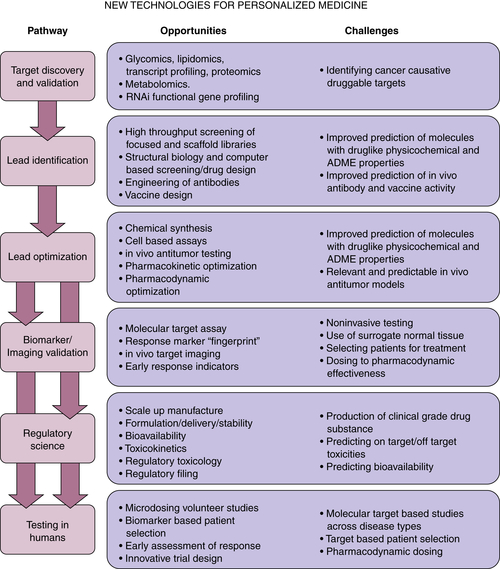
The Second Phase
Table 45-1
Rationale for Targeting EGF Receptors, 1980

Next Steps in Drug Development
Scientific Knowledge and Technology
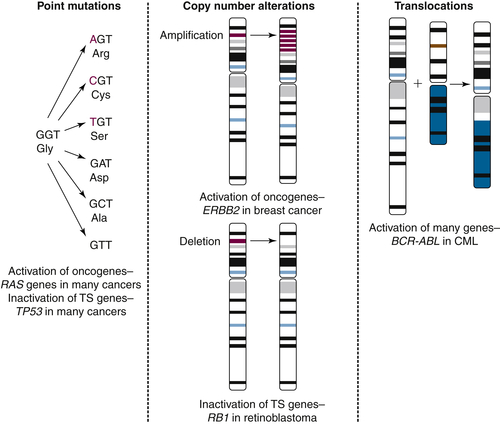
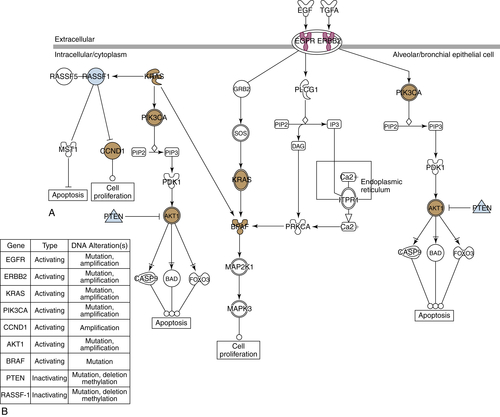
Table 45-2
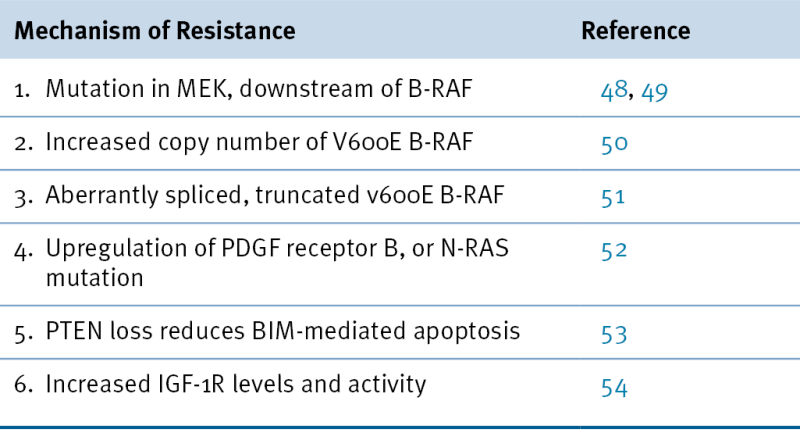
Mechanisms of Resistance to Vemurafenib Treatment in Patients with V600E B-RAF Malignant Melanoma
• Expression of genes. Data are being reported suggesting that a considerable fraction of mutated genes are not expressed. In the case of suppression genes, this is expected, because this is the basis for their impact on the cell’s phenotype. For potential oncogenes, however, this prevents the genetic aberrancy from having an impact. Furthermore, the level of expression of a gene—unrelated to the presence of a mutation—may alter cell behavior and phenotype. Again, the EGF receptor provides an example. This receptor is overexpressed (10- to 1000-fold) on a large fraction of epithelial cancers and is likely to create a “non-oncogene addiction” in these cells. 56 Thus an important area of cancer research will involve closely examining the expression of RNA and proteins coded for by aberrantly functioning genes, to assess the importance of their expression levels in identifying a biomarker signature useful for precision cancer care.
