[level-membership-for-basic-science-category]
Chapter 32 Ethanol, Other Alcohols, and Drugs for Alcohol Dependence
| Abbreviations | |
|---|---|
| ADH | Alcohol dehydrogenase |
| ALDH | Aldehyde dehydrogenase |
| BAC | Blood alcohol concentration |
| CNS | Central nervous system |
| GABA | γ-Aminobutyric acid |
| GI | Gastrointestinal |
| 5-HT | Serotonin |
| NAD+ | Nicotinamide adenine dinucleotide |
| NADH | Nicotinamide adenine dinucleotide, reduced |
| NADD+ | Nicotinamide adenine dinucleotide phosphate |
| NADPH | Nicotinamide adenine dinucleotide phosphate, reduced |
| NE | Norepinephrine |
| NMDA | N-methyl-D-aspartate |
| TNF | Tumor necrosis factor |
| VTA | Ventral tegmental area |
Therapeutic Overview
| Therapeutic Overview |
|---|
| Ethanol is used: |
| Topically to reduce body temperature and as an antiseptic |
| By injection to produce irreversible nerve block by protein denaturation |
| By inhalation to reduce foaming in pulmonary edema |
| In treatment of methanol and ethylene glycol poisoning |
| Ethanol dependence may be treated with psychosocial/behavioral therapy and an: |
| Aldehyde dehydrogenase inhibitor (Disulfiram) |
| Glutamate receptor antagonist (Acamprosate) |
| Opioid receptor antagonist (Naltrexone) |
Mechanisms of Action
Studies suggest that the effects of ethanol may be attributed to its direct binding to lipophilic areas either near or in ion channels and receptors. The ion channels influenced by ethanol are listed in Table 32-1. Ethanol may have either inhibitory or facilitatory effects, depending on the channel, but its resultant action is CNS depression. Because the barbiturates and benzodiazepines exhibit cross-tolerance to ethanol, and their CNS depressant effects are additive with those of ethanol, they may share a common mechanism, perhaps through the γ-aminobutyric acid (GABA) type A receptor (see Chapter 31). Ethanol may also exert some of its effects by actions at glutamate N-methyl-D-aspartate (NMDA) receptors or serotonin (5-HT) receptors.
| Channel | Effects | Ethanol Concentration (mM) |
|---|---|---|
| Na+ (voltage-gated) | Inhibition | 100 and higher* |
| K+ (voltage-gated) | Facilitation | 50-100 |
| Ca++ (voltage-gated) | Inhibition | 50 and higher |
| Ca++ (glutamate receptor-activated) | Inhibition | 20-50 |
| Cl− (GABAA receptor-gated) | Facilitation | 10-50 |
| Cl− (glycine receptor-gated) | Facilitation | 10-50 |
| Na+/K+ (5HT3 receptor-gated) | Facilitation | 10-50 |
* 100 mM ethanol is 460 mg/dL.
The reinforcing actions of ethanol are complex but are mediated in part through its ability to stimulate the dopaminergic reward pathway in the brain (see Fig. 27-8). Evidence has indicated that ethanol increases the synthesis and release of the endogenous opioid β-endorphin in both the ventral tegmental area (VTA) and the nucleus accumbens. Increased β-endorphin release in the VTA dampens the inhibitory influence of GABA on the tonic firing of VTA dopaminergic neurons, whereas increased β-endorphin release in the nucleus accumbens stimulates dopaminergic nerve terminals to release neurotransmitter. Both of these actions to increase dopamine release may be involved in the rewarding effects of ethanol.
