Immunotherapy for Metastatic Solid Cancers
Background of cancer immunology
The first cancer antigen to be characterized at a genetic and molecular level and recognized by T cells was MAGE-1 [1]. Since then, hundreds of peptides derived from tumors have been identified and shown to be expressed by solid tumors of various histologies, and restricted to presentation on different subclasses of MHC molecules [2]. Tumor-associated antigens fall into several major categories: (1) overexpressed normal proteins (eg, carcinoembryonic antigen [CEA] or nonmutated p53); (2) nonmutated differentiation antigens (eg, MART-1, overexpressed in melanoma and found in normal melanocytes); (3) cancer-testis antigens (CTA), consisting of nonmutated genes expressed during fetal development, then silent in normal adult tissues and reactivated in cancer cells across multiple malignancies (eg, MAGE and NY-ESO); and (4) mutated antigens, unique to a single tumor or shared by a group of tumors (eg, BRAF with the V600E mutation in melanoma and other solid tumors, or EGFRvIII in glioblastoma).
Despite the fact that tumor-associated antigens recognized by T cells have been described, solid cancers in humans grow and disseminate in immune competent hosts. Two main reasons, not mutually exclusive, explain this reality: (1) most cancers are weakly or not immunogenic, hence the frequency of tumor-reactive lymphocytes is low or null in most patients; and (2) immunologic mechanisms of T-cell anergy or tolerance, or immunosuppressive factors, either systemic or in the tumor microenvironment, thwart antitumor immune reactions. Box 1 summarizes general mechanisms described to result in cancer progression despite a competent immune system. Because cancer antigens are commonly nonmutated self-proteins, the naturally occurring pool of T cells able to recognize those self-antigens do so with low avidity, otherwise they would have been deleted during negative selection in the thymus during development. If a T cell is capable of reacting to a self-antigen in tissues and tumors, mechanisms involved in the prevention of autoimmunity are at play and referred to as peripheral tolerance. The fate of T cells able to strongly recognize altered self-antigens, such as mutated cancer antigens, is less clear. However, because the tumor can participate in immune suppression and tolerance at the tumor site in multiple ways, anticancer cytotoxic T cells are expected to lose cytotoxic functions and proliferative capacity, and may be driven to apoptosis. The lack of appropriate costimulation signals provided to naïve T cells by antigen-presenting cells found in an immature or inactivated state has also been proposed as a mechanism to explain the poor lytic and proliferative capacity of T cells on second encounters with tumor antigens.
Box 1 General mechanisms that can inhibit antitumor immune reaction in cancer patients
Multiple tumor mechanisms responsible for T-cell inhibition have been described, and include the secretion of soluble molecules able to suppress T-cell proliferation and functions (eg, transforming growth factor-β [TGF-β], interleukin-10, arginase-1, nitric oxide synthase 2), the competition for molecules essential for T-cell metabolism (eg, glucose, tryptophan), and the expression of surface molecules that inhibit immune cell activation (eg, programmed death ligand 1 [PD-L1, also called B7H1]) [3]. In addition, cancer cells can go unrecognized by T cells simply by downregulation of MHC molecules and by downmodulation of proteins involved in tumor antigen processing and presentation at the cell surface (eg, transporter associated with antigen processing 1 [TAP1]). The tumor microenvironment is also enriched in immunosuppressive cells, such as regulatory T cells (Treg) [4]. Recent studies also suggest that inhibition of T cells can be mediated by innate immune cells, granulocytic, monocytic, or their precursors, and are now generally referred to as myeloid-derived suppressor cells (MDSC) [5,6]. The potency of these regulatory immune cell subsets to impair antitumor cytotoxicity by T cells has been established mainly in mouse models, and the specific mechanisms by which T-cell inhibition occurs remains to be elucidated. In humans, in vitro assays have suggested the existence of Treg and MDSC in cancer patients, but in vivo studies are in their infancy. The heterogeneity of the transformed cells that constitute a tumor mass also contributes to tumor progression, because many cancer cells may go unrecognized by the immune system while partial tumor destruction by T cells occurs. This “natural selection” of less immunogenic tumor cells over time has been coined “tumor immunoediting” and is mainly supported in animal models [7,8].
