[level-membership-for-basic-science-category]
Gastrointestinal tract – physiology and drug absorption
Marianne Ashford
Chapter contents
Physiological factors influencing oral drug absorption
Physiology of the gastrointestinal tract
Transit of pharmaceuticals in the gastrointestinal tract
Key points
Introduction
The factors that influence the rate and extent of absorption depend upon the route of administration. As stated in Chapter 18, the intravenous route offers direct access to the systemic circulation and the total dose administered via this route is available in the plasma for distribution into other body tissues and the site(s) of action of the drug. Other routes will require an absorption step before the drug reaches the systemic circulation. Factors affecting this absorption will depend on the physiology of the administration site(s) and the membrane barriers present at those site(s) that the drug needs to cross in order to reach the systemic circulation. A summary of some of the properties of each route of administration is given in Chapter 1.
The gastrointestinal tract is discussed in detail in this chapter and a detailed description of the physiology of some of the other more important routes of administration is given in the relevant chapters of Part 5 of this book. The oral route of delivery is by far the most popular, with over 80% of medicines being given by mouth, mainly because it is natural and convenient for the patient and because it is relatively easy to manufacture oral dosage forms. Oral dosage forms do not need to be sterilized, are compact, and can be produced cheaply in large quantities by automated machines. This chapter and the next will therefore be confined to discussing the biopharmaceutical factors (that is, physiological, dosage form and drug factors) that influence oral drug absorption.
Physiological factors influencing oral drug absorption
The gastrointestinal tract is complex. Figure 19.1 outlines some of the main structures involved in and key physiological parameters that affect oral drug absorption. In order to gain an insight into the numerous factors that can potentially influence the rate and extent of drug absorption into the systemic circulation, a schematic illustration of the steps involved in the release and absorption of a drug from a tablet dosage form is presented in Figure 19.2. It can be seen from this that the rate and extent of appearance of intact drug in the systemic circulation depend on a succession of kinetic processes.
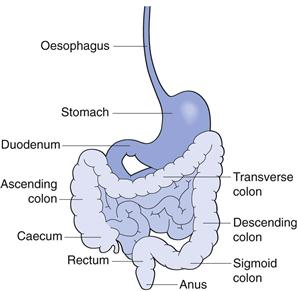
Fig. 19.1 The gastrointestinal tract.
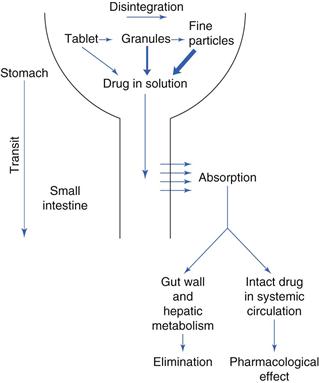
The slowest step in this series, which is the rate-limiting step, controls the overall rate and extent of appearance of intact drug in the systemic circulation. The rate-limiting step will vary from drug to drug. For a drug which has a very poor aqueous solubility, the rate at which it dissolves in the gastrointestinal fluids is often the slowest step and the bioavailability of that drug is said to be dissolution-rate limited. In contrast, for a drug that has a high aqueous solubility, its dissolution will be rapid and the rate at which the drug crosses the gastrointestinal membrane may be the rate-limiting step termed permeability limited.
Other potential rate-limiting steps include the rate of drug release from the dosage form (this can be by design, in the case of controlled-release dosage forms), the rate at which the stomach empties the drug into the small intestine, the rate at which the drug is metabolized by enzymes in the intestinal mucosal cells during its passage through them into the mesenteric blood vessels, and the rate of metabolism of drug during its initial passage through the liver, often termed the ‘first-pass’ effect.
Physiology of the gastrointestinal tract
The gastrointestinal tract is a muscular tube, approximately 6 m in length with varying diameters. It stretches from the mouth to the anus and consists of four main anatomical areas; the oesophagus, the stomach, the small intestine and the large intestine or colon. The luminal surface of the tube is not smooth but very rough, thereby increasing the surface area for absorption.
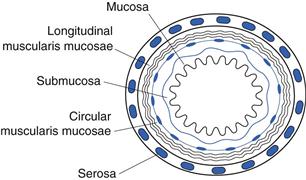
The wall of the gastrointestinal tract is essentially similar in structure along its length, consisting of four principal histological layers (Fig. 19.3):
The majority of the gastrointestinal epithelium is covered by a layer of mucus. This is a viscoelastic translucent aqueous gel that is secreted throughout the gastrointestinal tract, acting as a protective layer and a mechanical barrier. Mucus is a constantly changing mix of many secretions and exfoliated epithelial cells. It has a large water component (~95%). Its other primary components, which are responsible for its physical and functional properties, are large glycoproteins called mucins. Mucins consist of a protein backbone approximately 800 amino acids long and oligosaccharide side chains that are typically up to 18 residues in length.
The mucous layer ranges in thickness from 5 µm to 500 µm along the length of the gastrointestinal tract, with average values of around 80 µm. The layer is thought to be continuous in the stomach and duodenum but may not be so in the rest of the small and large intestines.
Mucus is constantly being removed from the luminal surface of the gastrointestinal tract through abrasion and acidic and/or enzymatic breakdown, and it is continually replaced from beneath. Turnover time has been estimated at 4–5 hours but this may well be an underestimate and is liable to vary along the length of the tract.
Oesophagus
The mouth is the point of entry for most drugs (so-called peroral – via the mouth – administration). At this point contact with the oral mucosa is usually brief. Linking the oral cavity to the stomach is the oesophagus. The oesophagus is composed of a thick muscular layer approximately 250 mm long and 20 mm in diameter. It joins the stomach at the gastrooesophageal junction, or cardiac orifice, as it is sometimes known.
The oesophagus, apart from the lowest 20 mm which is similar to the gastric mucosa, contains a well-differentiated squamous epithelium of non-proliferative cells. Epithelial cell function is mainly protective: simple mucous glands secrete mucus into the narrow lumen to lubricate food and protect the lower part of the oesophagus from gastric acid. The pH of the oesophageal lumen is usually between 5 and 6.
Materials are moved down the oesophagus by the act of swallowing. After swallowing, a single peristaltic wave of contraction, its amplitude linked to the size of the material being swallowed, passes down the length of the oesophagus at the rate of 20–60 mm per second, speeding up as it progresses. When swallowing is repeated in quick succession, the subsequent swallows interrupt the initial peristaltic wave and only the final wave proceeds down the length of the oesophagus to the gastrointestinal junction, carrying material within the lumen with it. Secondary peristaltic waves occur involuntarily in response to any distension of the oesophagus and serve to move sticky lumps of material or refluxed material to the stomach. In the upright position, the transit of materials through the oesophagus is assisted by gravity. The oesophageal transit of dosage forms is extremely rapid, usually of the order of 10–14 seconds.
Stomach
The next part of the gastrointestinal tract to be encountered by both food and pharmaceuticals is the stomach. The two major functions of the stomach are:
Another, perhaps less obvious, function of the stomach is its protective role in reducing the risk of noxious agents reaching the intestine.
The stomach is the most dilated part of the gastrointestinal tract and is situated between the lower end of the oesophagus and the small intestine. Its opening to the duodenum is controlled by the pyloric sphincter. The stomach can be divided into four anatomical regions (Fig. 19.4); the fundus, the body, the antrum and the pylorus.
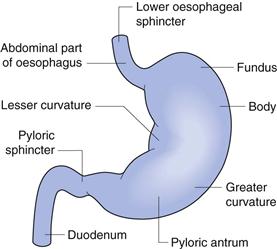
Fig. 19.4 The anatomy of the stomach.
The stomach has a capacity of approximately 1.5 L, although under fasting conditions it usually contains no more than 50 mL of fluid, which are mostly gastric secretions. These include:
Contrary to popular belief, very little drug absorption occurs in the stomach owing to its small surface area compared to the small intestine. The rate of gastric emptying can be a controlling factor in the onset of drug absorption from the major absorptive site, the small intestine. Gastric emptying will be discussed under gastrointestinal transit later in this chapter.
