[level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 13
Evaluation and Management of Childhood Hypothalamic And Pituitary Tumors
Epidemiology
Intracranial and spinal cord tumors are the second most frequent type of childhood malignancy after leukemia, accounting for approximately 20% of cases.1 While much is known about the epidemiology of malignant intracranial tumors in childhood, there is a paucity of information about benign tumors. The incidence of brain tumors in childhood is 3 per 100,000. The highest age-adjusted incidence, 31.4 per million, was observed in the Nordic countries, and rates between 24 and 27 per million were found in most other predominantly white populations. In the United States, the age-adjusted incidence rate was 36% higher in males and 68% higher in females than the rate based on malignant tumors alone. Black children had a significantly lower incidence than white children. Lower rates were seen in South America, Africa, and Asia; the lowest rates were for Chinese populations and for blacks in Africa, both below 15 per million. Among white populations, astrocytoma was the most common histologic type, often with an incidence of at least 10 per million, followed by medulloblastoma, 5 to 6 per million, and ependymoma, 2 to 4 per million. In other regions with lower incidence rates, these three types accounted for similar proportions of the total. Black children in the United States had a higher incidence of craniopharyngioma than white children, and there was an unusually high incidence of pineal tumors in Japan, 0.9 per million compared with 0.3 to 0.4 in many other countries. An incidence rate of 2.76 per 100,000 people younger than 18 years of age was found. Tumors in the suprasellar/hypothalamic region are unusual, the most common being craniopharyngiomas, which are approximately 9% of childhood intracranial tumors; other tumors are much rarer. The incidence of intracranial germinoma is only 0.26 cases per million children per year.1,2 Considerable progress has been made toward improving survival for children with brain tumors, and yet there is still relatively little known regarding the molecular genetic events that contribute to tumor initiation or progression. Nonrandom patterns of chromosomal deletions in several types of childhood brain tumors suggest that the loss or inactivation of tumor suppressor genes is a critical event in tumorigenesis. Deletions of chromosomal regions 10q, 11, and 17p, for example, are frequent events in medulloblastoma, whereas loss of a region within 22q11.2, which contains the INI1 gene, is involved in the development of atypical teratoid and rhabdoid tumors.
Classification
Intracranial tumors are most commonly situated in the posterior fossa in 70% of cases, in the supratentorial region in 30%, and can occur at any age, although the most frequent age is between 2 and 5 years. The classification can be made either on the basis of histology or on the location of tumor site (Table 13-1). Many sellar and suprasellar tumors in childhood, such as craniopharyngiomas and Rathke’s cysts, do not originate from the central nervous system and are not “brain tumors.” Hypothalamic tumors are usually hypothalamic hamartoma, low-grade astrocytoma, Langerhans’ cell histiocytosis (LCH), and dermoid and epidermoid tumors. Tumors such as craniopharyngiomas and germinomas tend to affect the hypothalamus indirectly, originating in the peripituitary or pituitary region and extending upward. The pituitary stalk is typically affected from lesions such as germinomas, LCH, and craniopharyngiomas. LCH commonly affects the middle of the pituitary stalk, in a similar appearance to tuberculosis and sarcoidosis, which may be related to LCH cells involving the cerebrospinal fluid. The anterior pituitary is frequently affected by benign pituitary adenoma, whereas the posterior pituitary is the common location of pilocytic astrocytoma and LCH. Malignant glioma, meningioma, Schwann cell, and pituitary tumors, as well as metastases (which are the most common intracranial tumors in adults), are comparatively uncommon in children.3 In contrast, benign glioma, primitive neuroectodermal tumors, and craniopharyngiomas account for a substantially higher percentage of intracranial tumors in children than in adults.4,5 Classification of primitive neuroepithelial cells is based on appearance of the tumor as determined by light microscopy, immunocytochemical techniques, and ultrastructural features without consideration for site of origin.6
Table 13-1
Histologic Classification of Intracranial Tumors
| Supratentorial midline tumors | Low-grade glioma |
| Craniopharyngioma | |
| Germ cell tumor | |
| Pineal cell tumors (pineocytoma/pineoblastoma) | |
| Supratentorial hemispheric tumors | |
| Infratentorial tumors |
Symptoms and Signs
The mode of presentation depends on the age of the child and the location of the tumor. Symptoms and signs can be usefully divided into those from raised intracranial pressure, focal neurologic signs, and endocrinopathy. Nonspecific symptoms of increased intracranial pressure are repeated and frequent headaches, especially if they are worsening and associated with nausea or vomiting, often occurring in the early morning; irritability; listlessness; vomiting; failure to thrive; macrocephaly; and loss of developmental milestones.7 Epilepsy may be the initial presenting feature of an intracranial tumor. This may be due to the structural abnormality caused by the space-occupying lesion but may be secondary to the associated endocrinopathies of hypoglycemia (secondary to growth hormone and/or cortisol insufficiency) or hyponatremia (from the syndrome of inappropriate antidiuretic hormone secretion). Although young children are more likely than infants to manifest localizing neurologic abnormalities, these are by no means uniformly present. In older children, a larger percentage of tumors manifest with localizing symptoms and signs that often suggest the location as well as the histologic identity of the tumor. Midline tumors often present in an insidious onset with various symptoms and signs: visual defects such as nystagmus, complete loss of vision, and diplopia because of paralysis of the lateral rectus muscles due to a sixth nerve palsy or due to raised intracranial pressure because of obstruction of the cerebrospinal fluid pathways; neuroendocrine dysfunction, behavioral and appetite disturbances, and regression of motor skills; or they may reflect the compression or infiltration of adjacent structures. Pineal region tumors typically manifest with eye movement abnormalities, such as Parinaud’s syndrome or hydrocephalus and alteration of consciousness.8
Endocrine Dysfunction
For both benign and malignant tumors, presenting symptoms usually reflect the age of the child and the position of the tumor. Growth failure in children with occult intracranial tumors is characteristic. In idiopathic (congenital) growth hormone deficiency (GHD), birth length is relatively short, but growth rate is normal until approximately 18 months of age, when a gradual deceleration occurs. Idiopathic GHD is usually easily distinguished from the growth failure associated with an occult intracranial tumor, in which growth is initially normal and height is appropriate for the parental percentiles, followed by a marked growth deceleration. This is usually due to GHD but may exceptionally be due to the presence of the intracranial tumor with normal endocrine function. Absence of puberty of more than 2 standard deviations (SD) will require neuroradiologic imaging, but delayed puberty with growth deceleration is usually due to constitutional delay of growth and puberty. Even a child with suspected constitutional delay who does not respond to sex-steroid therapy should be investigated endocrinologically and neuroradiologically. Craniopharyngiomas commonly present with failure to enter puberty or arrested puberty associated with an abnormal growth spurt; they usually demonstrate an absence of the normal consonance of puberty.9