Nonspecific immunotherapy
Stimulation of effector T cells
The T-cell growth factor interleukin (IL)-2 can activate endogenous tumor-reactive cells, and reproducibly mediate the regression of advanced metastatic melanoma and kidney cancer. In a consecutive series of 409 patients treated at the Surgery Branch of the National Cancer Institute (NCI), between 1985 and 1996, high-dose bolus intravenous IL-2 administration produced complete and durable regressions of metastatic melanoma and renal cell cancer in 6.6% and 9.3% of patients, respectively [9,10]. Responses were seen at all sites of disease, and more than 80% of complete responses appeared durable and were ongoing after a median follow-up of 7 years at the time of publication. IL-2 alone does not appear sufficient to induce regression of other solid cancers. The US Food and Drug Administration (FDA) approved the use of IL-2 for the treatment of metastatic renal cancer in 1992 and metastatic melanoma in 1998, based on the ability of IL-2 to mediate durable complete responses. For the same reason, IL-2 therapy should be the first-line treatment for patients with metastatic renal cancer and metastatic melanoma.
Experience with the administration of IL-2 has resulted in treatment-related mortalities of less than 1%. Toxicities occur owing to a capillary leak syndrome, and can be safely treated with appropriate monitoring and judicious fluid resuscitation [11]. Organ-specific autoimmunity seen in melanoma patients treated with IL-2, such as delayed-onset vitiligo in approximately 20% of patients and the development of autoimmune thyroiditis in approximately 55%, has been associated with the likelihood of cancer regression [12]. Of interest, patients with renal cell carcinoma do not develop vitiligo with IL-2, and autoimmune thyroid is seen less commonly in these patients, arguing for activation of a different subset of T cells in melanoma patients, able to recognize antigens expressed on tumor and on normal melanocytes (mainly MART-1, gp100, and Tyrosinase) [10].
Interferon-α2b (IFN-α2b), an important mediator of antiviral immunity, has also been used for the treatment of patients with melanoma and renal cell cancer. The role of IFN-α2b for the adjuvant treatment of patients at high risk of recurrence after definitive surgery is controversial, since improvements in recurrence-free survival have translated into little impact on overall survival. Long-term administration of a pegylated formulation, expected to maintain maximum exposure to IFN-α2b with less frequent subcutaneous injections than with the unpegylated formulation, has recently led to similar results in a large randomized control trial of patients with node-positive melanoma (stage III) [13,14]. In this trial, the relapse-free survival was improved by 9.3 months (34.8 months vs 25.5 months), without benefit in overall survival. The FDA approved pegylated IFN-α2b (Sylatron) on March 29, 2011 for the treatment of melanoma patients with microscopic or gross nodal involvement.
Blockade of negative regulators of T-cell function
Activated T cells express surface inhibitory molecules that, when bound by their ligands, are capable of inhibiting T-cell activity. Monoclonal antibodies directed against these surface inhibitory molecules have been studied as immunotherapeutic regents. The cytotoxic T-lymphocyte–associated 4 molecule (CTLA-4, CD152), part of the immunoglobulin-like family of surface proteins, is one of the inhibitory molecules expressed at the surface of activated T cells. Two fully human IgG monoclonal antibodies recognizing CTLA-4, ipilimumab (MDX-010) and tremelimumab (CP-675,206), have been tested, alone or in combination, in phase 2/3 trials. These antibodies are designed to prevent the binding of CTLA-4 to its ligand B7, mainly expressed on immature antigen-presenting cells and tumor cells [15].
The first demonstration of the ability of ipilimumab to mediate tumor regression in 2003 reported objective regressions in 3 of 13 patients with metastatic melanoma [16]. An updated summary of consecutive cohorts of 179 patients with metastatic melanoma revealed an overall response rate ranging from 13% to 25%, including 6% to 17% durable complete responses [17]. The highest response rates were seen in patients given ipilimumab in combination with high-dose IL-2. In this cohort, 6 of 36 (17%) patients enjoy ongoing complete responses after 7 years of median follow-up. It is interesting that prior response to IL-2 was not correlated with the likelihood of response to ipilimumab, pointing to a different quality of interaction between melanoma and the host immune system using these two agents alone. Important but delayed responses to treatment were observed in some patients.
