Gastroenterology and Nutrition
Development of the Gastrointestinal System
Folding occurs along the embryo in a cephalocaudal progression that leads to the incorporation of some of the endodermal-lined yolk sac into the embryo, which in turn results in the creation of the primitive gut. The primitive gut is composed of the foregut, midgut, and hindgut. The foregut is most cephalic and will become the esophagus and stomach. The midgut becomes the small intestines, and the hindgut becomes the colon ( Fig. 10-1).
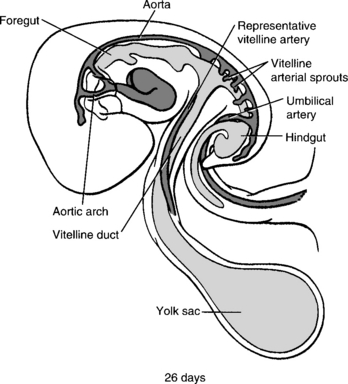
Figure 10-1 The foregut, midgut, and hindgut of the primitive gut tube are formed by the combined action of differential growth and lateral and cephalocaudal folding. The foregut and hindgut are blind-ending tubes that terminate at the buccopharyngeal and cloacal membranes, respectively. The midgut is at first completely open to the cavity of the yolk sac. (From Larsen WJ, Sherman LS, Potter SS, Scott WJ, editors. Human embryology. 3rd ed. New York: Churchill Livingstone; 2001. p. 237.)
The liver forms at about the third week of gestation as an outgrowth, known as the hepatic diverticulum or liver bud, of the endodermal epithelium of the foregut. This connection grows and narrows to form the bile duct to connect the developing liver to the foregut. A small ventral outgrowth forms that will develop into the gallbladder and connecting cystic duct. The intrauterine failure to develop a complete biliary tree can lead to extrahepatic biliary atresia of embryonic or fetal form, which occurs in 10% to 35% of all cases. 1
The pancreas develops in two separate locations as a bud from the endodermal-lined foregut. The dorsal pancreas develops from a bud on the dorsal surface opposite the developing biliary tree. The dorsal pancreatic bud is located within the dorsal mesentery and grows with a central dorsal pancreatic duct draining to the foregut through the minor papilla. The ventral pancreatic bud develops close to the developing bile duct. When the duodenum rotates to become C-shaped, the bud is rotated onto the dorsal surface along the dorsal pancreas in a position immediately below and behind it. The two developing pancreas parts grow together, and the dorsal pancreatic duct fuses with the ventral pancreas to form the main pancreatic duct (of Wirsung) draining through the major papilla into the duodenum ( Fig. 10-2).
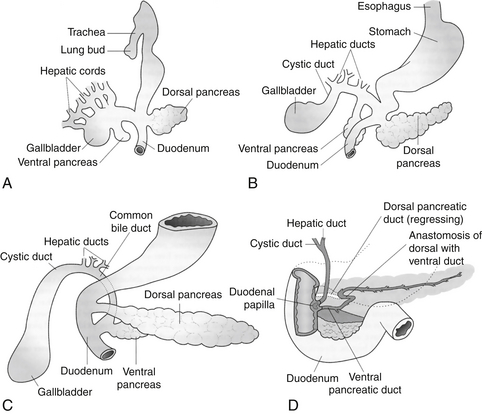
Figure 10-2 Development of the primordial pancreas from the ventral aspect. A, Fifth week of development. B, Sixth week of development. C, Seventh week of development. D, The late fetus, showing fusion of the dorsal and ventral pancreatic ducts and regression of the distal portion of the dorsal duct. (From Carlson BM. Human embryology and developmental biology. 2nd ed. St Louis: Mosby; 1999. p. 338.)
During the sixth week of gestation the small intestines and the colon herniate into the umbilical cord as a result of the rapid growth of the liver. The intestine then rotates around a central axis formed by the superior mesenteric artery. This counterclockwise rotation is completed, and the intestine migrates back into the abdominal cavity to be fixed in position. This rotation results in the colon being located anterior to the small intestines, with the cecum being located in the right lower quadrant. An interruption during this physiologic herniation and rotation will result in abnormalities. When the gut fails to return to the abdominal cavity, an omphalocele is formed. This abnormality occurs in approximately 2.5 in 10,000 births. There is a high rate of associated developmental defects, such as cardiac abnormalities, spinal defects, and chromosomal abnormalities. Malrotation is another abnormality that occurs when the midgut fails to rotate completely. Malrotation can cause the inappropriately positioned small bowel to twist on the superior mesenteric artery and lead to vascular insufficiency and volvulus. The gold standard for diagnosis of malrotation remains the upper gastrointestinal tract series that shows the duodenal C-loop crossing to the left of midline at a level equal to or greater than the pylorus. 2
The hindgut forms the most distal part of the primitive gut. It develops into the distal third of the transverse colon and the upper part of the rectal canal. Initially the urogenital system and the hindgut join together in the cloaca. The two systems separate from each other, and the rectal canal fuses with the surface to form an open pathway that will form the anus and rectum. Any abnormalities with this development can result in a continued connection, or urorectal fistula, between the urologic and gastrointestinal tracts. When the anorectal canal fails to fuse with the surface, a rectoanal atresia occurs with resulting imperforate anus. Imperforate anus occurs in 1 in 50,000 live births and has a high incidence of other associated birth defects. 3
The ENS is the nervous system that regulates intestinal smooth muscle to control gastrointestinal motility. The ENS is composed of a complex network of ganglia that function independently from the central nervous system. Although independent, the ENS can be influenced by vagal and pelvic nerves of the parasympathetic nervous system and spinal nerves. Within the ENS the interstitial cells of Cajal are the pacemaker cells and are responsible for the coordinated smooth muscle contractions within the gut.
 KEY POINTS: GASTROINTESTINAL DEVELOPMENT
KEY POINTS: GASTROINTESTINAL DEVELOPMENT
1. The most common form of transesophageal fistula is proximal esophageal atresia with a distal tracheoesophageal fistula.
2. Although most cases of biliary atresia are caused by a destructive, perinatal inflammatory process, a subset appears to be caused by a prenatal developmental abnormality of the extrahepatic biliary tree that is associated with other congenital anomalies, such as polysplenia.
3. Rotational abnormalities of pancreas development can be observed either as an annular pancreas presenting with obstruction or as ductal abnormalities presenting with pancreatitis later in childhood.
4. Delayed passage of meconium should raise consideration of both anatomic abnormalities (e.g., variants of imperforate anus) and motility disorders (e.g., Hirschsprung disease).
Meconium
When meconium is not passed by 48 hours of life, the possibility of an anatomic or neuromuscular abnormality must be considered, such as Hirschsprung disease. 4
Fetal Growth and Assessment
13. What do the terms low birth weight (LBW), very low birth weight (VLBW), and extremely low birth weight (ELBW) indicate?















Medical Problems of the Growth-Restricted Infant
17. What are the long-term risks of IUGR?
 Development: Because this group is heterogeneous, the outcome depends on perinatal events, the etiology of growth retardation, and the postnatal socioeconomic environment. In general, the asymmetric growth-retarded baby does not show significant differences in intelligence or neurologic sequelae but does demonstrate differences in school performance related to abnormalities in behavior and learning.
Development: Because this group is heterogeneous, the outcome depends on perinatal events, the etiology of growth retardation, and the postnatal socioeconomic environment. In general, the asymmetric growth-retarded baby does not show significant differences in intelligence or neurologic sequelae but does demonstrate differences in school performance related to abnormalities in behavior and learning.


Caloric Requirements
Energy, being neither created nor destroyed, conforms to classic balance relationships. Energy balance is a state of equilibrium when energy intake equals expenditure plus losses. If energy intake exceeds expenditure plus losses, the infant is in positive balance, and excess calories are stored. If energy intake is less than expenditure plus losses, the infant is in negative balance, and calories are mobilized from existing body stores. Maintenance, or basal, energy requirements are the energy needs required to cover basal metabolic rate or resting energy expenditure; total energy expenditure in infants is the sum of the energy required for basal metabolic rate, activity, thermoregulation, diet-induced thermogenesis, and growth. The energy balance equation may be stated as follows:
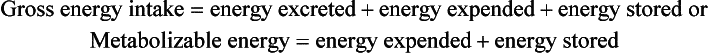
LBW infants require at least 120 cal/kg/day, partitioned to approximately 75 cal/kg/day for resting expenditure and the remainder for specific dynamic action (10 cal/kg/day), replacement of inevitable stool losses (10 cal/kg/day), and growth (25 cal/kg/day) ( Table 10-1).
TABLE 10-1
CALORIC REQUIREMENTS OF LOW-BIRTH-WEIGHT INFANTS
| REQUIREMENTS (kcal/kg/day) | |
| Resting ∗ | 50 to 75 |
| Specific dynamic action | 5% to 8% of total intake |
| Stool losses | 10% of total intake |
| Growth | 25 to 45 |
| Total † | 85 to 142 |
∗Estimate includes caloric expenditure for maintenance of basal metabolism plus activity and response to cold stress.
†Includes sum of resting and growth requirements for specific dynamic action and replacement of stool losses plus an increment of 15% to 18%.
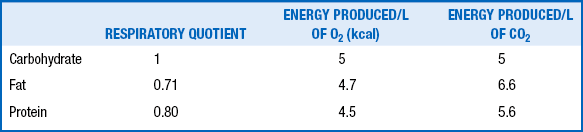
The RQ is the ratio of the volume of carbon dioxide (CO2) produced to the volume of oxygen (O2) consumed per unit of time (Vco2/VO2). This ratio varies with the type of nutrient oxidized. In addition, the energy produced varies with the type of substrate burned. Thus various substrates have different RQs, and varying proportions of different nutrients result in different energy production per liter of O2 consumption or CO2 production. The RQs and caloric equivalents of O2 and CO2 for carbohydrate, fat, and protein are shown in Table 10-2.
The energy cost of growth includes the energy used for synthesis of new tissues (e.g., absorption, metabolism, and assimilation of fat and protein) and the energy stored in these new tissues. The energy cost of growth varies with the type of tissue added during growth. The precise caloric requirements for growth are unknown. A wide range of values for energy cost of growth in neonates has been determined (1.2 to 6 kcal/g of weight gain). Separate evaluations of energy expenditure requirement for fat and protein deposition in premature newborns estimate that 1 g of protein deposition requires 7.8 kcal, and 1 g of fat requires 1.6 kcal.
Carbohydrate Requirements



