Published on 19/03/2015 by admin
Filed under Dermatology
Last modified 22/04/2025
This article have been viewed 2339 times
Lori D. Prok and James E. Fitzpatrick
Evidence Levels: A Double-blind study B Clinical trial ≥ 20 subjects C Clinical trial < 20 subjects D Series ≥ 5 subjects E Anecdotal case reports
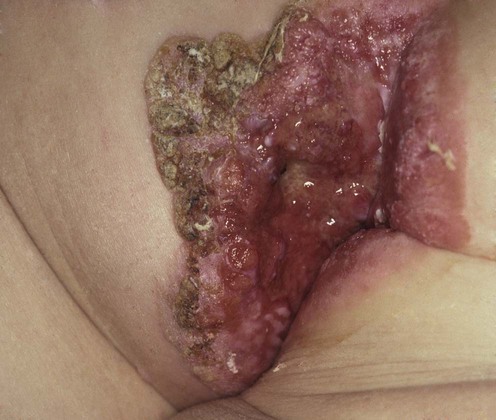
Extramammary Paget disease (EMPD) was first described by Crocker in 1889, when he noted skin lesions affecting the penis and scrotum of a male patient that were identical to the nipple disease described by Paget in 1874. Although an uncommon malignancy, EMPD should be included in the clinical differential diagnosis of any chronic dermatitis of the groin or perineum. EMPD most commonly affects postmenopausal Caucasian women, but can also be seen in men of all ethnicities. It typically presents as chronic, often sharply demarcated, erythematous scaling plaques of apocrine gland-bearing skin, including the genitalia, axillae, umbilicus, and external auditory canal. Pruritus is the most common presenting symptom. Primary EMPD results from epidermal infiltration of neoplastic glandular cells. Recent evidence supports the role of Toker cells (clear cells found in 10% of normal nipples, and recently identified in tissue of the milk line and the vulva) as the pathologic cell in this disease. Secondary EMPD accounts for approximately 25% of cases and is the result of direct cutaneous extension of an underlying adenocarcinoma, most commonly of the genitourinary system or from the anal canal.
Clinical suspicion of EMPD should prompt an immediate skin biopsy. Histologically, neoplastic cells are characterized by pale vacuolated cytoplasm and large pleomorphic nuclei, which can be seen infiltrating all levels of the epidermis. Extension into adnexal structures is common. Historically the diagnosis was established by demonstrating the presence of sialomucin with positive staining with mucicarmine, colloidal iron, periodic acid–Schiff, and Alcian blue at pH 2.5. While these studies can still be utilized, the diagnosis is now usually established by immunoperoxidase studies to demonstrate positivity for carcinoembryonic antigen (CEA), epithelial membrane antigen, CK7, gross cystic disease fluid protein-15 and/or Ber-EP4 to differentiate EMPD from pagetoid variants of squamous cell carcinoma in situ and melanoma in situ. CK7 positivity is also supportive of a diagnosis of primary EMPD, since it is less commonly expressed in secondary EMPD. In select cases it is used in tandem with CK20 since CK20 positivity is more commonly seen in secondary EMPD. Recent studies have also demonstrated that HER2/neu over-expression is useful both in helping to establish a diagnosis of EMPD but also in identifying invasive cases that might be responsive to trastuzumab.
A full-body skin examination and lymph node evaluation should be performed in all patients with EMPD. Patients should then have appropriate evaluation for underlying malignancy, including age- and gender-appropriate screening (Papanicolaou smear, fecal occult blood, colonoscopy, cystoscopy, and prostate-specific antigen). Additional investigations (imaging, colposcopy, etc.) are guided by screening results and the anatomic location of cutaneous lesions. Patients who have invasive EMPD (extension into the dermis and/or lymphatic vessels) should be considered for sentinel lymph node biopsy.
EMPD is treated locally with surgical excision, with adjuvant therapies in selected cases. Mohs micrographic surgery is the preferred technique, offering the most reliable margin control, maximal tissue preservation, and lowest recurrence rates. However, this technique is still limited by non-contiguous tumor spread and the high likelihood of EMPD involving clinically normal-appearing skin.
Skin biopsy
Full-body skin examination and lymph node evaluation
Cancer screening appropriate for age and gender (Papanicolaou smear, fecal occult blood, colonoscopy, cystoscopy, and prostate-specific antigen)
Sentinel lymph node biopsy in patients with dermal and/or lymphatic extension
Hatta N, Yamada M, Hirano T, Fujimoto A, Morita R. Br J Dermatol 2008; 158: 313–18.
Retrospective review of 76 patients with EMPD. Surgical margin was not correlated with local recurrence. Seventeen percent developed systemic metastases; 10 patients died. Nodules in the primary tumor, clinical lymph node swelling, elevated CEA levels, depth of tumor invasion, and lymph node metastasis were significant prognostic factors. Depth of tumor invasion and CEA level were associated with reduced survival.
Siesling S, Elferink M, van Dijck J, Pierie JP, Blokx WA. Eur J Surg Oncol 2007; 33: 951–5.
