human hemoglobin β-chain. Although only a small fraction of this region encodes the β-globin polypeptide, the adjacent regulatory se-quences are crucial for proper expression of β-globin. Genetic defects resulting in decreased β-globin production are called β-thalassemias. Such mutations can occur either in the coding region, resulting in an unstable or truncated polypeptide, or in the adjacent control regions, leading to low levels of transcription or aberrant processing of the newly synthesized RNA (see Chapter 16). Thus, the transcription unit can be thought of as a linked series of modules, all of which must be functional for the gene to be transcribed at the correct level.
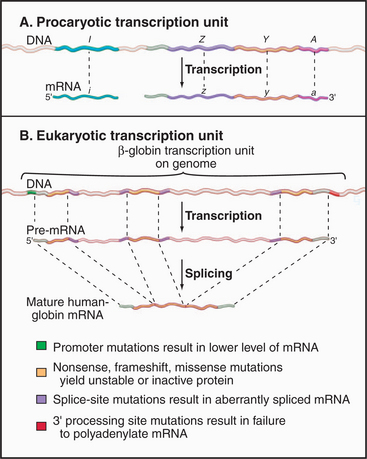
Biogenesis of RNA
Transcription of eukaryotic DNA in the nucleus is linked to subsequent steps that process the nascent transcript in preparation for its eventual function (see Chapter 16 for a complete discussion of these steps). For mRNA precursors, this includes capping and methylation of the 5′ end of the nascent transcript. Most messages are also spliced to remove introns; the 3′ end of the message is then cleaved, and a stretch of adenosine residues is added. The mRNA is then transported to the cytoplasm, where it serves as the template for protein synthesis.
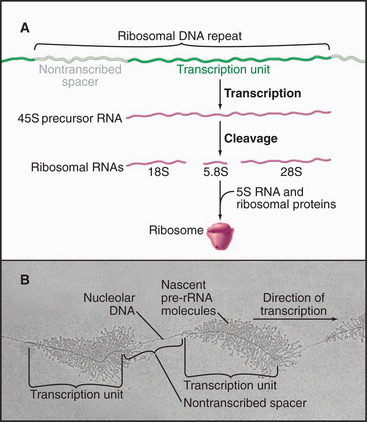
Eukaryotic ribosomal RNA is synthesized from a set of tandemly repeated genes as a single molecule, which is cleaved and modified to give the final 28S, 5.8S, and 18S RNAs (Fig. 15-3). These are assembled, together with 5S RNA and about 80 proteins, into ribosomes in the nucleolus. Transfer RNA is synthesized in the nucleus and transported to the cytoplasm, where it is charged with amino acids prior to participating in protein synthesis (see Chapter 17). snRNAs are synthesized and processed in the nucleus. From there, they migrate to the cytoplasm, where they acquire essential proteins, and then return to the nucleus, where they function in the enzymatic reactions of RNA processing (splicing; see Chapter 16). The postsynthetic processing pathway that a particular transcript follows is dictated, in part, by the transcription machinery that is used to initiate and elongate the transcript and by certain features of the nascent RNA.
RNA Polymerases
RNA polymerases synthesize a new strand of nucleic acid that is complementary to one of the chromosomal DNA strands. While the enzymatic reaction is similar to DNA replication (see Chapter 42), there are several important differences. First, RNA polymerases synthesize a strand of ribonucleotides. Second, unlike DNA polymerase, RNA polymerases can initiate transcription without a primer. Finally, unlike replication, the newly transcribed sequences do not remain base-paired with the template but are displaced after reaching a length of about 10 nucleotides. These properties are common to RNA polymerases in all cells; therefore, it is not surprising that all cellular RNA polymerases share common structural features.
Most eukaryotes have three different RNA polymerases (some species of plants contain four). The largest subunits of the three eukaryotic RNA polymerases are closely related to the bacterial β and β′ subunits. RNA polymerases I, II, and III have up to 10 additional subunits, most of which are unique to each enzyme (Fig. 15-4A). The subunits of both prokaryotic and eukaryotic enzymes assemble into a structure that is roughly spherical, with a diameter of approximately 150 Å and a 25-Å-wide cleft, large enough to accommodate the DNA template (Fig. 15-4B). The site of nucleotide addition is located on the back wall of the cleft. The framework of this structure is provided by the two largest subunits, which make up the two lobes that clamp down on the template DNA.
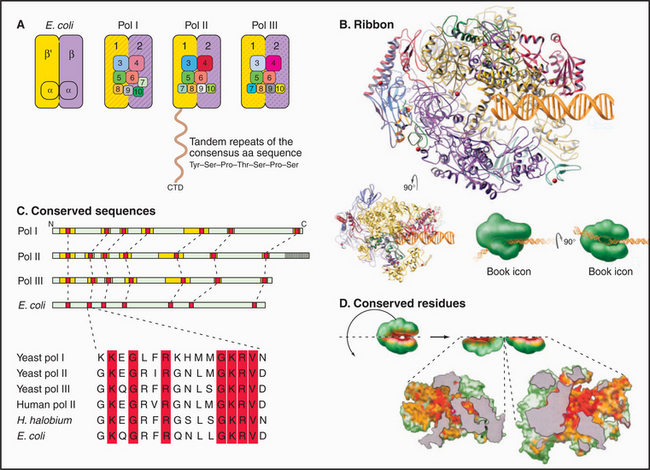
The eukaryotic polymerases can be distinguished experimentally on the basis of their sensitivity to the fungal toxin α-amanitin, RNA polymerase II being the most sensitive and RNA polymerase I being the most resistant. RNA polymerase I localizes to the nucleolus, where it synthesizes rRNA. RNA polymerase II synthesizes mRNA and several snRNAs involved in RNA splicing in the nucleoplasm. RNA polymerase III synthesizes tRNA, 5S rRNA, and the 7S RNA of the signal recognition particle (see Fig. 20-5). The newly described RNA polymerase IV is present in plants, where it is involved in heterochromatin formation and gene silencing.
Specialization has been balanced, however, by the need to retain the structural elements required for RNA synthesis. In each eukaryotic RNA polymerase, the largest subunits are homologous to the bacterial β′ – and β-subunits that make up the catalytic core of prokaryotic RNA polymerases (Fig. 15-4C). The structure of a bacterial RNA polymerase reveals that the most conserved residues are located on the inner surfaces of the enzymes, where they are likely to be involved in the synthesis of RNA (Fig. 15-4D).
RNA Polymerase Promoters
Initiation of transcription requires RNA polymerase loading onto the chromosome at the promoter of a gene or operon. The promoter can be loosely defined as the sum of DNA sequences necessary for transcription initiation. This definition is not sufficient, however, as most genes are regulated (positively or negatively) at the transcription initiation level. In eukaryotic cells, packaging into chromatin represses most promoters, and activator proteins are required for recruiting RNA polymerase to the site of initiation. In prokaryotes, both activators and repressors modulate the frequency of initiation at promoters. Strong promoters drive the expression of genes whose products are required in abundance, whereas weaker promoters are selected for expression of rare proteins or RNAs. In multicellular organisms, a promoter may direct expression at an intermediate level in some cells, at an activated level in others, and at a repressed level in yet others.
Promoters in bacteria are recognized by direct interactions between specific DNA sequences and the RNA polymerase σ factor. The most common σ factor in E. coli (σ 70) recognizes two conserved six-base sequences located 10 bases (minus 10) and 35 (minus 35) upstream of the transcription start site (Fig. 15-5A). Once initiation has occurred, σ is no longer required and can dissociate from the core enzyme. Bacterial cells have several distinct σ factors, each of which binds the core enzyme and direct RNA polymerase to a subset of promoters that contain different recognition sequences, thereby promoting transcription of genes with related functions.