23. The rate of endogenous glucose production in neonates has been estimated to range from 4 to 6 mg/kg/min. Do these values represent the ideal carbohydrate intake in neonates?
Excessive intake of carbohydrate in infant feedings may lead to delayed gastric emptying, emesis, diarrhea, and abdominal distention caused by excessive gas formation as colonic bacteria digest the extra carbohydrates. The excessive administration of intravenous glucose, at rates exceeding 13.8 mg/kg/min, may be associated with metabolic complications such as hyperglycemia, glycosuria, and osmotic diuresis. In addition, the excessive glucose metabolized is stored mainly as fat. Early overfeeding may be an important factor in later childhood and adult obesity, though more recent work suggests that genetic factors may be as important. 5
25. Why do infant formulas contain comparable amounts of lactose and glucose polymers?



The malabsorbed lactose is fermented in the colon, forming various gases such as CO2, methane, and hydrogen and short-chain fatty acids such as acetate, propionate, and butyrate. These short-chain fatty acids are absorbed in the colon, reducing energy losses in the stools and maintaining the nutrition and function of the colon. Despite these putative benefits of lactose fermentation, metabolic concerns that result from the reduced digestion and absorption of lactose in the small intestine include the following:



Protein Requirements
The amino acids that cannot be synthesized in the body are regarded as essential amino acids:









The whey-to-casein ratio of cow’s milk protein is 18:82 and that of human milk protein is 60:40. In total, most formulas contain up to 1.5 times more protein than human milk in order to approximate the protein quality of human milk. 6
32. What are the non-nutritive roles of protein in human milk?
 Whey proteins are known to be involved in the immune response (immunoglobulins), lactose synthesis (alpha-lactalbumin), and other host defenses (lactoferrin).
Whey proteins are known to be involved in the immune response (immunoglobulins), lactose synthesis (alpha-lactalbumin), and other host defenses (lactoferrin).
 Casein phosphopeptides are believed to enhance the absorption of minerals.
Casein phosphopeptides are believed to enhance the absorption of minerals.
 Casein fragments are thought to increase intestinal motility.
Casein fragments are thought to increase intestinal motility.
 Glycoproteins may promote the growth of certain beneficial bacteria.
Glycoproteins may promote the growth of certain beneficial bacteria.
33. Name the methods used for determining protein requirements.
 Factorial method (based on reference data of infant body composition)
Factorial method (based on reference data of infant body composition)
 Balance method (protein intake = protein retention − inevitable protein losses)
Balance method (protein intake = protein retention − inevitable protein losses)
 Indices of protein nutritional status (e.g., plasma albumin and transthyretin concentrations; protein intake required to maintain these indices within an acceptable range)
Indices of protein nutritional status (e.g., plasma albumin and transthyretin concentrations; protein intake required to maintain these indices within an acceptable range)





 Milk-based formulas (e.g., Similac Advance, Enfamil LIPIL, Good Start Supreme): 2.1 to 2.8 g/100 kcal or about 1.5 to 1.8 g/100 mL
Milk-based formulas (e.g., Similac Advance, Enfamil LIPIL, Good Start Supreme): 2.1 to 2.8 g/100 kcal or about 1.5 to 1.8 g/100 mL

Preterm formulas (e.g., Similac Special Care, Enfamil Premature LIPIL):

Follow-up formulas for LBW weight infants (e.g., Similac NeoSure Advance, EnfaCare LIPIL):



Lipid Requirements
42. What are the beneficial effects of lipid emulsions in a premature infant?


43. What is the percentage of calories provided by fat in human milk?
The percentage of fat calories in human milk is between 40% and 55%.
45. What structural features of fatty acids improve enteral absorption?
 Shorter-chain-length to medium-chain triglycerides are absorbed more efficiently than long-chain triglycerides.
Shorter-chain-length to medium-chain triglycerides are absorbed more efficiently than long-chain triglycerides.
 Fatty acids with double bonds are absorbed more efficiently.
Fatty acids with double bonds are absorbed more efficiently.
46. What are the energy contents of long-and medium-chain triglycerides?


47. What is the energy cost of synthesizing fat from carbohydrate?
50. What are the side effects of LCPUFA depletion?


51. What is the advantage of supplying calories as lipid rather than carbohydrate in infants with chronic lung disease?
The RQ of lipids is lower than that of carbohydrate. Therefore the use of lipid infusions should theoretically decrease CO2 production in infants with bronchopulmonary dysplasia, one of the cardinal problems of infants with chronic lung disease in the neonatal period.
52. What is the advantage of using a 20% lipid emulsion versus a 10% lipid emulsion in newborn infants?
Total Parenteral Nutrition: Monitoring and Complications
54. What is the usual distribution of nutrients in total parenteral nutrition (TPN) solutions used for neonates?
55. What are the metabolic advantages of using different regimens containing high carbohydrate (67%) and low fat (5%) or low carbohydrate (34%) and high fat (58%)?
There are none. The administration of TPN solutions containing a moderate carbohydrate (60%) to fat (32%) ratio has been shown to result in a higher nitrogen retention rate than that of the unbalanced regimens. 7
56. Hyperglycemia is a common complication observed in ELBW infants receiving parenteral nutrition. Should insulin infusions be provided routinely to these infants?
In most infants hyperglycemia is a transient problem and resolves when the rate of glucose or lipid administration is reduced. Insulin infusions have been used for infants weighing less than 1000 g who develop hyperglycemia (serum glucose level in excess of 150 mg/dL) and glycosuria during the course of parenteral nutrition, providing low glucose infusion rates (<12 mg/kg/min). In these infants insulin infusions at rates of 0.04 to 0.1 U/kg/h have been shown to improve glucose tolerance and promote weight gain, compared with infants in a control group. 89
57. The clearance of intravenous fat emulsions in neonates is improved by all the following measures except for which of the following? (A) Increasing the period of infusion from 8 to 24 hours; (B) adding a low dose of heparin to the TPN solutions (1 U/mL); (C) exposing the fat emulsions to ambient light or to phototherapy lights; (D) using 20% instead of 10% lipid emulsions.
The answer is (C). Exposure of lipid emulsions to ambient or phototherapy lights increases the formation of triglyceride hydroperoxide radicals but does not enhance lipid clearance. Lipid clearance in neonates is improved by prolonging the infusion period; by adding heparin to TPN solutions (which releases lipoprotein lipase from capillary endothelial cells); and by using 20% lipid emulsions, which contain a lower phospholipid content than 10% lipid emulsions.
58. Why do premature infants who receive prolonged courses of parenteral nutrition develop osteopenia resulting in pathologic bone fractures?
59. Which of the trace elements in TPN solutions can be potentially toxic for patients with cholestatic liver disease?
Enteral Nutrition
Lactose is the major source of carbohydrate in human milk and in formulas for term infants. The preterm formulas contain a mixture of lactose and glucose polymers to compensate for the developmental lag and lower concentration of lactase in the intestinal mucosa. Lactose, however, remains important both in calcium absorption and as a prebiotic. Glycosidase enzymes involved in the digestion of glucose polymers are active in preterm infants.
Soy formulas are recommended for the following:



69. Why are early minimal enteral feedings recommended for preterm infants receiving parenteral nutrition?
Gastrointestinal hormones such as gastrin, enteroglucagon, and pancreatic polypeptide may have a trophic effect on the gut. Postnatal surges of these hormones occur in preterm infants receiving minimal enteral feedings. Minimal enteral feeding has also been reported to produce more mature small intestinal motor activity patterns in preterm infants. Thus early minimal enteral feedings given along with parenteral nutrition may improve subsequent enteral feeding tolerance and may shorten the time to achieve full enteral intake. Furthermore, enteral feedings stimulate the enterohepatic circulation and are known to lessen parenteral nutrition–associated liver disease. The most recent Cochrane Review, however, suggests that the evidence for this effect is unclear, at best. 10
70. What are the reported advantages of feeding human milk to preterm infants over the commercially available infant formulas?