The idiopathic form of central precocious puberty (CPP) occurs in 74% of affected girls, and in 60% of affected boys, who are more likely to have an occult intracranial tumor than girls.10,11 Although it is commonly recognized that gonadotropin-dependent precocious puberty (or CPP) in boys is usually caused by an intracranial lesion, it used to be believed that girls had an idiopathic etiology and did not require neuroradiologic imaging. Recent large series of girls with gonadotropin-dependent precocious puberty have shown that both girls and boys should have neuroradiologic imaging. Although in girls with CPP, hypothalamic hamartoma is the most common lesion, other tumors such as astrocytomas may present in this fashion; it is important not to miss the opportunity for an early diagnosis. Intracranial tumors causing CPP in girls are histologically specific, despite being in the same anatomic site involving the hypothalamus between the mamillary bodies and the median eminence, and may be related to the secretion of specific local growth factors. CPP may be caused by hypothalamic hamartomas, astrocytomas, optic gliomas, pineal tumors, and arachnoid cysts. Interestingly, some other suprasellar tumors such as craniopharyngiomas, germinomas, and LCH are only rarely associated with the development of gonadotropin-dependent precocious puberty, despite the lesion being in the same anatomic site. High-risk factors for the presence of an intracranial tumor in children with CPP are: a young age of onset (under age 3), high serum luteinizing hormone concentrations not associated with the development of a luteinizing hormone surge, and high serum leptin concentrations. However, it is impossible to exclude an intracranial lesion in a child with CPP without performing a magnetic resonance imaging (MRI) scan.12,13 Diencephalic syndrome is a rare cause of failure to thrive in infancy and early childhood. The syndrome is characterized by profound emaciation despite normal or increased caloric intake, absence of cutaneous adipose tissue, locomotor hyperactivity, euphoria, and alertness. It commonly occurs in association with chiasmatic and hypothalamic gliomas. It has also been described in association with other lesions, such as midline cerebellar astrocytomas, suprasellar ependymomas, suprasellar spongioblastomas, and thalamic tumors.14 Such children may even present to an eating disorder clinic, their growth failure attributed to an anorexic illness.15
The onset of diabetes insipidus (DI) with or without an evolving anterior pituitary endocrinopathy is suspicious of a space-occupying lesion. DI followed by an evolving anterior pituitary deficiency, including growth failure from GHD, is usually due to a sella/suprasellar tumor. Although DI is also common in midline cerebral malformations (such as septo-optic dysplasia), this usually follows or is contemporaneous with anterior pituitary failure.16
The most common endocrine presentation of macroprolactinoma (more common in children and adolescents than microprolactinoma) is delayed/absent puberty due to prolactin (PRL) suppression of gonadotropin pulsatility, combined with gynecomastia in boys and galactorrhea in girls. The presentation may be part of multiple endocrine neoplasia type 1. Macroprolactinomas usually extend upward and encroach on the visual pathway and are often accompanied by visual field defects. It is important to measure the serum PRL in every child with pituitary enlargement, particularly before any surgery is contemplated.17
Isolated adrenocorticotropic hormone insufficiency may occur in lymphocytic hypophysitis, although this condition is not a malignant tumor but presents as the differential diagnosis of a central pituitary mass. This tumor usually occurs in the puerperium and is extremely rare in childhood.18
Diagnosis
MRI scans have become the preferred diagnostic study for pediatric intracranial tumors. MRI is preferred under most circumstances, providing superior resolution and multiplanar imaging capabilities and avoiding the “spray” artifact from the petrous ridge that may obscure computed tomographic images of the base of the brain, without a radiation burden to the child. The administration of gadolinium diethylenetriamine-pentaacetic acid appears to be a safe and effective contrast agent for MRI and provides a more accurate method of imaging in the follow-up of brain tumors in pediatric patients. Where clinical suspicion remains (normal neuroradiologic imaging in patients with DI), scans reported as normal should be sent for expert review and consideration of repeat imaging with time. The intervals for scanning should also be guided clinically, since any set interval is empirical. MRI scan should be performed at a minimum of yearly intervals.19 For lesions with a high frequency of cerebrospinal fluid dissemination, such as primitive neuroectodermal tumors and germ cell tumors, a neuraxis staging evaluation by spinal MRI, if not obtained preoperatively, should be performed approximately 2 weeks after surgery. In children with pineal region tumors, measurement of α-fetoprotein and β–human chorionic gonadotropin in the blood is useful for the diagnosis of malignant germ cell tumors (pineoblastomas); however, cerebrospinal fluid markers are of limited assistance. Placental alkaline phosphatase is a clinically useful tumor marker for primary intracranial germinoma.20
Thyroid function tests (as well as serum PRL concentration) are always required prior to surgery of a suspected pituitary tumor.17 An elevated serum PRL concentration requires the distinction between stalk compression with moderate rise in PRL from the very high PRL concentrations associated with a PRL-secreting tumor. Macroprolactinomas usually have very high PRL secretion, and there is little ambiguity about the diagnosis. It is important to distinguish thyroid-stimulating hormone–secreting adenomas, which are extremely rare, from the pituitary hyperplasia associated with longstanding primary hypothyroidism. After prolonged, severe primary hypothyroidism with increased secretion of thyroid-stimulating hormone, the pituitary gland is usually increased in size and may attain a suprasellar extension and compression of the optic chiasm/nerves. This may be accompanied by a gonadal form of premature sexual maturation which is not true puberty (isolated breast development in girls and large testicular volumes with minimal virilization in boys).21 These signs of premature maturation and pituitary enlargement decrease or resolve following the decrease in secretion of thyroid-stimulating hormone within 6 months of commencing appropriate thyroxin replacement.
Therapy
General Principles of Treatment
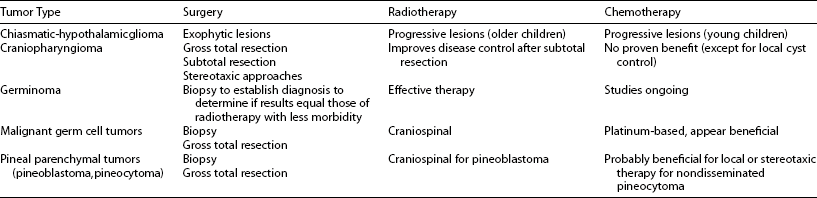
In general, the aim of therapy is to eradicate the tumor, with minimal morbidity and mortality, since prognosis is correlated with the extent of resection. If no biopsy has been obtained, histology will be undertaken postoperatively. Because the details of treatment for many types have evolved over time and likely will continue to evolve, treatment decisions for individual patients are best made in the context of a multidisciplinary “team” approach (Table 13-2). The neurosurgeons estimate the extent of resection, but it must be confirmed by computed tomography or MRI examination, preferably within the first 48 hours postoperatively. Postoperatively, computed tomography or MRI provides an objective assessment of the volume of residual tumor. These studies should be performed within 48 hours of surgery if possible to minimize the confounding effect of postsurgical enhancement around the operative bed. After diagnosis and initiation of treatment, follow-up imaging studies in children with malignant tumors are generally obtained every 3 months for 1 year, every 6 months for up to 5 years, and periodically thereafter. Imaging in benign tumors is typically obtained 3 to 6 months postoperatively and every 12 to 24 months thereafter for at least 5 years, the frequency largely depending on whether or not a complete resection has been confirmed on the initial postoperative scan. However, the optimal frequency of follow-up studies remains uncertain.