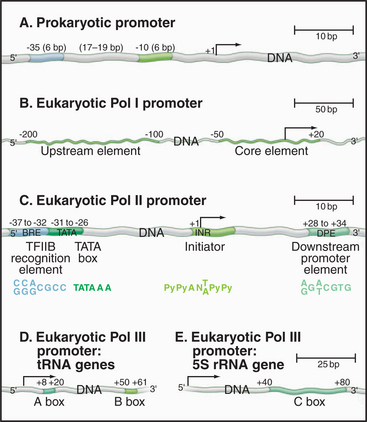
Eukaryotic RNA polymerase I and II promoter sequences are also situated upstream of the transcription start site. In contrast, RNA polymerase III promoters contain key promoter elements within the transcribed sequences. RNA polymerase I recognizes a single type of promoter located upstream of each copy of the long tandem array of pre-rRNA coding sequences (Fig. 15-5B). The core element of this promoter overlaps the transcription start site, while an upstream control element located approximately 100 base pairs (bp) from the start site stimulates transcription. RNA polymerase I is not required in yeast cells that contain a pre-rRNA gene under control of an RNA polymerase II promoter. Therefore, if RNA polymerase I does recognize other promoters, these transcripts are not required for viability.
Comparison of the first eukaryotic protein-coding gene sequences revealed a conserved consensus se-quence located approximately 30 bp upstream of the transcription start site of many RNA polymerase II–transcribed genes (Fig. 15-5C). This consensus sequence—TATAAAA—called a TATA box, shows some similarity to the bacterial -10 sequence. In addition to the TATA box, a less conserved promoter element, the initiator, is found in the vicinity of the transcription start site of many genes. RNA polymerase II–transcribed genes that do not contain TATA boxes often contain strong initiator elements. Together, these two elements account for the basal promoter activity of most protein-coding genes.
Both types of RNA polymerase III promoters have key elements within the transcribed sequences (Fig. 15-5D–E). tRNA genes contain two 11-bp elements, the A box and B box, centered about 15 bp from the 5′ and 3′ ends of the coding sequence, respectively. The 5S-rRNA gene contains a single internal element, the C box, located in the center of the coding region. Given the differences in classes of eukaryotic promoters, it is not surprising that different polymerases use different proteins to recognize the promoter sequences.
Transcription Initiation
The loading of RNA polymerase onto the double-stranded genomic DNA at a promoter sequence is best understood in prokaryotes and is discussed first before the discussion of eukaryotes. Initiation takes place in a series of defined steps (Fig. 15-1). First, holoenzyme binds to the double-stranded promoter, forming what is called the closed complex. The specificity and strength of this interaction are dictated by sequence-specific contacts between the σ factor and the bases in the -10 and -35 elements of the promoter (Fig. 15-6). The second step in initiation is the formation of an open complex in which a 14-bp region around the transcription start site is unpaired producing a transcription bubble. This unpairing is accompanied by a conformational change in the polymerase that positions the single-strand DNA template in the active site and narrows the DNA-binding cleft, effectively closing the polymerase clamp. In the next step, the DNA template in the active site base-pairs with the first two ribonucleotides, and the first phosphodiester bond is catalyzed. This process is repeated until the nascent RNA reaches a length of eight to nine bases, at which point addition of bases to the growing RNA chain results in the unpairing of one base of the RNA-DNA hybrid, and the nascent RNA begins to exit through a channel on the surface of the polymerase. The resulting conformational change in polymerase leads to the release of σ factor and formation of a stable ternary (three-way) complex containing RNA polymerase, the DNA template, and the nascent RNA.
General Eukaryotic Transcription Factors
Purified eukaryotic RNA polymerase on its own cannot initiate transcription from promoters in vitro. Specific transcription can be obtained in vitro using extracts from nuclei, and fractionation of such extracts has led to the identification of additional factors necessary for specific transcription by purified RNA polymerase in vitro. Rather than a σ factor, eukaryotic RNA polymerases require multiple initiation factors. Most of these factors are unique to each RNA polymerase, and because they are required for transcription of most promoters (within each class), they are termed general transcription factors (GTFs). GTFs are remarkably conserved among different eukaryotes. Although most factors required for transcription by each class of polymerase are distinct, one of them, first identified as the TATA box–binding protein, participates in different protein complexes involved in each of the three polymerase systems. The next sections compare transcription by the three forms of eukaryotic RNA polymerase.
RNA Polymerase II Factors
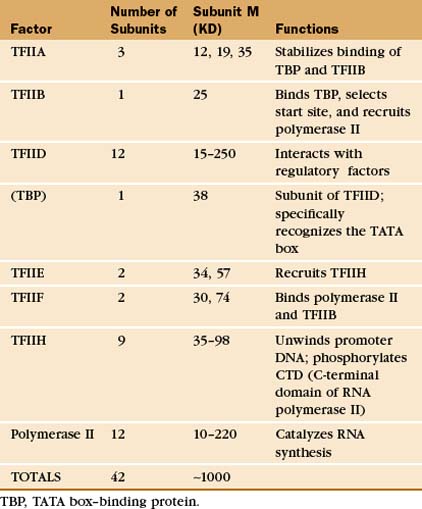
The RNA polymerase II GTFs comprise more than 20 polypeptides with an aggregate molecular weight of more than 106 D (Table 15-1). Before RNA polymerase II can initiate transcription in vitro, an ordered assembly of factors at the promoter must occur. Assembly of the RNA polymerase II preinitiation complex begins with the binding of TFIID, a large factor (˜700 kD) consisting of TATA box–binding protein (TBP) and a set of TBP-associated factors called TAFIIs (Fig. 15-7A). TBP alone is sufficient for basal transcription, while TAFs apparently serve as targets for further activation of transcription (see subsequent sections). TBP is the first polypeptide in the basal transcription machinery to recognize a specific DNA sequence during the initiation pro-cess. DNA binding is provided by a highly conserved C-terminal 180-amino-acid domain, which forms a saddle-shaped monomer with an axis of dyad symmetry (Fig. 15-7B). The underside of the TBP “saddle” binds to the minor groove of the TATA sequence, which is splayed open in the process. A pronounced DNA bend is produced at each end of the TATAAA element by the intercalation of phenylalanine side chains (Fig. 15-7C).
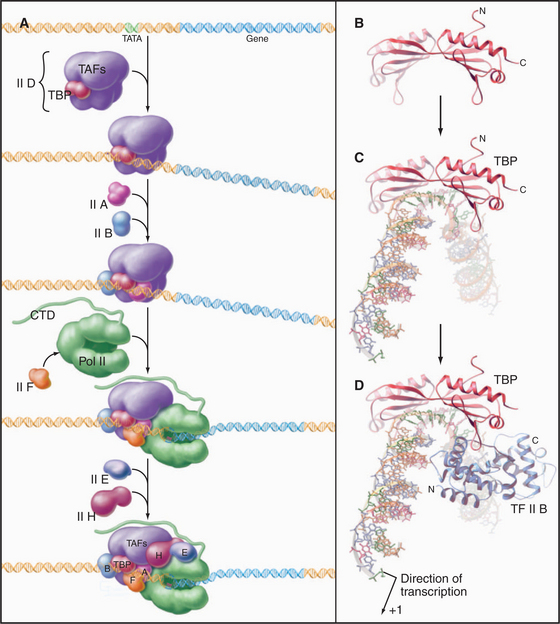
The TFIID-TATA box complex serves as a binding site for additional positive and negative regulators. TFIIA binding stabilizes the TBP-DNA interaction and prevents the binding of repressors that arrest further initiation complex formation.