71. Does human milk completely meet the nutritional requirements of VLBW preterm infants (birth weight below 1500 g)?
Breastfeeding















In studies of AGA gavage-fed infants, there was significantly lower energy expenditure in the infants fed human milk compared with those fed formula. 11
76. A mother has breastfed her 5-week-old infant exclusively. She now calls with the concern that she has recently noticed a burning pain in her nipple during breastfeeding. You examine the mother and note some erythema of her areola. You diagnose a fissure and advise her to use dry heat and a few drops of milk on her areola after breastfeeding. She calls back in a few days to report that the pain is increasing. What other diagnosis should you consider?
77. A mother calls you and explains that she is worried because her 4-day-old baby is not receiving enough breast milk. How do you assess whether a newborn is receiving sufficient amounts of breast milk during the first week after birth?
78. You see a 5-day-old male infant in the office for a routine check after early hospital discharge. The mother reports no particular problems; he is much easier to manage than she thought a newborn would be. She is breastfeeding every 3 hours but lets him sleep at night (last night he slept for 6 hours). About once a day she notes that he has dark yellow urine in his diaper. He had a dark-green, tarry stool yesterday. The mother thinks her milk has “come in,” but she acknowledges no signs of engorgement. You examine the infant and note jaundice to the level of the umbilicus and dry skin but moist mucous membranes. He is responsive and alert. You examine the mother and note that her breasts are moderately engorged. The infant’s body weight is 11 ounces below his birth weight of 7 pounds, 8 ounces. You check his serum bilirubin concentration, which is 11 mg/dL. There is no blood group incompatibility. How would you manage this case, and what would you advise the mother?
80. Breastfeeding a premature infant can be a challenge. How do you advance from tube-feeding to breastfeeding in a premature infant?
Note the sucking and swallowing ability of the infant. Parental skills, infant feeding cues, and timing of feedings should also be considered. Begin one breastfeeding in place of a tube feeding or in addition to the tube feeding. If the latch-on is good and clinical signs of sucking, swallowing, and some drooling of milk are noted, then continue the process each day. Withdrawing milk from an indwelling feeding tube to assess milk intake from breastfeeding will not yield accurate results because gastric emptying from the stomach occurs rapidly after a human milk feeding. Furthermore, clinical signs of feeding activity and maternal assessment of breast emptying are inexact measures of milk intake and may not reflect small amounts consumed. Weighing the infant before and after breastfeeding is the most accurate way to assess milk intake. 12
Postpartum weight loss and uterine involution may be more rapid with breastfeeding. The postpartum amenorrhea during lactation is an acknowledged method of child spacing, especially for 4 to 6 months. This technique is most reliable if breastfeeding is practiced around the clock. Several reports now suggest that women who breastfed their infants had a decreased incidence of premenopausal breast cancer and ovarian cancer. Women who breastfed their infants also may have a decreased incidence of osteoporosis. 13
Vitamins and Trace Nutrients
82. A 2-month-old preterm infant (with an estimated gestational age of 26 weeks) develops osteopenia of prematurity and fractures of both humeri. The infant is receiving 400 units of vitamin D daily. Should the dose of vitamin D be increased?
No. Contrary to an earlier theory, osteopenia of prematurity results primarily from inadequate intake of mineral substrate (calcium and phosphorus) and not vitamin D. High doses of vitamin D do not appear to aid in the prevention or treatment of osteopenia of prematurity. Infants born prematurely are at risk for developing osteopenia because of limited accretion of bone mass in utero (fetal accretion rates for calcium and phosphorous range from 92 to 119 mg/kg/day and 59 to 74 mg/kg/day, respectively). Preterm infants often cannot receive the ideal amount of calcium by way of parenteral nutrition and thus do not receive the daily goal of calcium until full enteral feedings are established. Diuretics, steroids, and physical inactivity have a negative effect on bone mineralization. To mimic fetal accretion, an enteral intake of 120 to 230 mg/kg/day of calcium and 60 to 140 mg/kg/day of phosphorus is recommended for preterm infants. This amount is provided by 150 cc/kg/day of premature infant formula or fortified breast milk.
83. A 6-week-old infant is recovering from necrotizing enterocolitis that necessitated resection of two thirds of the jejunum and placement of an ileostomy. When enteral feedings are restarted, the drainage from the ileostomy becomes excessive. The infant is growing poorly (despite an adequate caloric intake) and develops vesiculobullous and eczematous lesions around the eyes, mouth, and genitals. What mineral deficiency should be considered?





 KEY POINTS: GROWTH AND NUTRITION
KEY POINTS: GROWTH AND NUTRITION
1. The parents of IUGR infants should be aware that significant catch-up growth will occur in the first year of life but that ultimate stature may be less than that of an AGA infant.
2. Lactose malabsorption is extremely uncommon in infants unless they have had a significant insult to the intestinal mucosa (e.g., infection, short gut syndrome) or have the very rare disorder of congenital lactase deficiency.
3. The infant with an ostomy should be carefully monitored for excessive sodium losses and zinc deficiency, both of which can impair growth.
4. A critical nutritional goal for the infant with short gut syndrome is to advance enteral feeds as clinically tolerated to enable intestinal adaptation and ultimate discontinuation of TPN.
87. What is the scientific rationale for administering vitamin A to prevent bronchopulmonary dysplasia?
 Lung differentiation is regulated in part by vitamin A.
Lung differentiation is regulated in part by vitamin A.

 Bronchopulmonary dysplasia has been associated with vitamin A deficiency in VLBW preterm infants.
Bronchopulmonary dysplasia has been associated with vitamin A deficiency in VLBW preterm infants.
 Premature birth deprives the newborn infant of the supply of retinol (vitamin A).
Premature birth deprives the newborn infant of the supply of retinol (vitamin A).

88. What are the concentrations of water-soluble vitamins in mature human milk, and how do they compare with the recommended dietary allowances for healthy term infants?
TABLE 10-3
WATER-SOLUBLE VITAMINS IN MATURE HUMAN MILK
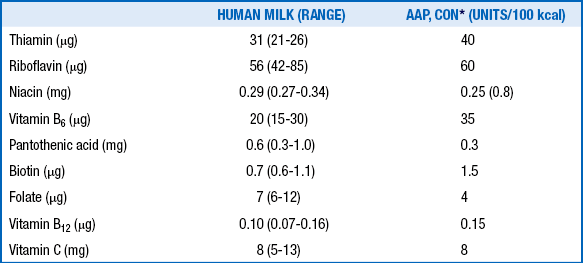
∗Committee on Nutrition of the American Academy of Pediatrics.
Adapted from Schanler RJ. Who needs water soluble vitamins? In: Tsang RC, Zlotkin SH, Nichols BL, Hansen JW, editors. Nutrition during infancy. 2nd ed. Cincinnati: Digital Educational Publishing; 1997.
Iron Requirements
89. How long can iron stores meet the needs of term, LBW, and preterm infants before supplementation is necessary?
Although the bioavailability of iron in breast milk is high (because of the presence of lactoferrin, which enhances iron absorption), the content is relatively low. Additional sources of iron are recommended for breastfed infants after 4 to 6 months of age.
Each gram of hemoglobin contains 3.4 mg of iron.
Growth-retarded infants and infants of diabetic mothers are at risk for reduced iron stores. Approximately 50% of IUGR infants and 65% of infants of diabetics have cord serum ferritin concentrations below the fifth percentile (60 ng/L). In growth-restricted infants the etiology is probably related to impaired placental transport of nutrients. The pathophysiology of low iron stores in infants of diabetic mothers is more complex. Chronic maternal hyperglycemia results in chronic fetal hyperglycemia and hyperinsulinemia, both of which increase the oxygen consumption of the fetus by approximately 30%. Chronic fetal hypoxia leads to increased erythropoietin secretion and secondary polycythemia, which in turn requires increased iron delivery. Each extra gram of hemoglobin synthesized by the fetus requires an additional 3.49 mg of elemental iron delivered by the placenta. The human placenta is not capable of upregulating placental transport to that extent, leaving the fetus of a diabetic mother dependent on its accreted iron stores to support its expanding fetal blood volume. The result is that iron is redistributed away from storage and nonstorage tissues and into the red cell mass. It does not appear that either group needs additional dietary iron postnatally, supporting the principle that the neonatal intestine avidly absorbs iron. 1617
96. What is the effect of recombinant human erythropoietin (rhEPO) on the iron needs of the premature infant?
Erythropoietin increases the need for iron by up to threefold to 6 mg/kg/day. A recent study suggested that once weekly dosing at 1200 units/Kg/dose was adequate to maintain hematocrit levels in premature neonates. Studies of erythropoietin given to sheep with varying degrees of iron sufficiency demonstrated that the degree of hemoglobin response is directly related to the iron sufficiency of the animal. In addition, some recent evidence suggests that EPO may have neuroprotective effects, including reduction of risk of retinopathy of prematurity. 1819
In pregnancies complicated by fetal iron deficiency, as indexed by a low cord serum ferritin concentration or decreased placental iron content, the expression of iron transport proteins such as the transferrin receptor is increased on the apical (maternal-facing) membrane of the syncytiotrophoblast. Studies have shown that this upregulation is most likely in response to the iron status of the syncytiotrophoblast. This upregulation is achieved by intracellular iron regulatory proteins that bind transferrin receptor mRNA, stabilizing it to produce more copies of the receptor and leading to greater iron transport. Thus the fetus appears to regulate its own iron accretion. A similar system has been described for the transport of certain amino acids by the placenta. 2021
Gastroesophageal Reflux
Gastroesophageal reflux is seen in up to 50% of infants with recurrent emesis.
During infancy GER is very common because of the immaturity of the lower esophageal sphincter. Recurrent vomiting is the most common manifestation of reflux in this age group and is usually effortless. It is clinically evident in 50% of infants in the first 3 months of life. Only 5% to 10% of children have reflux at the age of 1 year. There is gradual resolution of vomiting by the age of 1 to 2 years. If regurgitation has not resolved by 24 months of age, further evaluation is recommended.



A key point is differentiating GERD from the causes of recurrent or persistent vomiting:


 Malrotation with intermittent volvulus ( Fig. 10-3)
Malrotation with intermittent volvulus ( Fig. 10-3)
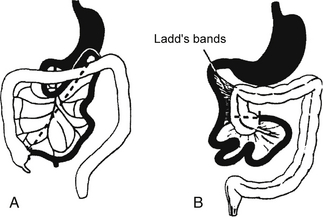
Figure 10-3 A, The cecum descends into the right lower quadrant. Note the normal broadness of the small bowel mesentery (dashed line). B, In malrotation the duodenal loop lacks 90 degrees of its normal 270-degree rotation such that the duodenojejunal flexure does not cross midline, and the cecocolic loop lacks 180 degrees of its normal rotation.