Retrospective review of 226 cases of EMPD in the Netherlands Cancer Registry, most of which were treated with surgical excision. Five-year survival for those with invasive disease was 72%. Patients had an increased risk of developing a second primary cancer (standard incidence ratio 1.7).
Tsutsumida A, Yamamoto Y, Minakawa H, Yoshida T, Kokubu I, Sugihara T. Dermatol Surg 2003; 29: 21–4.
A prospective study of 34 patients with genital or perineal EMPD treated with wide local excision. Patients with clinical or histologic evidence of metastatic disease underwent lymph node dissection. No patients with carcinoma in situ or microscopic papillary dermal invasion had lymph node metastasis; all had 100% 5-year survival. Tumor invasion into the reticular dermis correlated with 33% 5-year survival. Tumor invasion into the subcutaneous tissue correlated with 100% lymph node metastasis and death.
Shiomi T, Noguchi T, Nakayama H, Yoshida Y, Yamamoto O, Hayashi N, Ohara K. J Eur Acad Dermatol Venereol Feb 25 2012; [Epub ahead of print].
Retrospective review of 51 surgical specimens of primary invasive EMPD. Cases were divided into subgroups according to invasion depth: dermal invasion ≤1 mm (minimal invasion) and dermal invasion >1 mm in depth. Lymph node metastases were detected in two (7.7%) of 26 patients with minimal invasion and nodal metastases in 22/25 (88%) of patients with invasion >1 mm in depth. The authors conclude there is evidence to suggest that a cut-off depth of 1 mm of invasion is useful in staging patients for prognosis and treatment.
Wang Z, Lu M, Dong G, Jiang YQ, Lin MS, Cai ZK, et al. BJU Int 2008; 102: 485–8.
Retrospective review of 130 Chinese patients with penoscrotal EMPD. All underwent wide local resection and reconstruction. Forty-five patients had frozen-section margin confirmation during surgery; five with positive margins required immediate extended resection. Mean follow-up was 3.2 years (81/130 patients). Tumor recurrence was documented in five of nine patients with positive margins and in three of 72 patients with negative margins. Five patients died from metastatic disease.
Zhu Y, Ye D, Chen Z, Zhang SL, Qin XJ. BJU Int 2007; 100: 1282–7.
Retrospective review of 38 patients with primary penoscrotal EMPD who received wide local excision with intraoperative frozen-section analysis. Thirty-two percent had positive frozen-section margins and required immediate extended excision. Forty percent of patients had positive surgical margins after traditional wide local excision with 2 cm margins. At a mean follow-up of 33 months 16% of patients had recurrent disease, and four had systemic involvement.
Hendi A, Brodland D, Zitelli J. J Am Acad Dermatol 2004; 51: 767–73.
Retrospective review of patients with EMPD treated with Mohs micrographic surgery. Patients treated with Mohs micrographic surgery had recurrence rate of 16% for primary EMPD and 50% for recurrent disease, and 5-year tumor free rates of 80% for primary tumors and 56% for recurrent tumors. Literature review found 33–60% recurrence after non-Mohs micrographic surgical excision.
O’Connor W, Lim K, Zalla M. Dermatol Surg 2003; 29: 723–7.
Retrospective review of 95 patients at the Mayo Clinic comparing tumor recurrence in patients treated with Mohs micrographic surgery (8%) compared to wide local excision (22%). Surgeons used intraoperative staining with CK7.
Raspagliesi F, Fontanelli R, Rossi G, Ditto A, Solima E, Hanozet F, et al. Gynecol Oncol 2006; 103: 581–6.
Pilot study using photodynamic therapy and methyl 5-aminolevulinate (an ester of 5-ALA with higher efficacy and fewer side effects) to treat recurrent vulvar EMPD. Seven patients were treated weekly for 3 weeks, and follow-up biopsies were obtained 1 month after treatment. Four patients had complete clinical response; this was histologically confirmed in two cases.
Nardelli AA, Stainski T, Menon D. BMC Dermatol 2011; 15: 11:13.
Following Cochrane guidelines, this study reviewed of all clinical studies and reports reporting the use of photodynamic therapy for mammary and EMPD. This study identified a total of 99 patients (total of 133 lesions) with EMPD treated with modality. Although the follow-up periods were typically 1 year or less, the reported complete response was 58% (77/133 lesions). Although this a higher recurrence rate than most studies utilizing surgical modalities, this treatment has the advantage that it can be repeated without functional or physical impairment.
Fujisawa Y, Umebayashi Y, Otsuka F. Br J Dermatol 2006; 154: 375–6.
Report of a patient with EMPD with bone and lung metastases whose tumor markers and metastatic lesions decreased after treatment with intravenous docetaxel. The patient is still being treated on an outpatient basis, with good function and tumor control.
Hanawa F, Inozume T, Harada K, Kawamura T, Shibagaki N, Shimada S. Case Rep Dermatol 2011; 3: 223–7.
Report of a single patient with EMPD of the vulva with lymph node metastases in which the tumor cells expressed strong staining with HER2/neu. The patient demonstrated regression in both the primary tumor and the metastatic lesion after four courses of trastuzumab and paclitaxel.
