CHAPTER 41 G1 Phase and Regulation of Cell Proliferation
During the G1 phase of the cell cycle, each cell makes a key decision: whether to continue through another cycle and divide or to remain in a nondividing state either temporarily or permanently. During development of metazoans, this is the time when cells exit the cell cycle as the first step toward forming differentiated tissues. In adults, strict regulation of the timing and location of cell proliferation is critical to avoid cancer.
Cells enter G1 phase at the end of a proliferation cycle, after completing mitosis. To make an unbiased decision whether to proliferate or differentiate, the cell must erase the bias toward proliferation that was carried over from the preceding cell cycle. This is accomplished by inactivating cyclin-dependent kinases (Cdks [see Chapter 40]) through proteolytic destruction of cyclin subunits and synthesis of inhibitory proteins. The absence of Cdk activity turns on a regulatory network that represses the transcription of many genes that promote cell-cycle progression. While this repressive network is active, the cell cannot proceed through the cell cycle. The repression can be switched off if the cell is continuously stimulated by growth-promoting signals from the surrounding medium, extracellular matrix, and other cells (see Chapters 27 and 30). These stimuli can trigger another round of DNA replication and mitosis, but first, the cell must pass a major decision point in G1 called the restriction point (Fig. 41-1). Factors that promote the growth of cell mass are referred to herein as growth factors, and factors that promote cell-cycle proliferation are referred to herein as mitogens.
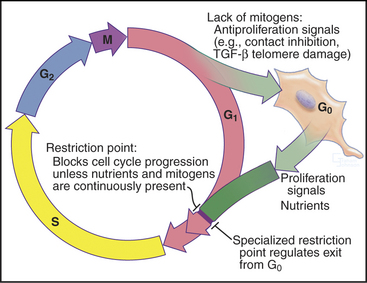
In metazoans, many cells cease cycling in the G1 phase, either temporarily or permanently, exiting the cell cycle into a state known as G0 (Fig. 41-1). This frequently accompanies their acquisition of specialized, differentiated characteristics. Occasionally, it is desirable in tissues for cells in G0 to reenter the cell cycle to replace cells lost through death. These cells reenter the cycle in G1 phase. During the G1 phase, cells also screen continuously for damage to their DNA. If damage is detected, the cell either stops cycling or undergoes apoptosis (see Chapter 46).
The G0 Phase and Growth Control
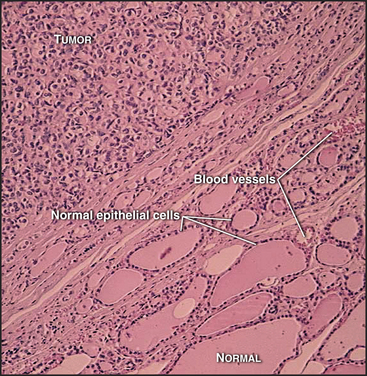
Most cells in multicellular organisms are differentiated (adapted to carry out specialized functions) and no longer divide. They typically form specialized tissues, each of which has a distinctive structural organization that is important for function. Unscheduled cell division can severely disrupt the organization of such tissues (Fig. 41-2). Accordingly, tissues strictly regulate both the location and the frequency of cell division. These divisions normally occur at a low rate, producing new cells in numbers just sufficient to replace those that die. Under special circumstances, however, such as in response to wounding (see Fig. 32-11), the rate of cell division increases dramatically. This highlights an important constraint on cell-cycle control in multicellular organisms: To make organized tissues, cells must exit from the cell cycle, but some cells must also retain the ability to reenter the active cell cycle when needed to repair injuries or replace worn-out cells.
Cells that stop cycling to differentiate normally do so in the G1 phase. Such cells are said to have left the cycle and entered a nonproliferating state called G0 (Fig. 41-1). G0 may last hours or days or even for the life of the organism, as it does for most neurons. It is important to note that nondividing cells are not dormant: G0 cells continue to expend energy for many ongoing processes. Because of turnover, all cells must continuously synthesize housekeeping proteins. They must also expend energy to maintain intracellular pH and ionic composition and to power intracellular motility. In addition, many specialized G0 cells consume large amounts of energy to synthesize and secrete protein products and generate action potentials. Energy metabolism is particularly dramatic in muscle cells that are responsible for all body movements. Thus, most G0 cells should be regarded as active cells that just happen no longer to be engaged in cell division.
How do cells stop cycling and enter the G0 phase? First, cells may receive external signals that stimulate withdrawal from the cell cycle. This often initiates differentiation of tissues. Second, cells may find themselves in an environment with insufficient mitogens to drive proliferation. Such conditions trigger many cell types to undergo suicide by apoptosis (see Chapter 46), but other cells enter a nondividing state. Third, at least in cell culture, cells that have divided more than a critical number of times or that have received certain types of unfavorable input from their environment undergo senescence, entering a viable but nondividing state. Senescence is a terminal G0 state from which cells normally cannot exit. One particularly interesting signal that can lead to senescence is stress caused by a critical shortening of the telomere regions of the chromosomes. In fact, overexpression of telomerase (see Fig. 12-15) can, when combined with suitable mitogenic stimuli, prevent cells from undergoing senescence in tissue culture.
