CHAPTER 16 Eukaryotic RNA Processing*
In all organisms, the genetic information is encoded in the sequence of the DNA. However, to be used, this information must be copied, or transcribed, into the related polymer, RNA. Eukaryotes synthesize many different types of RNA, but none of these RNAs is simply transcribed as a finished product. The mature, functional forms of all eukaryotic RNA species are generated by posttranscriptional processing, and these processing reactions are the major topic of this chapter.
The major RNAs can be assigned to three major classes: (1) The cytoplasmic messenger RNAs (mRNAs) and their nuclear precursors (pre-mRNAs) carry the information that is used to specify the sequence, and therefore the structure, of all proteins in the cell. (2) Other RNAs do not encode protein but function directly, playing major roles in several metabolic pathways, including protein synthesis. These include the ribosomal RNAs (rRNAs) and transfer RNAs (tRNAs), which are the key components of the protein synthesis machinery; the small nuclear RNAs (snRNAs), which form the core of the pre-mRNA splicing system; and the small nucleolar RNAs (snoRNAs), which are important factors in ribosome biogenesis. These RNAs are generally much longer-lived than are the mRNAs and therefore often are referred to as stable or nonprotein coding RNAs (ncRNAs). (3) The third and most recently identified class of RNA comprises several structurally related groups of very small (21 to 25 nucleotides) RNA species that play important roles in regulating gene expression. Base pairing between endogenous micro-RNAs (miRNAs) and target mRNAs in the cytoplasm represses their translation into protein. The packaging of DNA into a nontranscribed form termed heterochromatin (see Fig. 13-9) is promoted by a class of nuclear, small heterochromatic RNAs (shRNAs). Finally, the introduction of small double-stranded RNAs into many cell types and organisms results in cleavage of the target mRNA and consequent silencing of gene expression. This phenomenon is described as RNA interference (RNAi), and the RNAs are referred to as small interfering RNAs (siRNAs).
Synthesis of mRNAs
An overview of mRNA synthesis and degradation is shown in Figure 16-1.
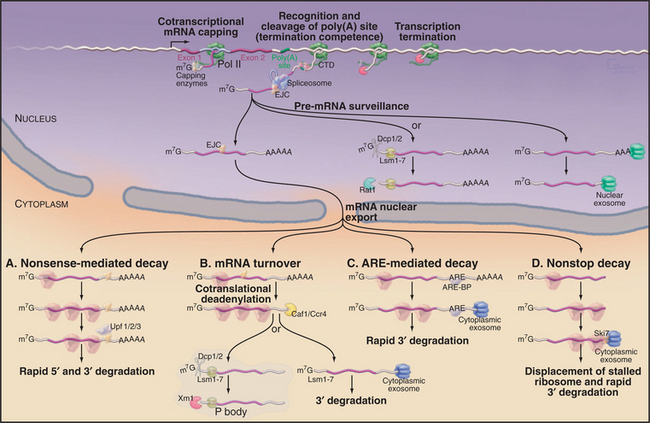
Figure 16-1 Synthesis and degradation of eukaryotic mRNAs. Nascent mRNA transcripts are transcribed by RNA polymerase II. Formation of the 5′ cap structure and cleavage and polyadenylation of the 3′ end of the mRNA both occur cotranscriptionally and involve factors that are recruited by the C-terminal domain (CTD) of the transcribing polymerase (see Fig. 15-4). The termination of transcription requires both the recognition of the site of polyadenylation and the activity of the 5′-exonuclease Rat1, which degrades the nascent RNA transcripts. Rat1 binds to the polymerase CTD via Rtt103. Pre-mRNA splicing can either be cotranscriptional or occur shortly after transcript release, and recruitment of splicing factors is not strongly dependent on the CTD. In human cells, the spliceosome deposits the exon-junction complex (EJC) around 24 nucleotides upstream of the site of splicing. Several steps in nuclear mRNA maturation also are subject to surveillance. In yeast, nuclear pre-mRNAs can be either 3′ degraded by the nuclear exosome complex or decapped and 5′ degraded by the exonuclease Rat1. Nuclear decapping requires the Lsm2–8 complex and is probably performed by the Dcp1/2 decapping complex. Once in the cytoplasm, the mRNA is translated into proteins and undergoes degradation. Several different mRNA degradation pathways have been identified. A, Nonsense-mediated decay (NMD). If the EJCs all lie within or very close to the ORF, they will be displaced by the translating ribosomes. However, if an EJC lies beyond the end of the ORF, it will remain on the translated mRNA. This is taken as evidence that translation has terminated prematurely and triggers the NMD pathway. Recognition of the EJC requires the Upf1/2/3 surveillance complex, which also interacts with the ribosomes as they terminate translation. In yeast, NMD triggers both rapid decapping and 5′ degradation, without prior deadenylation, and 3′ degradation by the exosome. B, General mRNA turnover. During translation, most mRNAs undergo progressive poly(A) tail shortening. Loss of the poly(A) tail leads to rapid degradation. As in the nucleus, cytoplasmic mRNAs can be degraded from either the 5′ or the 3′ end. 5′ degradation occurs largely in a specialized cytoplasmic region termed the P body in yeast or cytoplasmic foci in human cells. Here, the mRNAs are decapped by the Dcp1/2 heterodimer and then degraded by the cytoplasmic 5′-exonuclease Xrn1. Both activities are strongly stimulated by the cytoplasmic Lsm1–7 complex. Alternatively, deadenylated mRNAs can be 3′ degraded by the cytoplasmic exosome. C, ARE-mediated decay. In this pathway, specific A+U rich elements (AREs) are recognized by ARE-binding proteins (ARE-BP) in the nucleus. These are transported to the cytoplasm in association with the mRNA and recruit the cytoplasmic exosome to rapidly degrade the RNA. D, Nonstop decay. If the mRNA lacks a translation termination codon, the first translating ribosome will stall and be trapped at the 3′ end of the RNA. The Ski7 protein, which is associated with the cytoplasmic exosome complex, is believed to release the stalled ribosome and target the RNA for 3′ degradation by the exosome. Note that this legend provides detail beyond the text.