The next step in assembly of the initiation complex is binding of TFIIB, which binds to one side of TBP and makes contacts with DNA upstream and downstream of the TATA box (Fig. 15-7D). Mutations in the yeast gene that encodes TFIIB show altered mRNA start-site selection, indicating that TFIIB establishes the spacing between the TATA box and the transcription start site. TFIIB interacts directly with TBP and RNA polymerase II and is thus essential for the next steps in initiation complex assembly.
RNA polymerase II enters into the preinitiation complex (see Fig. 15-7A) in association with TFIIF. This factor is related to bacterial σ factor and acts to stabilize the interaction of RNA polymerase II with TFIIB and TBP. In addition, TFIIF binds to free polymerase and prevents interactions with nonpromoter DNA sites.
TFIIH also contains a protein kinase that phosphorylates the CTD. This is Cdk-activating kinase, itself a Cdk-cyclin complex that phosphorylates and activates other cyclin-dependent kinases (see Fig. 40-14). In the initiation complex, phosphorylation of the CTD is thought to release it from interactions with GTFs and allow the transition to the transcription elongation phase. Other TFIIH subunits have been identified as components of the DNA repair machinery. Several genes encoding TFIIH subunits are mutated in the human DNA excision repair disease xeroderma pigmentosa, suggesting that TFIIH might serve to link transcription to DNA repair (see later section).
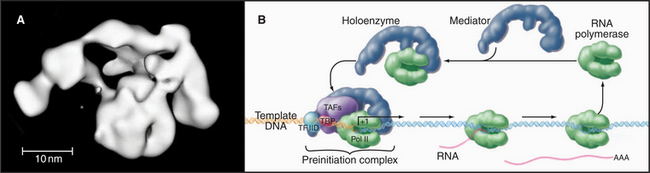
Mediator and the Holoenzyme
In vivo, many of the steps described previously involve the assembly of large macromolecular complexes containing RNA polymerase II, several of the GTFs, other factors that alter chromatin structure, and various additional transcription factors. One of these complexes, the mediator, contains over 20 polypeptides (many with unknown function) but lacks RNA polymerase II and the GTFs. Mediator reversibly interacts with RNA polymerase II and other factors to form a “holoenzyme,” which requires additional factors to be competent for initiation (Fig. 15-8). RNA polymerase II holoenzyme responds to transcription activators (de-scribed in a subsequent section) in vitro, suggesting that one role for the multitude of proteins in this complex is to offer multiple interaction sites for recruitment of holoenzyme to the promoter. Alternatively, a mediator lacking RNA polymerase II can be recruited to the promoter, where it subsequently attracts the polymerase. Thus, the mediator links DNA-bound activators to the basal transcription machinery. In this sense, the mediator acts as a coactivator. Other coactivators present in the holoenzyme act as chromatin remodeling factors (see subsequent section) that act to control access of the transcription machinery to the DNA template.
RNA Polymerase I Factors
Initiation at RNA polymerase I promoters can also proceed through an ordered assembly of transcrip-tion factors (Fig. 15-9). The upstream binding factor binds to the upstream control element and to part of the core element. This initial complex is stabilized by the SL1 complex of TBP with three RNA polymerase I–specific TAFs.
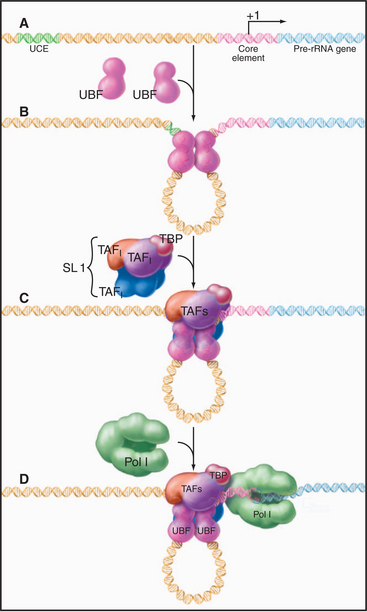
RNA Polymerase III Factors
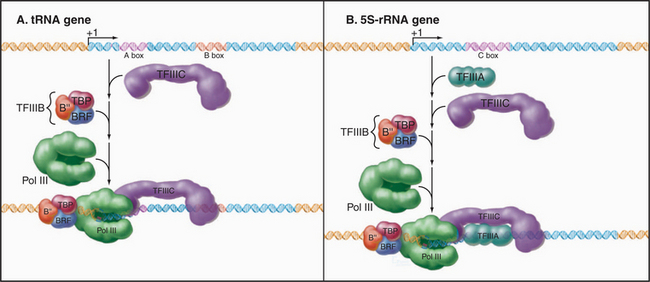
The assembly of RNA polymerase III initiation complexes differs at various promoters (Fig. 15-10). Initiation at tRNA genes begins with the binding of TFIIIC to the A and B boxes. TFIIIB then binds upstream of the A box at a sequence determined both by an interaction with TFIIIC and through the DNA-binding capacity of TBP. Once the TFIIIC-TFIIIB complex has been assembled, RNA polymerase III can initiate transcription. Multiple rounds of initiation can occur on the stable tDNA-TFIIIC-TFIIIB complex.
Summary of the Eukaryotic Basal Transcription Machinery
Why are so many factors required to make a transcript? Part of the complexity might be necessary to generate multiple sites for interaction with regulatory factors that could either activate or repress the assembly or function of the preinitiation complex. A second role for the complex set of factors could be to target polymerases to specific sites in the nucleus. Finally, some factors could help load elongation, splicing, or termination factors onto the RNA polymerases.
Transcription Elongation and Termination
The Catalytic Cycle
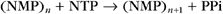
where (NMP)n is the RNA polymer; NTP is ATP, UTP, CTP, or GTP; and PPi is pyrophosphate. Polymerase extends the RNA chain in the 5′ to 3′ direction by adding ribonucleotide units to the chain’s 3′ end. Selection of the incoming NTP is directed by the DNA template and takes place at the transcription bubble, an unpaired segment of the DNA template (Fig. 15-11). The 3′ hydroxyl group acts a nucleophile, attacking the a-phosphate of the incoming NTP in a reaction similar to that seen in DNA replication (see Fig. 42-1). This reaction proceeds in vivo at a rate of 30 to 100 nucleotides per second.
Pausing, Arrest, and Termination
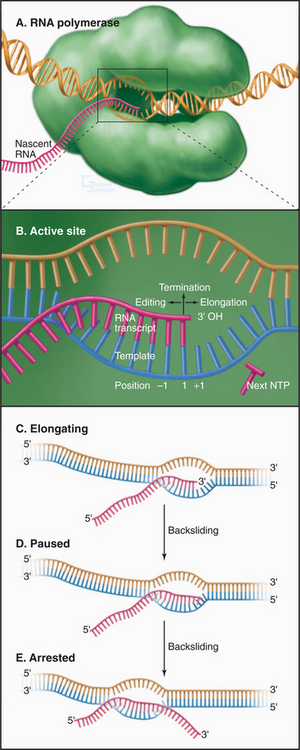
Following the addition of each nucleotide, RNA polymerase may add an additional nucleotide, pause, move in reverse, or terminate (Fig. 15-11B). The relative probabilities of these alternative reactions depend on interactions between the transcription complex and the template, the nascent RNA transcript, and regulatory transcription factors.