 Eosinophilic esophagitis or gastroenteritis
Eosinophilic esophagitis or gastroenteritis



 Neurologic problems: Increased intracranial pressure
Neurologic problems: Increased intracranial pressure









 Hereditary fructose intolerance
Hereditary fructose intolerance

 Congenital adrenal hyperplasia
Congenital adrenal hyperplasia








TABLE 10-4
DIAGNOSTIC TESTS FOR THE EVALUATION OF GASTROESOPHAGEAL REFLUX
| TEST | ADVANTAGES | DISADVANTAGES |
| Upper gastrointestinal tract series (UGIS) | Evaluates for structural abdnormalities | Short duration (<1 h) that may miss a reflux episode Manipulation during study can produce a reflux event in an otherwise healthy infant Low sensitivity and specificity for GER |
| Gastroesophageal scintigraphy | Evaluates for gastric emptying time, reflux, and aspiration in lungs Longer study period than UGIS (1 to 2 h) to better assess frequency and degree of aspiration |
Study limited to time period of feeding bolus May underestimate frequency of daily events if volume or composition of feed is altered for the study Lacks standardized technique and age-specific normative data. |
| 24-hour pH probe monitoring | Evaluates pH changes over extended period of time, ideally 24 h, to better assess frequency of reflux Can correlate with symptom scales No activity restriction during testing |
Must be on bolus feeds and off acid suppression medications for 72 h for an accurate study Invasive Does not evaluate for alkaline reflux |
| Impedance probe | Evaluates for pH changes as well as impedance Extended study, ideally 24 h, as with pH probe Can detect non–acid reflux episodes Can correlate with symptom scales No activity restriction during testing |
Must be on bolus feeds and off acid suppression medications for 72 h for an accurate study. Invasive |
| Esophago-gastroduodenoscopy | Evaluates gross and histologic mucosal changes of the esophagus, stomach, and duodenum | Most invasive test; often requires anesthesia |














Eosinophilic Gastrointestinal Disorders
This disease process can present as failure to thrive, diarrhea, malabsorption, regurgitation and irritability (identical to GER), and colitis. In neonates the protein found in cow’s milk or soy protein may be the offending antigen for the inflammatory process. When this occurs, the process is called dietary protein–induced colitis, milk protein colitis, enteritis, and so forth. A common presentation is an infant who has a history of irritability, diarrhea with mucus and blood, poor weight gain or failure to thrive, and some degree of anemia. It is the most common cause of bloody stools in the first year of life. The second common presentation in neonates is that of reflux that does not respond to therapeutic management. In this clinical picture the neonate has symptoms of GER and irritability that do not improve as expected despite appropriate medical and nonmedical therapeutic interventions.
 KEY POINTS: INFLAMMATORY GASTROINTESTINAL DISORDERS
KEY POINTS: INFLAMMATORY GASTROINTESTINAL DISORDERS
1. Features of gastroesophageal reflux and allergic esophagitis may be clinically similar; the latter should be considered when standard antireflux therapy is ineffective.
2. Allergic colitis is a relatively common cause of rectal bleeding and bloody stools in the otherwise healthy-appearing infant.
3. The presence of Clostridium difficile toxin in stools is a common finding in healthy infants.
4. Hirschsprung disease can be associated with an inflammatory enterocolitis that can be quite severe and life-threatening.
Malabsorption
Almost any process damaging the mucosa of the small intestine can result in malabsorption of lactose owing to secondary lactase deficiency. The most common cause of secondary lactase deficiency is mucosal damage resulting from infection (e.g., postviral damage). Lactase enzyme has the lowest activity of any brush border disaccharidases and is localized at the tip of the villus, thus making it most vulnerable to brush border injury at the time of infection. It is the first enzyme to be affected and the last one to recover after mucosal damage.
If other causes of diarrhea, such as that resulting from an infectious source, are excluded, congenital glucose-galactose malabsorption is high on the differential diagnosis because the carbohydrate in Pedialyte is dextrose (a form of glucose monohydrate). Glucose or galactose malabsorption is an autosomal recessive disease caused by a missense mutation in the SGLT1 gene resulting in a complete loss of the Na-dependent glucose transporter, which mediates glucose absorption in the brush border of the intestine. The treatment is elimination of glucose and galactose from the diet with resolution of the diarrhea.
122. A 21-year-old pregnant woman was diagnosed with polyhydramnios. A prenatal ultrasound study demonstrated distended loops of small intestine. The baby was delivered at 33 weeks’ gestation by cesarean section, and at the time of delivery the amniotic fluid was noted to contain yellow-green stool. On day 2 of life the infant developed a hypochloremic metabolic alkalosis and loose stools. A stool sample contained high concentrations of chloride. What is the most likely diagnosis in this case?
The following features of this case suggest a diagnosis of congenital chloride diarrhea:
 High concentrations of fecal chloride (exceeding the sum of sodium and potassium)
High concentrations of fecal chloride (exceeding the sum of sodium and potassium)










In 1957 Bishop and Koop described the technique of resection of the dilated ileal segment and proximal end-to-distal side ileal anastomosis with distal ostomy, also known as the Bishop–Koop ileostomy. This procedure minimizes contamination, allows for anastomosis between appropriately sized bowel segments, provides access to the distal bowel for decompression and irrigation, and allows for bedside closure of the stoma once the obstruction has resolved. Various irrigating solutions have been used, including normal saline, Gastrografin, hydrogen peroxide, and 2% to 4% solutions of N-acetylcysteine. Figures 10-4 and 10-5 illustrate the typical findings of meconium ileus with obstruction and the Bishop–Koop ileostomy technique.
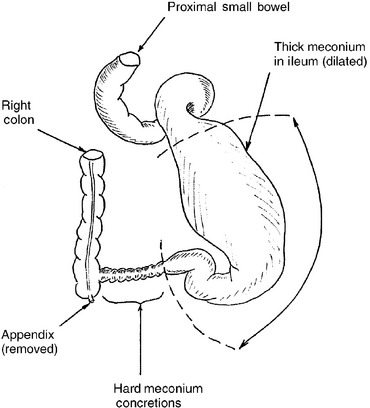
Figure 10-4 Typical appearance, at the time of operative exploration, of a neonate with meconium ileus that failed nonoperative management. Note the dilated ileum proximal to the point of obstruction. Thick, viscous meconium is found in the dilated segment, and hard meconium pellets are found in the segment of ileum that is causing the complete mechanical obstruction. For this degree of disease the massively dilated bowel must be resected.
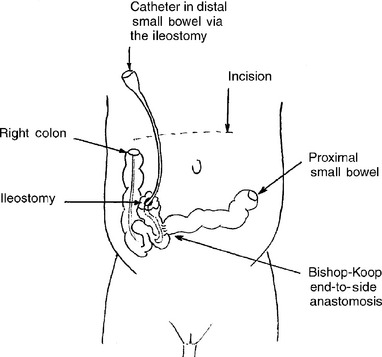
Figure 10-5 Creation of the Bishop–Koop ileostomy after segmental ileal resection for management of meconium ileus. Note that the distal loop of bowel forms the ostomy and the more proximal end forms the end-to-side anastamosis. A catheter can be placed in the ileostomy for postoperative irrigation of the distal ileum and colon to clear the remaining bowel of partially obstructing thick meconium.
125. What is the operative approach if the patient has meconium ileus with suspected intestinal perforation?
If the infant has had a perforation with peritonitis, the clinician must determine the degree of peritonitis. If perforation occurs just before delivery, meconium ascites without calcification is usually present, whereas if it occurs several weeks or months before delivery, calcification and dense adhesions will develop. Occasionally, a fibrous wall forms around the meconium, leading to a pseudocyst, often referred to as giant cystic meconium peritonitis. Operative repair of the obstruction can be difficult because the adhesions are usually quite vascular, carrying a high risk of intraoperative mortality. The goal is relief of the obstruction and, if possible, restoration of bowel continuity or creation of a temporary Bishop–Koop ileostomy. Ostomy closure is usually safe 6 to 8 weeks later. TPN may be necessary if inadequate bowel length is available for feeding.
 KEY POINTS: INHERITED DISORDERS OF ABSORPTION AND MOTILITY
KEY POINTS: INHERITED DISORDERS OF ABSORPTION AND MOTILITY
1. An infant who continues to produce significant amounts of stool in the absence of oral intake should be evaluated for an inherited or acquired disease of secretory diarrhea.
2. Congenital disorders of carbohydrate malabsorption that cause significant diarrhea in the infant are extremely rare.
3. The diagnosis of cystic fibrosis should be considered in any infant with meconium ileus.
4. An abnormal stooling pattern in an infant with Down syndrome should raise the possibility of Hirschsprung disease.
126. In newborns what are the three most common gastrointestinal manifestations of cystic fibrosis?
 Meconium ileus is the earliest clinical manifestation of cystic fibrosis. Between 10% and 20% of patients with cystic fibrosis develop intestinal obstruction in utero during the last trimester of development. Abdominal distention is marked, with no passage of meconium. The obstruction is secondary to a mass of extremely thick, tenacious meconium, which adheres to the wall of the distal small bowel and impacts the lumen.
Meconium ileus is the earliest clinical manifestation of cystic fibrosis. Between 10% and 20% of patients with cystic fibrosis develop intestinal obstruction in utero during the last trimester of development. Abdominal distention is marked, with no passage of meconium. The obstruction is secondary to a mass of extremely thick, tenacious meconium, which adheres to the wall of the distal small bowel and impacts the lumen.