Aldehyde Dehydrogenase Inhibitor
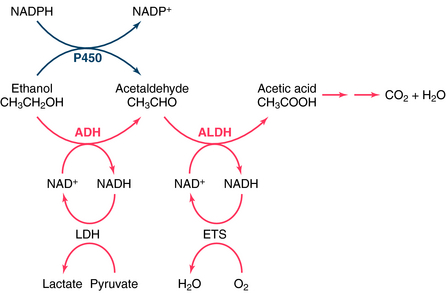
Disulfiram, used for the treatment of alcoholism since the 1940s, is an inhibitor of the enzyme aldehyde dehydrogenase (ALDH), a major enzyme involved in the metabolism of ethanol (Fig. 32-1). Inhibiting the catabolism of acetaldehyde produced by the oxidation of ethanol, leads to the accumulation of acetaldehyde in the plasma, resulting in aversive effects.
Naltrexone is an opioid receptor antagonist at both κ and μ opioid receptors (see Chapter 36). Its ability to inhibit alcohol consumption has been attributed to blockade of μ receptors in both the VTA and nucleus accumbens, thereby decreasing the ethanol-induced activation of the dopamine reward pathway.
Pharmacokinetics
Alcohol taken orally is absorbed throughout the gastrointestinal (GI) tract. Absorption depends on passive diffusion and is governed by the concentration gradient and the mucosal surface area. Food in the stomach will dilute the alcohol and delay gastric emptying time, thereby retarding absorption from the small intestine (where absorption is favored because of the large surface area). High ethanol concentrations in the GI tract cause a greater concentration gradient and therefore hasten absorption. Absorption continues until the alcohol concentration in the blood and GI tract are at equilibrium. Because ethanol is rapidly metabolized and removed from the blood, eventually all the alcohol is absorbed.
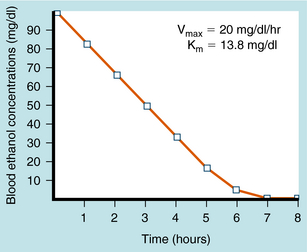
Ethanol undergoes significant first-pass metabolism. Most (>90%) of the ethanol ingested is metabolized in the liver, with the remainder excreted through the lungs and in urine. Alcohol dehydrogenase (ADH) catalyzes the oxidation of ethanol to acetaldehyde, which is oxidized further by ALDH to acetate (see Fig. 32-1). Acetate is oxidized primarily in peripheral tissues to CO2 and H2O. Both ADH and ALDH require the reduction of nicotinamide adenine dinucleotide (NAD+), with 1 mol of ethanol producing 2 mol of reduced NAD+ (NADH). The NADH is reoxidized to NAD+ by conversion of pyruvate to lactate by lactate dehydrogenase (LDH) and the mitochondrial electron transport system (ETS). During ethanol oxidation the concentration of NADH can rise substantially, and NADH product inhibition can become rate-limiting. Similarly, with large amounts of ethanol, NAD+ may become depleted, limiting further oxidation through this pathway. At typical blood alcohol concentrations (BACs), the metabolism of ethanol exhibits zero-order kinetics; that is, it is independent of concentration and occurs at a relatively constant rate (Fig. 32-2). Fasting decreases liver ADH activity, decreasing ethanol metabolism.
Ethanol may also be metabolized to acetaldehyde in the liver by cytochrome P450, a reaction that requires 1 mol of reduced nicotinamide adenine dinucleotide phosphate (NADPH) for every ethanol molecule (see Fig. 32-1). Although P450-mediated oxidation does not normally play a significant role, it is important with high concentrations of ethanol (≥100 mg/dL), which saturate ADH and deplete NAD+. Because this enzyme system also metabolizes other compounds, ethanol may alter the metabolism of many other drugs. In addition, this system may be inhibited or induced (Chapter 2), and induction by ethanol may contribute to the oxidative stress of chronic alcohol consumption by releasing reactive O2 species during metabolism.