Transforming growth factor–β (TGF-β) is an example of an external signal that arrests progress through the cycle and regulates differentiation and tissue morphogenesis (Fig. 41-3). TGF-β acts through a receptor serine/threonine kinase that activates the Smad transcription factors (see Fig. 27-10) and increases the expression of the Ink4 class Cdk inhibitor, p15Ink4b, by up to 30-fold (see Appendix 40-1). Like other Ink4 family members, p15Ink4B specifically inactivates Cdk4–cyclin D and Cdk6-cyclin D complexes. p15Ink4B binding also displaces CKI class inhibitors from the Cdk4–cyclin D complexes, permitting them to transfer to Cdk2–cyclin E complexes in the nucleus and further inhibit cell-cycle progression.
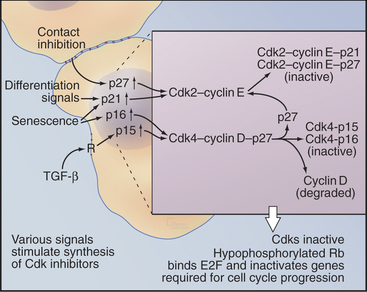
In addition to its role in stopping cell-cycle progression in response to TGF-β, the CKI inhibitor p27Kip1 helps to arrest the cell cycle when normal cells become crowded by neighboring cells (contact inhibition; see Fig. 30-8) or when the environment lacks mitogens. Genetic analysis in mice indicates that p27Kip1 regulates cell-cycle progression during development. Indeed, mice that lack p27Kip1 are 30% larger than their normal littermates by several weeks of age. This increase in size occurs at least partly because cells in many organs undergo extra rounds of division.
An analogous mechanism appears to arrest the cell cycle during the differentiation of muscle cells. The transcription factor MyoD is a master regulator of muscle differentiation. MyoD activates transcription of the CKI inhibitor p21Cip1, which helps to arrest proliferation and start muscle differentiation (Fig. 41-3). p21Cip1 stops cell-cycle progression in at least two ways. First, by binding Cdk-cyclin complexes, it blocks them from promoting cell-cycle progression. Second, p21Cip1 binds to the DNA replication factor proliferating cell nuclear antigen (PCNA [see Chapter 42]) in a way that blocks chromosomal replication but not repair of DNA damage.
Several of these mechanisms, including increased expression of both p16Ink4a and p21Cip1, are responsible for permanent cell-cycle arrest of aged cells (senescence). Furthermore, formation of heterochromatin permanently inactivates some genes required for proliferation. This is accomplished when inhibitory proteins of the Rb and E2F families (see later) bind to promoters and recruit histone methyltransferases (see Fig. 13-9).
Exit from the G0 Phase
Cells in the G0 phase may reenter the growth cycle in response to specific stimulation, often induced by injury or normal cell turnover. Cultured fibroblasts are favored for studies of this process, as they readily enter the G0 phase when deprived of serum (i.e., mitogens and growth factors) and rapidly reenter the cell cycle when serum is restored. The pattern of gene expression induced by serum in culture reproduces that found in wounded tissues. When a living tissue is wounded (see Fig. 32-11), fibroblasts are exposed to serum. In response, they divide and colonize the wound, where they lay down new extracellular matrix to repair the damage.
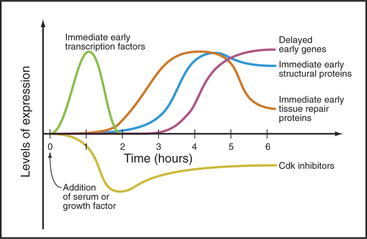
Serum stimulates three waves of gene expression in cultured fibroblasts in G0 (Fig. 41-4). The first wave includes more than 100 “immediate early” genes, including transcription factors of the Jun, fos, myc, and zinc finger families (see Chapter 15) that activate numerous downstream genes required for cell growth and division. Other immediate early genes encode tissue remodeling factors, cytokines (growth factors), extracellular matrix components (fibronectin), plasma membrane receptors (integrins), and cytoskeletal proteins (actin, tropomyosin, vimentin), as well as activities involved in angiogenesis (blood vessel formation), inflammation, and coagulation. These proteins facilitate the movement of fibroblasts into wounds and initiate the repair of tissue damage.