mRNA Capping and Polyadenylation
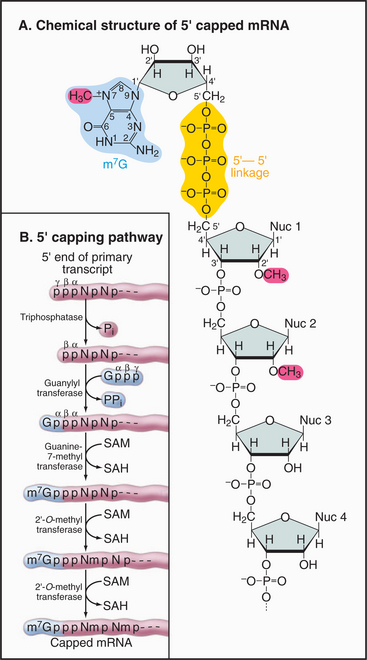
The mRNA cap is an unusual structure. It consists of an inverted 7-methylguanosine residue, which is joined onto the body of the mRNA by a 5′-triphosphate-5′ linkage (Fig. 16-2). Cap addition involves three enzymatic activities: A 5′ RNA triphosphatase cleaves the 5′ triphosphate on the nascent transcript to a diphosphate; RNA guanylyltransferase forms a covalent enzyme–GMP complex and then caps the RNA by transferring this to the diphosphate; and RNA (guanine-7) methyltransferase covalently alters the guanosine base by methylation, generating m7G. In addition, the first encoded nucleotides are frequently modified by methylation of the 2′ hydroxyl position on the ribose group, but the functional significance of these internal modifications is currently unclear.
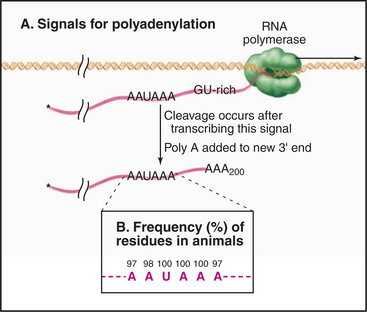
During 3′ processing, the nascent pre-mRNA is cleaved by an endonuclease, and a tail of adenosine residues is added by poly(A) polymerase. Around 200 to 250 A residues are added to mRNAs in human cells, and around 70 to 90 are added in yeast. Cleavage and polyadenylation are performed by a large complex containing approximately 20 proteins that recognizes sequences in the mRNA, of which the best defined is a highly conserved AAUAAA motif located upstream of the site of polyadenylation (Fig. 16-3).
Links between mRNA Processing and Transcription
The processes of cap addition and 3′ cleavage and polyadenylation are both linked to transcription of the mRNA by RNA polymerase II and occur cotranscriptionally on the nascent RNA (Fig. 16-1). The C-terminal domain (CTD) of the largest subunit of RNA polymerase II (RNA pol II) consists of many copies of a seven-amino-acid repeat (YSPTSPS), which undergo reversible modification by phosphorylation (see Fig. 15-4). A pronounced change in the CTD phosphorylation pattern coincides with the release of the polymerase from initiation mode into processive elongation mode. Immediately following transcription initiation, the repeats are largely phosphorylated on the serine residue at position 5. This modification is lost, while serine 2 phosphorylation increases, as the polymerase moves along the transcript. Capping of the 5′ end of the mRNA occurs by the time the transcript is approximately 25 to 30 nucleotides long, and the capping enzyme interacts with the serine 5 phosphorylated CTD. This and other interactions with the polymerase result in strong allosteric activation of capping activity. In contrast, the cleavage and polyadenylation factors involved in 3′ end processing are recruited by interaction with the CTD phosphorylated at serine 2.
Pre-mRNA Splicing
Signals for Splicing
Three conserved sequences within introns play key roles in their accurate recognition by the splicing machinery (Fig. 16-4). These lie immediately adjacent to the 5′ splice site and the 3′ splice site and surrounding an internal region that will form the intron branch point during the splicing reaction. The U1 and U6 snRNAs have sequences that are complementary to the 5′ splice site, while U2 is complementary to the branch point region.
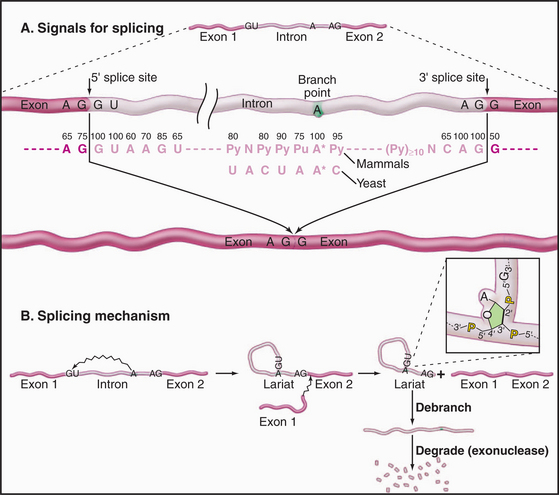
No sequences in the exons are strictly required for splicing, but there are important stimulatory elements termed exonic splicing enhancers (ESEs), which generally bind members of the SR-protein family. The ESEs have two major functions: They stimulate the use of the flanking 5′ and 3′ splice sites, promoting exon definition, and they prevent the exon in which they are located from being included in an intron. This latter function is particularly important in ensuring that all introns are spliced out without the splicing machinery skipping from the 5′ end of one intron to the 3′ end of a downstream intron.
The Pre-mRNA Splicing Reaction
The splicing reaction proceeds in two steps (Fig. 16-4). In the first, the 5′–3′ phosphate linkage that joins the 5′ exon to the first nucleotide of the intron—at the 5′ splice site—is attacked and broken. This reaction leaves the 5′ end of the intron attached to the adenosine residue via an unusual 5′–2′ phosphate linkage. Since this adenosine remains attached to the flanking nucleotides by conventional 5′ and 3′ phosphodiester bonds, this creates a circular molecule with a tail that includes the 3′ exon. This structure is termed the intron lariat, and the adenosine to which the 5′ end of the intron is attached is termed the branch point, because it has a branched structure. In the second step of splicing, the free 3′ hydroxyl on the 5′ exon is used to attack and break the linkage between the last nucleotide of the intron and the 3′ exon—at the 3′ splice site. This leaves the 5′ and 3′ exons joined by a conventional 5′–3′ linkage and releases the intron as a lariat. This is linearized by the debranching enzyme and is probably rapidly degraded from both ends by exonucleases.