• Epigenetics. Current technologies and computational methods in this area are not adequate to inform clinical decisions. Many investigators believe that epigenetic changes play a major role in oncogenesis and the malignant phenotype. 57 This is a topic that will likely need to be visited more thoroughly in the future, as advancements are made in knowledge and assay techniques.
• Immunologic profile. As discussed in Chapters 50-52, immunological interventions are beginning to affect cancer therapy, leveraging recent gains in understanding the functioning of the human immune system. The patient’s immune system is likely to play an important role in controlling malignancy, as was postulated five decades ago. With increased understanding of the role of cytokines derived from the patient’s lymphocytes and leukocytes in promoting or inhibiting cancer cell growth, a profile of the patient’s immune functions is likely to become a component of the biomarker profile informing precision cancer care. 58
• Proteomics. Evaluation of the functional status of proteins in a patient’s cancer (e.g., phosphorylation status) and detection of circulating protein markers derived from cancers (e.g., HCG levels) are promising areas for the clinic and should expand in value as biomarkers, as mass spectrometry techniques and the use of reverse phase protein arrays are developed into more useful clinical tests. 59
Predicting Drug Efficacy and Toxicity
• Growth of primary human tumor specimens in direct xenografts, without an intermediate step of culture on plastic surfaces, has produced models of human cancer in mice that are more closely comparable to primary human cancers. 61
• GEMMs with inducible promoters on mutated genes permit activation of the aberrant gene in specific organ sites and at specific time points, creating murine models that can more closely mimic human cancers. 21
Clinical Trials
Table 45-3
Challenges for Personalized Cancer Therapy

CLIA, Clinical Laboratory Improvement Amendment; CMS, Centers for Medicare and Medicaid Services; FDA, U.S. Food and Drug Administration.
Conclusion
1. Microbe Hunters . New York, NY : Harcourt, Brace & Co ; 1926 : 326 .
2. The biological actions and therapeutic applications of the B-chloroethyl amines and sulfides . Science . 1946 ; 103 : 409 – 436 .
3. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid (Aminopterin) . N Engl J Med . 1948 ; 238 : 787 – 793 .
4. Clinical evaluation of a new antimetabolite, 6-mercaptopurine, in the treatment of leukemia and allied diseases . Blood . 1953 ; 8 : 965 – 999 .
5. From bench to bedside with targeted therapies . In: Mendelsohn J. , Howley P.M. , Israel M.A. , Gray J.W. , Thompson C.B. , eds. The Molecular Basis of Cancer . 3rd ed. Philadelphia, Pa : WB Saunders Co ; 2008 : 521 – 530 .
6. Progress and perspectives in the chemotherapy of acute leukemia . Adv Chemother . 1965 ; 2 : 269 – 298 .
7. Effects of combined drug therapy on metastatic cancer of the testis . JAMA . 1960 Nov 5 ; 174 : 1291 – 1299 .
8. Continuous cultures of fused cells secreting antibody of predefined specificity . Nature . 1975 ; 256 : 495 – 497 .
9. Treatment of B-cell lymphoma with monoclonal anti-idiotype antibody . N Engl J Med . 1982 Mar 4 ; 306 : 517 – 522 .
10. Endocrine-induced regression of cancers . Science . 1967 ; 156 : 1050 – 1054 .
11. Growth stimulation of A431 cells by EGF: Identification of high affinity receptors for epidermal growth factor by an anti-receptor monoclonal antibody . Proc Natl Acad Sci USA . 1983 ; 80 : 1337 – 1341 .