Short Gut Syndrome
The healthy newborn intestine is approximately 200 to 300 cm in length. Traditionally, the definition of short gut was less then 75 cm of total small bowel, thus an approximate loss of about half of the small bowel. Short gut syndrome is presently defined on the basis of the constellation of symptoms, signs, and metabolic and nutritional alterations associated with a physiologically significant loss of gut. The overall prognosis would depend on what specific sections were lost and the overall remaining bowel function, including the following:
 Amount of remaining small intestine
Amount of remaining small intestine
 Whether it is proximal (jejunal) or distal (ileal)
Whether it is proximal (jejunal) or distal (ileal)
 Whether the ileocecal valve is resected
Whether the ileocecal valve is resected



128. What are the mechanisms responsible for diarrhea in short gut syndrome?
 Decreased absorptive surface area
Decreased absorptive surface area


 Hypersecretion and impaired regulation of gut motility
Hypersecretion and impaired regulation of gut motility

 Steatorrhea: secondary to decreased availability of bile salts necessary for fat absorption
Steatorrhea: secondary to decreased availability of bile salts necessary for fat absorption

 Colonic resection: The colon is where most fluid reabsorption occurs.
Colonic resection: The colon is where most fluid reabsorption occurs.
129. What problems may be associated with enteral feeding in patients with short gut syndrome?









Imperforate Anus
Imperforate anus is often diagnosed in the nursery as the nursing staff attempts to obtain a rectal temperature from the neonate or during the newborn examination. Rectal atresia might be missed during the examination because the anal opening can appear normal. However, failure to pass meconium and increasing abdominal distention should warrant further evaluation.


Hirschsprung Disease
Approximately 10% of children have a family history, especially with longer-segment Hirschsprung disease. A higher incidence occurs in children with Down syndrome and other genetic abnormalities. Recent studies indicate the presence of mutations in the RET proto-oncogene in 17% to 38% of children with short-segment disease and in 70% to 80% of those with long-segment disease. Additional genes linked to the RET activation pathway and other mechanisms have now been identified. 22