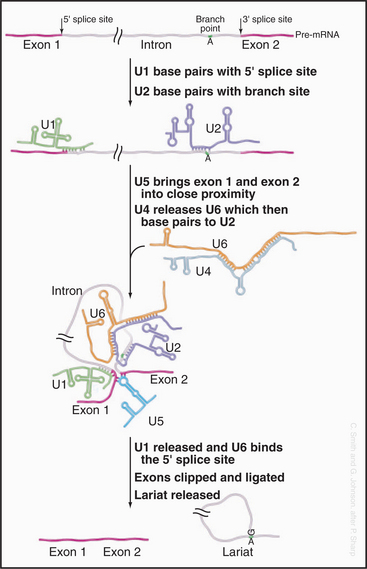
The initial steps in splicing are the recognition of the 5′ splice site by the U1 snRNA and the binding of U2 snRNA to the branch-point region, assisted by SR-proteins (Fig. 16-5). Base pairing between U2 and the pre-mRNA leaves a single adenosine bulged out of a helix and available for interaction with the 5′ splice site. The U4 and U6 snRNAs then join the spliceosome as a base-paired duplex, within a large complex that also contains the U5 snRNA. The U4 and U6 base pairing is opened, and the liberated U6 sequences displace U1 at the 5′ splice site. They also bind to U2—bringing the 5′ splice site and branch point into close proximity. At this point, the first enzymatic step of splicing occurs. This reaction is believed to be directly catalyzed by the intricate structure of the snRNA/pre-mRNA interactions rather than by the protein components of the spliceosome. The 5′ splice site is attacked and broken by the ribose 2′ hydroxyl group of the adenosine residue that is bulged out of the U2-intron duplex. The U5 snRNA and its associated proteins are responsible for holding onto the now free 5′ exon and correctly aligning it with the 3′ exon for the second catalytic step of splicing.
AT-AC Introns
The large majority of human mRNA splice sites have a GU dinucleotide at the 5′ splice site and AG at the 3′ splice site (Fig. 16-4). However, a minor group of introns contain different consensus splicing signals and are termed AT-AC (pronounced “attack”) introns because of the identities of the nucleotides located at the 5′ and 3′ splice sites. The splicing of the AT-AC introns involves a distinct set of snRNAs—U11, U12, U4ATAC, and U6ATAC—which replace U1, U2, U4, and U6, respectively. Only U5 is common to both spliceosomes. However, the underlying splicing mechanism is believed to be the same for both classes of intron.
Alternative Splicing
A surprising finding from the human genomic sequencing project was the relatively low number of predicted protein-coding genes, currently estimated at around 30,000. This result has caused increased interest in the phenomenon of alternative splicing, which allows the production of more than one mRNA, and therefore more than one protein product, from a single gene. Several general forms of alternative splicing are commonly found. Exons can be excluded from the mRNAs, or introns can be included. Some genes have arrays of multiple alternative exons, only one of which is included in each mRNA. In addition, the use of alternative splice sites can generate longer or shorter forms of individual exons (Fig. 16-6).
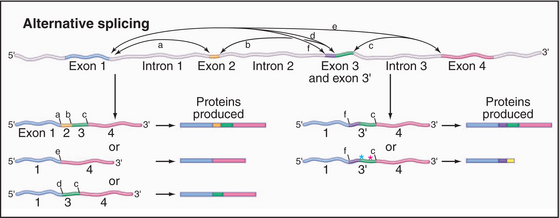
Figure 16-6 alternative splicing can generate multiple different proteins from a single gene. Here are some of the possible mRNA and protein products of a gene whose pre-mRNA is subject to alternative splicing. Left, Examples show the other effects of skipping one or more internal exons, which produces a set of related proteins with different combinations of “modules.” Right, Examples show the effects of alternative splice sites. In the case shown, the use of alternative 3′ splice sites redefines the 5′ end of the downstream exon. This can lead to the inclusion of additional amino acids in the protein product. Use of an alternative splice site can also cause the exon to be read in a different reading frame (green asterisk), changing the amino acid sequence. If the alternative reading frame contains a translation stop codon (red asterisk), a truncated protein will be produced, and the mRNA will generally be targeted for rapid degradation by the NMD pathway (Fig. 16-1).
Current estimates for the proportion of human genes that are subject to alternative splicing range from 30% to 75%. In some cases, this could potentially give rise to a very large number of different protein isoforms. In other cases, alternatively spliced proteins can have antagonistic functions, such as transcription activation versus transcription repression. For the vast majority of human genes, no information is available on the relative activities of different spliced isoforms. Compounding the difficulty in understanding is the fact that many genes show tissue-specific splicing. Thus, a gene could be transcribed in, say, both the liver and brain but generate products with substantially different functions in each tissue. In addition to generating protein diversity, alternative splicing can generate mRNAs with premature translation termination codons—“nonsense” co-dons. These are subject to rapid degradation by the nonsense-mediated decay (NMD) surveillance pathway (see later). Switching splicing into a pathway that generates an NMD target is therefore a means of downregulating gene expression.
Localization of Pre-mRNA Splicing
The location of the splicing reaction within the nucleus was long a contentious topic. The snRNAs can be detected dispersed in the nucleoplasm but concentrate in small structures referred to as nuclear speckles or interchromatin granules, as well as in discrete larger structures known as Cajal bodies (see Fig. 14-2). It is now widely accepted that most splicing is performed by the dispersed snRNA population and can occur either cotranscriptionally or immediately following transcript release. Consistent with this, there is evidence that the recruitment of some splicing factors is promoted by association with the CTD of the transcribing polymerase. The speckles are likely to represent sites at which splicing factors are stockpiled ready for use. The Cajal bodies, in contrast, represent sites of maturation in which the snRNAs undergo site-specific nucleotide modification and perhaps assembly with specific proteins.
Editing of mRNAs
C-to-U Editing
Deamination of cytosine to uracil is performed by an editing complex, sometimes referred to as the editosome, which includes the deaminase Apobec-1 (Fig. 16-7). Only a small number of nuclear-encoded targets have been identified to date, and in these, editing generates translation termination codons, producing shorter forms of the encoded proteins. The best-characterized example of C-to-U RNA editing involves the mRNA encoding intestinal apolipoprotein B (ApoB), where CAA-to-UAA editing in the loop of a specific stem-loop structure generates a stop codon. The truncated protein, ApoB48, has an important role in lipoprotein metabolism. In other cases editing may generate mRNAs that are targets for NMD (see later), leading to down-regulation of protein expression.