Chemotherapy
A radiosensitizing effect of certain drugs is often postulated. Among patients with malignant brain tumors, infants and very young children have the worst prognosis and the most severe treatment-related neurotoxic effects. Chemotherapy appears to be an effective primary postoperative treatment for many malignant brain tumors in young children. Disease control for 1 or 2 years in a large minority of patients permits a delay in the delivery of radiation and, on the basis of preliminary results, a reduction in neurotoxicity. For patients who had undergone total surgical resection or who had a complete response to chemotherapy, the results are sufficiently encouraging to suggest that radiation therapy may not be needed in this subgroup of children after at least 1 year of chemotherapy.22 Also, a significant proportion of children with malignant brain tumors can avoid radiotherapy and prolonged maintenance chemotherapy yet still achieve durable remission by administering myeloablative consolidation chemotherapy with autologous bone marrow reconstitution after maximal surgical resection and conventional induction chemotherapy.23 No long-term side effects on height, bone mineral density, body composition, and bone maturation were found in patients with leukemia treated with chemotherapy alone. It causes growth retardation, but catch-up growth occurs after cessation of treatment.24 Gonadal damage after cyclophosphamide (dose related; may be reversible) and busulfan (the association may cause permanent ovarian failure) is well documented in adults; it seems that prepubertal and pubertal ovaries are more resistant than ovaries of adults. Ovaries are more resistant than testes, and seminiferous tubules are more sensitive.25
Radiotherapy
The indications for radiotherapy of pediatric intracranial tumors and the parameters for radiation delivery have evolved in several ways during the last decade. Tumors have conventionally been treated with 5000 to 6000 cGy in 180 to 200 cGy/day fractions using multiple portals. Newer approaches, such as hyperfractionated irradiation and interstitial irradiation (stereotactic radiosurgery and interstitial brachytherapy) attempt to improve therapeutic efficacy while minimizing irradiation of surrounding brain and correspondingly reduce toxicity. Nevertheless, because more children are surviving brain tumors following surgery and radiation therapy, the price of the successful therapy is being increasingly realized in terms of adverse effects, particularly in the very young child. Chemotherapy is increasingly used to delay or avoid using irradiation in children younger than 3 years of age with high-grade and incompletely resected low-grade tumors. Improvements in imaging and dose-delivery techniques have allowed radiotherapy administration to be tailored to the geometry of the tumor. Hyperfractionated irradiation technique is based on the premise that normal cells are better able than tumor cells to repair sublethal damage between doses and that multiple fractions are more likely to irradiate proliferating cells in a sensitive part of the cell cycle.26 Finally, novel approaches for focal irradiation, such as stereotactic radiosurgery and interstitial brachytherapy, are increasingly being employed in selected unresectable lesions to provide high doses of radiation to the tumor while minimizing irradiation of surrounding brain.27,28 Radiosurgery is ideally suited to the treatment of small foci of unresectable disease and has led to long-term disease control in well-circumscribed benign lesions. In addition, ongoing studies in older children with selected lesions, such as “standard-risk” medulloblastoma and germinoma, use reduced doses of radiotherapy in conjunction with chemotherapy to minimize radiation-induced neurotoxicity. For many low-grade gliomas that have been extensively resected, adjuvant therapy often is deferred because these tumors may remain quiescent for extended periods.
Sequelae of Treatment
The small increase in incidence noted over the past 2 decades most likely represents advancements in diagnostic technology rather than true changes in disease frequency, though this is controversial. Survivors of childhood intracranial tumors are 13 times more likely to die than healthy age- and sex-matched peers.29 Disease recurrence remains the single most common cause of late deaths. The sequelae of surgical treatment are evident soon after the operation, but the sequelae of irradiation and chemotherapy become apparent over many decades. Neurologic, neurocognitive, and endocrine disturbances are the most prevalent disabilities observed among the long-term survivors of pediatric intracranial tumors. Maximum quality of life for the individual patient can only be achieved by long-term care and close cooperation of specialists in the different medical disciplines involved. It has been demonstrated that cranial irradiation has been implicated as the major cause for cognitive dysfunction. In that study, intellectual functioning was significantly lower in children whose treatment included cranial irradiation than in those treated without cranial irradiation, and this effect was more pronounced in nonverbal than in verbal intellectual abilities.30 Some authors also showed that children younger than 7 years at diagnosis had a mean IQ loss of 27 points, whereas children older than 7 years at diagnosis showed no significant decrease in IQ. They also demonstrated that decline in IQ occurred between baseline and year 2 of follow-up; none could be documented between years 2 and 4. All children younger than 7 years at diagnosis were receiving special education at follow-up; 50% of the children older than 7 years at diagnosis were receiving supplemental educational services.30 In Packer’s study, children demonstrated a wide range of dysfunction, including deficits in fine-motor, visual-motor, and visual-spatial skills and memory difficulties; although not retarded, they had a multitude of neurocognitive deficits that detrimentally affected school performance after 2 years from treatment. The younger the child is at the time of treatment, the greater is the likelihood and severity of damage.31 Reimers and colleagues tried to identify subgroups of children who are at increased risk for cognitive deficits; they showed that younger age at diagnosis, tumor site in the cerebral hemisphere, hydrocephalus treated with a shunt, and treatment with radiation therapy were found to be significant predictors of lower cognitive function. Radiation therapy was the most important risk factor for impaired intellectual outcome. The mean observed full-scale IQ was 97 for the nonirradiated patients and 79 for the irradiated patients. Verbal IQ, but not performance and full-scale IQ, had a significant negative correlation to biological effective dose of irradiation to the tumor site.32 Tumor involving the hypothalamic-pituitary area often produces a loss of endocrine function during a characteristic sequence in time, an evolving endocrinopathy. The risk of developing these adverse events is related to the underlying disease and its treatment with cytotoxic drugs and radiation therapy. The incidence and time course of disorders and the number of anterior pituitary hormones that are deficient depend on the sensitivity of the hormone itself to such therapy, on the dose, fractionation, and time elapsed since irradiation. Early detection and appropriate replacement therapy before clinical manifestations occur may carry important benefits in terms of normal pubertal and social development, growth, fertility, and bone mineralization.33 The GH axis is the most sensitive and the adrenal axis the most resistant to the effects of direct irradiation to the hypothalamic-pituitary region. Patients who have received high doses (>30 Gy) of cranial, craniospinal, or total body irradiation are likely to develop GHD within 2 to 5 years from cessation of treatment.34 Growth may be further impaired by spinal irradiation which directly interferes with spinal growth and is not due to an endocrinopathy.