Small intestine
The small intestine is the longest (4–5 m) and most convoluted part of the gastrointestinal tract, extending from the pyloric sphincter of the stomach to the ileocaecal junction where it joins the large intestine. It is approximately 25 to 30 mm in diameter. Its main functions are:
The small intestine is divided into the duodenum, which is 200–300 mm in length, the jejunum, which is approximately 2 m in length, and the ileum, which is approximately 3 m in length.
The wall of the small intestine has a rich network of both blood and lymphatic vessels. The gastrointestinal circulation is the largest systemic regional vasculature and nearly a third of the cardiac output flows through the gastrointestinal viscera. The blood vessels of the small intestine receive blood from the superior mesenteric artery via branched arterioles. The blood leaving the small intestine flows into the hepatic portal vein that carries it via the liver to the systemic circulation. Drugs that are metabolized by the liver are degraded before they reach the systemic circulation; this is termed hepatic presystemic clearance or first-pass metabolism.
The wall of the small intestine also contains lacteals, which contain lymph and are part of the lymphatic system. The lymphatic system is important in the absorption of fats from the gastrointestinal tract. In the ileum there are areas of aggregated lymphoid tissue close to the epithelial surface which are known as Peyer’s patches (named after the 17th century Swiss anatomist Johann Peyer). These cells play a key role in the immune response as they transport macromolecules and are involved in antigen uptake.
The surface area of the small intestine is increased enormously, by about 600 times that of a simple cylinder, to approximately 200 m2 in an adult, by several adaptations which make the small intestine such a good absorption site:
• Villi – these have been described as finger-like projections into the lumen (approximately 0.5–1.5 mm in length and 0.1 mm in diameter). They are well supplied with blood vessels. Each villus contains an arteriole, a venule and a blind-ending lymphatic vessel (lacteal). The structure of a villus is shown in Figure 19.5.
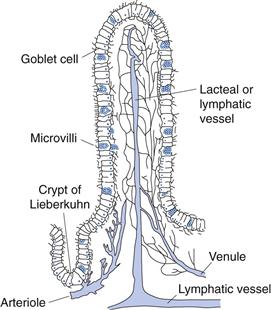
Fig. 19.5 Structure of a villus.
The luminal pH of the small intestine increases to between 6 and 7.5. Sources of secretions that produce these pH values in the small intestine are:
Colon
The colon is the final major part of the gastrointestinal tract. It stretches from the ileocaecal junction to the anus and makes up approximately the last 1.5 m of the 6 m of the gastrointestinal tract. It is composed of the caecum (~85 mm in length), the ascending colon (~200 mm), the hepatic flexure, the transverse colon (usually greater than 450 mm), the splenic flexure, the descending colon (~300 mm), the sigmoid colon (~400 mm) and the rectum, as shown in Figure 19.6. The ascending and descending colons are relatively fixed, as they are attached via the flexures and the caecum. The transverse and sigmoid colons are much more flexible.
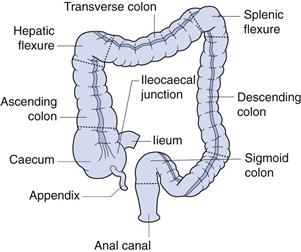
Fig. 19.6 The anatomy of the colon.
The colon, unlike the small intestine, has no specialized villi. However, the microvilli of the absorptive epithelial cells, the presence of crypts and the irregularly folded mucosae serve to increase the surface area of the colon by 10–15 times that of a simple cylinder. The surface area nevertheless remains approximately l/30th that of the small intestine.
The main functions of the colon are:
The colon is permanently colonized by an extensive number (about 1012 per gram of contents) and variety of bacteria. This large bacterial mass is capable of several metabolic reactions, including hydrolysis of fatty acid esters and the reduction of inactive conjugated drugs to their active form. The bacteria rely upon undigested polysaccharides in the diet and the carbohydrate components of secretions such as mucus for their carbon and energy sources. They degrade the polysaccharides to produce short-chain fatty acids (acetic, proprionic and butyric acids), which lower the luminal pH, and the gases hydrogen, carbon dioxide and methane. Thus, the pH of the caecum is around 6–6.5. This increases to around 7–7.5 towards the distal parts of the colon.
Recently there has been much interest in the exploitation of the enzymes produced by these bacteria with respect to targeted drug delivery to this region of the gastrointestinal tract.
Transit of pharmaceuticals in the gastrointestinal tract
As the oral route is the one by which the majority of pharmaceuticals are administered, it is important to know how these materials behave during their passage through the gastrointestinal tract. It is known that the small intestine is the major site of drug absorption, and thus the time a drug is present in this part of the gastrointestinal tract is extremely important. If sustained- or controlled-release drug delivery systems are being designed, it is important to consider factors that will affect their behaviour and, in particular, their transit times through certain regions of the gastrointestinal tract.
In general, most dosage forms, when taken in an upright position, transit through the oesophagus quickly, usually in less than 15 seconds. Transit through the oesophagus is dependent upon both the dosage form and posture.
Tablets/capsules taken in the supine (lying down) position, especially if taken without water, are liable to lodge in the oesophagus. Adhesion to the oesophageal wall can occur as a result of partial dehydration at the site of contact and the formation of a gel between the formulation and the oesophagus. The chances of adhesion will depend on the shape, size and type of formulation. Transit of liquids, for example, has always been observed to be rapid, and in general faster than that of solids. A delay in reaching the stomach may well delay a drug’s onset of action or cause damage or irritation to the oesophageal wall, e.g. potassium chloride tablets.
Gastric emptying
The time a dosage form takes to traverse the stomach is usually termed the gastric residence time, gastric emptying time or gastric emptying rate.
Gastric emptying of pharmaceuticals is highly variable and is dependent on the dosage form and the fed/fasted state of the stomach. Normal gastric residence times usually range between 5 minutes and 2 hours, although much longer times (over 12 hours) have been recorded, particularly for large single dosage units.
In the fasted state, the electrical activity in the stomach – the interdigestive myoelectric cycle or migrating myoelectric complex (MMC), as it is known – governs its activity and hence the transit of dosage forms. It is characterized by a repeating cycle of four phases. Phase I is a relatively inactive period of 40–60 minutes with only rare contractions occurring. Increasing numbers of contractions occur in phase II, which has a similar duration to phase I. Phase III is characterized by powerful peristaltic contractions which open the pylorus at the base and clear the stomach of any residual material. This is sometimes called the housekeeper wave. Phase IV is a short transitional period between the powerful activity of phase III and the inactivity of phase I.
The cycle repeats itself every 2 hours until a meal is ingested and the fed state or motility is initiated. In this state, two distinct patterns of activity have been observed. The proximal stomach relaxes to receive food and gradual contractions of this region move the contents distally. Peristalsis – contractions of the distal stomach – serves to mix and break down food particles and move them towards the pyloric sphincter. The pyloric sphincter allows liquids and small food particles to empty while other material is retropulsed into the antrum of the stomach and is caught up by the next peristaltic wave for further size reduction before emptying.
Thus, in the fed state, liquids, pellets and disintegrated tablets will tend to empty with food, yet large sustained-or controlled-release dosage forms can be retained in the stomach for long periods of time. In the fasted state, the stomach is less discriminatory between dosage form types, with emptying appearing to be an exponential process and being related to the point in the MMC at which the formulation is ingested.
Many factors influence gastric emptying, as well as the type of dosage form and the presence of food. These include posture, the composition of the food and the effect of drugs and disease state. In general, food, particularly fatty foods, delays gastric emptying and hence the absorption of drugs. Therefore, a drug is likely to reach the small intestine most rapidly if it is administered with water to a patient whose stomach is empty.