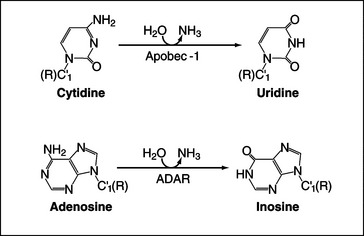
A-to-I Editing
The enzyme ADAR (adenosine deaminase acting on RNA) can convert adenine residues to inosine by deamination of the base (Fig. 16-7). Inosine acts like guanosine and base-pairs with cytosine rather than uracil, potentially altering the protein encoded by the mRNA. Most of the transcripts that are edited by ADAR encode receptors of the central nervous system, and RNA editing is required to create the full receptor repertoire. The amino acid substitutions that result from editing of the mRNAs can greatly alter the properties of ion channels, and aberrant editing occurs in various disorders ranging from epilepsy to malignant brain gliomas. ADAR binds as a dimer to imperfect double-stranded RNA duplexes, which are formed between the target site and sequences in a flanking intron. Editing is generally not 100% efficient, so heterogeneous populations of proteins are generated.
mRNA Degradation and Surveillance
Degradation of mRNA
Different pathways of mRNA degradation can be classified as (1) the default pathway (i.e., when we do not yet know of any specific activator or repressor of degradation), (2) regulated degradation pathways that respond to developmental or other signals, and (3) surveillance pathways that identify and rapidly degrade aberrant mRNAs or pre-mRNAs. A theme emerging from studies of all mRNA decay pathways is that RNA-binding proteins, which associate with the newly transcribed precursor in the nucleus, can be retained when the mRNA is exported to the cytoplasm. These proteins maintain a record of the nuclear history of the RNA that can be “read” by the cytoplasmic degradation machinery, and this plays a key role in determining the cytoplasmic fate of the mRNA.
A key step in the timing of degradation of most mRNA is the slow, stepwise removal of the poly(A) tail by enzymes called deadenylases. The intact poly(A) tail is bound by multiple copies of the poly(A)-binding protein (PABP), at a stoichiometry of around one molecule per 10 to 20 A residues. Surprisingly, PABP antagonizes 5′ cap removal, probably via interactions with the translation initiation factor eIF4G, which in turn stabilizes the cap-binding protein eIF4E. These interactions effectively circularize the mRNA and strongly stimulate translation initiation (see Fig. 17-9). When the tail becomes too short for the last PABP molecule to bind, these interactions are lost. The cap can then be removed by a decapping complex, which cleaves the triphosphate linkage to the body of the mRNA, releasing m7GDP. Cap removal allows rapid 5′ to 3′ degradation of the mRNA by the 5′ exonuclease Xrn1. In addition, loss of the PABP/poly(A) complex allows 3′-degradation of the mRNA by the cytoplasmic exosome.
ARE-Mediated Degradation
The degradation of many mRNA species in human cells is triggered by the presence of sequence motifs referred to as A+U-rich elements (AREs) (Fig. 16-1C). These are generally located in the 3′ UTR of the mRNA, where bound proteins will not be displaced by the translating ribosomes. This pathway plays an important regulatory role in gene expression, as it targets for rapid turnover mRNAs that encode proteins such as cytokines, growth factors, oncogenes, and cell-cycle regulators, for which limited and transient expression is important. Computational analyses indicate that up to 8% of human mRNAs carry AREs, and there is evidence that alterations in the activity of this pathway are associated with both developmental decisions and cancer. ARE-binding proteins associate with the nuclear pre-mRNAs and are exported to the cytoplasm, where they can either activate or repress ARE-mediated decay. Some ARE-binding proteins that activate degradation function by directly recruiting the exosome complex to degrade the mRNA from the 3′ end.
Surveillance of mRNAs
Nonsense-Mediated Decay
The surveillance of mRNA integrity is important because defective molecules can encode truncated proteins, which are frequently toxic to the cell. The presence of a premature translation termination signal (or nonsense codon) strongly destabilizes mRNA via the nonsense-mediated decay (NMD) pathway (Fig. 16-1A). In human cells, termination codons are identified as being located in a premature position by reference to the sites of pre-mRNA splicing. Normal termination codons are within, or very close to, the 3′ exon, so no former splice sites lie far downstream. If any former splice site is located more than about 50 nucleotides downstream of the site of translation termination, the mRNA is targeted for degradation. The sites of former splicing events can be identified in the spliced mRNA product, because the spliceosome deposits a specific protein complex on the mRNA during the splicing reaction (Fig. 16-1). This is called the exon-junction complex (EJC), and it binds to the 5′ exon sequence ˜24 nucleotides upstream of the splice site. Several of the EJC components remain associated with the mRNA following its export to the cytoplasm. In normal mRNAs, the EJCs will all be displaced by the first translating ribosome, so if one (or more) remains on the mRNA, then translation has terminated too soon.
Nonstop Decay
Some mRNAs lack any translation termination codon, because they have been inappropriately polyadenylated, inaccurately spliced, or partially 3′-degraded. Translating ribosomes efficiently stall at the ends of such nonstop mRNAs, and this inhibits the repeated synthesis of truncated proteins (Fig. 16-1D). The cytoplasmic form of the exosome complex is associated with Ski7p, which is homologous to the GTPases that function in translation. The interaction of Ski7p with the stalled ribosome is believed to both release the ribosome and target the mRNA for rapid degradation.
Synthesis of Stable RNAs
Transfer RNA Synthesis
All tRNAs are processed from precursors (pre-tRNAs) that are extended at their 5′ and 3′ ends (Fig. 16-8). Some pre-tRNAs are polycistronic, with two or more tRNAs excised from the same precursor. In yeast, at least, the genes that encode tRNAs cluster around the surface of the nucleolus, and pre-tRNA processing appears to occur largely within the nucleolus.