Immune-related adverse events were more frequently observed in responders than in nonresponders, and could be severe. Approximately 35% of patients developed Grade 3 and 4 immune-related toxicities, the most common being enterocolitis in 17%, followed by hypophysitis and dermatitis in 9% and 6%, respectively. Other less common side effects included hepatitis, nephritis, uveitis, and arthritis. With the exception of hypophysitis with hypopituitarism, immune-related complications were usually reversible with systemic and topical corticosteroid treatment. The addition of anti–tumor necrosis factor α (infliximab) monoclonal antibodies to systemic corticosteroids has been successfully used to treat patients with severe colitis. As reported in a recent literature review, a colectomy may be life-saving for some patients—as much as 12% in one series—who develop bleeding or perforation from colitis unresponsive to medical therapy [18].
The effectiveness of ipilimumab as second-line treatment for patients with advanced melanoma has now been confirmed in a large double-blinded, randomized, multi-institutional phase 3 trial [19]. A total of 676 HLA-A*0201 patients were randomized 3:1:1 to receive ipilimumab plus a gp100 vaccine, ipilimumab alone, or the gp100 vaccine alone. The overall median survival was equivalent in both groups of patients who received ipilimumab, approximately 10 months versus 6.4 months for the group who received the gp100 vaccine alone. Objective responses were seen in 38 patients among the 540 who received ipilimumab (7%), with 3 complete responses (0.5%). Immune-related side effects were seen at comparable rates to what has been described above, and 4 patients died following bowel perforation, due to ipilimumab-induced colitis. The addition of the vaccine did not confer a survival benefit or lead to unexpected side effects. Based on the results of this trial, the FDA approved ipilimumab (Yervoy) for the treatment of unresectable or metastatic melanoma on March 25, 2011. Ongoing trials are now assessing the efficacy anti–CTLA-4 in combination with other biological and cytotoxic agents, notably dacarbazine [20].
The efficacy of ipilimumab has been tested for other solid malignancies. In a nonrandomized phase 2 study of 40 patients with metastatic renal cell cancer treated with ipilimumab alone, 5 patients had a partial response [21]. A single or 2 doses of ipilimumab has been associated with a decrease of 50% or more of the prostate-specific antigen (PSA) level in 2 of 12 patients with metastatic hormone-refractory prostate cancer patients [22]. Of 27 patients with unresectable or metastatic pancreatic adenocarcinoma treated with ipilimumab, only one experienced a mixed response [23].
Tremelimumab, the second fully humanized anti–CTLA-4 monoclonal antibody, has been less studied than ipilimumab. In the most recent and largest phase 2 trial in patients with advanced melanoma treated with tremelimumab, a response rate of 6.6% in 246 patients was reported, without complete responses, 2 treatment-related deaths, and a median survival of 10 months [24]. In heavily pretreated metastatic colorectal cancer patients, 1 of 45 partial but sustained response was reported [25].
Programmed death 1 (PD-1, CD279) is another inhibitory molecule expressed at the surface of activated T cells after repeated encounter with antigen. It belongs to the same family of surface molecules as CTLA-4, but binds different ligands and provides distinct intracellular signaling that leads to shutdown of T-cell effector function. Tumor cells and antigen-presenting cells found in tumors can express a high level of PD-L1, the main PD-1 ligand (also called B7-H1), and this has been associated with poor prognosis in renal and ovarian cancer [26,27]. Interrupting the interaction between PD-1 and its ligand using the fully human IgG4 monoclonal antibody MDX-1106 has been reported to mediate cancer regression in a phase 1, dose-escalation trial [28]. Objective response was documented in 3 of 39 patients, one each with melanoma, renal cancer, and colorectal cancer.
Summary and new directions
Overall, nonspecific modulation of immunity, either to promote activation or to block inhibition of effector T cells, can mediate tumor regression mainly in a subset of patients with metastatic melanoma and renal cancer. Although occasional tumor responses have been observed for other solid cancers, melanoma and renal cancer appear exceptional in their ability to harbor endogenous antitumor cells of sufficient avidity and in sufficient numbers to respond to nonspecific immunomodulators. It remains to be seen if combination of standard chemotherapy with immunomodulators such as anti–CTLA-4 and anti-PD1, strategies currently being tested, will expand the use of these agents to other solid tumors. Investigations are under way to evaluate other general immune modulators for solid cancer treatment. IL-15 is under investigation at the NCI for patients with melanoma. The rationale behind using IL-15 instead of IL-2 is to promote the expansion of a pool of effector-memory T cells and to avoid preferential expansion of regulatory T cells, because the later constitutively express the high-affinity receptor chain for IL-2. IL-21, another T-cell–stimulating cytokine, was reported to mediate objective response in 22% of patients with metastatic melanoma [29]. IL-12, a cytokine that activates APCs, T cells, and the natural killer subset of lymphocytes, had shown good tumor effect in animal models; however, attempts to give a therapeutic dose of IL-12 systemically in humans have been limited by toxicities [30]. Current efforts are focused on strategies to deliver IL-12 at the tumor site in order to avoid systemic toxicities [31].