Gastrointestinal Hemorrhage
139. How does one determine whether swallowed maternal blood is the cause for gastrointestinal bleeding in the neonate?
TABLE 10-5
SOURCES OF NEONATAL GASTROINTESTINAL BLEEDING
| HEMATEMESIS/MELENA | HEMATOCHEZIA |
| Swallowed maternal blood | Swallowed maternal blood |
| Gastritis or stress ulcers | Dietary protein intolerance |
| Duplication cyst | Duplication cyst |
| Coagulopathy: Vitamin K deficiency or DIC | Coagulopathy: Vitamin K deficiency or DIC |
| Maternal NSAID use | Maternal NSAID use |
| Maternal idiopathic thrombocytopenic purpura | Maternal idiopathic thrombocytopenic purpura |
| Vascular malformation | Colitis: infectious, NEC, Hirschsprung disease with entercolitis |
| Hemophilia | Hemophilia |
| Esophagitis | Rectal fissure, tear, or hemorrhoids |
141. What are some of the risk factors and clinical features that help distinguish necrotizing enterocolitis (NEC) from other causes of gastrointestinal bleeding in the neonate?
NEC tends to be more common in premature infants and often occurs in those who have experienced some type of perinatal stress, such as hypoxia, need for mechanical ventilation, or sepsis. The addition of gross or occult blood in stools, feeding intolerance, abdominal distention or discoloration, bilious emesis, and lethargy should all lead to the consideration of NEC in the differential diagnosis.
Necrotizing Enterocolitis
Breast milk may reduce the risk of NEC. Breast milk offers many nutritive advantages in addition to protective immunologic substances. Milk macrophages and phagocytes, immunoglobulins A and G, and immunocompetent T and B lymphocytes may offer a protective advantage to the mucosa. These components potentiate the effect of the complement components C3 and C4, lysozyme, lactoferrin, and secretory immunoglobulin A. Furthermore, breast milk contains hormones (e.g., thyroid, thyroid-stimulating hormone, prolactin, steroid), enzymes (e.g., amylase, lipase), and growth factors (endothelial growth factor). Breast milk also favors the growth of Lactobacillus bifidus and promotes the development of a healthy gut microbiome. 23
Large-volume milk feedings that are increased too rapidly during the feeding schedule may place undue stress on a previously injured or immature intestine. Two studies have shown that volume increments in excess of 20 to 25 mL/kg/day have been associated with NEC, whereas another two studies have shown the safety of 30 to 35 mL/kg/day increments. The evidence therefore is unclear as to the role of rapid feeding advancement in the development of NEC. Volume increments probably should not be more than 20 to 35 mL/kg/day and should be advanced on the basis of the clinical examination, physiologic stability, and feeding tolerance. 24
Recent prospective randomized trials have looked at the effects of probiotics and their ability to prevent NEC. Studies have shown that the use of probiotics decreases the incidence of NEC but not the mortality rates among those patients that do develop NEC. However, a higher incidence of sepsis was reported in those infants receiving probiotics. Thus probiotics can be considered but should be used with caution, based on current data. To date, no large-scale trial of probiotics has been successfully carried out, and there are currently many different bacterial components in available probiotics. No probiotic is currently approved by the Food and Drug Administration for neonatal use. 25
NEC is a common cause of systemic inflammatory response syndrome in neonates. Based on systemic signs, intestinal signs, and radiologic signs, staging of NEC is performed as shown in Table 10-6.
TABLE 10-6
STAGING OF NECROTIZING ENTEROCOLITIS
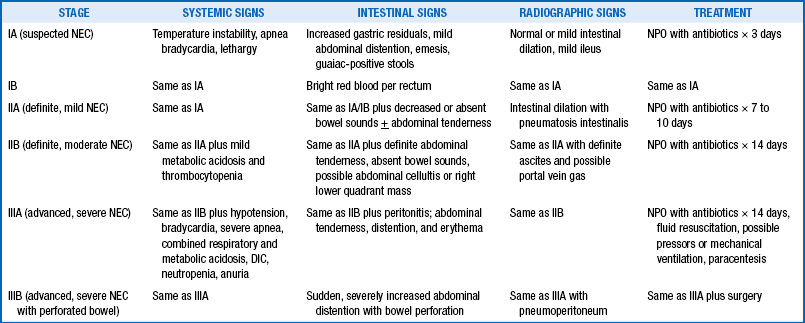
DIC, Disseminated intravascular coagulopathy; NEC, necrotizing enterocolitis; NPO, nothing by mouth.
151. A 1000-g infant, born at 28 weeks’ gestation, had an initial course characterized by respiratory distress syndrome and suspected sepsis. He was initially treated with surfactant and mechanical ventilation. The antibiotics were stopped after 3 days because the blood culture results were negative. On day 5 he was placed on nasal continuous positive airway pressure until day 18. He began enteral gavage feeds on day 5, at 20-cc/kg/day increments, and finally achieved “full feeds” (150 cc/kg/day) by day 20. He then developed an increased frequency of apnea and bradycardia associated with temperature instability. The gavage feeds were held because of increasing gastric residuals, presence of blood in stools, and abdominal distention. What should be done next?
152. One day after beginning appropriate management, the same infant (see Question 151) develops persistent abdominal distention, right lower quadrant tenderness, and diminished bowel sounds. The abdominal radiographs are shown in Figures 10-6 and 10-7. How do you interpret these signs? What should be done next?
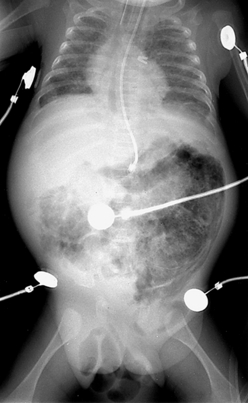
Figure 10-6 Abdominal radiograph 1 day after treatment for necrotizing enterocolitis stage IB. (Courtesy Dr. Jack Sty, Department of Pediatric Radiology, Children’s Hospital of Wisconsin, Medical College of Wisconsin, Milwaukee, Wisconsin.)
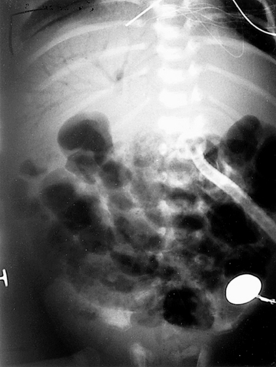
Figure 10-7 Abdominal radiograph of the infant shown in Figure 10-6 taken 8 hours later. (Courtesy Dr. Jack Sty, Department of Pediatric Radiology, Children’s Hospital of Wisconsin, Medical College of Wisconsin, Milwaukee, Wisconsin.)
At this stage the infant is showing definite signs of NEC (stage II), as demonstrated by the failure to recover from stage IB and worsening intestinal signs, such as diminished bowel sounds, guarding, and abdominal distention and tenderness. These clinical findings may herald the beginning of dilated viscus, submucosal or subserosal dissection of air, and peritonitis. Such an infant should be regarded as having NEC and is moderately ill. The radiograph in Figure 10-6 shows grossly dilated bowel loops and submucosal and subserosal pneumatosis intestinalis. Management based on this radiograph includes careful monitoring for worsening of clinical status (e.g., metabolic acidosis and thrombocytopenia) and a course of antibiotics for a minimum of 7 to 10 days. However, the second radiograph in Figure 10-7 shows worsening disease, as manifested by portal vein gas (within the liver). At this stage it is imperative to anticipate the possibility of intestinal perforation. Management should now include correction of hypovolemia and metabolic acidosis using colloids and sodium bicarbonate, respectively. If NEC does not progress further, 2 weeks of appropriate antibiotics would suffice, with hopes to avoid surgical intervention.
153. The infant’s condition suddenly deteriorates 24 hours later. He develops generalized abdominal tenderness and periumbilical erythema. An arterial blood gas determination shows a pH of 7.10, PCO2 of 80 mmHg, PO2 of 32 mmHg,  of 12 mEq/L, and a base deficit of 16. The blood count is remarkable for a platelet count of 22,000/µL. The abdominal radiography is shown in Figure 10-8. How do you interpret these signs? What should be the approach to the management of this infant?
of 12 mEq/L, and a base deficit of 16. The blood count is remarkable for a platelet count of 22,000/µL. The abdominal radiography is shown in Figure 10-8. How do you interpret these signs? What should be the approach to the management of this infant?
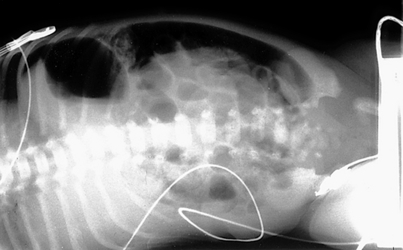
The infant at this stage has advanced NEC and is severely ill. This condition is characterized by worsening hypotension, a combined respiratory and metabolic acidosis, thrombocytopenia, and anuria. Thrombocytopenia usually represents a consumptive thrombocytopenia with or without intestinal perforation. The sudden deterioration is ominous for a bowel perforation, and progressive abdominal distention with erythema signifies worsening peritonitis and pneumoperitoneum. The lateral decubitus abdominal radiograph is remarkable for worsening pneumatosis and free air (see Figure 10-8).
154. When is surgery indicated for an infant with NEC? What are the complications of performing surgery on an infant with advanced NEC with bowel perforation?
155. A 30-week-gestation male infant had been diagnosed with stage IIA NEC and was appropriately managed medically for 10 days. He subsequently tolerated feeds poorly. The stooling pattern was reported as normal (small green stools). Different formulas and prokinetics were tried without any positive result. An abdominal x-ray revealed what was reported as a “gassy abdomen.” Treatment with antibiotics was begun again, and feedings were held for 3 days. A sepsis work-up yielded negative results at 3 days. Feedings were then resumed with an elemental formula. The same feeding-intolerance pattern prevailed. What are the diagnostic considerations in this infant?
156. A 3500-g term female infant born after an uncomplicated pregnancy was discharged home from the newborn nursery after a normal transition. She was fed exclusively with breast milk. On her seventh day of life she presented acutely with bilious emesis. The clinical examination was remarkable for a pulse rate of 180 bpm, respiratory rate of 70/min, mean blood pressure of 30 mmHg, abdominal distention, and marked tenderness with diminished bowel sounds. She passed a dark, bloody stool. Laboratory study results were notable for an arterial blood gas of pH 7.15, PCO2 level of 30 mmHg, PO2 level of 120 mmHg, and  level of 10 mEq/L. The complete blood count was remarkable for a hematocrit level of 24 and platelet count of 400,000/µL. What is the approach to management in this infant? How would you establish a diagnosis in this infant?
level of 10 mEq/L. The complete blood count was remarkable for a hematocrit level of 24 and platelet count of 400,000/µL. What is the approach to management in this infant? How would you establish a diagnosis in this infant?
The infant’s examination is consistent with an acute abdomen. She also has signs of hypovolemia and shock. She needs immediate fluid resuscitation and correction of metabolic acidosis. Because sepsis is common in the neonatal period, antibiotics are indicated after blood cultures are obtained. A nasogastric tube should be placed and the stomach decompressed. An abdominal x-ray reveals gassy distended bowel loops with air-fluid levels. An upper gastrointestinal contrast radiograph is shown in Figure 10-9. The contrast fails to flow distally, suggesting intestinal obstruction. Furthermore, the pig-tail appearance of the contrast is classic for a diagnosis of volvulus, and surgical exploration should be considered.
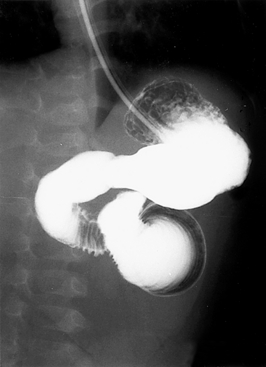
Figure 10-9 Upper gastrointestinal contrast radiograph. (Courtesy Dr. Jack Sty, Department of Pediatric Radiology, Children’s Hospital of Wisconsin, Medical College of Wisconsin, Milwaukee, Wisconsin.)
157. How do you differentiate NEC from volvulus? In what conditions is pneumatosis intestinalis seen?
Table 10-7 summarizes the features that differentiate NEC from volvulus. Apart from NEC, pneumatosis intestinalis is also seen in midgut volvulus, acute or chronic diarrhea, postoperative gastrointestinal surgery, Hirschsprung disease, short gut syndrome, mesenteric thrombosis, postcardiac catheterization, structural disease of the hindgut (colonic atresias and stricture, imperforate anus), and intestinal malignancies.
TABLE 10-7
NECROTIZING ENTEROCOLITIS DIFFERENTIATED FROM VOLVULUS
| CHARACTERISTICS | NEC | VOLVULUS |
| Preterm | 90% | 30% |
| Onset by 2 weeks | 90% | 60% |
| Male:female ratio | 1:1 | 2:1 |
| Associated anomalies | Rare | 25% to 40% |
| Bilious emesis | Unusual | 75% |
| Grossly bloody stools | Common | Less common |
| Pneumatosis intestinalis | 90% | 2% |
| Marked proximal obstruction | Rare | Common |
| Thrombocytopenia without DIC | Common | Rare |
DIC, Disseminated intravascular coagulopathy; NEC, necrotizing enterocolitis.
Modified from Kliegman R. Necrotizing enterocolitis: differential diagnosis and management. In: Polin RA, Yoder MC, Burg FD, editors. Workbook in practical neonatology. 2nd ed. Philadelphia: Saunders; 1993. p. 449–70.
Neonatal Hepatitis
158. What are the common causes of neonatal liver failure, and what are the diagnostic tests for each?
TABLE 10-8
NEONATAL CAUSES OF ACUTE LIVER FAILURE AND DIAGNOSTIC TESTS
| DISEASE | DIAGNOSTIC FINDINGS |
| Galactosemia Tyrosinemia Neonatal hemochromatosis Hemophagocytic lymphohistiocytosis and congenital leukemia Sepsis, shock Giant cell hepatitis with hemolytic anemia HHV-6, Hepatitis b, adenovirus, parvovirus Mitochondrial hepatopathy Vascular malformations and congenital heart disease Maternal overdose Hypocortisolism |
Red cell galactose-1-phosphate uridyl transferase Urine succinyl acetone Elevated ferritin, extrahepatic iron deposition Bone marrow findings Positive blood cultures |
ACTH, Adrenocorticotropic hormone; HHV-6, human herpesvirus 6.
Conjugated bilirubin greater than 2 mg/dL or exceeding 15% of the total bilirubin is referred to as direct hyperbilirubinemia and is a clinical indicator of cholestatic jaundice. Unlike indirect hyperbiloirubinemia, cholestatic jaundice is always physiologically abnormal and warrants a medical evaluation. Note that biliary disease can present with or without cholestatic jaundice.
The mechanisms include the following:
 Impaired bilirubin metabolism secondary to parenchymal disease of the liver
Impaired bilirubin metabolism secondary to parenchymal disease of the liver
 Inherited disorders of bilirubin excretion
Inherited disorders of bilirubin excretion
 Mechanical obstruction to biliary flow, either intrahepatic or extrahepatic
Mechanical obstruction to biliary flow, either intrahepatic or extrahepatic
 Excessive bilirubin loads, such as may occur in massive hemolysis
Excessive bilirubin loads, such as may occur in massive hemolysis
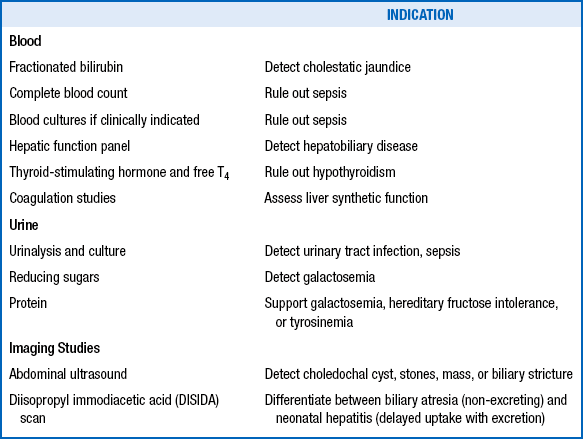
The clinician should initiate further evaluation to promptly identify clinical conditions amenable to therapy ( Table 10-9), particularly those in which any delay in treatment could be tragic (e.g., sepsis; urinary tract infection; hypothyroidism; biliary atresia; and congenital metabolic disorders requiring special diets such as galactosemia, hereditary fructose intolerance, and tyrosinemia).
TABLE 10-9
TREATABLE CAUSES OF NEONATAL CHOLESTASIS
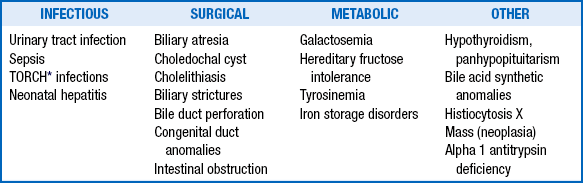
∗TORCH is an acronym for Toxoplasmosis, Other infections, Rubella, Cytomegalovirus, and Herpes simplex virus 2.
Adapted from Dhawah A, Mieli-Vergani G. Acute liver failure in neonates. Early Hum Dev 2005;81:1005–10.
As soon as cholestatic jaundice is diagnosed and sepsis ruled out, a gastroenterologist should be consulted. The tests mentioned in the previous question can be scheduled, but the clinician should not wait for the results before making the referral. In most cases a liver biopsy is needed to help make the final diagnosis. The hepatologist will also conduct a broad laboratory evaluation to make a diagnosis and initiate therapy. In addition to medical therapy, preventive therapy can be provided through genetic counseling. Time is of the essence to identify treatable causes of cholestasis and intervene early in such cases as biliary atresia for better outcomes.
Spontaneous perforation of the bile ducts is a rare occurrence but has been documented in infants between 4 and 12 weeks of age. The cause is currently unknown. It most often occurs at the point at which the cystic duct is joined to the common bile duct. Infants can present with lethargy, nonbilious vomiting, acholic stools, mild jaundice, dark urine, abdominal distention, and a mildly elevated conjugated hyperbilirubinemia. Definitive diagnosis can be made with a hepatoiminodiacetic acid scan or abdominal paracentesis.
This group of conditions is collectively known as progressive familial intrahepatic cholestasis. They typically present as neonatal cholestasis but individually have distinct clinical, laboratory, and histologic features that differentiate them ( Table 10-11).
TABLE 10-11
CHARACTERISTIC FEATURES IN PROGRESSIVE FAMILIAL INTRAHEPATIC CHOLESTASIS
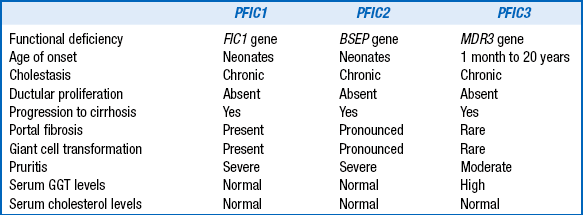
GGT, Gamma-glutamyl transferase.
Adapted from Hori T, Nguyen JH, Uemoto S. Progressive familial intrahepatic cholestasis. Hepatobiliary Pancreat Dis Int 2010;9(6): 570–8.
Bile Duct and Biliary Atresia
168. A 6-week-old healthy term breast-fed infant was noted to be jaundiced at the routine well-baby visit. She was growing well. Examination of the abdomen revealed a palpable liver (1 cm below right costal margin) and spleen (2 cm below left costal margin). Her history revealed that she had pigmented stools since birth. Total and direct bilirubin levels were 6.9 and 4.3 mg/dL, respectively. Other findings include alanine aminotransferase (ALT), 138 U/L; aspartate aminotransferase (AST), 120 U/L; alkaline phosphatase (ALK), 205 U/L; GGT, 420 U/L; albumin, 3 g/dL; and prothrombin time (PT) 13.9 sec. Calcium, phosphate, and magnesium levels were normal; complete blood count, urinalysis, and culture had normal results. What do the laboratory results suggest, and which further tests need to be performed?
 Ultrasound: This is a quick, noninvasive test useful for detecting causes of extrahepatic cholestasis (e.g., choledochal cysts, biliary stones, tumors). Finding a gallbladder on ultrasound does not rule out biliary atresia, although the absence of a gallbladder would rasie the suspicion of biliary atresia.
Ultrasound: This is a quick, noninvasive test useful for detecting causes of extrahepatic cholestasis (e.g., choledochal cysts, biliary stones, tumors). Finding a gallbladder on ultrasound does not rule out biliary atresia, although the absence of a gallbladder would rasie the suspicion of biliary atresia.