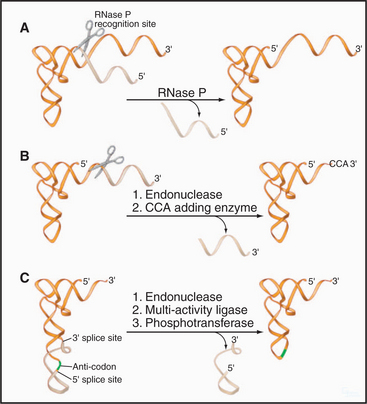
In addition, tRNAs are subject to a bewildering array of covalent nucleotide modifications. Almost 100 different modified nucleotides have been identified in tRNAs, ranging from simple methylation to the addition of very elaborate molecules. All are added without breaking the phosphate backbone of the RNA. The structures of all mature tRNAs are very similar, since each must fit exactly into the A, P, and E sites of the translating ribosome (see Fig. 17-7). It is likely that the modifications help the tRNAs fold into precisely the correct shape. They also aid accurate recognition of different tRNAs by the aminoacyl-tRNA synthases, which are responsible for charging each species of tRNA with the correct amino acid.
Ribosome Synthesis
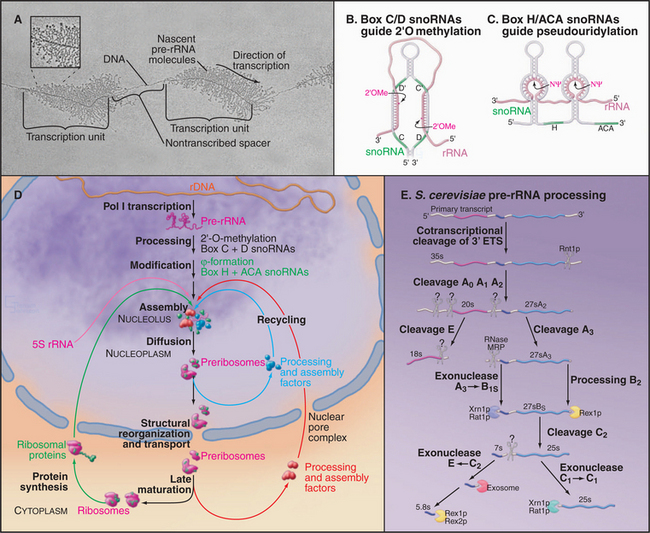
The synthesis of ribosomes is a major activity of any actively growing cell. Three of the four rRNAs—the 18S, 5.8S, and 25S/28S rRNAs—are cotranscribed by RNA polymerase I as a polycistronic transcript. This pre-rRNA is the only RNA synthesized by RNA polymerase I (RNA pol I) and is transcribed from arrays of the ribosomal DNA (rDNA) repeated in tandem. In humans, approximately 300 to 400 rDNA repeats are present in five clusters (on chromosomes 13, 14, 15, 21, and 22). These sites often are referred to as nucleolar organizer regions, reflecting the fact that nucleoli assemble at these locations in newly formed interphase nuclei. The pre-rRNAs are very actively transcribed and can be visualized as “Christmas trees” in electron micrographs taken following spreading of the chromatin using low-salt conditions and detergent (Fig. 16-9A). The 5S rRNA is independently transcribed by RNA polymerase III. In the majority of eukaryotes, the 5S rRNA genes are present in separate repeat arrays.
The Nucleolus
Most steps in ribosome synthesis take place within a specialized nuclear substructure, the nucleolus (see Fig. 14-3). In micrographs, the nucleolus appears to be a very large and stable structure, but kinetic experiments indicate that it is in fact highly dynamic, with most nucleolar proteins rapidly exchanging with nucleoplasmic pools. There is little evidence that signals for the localization of proteins or mature snoRNAs to the nucleolus are distinct from the features that allow them to function there. A current view of the nucleolus is that its assembly is the consequence of many relatively weak and transient interactions between the nucleolar proteins. The result is a self-assembly process that greatly increases the local concentration of ribosome synthesis factors. This is envisaged to promote efficient preribosome assembly and maturation while allowing the rapid and dynamic changes in preribosome composition in-volved in this pathway. Similar mechanisms may generate other subnuclear structures such as Cajal bodies.
The key steps in ribosome synthesis are (1) transcription of the pre-rRNA, (2) covalent modification of the mature rRNA regions of the pre-rRNA, (3) processing of the pre-rRNA to the mature rRNAs, and (4) assembly of the rRNAs with the ribosomal proteins (Fig. 16-9D). During ribosome synthesis, the maturing preribosomes move from their site of transcription in the dense fibrillar component of the nucleolus, through the granular component of the nucleolus. They are then released into the nucleoplasm prior to transport through the nuclear pores to the cytoplasm. Here, the final maturation into functional 40S and 60S ribosomal subunits takes place.
Pre-rRNA Processing
The posttranscriptional steps in ribosome synthesis are very complex, involving approximately 200 proteins and approximately 100 snoRNA species, in addition to the four rRNAs and approximately 80 ribosomal proteins. Ribosome synthesis is best understood in budding yeast, but all available evidence indicates that it is highly conserved throughout eukaryotes. Many pre-rRNA processing enzymes have been identified, although others remain to be found (Fig. 16-9E). A combination of endonuclease cleavages and exonuclease digestion steps generates the mature rRNAs in a complex, multistep processing pathway. The remaining species, 5S rRNA, is independently transcribed and undergoes only 3′-trimming. Notably, all of the nucleases indicated in Figure 16-9E process other RNAs in addition to the pre-rRNAs. It seems very probable that when the enzymes responsible for the remaining processing activities are identified, they too will be found to process other substrates.
Modification of the Pre-rRNA
The rRNAs are subject to covalent nucleotide modification at many sites. Modification takes place on the pre-rRNA, either on the nascent transcript or shortly following transcript release. The majority of modifications are methylation of the 2′-hydroxyl group on the sugar ring (2′-O-methylation) and conversion of uracil to pseudouridine by base rotation. The sites of these modifications are selected by base pairing with two groups of small nucleolar RNAs (snoRNAs). The box C/D snoRNAs direct sites of 2′-O-methylation and carry the methyltransferase (called fibrillarin in humans and Nop1 in yeast) (Fig. 16-9B). The box H/ACA snoRNAs select sites of pseudouridine formation and carry the pseudouridine synthase (called dyskerin in humans and Cbf5 in yeast [Fig. 16-9C]).