Small intestinal transit
There are two main types of intestinal movement – propulsive and mixing. The propulsive movements primarily determine the intestinal transit rate and hence the residence time of the drug or dosage form in the small intestine. As this is the main site of absorption in the gastrointestinal tract for most drugs, the small intestinal transit time (that is, the time of transit between the stomach and the caecum) is an important factor with respect to drug bioavailability.
Small intestinal transit is normally considered to be between 3 to 4 hours although both faster and slower transit have been measured. In contrast to the stomach, the small intestine does not discriminate between solids and liquids, and hence between dosage forms, or between the fed and the fasted state.
Small intestinal residence time is particularly important for:
Colonic transit
The colonic transit of pharmaceuticals is prolonged and variable, and depends on the type of dosage form, diet, eating pattern, defaecation pattern and frequency and disease state.
Contractile activity in the colon can be divided into two main types:
Colonic transit is thus characterized by short bursts of activity followed by long periods of stasis. Movement is mainly aboral, i.e. towards the anus. Motility and transit is highly influenced by defaecation time; both frequency of defaecation and likelihood of being included in a defaecation event. Colonic transit can vary from anything between 2 and 48 hours. In most individuals, total transit times (i.e. mouth to anus) are between 12 and 36 hours however they can range from several hours to several days.
Barriers to drug absorption
Figure 19.7 shows some of the barriers to absorption that a drug may encounter once it is released from its dosage form and has dissolved into the gastrointestinal fluids. The drug needs to remain in solution, not become bound to food or other material within the gastrointestinal tract and not precipitate. It needs to be chemically stable in order to withstand the pH of the gastrointestinal tract and it must be resistant to enzymatic degradation in the lumen. The drug then needs to diffuse across the mucous layer without binding to it, across the unstirred water layer and subsequently across the gastrointestinal membrane, its main cellular barrier. After passing through this cellular barrier, the drug encounters the liver and all its metabolizing enzymes before it reaches the systemic circulation. Any of these barriers can prevent some or all of the drug reaching the systemic circulation and can therefore have a detrimental effect on its bioavailability.
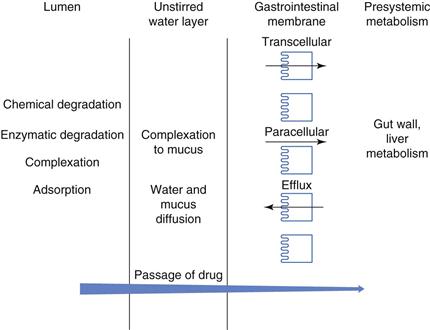
Fig. 19.7 Barriers to absorption.
Environment within the lumen
The environment within the lumen of the gastrointestinal tract has a major effect on the rate and extent of drug absorption.
Gastrointestinal pH
The pH of fluids varies considerably along the length of the gastrointestinal tract. Gastric fluid is highly acidic, normally exhibiting a pH within the range 1–3.5 in healthy people in the fasted state. Following the ingestion of a meal, the gastric juice is buffered to a less acidic pH that is dependent on meal composition. Typical gastric pH values following a meal are in the range 3–7. Depending on the size of the meal, the gastric pH returns to the lower fasted-state values within 2–3 hours. Thus, only a dosage form ingested with or soon after a meal will encounter these higher pH values. This may be an important consideration in terms of the chemical stability of a drug or in achieving drug dissolution or absorption.
Intestinal pH values are higher than gastric pH values owing to the neutralization of the gastric acid with bicarbonate ions secreted by the pancreas into the small intestine. There is a gradual rise in pH along the length of the small intestine from the duodenum to the ileum. Table 19.1 summarizes some of the literature values recorded for small intestinal pH in the fed and fasted states. The pH drops again in the colon as the bacterial enzymes, which are localized in the colonic region, break down undigested carbohydrates into short-chain fatty acids; this lowers the pH in the colon to around 6.5.
Table 19.1
pH in the small intestine in healthy humans in the fasted and fed states
| Location | Fasted state pH | Fed state pH |
| Mid-distal duodenum | 4.9 | 5.2 |
| 6.1 | 5.4 | |
| 6.3 | 5.1 | |
| 6.4 | ||
| Jejunum | 4.4–6.5 | 5.2–6.0 |
| 6.6 | 6.2 | |
| Ileum | 6.5 | 6.8–7.8 |
| 6.8–8.0 | 6.8–8.0 | |
| 7.4 | 7.5 |
Data from Gray & Dressman (1996)
The gastrointestinal pH may influence the absorption of drugs in a variety of ways. If the drug is a weak electrolyte, pH may influence the drug’s chemical stability in the lumen, its rate and extent of dissolution or its absorption characteristics. Chemical degradation due to pH-dependent hydrolysis can occur in the gastrointestinal tract. The result of this instability is incomplete bioavailability, as only a fraction of the administered dose reaches the systemic circulation in the form of intact drug. The extent of degradation of penicillin G (benzylpenicillin), the first of the penicillins, after oral administration, depends on its residence time in the stomach and the gastric pH. This gastric instability has tended to preclude its oral use. The antibiotic erythromycin and proton pump inhibitors (e.g. omeprazole) degrade rapidly at acidic pH values and therefore have to be formulated as enteric-coated dosage forms to ensure good bioavailability (Chapter 20). The effects of pH on the drug dissolution and absorption processes are also discussed in Chapter 20.
Luminal enzymes
The primary enzyme found in gastric juice is pepsin. Lipases, amylases and proteases are secreted from the pancreas into the small intestine in response to ingestion of food. These enzymes are responsible for most nutrient digestion. Pepsins and the proteases are responsible for the degradation of protein and peptide drugs in the lumen. Other drugs that resemble nutrients, such as nucleotides and fatty acids, may also be susceptible to enzymatic degradation. The lipases may also affect the release of drugs from fat/oil-containing dosage forms. Drugs that are esters can also be susceptible to hydrolysis in the lumen.
Bacteria, which are mainly localized within the colonic region of the gastrointestinal tract, secrete enzymes that are capable of a range of reactions. These enzymes have been utilized when designing drugs or dosage forms to target the colon. Sulfasalazine, for example, is a prodrug of 5-aminosalicylic acid linked via an azo bond to sulfapyridine. The sulfapyridine moiety makes the drug too large and hydrophilic to be absorbed in the upper gastrointestinal tract, and thus permits its transport intact to the colonic region. Here the bacterial enzymes reduce the azo bond in the molecule and release the active drug, 5-aminosalycylic acid, for local action in colonic diseases such as inflammatory bowel disease.
Influence of food in the gastrointestinal tract
The presence of food in the gastrointestinal tract can influence the rate and extent of absorption, either directly or indirectly via a range of mechanisms.
Complexation of drugs with components in the diet.
Drugs are capable of binding to components within the diet. In general this only becomes an issue (with respect to bioavailability) where an irreversible or an insoluble complex is formed. In such cases the fraction of the administered dose that becomes complexed is unavailable for absorption. Tetracycline, for example, forms non-absorbable complexes with calcium and iron, and thus patients are advised not to take products containing calcium or iron, such as milk, iron preparations or indigestion remedies, at the same time of day as the tetracycline. However, if the complex formed is water soluble and readily dissociates to liberate the ‘free’ drug then there may be little effect on drug absorption.
Alteration of pH.
In general, food tends to increase stomach pH by acting as a buffer. This is liable to decrease the rate of dissolution and subsequent absorption of a weakly basic drug and increase that of a weakly acidic one.
Alteration of gastric emptying.
As already mentioned, some foods, particularly those containing a high proportion of fat, and some drugs tend to reduce gastric emptying and thus delay the onset of action of certain drugs. Food slows the rate of absorption, due to delayed gastric emptying, of the antiretroviral nucleoside analogues lamivudine and zidovudine; however this is not considered to be clinically significant.
Stimulation of gastrointestinal secretions.