169. In the infant described in the preceding question, the ultrasound revealed hepatosplenomegaly, and no gallbladder was seen. The DISIDA scan showed normal uptake but no excretion at 24 hours. A liver biopsy specimen was obtained, which showed intrahepatic cholestasis with proliferation of the bile ducts. Is the evaluation now complete for making a definitive diagnosis?





TABLE 10-12
CAUSES OF PEDIATRIC ELEVATIONS OF GAMMA-GLUTAMYL TRANSFERASE
| DISEASE | VARIATION OF GGT |
| Extrahepatic biliary atresia Alagille syndrome Sclerosing cholangitis PFIC types I and II PFIC type III Bile acid disorders |
Increased up to 10 times upper limit Increased 3 to 20 times upper limit Increased 50 to 100 times upper limit Normal or decreased Increased Normal |
GGT, Gamma-glutamyl transferase; PFIC, progressive familial cholestasis.
173. A 10-week-old, former 34-week premature, breastfed boy was referred for evaluation of jaundice and elevated liver enzymes. His test results indicated a conjugated bilirubin, 3.8 mg/dL; ALK 650 U/L; AST, 120 U/L; ALT, 138 U/L; and GGT, 1200 U/L. During the newborn period he had mild respiratory distress syndrome, was treated for sepsis, and received TPN for 7 days. He was discharged home on breast-milk feeds at the age of 3 weeks. How should the evaluation proceed?
The laboratory results show a disproportionately elevated serum GGT and ALK as well as a high conjugated bilirubin level, all of which suggest biliary disease. However, because the clinical manifestations of neonatal cholestasis are independent of the etiology, the initial basic evaluation should be broad, as previously described in Table 10-10.
174. A careful physical examination revealed that the patient in Question 173 had a prominent forehead, small chin, and a systolic heart murmur consistent with peripheral pulmonary stenosis. The ultrasound yielded normal results. The DISIDA scan showed excretion at 24 hours. Is this sufficient for making the diagnosis of Alagille syndrome?
 Chronic cholestasis: associated with hypercholesterolemia and paucity of intralobular bile ducts
Chronic cholestasis: associated with hypercholesterolemia and paucity of intralobular bile ducts
 Congenital heart disease: most commonly peripheral pulmonic stenosis
Congenital heart disease: most commonly peripheral pulmonic stenosis
 Bone defects: commonly butterfly vertebrae
Bone defects: commonly butterfly vertebrae
 Eye findings: posterior embryotoxon
Eye findings: posterior embryotoxon
 Typical facies: frontal bossing, deep-set eyes, bulbous tip of nose, and pointed chin 26
Typical facies: frontal bossing, deep-set eyes, bulbous tip of nose, and pointed chin 26
 KEY POINTS: LIVER DISEASE
KEY POINTS: LIVER DISEASE
1. Direct hyperbilirubinemia is always abnormal.
2. The key to the evaluation of an infant with cholestatic jaundice requires early assessment for treatable causes and surgical intervention if biliary atresia is confirmed.
3. Significant hypoglycemia or coagulopathy in an infant with cholestatic jaundice may be an important sign of significant hepatocellular disease.
4. Do not forget silent urinary tract infection as an important, treatable cause of cholestatic jaundice in the infant.
175. What clinical conditions are associated with cholelithiasis?






176. What are common mistakes in the evaluation of an infant with neonatal cholestasis?





The embryonic/fetal type of biliary atresia occurs in 10% to 35% of cases:



The perinatal type of biliary atresia occurs in 65% to 90% of cases:
 Direct hyperbilirubinemia occurs at 4 to 8 weeks of age.
Direct hyperbilirubinemia occurs at 4 to 8 weeks of age.
 There is a jaundice-free period after physiologic jaundice.
There is a jaundice-free period after physiologic jaundice.
 Bile duct remnants are often seen at the time of surgery. 27
Bile duct remnants are often seen at the time of surgery. 27
179. What are the typical presenting clinical features of an infant with extrahepatic biliary atresia?
180. What are the typical radiographic findings in extrahepatic biliary atresia?
 Ultrasound: Hepatic parenchyma is often normal, and the gallbladder and common bile duct are generally not visualized. It is important to note that the gallbladder may not be visualized in healthy infants because of contraction; failure to visualize the gallbladder should not be considered evidence of biliary atresia. The main purpose of ultrasound in this setting is to rule out an obstructing choledochal cyst.
Ultrasound: Hepatic parenchyma is often normal, and the gallbladder and common bile duct are generally not visualized. It is important to note that the gallbladder may not be visualized in healthy infants because of contraction; failure to visualize the gallbladder should not be considered evidence of biliary atresia. The main purpose of ultrasound in this setting is to rule out an obstructing choledochal cyst.