A small number of snoRNAs do not direct RNA modification but are required for pre-rRNA processing. The best characterized is the U3 snoRNA, which binds cotranscriptionally to the 5′–external transcription factor (ETS) region of the pre-rRNA. Base pairing between U3 and the pre-rRNA is required for the early processing reactions on the pathway of 18S rRNA synthesis and directs the assembly of a large pre-rRNA processing complex called the small subunit processome. This complex can be visualized as a “terminal knob” in micrographs of spread pre-rRNA transcripts (Fig. 16-9A).
Small Nuclear RNA Maturation
The U1, U2, U4, and U5 snRNAs are encoded by individual genes transcribed by RNA polymerase II (Fig. 16-10C). Like mRNAs, the snRNA precursors undergo cotranscriptional capping with 7-methylguanosine, but they are not polyadenylated. In human cells, the newly synthesized precursors to these snRNAs are then exported to the cytoplasm. Once in the cytoplasm, the snRNAs form complexes with the Sm-proteins. This set of seven different, but closely related, proteins assembles into a heptameric ring structure. Sm-proteins are named after the human autoimmune serum that was initially used in their identification. On their own, the Sm-proteins show low substrate specificity in RNA binding. However, in human cells, the assembly of the snRNAs with the Sm-proteins is highly specific and is mediated by a large protein complex. This complex includes the SMN protein (survival of motor neurons), which is the target of mutations in the relatively common genetic disease spinal muscular atrophy. While in the cytoplasm the snRNAs are further processed; the 3′ end of the RNA is trimmed, and the cap structure undergoes additional methylation to generate 2,2,7-trimethylguanosine. This hypermethylated cap structure is also present on small nucleolar RNAs (see later) and might be important to allow resident nuclear RNAs to be distinguished from mRNA precursors.
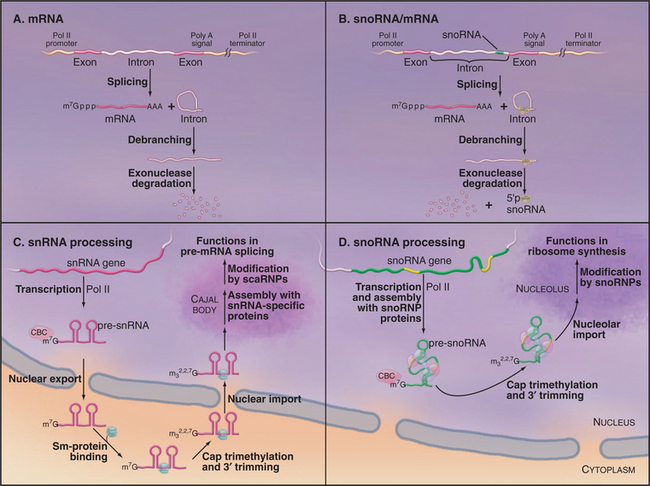
Once the cap is trimethylated and bound by the Sm-proteins, the snRNAs can be reimported into the nucleus, where they initially localize to discrete subnuclear structures termed Cajal bodies (see Fig. 14-2). Within the Cajal bodies, specific nucleotides in the snRNAs are modified by 2′-O-methylation and pseudouridine formation. The sites of these modifications are selected by base pairing with a group of resident small Cajal body RNAs (scaRNAs), which carry the RNA-modifying enzymes. The scaRNAs closely resemble the snoRNAs except that single scaRNAs can frequently direct both 2′-O-methylation and pseudouridine for-mation.
Maturation of U6 snRNA is quite different from that of the other snRNAs. U6 is transcribed by RNA polymerase III and is not exported to the cytoplasm. Mature U6 retains the 5′ triphosphate and 3′ poly(U) tract that are characteristic of primary transcripts made by RNA pol III (see Chapter 15). However, the 5′ triphosphate is methylated on the g-phosphate (i.e., the position furthest from the nucleotide), while the terminal U of the poly(U) tract carries a 2′ to 3′ cyclic phosphate. Both of these modifications may help to protect the RNA against degradation. U6 does not bind the Sm-proteins but instead associates with a related heptameric ring structure that is comprised of seven Lsm-proteins (“like sm”). Two distinct but related heptameric Lsm-complexes are present in the nucleus and cytoplasm. The nuclear Lsm2-8 complex binds to the U6 snRNA and also participates in the decapping of mRNA precursors that are destined for degradation in the nucleus (Fig. 16-1). In contrast, the Lsm1-7 complex participates in mRNA decapping and 5′ degradation in the cytoplasm. Nucleotides within the U6 snRNA are also modified at positions that are selected by guide RNAs, but this modification occurs in the nucleolus rather than the Cajal body.
Small Nucleolar RNA Maturation
The small nucleolar RNAs (snoRNAs) are all transcribed by RNA polymerase II (except in some plants in which pol III-transcribed snoRNAs can be found). However, the genes encoding snoRNAs can have a surprising variety of different organizations. In human cells, most snoRNAs are excised from the introns of genes that also encode proteins in their exons (Fig. 16-10B). The introns that encode snoRNAs are released by splicing and then linearized by debranching. The mature snoRNA is then generated by controlled exonuclease digestion. In contrast, most characterized snoRNAs in higher plants and several yeast snoRNAs are processed from polycistronic precursors that encode multiple snoRNA species. Individual pre-snoRNAs are liberated by cleavage of the precursor by the double–strand-specific endonuclease RNase III (Rnt1 in yeast) and then trimmed at both the 5′ and 3′ ends. SnoRNAs can also be processed from single transcripts, and these have many features in common with snRNA transcripts. Like snRNAs, these individually transcribed snoRNAs carry trimethylguanosine cap structures (Fig. 16-10D). However, unlike snRNAs, which have a cytoplasmic phase, the maturation of snoRNAs and assembly of snoRNPs take place entirely within the nucleus, most steps probably occurring in the nucleolus.