Gastrointestinal secretions (e.g. pepsin) produced in response to food may result in the degradation of drugs that are susceptible to enzymatic metabolism and hence in a reduction in their bioavailability. The ingestion of food, particularly fats, stimulates the secretion of bile. Bile salts are surface-active agents and can increase the dissolution of poorly soluble drugs, thereby enhancing their absorption. However, bile salts have been shown to form insoluble and hence non-absorbable complexes with some drugs such as neomycin, kanamycin and nystatin.
Competition between food components and drugs for specialized absorption mechanisms.
In the case of those drugs that have a chemical structure similar to nutrients required by the body for which specialized absorption mechanisms exist, there is a possibility of competitive inhibition of drug absorption.
Increased viscosity of gastrointestinal contents.
The presence of food in the gastrointestinal tract provides a viscous environment which may result in a reduction in the rate of drug dissolution. In addition, the rate of diffusion of a drug in solution from the lumen to the absorbing membrane lining the gastrointestinal tract may be reduced by an increase in viscosity. Both of these effects tend to decrease the bioavailability of a drug.
Food-induced changes in presystemic metabolism.
Certain foods may increase the bioavailability of drugs that are susceptible to presystemic intestinal metabolism by interacting with the metabolic process. Grapefruit juice, for example, is capable of inhibiting the intestinal cytochrome P450 (CYP3A family) and thus, when taken with drugs that are susceptible to CYP3A metabolism, is likely to result in their increased bioavailability. Clinically relevant interactions exist between grapefruit juice and the antihistamine terfenadine, the immunosuppressant ciclosporin, the protease inhibitor saquinavir and the calcium channel blocker verapamil.
Food-induced changes in blood flow.
Blood flow to the gastrointestinal tract and liver increases shortly after a meal, thereby increasing the rate at which drugs are presented to the liver. The metabolism of some drugs (e.g. propranolol) is sensitive to their rate of presentation to the liver; the faster the rate of presentation, the larger the fraction of drug that escapes first-pass metabolism. This is because the enzyme systems responsible for their metabolism become saturated by the increased rate of presentation of the drug to the site of biotransformation. For this reason, the effects of food serve to increase the bioavailability of some drugs that are susceptible to first-pass metabolism.
It is evident that food can influence the absorption of many drugs from the gastrointestinal tract by a variety of mechanisms. Drug-food interactions are often classified into five categories: those that cause reduced, delayed, increased or accelerated absorption, and those on which food has no effect. The reader is referred to reviews by Custodio et al (2008), Davit & Conner (2008) and Fleisher et al (2010) for more detailed information on the effect of food on the rate and extent of drug absorption.
Disease state and physiological disorders
Disease states and physiological disorders associated with the gastrointestinal tract are likely to influence the absorption and hence the bioavailability of orally administered drugs. Local diseases can cause alterations in gastric pH that can affect the stability, dissolution and/or absorption of the drug. Gastric surgery can cause drugs to exhibit differences in bioavailability from that in normal individuals. For example, partial or total gastrectomy results in drugs reaching the duodenum more rapidly than in normal individuals and significant changes in fluid composition and volumes can significantly affect drug bioavailability. AIDS patients often have over-secretion of gastrin and thus low pH, which can adversely affect the dissolution and hence bioavailability of weakly basic drugs such as the anti-fungal ketoconazole. Lower pH values are often seen in disease states of the colon such as Crohn’s disease and ulcerative colitis.
Mucus and the unstirred water layer
Before drugs can permeate across the epithelial surface the mucous layer and unstirred water layer need to be crossed. The mucus layer, whose thickness and turnover rates can vary along the length of the gastrointestinal tract, can hinder drug diffusion. The unstirred water layer or aqueous boundary layer is a more or less stagnant layer of water, mucus and glycocalyx adjacent to the intestinal wall. It is thought to be created by incomplete mixing of the luminal contents near the intestinal mucosal surface. This layer, which is around 30–100 µm in thickness, can provide a diffusion barrier to drugs. Some drugs are also capable of complexing with mucus, thereby reducing their availability for absorption.
Gastrointestinal membrane
Structure of the membrane
The gastrointestinal membrane separates the lumen of the stomach and intestines from the systemic circulation. It is the main cellular barrier to the absorption of drugs from the gastrointestinal tract. The membrane is complex in nature, being composed of lipids, proteins, lipoproteins and polysaccharides. It has a bilayer structure, as shown in Figure 19.8. The barrier has the characteristics of a semipermeable membrane, allowing the rapid transit of some materials and impeding or preventing the passage of others. It is permeable to amino acids, sugars, fatty acids and other nutrients and is impermeable to plasma proteins. The membrane can be viewed as a semipermeable lipoidal sieve, which allows the passage of lipid-soluble molecules across it and the passage of water and small hydrophilic molecules through its numerous aqueous pores. In addition, there are a number of transporter proteins or carrier molecules that exist in the membrane and which, with the help of energy, transport materials back and forth across it.
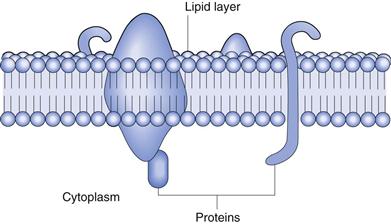
Fig. 19.8 Structure of the membrane.
Mechanisms of transport across the membrane
There are two main mechanisms of drug transport across the gastrointestinal epithelium: transcellular (i.e. across the cells) and paracellular (i.e. between the cells). The transcellular pathway is further divided into simple passive diffusion, carrier-mediated transport (active transport and facilitated diffusion) and endocytosis. These pathways are illustrated in Figure 19.9.
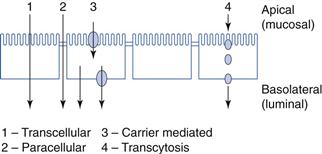
Fig. 19.9 Mechanisms of permeability (absorptive).
Transcellular
Passive diffusion
This is the preferred route of transport for relatively small lipophilic molecules and thus many drugs. In this process, drug molecules pass across the lipoidal membrane via passive diffusion from a region of high concentration in the lumen to a region of lower concentration in the blood. This lower concentration is maintained primarily by blood flow. The rate of transport is determined by the physicochemical properties of the drug, the nature of the membrane and the concentration gradient of the drug across the membrane. The process initially involves the partitioning of the drug between the aqueous fluids within the gastrointestinal tract and the lipoidal-like membrane of the lining of the epithelium. The drug in solution in the membrane then diffuses across the epithelial cell/cells within the gastrointestinal barrier to blood in the capillary network in the lamina propria. Upon reaching the blood, the drug will be rapidly distributed, so maintaining a much lower concentration than that at the absorption site. If the cell membranes and fluid regions making up the gastrointestinal-blood barrier can be considered as a single membrane, then the stages involved in gastrointestinal absorption could be represented by the model shown in Figure 19.10.
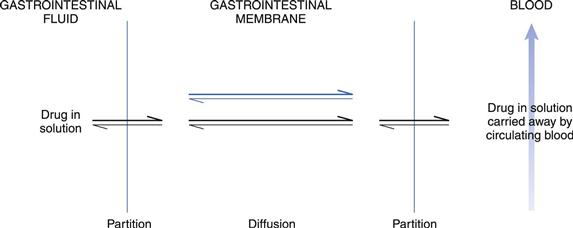
Passive diffusion of drugs across the gastrointestinal-blood barrier can often be described mathematically by Fick’s First Law of Diffusion (see Chapter 2). When considered in the context of bioavailability, this indicates that the rate of diffusion across a membrane (dC/dt) is proportional to the difference in concentration on each side of that membrane. Therefore, the rate of appearance of drug in the blood at the absorption site is given by:
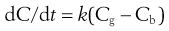 (19.1)
(19.1)
where dC/dt is the rate of appearance of drug in the blood at the site of absorption, k is the proportionality constant, Cg is the concentration of drug in solution in the gastrointestinal fluid at the absorption site and Cb is the concentration of drug in the blood at the site of absorption.
The proportionality constant k incorporates the diffusion coefficient of the drug in the gastrointestinal membrane (D), and the thickness (h) and surface area of the membrane (A).