Significant genetic differences exist for both ADH and ALDH that affect the rate of ethanol metabolism. Several forms of ADH exist in human liver, with differing affinities for ethanol. Whites, Asians, and African-Americans express different relative percentages of the genes and their respective alleles that encode subunits of ADH, contributing to ethnic differences in the rate of ethanol metabolism. Similarly, there are genetic differences in ALDH. Approximately 50% of Asians have an inactive ALDH, caused by a single base change in the gene that renders them incapable of oxidizing acetaldehyde efficiently, especially if they are homozygous. When these individuals consume ethanol, high concentrations of acetaldehyde are achieved, leading to flushing and other unpleasant effects. People with this condition rarely become alcoholic. As discussed, the unpleasant effects of acetaldehyde accumulation form the basis for the aversive treatment of chronic alcoholism with disulfiram. The pharmacokinetics of ethanol are summarized in Table 32-2.
| Pharmacokinetic Parameter | Considerations |
|---|---|
| Route of administration | Topically, orally, inhalation, by injection into nerve trunks, or intravenously for poison management |
| Absorption |
Naltrexone is available both orally and as depot injections for the treatment of alcohol dependence. The pharmacokinetics of oral naltrexone are discussed in Chapter 36. The depot microsphere preparations are administered intramuscularly once per month and release naltrexone steadily to maintain constant plasma levels.
Relationship of Mechanisms of Action to Clinical Response
Anesthesia occurs when BAC increases to 0.25% to 0.30%. Ethanol shares many properties with general anesthetics but is less safe because of its low therapeutic index (see Chapter 3). It is also a poor analgesic. Coma in humans occurs with BAC above 0.3%, and the lethal range for ethanol, in the absence of other CNS depressants, is 0.4% to 0.5%, though people with much higher concentrations have survived. Death from acute ethanol overdose is relatively rare compared with the frequency of death resulting from combinations of alcohol with other CNS depressants, such as barbiturates and benzodiazepines. Death is due to a depressant effect on the medulla, resulting in respiratory failure.
Physiological and behavioral changes as a function of BAC are summarized in Table 32-3. Measures of BAC are important for providing adequate medical care to intoxicated individuals. BAC is calculated based on the amount of ethanol ingested, the percentage of alcohol in the beverage (usually volume/volume, with 100-proof equivalent to 50% ethanol by volume), and the density of 0.8 g/mL of ethanol. BACs are expressed in a variety of ways. The legal limit for operating a motor vehicle in most states is 80 mg/dL, or 0.08%. An example of a typical calculation for a 70 kg person ingesting 1 oz, or 30 mL, of 80-proof distilled spirits is as follows:
TABLE 32–3 Physiological and Behavioral States as a Function of Blood Ethanol Concentrations
| Blood Ethanol Concentrations | ||
|---|---|---|
| (mg/dL) | % | Reactions |
| 0-50 | 0-0.05 | Loss of inhibitions, excitement, incoordination, impaired judgment, slurred speech, body sway |
| 50-100 | 0.05-0.1 | Impaired reaction time, further impaired judgment, impaired driving ability, ataxia |
| 100-200 | 0.1-0.2 | Staggering gait, inability to operate a motor vehicle |
| 200-300 | 0.2-0.3 | Respiratory depression, danger of death in presence of other CNS depressants, blackouts |
| >300 | >0.3 | Unconsciousness, severe respiratory and cardiovascular depression, death |
| >1200 | >1.2 | Highest known blood concentration with survival in a chronic alcoholic |
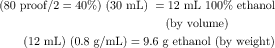
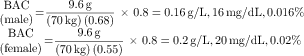
Figure 32-3 shows approximate maximum BACs in men of various body weights ingesting one to five drinks in 1 hour. Rapid absorption is assumed. This figure emphasizes how little consumption is required to impair motor skills and render a person unable to drive safely.