In the rare syndrome of “growth without GH,” normal or accelerated growth continues despite the patient having GHD, and this occurs at the expense of hyperphagia and rapid weight gain. It is considered that the etiology of this condition is related to insulin and insulin-like peptides, which allow growth in the presence of GH insufficiency. This phenomenon usually occurs after craniopharyngioma surgery. Indeed, the first sign of a recurrence of a treated intracranial tumor while on GH therapy may be growth deceleration.35 GHD newborns can have a length within the normal range, which suggests that other growth factors dominate longitudinal gain during gestation. Obese children grow at a normal rate despite their low serum GH levels and reduced response to pharmacologic stimulation tests. Children with hypopituitarism secondary to craniopharyngioma resection may continue to grow and may even show growth rate acceleration if their weight increases significantly. Several possible mechanisms might underlie the growth stimulation in obese children, such as elevated levels of insulin and reduced levels of insulin-like growth factor binding protein 11. Recently, elevated sex hormone levels and elevated leptin levels in obese children were found to affect epiphyseal growth, and it may be that leptin also participates in the growth without GH observed in obesity, especially after craniopharyngioma removal. In the absence of GH, the sex hormones stimulate growth through a direct GH-independent effect on the epiphyses. Leptin, insulin, and sex hormones locally activate the insulin-like growth factor system in the epiphyseal growth plate.36
There is a correlation between the age at diagnosis (the immature hypothalamus may be more sensitive to irradiation), the dose of radiation given, different regimens, fractionation of irradiation, and pubertal development. Gonadal dysfunction can be induced by a direct injury to the gonads (hypergonadotropic hypogonadism) and less frequently by neuroendocrine injury to the hypothalamic-pituitary axis (hypogonadotropic hypogonadism).37 Low doses of cranial irradiation (18 to 24 Gy) can cause precocious puberty, especially in girls, with a compromised growth spurt leading to a loss in final height, whereas delayed puberty has been reported after high doses (>40 Gy) used to treat solid tumors adjacent to the hypothalamus. Either low-dose cranial irradiation given as prophylaxis in the treatment of acute lymphoblastic leukemia, or high-dose irradiation for tumors distant from the hypothalamic-pituitary axis can cause hypogonadism.38 The irradiation to the gonads from the spinal irradiation could potentially cause oligo/azoospermia with total doses of 6 Gy; the Leydig cell damage is common after total doses greater than 20 Gy.39,40 Early menopause has been reported as well.41 The possibility of using gonadotropin-releasing hormone agonists to prevent ovarian damage has been proposed. A number of treatment options for preserving fertility are available for cancer survivors. Sperm banking should be offered even to young adolescent boys. Sperm are present in urine from the early teens onward and can be obtained and banked. Ovary banking will be a technique in the future as some centers develop the procedures. Concern that residual cancer cells are not also banked with the ovarian cells is an issue still to be addressed. Cryopreservation of embryos is part of current practice and is useful in cases when couples desire it.42 Further information on the ability of both ovary and uterus to sustain a pregnancy is crucial in deciding which treatment option to pursue. Pregnancy presents a cardiorespiratory stress; peripartum heart failure in women treated as children with anthracycline chemotherapy is a known complication.43 Survivors who have been exposed to anthracycline therapy with or without radiation to the heart and those who received therapy known to induce pulmonary fibrosis or cardiopulmonary radiation therapy may benefit from a cardiac evaluation or pulmonary function test before pregnancy.44 Delayed puberty development was reported in boys and girls after a total body irradiation (TBI) containing conditioning regimen, whereas patients given bone marrow transplantation for severe aplastic anemia (without total body irradiation) presented a normal puberty.45 Other authors demonstrated that children who have been treated with a dose of 25 Gy for acute lymphoblastic leukemia at an early age (<7 years) had normal pubertal development. Girls who had a late presentation of acute lymphoblastic leukemia and a late treatment had delayed puberty.37 Deficiency of thyroid-stimulating hormone, adrenocorticotropic hormone, and hyperprolactinemia can be seen following high-dose radiotherapy (>40 Gy) of the hypothalamic-pituitary axis, especially among young women.46,47
The incidence of second malignancies ranges from 1% to more than 3%. The majority of second tumors are thyroid cancer, malignant gliomas, meningiomas, and sarcomas that occur within radiotherapy treatment fields 10 to 20 years after irradiation. An increased incidence of hematologic malignancies has been noted after chemotherapy.48 Thyroid ultrasound scan should be performed once a year. Cranial radiation has also been associated with carotid occlusive disease, which often manifests as Moyamoya syndrome with progressive ischemic cerebrovascular symptoms. This syndrome is particularly common in patients irradiated for parasellar lesions such as chiasmatic-hypothalamic gliomas.49
Treatment of Specific Tumors
The treatment of hypothalamic hamartoma is generally pharmacologic; surgical intervention is difficult and does not usually lead to resolution of the early puberty. The hamartoma usually remains the same size. Surgery would be very unlikely to be successful for the resolution of CPP. However, surgical resection, especially if the hamartoma is pedunculated, would be an option for frequent and inadequately controlled gelastic seizures.50
OPTIC CHIASMATIC–HYPOTHALAMIC GLIOMAS
Optic chiasmatic–hypothalamic gliomas have been considered benign tumors and self-limiting in growth potential because of their histologic appearance. Chiasmatic and chiasmatic-hypothalamic tumors are different entities. Most clinical series have reported significant morbidity and mortality, especially with the more extensive, posteriorly positioned tumors. The biological behavior of optic chiasmatic–hypothalamic gliomas is age dependent, with patients younger than 5 years and older than 20 years typically having tumors that exhibit aggressive growth. There are no specific pathologic features to help differentiate the clinical behavior of such tumors. The emergence of modern imaging techniques, including MRI, has facilitated the monitoring of the natural history of the disease and the determination of the effects of therapy. Most patients with optic chiasmatic–hypothalamic gliomas survive for many years. Management is controversial, partly related to failure to separate out those tumors involving the optic chiasm only (chiasmatic tumors) from those also involving the hypothalamus (chiasmatic-hypothalamic tumors). Some authors suggested a conservative treatment for patients with optic chiasmatic–hypothalamic gliomas in the context of neurofibromatosis type 1 (NF-1) without visual failure, with cerebrospinal fluid shunting for hydrocephalus, if present, and medical therapy for endocrine dysfunction.51,52 For the chiasmatic-hypothalamic tumors, there was more morbidity and no prolongation of time to progression when radical resections were compared to more limited resections. However, over 90% of children with optic glioma without NF-1 will require some form of therapy.53 Therefore, if surgery is performed, it may be appropriate to do a surgical procedure that strives only to provide a tissue diagnosis and to decompress the optic apparatus and/or ventricular system.54 After tumor resection, patients whose vision is significantly compromised or who show progression of their disease on serial neuroimaging scans receive chemotherapy. A variety of regimens have been employed (e.g., carboplatinum, vincristine; and 6-thioguanine, procarbazine, dibromodulcitol), with response or stabilization rates of 75% to 100%.55 Radiation therapy is effective in stabilization or improvement of vision and prevention of tumor progression in both optic pathway and chiasmatic-hypothalamic gliomas to children older than 5 years.56 Optic chiasmatic–hypothalamic gliomas have an excellent long-term prognosis, with a 10-year survival of over 85%.57
Cystic Sella Tumors