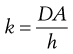 (19.2)
(19.2)
These equations indicate that the rate of gastrointestinal absorption of a drug by passive diffusion depends on the surface area of the membrane that is available for drug absorption. Thus the small intestine, primarily the duodenum, is the major site of drug absorption, owing principally to the presence of villi and microvilli which provide such a large surface area for absorption (discussed earlier in this chapter).
Equation 19.1 also indicates that the rate of drug absorption depends on a large concentration gradient of drug existing across the gastrointestinal membrane. This concentration gradient is influenced by the apparent partition coefficients exhibited by the drug with respect to the gastrointestinal membrane-fluid interface and the gastrointestinal membrane-blood interface. It is important that the drug has sufficient affinity (solubility) for the membrane phase so that it can partition readily into the gastrointestinal membrane. In addition, after diffusing across the membrane, the drug should exhibit sufficient solubility in the blood such that it can partition readily out of the membrane phase into the blood.
On entering the blood in the capillary network in the lamina propria, the drug will be carried away from the site of absorption by the rapidly circulating gastrointestinal blood supply. It will then become diluted by distribution into a large volume of blood (i.e. the systemic circulation), by distribution into body tissues and other fluids, and by subsequent metabolism and excretion. In addition, the drug may bind to plasma proteins in the blood which will further lower the concentration of free (i.e. diffusible) drug in the blood. Consequently, the blood acts as a ‘sink’ for absorbed drug and ensures that the concentration of drug in the blood at the site of absorption is low in relation to that in the gastrointestinal fluids at the site of absorption, i.e. Cg » Cb. The ‘sink’ conditions provided by the systemic circulation ensure that a large concentration gradient is maintained across the gastrointestinal membrane during the absorption process.
The passive absorption process is driven solely by the concentration gradient of the diffusible species of the drug that exists across the gastrointestinal-blood barrier. Thus Equations 19.1 and 19.2 can be combined and written as:
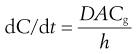 (19.3)
(19.3)
and because for a given membrane D, A and h can be regarded as constants, Equation 19.3 becomes:
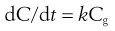 (19.4)
(19.4)
Equation 19.4 is an expression for a first-order kinetic process (discussed in Chapter 7) and indicates that the rate of passive absorption will be proportional to the concentration of absorbable drug in solution in the gastrointestinal fluids at the site of absorption and therefore that the gastrointestinal absorption of most drugs follows first-order kinetics.
It has been assumed in this description that the drug exists solely in one single absorbable species. Many drugs, however, are weak electrolytes that exist in aqueous solution as two species, namely the unionized species and the ionized species. Because it is the unionized form of a weak electrolyte drug that exhibits greater lipid solubility compared to the corresponding ionized form, the gastrointestinal membrane is more permeable to the unionized species. Thus, the rate of passive absorption of a weak electrolyte is related to the fraction of total drug that exists in the unionized form in solution in the gastrointestinal fluids at the site of absorption. This fraction is determined by the dissociation constant of the drug (i.e. its pKa value) and by the pH of the aqueous environment, in accordance with the Henderson-Hasselbalch equations for weak acids and bases (discussed in Chapter 3). The gastrointestinal absorption of a weak electrolyte drug is enhanced when the pH at the site of absorption favours the formation of a large fraction of the drug in aqueous solution that is unionized. This forms the basis of the pH-partition hypothesis (see Chapter 20).
Carrier-mediated transport
As already stated, the majority of drugs are absorbed across cells (i.e. transcellularly) via passive diffusion. However, certain compounds and many nutrients are absorbed transcellularly by a carrier-mediated transport mechanism of which there are two main types: active transport and facilitated diffusion or facilitated transport.
Active transport
In contrast to passive diffusion, active transport involves the active participation by the apical cell membrane of the columnar absorption cells. A carrier or membrane transporter is responsible for binding a drug and transporting it across the membrane by a process illustrated in Figure 19.11.
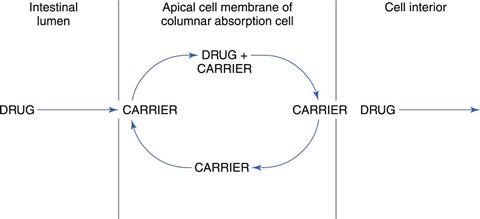
Carrier-mediated absorption is often explained by assuming a shuttling process across the epithelial membrane. The drug molecule or ion forms a complex with the carrier/transporter in the surface of the apical cell membrane of a columnar absorption cell. The drug-carrier complex then moves across the membrane and liberates the drug on the other side of the membrane. The carrier (now free) returns to its initial position in the surface of the cell membrane adjacent to the gastrointestinal tract to await the arrival of another drug molecule or ion.
Active transport is a process whereby materials can be transported against a concentration gradient across a cell membrane, i.e. transport can occur from a region of lower concentration to one of higher concentration. Therefore, active transport is an energy-consuming process. The energy arises either from the hydrolysis of ATP or from the transmembranous sodium gradient and/or electrical potential.
There are a large number of carrier-mediated active transport systems or membrane transporters in the small intestine. These can be present either on the apical (brush border) or on the basolateral membrane. They include the peptide transporters, the nucleoside transporters, the sugar transporters, the bile acid transporters, the amino acid transporters, the organic anion transporters and the vitamin transporters.
Many nutrients, such as amino acids, sugars, electrolytes (e.g. sodium, potassium, calcium, iron, chloride, bicarbonate), vitamins (thiamine (B1), nicotinic acid, riboflavin (B2), pyroxidine (B6) and cobalamin (B12)) and bile salts, are actively transported. Each carrier system is generally concentrated in a specific segment of the gastrointestinal tract. The substance that is transported by that carrier will thus be absorbed preferentially in the location of highest carrier density. For example, the bile acid transporters are only found in the lower part of the small intestine, the ileum. Each carrier/transporter has its own substrate specificity with respect to the chemical structure of the substance that it will transport. Some carriers/transporters have broader specificity than others. Thus if a drug structurally resembles a natural substance which is actively transported then the drug is also likely to be transported by the same carrier mechanism.
Many peptide-like drugs, such as the penicillins, cephalosporins, angiotensin-converting enzyme (ACE) inhibitors and renin inhibitors, rely on the peptide transporters for their efficient absorption. Nucleosides and their analogues for antiviral and anticancer drugs depend on the nucleoside transporters for their uptake. L-dopa (levodopa) and α-methyldopa are transported by the carrier-mediated process for amino acids. L-dopa has a much faster permeability rate than methyldopa, which has been attributed to the lower affinity of methyldopa for the amino acid carrier.
Unlike passive absorption, where the rate of absorption is directly proportional to the concentration of the absorbable species of the drug at the absorption site, active transport proceeds at a rate that is proportional to the drug concentration only at low concentrations. At higher concentrations, the carrier mechanism becomes saturated and further increases in drug concentration will not increase the rate of absorption, i.e. the rate of absorption remains constant. Absorption rate-concentration relationships for active and passive processes are compared in Figure 19.12.
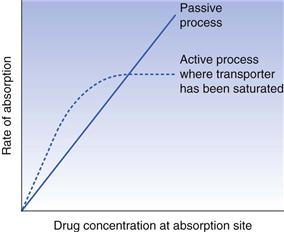
Competition between two similar substances for the same transfer mechanism, and the inhibition of absorption of one or both compounds, are other characteristics of carrier-mediated transport. Inhibition of absorption may also be observed with agents that interfere with cell metabolism. Some substances may be absorbed by simultaneous carrier-mediated and passive transport processes. The contribution of the carrier-mediated process to the overall absorption rate decreases with concentration, and at a sufficiently high concentration is negligible.
In summary, active transport mechanisms:
• must have a carrier molecule
• must have a source of energy
• can be inhibited by metabolic inhibitors such as dinitrophenol
Active transport also plays an important role in the intestinal, renal and biliary excretion of many drugs.