Active immunization approaches (cancer vaccines)
With rare exceptions, therapeutic cancer vaccines have not been effective in the treatment of cancers in animal models or in humans. Successful vaccines for infectious disease are preventive and designed to initiate protective humoral immune responses mediated by antibodies. These approaches have thus far been largely unsuccessful in generating the highly avid T cells required to destroy cancer cells. Vaccines to prevent cervical cancer or hepatoma target the human papilloma viruses 16 and 18 or hepatitis B viruses, respectively, that are involved in the etiology of those cancers [32,33]. Most solid malignancies, however, do not have known viral etiology. The challenge of therapeutic cancer vaccines is to rely on rare spontaneous antitumor T-cell precursors to mount an immune response in vivo against weakly immunogenic tumor antigens and, if successful, to deliver antitumor T cells to a tumor microenvironment that is overwhelmingly immunosuppressive.
The efficacy of these approaches, assessed by tumor shrinkage defined by standard response criteria, has been consistently low. In a review of cancer vaccines published in 2004, the mean objective response rate when calculated from 765 patients with metastatic cancers treated in 35 trials using the aforementioned type of vaccines was estimated to be 3.8% [34]. At the Surgery Branch, NCI, an overall objective response rate of 2.6% was observed in 440 patients with metastatic cancers treated with a variety of vaccines in sequential trials. This lack of objective response occurred even when functional T cells specific to the antigen used for vaccination were generated [35]. A recent comprehensive review of nonrandomized cancer vaccine trials published since 2004 including 936 patients treated in 41 trials confirmed a low overall response rate of 3.6% [36].
The previously cited vaccine trials were not powered to formally test patient survival. However, 8 recent prospective randomized therapeutic cancer vaccine trials for melanoma, and kidney, lung and prostate cancers did not show efficacy of different agents that looked promising in earlier uncontrolled trials [37–44]. Although various vaccination strategies were tested in these large trials, 3 of them used allogeneic cancer cell lines in metastatic melanoma (Canvaxin) and in prostate cancer (GVAX), suggesting major limitations with this approach.
Interest in cancer vaccines has nonetheless been spurred by two recent studies. High-dose IL-2 with or without immunization with a melanoma differentiation antigen (gp100 peptide) in incomplete Freund adjuvant has been evaluated in a prospective randomized phase 3 trial conducted in 21 centers for patients with stage IV melanoma [45]. In the 86 evaluable patients who received the peptide vaccine in conjunction with IL-2, the response rate was of 22.1% versus 9.7% among the 91 patients who received IL-2 alone (P = .02). Progression-free survival was prolonged in the vaccine plus IL-2 group compared with IL-2-alone group (2.9 months vs 1.6 months; P = .01). The difference in overall survival (17.6 vs 12.8 months, P = .096), favoring the combination regimen was only suggestive.
In metastatic castration-resistant prostate cancer, a double-blinded, multicenter, placebo-controlled phase 3 trial comparing the efficacy of an autologous activated leukocyte-based product (Sipuleucel-T) versus a non-antigen containing leukocyte product in 512 men showed improvement in median survival by 4.1 months in the vaccinated arm (25.8 vs 21.7 months, P = .032) [46]. The Sipuleucel-T product was constituted in each patient from autologous mononuclear cells collected by leukapheresis. The leukocytes were stimulated in vitro by a fusion protein consisting of prostatic acid phosphatase (PAP) and GM-CSF. The activated cellular product, tailored for each patient in a central facility, was then reinfused into the autologous patient in 3 biweekly doses. It is not clear which component of the infused product mediated the anticancer effect. Given the consistency of the survival results with a previous smaller randomized trial (n = 127) and the favorable toxicity profile, Sipuleucel-T became the first nonviral-related cancer vaccine approved by the FDA on April 29, 2010 for minimally symptomatic metastatic hormone-refractory prostate cancer. These results remain nonetheless surprising because there was no difference among the groups in progression-free survival, minimal tumor responses using Response Evaluation Criteria in Solid Tumors (RECIST) criteria, and no significant decline in PSA value in 97% of patient receiving Sipuleucel-T [47].