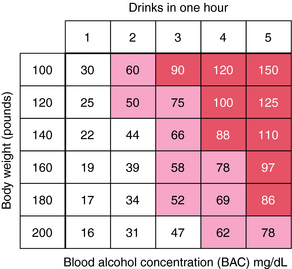
BACs can also be calculated from the weight and sex of the person if the amount of ethanol consumed orally is known. However, this estimate is somewhat higher than actual concentrations because of rapid first-pass metabolism after oral administration. BACs are higher in women than in men after consumption of comparable amounts of ethanol, even after correcting for differences in body weight. This can be attributed to both the volume of distribution and first-pass metabolism of ethanol in women. Women have a smaller volume of distribution than men because, on average, they have a greater percentage of adipose tissue that does not contain as much H2O as do other tissues. In addition, the first-pass metabolism of ethanol, which occurs primarily in gastric tissue, is less in women than men because ADH activity in the female gastric mucosa is less than that in the male. Thus with low doses of ethanol, first-pass metabolism is lower in women, leading to higher BACs; at higher doses, the percentage of ethanol that undergoes first-pass metabolism is relatively small. Women are also more susceptible to alcoholic liver disease for this reason as well as a consequence of interactions with estrogen. This applies to both nonalcoholic and alcoholic women and partially explains the increased vulnerability of women to the deleterious effects of acute and chronic alcoholism. It was once assumed that higher BACs in women were entirely the result of differences in apparent volumes of distribution between men and women; however, they do not account entirely for this difference.
Pharmacovigilance: Side Effects, Clinical Problems, and Toxicity
Large amounts of ethanol decrease testosterone concentrations in males and cause a loss of secondary sex characteristics and feminization. Ovarian function may be disrupted in premenopausal females who abuse alcohol, and this may be manifest as oligomenorrhea, hypomenorrhea, or amenorrhea. Ethanol also stimulates release of adrenocortical hormones by increasing secretion of adrenocorticotropic hormone.
Other studies have categorized alcoholics into several subgroups. One is the alcoholism most frequently seen in males and is associated with criminality; the second is a subtype observed in both sexes and influenced by the environment. Genetic predisposition, however, is merely one of several factors leading to alcoholism. Studies are attempting to reveal biological markers with which to identify potential alcoholics (e.g., differences in blood proteins, enzymes involved in ethanol degradation, and enzymes concerned with brain neurotransmitters and signaling components, including G proteins) to encourage such people to seek assistance sooner.
Aldehyde Dehydrogenase Inhibitor
Disulfiram causes a rise in blood acetaldehyde concentrations, producing flushing, headache, nausea and vomiting, sweating, and hypotension. Disulfiram also inhibits dopamine β-hydroxylase, the enzyme that converts dopamine to norepinephrine (NE) in sympathetic neurons (see
Chapter 9). Thus, in an alcoholic taking disulfiram, there is an altered ability to synthesize NE, possibly contributing to the hypotension when alcohol is taken in conjunction with disulfiram.
New Horizons
Although alcohol dependence has been viewed as a social problem for many years, it is finally beginning to be accepted as a medical problem much like other chronic illnesses such as asthma, type 2 diabetes, and hypertension. To this end, drugs are being investigated for both the treatment of alcohol craving and the prevention of relapse. Several nonapproved medications being studied include other NMDA receptor antagonists such as memantine (see Chapter 28), the anticonvulsant topiramate (see Chapter 34), the GABAB receptor agonist baclofen (see Chapter 12), and the dopamine receptor antagonists such as aripiprazole and quetiapine (see Chapter 29). In addition, based on data from animal studies and limited clinical trials, agents affecting 5-HT transmission are being studied including 5-HT reuptake inhibitors, 5-HT1 receptor partial agonists, and 5-HT2/3 receptor antagonists.
Johnson BA. Update on neuropharmacological treatments for alcoholism: Scientific basis and clinical findings. Biochem Pharmacol. 2008;751:34-56.
McLellan AT, Lewis DC, O’Brien CP, Kleber HD. Drug dependence, a chronic medical illness. JAMA. 2000;284:1689-1695.
Mukamal KJ, Conigrave KM, Mittleman MA, et al. Roles of drinking pattern and type of alcohol consumed in coronary heart disease in men. N Engl J Med. 2003;348:109-118.
Spanagel R, Kiefer F. Drugs for relapse prevention of alcoholism: Ten years of progress. Trends Pharmacol Sci. 2008;29:109-115.