Facilitated diffusion or transport
This carrier-mediated process differs from active transport in that it cannot transport a substance against a concentration gradient of that substance. Therefore, facilitated diffusion does not require an energy input but does require a concentration gradient for its driving force like passive diffusion. When substances are transported by facilitated diffusion, they are transported down the concentration gradient but at a much faster rate than would be anticipated based on the molecular size and polarity of the molecule. The process, like active transport, can be saturated and is subject to inhibition by competitive inhibitors. In terms of drug absorption, facilitated diffusion seems to play a very minor role.
Endocytosis
Endocytosis is the process by which the plasma membrane of the cell invaginates and the invaginations become pinched off, forming small intracellular membrane-bound vesicles that enclose a volume of material. Thus, material can be transported into the cell. After invagination, the material is often transferred to other vesicles or lysosomes and digested. Some material will escape digestion and migrate to the basolateral surface of the cell where it is exocytosed. This uptake process is energy dependent. Endocytosis can be further subdivided into four main processes: fluid-phase endocytosis or pinocytosis; receptor-mediated endocytosis; phagocytosis; and transcytosis. Endocytosis is thought to be the primary mechanism of transport of macromolecules. The process and pathways of endocytosis are complex.
Pinocytosis
Fluid-phase endocytosis or pinocytosis is the engulfment of small droplets of extracellular fluid by membrane vesicles. The cell will internalize material regardless of its metabolic importance to that cell. The efficiency of this process is low. The fat-soluble vitamins A, D, E and K are absorbed via pinocytosis.
Receptor-mediated endocytosis
Many cells within the body have receptors on their cell surfaces that are capable of binding with suitable ligands to form ligand-receptor complexes. These complexes cluster on the cell surface and then invaginate and break off from the membrane to form coated vesicles. The binding process between the ligand and the receptor on the cell surface is thought to trigger a conformational change in the membrane to allow this process to occur. Once within the cytoplasm of the cell, the coated vesicles rapidly lose their coat and the resulting uncoated vesicles will promptly deliver their contents to early endosomes. Within the endosomes, the ligands usually dissociate from their receptors, many of which are then recycled to the plasma membrane. The dissociated ligands and solutes are next delivered to prelysosomes and finally to lysosomes, the end-stage of the endocytic pathway. Lysosomes are spherical or oval cell organelles surrounded by a single membrane. They contain digestive enzymes which break down bacteria and large molecules, such as proteins, polysaccharides and nucleic acids, which have entered the cell via endocytosis.
Phagocytosis
Phagocytosis can be defined as the engulfment by the cell membrane of particles larger than 500 nm. This process is important for the absorption of polio and other vaccines from the gastrointestinal tract.
Transcytosis
Transcytosis is the process by which the material internalized by the membrane domain is transported through the cell and secreted on the opposite side.
Paracellular pathway
The paracellular pathway differs from all the other absorption pathways as it is the transport of materials in the aqueous pores between the cells rather than across them. The cells are joined together via closely fitting tight junctions on their apical side. The intercellular spaces occupy only about 0.01% of the total surface area of the epithelium. The tightness of these junctions can vary considerably between different epithelia in the body. In general, absorptive epithelia, such as that of the small intestine, tend to be leakier than other epithelia. The paracellular pathway decreases in importance down the length of the gastrointestinal tract and as the number and size of pores between the epithelial cells decrease.
The paracellular route of absorption is important for the transport of ions such as calcium and for the transport of sugars, amino acids and peptides at concentrations above the capacity of their carriers. Small hydrophilic charged drugs that do not distribute into cell membranes cross the gastrointestinal epithelium via the paracellular pathway. The molecular weight cut-off for the paracellular route is usually considered to be 200 Da, although some larger drugs have been shown to be absorbed via this route.
The paracellular pathway can be divided into convective (‘solvent drag’) and diffusive components. The convective component is the rate at which the compound is carried across the epithelium via the water flux.
In celiac disease there is an increase in intestinal permeability due to a ‘loosening’ of the tight junctions. An approach to improving the absorption of poorly permeable drugs is aimed at making the intestine more ‘leaky’ by opening up the tight junctions.
Efflux of drugs from the intestine
Efflux proteins or transporters that expel specific drugs back into the lumen of the gastrointestinal tract after they have been absorbed, can play a key role on the bioavailability of drugs. One of the key counter-transport proteins is P-glycoprotein. P-glycoprotein is expressed at high levels on the apical surface of columnar cells (brush border membrane) in the jejunum. It is also present on the surface of many other epithelia and endothelia in the body, and on the surface of tumour cells. P-glycoprotein expression tends to be significantly higher in the small intestine than colon. P-glycoproteins were discovered because of their ability to cause multidrug resistance in tumour cells by preventing the intracellular accumulation of many cytotoxic cancer drugs by pumping the drugs back out of the tumours. Certain drugs with wide structural diversity (Table 19.2) are susceptible to efflux from the intestine via P-glycoprotein. Such efflux may have a detrimental effect on drug bioavailability. These countertransport efflux proteins pump drugs out of cells in a similar way to which nutrients, and drugs are actively absorbed across the gastrointestinal membrane. This process therefore requires energy, can work against a concentration gradient, can be competitively inhibited by structural analogues or by inhibitors of cell metabolism, and is a saturable process.
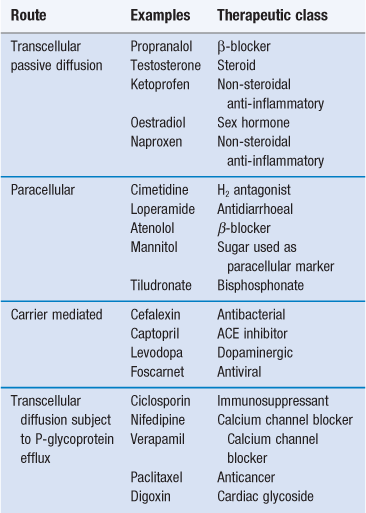
Transporters in the gastrointestinal tract
As discussed earlier in this chapter there are a number of transporters in the gastro-intestinal tract. These can be classified as either efflux or uptake (or influx) transporters depending on the direction of the transport. Both these transporters can be also classified as unitransporters, symporters and antiporters. Uniporters bind and transport only one type of substrate at a time. There are both passive and active uniporters for example the glucose and nucleoside transporters which are driven by an electrochemical gradient and P-glycoproteins, the breast cancer resistance protein (BRRP), the multidrug resistance protein proteins (MRPs) and sodium, potassium and ATPase which are driven by ATP. In contrast, symporters and antiporters are active transporters which can move more than one type of substrate at once, usually a drug molecule and a metal ion. Symporters (or cotransporters) transport ions and substrates simultaneously in the same direction while antiporters (or counter transporters) simultaneously transport ions in one direction and substrates in the opposite direction. As the driving force for symporters and antitransporters are voltage or ion gradients (usually sodium), they are also called ion-couple solute transporters, however as the driving force for these transporters is voltage (H+) or sodium they can also be known as secondary active transporters. A number of substrates can usually bind to a transporter and thus different drugs can compete for the same transporter. Thus, the transporter can be inhibited; competitively, non-competitively or uncompetitively. Competitive inhibition occurs when both the substrate and inhibitor compete for the same binding site. Non-competitive inhibition occurs when the inhibitor does not bind to the transporter active site but an allosteric site which lowers the affinity of the transporter for the substrate due to changing the conformation of the transporter. Uncompetitive binding occurs when the inhibitor binds to the intermediate of the substrate-transporter complex to terminate the translocation step.
In summary, drugs can be absorbed via passive diffusion and carrier mediated pathways. A drug can cross the intestinal epithelium via one pathway or a combination of pathways. The relative contribution of these pathways depends on the drug’s location within the gastrointestinal tract, the formulation and the physicochemical properties of the drug which are discussed in Chapter 20. Table 19.2 summarizes the main mechanisms of drug transport across the gastrointestinal epithelia for a number of commonly used drugs.