RNA polymerase does not elongate at a constant rate but rather synthesizes RNA in short spurts between pauses. A pause of short duration can be caused by low NTP concentrations or alternatively by the transient unpairing of the 3′-end of the nascent transcript and template. Longer pauses are provoked by the formation, in the nascent RNA, of short (˜20 base) self-complementary sequences that can fold to form a stem-loop or hairpin, or the presence of a weak RNA-DNA hybrid. The presence of an unstable RNA-DNA hybrid can arise from the misincorporation of an NTP leading to an unpaired base in the hybrid. In this case, the RNA polymerase can backtrack or slide backward on the template (Fig. 15-11C). This backward movement of the transcription bubble is accompanied by a zippering movement of the RNA-DNA hybrid in which the nascent RNA in the exit channel rehybridizes with upstream template sequences while the 3′ end of the transcript unpairs from the hybrid and is extruded through the same channel that NTPs use to enter the active site. The backtracked transcription complex is said to be arrested. Transcription elongation factors bind in the NTP channel of arrested complexes and activate the RNA polymerase to cleave the backtracked RNA. The new 3′ terminal residue is correctly positioned for incorporation of the next complementary NTP. This editing process increases the fidelity of transcription. Pausing also occurs following transcription of U-rich sequences, and this is often associated with transcription termination.
Termination
When elongating RNA polymerase reaches the end of a gene or operon, specific sequences in the RNA trigger the release of the transcript and dissociation of the RNA polymerase. Bacteria have two types of termination signals, called terminators. The first are called intrinsic (or rho-independent) terminators, because they function in the absence of any protein factors (Fig. 15-12A). Intrinsic terminators consist of two sequence elements: a stable GC-rich hairpin and a run of about eight consecutive U residues. As the first of these elements is synthesized, it forms a hairpin, causing polymerase to pause with unstable U: A bps (with only two H-bonds [see Fig. 3-14]) in the hybrid. The nascent transcript is released from this unstable transcription complex. The second type of prokaryotic termination requires a protein factor called rho (Fig. 15-12B). Rho is a hexameric protein that binds cytosine-rich sequences and uses ATP hydrolysis to translocate along the nascent transcript in the 5′ to 3′ direction, essentially chasing the RNA polymerase. When polymerase pauses, rho can catch up and use the energy derived from ATP hydrolysis to pull the RNA out of the transcription elongation complex.
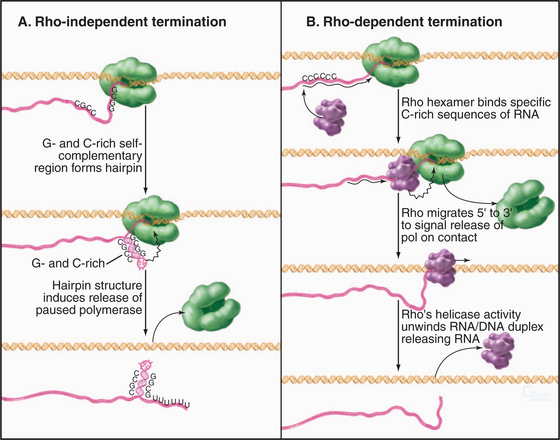
Eukaryotic RNA polymerases have evolved distinct mechanisms for termination. RNA polymerase III requires no protein factors but terminates efficiently after transcribing four to six consecutive U residues, presumably owing to instability of the RNA-DNA hybrid in the enzyme active site. RNA polymerase I terminates in response to a protein factor that blocks further elongation by binding to a DNA sequence downstream of the termination site, leaving an inherently unstable U-rich RNA-DNA hybrid in the active site. The RNA polymerase II termination mechanism is more complex, requiring a large multiprotein complex that recognizes the poly(A) addition in the nascent transcript (see Fig. 16-3 for pre-mRNA processing). Deletion or mutation of the poly(A) signal results in a failure to terminate messages at the appropriate site, indicating that RNA polymerase II termination is coupled to 3′-end processing.
Gene-Specific Transcription
Regulation of Transcription Initiation in Prokaryotes
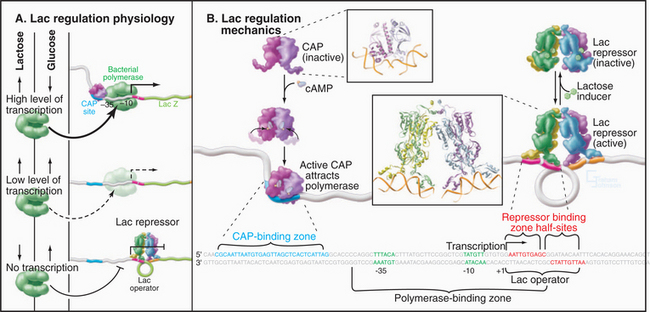
Prokaryotes typically regulate gene expression in response to signals that are produced in response to the internal metabolic state and to environmental cues such as the presence of nutrients in the growth medium (see Fig. 27-11). These signals inside the organism are transmitted to the appropriate genes through transcription regulatory proteins that bind to specific sequences near the genes they control to either activate or repress transcription. Both of these regulatory mechanisms come into play in regulation of the E. coli lactose (lac) operon (Fig. 15-2A). The genes expressed from this operon are required for cells to metabolize lactose but are not expressed in the absence of lactose. Genetic studies in the 1960s showed that the gene upstream of the lac operon (I in Fig. 15-2A) encodes a repressor (lac repressor) that blocks expression of the lac operon in the absence of lactose (Fig. 15-13). The lac repressor binds to a site called an operator that overlaps the RNA polymerase binding site in the lac promoter. In the presence of lactose, the repressor undergoes a conformational change that eliminates DNA binding allowing the recruitment of RNA polymerase to the promoter. Full expression of the lac operon requires the catabolite activator protein (CAP), which is also an allosteric DNA-binding protein that binds just upstream of the lac promoter. If cellular glucose levels diminish, the cAMP concentration rises, and CAP binds cAMP. This induces a structural alteration in CAP, allowing it to dimerize and bind specific DNA sequences. CAP bound to its site stabilizes the otherwise weak interaction of RNA polymerase with the promoter. The resulting activation allows maximum expression of the lac operon in the presence of lactose and the absence of glucose.
Eukaryotic Promoter Proximal and Enhancer Elements
In vivo techniques for analyzing eukaryotic promoter function led to the discovery of a number of regulatory elements in addition to the basal promoter elements. In these experiments, transgenes containing a promoter or its mutated derivative are introduced into eukaryotic cells by transfection or microinjection. Transcription directed by the cloned promoter is detected by various approaches that allow the transgene product to be identified from among the background of cellular transcripts. In one approach (Fig. 15-14A), the promoter drives expression of a bacterial reporter gene such as chloramphenicol acetyl-transferase (CAT), β-galactosidase, or luciferase. Eukaryotes lack these enzymes, so their expression can be assayed in extracts of transfected cells with little or no background activity. This approach applies only to RNA polymerase II, which produces translatable mRNAs. A more direct analysis, applicable to transcription by all three RNA polymerases, makes use of specific RNA or DNA probes to quantify RNAs transcribed from the transgene.