The distinction among craniopharyngioma, Rathke’s cleft cyst, and intrasellar arachnoid cyst remains a difficult preoperative problem, although the presence of calcification makes the diagnosis of the former more likely. Accurate diagnosis of these rare pituitary lesions is important to determine the type of treatment and predict prognostic outcome. Only 10% of craniopharyngiomas are completely solid. The treatment of craniopharyngioma remains controversial. Although craniopharyngioma is a benign tumor, its location makes even advanced microsurgical techniques difficult to perform because of its adherence to the optic chiasm, hypothalamus, and vessels of the circle of Willis. Despite advances in microsurgical techniques, the complete removal of the tumor is possible in only 66% to 90% of patients.58 Radiosurgery avoids the shortcoming of surgical resection near the hypothalamic-pituitary axis, without the morbidity of open surgery.59–61 In tumors with a large cystic component, stereotactic drainage or instillation of radioactive and/or chemotherapeutic agents have been used. Only several authors have reported the use of bleomycin for the treatment of recurrent cystic craniopharyngioma, although there is not an established protocol for using it. However, the risk of local complications after the administration of intratumoral bleomycin in these patients is around 10%, and some fatal toxic reactions have been recently reported.62 Intracystic administration of bleomycin is a valid option as adjuvant therapy for craniopharyngioma in patients with recurrences that are not surgical candidates because of the high risk of complications. Other authors suggest that cystic lesions may be treated with intracavitary instillation of phosphorus-32 to deliver a cyst wall radiation dose of approximately 20,000 cGy.63 If a treatment algorithm has been devised and followed that combines both surgery (radical and conservative) and radiotherapy (both external fractionated and intracyst instillations), long-term tumor control and minor disability are achieved. Endocrinologic deficits had the worst prognosis after surgery, especially DI combined with absent thirst. Tumor recurrence occurred both radiographically and clinically, typically in the first 3 to 4 years after surgery; this suggests a need for close surveillance initially with neuroimaging, particularly in younger children, and also clinical examination. Lack of calcification at diagnosis is associated with a tendency to remain free of relapse. Predictors of high morbidity included severe hydrocephalus, intraoperative adverse events, and age younger than 5 years at presentation. Large tumor size, young age, and severe hydrocephalus were predictors of tumor recurrence, whereas complete tumor resection (as determined by postoperative neuroimaging) and radiotherapy given electively after subtotal excision were less likely to be associated with recurrent disease. However, patients treated with surgery alone have a significantly worse freedom from progression when compared to patients treated with surgery and radiation therapy or radiation therapy alone.58 In the extensive experience of Yasargil and colleagues from the University of Zurich, using an aggressive surgical approach, total resection was achieved in 90% of 144 patients, with an operative mortality rate of 16% and a good functional outcome in 67%.59 In general, if total resection can be obtained, long-term control rate is between 50% and 80%; however, after subtotal resection, 50% to 100% of children experience local recurrence.64,65 Since craniopharyngioma is potentially radioresponsive, external beam radiation therapy has long been used in the treatment of craniopharyngioma following incomplete surgical resection. Regine and Kramer reported a 60% 20-year survival rate in these patients.66 In the series from the Royal Marsden Hospital, in 25 patients treated with salvage radiotherapy, the 15-year progression-free survival rate was 72%.67 Recommended doses have ranged from 50 to 60 Gy in 180 to 200 cGy daily fractions.68 Although the addition of fractionated postoperative radiation therapy has shown to reduce the recurrence rate, the incidence of hypothalamic and pituitary disorders is increased as seen with radical surgery.58,69 Merchant and colleagues concluded that DI was the only endocrine deficiency that differed substantially in frequency between the groups treated with surgery and with radiotherapy, respectively.70 Other complications of conventional radiation therapy include radiation necrosis, optic neuritis, and dementia. Obesity and the metabolic syndrome secondary to hypothalamic dysfunction seem to be important complications of craniopharyngioma treatment.71
Pineal Tumors
It is now recognized that the wide variety of tumor types found in the pineal region necessitates different modes of treatment; improved microsurgical and stereotactic surgical techniques have made mortality and morbidity rates acceptably low. The secondary deposits from pineal tumors, especially germinomas, are common in the suprasellar region. Benign teratomas, if resected totally, may require no further therapy; lesions that have been resected subtotally are treated with local radiotherapy. Nondisseminated germinomas are highly radiosensitive. Nongerminomatous germ cell tumors are treated with craniospinal radiation and chemotherapy, but even with aggressive therapy, 5-year survival is less than 50%.72–74 Pineoblastomas are considered to be primitive neuroectodermal tumors, and their treatment and outcome are comparable to those of high-risk medulloblastomas. Chemotherapy without radiotherapy appears to be ineffective therapy for young children and for children older than 18 months of age at diagnosis treated with craniospinal radiation therapy and chemotherapy.75,76 Some authors concluded that most pineal cysts are clinically benign, and they should be followed up for many years.77 Pineocytomas, although benign histologically, show a propensity for local recurrence and cerebrospinal fluid dissemination; these lesions have been treated with local or craniospinal radiotherapy and, in some cases, chemotherapy.
References
1. Stiller, CA, Nectoux, J. International incidence of childhood brain and spinal tumours. Int J Epidemiol. 1994;23:458–464.
2. Keene, DL, Hsu, E, Ventureyra, E. Brain tumors in childhood and adolescence. Pediatr Neurol. 1999;20:198–203.
3. Reed, UC, Rosemberg, S, Gherpelli, JL, et al. Brain tumors in the first two years of life: a review of forty cases. Pediatr Neurosurg. 1993;19:180–185.
4. Rickert, CH, Probst-Cousin, S, Gullotta, F. Primary intracranial neoplasms of infancy and early childhood. Childs Nerv Syst. 1997;13:507–513.
5. Pollack, IF. Brain tumors in children [Review]. N Engl J Med. 1994;331:1501–1507.
6. Rorke, LB. The cerebellar medulloblastoma and its relationship to primitive neuroectodermal tumors. J Neuropathol Exp Neurol. 1983;42:1–15.
7. Albright, AL. Pediatric brain tumors. CA Cancer J Clin. 1993;43:272–288.
8. Pollack, IF. Pediatric brain tumors [Review]. Semin Surg Oncol. 1999;16:73–90.
9. Stanhope, R, Adams, J, Brook, CG. Disturbances of puberty. Clin Obstet Gynaecol. 1985;12:557–577.
10. De Sanctis, V, Corrias, A, Rizzo, V, et al. Etiology of central precocious puberty in males: The results of the Italian Study Group for Physiopathology of Puberty. J Pediatr Endocrinol Metab. 2000;13:687–693.
11. Cisternino, M, Arrigo, T, Pasquino, AM, et al. Etiology and age incidence of precocious puberty in girls: a multicentric study. J Pediatr Endocrinol Metab. 2000;13:695–701.
12. Chemaitilly, W, Trivin, C, Adan, L, et al. Central precocious puberty: clinical and laboratory features. Clin Endocrinol (Oxf). 2001;54:289–294.
13. Stanhope, R. Central precocious puberty and occult intracranial tumours. Clin Endocrinol (Oxf). 2001;54:287–288.
14. Russell, A. A diencephalic syndrome of emaciation in infancy and childhood. Arch Dis Child. 1951;26:274.
15. De Vile, CJ, Sufraz, R, Lask, BD, et al. Occult intracranial tumours masquerading as early onset anorexia nervosa. Br Med J. 1995;311:1359–1360.
16. Mootha, SL, Barkovich, AJ, Grumbach, MM, et al. Idiopathic hypothalamic diabetes insipidus, pituitary stalk thickening, and the occult intracranial germinoma in children and adolescents. J Clin Endocrinol Metab. 1997;82:1362–1367.
17. Torpiano, J, Vanderpump, M, Stanhope, R. The management of sellar masses: not all pituitary tumours require surgery for diagnosis and/or therapy. J Pediatr Endocrinol Metab. 2004;17:663–664.
18. Cemeroglu, AP, Blaivas, M, Muraszko, KM, et al. Lymphocytic hypophysitis presenting with diabetes insipidus in a 14-year-old girl: case report and review of the literature. Eur J Pediatr. 1997;156:684–688.
19. Sherwood, MC, Stanhope, R, Preece, MA, et al. Diabetes insipidus and occult intracranial tumours. Arch Dis Child. 1986;61:1222–1224.
20. Shinoda, J, Yamada, H, Sakai, N, et al. Placental alkaline phosphatase as a tumor marker for primary intracranial germinoma. J Neurosurg. 1988;68:710–720.
21. Pringle, PJ, Stanhope, R, Hindmarsh, P, et al. Abnormal pubertal development in primary hypothyroidism. Clin Endocrinol (Oxf). 1988;28:479–486.
22. Duffner, PK, Horowitz, ME, Krischer, JP, et al. Postoperative chemotherapy and delayed radiation in children less than three years of age with malignant brain tumors. N Engl J Med. 1993;328:1725–1731.
23. Mason, WP, Grovas, A, Halpern, S, et al. Intensive chemotherapy and bone marrow rescue for young children with newly diagnosed malignant brain tumors. J Clin Oncol. 1998;16:210–221.
24. Van der Sluis, I, van der Heuvel-Eibrink, MM, Hahlen, K, et al. Bone mineral density, body composition, and height in long term survivors of acute lymphoblastic leukemia in childhood. Med Pediatr Oncol. 2000;35:415–420.
25. Kumar, R, Biggart, JD, McEvoy, J, et al. Cyclophosphamide and reproductive function. Lancet. 1972;1:1212–1214.
26. Packer, RJ, Boyett, JM, Zimmerman, RA, et al. Hyperfractionated radiotherapy (72 Gy) for children with brain stem gliomas. A Children’s Cancer Group Phase I/II Trial. Cancer. 1993;72:1414–1421.