Presystemic metabolism
As well as having the ability to cross the gastrointestinal membrane by one of the routes described, drugs also need to be resistant to degradation/metabolism during this passage. All drugs that are absorbed from the stomach, small intestine and upper colon pass into the hepatic portal system and are presented to the liver before reaching the systemic circulation. Therefore, if the drug is going to be available to the systemic circulation, it must also be resistant to metabolism by the liver. Hence, an oral dose of drug could be completely absorbed but incompletely available to the systemic circulation because of first-pass or presystemic metabolism by the gut wall and/or liver.
Gut wall metabolism
The gut walls contain a number of metabolizing enzymes that can degrade drugs before they reach the systemic circulation. For example, the major cytochrome P450 enzyme CYP3A, present in the liver and responsible for the hepatic metabolism of many drugs, is present in the intestinal mucosa and intestinal metabolism may be important for substrates of this enzyme. This effect can also be known as first-pass metabolism by the intestine. CYP levels tend to be higher in the intestine than in the colon.
Hepatic metabolism
The liver is the primary site of drug metabolism and thus acts as a final barrier for oral absorption. The first pass of absorbed drug through the liver may result in extensive metabolism of the drug, and a significant portion may never reach the systemic circulation, resulting in a low bioavailability of those drugs which are rapidly metabolized by the liver. The bioavailability of a susceptible drug may be reduced to such an extent as to render the gastrointestinal route of administration ineffective, or to necessitate an oral dose which is many times larger than the intravenous dose, e.g. propranolol. Although propranolol is well absorbed, only about 30% of an oral dose is available to the systemic circulation owing to the first-pass effect. The bioavailability of sustained-release propranolol is even less as the drug is presented via the hepatic portal vein more slowly than from an immediate-release dosage form, and the liver is therefore capable of extracting and metabolizing a larger portion. Other drugs which are susceptible to a large first-pass effect are the cholesterol lowering agent, atorvastatin, the anaesthetic lidocaine (lignocaine), the tricyclic antidepressant imipramines and diazepam and the analgesics pentazocine and morphine.
First-pass metabolism can be avoided by drug administration to the mouth (buccal or sublingual; see Chapter 30) or to the rectum (see Chapter 42). The arrangement of the blood vessels in these regions means that absorbed drug does not pass through the liver first, prior to entering the systemic circulation.
Summary
There are many physiological factors that influence the rate and extent of drug absorption; these are initially dependent on the route of administration. For the oral route, the physiological and environmental factors of the gastrointestinal tract, the gastrointestinal membrane and presystemic metabolism can all influence drug bioavailability.
References
1. Custodio JM, Wu Chi-Yuan, Benet L. Predicting drug disposition, absorption/elimination/transporter interplay and the role of food on drug absorption. Advanced Drug Delivery Reviews. 2008;60:717–733.
2. Davit B, Conner D. Food effects on drug bioavailability: Implications for new and generic drug development. In: Krishna R, Yu L, eds. New York: Springer; 2008.
3. Fleisher D, Sweet BV, Parekh A, Boullata JI. Drug Absoprtion with Food In: Handbook of Drug Nutrient Interactions, Nutrition and Health. Part 3, 209–241 Springer: Humana Press; 2010.
4. Gray V, Dressman J. Change of pH requirements for simulated intestinal fluid TS. Pharmacopeial Forum. 1996;22:1943–1945.
Bibliography
1. Dobson PD, Kell DB. Carrier-mediated cellular uptake of pharmaceutical drugs: an exception or the rule? Nature Drug Discovery Reviews. 2008;7:205–220.
2. Hu M, Li X, eds. Bioavailability: Basic Principles, Advanced Concepts, and Applications. New Jersey: John Wiley & Sons; 2011.
3. McConnell EL, Fadda HM, Basit AW. Gut instincts: Explorations in intestinal physiology and drug delivery. International Journal of Pharmaceutics. 2008;34:213–226.
4. Sugano K, Kansy M, Artursson P, et al. Coexistence of passive and carrier-mediated processes in transport. Nature Reviews Drug Discovery. 2010;9:597–614.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Gastrointestinal tract – physiology and drug absorption
Marianne Ashford
Chapter contents
Physiological factors influencing oral drug absorption
Physiology of the gastrointestinal tract
Transit of pharmaceuticals in the gastrointestinal tract
Key points
Introduction
The factors that influence the rate and extent of absorption depend upon the route of administration. As stated in Chapter 18, the intravenous route offers direct access to the systemic circulation and the total dose administered via this route is available in the plasma for distribution into other body tissues and the site(s) of action of the drug. Other routes will require an absorption step before the drug reaches the systemic circulation. Factors affecting this absorption will depend on the physiology of the administration site(s) and the membrane barriers present at those site(s) that the drug needs to cross in order to reach the systemic circulation. A summary of some of the properties of each route of administration is given in Chapter 1.
The gastrointestinal tract is discussed in detail in this chapter and a detailed description of the physiology of some of the other more important routes of administration is given in the relevant chapters of Part 5 of this book. The oral route of delivery is by far the most popular, with over 80% of medicines being given by mouth, mainly because it is natural and convenient for the patient and because it is relatively easy to manufacture oral dosage forms. Oral dosage forms do not need to be sterilized, are compact, and can be produced cheaply in large quantities by automated machines. This chapter and the next will therefore be confined to discussing the biopharmaceutical factors (that is, physiological, dosage form and drug factors) that influence oral drug absorption.
Physiological factors influencing oral drug absorption
The gastrointestinal tract is complex. Figure 19.1 outlines some of the main structures involved in and key physiological parameters that affect oral drug absorption. In order to gain an insight into the numerous factors that can potentially influence the rate and extent of drug absorption into the systemic circulation, a schematic illustration of the steps involved in the release and absorption of a drug from a tablet dosage form is presented in Figure 19.2. It can be seen from this that the rate and extent of appearance of intact drug in the systemic circulation depend on a succession of kinetic processes.
Fig. 19.1 The gastrointestinal tract.
The slowest step in this series, which is the rate-limiting step, controls the overall rate and extent of appearance of intact drug in the systemic circulation. The rate-limiting step will vary from drug to drug. For a drug which has a very poor aqueous solubility, the rate at which it dissolves in the gastrointestinal fluids is often the slowest step and the bioavailability of that drug is said to be dissolution-rate limited. In contrast, for a drug that has a high aqueous solubility, its dissolution will be rapid and the rate at which the drug crosses the gastrointestinal membrane may be the rate-limiting step termed permeability limited.
Other potential rate-limiting steps include the rate of drug release from the dosage form (this can be by design, in the case of controlled-release dosage forms), the rate at which the stomach empties the drug into the small intestine, the rate at which the drug is metabolized by enzymes in the intestinal mucosal cells during its passage through them into the mesenteric blood vessels, and the rate of metabolism of drug during its initial passage through the liver, often termed the ‘first-pass’ effect.
Physiology of the gastrointestinal tract
The gastrointestinal tract is a muscular tube, approximately 6 m in length with varying diameters. It stretches from the mouth to the anus and consists of four main anatomical areas; the oesophagus, the stomach, the small intestine and the large intestine or colon. The luminal surface of the tube is not smooth but very rough, thereby increasing the surface area for absorption.
The wall of the gastrointestinal tract is essentially similar in structure along its length, consisting of four principal histological layers (Fig. 19.3):
The majority of the gastrointestinal epithelium is covered by a layer of mucus. This is a viscoelastic translucent aqueous gel that is secreted throughout the gastrointestinal tract, acting as a protective layer and a mechanical barrier. Mucus is a constantly changing mix of many secretions and exfoliated epithelial cells. It has a large water component (~95%). Its other primary components, which are responsible for its physical and functional properties, are large glycoproteins called mucins. Mucins consist of a protein backbone approximately 800 amino acids long and oligosaccharide side chains that are typically up to 18 residues in length.