The Restriction Point: A Critical G1 Decision Point
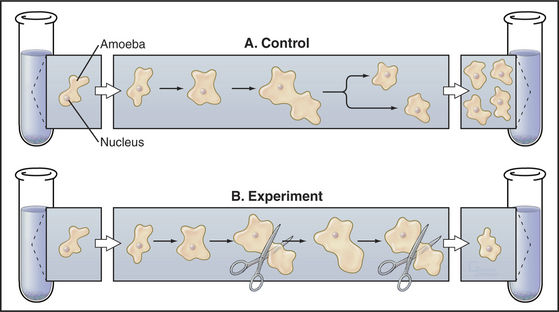
The influence of cell size on the division cycle was first demonstrated in an elegant microsurgery experiment (Fig. 41-5). Two Amoeba proteus cells were grown under identical conditions in parallel cultures. Each day, a portion of the cytoplasm was amputated from one amoeba, and the other was left untouched as a control. Under those circumstances, the cell that suffered the amputations did not divide for 20 days. During this time, the control amoeba divided 11 times. When the amputations were stopped, the amoeba that had been operated on divided within 38 hours. The interpretation of this experiment was that the repeated amputations prevented the experimental amoeba from ever attaining a size sufficient to undergo division. Evidence suggests that some types of human cells have a similar size control while others do not.
An essential aspect of growth control during the G1 phase involves monitoring the external environment for nutrient availability and for signals to proliferate (mitogenic signals) coming from other cells and from the extracellular matrix. In a classic experiment, when three populations of cells proliferating in culture were starved by deprivation of amino acids, serum, or phosphate, they stopped cycling in G1. When the missing ingredients were restored, all three populations of treated cells resumed the cell cycle and entered the S phase at about the same time. This was surprising because amino acids are needed to make protein, serum provides growth factors and mitogens, and phosphate is needed for synthesis of DNA phospholipids (needed to make membranes). This experiment was interpreted as evidence that all three types of starvation caused cells to arrest at an equivalent point in the G1 phase, termed the restriction point. The restriction point is defined as the point after which the cell cycle will proceed even if mitogenic factors are withdrawn (Fig. 41-6). This supremely important aspect of cell-cycle control prevents cells from dividing at inappropriate times and in inappropriate places. Defects in restriction point control are among the most common causes of cancer.
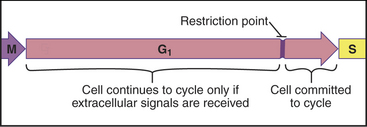
Regulation of Cell Proliferation by the Restriction Point
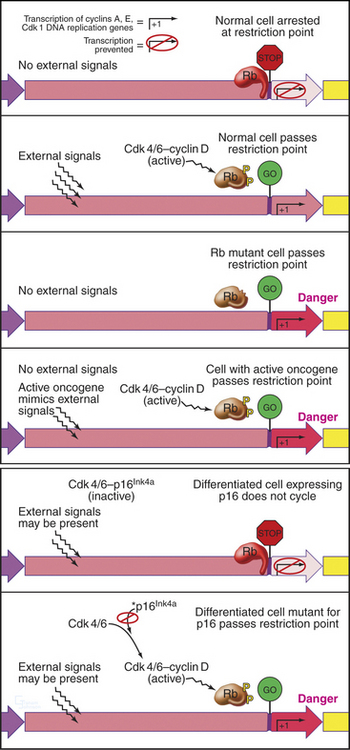
Rb regulates the ability of E2F to activate genes required for cell-cycle progression. E2F forms a heterodimer with one of two DP family members and associates with the promoter region of its target cell-cycle genes (Fig. 41-7A). Rb binding blocks the ability of E2F to activate transcription. In addition, Rb recruits histone deacetylases, enzymes that remove acetyl groups from the amino-terminal tails of histones (see Fig. 13-9). This causes compaction of chromatin structure and represses genes required for cell-cycle progression.
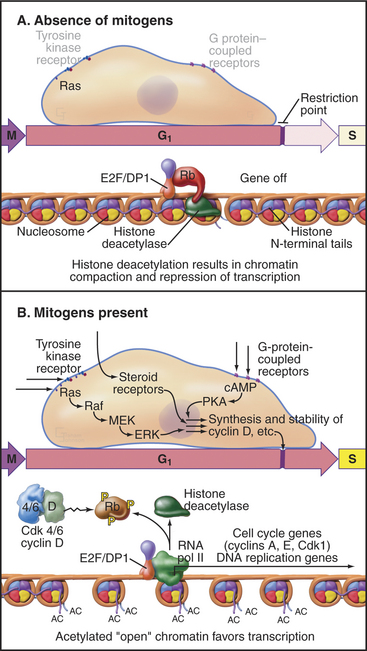
Figure 41-7 Regulation of cell-cycle progression by the E2F/DP/Rb complex. A, The E2F/DP/Rb complex recruits histone deacetylases (see Chapter 13) and represses specific genes that are required for cell-cycle progression. This blocks cell-cycle progression at the restriction point. B, Phosphorylation of Rb by Cdks alleviates this block and permits passage of the restriction point. cAMP, cyclic adenosine monophosphate; MEK, mitogen-activated protein kinase kinase; PKA, protein kinase A.