A different vaccine has also demonstrated a benefit in overall survival without evidence of tumor shrinkage and change in progression-free survival in patients with castration-resistant prostate cancer. This vaccine uses vaccinia and fowlpox viruses encoding PSA along with 3 costimulatory molecules: B7.1, intercellular adhesion molecule 1 (ICAM-1), and lymphocyte function-associated antigen 3 (LFA-3). In a 43-center randomized phase 2 trial (n = 125), this “PSA-TRICOM” vaccine was associated with a median survival of 25.1 months, which was 8.5 months longer than for patients receiving the placebo (P = .015). The ongoing phase 3 trial with this vaccine should provide new data to resolve the apparent paradox of improved survival without objective tumor response [48,49]. Clinical trials testing the efficacy of this type of vaccine for patients with advanced cancers of the gastrointestinal tract are under way [50].
Adoptive cell transfer of activated immune cells with antitumor activity
Adoptive cell transfer using tumor-infiltrating lymphocytes
In 1986 it was demonstrated that murine sarcoma and colon adenocarcinoma transplanted in non-immunized syngeneic mice harbored TIL that could be expanded in vitro with IL-2 and would mediate regression of disseminated tumors after adoptive transfer (Rosenberg SA and colleagues, Science 1986 [51]). The adoptive transfer of TIL obtained from human melanomas was first shown to mediate regression of autologous metastatic melanoma in 1988 [52], but decisive improvement in efficacy came in 2002 with the introduction of an immunodepleting preparative regimen given before TIL infusion [53]. This approach could result in clonal repopulation of patients’ circulating lymphocytes with antitumor activity [54]. This lymphodepleting preparative regimen was shown to contribute to the antitumor activity of the transferred TIL primarily by depleting endogenous regulatory cells and by depleting endogenous lymphocytes that competed with the transferred cells for growth-promoting homeostatic cytokines such as IL-7 and IL-15. A schematic description of TIL therapy is illustrated in Fig. 1.
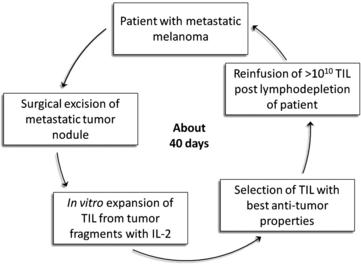
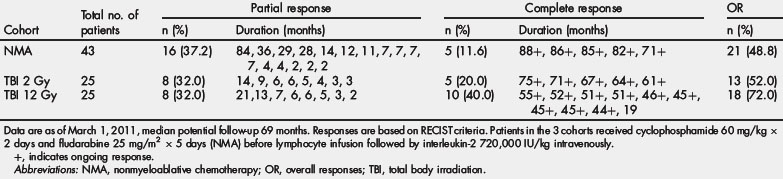
Three sequential ACT trials performed in the Surgery Branch, NCI, on 93 patients using autologous TIL harvested from metastatic melanoma patients infused with IL-2 after preconditioning immune suppression, are summarized in Table 1. The median potential follow-up of these trials was 69 months. Increasing the level of immune suppression using total body irradiation combined with lymphodepleting chemotherapy before TIL infusion was associated with a higher overall and complete response rate. The objective response rate by RECIST criteria reached 72% with maximum immune suppression, including 40% of patients with complete tumor eradication. Of the 20 complete responders enrolled in these 3 trials only one has relapsed, with all others in ongoing complete response beyond 3 to 7 years. Nonhematological grade 3 and 4 toxicities observed in the cohort of patients receiving the nonmyeloablative regimen were febrile neutropenia in 37% and intubation for dyspnea in 9%. Adding total body irradiation at high dose led to more intubation for somnolence, but comparable rate of adverse events otherwise. In 93 patients treated, one mortality was observed consequent to an unrecognized diverticular abscess.