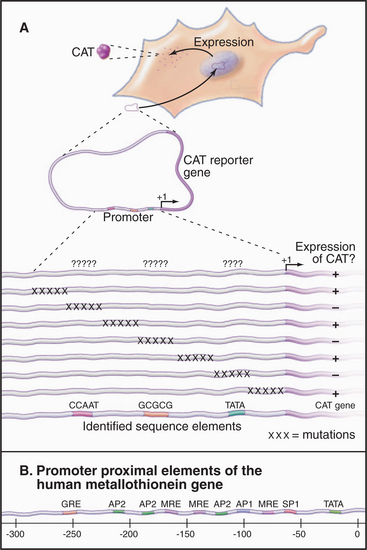
Promoter proximal elements are short (˜10 bp) sequences located within a few hundred bp upstream of the TATA box. One example of a promoter proximal element is the CCAAT box in the promoter of the herpes simplex virus thymidine kinase gene. This site was identified by a technique called linker-scanning, in which clustered mutations are introduced at regular intervals in the promoter (Fig. 15-14A). Mutations that result in a decrease in transcription define important sequences. In the case of the thymidine kinase promoter, the CCAAT and TATAAA sequences are required for full transcription. Thymidine kinase expression also requires the sequence GGCGCC, which serves as the binding site for SP1, a transcription factor involved in expression of a number of so-called housekeeping genes, whose products are involved in normal cellular functions. These promoter proximal elements are present in many different genes, where they are necessary for constitutive expression. Other promoter proximal elements are involved in regulated expression, for example, in re-sponse to cellular stress or exposure to heavy metals. Most promoters contain several different promoter proximal elements. This allows for combinatorial regulation of transcription levels by varying the relative abundance or activity of the various factors. The location of numerous regulatory elements directly upstream of the human metallothionein gene, whose product protects cells from the toxic effects of metals (Fig. 15-14B) suggests that a variety of different mechanisms regulate this gene.
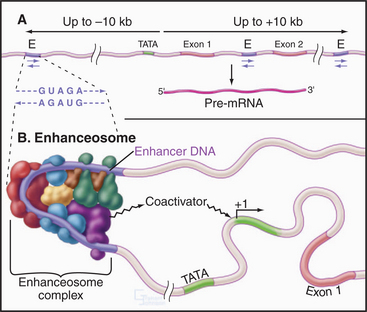
Enhancers are clusters of regulatory elements in the DNA similar to promoter proximal elements, but they are considerably more complicated and have several distinguishing features. First, an enhancer increases the rate of initiation from a basal promoter even if it is located up to 10 kb away from the promoter. Second, enhancers work even if located internal to or downstream of the promoter. Finally, the enhancer element will work in either orientation relative to the promoter (Fig. 15-15A). Figure 15-15B shows an example of an enhancer sequence with a number of transcription factors (see the following section) bound, forming a complex called an enhanceosome. Enhancer elements are found in the vicinity of many but not all genes. In most cases, the enhancer works in a cell type–specific fashion. An example is a sequence in an intron of the immunoglobulin heavy chain gene that enhances transcription in lymphocytes but not in other cells. This regulation of enhancer function is likely to be accomplished by changes in the levels of various enhancer-binding factors in different tissues.
Gene-Specific Eukaryotic Transcription Factors
Eukaryotic transcription factors bind specific DNA sequences located near the genes they regulate. This binding leads to activation or repression of expression by mechanisms more varied than in prokaryotes. In the simplest cases, the transcription factor interacts directly with the basal machinery. In more complex cases, this interaction may involve a coactivator or corepressor. Transcription factors may also act on the chromatin template rather than the basal transcription machinery. The 1990s witnessed the identification and characterization of hundreds of eukaryotic gene-specific transcription factors. Current estimates indicate that approximately 6% of the coding capacity of the human genome is devoted to transcription factors that recognize specific DNA sequences. The following sections discuss the identification of transcription factors, the functional organization of these proteins, and regulation of the basal transcription machinery and the chromatin template by transcription factors.
Methods for Identifying, Isolating, and Localizing Transcription Factors
Identifying and characterizing transcription factors requires techniques to detect and characterize specific DNA-protein complexes. In one such technique, DNA footprinting, protein is mixed with DNA that is radioactively labeled at one end (Fig. 15-16A–B). The resulting DNA-protein complex is then lightly digested with deoxyribonuclease to give, on average, one random cut per DNA molecule. The population of cleaved DNA molecules thus produced is then stripped of protein and separated by gel electrophoresis. The area protected from cleavage by a specific DNA-binding protein appears as a blank area or “footprint” that results from the protein’s blocking access to the nuclease, thus leaving a gap in the family of digestion products of differing lengths. A less precise but more versatile method of visualizing protein-DNA complexes is the DNA mobility shift assay (Fig. 15-16C). The principle of this technique is that fragments of DNA with a bound protein move more slowly during gel electrophoresis than the same DNA fragments without bound protein.
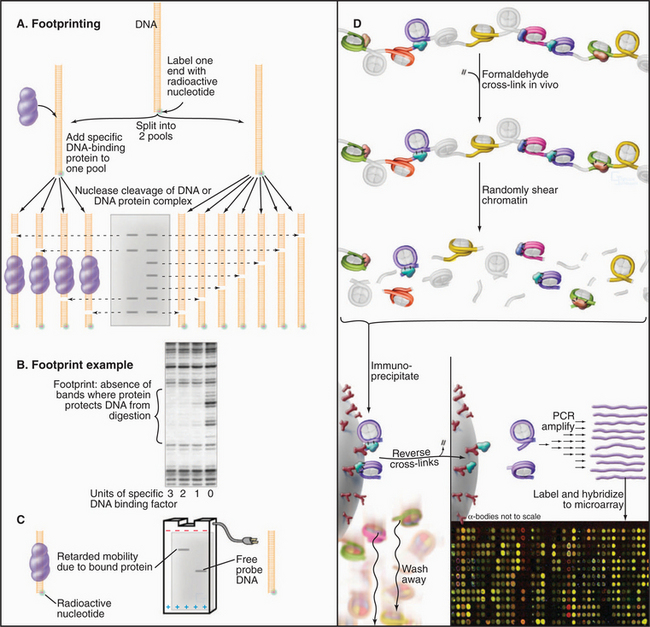
(D, Based on data from Stephen Hartman and Michael Snyder, Yale University, New Haven, Connecticut.)
The DNA sites that bind known transcription factors in vivo can be determined by using a technique called chromatin immunoprecipitation (ChIP; Fig. 15-16D). By using this approach, a transcription factor can be localized to a specific promoter at a specific time. The combination of ChIP with microarray approaches allows the distribution of the factor across the genome to be determined.
DNA-Binding Domains
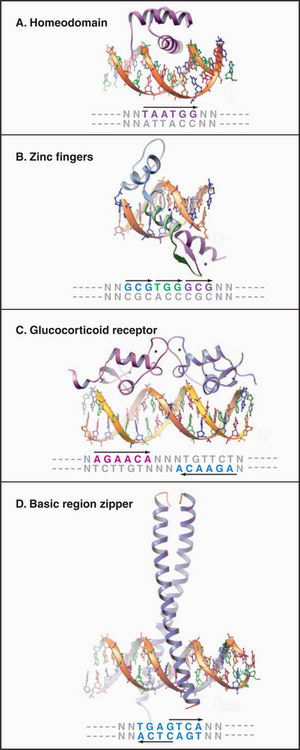
DNA-binding proteins can be grouped into families based on the structure of the domains used for DNA sequence recognition (Fig. 15-17 and Table 15-2). These include the helix-turn-helix (HTH) proteins, homeodomains, zinc finger proteins, steroid receptors, leucine zipper proteins, and helix-loop-helix proteins. Although these families include most of the known transcription factors, there remain other, uncharacterized recognition domains. Within a given family, the recognition domain of each transcription factor has an amino acid sequence that targets the protein to a particular DNA sequence. Conversely, different families of transcription factors can recognize the same promoter element. The following sections discuss some of the more common eukaryotic DNA-binding domains.