The mucous layer ranges in thickness from 5 µm to 500 µm along the length of the gastrointestinal tract, with average values of around 80 µm. The layer is thought to be continuous in the stomach and duodenum but may not be so in the rest of the small and large intestines.
Mucus is constantly being removed from the luminal surface of the gastrointestinal tract through abrasion and acidic and/or enzymatic breakdown, and it is continually replaced from beneath. Turnover time has been estimated at 4–5 hours but this may well be an underestimate and is liable to vary along the length of the tract.
Oesophagus
The mouth is the point of entry for most drugs (so-called peroral – via the mouth – administration). At this point contact with the oral mucosa is usually brief. Linking the oral cavity to the stomach is the oesophagus. The oesophagus is composed of a thick muscular layer approximately 250 mm long and 20 mm in diameter. It joins the stomach at the gastrooesophageal junction, or cardiac orifice, as it is sometimes known.
The oesophagus, apart from the lowest 20 mm which is similar to the gastric mucosa, contains a well-differentiated squamous epithelium of non-proliferative cells. Epithelial cell function is mainly protective: simple mucous glands secrete mucus into the narrow lumen to lubricate food and protect the lower part of the oesophagus from gastric acid. The pH of the oesophageal lumen is usually between 5 and 6.
Materials are moved down the oesophagus by the act of swallowing. After swallowing, a single peristaltic wave of contraction, its amplitude linked to the size of the material being swallowed, passes down the length of the oesophagus at the rate of 20–60 mm per second, speeding up as it progresses. When swallowing is repeated in quick succession, the subsequent swallows interrupt the initial peristaltic wave and only the final wave proceeds down the length of the oesophagus to the gastrointestinal junction, carrying material within the lumen with it. Secondary peristaltic waves occur involuntarily in response to any distension of the oesophagus and serve to move sticky lumps of material or refluxed material to the stomach. In the upright position, the transit of materials through the oesophagus is assisted by gravity. The oesophageal transit of dosage forms is extremely rapid, usually of the order of 10–14 seconds.
Stomach
The next part of the gastrointestinal tract to be encountered by both food and pharmaceuticals is the stomach. The two major functions of the stomach are:
Another, perhaps less obvious, function of the stomach is its protective role in reducing the risk of noxious agents reaching the intestine.
The stomach is the most dilated part of the gastrointestinal tract and is situated between the lower end of the oesophagus and the small intestine. Its opening to the duodenum is controlled by the pyloric sphincter. The stomach can be divided into four anatomical regions (Fig. 19.4); the fundus, the body, the antrum and the pylorus.
Fig. 19.4 The anatomy of the stomach.
The stomach has a capacity of approximately 1.5 L, although under fasting conditions it usually contains no more than 50 mL of fluid, which are mostly gastric secretions. These include:
Contrary to popular belief, very little drug absorption occurs in the stomach owing to its small surface area compared to the small intestine. The rate of gastric emptying can be a controlling factor in the onset of drug absorption from the major absorptive site, the small intestine. Gastric emptying will be discussed under gastrointestinal transit later in this chapter.
Small intestine
The small intestine is the longest (4–5 m) and most convoluted part of the gastrointestinal tract, extending from the pyloric sphincter of the stomach to the ileocaecal junction where it joins the large intestine. It is approximately 25 to 30 mm in diameter. Its main functions are:
The small intestine is divided into the duodenum, which is 200–300 mm in length, the jejunum, which is approximately 2 m in length, and the ileum, which is approximately 3 m in length.
The wall of the small intestine has a rich network of both blood and lymphatic vessels. The gastrointestinal circulation is the largest systemic regional vasculature and nearly a third of the cardiac output flows through the gastrointestinal viscera. The blood vessels of the small intestine receive blood from the superior mesenteric artery via branched arterioles. The blood leaving the small intestine flows into the hepatic portal vein that carries it via the liver to the systemic circulation. Drugs that are metabolized by the liver are degraded before they reach the systemic circulation; this is termed hepatic presystemic clearance or first-pass metabolism.
The wall of the small intestine also contains lacteals, which contain lymph and are part of the lymphatic system. The lymphatic system is important in the absorption of fats from the gastrointestinal tract. In the ileum there are areas of aggregated lymphoid tissue close to the epithelial surface which are known as Peyer’s patches (named after the 17th century Swiss anatomist Johann Peyer). These cells play a key role in the immune response as they transport macromolecules and are involved in antigen uptake.
The surface area of the small intestine is increased enormously, by about 600 times that of a simple cylinder, to approximately 200 m2 in an adult, by several adaptations which make the small intestine such a good absorption site:
• Villi – these have been described as finger-like projections into the lumen (approximately 0.5–1.5 mm in length and 0.1 mm in diameter). They are well supplied with blood vessels. Each villus contains an arteriole, a venule and a blind-ending lymphatic vessel (lacteal). The structure of a villus is shown in Figure 19.5.
Fig. 19.5 Structure of a villus.
The luminal pH of the small intestine increases to between 6 and 7.5. Sources of secretions that produce these pH values in the small intestine are:
Colon
The colon is the final major part of the gastrointestinal tract. It stretches from the ileocaecal junction to the anus and makes up approximately the last 1.5 m of the 6 m of the gastrointestinal tract. It is composed of the caecum (~85 mm in length), the ascending colon (~200 mm), the hepatic flexure, the transverse colon (usually greater than 450 mm), the splenic flexure, the descending colon (~300 mm), the sigmoid colon (~400 mm) and the rectum, as shown in Figure 19.6. The ascending and descending colons are relatively fixed, as they are attached via the flexures and the caecum. The transverse and sigmoid colons are much more flexible.
Fig. 19.6 The anatomy of the colon.
The colon, unlike the small intestine, has no specialized villi. However, the microvilli of the absorptive epithelial cells, the presence of crypts and the irregularly folded mucosae serve to increase the surface area of the colon by 10–15 times that of a simple cylinder. The surface area nevertheless remains approximately l/30th that of the small intestine.
The main functions of the colon are:
The colon is permanently colonized by an extensive number (about 1012 per gram of contents) and variety of bacteria. This large bacterial mass is capable of several metabolic reactions, including hydrolysis of fatty acid esters and the reduction of inactive conjugated drugs to their active form. The bacteria rely upon undigested polysaccharides in the diet and the carbohydrate components of secretions such as mucus for their carbon and energy sources. They degrade the polysaccharides to produce short-chain fatty acids (acetic, proprionic and butyric acids), which lower the luminal pH, and the gases hydrogen, carbon dioxide and methane. Thus, the pH of the caecum is around 6–6.5. This increases to around 7–7.5 towards the distal parts of the colon.
Recently there has been much interest in the exploitation of the enzymes produced by these bacteria with respect to targeted drug delivery to this region of the gastrointestinal tract.
Transit of pharmaceuticals in the gastrointestinal tract
As the oral route is the one by which the majority of pharmaceuticals are administered, it is important to know how these materials behave during their passage through the gastrointestinal tract. It is known that the small intestine is the major site of drug absorption, and thus the time a drug is present in this part of the gastrointestinal tract is extremely important. If sustained- or controlled-release drug delivery systems are being designed, it is important to consider factors that will affect their behaviour and, in particular, their transit times through certain regions of the gastrointestinal tract.
In general, most dosage forms, when taken in an upright position, transit through the oesophagus quickly, usually in less than 15 seconds. Transit through the oesophagus is dependent upon both the dosage form and posture.
Tablets/capsules taken in the supine (lying down) position, especially if taken without water, are liable to lodge in the oesophagus. Adhesion to the oesophageal wall can occur as a result of partial dehydration at the site of contact and the formation of a gel between the formulation and the oesophagus. The chances of adhesion will depend on the shape, size and type of formulation. Transit of liquids, for example, has always been observed to be rapid, and in general faster than that of solids. A delay in reaching the stomach may well delay a drug’s onset of action or cause damage or irritation to the oesophageal wall, e.g. potassium chloride tablets.
Gastric emptying
[/not-level-membership-for-basic-science-category]