12. Monoclonal anti-epidermal growth factor receptor antibodies which are inhibitors of epidermal growth factor binding and antagonists of epidermal growth factor-stimulated tyrosine protein kinase activity . J Biol Chem . 1984 ; 259 : 7755 – 7760 .
13. Growth inhibition of human tumor cells in athymic mice by anti-EGF receptor monoclonal antibodies . Cancer Res . 1984 ; 44 : 1002 – 1007 .
14. p185HER2 monoclonal antibody has antiproliferative effects in vitro and sensitizes human breast tumor cells to tumor necrosis factor . Mol Cell Biol . 1989 Mar ; 9 : 1165 – 1172 .
15. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2 . N Engl J Med . 2001 ; 344 : 783 – 792 .
16. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia . N Engl J Med . 2001 ; 344 : 1031 – 1037 .
17. Phase I and imaging trial of indium-111 labeled anti-EGF receptor monoclonal antibody 225 in patients with squamous cell lung carcinoma . J Natl Cancer Inst . 1991 ; 83 : 97 – 104 .
18. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer . N Engl J Med . 2004 Jul 22 ; 351 3373-45 .
19. Cellular oncogenes and retroviruses . Annu Rev Biochem . 1983 ; 52 : 301 – 354 .
20. Towards systematic functional characterization of cancer genomes . Nat Rev Genet . 2011 Jun 17 ; 12 : 487 – 498 .
21. How genetically engineered mouse tumor models provide insights into human cancers . J Clin Oncol . 2011 Jun 1 ; 29 : 2273 – 2281 .
22. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib . N Engl J Med . 2004 May 20 ; 350 : 2129 – 2139 .
23. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy . Science . 2004 Jun 4 ; 304 : 1497 – 1500 .
24. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib . Proc Natl Acad Sci U S A . 2004 Sep 7 ; 101 : 13306 – 13311 .
25. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma . N Engl J Med . 2009 Sep 3 ; 361 : 947 – 957 .
26. Pilot study using molecular profiling of patients’ tumors to find potential targets and select treatments for their refractory cancers . J Clin Oncol . 2010 Nov 20 ; 28 : 4877 – 4883 .
27. , et al. The BATTLE Trial: personalizing therapy for lung cancer . Cancer Discov . 2011 ; 1 : 44 – 53 .
28. PIK3CA mutations in patients with advanced cancers treated with PI3K/AKT/mTOR axis inhibitors . Mol Cancer Ther . 2011 ; 10 : 558 – 565 .
29. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer . N Engl J Med . 2010 ; 363 : 1693 – 1703 .
30. Inhibition of mutated, activated BRAF in metastatic melanoma . N Engl J Med . 2010 ; 363 : 809 – 819 .
31. Exploring the genomes of cancer cells: progress and promise . Science . 2011 Mar 25 ; 331 : 1553 – 1558 .
32. Clinical implications of the cancer genome . J Clin Oncol . 2010 Dec 10 ; 28 : 5219 – 5228 .
33. Personalized oncology through integrative high-throughput sequencing: a pilot study . Sci Transl Med . 2011 Nov 30 ; 3 : 111 – 121 .
34. Molecular prescreening to select patient population in early clinical trials . Nat Rev Clin Oncol . 2012 Apr 3 ; 9 : 359 – 366 .
35. President’s Council of Advisors on Science and Technology. Report to the President Propelling Innovation in Drug Discovery, Development and Evaluation, September 2012
36. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses . Science . 2008 Sep 26 ; 321 : 1801 – 1806 .
37. Unbiased reconstruction of a mammalian transcriptional network mediating pathogen responses . Science . 2009 Oct 9 ; 326 : 257 – 263 .
38. Translating cancer ‘omics’ to improved outcomes . Genome Res . 2012 ; 22 : 188 – 195 .
39. Cancer. Addiction to oncogenes—the Achilles heal of cancer . Science . 2002 Jul 5 ; 297 : 63 – 64 .
40. A blueprint for advancing genetics-based cancer therapy . Cell . 2011 Sep 30 ; 147 : 26 – 31 .
41. PLX4032 in metastatic colorectal cancer patients with mutant BRAF tumors . J Clin Oncol . 2010 ; 28 ( suppl ) 15s (abstr 3534) .
42. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR . Nature . 2012 Jan 26 ; 483 : 100 – 103 .
43. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling . Science . 2007 May 18 ; 316 : 1039 – 1043 .
44. Basal subtype and MAPK/ERK kinase (MEK)-phosphoinositide 3-kinase feedback signaling determine susceptibility of breast cancer cells to MEK inhibition . Cancer Res . 2009 Jan 15 ; 69 : 565 – 572 .
45. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status . J Clin Oncol . 2011 ; 29 : 2011 – 2019 .
46. Genomics, health care, and society . N Engl J Med . 2011 ; 365 : 1033 – 1041 .
47. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers . Nature . 2012 Jun 28 ; 486 : 537 – 540 .
48. MEK1 mutations confer resistance to MEK and B-RAF inhibition . Proc Natl Acad Sci U S A . 2009 ; 106 : 20411 – 20416 .
49. Dissecting THERAPEUTIC Resistance to RAF inhibition in melanoma by tumor genomic profiling . J Clin Oncol . 2011 Aug 1 ; 29 : 3085 – 3096 .
50. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance . Nat Commun . 2012 Mar 6 ; 3 : 1 – 8 .
51. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF (V600E) . Nat Lett . 2011 Dec 15 ; 480 : 387 – 391 .
52. Melanomas require resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation . Nat Lett . 2010 Dec 16 ; 468 : 973 – 979 .
53. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression . Cancer Res . 2011 ; 71 : 2750 – 2760 .
54. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K . Cancer Cell . 2010 Dec 14 ; 18 : 683 – 695 .
55. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations . N Engl J Med . 2012 ; 367 : 1694 – 1703 .
56. Principles of cancer therapy: oncogene and non-oncogene addiction . Cell . 2009 Mar 6 ; 136 : 823 – 837 .
57. Cancer epigenetics reaches mainstream oncology . Nat Med . 2011 Mar ; 17 : 330 – 339 .
58. Interactions between lymphocytes and myeloid cells regulate pro- versus anti-tumor immunity . Cancer Metastasis Rev . 2010 Jun ; 29 : 309 – 316 .
59. The grand challenge to decipher the cancer proteome . Nat Rev Cancer . 2010 Sep ; 10 : 652 – 660 .
60. The concept of synthetic lethality in the context of anticancer therapy . Nat Rev Cancer . 2005 Sep ; 5 : 689 – 698 Review .
61. A pilot clinical study of treatment guided by personalized tumorgrafts in patients with advanced cancer . Mol Cancer Ther . 2011 Aug ; 10 : 1311 – 1316 .
62. Bayesian clinical trials at the University of Texas M . D. Anderson Cancer Center. Clin Trials . 2009 Jun ; 6 : 205 – 216 .
63. I-SPY 2: an adaptive breast cancer trial design in the setting of neoadjuvant chemotherapy . Clin Pharmacol Ther . 2009 Jul ; 86 : 97 – 100 .
64. Utilizing targeted cancer therapeutic agents in combination: novel approaches and urgent requirements . Nat Rev Drug Discov . 2010 ; 9 : 843 – 856 .
65. Combinatorial drug therapy for cancer in the post-genomic era . Nat Biotechnol . 2012 Jul 10 ; 30 : 679 – 692 .
66. Guidance for Industry Codevelopment of Two or More Unmarketed Investigational Drugs for Use in Combination. http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htm
67. . Informatics Needs and Challenges in Cancer Research: Workshop Summary . Washington, DC : The National Academies Press ; 2012 : 1-129 .
68. Rapid-learning system for cancer care . J Clin Oncol . 2010 Sep 20 ; 28 : 4268 – 4274 .
69. Molecular imaging: current status and emerging strategies . Clin Radiol . 2010 ; 65 : 500 – 516 .
70. Circulating mutant DNA to assess tumor dynamics . Nat Med . 2008 Sep ; 14 : 985 – 990 .
71. Circulating tumor cells: a window into cancer biology and metastasis . Curr Opin Genet Dev . 2010 Feb ; 20 : 96 – 99 .