For further information on alcohol, see: http://www.niaaa.nih.gov.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Chapter 32 Ethanol, Other Alcohols, and Drugs for Alcohol Dependence
| Abbreviations | |
|---|---|
| ADH | Alcohol dehydrogenase |
| ALDH | Aldehyde dehydrogenase |
| BAC | Blood alcohol concentration |
| CNS | Central nervous system |
| GABA | γ-Aminobutyric acid |
| GI | Gastrointestinal |
| 5-HT | Serotonin |
| NAD+ | Nicotinamide adenine dinucleotide |
| NADH | Nicotinamide adenine dinucleotide, reduced |
| NADD+ | Nicotinamide adenine dinucleotide phosphate |
| NADPH | Nicotinamide adenine dinucleotide phosphate, reduced |
| NE | Norepinephrine |
| NMDA | N-methyl-D-aspartate |
| TNF | Tumor necrosis factor |
| VTA | Ventral tegmental area |
Therapeutic Overview
| Therapeutic Overview |
|---|
| Ethanol is used: |
| Topically to reduce body temperature and as an antiseptic |
| By injection to produce irreversible nerve block by protein denaturation |
| By inhalation to reduce foaming in pulmonary edema |
| In treatment of methanol and ethylene glycol poisoning |
| Ethanol dependence may be treated with psychosocial/behavioral therapy and an: |
| Aldehyde dehydrogenase inhibitor (Disulfiram) |
| Glutamate receptor antagonist (Acamprosate) |
| Opioid receptor antagonist (Naltrexone) |
Mechanisms of Action
Studies suggest that the effects of ethanol may be attributed to its direct binding to lipophilic areas either near or in ion channels and receptors. The ion channels influenced by ethanol are listed in Table 32-1. Ethanol may have either inhibitory or facilitatory effects, depending on the channel, but its resultant action is CNS depression. Because the barbiturates and benzodiazepines exhibit cross-tolerance to ethanol, and their CNS depressant effects are additive with those of ethanol, they may share a common mechanism, perhaps through the γ-aminobutyric acid (GABA) type A receptor (see Chapter 31). Ethanol may also exert some of its effects by actions at glutamate N-methyl-D-aspartate (NMDA) receptors or serotonin (5-HT) receptors.
| Channel | Effects | Ethanol Concentration (mM) |
|---|---|---|
| Na+ (voltage-gated) | Inhibition | 100 and higher* |
| K+ (voltage-gated) | Facilitation | 50-100 |
| Ca++ (voltage-gated) | Inhibition | 50 and higher |
| Ca++ (glutamate receptor-activated) | Inhibition | 20-50 |
| Cl− (GABAA receptor-gated) | Facilitation | 10-50 |
| Cl− (glycine receptor-gated) | Facilitation | 10-50 |
| Na+/K+ (5HT3 receptor-gated) | Facilitation | 10-50 |
* 100 mM ethanol is 460 mg/dL.
The reinforcing actions of ethanol are complex but are mediated in part through its ability to stimulate the dopaminergic reward pathway in the brain (see Fig. 27-8). Evidence has indicated that ethanol increases the synthesis and release of the endogenous opioid β-endorphin in both the ventral tegmental area (VTA) and the nucleus accumbens. Increased β-endorphin release in the VTA dampens the inhibitory influence of GABA on the tonic firing of VTA dopaminergic neurons, whereas increased β-endorphin release in the nucleus accumbens stimulates dopaminergic nerve terminals to release neurotransmitter. Both of these actions to increase dopamine release may be involved in the rewarding effects of ethanol.
Aldehyde Dehydrogenase Inhibitor
Disulfiram, used for the treatment of alcoholism since the 1940s, is an inhibitor of the enzyme aldehyde dehydrogenase (ALDH), a major enzyme involved in the metabolism of ethanol (Fig. 32-1). Inhibiting the catabolism of acetaldehyde produced by the oxidation of ethanol, leads to the accumulation of acetaldehyde in the plasma, resulting in aversive effects.