Homeodomain
This 60-amino-acid motif was discovered in Drosophila proteins that regulate development and has been found in a wide range of eukaryotic transcription factors, including more than 150 in the human genome. Recognition is provided by a helix-turn-helix (HTH) motif composed of two helices, one of which sits in the major groove of the DNA-binding site contacting a recognition sequence of 6 bp (Fig. 15-17A). The HTH structure is not a stable domain on its own but exists as part of a larger DNA-binding domain, such as the homeodomain. Additional binding affinity is provided in the homeodomain by a flexible arm that interacts with the minor groove.
Zinc Finger Proteins
The zinc finger protein sequence motif (Fig. 15-17B), first identified in the RNA polymerase III basal factor TFIIIA, has since been found in a variety of different RNA polymerase II factors, including more than 600 human transcription factors. Each “finger” consists of a 30-residue sequence with conserved pairs of cysteines and histidines that bind a single zinc ion. The tip of the finger sticks into the DNA major groove, where it contacts three bases. Most zinc finger proteins contain multiple fingers, allowing longer sequences to be recognized to increase specificity. A related structure is present in the steroid hormone receptor family, although in this case, four cysteine residues coordinate the zinc ion and the finger is composed of two helices rather than one. Steroid hormone receptors also contain a dimerization domain, allowing recognition of sequences with dyad symmetry (Fig. 15-17C).
Leucine Zipper Proteins
Leucine zipper domains are made up of two motifs: a basic region that recognizes a specific DNA sequence and a series of repeated leucine residues (leucine zipper) that mediate dimerization. These motifs form a continuous a-helix that can dimerize through formation of a coiled-coil structure involving specific contacts between hydrophobic leucine zipper domains (Fig. 15-17D; also see Fig. 3-10). CAAT/enhancer-binding protein, the factor that recognizes the CCAAT sequence, was the first member of this family to be discovered. Dimers of leucine zipper proteins recognize short, inverted, repeat sequences. The zipper family comprises many members, some of which can cross-dimerize and recognize asymmetrical sequences. Another family of factors comprises the helix-loop-helix proteins, which have the same type of basic region but differ in that they have two helical dimerization domains separated by a loop region.
Factor Interactions
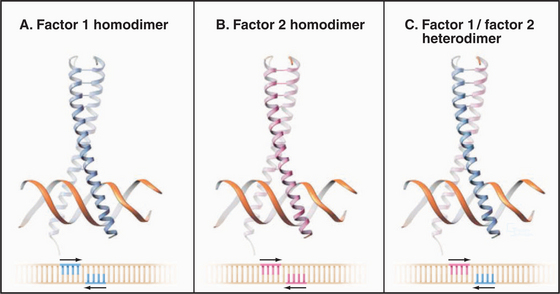
An important aspect of transcription factor function is the ability to associate with other factors. Such associations can expand the repertoire of DNA sequences that can be specifically recognized. In the case of the leucine zipper proteins, formation of a heterodimer leads to recognition of a site that is different from either of the sites recognized by the two homodimers (Fig. 15-18). Thus, a diverse set of binding sites can be recognized by using a relatively small set of interacting factors. Such interactions need not be limited to related proteins, and small interactions surfaces involving only a few specific contacts often suffice.
Transcription Factors as Modular Proteins
In addition to interaction with specific DNA sequences, transcription factors may also interact with regulatory molecules and/or the basal transcription machinery. Functional domains in transcription factors have been mapped by testing various domains in vivo (Fig. 15-19). Such chimeric factors will stimulate transcription as long as two functions are maintained: specific DNA binding and transcription activation. The surprising result of this type of analysis is that many transcription factors are modular proteins with discrete functional domains that can be exchanged without impairing activity. For example, exchanging the DNA-binding domains of the glucocorticoid and estrogen receptors creates a hybrid factor that recognizes estrogen-responsive promoters but activates transcription in response to glucocorticoid hormone.
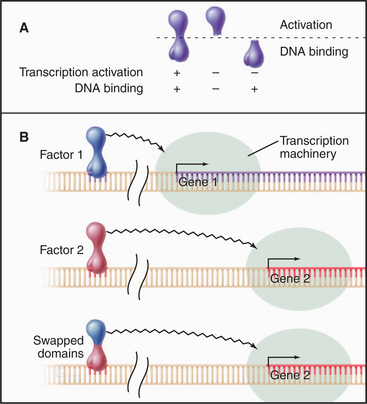
Transcriptional Activation
Binding of a transcription factor to DNA per se does not activate transcription (Fig. 15-19). A separate domain provides this function by interacting directly or indirectly with the basal transcription machinery to elevate the rate of transcription. The best-characterized activation domain is an acidic region derived from the herpesvirus VP16 protein. Acidic activation domains are generally disordered segments of polypeptide consisting of multiple acidic residues dispersed among a few key hydrophobic residues. Such domains activate transcription when experimentally grafted to a wide variety of different DNA-binding domains in a number of different cell types. Other types of activator domains have been characterized as being proline-rich or glutamine-rich.
The diverse activation domains use several mechanisms to promote transcription (Fig. 15-20). The most direct mechanism is recruitment of the basal transcription machinery. Recall that RNA polymerase II requires a number of additional factors for specific transcription. TFIID binds the TATA box and recruits the polymerase in a complex with the mediator. Interactions between activation domains and mediator or TFIID components in these complexes stabilize the preinitiation complex and produce higher rates of RNA polymerase II initiation. The chromatin immunoprecipitation technique (Fig. 15-16D) has been used to demonstrate recruitment of the transcription machinery to specific genes.
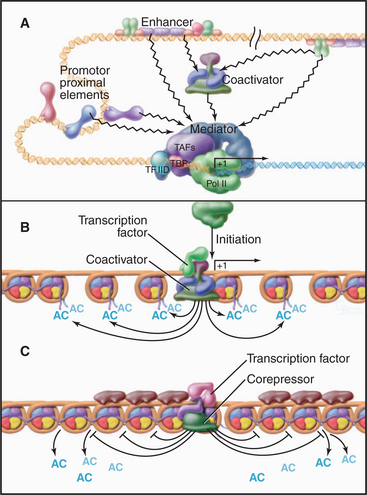
Transcriptional Repressors
As in prokaryotes, some eukaryotic transcription factors repress transcription. Unlike the lac repressor, however, the eukaryotic repressors generally do not act by blocking binding of RNA polymerase. Some eukaryotic repressors act by competing with activators for the same DNA sequence. Often, these repressors are related to the activator they block but simply lack the activation domain. Another type of eukaryotic repressor binds near the activator and interacts with the activator to mask its activation domain. Some repressors bind to specific sites on DNA and interact with coactivators in a manner that blocks their function. Finally, some repressors bind corepressors that can alter chromatin structure in a way that the transcription machinery is denied access.
Chromatin and Transcription
DNA in eukaryotic cells associates with an equal mass of protein to form chromatin (see Chapter 13). Packaging DNA in arrays of nucleosomes compacts the DNA, and the most obvious influence of chromatin on transcription is the ability of nucleosomes to restrict access of transcription proteins to the DNA template. Thus, if histone synthesis is artificially shut off, there is an increase in the basal expression of many genes. Additional evidence of the repressive nature of chromatin is seen in the resistance to nuclease digestion of unexpressed genes and the localization of unexpressed genes in highly condensed heterochromatin.