Phosphorylation of Rb by Cdks causes it to dissociate from E2F, allowing E2F/DP to activate, rather than repress, transcription of the target genes. Cells that can phosphorylate Rb pass the restriction point and complete a cell cycle, whereas cells that cannot phosphorylate Rb remain arrested in the G1 or G0 phase. The E2F/DP heterodimer remains bound to its target promoter regions after phosphorylated Rb dissociates from E2F. If activated as a result of DNA damage, E2F can also act as a potent inducer of cell death by apoptosis (see Chapter 46).
E2F/DP that is free of Rb is a potent transcription factor, promoting expression of genes that stimulate both reentry of G0 cells back into the cycle and their subsequent passage through the cell cycle. These target genes encode proteins that are required for DNA synthesis (DNA polymerase a1, accessory factors, and enzymes that synthesize nucleotide precursors; see Chapter 42), proteins that promote cell-cycle progression (cyclins E and A, Cdk1 and Cdc25), and proteins that regulate cell-cycle progression (pRb, p107, Emi1).
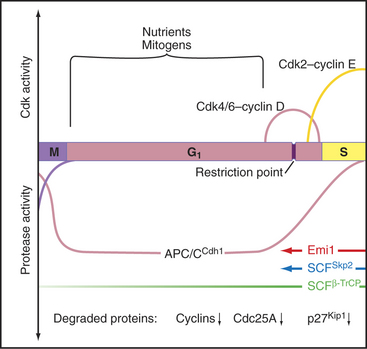
Passage through the restriction point therefore hinges on Cdk activation (Fig. 41-7B; see also Chapter 40 and Appendix 40-1), which leads to Rb phosphorylation and activation of E2F/DP dimers on the promoters of essential cell-cycle genes. The normal pathway of Rb phosphorylation is started by Cdk4–cyclin D and Cdk6–cyclin D (referred to hereafter as Cdk4/6–cyclin D) and is carried forward by Cdk2–cyclin E and Cdk2–cyclin A. Cdk activity in early G1 is regulated by adjusting the relative levels of the three D-type cyclins and the CKI class Cdk2 inhibitors p27Kip1 and p21Cip1. Regulation of cyclin D levels provides the crucial link between extracellular mitogens and the cell cycle.
Nonproliferating cells control cyclin levels in G1 cells in two ways. First, cyclin D mRNA levels are so low that little protein is made. Second, the little cyclin D that is made is kept in the cytoplasm, where it is phosphorylated by glycogen synthase kinase-β (GSK [see Fig. 30-8]) and degraded by SCF (see Fig. 40-17). E2F controls the expression of the genes for cyclins E and A, so these cyclins are present at only low levels, while E2F/Rb/DP acts as an inhibitor. Furthermore, levels of the CKI class Cdk2 inhibitor p27Kip1 are high in prerestriction point G1 cells. Thus, any Cdk2–cyclin E or Cdk2–cyclin A that happens to be present is inactive (see Figs. 40-14 and 40-15).
How do cells convert signals from mitogens and the extracellular matrix into a decision to open the restriction point gate? Stimulation of receptor tyrosine kinases (see Chapters 25 and 27) or integrins (see Chapter 30) initiates a signal transduction pathway starting with Ras activation of Raf and leading to activation of the mitogen-activated protein (MAP) kinase/extracellular signal–regulated kinase (ERK) cascade (see Fig. 27-6). The output of this cascade stimulates transcription of D-type cyclins (Figs. 41-7 and 41-8) and also inactivates GSK. This allows cyclin D to accumulate in nuclei.
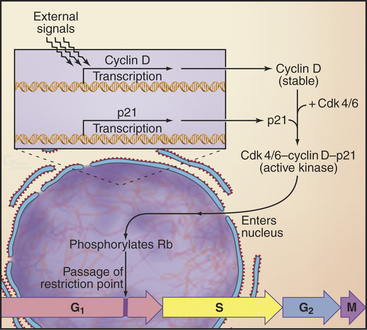
In addition to promoting the accumulation of cyclin D, mitogens stimulate transcription of the CKI class Cdk2 inhibitor p21Cip1. This protein and p27Kip1 actually promote the activation of Cdk4/6–cyclin D complexes in two ways. First, they enhance assembly of complexes of cyclin D with Cdk4 and Cdk6. Second, they promote the nuclear import of Cdk4/6–cyclin D, thereby leading to activation of Cdks by Cdk-activating kinase (a nuclear enzyme; see Chapter 40), as well as an increase in the stability of cyclin D. All of this depends on the continuous presence of mitogenic signals; if these cease, then cyclin D stability rapidly declines again.
The Restriction Point and Cancer
Cancer is a complex class of diseases in which genetic changes within clones of cells lead to production of cell populations whose uncontrolled growth can disrupt tissue function and can ultimately kill the individual. Two in five Americans will be affected by cancer during their lifetimes. This sounds very high, but considering the number of cell cycles that are required to produce a human composed of about 1014 cells, the disease is actually remarkably rare on a per cell basis. Why is this so? One reason is that multiple genetic alterations are required to transform a normal cell into a cancer cell. This is because the cell cycle is highly regulated, and activities that tend to drive cellular proliferation are held in check by a web of negative feedback pathways. As a result, some cancer-causing mutations are actually deleterious in normal cells. In fact, most cells with disturbed growth control pathways are eliminated by backup mechanisms that cause them to commit suicide by apoptosis (see Chapter 46).