Synthesis and Function of miRNAs
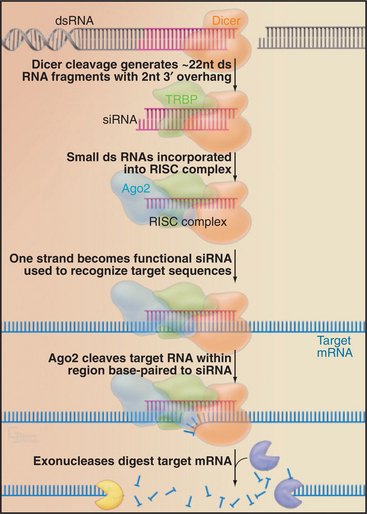
Endogenous micro-RNAs (miRNAs) are encoded in the genomes of many eukaryotes, including humans (Fig. 16-11). These are frequently transcribed as polycistronic precursors called pri-miRNAs. Within the pre-miRNA, the precursors to the individual miRNAs (pre-miRNAs) form stem-loop structures. The stems are first cleaved by a nuclear double-strand-specific endonuclease called Drosha, releasing the individual pre-miRNAs. These are then exported to the cytoplasm, where cleavage by a second double-strand-specific endonuclease, Dicer, releases the miRNA in the form of a duplex with characteristic 2-nucleotide 3′ overhangs and 5′ phosphate groups. These duplexes are incorporated into the RISC complex, where one of the strands becomes the functional miRNA. If the target mRNA sequence is incompletely complementary to the miRNA, its translation is repressed (Fig. 16-11). This is likely to be the normal function of most endogenous miRNAs. It has recently been estimated that 30% or more of human mRNAs are targets of miRNA regulation. miRNAs show tissue-specific patterns of expression and dynamic changes in expression during differentiation. Individual miRNAs can modulate the expression of many different mRNAs.
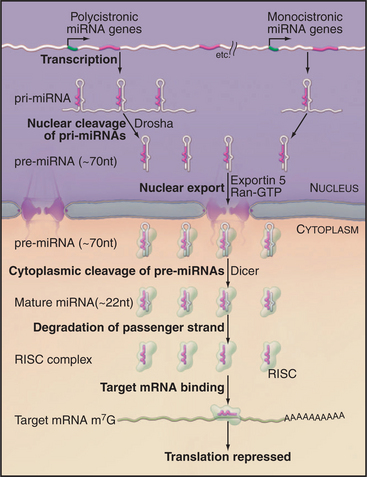
Figure 16-11 mrna maturation. The polycistronic miRNA precursors (termed primary-miRNAs, or pri-miRNAs) are cleaved by the double-strand-specific endonuclease Drosha within the nucleus. The individual pre-miRNAs are then exported to the cytoplasm by the export factor Exportin 5 in complex with Ran-GTP (see Fig. 14-17). Once in the cytoplasm, the pre-miRNAs are cleaved by the double-strand-specific endonuclease Dicer. One strand of the resulting duplex is then incorporated into the RNA-induced silencing complex (RISC) and becomes the functional miRNA. Imperfect duplexes are formed between the miRNA and target mRNAs; this results in the inhibition of the mRNA translation.
If a target RNA sequence is found that is perfectly complementary to the miRNA, it is cleaved by a component of the RISC complex, Ago2 (“Slicer”). Target RNA cleavage occurs within the miRNA: mRNA duplex at a fixed distance (between nucleotides 10 and 11) from the 5′ end of the miRNA, which is specifically bound and used to precisely position the duplex relative to the catalytic site.
This pathway can be exploited in techniques for the specific inactivation of target mRNAs, termed RNA interference (RNAi [Fig. 16-12]). The technique uses exogenously provided RNAs that are generally fully complementary to the target, typically provided as 22-nucleotide RNAs termed small interfering RNAs (siRNAs). In many organisms (e.g., in Drosophila or the nematode Caenorhabditis elegans), RNAi can be performed by introducing long double-stranded RNAs. These are cleaved in vivo by Dicer into 22-base-pair fragments, which are then incorporated into the RISC complex. In mammals, including human cells, long double-stranded RNAs cannot be used for RNAi, as they trigger an antiviral response and cell death. RNAi can, however, be performed in human cells by the introduction of precleaved 22-bp RNA fragments. Alternatively, small hairpin structures can be expressed that resemble endogenous pre-miRNAs and are processed into functional 22-nucleotide siRNAs in vivo. The small size, ease of use, and potent function of siRNAs have made RNAi the method of choice for many analyses of eukaryotic gene function.
In the nucleus, a closely related system is used to establish transcriptional silencing of RNA synthesis (Fig. 16-13). Although important gaps remain in our understanding, it appears that transcription of a region of the chromosomal DNA on both strands, generating a double-stranded RNA, may be sufficient to induce its silencing. The double-stranded RNA is likely to be cleaved by Dicer and/or Drosha to generate 22-nucleotide fragments, in this case termed small heterochromatic RNAs (shRNAs). These associate with a nuclear complex called RITS (RNA-induced transcriptional silencing [see Fig. 16-13]), which is related to the cytoplasmic RISC complex. These shRNAs identify the corresponding gene, possibly by binding to nascent RNA transcripts and, together with the RITS complex components, recruit a protein methyltransferase. This methylates histone H3 on lysine 9, a hallmark of repressive heterochromatin, which in turn recruits other heterochromatin proteins such as HP1 (see Fig. 13-9). The RITS complex includes an RNA-dependent RNA polymerase, and this may be able to generate new shRNAs, allowing the spreading of the heterochromatin into flanking sequences. The tendency of heterochromatin to spread into the flanking euchromatin has long been recognized and gives rise to the phenomenon of position effect variegation (see Fig. 13-9). In some eukaryotes, the methylated histone H3 can also recruit DNA methyltransferases that modify cytosine residues to 5′-methylcytosine. This reinforces heterochromatin formation and makes it heritable by daughter cells. It is likely that this system is important for the establishment of heterochromatin domains, such as those surrounding the centromeres in higher eukaryotes. It might also function as a defense system against the amplification of transposable elements.
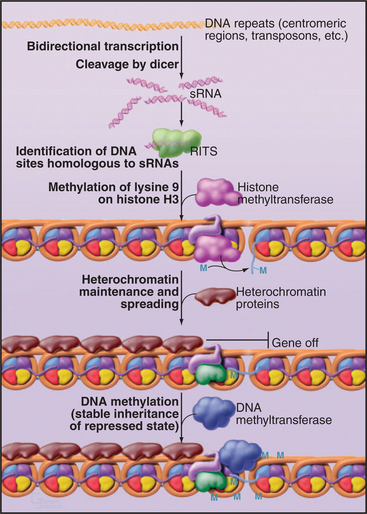
Figure 16-13 shrna function in heterochromatin formation. The targets of miRNAs and siRNAs are cytoplasmic mRNAs. However, sRNAs can also function in the nucleus. Small double-stranded RNAs in the nucleus can associate with the RNA-induced transcriptional silencing (RITS) complex. The sRNA-RITS complex then identifies the genomic site of transcription, possibly by recognition of the nascent transcripts. This leads to the establishment of heterochromatin at this location, via the recruitment of protein methyltransferases that methylate lysine 9 on histone H3, a hallmark of repressive heterochromatin (see Fig. 13-9). In some organisms, this is followed by methylation of the DNA, which makes the repressed heterochromatic state more stable and heritable.