Naltrexone is an opioid receptor antagonist at both κ and μ opioid receptors (see Chapter 36). Its ability to inhibit alcohol consumption has been attributed to blockade of μ receptors in both the VTA and nucleus accumbens, thereby decreasing the ethanol-induced activation of the dopamine reward pathway.
Pharmacokinetics
Alcohol taken orally is absorbed throughout the gastrointestinal (GI) tract. Absorption depends on passive diffusion and is governed by the concentration gradient and the mucosal surface area. Food in the stomach will dilute the alcohol and delay gastric emptying time, thereby retarding absorption from the small intestine (where absorption is favored because of the large surface area). High ethanol concentrations in the GI tract cause a greater concentration gradient and therefore hasten absorption. Absorption continues until the alcohol concentration in the blood and GI tract are at equilibrium. Because ethanol is rapidly metabolized and removed from the blood, eventually all the alcohol is absorbed.
Ethanol undergoes significant first-pass metabolism. Most (>90%) of the ethanol ingested is metabolized in the liver, with the remainder excreted through the lungs and in urine. Alcohol dehydrogenase (ADH) catalyzes the oxidation of ethanol to acetaldehyde, which is oxidized further by ALDH to acetate (see Fig. 32-1). Acetate is oxidized primarily in peripheral tissues to CO2 and H2O. Both ADH and ALDH require the reduction of nicotinamide adenine dinucleotide (NAD+), with 1 mol of ethanol producing 2 mol of reduced NAD+ (NADH). The NADH is reoxidized to NAD+ by conversion of pyruvate to lactate by lactate dehydrogenase (LDH) and the mitochondrial electron transport system (ETS). During ethanol oxidation the concentration of NADH can rise substantially, and NADH product inhibition can become rate-limiting. Similarly, with large amounts of ethanol, NAD+ may become depleted, limiting further oxidation through this pathway. At typical blood alcohol concentrations (BACs), the metabolism of ethanol exhibits zero-order kinetics; that is, it is independent of concentration and occurs at a relatively constant rate (Fig. 32-2). Fasting decreases liver ADH activity, decreasing ethanol metabolism.
Ethanol may also be metabolized to acetaldehyde in the liver by cytochrome P450, a reaction that requires 1 mol of reduced nicotinamide adenine dinucleotide phosphate (NADPH) for every ethanol molecule (see Fig. 32-1). Although P450-mediated oxidation does not normally play a significant role, it is important with high concentrations of ethanol (≥100 mg/dL), which saturate ADH and deplete NAD+. Because this enzyme system also metabolizes other compounds, ethanol may alter the metabolism of many other drugs. In addition, this system may be inhibited or induced (Chapter 2), and induction by ethanol may contribute to the oxidative stress of chronic alcohol consumption by releasing reactive O2 species during metabolism.
Significant genetic differences exist for both ADH and ALDH that affect the rate of ethanol metabolism. Several forms of ADH exist in human liver, with differing affinities for ethanol. Whites, Asians, and African-Americans express different relative percentages of the genes and their respective alleles that encode subunits of ADH, contributing to ethnic differences in the rate of ethanol metabolism. Similarly, there are genetic differences in ALDH. Approximately 50% of Asians have an inactive ALDH, caused by a single base change in the gene that renders them incapable of oxidizing acetaldehyde efficiently, especially if they are homozygous. When these individuals consume ethanol, high concentrations of acetaldehyde are achieved, leading to flushing and other unpleasant effects. People with this condition rarely become alcoholic. As discussed, the unpleasant effects of acetaldehyde accumulation form the basis for the aversive treatment of chronic alcoholism with disulfiram. The pharmacokinetics of ethanol are summarized in Table 32-2.
| Pharmacokinetic Parameter | Considerations |
|---|---|
| Route of administration | Topically, orally, inhalation, by injection into nerve trunks, or intravenously for poison management |
| Absorption |