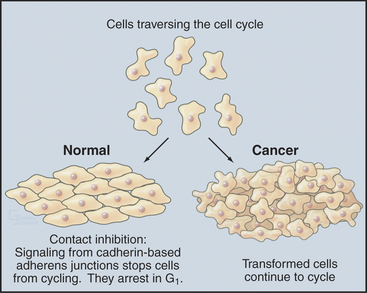
Almost all types of cancer are caused by a disregulation of cell proliferation in the G1 phase. This is readily seen in the laboratory when cells are grown on plastic tissue culture dishes. Most normal cells proliferate until they cover the surface completely, forming a monolayer. When the monolayer is confluent (i.e., when cells are touched by other cells on all sides), signaling initiated by cadherin proteins (see Fig. 30-8) causes cells to arrest their cell-cycle progression in G1. This is called contact inhibition of growth (see Chapter 30, in the section titled “Cadherin Family of Adhesion Receptors”). Cancer cells lack this control, so they keep proliferat-ing and piling up on top of one another as long as nutrient and mitogen supplies last (Fig. 41-9). Cells that lose this aspect of growth regulation are said to be transformed.
More than 100 oncogenes have been identified thus far. Most normally function in signal transduction pathways that lie downstream of signals that stimulate cell-cycle progression. Their inappropriate activation can mimic the effects of persistent mitogenic stimulation, thereby uncoupling cells from normal environmental controls and leading to uncontrolled proliferation and cancer. For example, Ras proteins are key components of signaling pathways that lead to activation of the MAP/ERK kinase cascade and accumulation of cyclin D (Fig. 41-7). They are mutated in about 15% of human cancers. Inappropriate activation of Ras tricks the cell into thinking that it is receiving mitogenic signals, leading it to express cyclin D, phosphorylate Rb, and proliferate. Luckily, in normal cells, this usually activates a checkpoint mechanism and leads to rapid cell-cycle arrest. Other proteins that are involved in restriction point control can also act as oncogenes if hyperactivated. These include E2F1, cyclin D (overexpressed in 50% of breast cancers), and Cdk4. In each case, activation of the protein causes inappropriate transcription of genes promoting cell-cycle progression, bypassing the restriction point, and leading to uncontrolled cell cycles and cancerous transformation (Fig. 41-10).
Rb is one of the best-characterized tumor suppressor genes. As was discussed earlier, a primary function of Rb is to block cell-cycle progression until mitogenic stimulation results in its inactivation. It is therefore not surprising that loss of Rb can lead to inappropriate cell-cycle progression and cancer. Rare individuals who inherit one defective Rb gene usually develop retinoblastomas as children and osteosarcomas as adults. The cancer arises when the “good” allele is inactivated in a proliferating cell (this is called a somatic mutation). Such cancers are rare and occur only later in life in individuals who inherit two good Rb genes, as two independent somatic mutations (two “hits”) are required in the same proliferating cell. Homozygous loss of pRb is lethal during embryogenesis. This is partly because under some circumstances, the unleashed E2F can act as a potent inducer of apoptotic cell death (see Chapter 46).
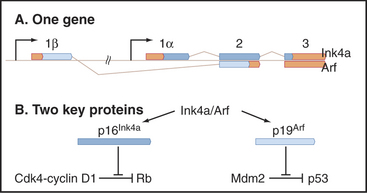
P16Ink4a is another important tumor suppressor that is involved in G1 growth control. Normally, it suppresses Cdk4/6 activity in nondividing cells (see next section; also see Chapter 40), thereby reinforcing the ability of Rb to maintain the growth arrest of G1 cells (Fig. 41-10). Mutations in the p16Ink4a gene are very commonly seen in cancer, but this is partly because this gene is fascinatingly complex (see Fig. 41-14). Mutations in other INK4 Cdk inhibitors and the CKI p27Kip1 are also found in cancer, though less frequently.
Proteolysis and G1 Cell Cycle Progression
Just as controlled destruction of proteins is key to the transition of cells from mitosis to the G1 phase (see Chapter 40), proteolysis also fulfills a number of key roles during progression through G1 into the S phase (Fig. 41-11). For example, when Cdk2–cyclin E is activated following synthesis of cyclin D, it phosphorylates its p27Kip1 inhibitor. This allows p27Kip1 to be recognized by a specific class of ubiquitin ligase (E3) called SCFSkp2 (see Figs. 40-16 and 40-17). The resulting destruction of p27Kip1 helps to produce a burst of Cdk2-cyclin E activation in a feedback loop that allows for rapid amplification of Cdk activity and contributes to initiation of the S phase. Later in the S phase, phosphorylation of the DP subunit of E2F causes its dissociation from DNA, recognition by SCF, and destruction. This is essential to complete the S phase. SCF also targets cyclins D1 and E for destruction, the former when mitogens are limiting and the latter during progression through the S phase.