Group I and Group II Self-splicing Introns
Two classes of introns can catalyze their own excision from precursor RNAs. These ribozymes are referred to as group I and group II self-splicing introns. Both classes of RNA fold into complex structures that catalyze splicing via two-step transesterification pathways (Fig. 16-14).
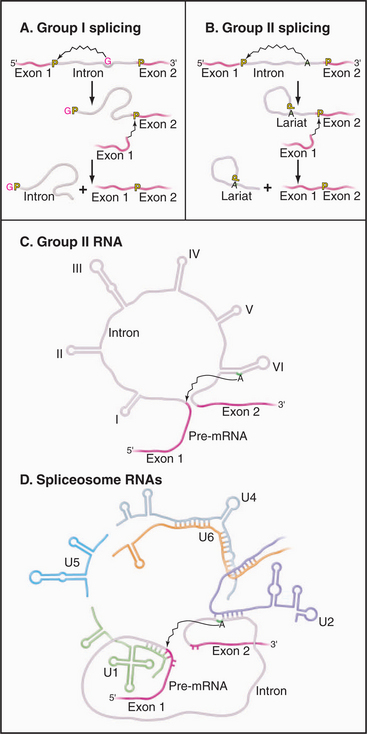
Figure 16-14 comparison of self-splicing with pre-mrna splicing. Groups I and II introns are catalytic RNAs or ribozymes that are able to excise themselves from precursor RNAs in the absence of proteins. A, The removal of group I introns is mechanistically distinct from nuclear pre-mRNA splicing and commences with the binding of an exogenous guanosine nucleotide (red G) within a pocket created by the intronic RNA structure. This G is used to attack and break the phosphate backbone at the 5′ splice site. Subsequently, the free 3′ end of exon 1 attacks the phosphodiester bond at the 3′ splice site, leading to exon ligation and the release of the linear intron. B, In contrast, the mechanism of splicing group II introns is very similar to pre-mRNA splicing. An adenine residue (A) near the 3′ end of the intron attacks the 5′ splice site, leading to the formation of a lariat intermediate. The subsequent attack of the free 3′ end of exon 1 on the phosphodiester bond at the 3′ splice site leads to exon ligation and the release of the intron lariat (compare to Fig. 16-4). C–D, Parallels can be drawn between structure and mechanism of group II self-splicing introns and pre-mRNA splicing. This suggested the model that group II introns gave rise to the nuclear pre-mRNA splicing system. The snRNAs may be derived from fragments of a group II intron, which developed the ability to function in trans (i.e., on other RNAs) rather than acting only in cis on its own sequence. Specifically, Domain VI of the group II introns functions like the U2-branch point duplex in activating the branch-point adenosine by bulging it out of a helix. Domain V acts like the U2-U6 duplex in bringing this adenosine to the 5′ splice site. Domain III resembles the U5 snRNA in base pairing to both the 5′ and 3′ exons at the splice sites.
Group II introns have been found in mitochondria of plants and fungi and in chloroplasts. The splicing mechanism of group II introns strikingly resembles nuclear pre-mRNA splicing (Fig. 16-14C–D). This led to the proposal that the nuclear pre-mRNA splicing system derived from ancestral group II introns. During early eukaryotic evolution, the catalytic center of the group II intron might have become fragmented and separated into the present spliceosomal snRNAs. This would have converted a system that could work only on its own transcript into a system that could process other RNAs, greatly increasing the potential range of spliced RNAs.
RNase P and RNase MRP
Eukaryotes also contain a second RNA-protein enzyme, called RNase MRP, which is closely related to RNase P. The RNA components share common structural features, and the complexes share eight common proteins. RNase MRP cleaves the preribosomal RNA between the small and large subunit rRNAs (Fig. 16-9E). Notably, in many Bacteria, RNase P can cleave the pre-rRNA at a similar position because of the presence of a tRNA within the pre-rRNA transcript. This suggests that RNase MRP arose in an early eukaryote as a specialized form of RNase P, with a specific function in pre-rRNA processing. By analogy to RNase P, cleavage by RNase MRP is predicted to be RNA catalyzed. RNase MRP also functions in mRNA turnover, at least in yeast, initiating the cell-cycle-regulated degradation of a small number of mRNAs.
Large Subunit rRNA
The most important ribozyme is the rRNA component of the large ribosomal subunit, which does not participate in RNA processing but catalyzes peptide bond formation (see Fig. 17-10). During translation elongation, the peptidyl-transferase reaction (the reaction by which amino acid residues are attached to each other to form proteins) is catalyzed by the rRNA itself. The peptidyl-transfer reaction is energetically favorable, and it is currently believed that the catalytic activity derives primarily from the precise spatial positioning of the A-site and P-site tRNAs by the rRNA. The ribosomal proteins act as chaperones in ribosome assembly and as cofactors to increase the efficiency and accuracy of translation.
Almeida R, Allshire RC. RNA silencing and genome regulation. Trends Cell Biol. 2005;15:251-258.
Fromont-Racine M, Senger B, Saveanu C, Fasiolo F. Ribosome assembly in eukaryotes. Gene. 2003;313:17-24.
Kiss T. Small nucleolar RNAs: An abundant group of noncoding RNAs with diverse cellular functions. Cell. 2002;109:145-148.
Parker R, Song H. The enzymes and control of eukaryotic mRNA turnover. Nat Struct Mol Biol. 2004;11:121-127.
Rodriguez MS, Dargemont C, Stutz F. Nuclear export of RNA. Biol Cell. 2004;96:639-655.
Sanford JR, Caceres JF. Pre-mRNA splicing: Life at the centre of the central dogma. J Cell Sci. 2004;117:6261-6263.
Wilusz CJ, Wilusz J. Bringing the role of mRNA decay in the control of gene expression into focus. Trends Genet. 2004;20:491-497.