181. What are the typical histopathologic findings in extrahepatic biliary atresia?
182. How do the radiologic and histopathologic findings in biliary atresia compare with those of neonatal hepatitis?
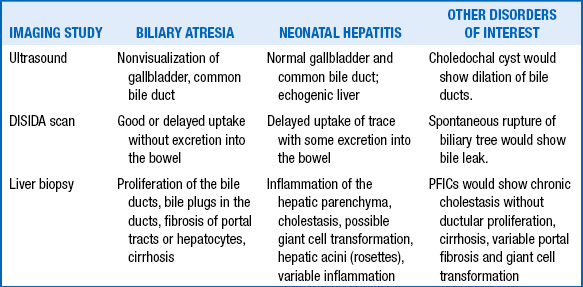
 Untreated biliary atresia is uniformly fatal within 2 years, with a median survival of 8 months. Untreated biliary atresia leads to biliary cirrhosis, portal hypertension, esophageal varices, failure to thrive, and liver failure with subsequent death from any of a number of complications.
Untreated biliary atresia is uniformly fatal within 2 years, with a median survival of 8 months. Untreated biliary atresia leads to biliary cirrhosis, portal hypertension, esophageal varices, failure to thrive, and liver failure with subsequent death from any of a number of complications.
184. What is appropriate surgical and medical therapy for biliary atresia, and how does therapy affect survival?
 Surgical: If the liver biopsy suggests biliary atresia, the infant undergoes exploratory laparotomy and an intraoperative cholangiogram. If biliary atresia is confirmed, an attempt to restore biliary drainage using the Kasai procedure (i.e., hepatic portoenterostomy) is made. During the Kasai procedure a loop of bowel is anastomosed directly to the hepatic capsule at the porta hepatis after resection of the fibrous biliary remnants. Bowel continuity is restored by formation of a Roux-en-Y intestinal anastomosis. The success of the procedure depends on the age of the infant at the time of the operation and the experience of the surgeon. Long-term survival rates may exceed 60% for infants younger than 2 months of age at the time of portoenterostomy, compared with only 25% for those older than 2 months of age. The first sign of a successful portoenterostomy is the passage of green (bile-stained) stools rather than the acholic stools seen preoperatively. A retrospective study of 81 patients in the United States noted a success rate of approximately 38% with the Kasai procedure alone.
Surgical: If the liver biopsy suggests biliary atresia, the infant undergoes exploratory laparotomy and an intraoperative cholangiogram. If biliary atresia is confirmed, an attempt to restore biliary drainage using the Kasai procedure (i.e., hepatic portoenterostomy) is made. During the Kasai procedure a loop of bowel is anastomosed directly to the hepatic capsule at the porta hepatis after resection of the fibrous biliary remnants. Bowel continuity is restored by formation of a Roux-en-Y intestinal anastomosis. The success of the procedure depends on the age of the infant at the time of the operation and the experience of the surgeon. Long-term survival rates may exceed 60% for infants younger than 2 months of age at the time of portoenterostomy, compared with only 25% for those older than 2 months of age. The first sign of a successful portoenterostomy is the passage of green (bile-stained) stools rather than the acholic stools seen preoperatively. A retrospective study of 81 patients in the United States noted a success rate of approximately 38% with the Kasai procedure alone.
 Medical: Any child with biliary atresia, regardless of the status of portoenterostomy, should be treated for chronic liver disease and its potential complications. Infants should receive fat-soluble vitamin supplementation (vitamins A, D, E, and K). Many infants require supplemental tube feedings, particularly if the portoenterostomy is unsuccessful. Good nutritional status will optimize the infant’s survival if liver transplantation becomes necessary. Medical treatment with steroids after the Kasai procedure has been found to be beneficial in many studies and is implemented in practice at various institutions. 28
Medical: Any child with biliary atresia, regardless of the status of portoenterostomy, should be treated for chronic liver disease and its potential complications. Infants should receive fat-soluble vitamin supplementation (vitamins A, D, E, and K). Many infants require supplemental tube feedings, particularly if the portoenterostomy is unsuccessful. Good nutritional status will optimize the infant’s survival if liver transplantation becomes necessary. Medical treatment with steroids after the Kasai procedure has been found to be beneficial in many studies and is implemented in practice at various institutions. 28
186. What are the therapeutic options for children who do not undergo portoenterostomy or in whom drainage is not achieved?
Choledochal cysts are much less common than biliary atresia, with estimates of incidence ranging from 1 in 13,000 to 1 in 2,000,000 live births. Girls are affected four times more frequently than boys. The classic triad of abdominal pain, mass, and jaundice occurs in fewer than 20% of cases. Choledochal cysts may present as jaundice, mass, vomiting, fever, and even pancreatitis. Fewer than half of choledochal cysts present in infancy.





Abdominal Masses
More than half of all abdominal masses in the neonate arise from the urinary tract.
191. List the two most common causes of abdominal masses of urologic origin in the neonate.


192. A pregnant woman has an antenatal ultrasound scan that reveals an intraabdominal mass in the fetus. Are any special arrangements necessary for the timing and mode of delivery?
193. How do the location and other physical examination characteristics of the common abdominal masses in newborn infants provide clues for their identification?
Physical examination may significantly narrow the diagnostic possibilities, even if it does not provide an absolute answer (Table 10-14). Of note:
TABLE 10-14
COMMON ABDOMINAL MASSES IN NEONATES
| MASS LOCATION | EXAMPLES | CHARACTERISTICS |
| Lateral | Renal cysts, hydronephrosis Renal tumor Neuroblastoma |
Smooth, moderate mobility, transilluminates
Smooth, minimally mobile, does not transilluminate |
| Midabdominal | Mesenteric cyst Gastrointestinal duplication cyst Ovarian cyst |
Smooth, mobile, transilluminates Smooth, mobile, does not transilluminate, associated with obstruction Smooth, mobile, transilluminates |
| Upper abdominal | Hepatic tumors Choledochal cyst |
Hard, immobile, do not transilluminate Smooth, immobile, does not transilluminate, associated with jaundice |
| Lower abdominal | Hydrometrocolpos
Urachal cyst |
Smooth, immobile, does not transilluminate, associated with imperforate hymen Smooth, fixed to abdominal wall, extends to umbilicus Hard, fixed, does not transilluminate, associated with external sacral component |



1Petersen C. Biliary atresia: the animal models. Semin Pediatr Surg 2012;21(3):185–91.
2Martin V, Shaw-Smith C. Review of genetic factors in intestinal malrotation. Pediatr Surg Int 2010;26(8):769–81.
3Juang D, Snyder CL. Neonatal bowel obstruction. Surg Clin North Am 2012;92(3):685–711.
4Kenny SE, Tam PK, Garcia-Barcelo M. Hirschsprung’s disease. Semin Pediatr Surg 2010;19(3):194–200.
5Zhao J, Grant SF. Genetics of childhood obesity. J Obes 2011;2011:845148.
6Martinez JA, Ballew MP. Infant formulas. Pediatr Rev 2011;32:179–89.
7Nose O, Tipton JR, Ament ME, et al. Effect of energy source on changes in energy expenditure, respiratory quotient and nitrogen balance during total parenteral nutrition in children. Pediatr Res 1987;21:538–41.
8Sinclair JC, Bottino M, Cowett RM. Interventions for prevention of neonatal hyperglycemia in very low birth weight infants. Cochrane Database Syst Rev. 2011 Oct 5;10:CD007615.
9Bottino M, Cowett RM, Sinclair JC. Interventions for treatment of neonatal hyperglycemia in very low birth weight infants. Cochrane Database Syst Rev. 2011 Oct 5;10:CD007453 [Review].
10Bombell S, McGuire W. Early trophic feeding for very low birth weight infants. Cochrane Database Syst Rev 2009;3:CD000504.
11Lubetzky R, Vaisman N, Mimouni F, et al. Energy expenditure in human milk– versus formula-fed preterm infants. J Pediatr 2003;143:750–3.
12Funkquist EL, Tuvemo T, Jonsson B, et al. Influence of test weighing before/after nursing on breastfeeding in preterm infants. Adv Neonatal Care 2010;10(1):33–9.
13Labbok MH: Health sequelae of breastfeeding for the mother. Clin Perinatol 1999;26:491–503.
14Adapted from Atkinson SA, Zlotkin S. Recognizing deficiencies and excesses of zinc, copper, and other trace elements. In: Tsang RC, Zlotkin SH, Nichols BL, Hansen JW, editors. Nutrition during infancy. 2nd ed. Cincinnati: Digital Educational Publishing; 1997.
15Shah MD, Shah SR. Nutrient deficiencies in the premature infant. Pediatr Clin North Am 2009;56(5):1069–83.
16Verner AM, Manderson J, Lappin TR, et al. Influence of maternal diabetes mellitus on fetal iron status. Arch Dis Child Fetal Neonatal Ed. 2007;92(5):F399–401.
17Siddappa AM, Rao R, Long JD, et al. The assessment of newborn iron stores at birth: a review of the literature and standards for ferritin concentrations. Neonatology. 2007;92(2):73–82.
18Ohls RK, Roohi M, Peceny HM, et al. A randomized, masked study of weekly erythropoietin dosing in preterm infants. J Pediatr 2012;160(5):790–5.
19Romagnoli C, Tesfagabir MG, Giannantonio C, et al. Erythropoietin and retinopathy of prematurity. Early Hum Dev 2011;87(Suppl 1):S39–42.
20Georgieff MK, Berry SA, Wobken JA, et al. Increased placental iron regulatory protein-1 expression in diabetic pregnancies complicated by fetal iron deficiency. Placenta 1999;20:87–93.
21Petry CD, Wobken JD, McKay H, et al. Placental transferrin receptor in diabetic pregnancies with increased fetal iron demand. Am J Physiol 1994;267:E507–E514.
22Carter TC, Kay DM, Browne ML, et al. Hirschsprung’s disease and variants in genes that regulate enteric neural crest cell proliferation, migration and differentiation. J Hum Genet 2012;57(8):485–93.
23Gephart SM, McGrath JM, Effken JA, et al. Necrotizing enterocolitis risk: state of the science. Adv Neonatal Care 2012;12(2):77–87.
24Morgan J, Young L, McGuire W. Slow advancement of enteral feed volumes to prevent necrotising enterocolitis in very low birth weight infants. Cochrane Database Syst Rev 2011 Mar 16;3:CD001241.
25Neu J, Walker WA. Necrotizing entercolitis. N Engl J Med 2011;364:255–64.
26Turnpenny PD, Ellard S. Alagille syndrome: pathogenesis, diagnosis and management. Eur J Hum Genet 2012;20(3):251–7.
27Hinds R, Davenport M, Mieli-Vergani G, et al. Antenatal presentation of biliary atresia. J Pediatr 2004;144:43–6.
28Wildhaber BE, Coran AG, Drongowski RA, et al. The Kasai portoenterostomy for biliary atresia: a review of a 27-year experience with 81 patients. J Pediatr Surg 2003;38:1480–5.