27. Grabb, PA, Lunsford, LD, Albright, AL, et al. Stereotactic radiosurgery for glial neoplasms of childhood. Neurosurgery. 1996;38:696–702.
28. McDermott, MW, Gutin, PH, Larson, DA, et al. Interstitial brachytherapy [Review]. Neurosurg Clin North Am. 1990;1:801–824.
29. Sklar, CA. Childhood brain tumors. J Pediatr Endocrinol Metab. 2002;15(Suppl):669.
30. Radcliffe, J, Packer, RJ, Atkins, TE, et al. Three- and four-year cognitive outcome in children with noncortical brain tumors treated with whole-brain radiotherapy. Ann Neurol. 1992;32:551–554.
31. Packer, RJ, Sutton, LN, Atkins, TE, et al. A prospective study of cognitive function in children receiving whole-brain radiotherapy and chemotherapy: 2-year results. J Neurosurg. 1989;70:707–713.
32. Reimers, TS, Ehrenfels, S, Mortensen, EL, et al. Cognitive deficits in long-term survivors of childhood brain tumors: identification of predictive factors. Med Pediatr Oncol. 2003;40:26–34.
33. Cohen, LE. Endocrine late effects of cancer treatment. Curr Opin Pediatr. 2003;15:3–9.
34. Livesey, EA, Hindmarsh, PC, Brook, CG, et al. Endocrine disorders following treatment of childhood brain tumours. Br J Cancer. 1990;61:622–625.
35. Locatelli, F, Giorgiani, G, Pession, A, et al. Late effects in children after bone marrow transplantation: a review. Haematologica. 1993;78:319–328.
36. Phillip, M, Moran, O, Lazar, L. Growth without growth hormone. J Pediatr Endocrinol Metab. 2002;15(Suppl):1267–1272.
37. Hughes, IA, Napier, A, Thompson, EN. Pituitary-gonadal function in children treated for acute lymphoblastic leukaemia. Acta Paediatr Scand. 1980;69:691–692.
38. Rappaport, R, Brauner, R, Czernichow, P, et al. Effect of hypothalamic and pituitary irradiation on pubertal development in children with cranial tumors. J Clin Endocrinol Metab. 1982;54:1164–1168.
39. Castillo, LA, Craft, AW, Kernahan, J, et al. Gonadal function after 12-Gy testicular irradiation in childhood acute lymphoblastic leukaemia. Med Pediatr Oncol. 1990;18:185–189.
40. Leiper, AD, Grant, DB, Chessells, JM. Gonadal function after testicular radiation for acute lymphoblastic leukaemia. Arch Dis Child. 1986;61:53–56.
41. Byrne, J. Infertility and premature menopause in childhood cancer survivors. Med Pediatr Oncol. 1999;33:24–28.
42. Edwards, RG, Morcos, S, Macnamee, M, et al. High fecundity of amenorrhoeic women in embryo-transfer programmes. Lancet. 1991;338:292–294.
43. Katz, A, Goldenberg, I, Maoz, C, et al. Peripartum cardiomyopathy occurring in a patient previously treated with doxorubicin. Am J Med Sci. 1997;314:399–400.
44. Collis, CH. Chemotherapy-related morbidity to the lungs. In: Plowman PN, McElwain TJ, Meadows AT, eds. Complications of Cancer Management. Guildford, England: Butterworth Scientific Ltd; 1991:250–271.
45. Sanders, JE. The impact of marrow transplant preparative regimens on subsequent growth and development. The Seattle Marrow Transplant team. Semin Hematol. 1991;28:244–249.
46. Ogilvy-Stuart, AL, Clark, DJ, Wallace, WH, et al. Endocrine deficit after fractionated total body irradiation. Arch Dis Child. 1992;67:1107–1110.
47. Wittert, G, Donald, RA, Espiner, EA, et al. The hormonal effects of pituitary surgery and irradiation: a review of 59 cases. N Z Med J. 1985;98:93–97.
48. Hawkins, MM, Draper, GJ, Kingston, JE. Incidence of second primary tumours among childhood cancer survivors. Br J Cancer. 1987;56:339–347.
49. Bitzer, M, Topka, H. Progressive cerebral occlusive disease after radiation therapy. Stroke. 1995;26:131–136.
50. de Brito, VN, Latronico, AC, Arnhold, IJ, et al. Treatment of gonadotropin dependent precocious puberty due to hypothalamic hamartoma with gonadotropin releasing hormone agonist depot. Arch Dis Child. 1999;80:231–234.
51. Alshail, E, Rutka, JT, Becker, LE, et al. Optic chiasmatic-hypothalamic glioma. Brain Pathol. 1997;7:799–806.
52. Wisoff, JH, Abbott, R, Epstein, F. Surgical management of exophytic chiasmatic-hypothalamic tumors of childhood. J Neurosurg. 1990;73:661–667.
53. Jenkin, D, Angyalfi, S, Becker, L, et al. Optic glioma in children: surveillance, resection or irradiation? Int J Radiat Oncol Biol Phys. 1993;25:215–225.
54. Steinbok, P, Hentschel, S, Almqvist, P, et al. Management of optic chiasmatic/hypothalamic astrocytomas in children. Can J Neurol Sci. 2002;29:132–138.
55. Gajjar, A, Heideman, RL, Kovnar, EH, et al. Response of pediatric low grade gliomas to chemotherapy [Review]. Pediatr Neurosurg. 1993;19:113–120.
56. Erkal, HS, Serin, M, Cakmak, A. Management of optic pathway and chiasmatic-hypothalamic gliomas in children with radiation therapy. Radiother Oncol. 1997;45:11–15.
57. Pollack, IF, Claassen, D, al-Shboul, Q, et al. Low-grade gliomas of the cerebral hemispheres in children: an analysis of 71 cases. J Neurosurg. 1995;82:536–547.
58. De Vile, CJ, Grant, DB, Kendall, BE, et al. Management of childhood craniopharyngioma: Can the morbidity of radical surgery be predicted? Neurosurg. 1996;85:73–81.
59. Yasargil, MG, Curcic, M, Kis, M, et al. Total removal of craniopharyngiomas. Approaches and long-term results in 144 patients. J Neurosurg. 1990;73:3–11.
60. Carmel, PW. Craniopharyngiomas. In: Wilkins RH, Rengachary SS, eds. Neurosurgery. New York: McGraw-Hill; 1985:905–916.
61. Pang, D. Surgical management of craniopharyngioma. In: Sekhar LN, Janecka IP, eds. Surgery of Cranial Base Tumors. New York: Raven Press; 1993:787–807.
62. Hader, WJ, Steinbok, P, Hukin, J, et al. Intratumoral therapy with bleomycin for cystic craniopharyngiomas in children. Pediatr Neurosurg. 2000;33:211–218.
63. Pollock, BE, Lunsford, LD, Kondziolka, D, et al. Phosphorus-32 intracavitary irradiation of cystic craniopharyngiomas: current technique and long-term results. Int J Radiat Oncol Biol Phys. 1995;33:437–446.
64. Kalapurakal, JA, Goldman, S, Hsieh, YC, et al. Clinical outcome in children with craniopharyngioma treated with primary surgery and radiotherapy deferred until relapse. Med Pediatr Oncol. 2003;40:214–218.
65. Kalapurakal, JA, Goldman, S, Hsieh, YC, et al. Clinical outcome in children with recurrent craniopharyngioma after primary surgery. Cancer J. 2000;6:388–393.