Integrity of Cellular DNA Monitored by a G1 Checkpoint
This quality control mechanism involves a checkpoint that operates throughout the G1 phase (Fig. 41-12). Checkpoints are biochemical circuits superimposed on the normal cell cycle. When activated, checkpoints block progression through the cycle, either temporarily or, in some cases, permanently. In certain cases, checkpoint activation leads to cell death by apoptosis. Checkpoints are activated by sensor proteins that detect problems—typically, DNA damage in the case of the G1 checkpoint. Sensor proteins activate protein kinases that modify target proteins, which then block cell-cycle progression (see Fig. 40-4).
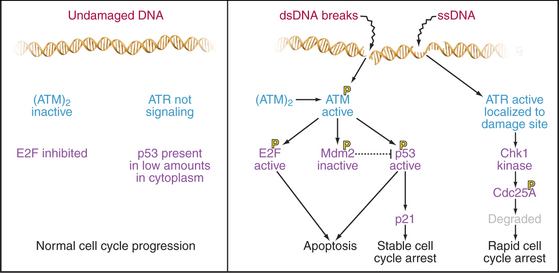
DNA damage checkpoints have fast and slow components: The former is analogous to applying the brakes in a car; the latter is analogous to removing the wheels and putting it up on blocks. Both components depend on the protein kinases, ATM and ATR (see Fig. 40-4). ATM and ATR are related to the lipid kinase phosphatidylinositol 3-kinase (see Chapters 25 and 27), but their only known substrates are proteins (see Chapter 40). People who lack ATM have the disease ataxia-telangiectasia, which is characterized by immunodeficiency, photosensitivity, cerebellar degeneration, and an elevated incidence of leukemias and lymphomas. ATR is essential for life.
When ATR is activated, primarily by DNA damage that disrupts ongoing DNA replication, it phosphorylates and activates a downstream kinase called Chk1, one of whose targets is the essential phosphatase CDC25A (Fig. 41-12). This phosphorylation signals CDC25A for destruction. Since CDC25A is required to remove inhibitory phosphate groups from inactive Cdks, its destruction applies a rapid brake to cell-cycle progression.
p53 is very powerful medicine for the cell cycle. If present in excessive amounts, it is extremely toxic. For this reason, p53 is regulated by a partner protein called Mdm2 (mouse double minute 2; the human ortholog of this is Hdm2), a ubiquitin ligase (E3) whose job is to keep p53 levels low when the cell cycle is running normally (Fig. 41-13A). Loss of the Mdm2 gene in mice is lethal unless the p53 gene is also lost. Mdm2 protein shuttles in and out of the nucleus (see Chapter 14). p53 also has a nuclear export signal, and when these two proteins associate in the cytoplasm, Mdm2 promotes the rapid degradation of p53 by the ubiquitin/proteasome system (see Chapter 23). Because p53 directly stimulates expression of Mdm2, a negative feedback loop keeps levels of p53 low.
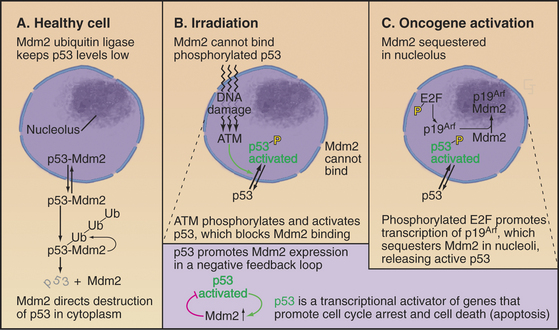
Both p53 and Mdm2 are phosphorylated following DNA damage (Fig. 41-13B). These phosphorylations prevent Mdm2 from binding, so p53 is stabilized, and its concentration in the nucleus increases dramatically. The phosphorylations also make p53 a more potent transcriptional activator. The result is a burst of transcription of p53-regulated genes.
A different mechanism is used to defend the cell against cancer caused by inappropriate activation of oncogenes that disrupt normal cell-cycle controls. Dysregulated cell-cycle progression allows E2F to stimulate expression of the tumor suppressor protein p19Arf, which binds and sequesters Mdm2 (but not p53) in the nucleolus (Fig. 41-13C). This allows p53 to accumulate in the nucleus, where it activates a pathway promoting apoptotic cell death by stimulating transcription of a number of genes involved in cell killing, including Bax, BH3-domain proteins, CD95 (Fas/Apo1) and Apaf-1 (Fig. 41-13, also discussed in Chapter 46). Thus, aberrantly proliferating cells are removed, and the body is protected.