Gene activation often involves disruption or displacement of nucleosomes located on specific genes. Before the discussion of specific mechanisms, it is useful to consider some aspects of nucleosome structure. The nucleosome consists of DNA wrapped in a left-handed helix around an octamer of histone subunits (see Fig. 13-1). The histone core makes numerous contacts with the DNA minor groove and phosphate backbone, leading to tight binding that is not sequence specific. This aspect of the nucleosome allows for a dynamic association with DNA because binding to one position along the DNA strand is as energetically favorable as another. The N-terminal histone “tails” are highly conserved and play multiple roles in chromatin structure and gene regulation. Histone tails are important sites of modifications that regulate chromatin structure and transcription. Both the nonspecific nature of interactions between histones and DNA and the ability to modify the histone tails are exploited to regulate the ability of nucleosomes to block access of the transcription machinery to the DNA template.
SWI/SNF complexes (see Chapter 13) are recruited to a specific subset of genes through interactions with transcription activators. The resulting remodeling of nucleosomes in the vicinity of the promoter is required for stable preinitiation complex formation at SWI/SNF-regulated genes. Other remodeling complexes are thought to regulate distinct sets of genes in a similar manner. SWI/SNF can also repress transcription at some promoters, presumably by repositioning nucleosomes to restrict access to the promoter.
Gene activation by nucleosome disruption can also be counteracted by factors that stabilize chromatin. In one example, broad regions of chromatin are silenced by recruitment of histone deacetylases that maintain heterochromatin domains (see Fig. 13-9).
Histone Modification and Chromatin Accessibility
The pattern of modification of the histone N-terminal tails forms the basis of a “histone code” that is read by the gene expression machinery. Modification of the histone tails is carried out by enzymes that are specific both for a particular modifying group and for specific residues within the tail of particular histones. For example, the histone methyl transferase SET1 is specific for lysine 4 in the histone H3 tail. The modifying enzymes are generally part of large complexes (Table 15-3) that are recruited to chromatin through interactions with gene regulatory proteins and thus, like the mediator, are considered coactivators (or corepressors).
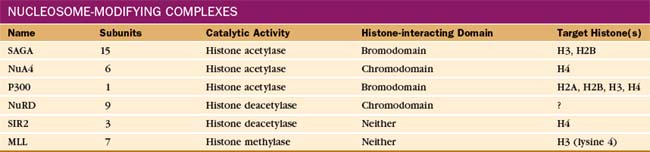
Proteins containing bromodomains or chromodomains interact with acetylated or methylated tails, respectively. Many of the protein complexes involved in gene regulation contain one or more of these domains. For example, the SAGA histone acetyltransferase complex contains a bromodomain that anchors the complex to chromatin, facilitating further modification of regions that are already acetylated. A subunit of TFIID also contains a bromodomain that can facilitate the binding of TFIID to acetylated nucleosomes associated with active chromatin. Similarly, a number of histone methyltransferases contain chromodomains and are therefore targeted to their substrates by preexisting histone methylation.
Modulation of Transcription Factor Activity
Regulation of transcription initiation is of fundamental importance in controlling gene expression. In many cases, the availability of factors that bind to specific sites in promoters is the switch that turns a gene on. Various strategies to control the binding of specific factors have been discovered (Fig. 15-21). One of the most straightforward is de novo synthesis of the specific factor (Fig. 15-21A). This requires an additional level of transcription regulation and translation of the mRNA that encodes the specific factor. All of these steps take some time; therefore, this regulatory scheme is not used in situations in which rapid responses are required. Instead, it is used more commonly in regulating developmental pathways.
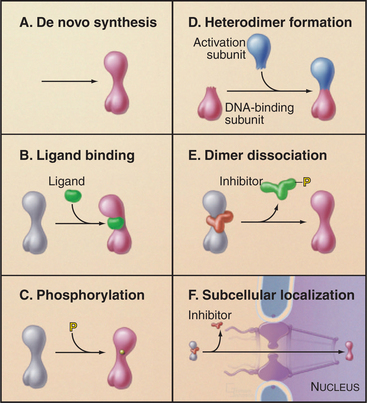
Several mechanisms are used for rapid regulation of the activity of existing transcription factors. One mechanism involves the formation of an active factor from two inactive subunits (Fig. 15-21D). This association can be regulated through synthesis or by modification of preexisting subunits, leading to their association. Binding of small-molecule ligands is another means of controlling transcription factor activity (Fig. 15-21B). In this case, the binding of the ligand induces a conformational change that leads to DNA binding and transcription activation. Interaction of transcription factors with inhibitory subunits is also used to regulate factor activity (Fig. 15-21E). The DNA binding or activation potential is held in check until the appropriate signal leads to dissociation of the inhibitory factor. Covalent modification—for example, by phosphorylation—is also used to convert inactive transcription factors to a functional form (Fig. 15-21C). Finally, the ability of transcription factors to bind DNA may be regulated by restricting their localization to the cytoplasm (Fig. 15-21F). These regulatory schemes are not mutually exclusive, and many regulatory pathways (see the examples that follow) employ several different levels of regulation.
Transcription Factors and Signal Transduction
One of the hallmarks of eukaryotic gene regulation is the ability of cells to respond to a wide range of external signals. Cells detect the presence of hormones, growth factors, cytokines, cell surface contacts, and many other signals. This information is then transmitted to the nucleus, where appropriate changes in expression of specific genes are executed. Transcription factors represent the final step in these signal transduction pathways; the following sections discuss several specific examples. Chapter 27 covers several other signaling pathways that regulate gene expression (see Fig. 27-4 for the three types of signaling pathways to the nucleus).
Steroid Hormone Receptors
Regulation of gene expression by steroid hormone receptors involves both ligand-binding and inhibitory subunits. This family of nuclear receptors includes transcription factors with a common sequence organization consisting of a specific DNA-binding domain, a ligand-binding domain that regulates DNA binding, and one or more transcription activation domains. The ligands that regulate these factors are small, lipid-soluble hormone molecules that diffuse through cell membranes and bind directly to the transcription factor (Fig. 15-22A). The steroid hormones, retinoids, thyroid hormone, and vitamin D bind to distinct members of the nuclear receptor family, enabling them to recognize sequences in the promoters of a range of target genes. The specific sites of action in promoter DNA, termed hormone response elements, are related to either AGAACA or AGGTCA (Fig. 15-17C). Specificity of the response is generated by the spacing and relative orientation of the binding sites. Steroid receptors usually bind to inverted repeats separated by three nucleotides, whereas some other related receptors prefer direct repeats of similar sites. The nuclear receptors can bind as homodimers, although recent evidence suggests that heterodimers actually prevail in vivo. In addition to heterodimerizing with other members of the nuclear receptor family, interactions with other types of transcription factors could serve to link the steroid response to other pathways that signal through cell surface receptors.
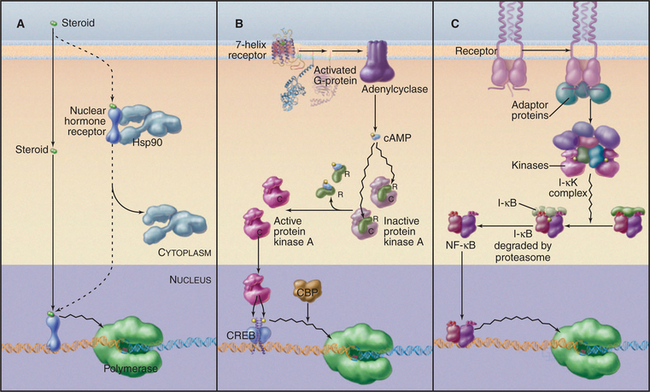
Inactive steroid hormone receptors are blocked from interacting with DNA by heat shock protein 90 (Hsp90; Fig. 15-22A). This protein is a molecular chaperone that keeps the receptor ligand-binding domain in a conformation ready to bind the ligand but unable to enter the nucleus. Hormone binding to the receptor dissociates Hsp90 and frees the receptor’s DNA-binding domain. The free ligand–bound receptor moves from the cytoplasm to the nucleus, where it binds its DNA target and activates transcription.