66. Regine, WF, Kramer, S. Pediatric craniopharyngiomas: long term results of combined treatment with surgery and radiation. Int J Radiat Oncol Biol Phys. 1992;24:611–617.
67. Bloom, HJG. Combined modality therapy for intracranial tumors. Cancer. 1975;35:111–120.
68. Tarbell, N, Barnes, P, Scott, RM, et al. Advances in radiation therapy for craniopharyngiomas. Pediatr Neurosurg. 1994;21(Suppl):101–107.
69. Honegger, J, Buchfelder, M, Fahlbusch, R. Surgical treatment of craniopharyngiomas: endocrinological results. J Neurosurg. 1999;90:251–257.
70. Merchant, TE, Kiehna, EN, Sanford, RA, et al. Craniopharyngioma: the St. Jude Children’s Research Hospital experience 1984–2001. Int J Radiat Oncol Biol Phys. 2002;53:533–542.
71. Harz, KJ, Muller, HL, Waldeck, E, et al. Obesity in patients with craniopharyngioma: assessment of food intake and movement counts indicating physical activity. J Clin Endocrinol Metab. 2003;88:5227–5231.
72. Regis, J, Bouillot, P, Rouby-Volot, F, et al. Pineal region tumors and the role of stereotactic biopsy: review of the mortality, morbidity, and diagnostic rate in 370 cases. Neurosurgery. 1996;39:907–914.
73. Edwards, MS, Hudgins, RJ, Wilson, CB, et al. Pineal region tumors in children. J Neurosurg. 1988;68:689–697.
74. Balmaceda, C, Heller, G, Rosenblum, M, et al. Chemotherapy without irradiation—a novel approach for newly diagnosed CNS germ cell tumors: results of an international cooperative trial. J Clin Oncol. 1996;14:2908–2915.
75. Jakacki, RI, Zeltzer, PM, Boyett, JM, et al. Survival and prognostic factors following radiation and/or chemotherapy for primitive neuroectodermal tumors of the pineal region in infants and children: a report of the Children’s Cancer Group. J Clin Oncol. 1995;13:1377–1383.
76. Dirks, PB, Harris, L, Hoffman, HJ, et al. Supratentorial primitive neuroectodermal tumors in children. J Neurooncol. 1996;29:75–84.
77. Mandera, M, Marcol, W, Bierzynska-Macyszyn, G, et al. Pineal cysts in childhood. Childs Nerv Syst. 2003;19:750–775.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 13
Evaluation and Management of Childhood Hypothalamic And Pituitary Tumors
Epidemiology
Intracranial and spinal cord tumors are the second most frequent type of childhood malignancy after leukemia, accounting for approximately 20% of cases.1 While much is known about the epidemiology of malignant intracranial tumors in childhood, there is a paucity of information about benign tumors. The incidence of brain tumors in childhood is 3 per 100,000. The highest age-adjusted incidence, 31.4 per million, was observed in the Nordic countries, and rates between 24 and 27 per million were found in most other predominantly white populations. In the United States, the age-adjusted incidence rate was 36% higher in males and 68% higher in females than the rate based on malignant tumors alone. Black children had a significantly lower incidence than white children. Lower rates were seen in South America, Africa, and Asia; the lowest rates were for Chinese populations and for blacks in Africa, both below 15 per million. Among white populations, astrocytoma was the most common histologic type, often with an incidence of at least 10 per million, followed by medulloblastoma, 5 to 6 per million, and ependymoma, 2 to 4 per million. In other regions with lower incidence rates, these three types accounted for similar proportions of the total. Black children in the United States had a higher incidence of craniopharyngioma than white children, and there was an unusually high incidence of pineal tumors in Japan, 0.9 per million compared with 0.3 to 0.4 in many other countries. An incidence rate of 2.76 per 100,000 people younger than 18 years of age was found. Tumors in the suprasellar/hypothalamic region are unusual, the most common being craniopharyngiomas, which are approximately 9% of childhood intracranial tumors; other tumors are much rarer. The incidence of intracranial germinoma is only 0.26 cases per million children per year.1,2 Considerable progress has been made toward improving survival for children with brain tumors, and yet there is still relatively little known regarding the molecular genetic events that contribute to tumor initiation or progression. Nonrandom patterns of chromosomal deletions in several types of childhood brain tumors suggest that the loss or inactivation of tumor suppressor genes is a critical event in tumorigenesis. Deletions of chromosomal regions 10q, 11, and 17p, for example, are frequent events in medulloblastoma, whereas loss of a region within 22q11.2, which contains the INI1 gene, is involved in the development of atypical teratoid and rhabdoid tumors.
Classification
Intracranial tumors are most commonly situated in the posterior fossa in 70% of cases, in the supratentorial region in 30%, and can occur at any age, although the most frequent age is between 2 and 5 years. The classification can be made either on the basis of histology or on the location of tumor site (Table 13-1). Many sellar and suprasellar tumors in childhood, such as craniopharyngiomas and Rathke’s cysts, do not originate from the central nervous system and are not “brain tumors.” Hypothalamic tumors are usually hypothalamic hamartoma, low-grade astrocytoma, Langerhans’ cell histiocytosis (LCH), and dermoid and epidermoid tumors. Tumors such as craniopharyngiomas and germinomas tend to affect the hypothalamus indirectly, originating in the peripituitary or pituitary region and extending upward. The pituitary stalk is typically affected from lesions such as germinomas, LCH, and craniopharyngiomas. LCH commonly affects the middle of the pituitary stalk, in a similar appearance to tuberculosis and sarcoidosis, which may be related to LCH cells involving the cerebrospinal fluid. The anterior pituitary is frequently affected by benign pituitary adenoma, whereas the posterior pituitary is the common location of pilocytic astrocytoma and LCH. Malignant glioma, meningioma, Schwann cell, and pituitary tumors, as well as metastases (which are the most common intracranial tumors in adults), are comparatively uncommon in children.3 In contrast, benign glioma, primitive neuroectodermal tumors, and craniopharyngiomas account for a substantially higher percentage of intracranial tumors in children than in adults.4,5 Classification of primitive neuroepithelial cells is based on appearance of the tumor as determined by light microscopy, immunocytochemical techniques, and ultrastructural features without consideration for site of origin.6
Table 13-1
Histologic Classification of Intracranial Tumors
| Supratentorial midline tumors | Low-grade glioma |
| Craniopharyngioma | |
| Germ cell tumor | |
| Pineal cell tumors (pineocytoma/pineoblastoma) | |
| Supratentorial hemispheric tumors | |
| Infratentorial tumors |
Symptoms and Signs
The mode of presentation depends on the age of the child and the location of the tumor. Symptoms and signs can be usefully divided into those from raised intracranial pressure, focal neurologic signs, and endocrinopathy. Nonspecific symptoms of increased intracranial pressure are repeated and frequent headaches, especially if they are worsening and associated with nausea or vomiting, often occurring in the early morning; irritability; listlessness; vomiting; failure to thrive; macrocephaly; and loss of developmental milestones.7 Epilepsy may be the initial presenting feature of an intracranial tumor. This may be due to the structural abnormality caused by the space-occupying lesion but may be secondary to the associated endocrinopathies of hypoglycemia (secondary to growth hormone and/or cortisol insufficiency) or hyponatremia (from the syndrome of inappropriate antidiuretic hormone secretion). Although young children are more likely than infants to manifest localizing neurologic abnormalities, these are by no means uniformly present. In older children, a larger percentage of tumors manifest with localizing symptoms and signs that often suggest the location as well as the histologic identity of the tumor. Midline tumors often present in an insidious onset with various symptoms and signs: visual defects such as nystagmus, complete loss of vision, and diplopia because of paralysis of the lateral rectus muscles due to a sixth nerve palsy or due to raised intracranial pressure because of obstruction of the cerebrospinal fluid pathways; neuroendocrine dysfunction, behavioral and appetite disturbances, and regression of motor skills; or they may reflect the compression or infiltration of adjacent structures. Pineal region tumors typically manifest with eye movement abnormalities, such as Parinaud’s syndrome or hydrocephalus and alteration of consciousness.8