The p19Arf gene (in humans, the protein is smaller and so is called p14Arf) is quite unusual, as it is encoded in a common gene with p16Ink4a (Fig. 41-14). In fact, the genes not only overlap but also share a common exon. Nevertheless, the two proteins have no common amino acid sequences because the shared exons are read in different frames in the mature messenger RNAs (mRNAs) for the two proteins. Thus, the p16Ink4a/p14Arf locus encodes two vital protective factors with different jobs. It is not surprising that mutations in this key locus are found in between 25% and 70% of human cancers.
Moving into and out of G0: Stem Cells
Some cells are professionals at moving back and forth between G0 and more active cell cycles. Without doubt, the champions at this are stem cells, one of whose roles is to replace worn-out parts of tissues as differentiated cells age or die as a result of various misadventures. Box 41-1 provides a brief introduction to the very topical world of stem cells.
BOX 41-1 Stem Cells
Most adult tissues set aside a few tissue stem cells that have the capacity to renew themselves and to produce daughter cells that differentiate into a limited range of specialized cells (see Figs. 28-1, 28-5, and 40-1). Adult stem cells have diverse patterns of cell-cycle regulation. Some of these tissue stem cells cycle continuously throughout life. For example, epithelial stem cells give rise to mature cells that continuously replace the skin and the lining of the gastrointestinal tract. Hematopoietic stem cells in bone marrow give rise to several different types of short-lived blood cells. Plant meristematic stem cells produce cells for roots and shoots. In other organs such as liver and skeletal muscle, tissue stem cells are held in reserve unless the tissue is damaged, when they produce daughter cells to repair the damage. Stem cells are present but largely inactive in organs such as the nervous system, which have limited capacity for renewal and regeneration. The potential for regeneration from stem cells has stimulated research to find ways of using embryonic or tissue stem cells to repair damaged or diseased organs in human patients. Stem cells have also been useful for production of transgenic animals for scientific research (e.g., knockout mice) or for production of therapeutically important proteins.
Discovery and Defining Features of Stem Cells
Pioneering work on blood cell development (see Fig. 28-5) established the existence of stem cells and defined many of the concepts that apply to all types of stem cells. The key experiment was to inject bone marrow cells from a normal mouse into a mouse that had been irradiated to kill all of the cells that produce blood cells. Transplantation of bone marrow cells rescued these irradiated mice from death from anemia, bleeding, and infections. The transplanted bone marrow contained precursor cells that formed colonies of proliferating cells that regenerated the full range of blood cells. The blood-forming colonies in the spleen, each of which formed from a single stem cell, contained either one or, infrequently, several types of differentiating blood cells.
This experimental system first revealed the existence of several different types of hematopoietic stem cells in bone marrow with the dual capacity to renew themselves and to give rise to differentiated cells (see Fig. 28-5). Very rare pluripotent hematopoietic stem cells can give rise to all types of blood cells, including themselves. Other committed stem cells with a more restricted capacity for self-renewal can give rise to specific subsets of blood cells, such as red blood cells, platelets, granulocytes, or lymphocytes. Antibodies for surface markers can now be used to distinguish and purify the various types of hematopoietic stem cells from mice and humans. Once separated from the far more numerous mature and differentiating cells in bone marrow, stem cells can be used for transplantation into patients with bone marrow defects.
Cytokines and other growth factors regulate proliferation and differentiation at every stage of blood cell production. The later stages are best understood. For example, the cytokine erythropoietin acts through a kinase-coupled receptor to activate a cytoplasmic transcription factor that stimulates the proliferation and differentiation of the red blood cell lineage. Other cytokines guide the differentiation of granulocytes and monocytes. Hematopoietic stem cells respond to the same families of growth factors that control other aspects of development, including Wnts (see Fig. 30-8), Notch (see Chapter 24), fibroblast growth factor (see Fig. 24-4), and insulin-like growth factor (see Fig. 24-4). However, too little is yet known about these regulatory mechanisms to grow hematopoietic stem cells in the laboratory.
Properties of Adult Stem Cells
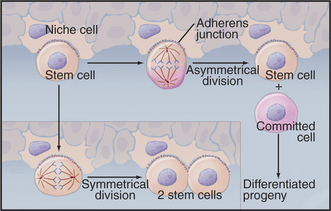
Years of detailed analysis in the laboratory and clinic established hematopoietic stem cells as a model for stem cells in other tissues. General features include the capacity for self-renewal and the production of daughters that proliferate and differentiate. This dichotomy is achieved by asymmetrical cell divisions guided by the same types of internal cues that control unequal divisions of cells in early embryos (Fig. 41-15). Symmetrical divisions (to give two daughter stem cells) can also expand the numbers of stem cells during growth to maturity and during regeneration of damaged tissues.