Cyclic Adenosine Monophosphate (cAMP) Signaling
One of the best-understood examples of transcriptional regulation through cell surface receptor signaling is the adenyl cyclase system. The binding of ligand to some seven-helix receptors results in an increase in synthesis of cAMP, which, in turn, activates protein kinase A (see Fig. 27-3). The promoters of cAMP-regulated genes contain a conserved DNA sequence element, called a cAMP response element, that mediates the transcriptional response to cAMP. A transcription factor, termed cAMP response element–binding (CREB) protein, binds this sequence specifically. CREB protein is a member of the leucine zipper family and binds the DNA as a dimer. The DNA-binding domain of CREB protein can be exchanged with other DNA-binding domains without loss of cAMP responsiveness. This indicates that cAMP does not work by altering the DNA binding of CREB protein; rather, it suggests that cAMP alters the transcription activation function. Recent experiments have identified a site in the activation domain of CREB protein that is phosphorylated by protein kinase A. Mutation of serine 133 to alanine results in a CREB protein that cannot be phosphorylated and cannot activate transcription of target genes. Phosphorylation of serine 133 leads to a conformational change in CREB protein that allows it to interact with a protein adaptor that recruits the transcription machinery leading to transcription of target genes. Thus, the signal generated by binding of a ligand to a cell surface receptor is transduced to a DNA-binding factor that activates transcription of genes containing the appropriate regulatory elements.
NF-kB Signaling
NF-kB proteins are a family of related transcription factors present in many cell types. These factors control a diverse set of cellular processes, including immune and inflammatory responses, development, cell growth, and apoptosis. The activity of NF-kB is normally tightly controlled as persistently active NF-kB is associated with cancer, arthritis, asthma, and heart disease. In most cells, NF-kB is held in an inactive form in the cytoplasm through interaction with an inhibitor called I-kB (see Figs. 14-18 and 15-22C). When B lymphocytes (see Fig. 28-9) are stimulated to produce antibody, NF-kB binds to an enhancer in the immunoglobulin k-chain gene and activates transcription. The stimulatory signal leading to NF-kB activity is transmitted through a protein kinase cascade that eventually phosphorylates I-kB, signaling its destruction by proteolysis. This event unmasks the NF-kB nuclear localization signal, leading to its transport to the nucleus, where it activates transcription of immunoglobulin genes.
Transcription Factors in Development
The exact program of transcription factor interaction during development is extremely complicated and is certainly beyond the scope of this chapter. The underlying principles of these pathways are worth considering, however. One important observation is that developmentally regulated transcription factors are often autoregulatory. For factors that activate their own expression, this form of regulation acts as a switch that leads to continued expression after the initial stimulus is gone. Another important property of developmentally regulated transcription factors is that they are, in turn, regulated by several different factors. This allows complicated combinatorial signals to dictate expression. For example, some transcription factors activate certain promoters while repressing others. The basis of this contradictory property is thought to be the ability of transcription factors to cooperate with each other when bound at the same promoter. This cooperation can be either positive or negative. This allows the expression of a target gene to be regulated both by external signals (e.g., proximity of an adjacent cell that expresses a signaling molecule) and by the preexistence of a given factor in the cell. In this way, only cells of a certain lineage that are located in a certain area of an embryonic segment are able to express the gene. As new transcription factors involved in development are discovered, the challenge will be to decipher the complicated combinatorial interactions among them.
Transcription Factors and Human Disease
TFIIH and Human Disease
Mutations in TFIIH subunits are associated with a set of rare human disorders (xeroderma pigmentosum, Cockayne’s syndrome, and trichothiodystrophy), each linked to defects in nucleotide excision repair of DNA damaged by ultraviolet light or chemical mutagens (see Box 43-1). Mutations in these diseases map to the genes encoding two different TFIIH helicase activities. Presumably, the alterations in these activities cause changes in DNA unwinding, either in the transcription initiation reaction or in the process of nucleotide excision repair. Some mutations are more selective for the DNA repair function, whereas other TFIIH mutations cause little or no DNA repair phenotype but rather seem to affect the TFIIH transcription function. The latter mutations cause wide-ranging defects, as might be expected for a defect in a general transcription factor.
Asturias FJ. RNA polymerase II structure, and organization of the preinitiation complex. Curr Opin Struct Biol. 2004;14:121-129.
Bai L, Santangelo TJ, Wang MD. Single-molecule analysis of RNA polymerase transcription. Annu Rev Biophys Biomol Struct. 2006;35:343-360.
Bracken AP, Ciro M, Cocito A, Helin K. E2F target genes: Unraveling the biology. Trends Biochem Sci. 2004;29:409-417.
Cramer P, Bushnell DA, Kornberg RD. Structural basis of transcription: RNA polymerase II at 2.8 angstrom resolution. Science. 2001;292:1863-1876.
Davidson I. The genetics of TBP and TBP-related factors. Trends Biochem Sci. 2003;28:391-398.
Dilworth FJ, Chambon P. Nuclear receptors coordinate the activities of chromatin remodeling complexes and coactivators to facilitate initiation of transcription. Oncogene. 2001;20(24):3047-3054.
Garvie CW, Wolberger C. Recognition of specific DNA sequences. Mol Cell. 2001;8:937-946.
Gnatt AL, Cramer P, Fu J, et al. Structural basis of transcription: An RNA polymerase II elongation complex at 3.3 Å resolution. Science. 2001;292:1876-1882.
Holmberg CI, Tran SEF, Eriksson JE, Sistonen L. Multisite phosphorylation provides sophisticated regulation of transcription factors. Trends Biochem Sci. 2002;27:619-627.
Kornberg RD. Mediator and the mechanism of transcriptional activation. Trends Biochem Sci. 2005;30:235-239.
Le Hir H, Nott A, Moore MJ. How introns influence and enhance eukaryotic gene expression. Trends Biochem Sci. 2003;28:215-220.
Lemon B, Tjian R. Orchestrated response: A symphony of transcription factors for gene control. Genes Dev. 2000;14:2551-2569.
Levsky JM, Singer RH. Gene expression and the myth of the average cell. Trends Cell Biol. 2003;13:4-6.
Pombo A. Cellular genomics: Which genes are transcribed, when and where? Trends Biochem Sci. 2003;28:6-9.
Ptashne M, Gann A. Transcriptional activation by recruitment. Nature. 1997;386:569-577.
Reinberg D, Orphanides G, Ebright R, et al. The RNA polymerase II general transcription factors: Past, present, and future. Cold Spring Harb Symp Quant Biol. 1998;63:83-103.
Roeder RG. Role of general and gene-specific cofactors in the regulation of eukaryotic transcription. Cold Spring Harb Symp Quant Biol. 1998;63:201-218.
Shilatifard A, Conaway RC, Conaway JW. The RNA polymerase II elongation complex. Annu Rev Biochem. 2003;72:693-715.
Spector DL. The dynamics of chromosome organization and gene regulation. Annu Rev Biochem. 2003;72:573-608.
Steinmetz ACU, Renaud J-P, Moras D. Binding of ligands and activation of transcription by nuclear receptors. Annu Rev Biophys Biomol Struct. 2001;30:329-359.
Steitz TA. The structural basis of the transition from initiation to elongation phases of transcription, as well as translocation and strand separation, by T7 RNA polymerase. Curr Opin Struct Biol. 2004;14:4-9.