Endocrine Dysfunction
For both benign and malignant tumors, presenting symptoms usually reflect the age of the child and the position of the tumor. Growth failure in children with occult intracranial tumors is characteristic. In idiopathic (congenital) growth hormone deficiency (GHD), birth length is relatively short, but growth rate is normal until approximately 18 months of age, when a gradual deceleration occurs. Idiopathic GHD is usually easily distinguished from the growth failure associated with an occult intracranial tumor, in which growth is initially normal and height is appropriate for the parental percentiles, followed by a marked growth deceleration. This is usually due to GHD but may exceptionally be due to the presence of the intracranial tumor with normal endocrine function. Absence of puberty of more than 2 standard deviations (SD) will require neuroradiologic imaging, but delayed puberty with growth deceleration is usually due to constitutional delay of growth and puberty. Even a child with suspected constitutional delay who does not respond to sex-steroid therapy should be investigated endocrinologically and neuroradiologically. Craniopharyngiomas commonly present with failure to enter puberty or arrested puberty associated with an abnormal growth spurt; they usually demonstrate an absence of the normal consonance of puberty.9
The idiopathic form of central precocious puberty (CPP) occurs in 74% of affected girls, and in 60% of affected boys, who are more likely to have an occult intracranial tumor than girls.10,11 Although it is commonly recognized that gonadotropin-dependent precocious puberty (or CPP) in boys is usually caused by an intracranial lesion, it used to be believed that girls had an idiopathic etiology and did not require neuroradiologic imaging. Recent large series of girls with gonadotropin-dependent precocious puberty have shown that both girls and boys should have neuroradiologic imaging. Although in girls with CPP, hypothalamic hamartoma is the most common lesion, other tumors such as astrocytomas may present in this fashion; it is important not to miss the opportunity for an early diagnosis. Intracranial tumors causing CPP in girls are histologically specific, despite being in the same anatomic site involving the hypothalamus between the mamillary bodies and the median eminence, and may be related to the secretion of specific local growth factors. CPP may be caused by hypothalamic hamartomas, astrocytomas, optic gliomas, pineal tumors, and arachnoid cysts. Interestingly, some other suprasellar tumors such as craniopharyngiomas, germinomas, and LCH are only rarely associated with the development of gonadotropin-dependent precocious puberty, despite the lesion being in the same anatomic site. High-risk factors for the presence of an intracranial tumor in children with CPP are: a young age of onset (under age 3), high serum luteinizing hormone concentrations not associated with the development of a luteinizing hormone surge, and high serum leptin concentrations. However, it is impossible to exclude an intracranial lesion in a child with CPP without performing a magnetic resonance imaging (MRI) scan.12,13 Diencephalic syndrome is a rare cause of failure to thrive in infancy and early childhood. The syndrome is characterized by profound emaciation despite normal or increased caloric intake, absence of cutaneous adipose tissue, locomotor hyperactivity, euphoria, and alertness. It commonly occurs in association with chiasmatic and hypothalamic gliomas. It has also been described in association with other lesions, such as midline cerebellar astrocytomas, suprasellar ependymomas, suprasellar spongioblastomas, and thalamic tumors.14 Such children may even present to an eating disorder clinic, their growth failure attributed to an anorexic illness.15
The onset of diabetes insipidus (DI) with or without an evolving anterior pituitary endocrinopathy is suspicious of a space-occupying lesion. DI followed by an evolving anterior pituitary deficiency, including growth failure from GHD, is usually due to a sella/suprasellar tumor. Although DI is also common in midline cerebral malformations (such as septo-optic dysplasia), this usually follows or is contemporaneous with anterior pituitary failure.16
The most common endocrine presentation of macroprolactinoma (more common in children and adolescents than microprolactinoma) is delayed/absent puberty due to prolactin (PRL) suppression of gonadotropin pulsatility, combined with gynecomastia in boys and galactorrhea in girls. The presentation may be part of multiple endocrine neoplasia type 1. Macroprolactinomas usually extend upward and encroach on the visual pathway and are often accompanied by visual field defects. It is important to measure the serum PRL in every child with pituitary enlargement, particularly before any surgery is contemplated.17
Isolated adrenocorticotropic hormone insufficiency may occur in lymphocytic hypophysitis, although this condition is not a malignant tumor but presents as the differential diagnosis of a central pituitary mass. This tumor usually occurs in the puerperium and is extremely rare in childhood.18
Diagnosis
MRI scans have become the preferred diagnostic study for pediatric intracranial tumors. MRI is preferred under most circumstances, providing superior resolution and multiplanar imaging capabilities and avoiding the “spray” artifact from the petrous ridge that may obscure computed tomographic images of the base of the brain, without a radiation burden to the child. The administration of gadolinium diethylenetriamine-pentaacetic acid appears to be a safe and effective contrast agent for MRI and provides a more accurate method of imaging in the follow-up of brain tumors in pediatric patients. Where clinical suspicion remains (normal neuroradiologic imaging in patients with DI), scans reported as normal should be sent for expert review and consideration of repeat imaging with time. The intervals for scanning should also be guided clinically, since any set interval is empirical. MRI scan should be performed at a minimum of yearly intervals.19 For lesions with a high frequency of cerebrospinal fluid dissemination, such as primitive neuroectodermal tumors and germ cell tumors, a neuraxis staging evaluation by spinal MRI, if not obtained preoperatively, should be performed approximately 2 weeks after surgery. In children with pineal region tumors, measurement of α-fetoprotein and β–human chorionic gonadotropin in the blood is useful for the diagnosis of malignant germ cell tumors (pineoblastomas); however, cerebrospinal fluid markers are of limited assistance. Placental alkaline phosphatase is a clinically useful tumor marker for primary intracranial germinoma.20
Thyroid function tests (as well as serum PRL concentration) are always required prior to surgery of a suspected pituitary tumor.17 An elevated serum PRL concentration requires the distinction between stalk compression with moderate rise in PRL from the very high PRL concentrations associated with a PRL-secreting tumor. Macroprolactinomas usually have very high PRL secretion, and there is little ambiguity about the diagnosis. It is important to distinguish thyroid-stimulating hormone–secreting adenomas, which are extremely rare, from the pituitary hyperplasia associated with longstanding primary hypothyroidism. After prolonged, severe primary hypothyroidism with increased secretion of thyroid-stimulating hormone, the pituitary gland is usually increased in size and may attain a suprasellar extension and compression of the optic chiasm/nerves. This may be accompanied by a gonadal form of premature sexual maturation which is not true puberty (isolated breast development in girls and large testicular volumes with minimal virilization in boys).21 These signs of premature maturation and pituitary enlargement decrease or resolve following the decrease in secretion of thyroid-stimulating hormone within 6 months of commencing appropriate thyroxin replacement.
Therapy
General Principles of Treatment
In general, the aim of therapy is to eradicate the tumor, with minimal morbidity and mortality, since prognosis is correlated with the extent of resection. If no biopsy has been obtained, histology will be undertaken postoperatively. Because the details of treatment for many types have evolved over time and likely will continue to evolve, treatment decisions for individual patients are best made in the context of a multidisciplinary “team” approach (Table 13-2
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