Kariya K, Tsuji T, Schwartz R. Dermatol Surg 2004; 30: 341–4.
Case report of penoscrotal EMPD and multiple visceral metastases treated with intravenous 5-fluorouracil and cisplatin for 6 weeks. The patient had resolution of cutaneous disease, with a significant reduction in tumor markers and radiographic evidence of metastatic disease.
Watanabe Y, Hoshial H, Ueda H. Int J Gynecol Cancer 2002; 12: 304–7.
Three patients with invasive vulvar Paget’s disease who declined surgery were treated with low-dose mitomycin, etoposide, and cisplatin. One achieved a complete response; two showed a partial response and ultimately underwent partial vulvectomy and inguinal lymph node dissection. No patients had recurrent disease at 10 months’ follow-up.
Hata M, Koike I, Wada H, Miyagi E, Odagiri K, Minagawa Y, et al. Anticancer Res 2012; 32: 3315–20.
Retrospective review of the outcomes of 14 patients with EMPD including three patients with regional lymph node metastases. Total doses of 52–80.2 Gy (median = 60.6 Gy) were utilized with a median of 33 fractions. The 5-year disease-free, cause-specific and overall survival rates were 46%, 100%, and 79%, respectively. The authors conclude that radiation therapy is effective and safe with this being a curative option for patients that are inoperable.
Hatch K, Davis J. J Low Genit Tract Dis 2008; 12: 90–4.
Two patients with vulvar EMPD achieved complete clinical clearance after application of topical imiquimod 5% cream. Histology confirmed tumor resolution.
Feldmeyer L, Kerl K, Kamarashev de Viragh P, French LE. J Dermatol Case Rep 2011; 21: 42–6.
This is a case report of a woman with primary EMPD of the vulva who had a complete clinical response to a course of topical imiquimod applied three times per week for 18 weeks with a 1-year follow-up. This paper is useful in that the authors reviewed and compiled the results in 17 patients in 12 publications utilizing this therapy. The total treatment duration varied from 6 to 24 weeks (median = 11.4 weeks) with a reported follow-up of 0.5 to 26 months (median = 10.5 months). The reported complete response rate in the reported cases was 88% (15/17).
Ye J, Rhew D, Yip F, Edelstein L. Cutis 2006; 7: 245–50.
Report of a patient with EMPD (recurrent after surgery and resistant to topical imiquimod alone) that resolved after treatment with combination imiquimod and topical 5-fluorouracil and retinoic acid.
Panasiti V, Bottoni U, Devirgilis V, Mancini M, Rossi M, Curzio M, et al. J Eur Acad Dermatol Venereol 2008; 22: 522–3.
Report of a patient with perianal EMPD who, after refusing surgical excision, was treated with intralesional IFN-α2b at a dose of 1 million IU three times per week for 3 weeks. At 7 weeks the tumor had decreased in diameter and surgical resection was performed. Patient had no clinical disease at 108 months’ follow-up.
Choi J, Yoon E, Yoon D, Kim DS, Kim JJ, Cho JH. BJU Int 2001; 88: 297–8.
Report of three patients with EMPD treated with CO2 laser guided by Wood’s lamp fluorescence to determine clinical margins. All three had disease recurrence within 6 months and required traditional surgical excision.
Iijima M, Uhara H, Ide Y, Sakai S, Onuma H, Muto M, et al. Dermatology 2006; 213: 144–6.
Case report of a patient with scrotal and penile EMPD expressing estrogen receptor-α and associated prostate cancer. The patient was treated systemically with both anti-estrogen (tamoxifen) and anti-androgen (bicalutamide). The patient’s performance status was well maintained at 17 months’ follow-up.
Yoneyama K, Kamada N, Kinoshita K. Br J Dermatol 2005; 153: 853–5.
Case report of EMPD with multiple bone metastases successfully suppressed with anti-androgen bicalutamide and LH-RH agonist leuprorelin. Tumor markers decreased and bone scintigraphy evidence of metastasis disappeared within 2 months. When tumor markers rose at day 70, other anti-androgens and systemic chemotherapy failed. Bone metastases reappeared, and the patient ultimately died 14 months after the start of anti-androgen therapy. The authors postulate that the rapid development of resistance to the androgen-deprivation therapy suggests that mutation or amplification in the androgen receptor gene occurred in this case, as seen in cases of prostate cancer.
Treatment of Skin Disease Comprehensive Therapeutic Strategies 4e
WhatsApp us




 Wide local excision, with or without lymph node dissection
Wide local excision, with or without lymph node dissection Frozen section-guided wide local excision
Frozen section-guided wide local excision Mohs micrographic surgery
Mohs micrographic surgery Photodynamic therapy
Photodynamic therapy Systemic chemotherapy
Systemic chemotherapy Radiation therapy
Radiation therapy Topical imiquimod
Topical imiquimod
 Topical 5-fluorouracil and retinoic acid
Topical 5-fluorouracil and retinoic acid Intralesional interferon-α2b
Intralesional interferon-α2b Laser therapy
Laser therapy Androgen receptor antagonist
Androgen receptor antagonist