Adoptive cell transfer using genetically modified autologous peripheral blood lymphocytes
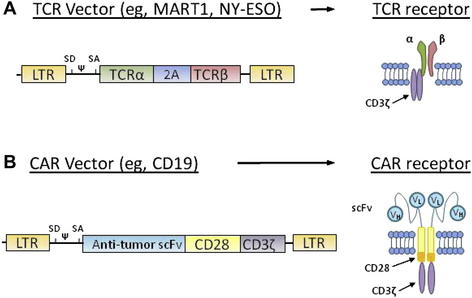
Two types of receptors can be introduced into T cells to redirect effector T-cell specificity to tumor antigens. The first is a conventional TCR comprising two chains (α and β) that recognize peptides presented by MHC molecules. Thus, these TCRs can recognize antigens only on specific human leukocyte antigen haplotypes (HLA). The first vectors were designed to recognize peptides presented by HLA-A*0201, one of the most commonly expressed HLA in humans. The second type of receptor that recognizes tumor antigens is called chimeric antigen receptor (CAR). A CAR is a fusion protein that links the variable portions of the heavy and light chains of an antibody to the intracellular signaling domains of a TCR. Introduced into a T cell, CAR enable the lymphocyte to recognize tridimensional proteins found at the surface of tumor cells without MHC restriction, rather than a short peptide nested in MHC molecules recognized by conventional TCR. By combining the antigen specificity of an antibody and the cytotoxic properties of a T cell in an HLA-unrestricted manner, CAR can be resistant to tumor-immune evasion mechanisms, such as downregulation of MHC molecules and failure to process antigens to the cell surface. Selected TCR and CAR expression vectors optimized at the Surgery Branch are summarized in Fig. 2. In the United States, all clinical trials using gene transfer technology are reviewed by the National Institutes of Health Office of Biotechnology (OBA) Activities. A list of gene therapy protocols can be found on the OBA Web site (http://oba.od.nih.gov), and on the Clinical Trials Web site (www.clinicaltrials.gov).
TCR-engineered T cells
The first report of ACT using TCR gene-engineered lymphocytes for the treatment of metastatic melanoma used a TCR isolated from a patient who had been administered TIL therapy with excellent clinical response [55]. The TCR genes were inserted into a gammaretroviral vector, and the transduced lymphocytes displayed a high level of antitumor activity in vitro [56]. Fifteen patients were treated with MART-1 TCR gene-engineered T cells after a preconditioning immunodepleting chemotherapy along with IL-2 administration. Sustained tumor regression was observed in 2 patients (13%). In an attempt to increase the response rate, a subsequent trial employed 2 more highly reactive TCRs, one against MART-1 and a second against gp100 [57]. Objective regression of tumor was observed in 6 of 20 (30%) and 3 of 16 (19%) patients, with the 2 TCRs respectively. Although the response rate was improved, significant toxicity to the skin, eye, and ear were documented and could be explained by the low expression of the targeted melanocyte antigens found at these sites. The on-target toxicities to the eye and ear were managed by steroid drops and transtympanic steroid injections, and disabling impairments were avoided.
Recently, a series of 3 patients with metastatic colorectal cancer refractory to standard treatment were treated with T lymphocytes engineered to express a murine TCR against human CEA [58]. For this study, the TCR gene was isolated by immunizing HLA-A*0201 transgenic mice with the immunogenic peptide CEA:691-699 [59]. The functional avidity of the TCR was enhanced by introducing a single amino acid substitution in the α chain [60]. One of 3 patients demonstrated an objective response. Although serum CEA levels dropped by 74% to 99% after ACT in all 3 patients, these decreases were transient, with a nadir at 3 to 4 months. All 3 patients experienced severe colitis approximately 1 week post cell transfer that recovered by 2 to 3 weeks post cell transfer. This trial emphasizes the need to identify and choose antigens most restricted to tumor targets, given the potency of the gene-engineered cells and the risks of autoimmune complications when even low levels of antigens are expressed on normal tissues.
The cancer-testis antigen family members appear to represent ideal tumor antigen candidates because they are expressed by a wide range of solid malignancies, found only in germ cell tissues but not in other normal tissues [61]. NY-ESO-1, discovered in 1997, is a cancer-testis antigen known to elicit spontaneous antibody and T-cell responses in cancer patients [62]. As recently reported, the adoptive transfer of NY-ESO-1 TCR-engineered T-cell mediated objective cancer regressions in 4 of 6 patients with metastatic synovial sarcoma, and in 5 of 12 patients with metastatic melanoma [63,64]. Encouraging tumor responses were seen in the absence of organ-specific toxicities. Other cancer-testis antigens, such as the MAGE family, presented by HLA-A2 and other class-I MHC subclasses, are also being targeted to increase the pool of eligible patients with diverse tumor types and to optimize the efficacy of this approach [65].