Stem cells depend on local environmental cues to maintain their status as stem cells. These special environments, called stem cell niches, are created by tissue cells and the extracellular matrix. Niche cells anchor stem cells with adherens junctions and provide cell surface and secreted proteins that activate the signaling pathways that regulate the cell cycle of the stem cell. Some of these factors stimulate division; others inhibit differentiation. The niches that are occupied by germ cells and neural stem cells from invertebrates are particularly well characterized. During asymmetrical divisions of germ stem cells, the renewed stem cell stays behind in the niche, while the daughter that is destined to differentiate into an egg, sperm, or neuron is released. In bone marrow, osteoblasts (see Fig. 32-5) and endothelial cells (see Fig. 30-13) provide niches for hematopoietic stem cells.
Epidermal Stem Cells
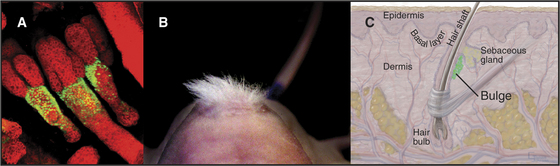
Skin is an example of a continuously renewing organ with a considerable capacity for regeneration (see Fig. 40-1). Multipotential and committed stem cells contribute to both renewal and regeneration. Committed stem cells reside in the basal layer of the epidermis. Asymmetrical cell divisions oriented at right angles to the basal lamina produce two daughter cells. The daughter on the basal lamina carries on as the stem cell. The apical daughter cell divides multiple times and differentiates into a column of cells, forming the superficial layers of the epidermis (see Fig. 35-6). Multipotential stem cells associated with hair follicles give rise to all of the cells of the hair follicle and also serve as a reserve for the committed epidermal stem cells in the event of injury (Fig. 41-16). Like other adult stem cells, the stem cells of the skin are relatively quiescent, are responsive to growth and differentiation factors, and are influenced by their local environments.
Skeletal Muscle Stem Cells
Small numbers of stem cells reside in a niche between the basal lamina and the giant multinucleated muscle cells. If the muscle is damaged, these quiescent “satellite cells” multiply and produce cells that regenerate the tissue. Positive signals for proliferation and differentiation come through receptor tyrosine kinases and the MAP kinase pathway (see Fig. 27-6) and other pathways. Restraining signals are provided by myostatin, a member of the TGF-β family (see Fig. 27-10). Inactivation of the myostatin pathway results in massive enlargement of muscles in mice and humans. Muscles are capable of regenerating multiple times, so the stem cell population renews itself during regeneration or is augmented by stem cells that migrate through the blood from bone marrow or other tissues.
ACKNOWLEDGMENTS
Thanks go to Jiri Bartek and Martin Raff for their suggestions on revisions to this chapter.
Blais A, Dynlacht BD. Hitting their targets: An emerging picture of E2F and cell cycle control. Curr Opin Genet Dev. 2004;14:527-532.
Blanpain C, Fuchs E. Epidermal stem cells of the skin. Annu Rev Cell Dev Biol. 2006;22:339-373.
Bryder D, Rossi DJ, Weissman IL. Hematopoietic stem cells: The paradigmatic tissue-specific stem cell. Am J Pathol. 2006;169:338-346.
Cardozo T, Pagano M. The SCF ubiquitin ligase: Insights into a molecular machine. Nat Rev Mol Cell Biol. 2004;5:739-751.
DeGregori J. The Rb network. J Cell Sci. 2004;117:3411-3413.
Ekholm SV, Reed SI. Regulation of G(1) cyclin-dependent kinases in the mammalian cell cycle. Curr Opin Cell Biol. 2000;12:676-684.
Fuchs E, Tumbar T, Gausch G. Socializing with the neighbors: Stem cells and their niche. Cell. 2004;116:769-778.
Hochedlinger K, Jaenisch R. Nuclear reprogramming and pluripotency. Nature. 2006;441:1061-1067.
Jackson PK, Eldridge AG. The SCF ubiquitin ligase: An extended look. Mol Cell. 2002;9:923-925.
Jorgensen P, Tyers M. How cells coordinate growth and division. Curr Biol. 2004;14:R1014-R1027.
Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316-323.
McGowan CH, Russell P. The DNA damage response: Sensing and signaling. Curr Opin Cell Biol. 2004;16:629-633.
Morrison SJ, Kimble J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature. 2006;441:1068-1074.
Muotri AR, Gage FH. Generation of neuronal variability and complexity. Nature. 2006;441:1087-1093.
Rando TA. Stem cells, ageing and the quest for immortality. Nature. 2006;441:1080-1086.
Scadden DT. The stem-cell niche as an entity of action. Nature. 2006;441:1075-1079.
Sherr CJ. The INK4a/ARF network in tumour suppression. Nat Rev Mol Cell Biol. 2001;2:731-737.
Shi X, Garry DJ. Muscle stem cells in development, regeneration and disease. Genes Dev. 2006;20:1692-1708.
Veit B. Stem cell signalling networks in plants. Plant Mol Biol. 2006;60:793-810.
Weissman IL, Anderson DJ, Gage F. Stem and progenitor cells: Origins, phenotypes, lineage commitments and transdifferentiation. Annu Rev Cell Dev Biol. 2001;17:387-403.