CAR-engineered T cells
Early studies of ACT using CAR used constructs made of a single-chain variable fragment of an antibody combined to the transmembrane and intracellular signaling domains of either CD3ζ or the gamma chain of antibody receptor Fc (FcRg) lymphodepletion. These studies resulted in short-term persistence of cells in vivo, no clinical benefit, and no overt toxicities. Carbonic-anhydrase-IX (CAIX), frequently overexpressed on clear cell renal cell carcinoma, was targeted with a CAR based on a murine monoclonal antibody at the Daniel den Hoed Cancer Center in Rotterdam, The Netherlands [66,67]. No objective clinical response was observed in 11 patients treated in 3 sequential cohorts. Grade 3 liver toxicity was observed in 3 patients, resulting from the recognition of CAIX on bile ducts. Limited persistence of the infused cells was a hallmark of this trial, as well as the development of antibody responses to the murine portion of the CAR in 6 out of 7 evaluable patients. At the Surgery Branch, NCI, CAR that targeted the α-folate receptor were administered to 14 patients with metastatic ovarian cancer without preconditioning lymphodepletion [68]. There was no evidence of clinical and biochemical response based on serum carbohydrate antigen–125 levels in this trial, and rapid disappearance of the engineered cells from the circulation was documented.
A strategy to improve first-generation CAR by providing costimulation to CAR-transduced T cells was tested at the Baylor College of Medicine in Houston, Texas. Epstein-Barr virus (EBV)-specific T lymphocytes found in the circulation of patients were transduced with CAR directed against diasialoganglioside GD2, a nonviral tumor antigen expressed by neuroblastoma [69]. The rationale behind this strategy was to provide costimulation to T cells after engagement of their native anti-EBV TCR, while allowing engagement of the CAR with GD2 on neuroblastoma cells. Persistence of infused CAR-EBV–specific T cells was indeed improved compared with standard bulk engineered T cells infused concurrently in all subjects. No adverse events attributed to the genetically modified T cells were reported, and 4 of the 8 patients with evaluable disease experienced tumor necrosis or regressions, one achieving a complete response.
Second-generation and third-generation CAR attempted to improve persistence and function of transduced cells by incorporating one or more costimulatory intracellular signaling molecules, such as CD28, OX40, and 4-1BB. The first successful treatment of a patient with a highly refractory lymphoma targeting the CD19 antigen using a CAR containing CD28 and CD3ζ intracellular signaling chains was recently reported [70]. The dangers of this approach, however, were emphasized by two recent reports of deaths following administration of CAR-transduced T cells, one in a patient who received CAR targeting ERBB2 (HER2/neu) [71] and the second in 1 of 6 patients with chronic lymphocytic leukemia treated with a second-generation CAR that recognized CD19 [72,73].
Summary and future directions
Adoptive transfer of TIL is the most effective therapy reported thus far for patients with metastatic melanoma. A simplified method of generating TIL, allowing for the treatment of additional patients in a shorter time frame, is currently being clinically evaluated [74]. Other institutions have now begun to treat patients with metastatic melanoma with adoptive transfer of TIL. At the M.D. Anderson Cancer Center, approximately a 50% objective rate was seen in 30 patients receiving TIL selected for tumor reactivity after nonmyeloablative lymphodepletion [75]. At the Sheba Medical Center in Israel, administration of TIL led to 10 objective responses out of 20 patients treated, with 2 complete responses [76]. At the Surgery Branch, NCI, the efficacy of TIL therapy is currently being tested for patients with metastatic digestive tract adenocarcinomas.
Gene engineering of peripheral blood T cells is capable of mediating regression of metastatic melanoma and other solid malignancies. The choice of target antigen is critical. Targeting differentiation tumor antigens, such as MART-1, gp100, and CEA, or overexpressed normal proteins such as HER2/neu, may be accompanied with significant toxicities. Targeting the family of cancer-testis antigens, such as NY-ESO, or mutated antigens expressed exclusively by cancer cells, appears promising. Targeting the tumor stroma with a CAR against the vascular endothelial growth factor receptor 2, overexpressed in the tumor vasculature and by some myeloid cells, is an effective strategy in mouse models [77]. Cells with antitumor reactivity could also be used as a “Trojan horse” to deliver other molecules at the tumor site, such as cytokines.