[level-membership-for-basic-science-category]
Chapter 40 Estrogens and Progestins
| Abbreviations | |
|---|---|
| CBG | Corticosteroid-binding globulin |
| ER | Estrogen receptor |
| ERT | Estrogen replacement therapy |
| FSH | Follicle-stimulating hormone |
| GI | Gastrointestinal |
| GnRH | Gonadotropin-releasing hormone |
| HDL | High-density lipoprotein |
| HRE | Hormone responsive element |
| IM | Intramuscular |
| IUD | Intrauterine devices |
| LDL | Low-density lipoprotein |
| LH | Luteinizing hormone |
| PGR | Progesterone receptor |
| SERM | Selective estrogen receptor modulator |
| SHBG | Sex hormone-binding globulin |
Therapeutic Overview
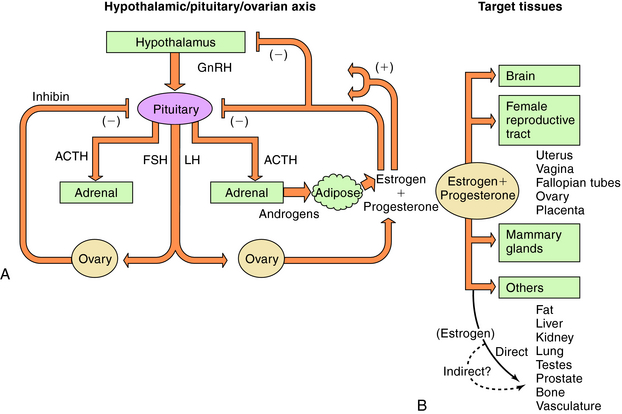
There are three endogenous nineteen-carbon steroids in humans that have estrogenic activity. The principal ovarian estrogens are 17β-estradiol, which is the primary circulating form, and its metabolite, estrone, which the primary postmenopausal estrogen. During pregnancy the placenta synthesizes estriol. Estrogens coordinate systemic responses during the ovulatory cycle, including regulation of the reproductive tract, pituitary, breasts, and other tissues. Also, some forms of cancer are estrogen-dependent for growth. The hypothalamic-pituitary-ovarian axis and target organs for the actions of estrogens are shown in Figure 40-1. Estrogens are also responsible for mediating development of secondary sex characteristics when females enter puberty, including progressive maturation of the fallopian tubes, uterus, vagina, and external genitalia. Upon estrogenic stimulation, more fat is deposited in the breast, buttocks, and thighs, leading to the normal adult female habitus. The following are characteristics promoted by estrogens.
The important endogenous progestin is progesterone, although 17α-, 20α-, and 20β-hydroxyprogesterones have weak progestational activities. Estrogen priming is necessary for progesterone receptor (PGR) expression in almost all progesterone-responsive tissues, including the uterus. Progesterone concentrations rise rapidly in the luteal phase of the menstrual cycle, resulting in a modulation of the action of estrogen on the uterus. Progesterone antagonizes estrogen-induced proliferation in the uterus and initiates secretory changes in preparation for embryo implantation. In the absence of pregnancy, plasma progesterone concentrations decrease, resulting in sloughing of the endometrial lining. Progesterone is responsible for causing the increased basal body temperature observed in the luteal phase. Progesterone is important in mammary glandular development and, unlike the uterus, probably stimulates breast cell proliferation. During pregnancy, progesterone can promote maintenance of pregnancy, inhibit uterine contraction, alter carbohydrate metabolism, decrease HDL, increase LDL, and increase Na+ and water elimination by competitive antagonism of aldosterone interaction with mineralocorticoid receptors. A variety of menstrual cycle disorders are treated with progestins, estrogens, or both.
| Therapeutic Overview |
|---|
| Fertility Control |
| Combination contraception (estrogens plus progestins) |
| Progestin-only contraception |
| Emergency contraception (estrogens plus progestins, progestins) |
| Contragestation (antiprogestin) |
| Infertility Treatment |
| Ovulation induction (SERMs, GnRH analogs, gonadotropins) |
| Replacement Therapy |
| Acute symptoms of menopause (estrogens plus progestins, estrogens) |
| Prevention of osteoporosis (SERMs, estrogens) |
| Ovarian failure (estrogens plus progestins) |
| Dysfunctional uterine bleeding (progestins, estrogens plus progestins) |
| Luteal phase dysfunction (progestins) |
| Cancer Chemotherapy |
| Breast cancer adjuvant treatment (SERMs, aromatase inhibitors, steroidogenesis inhibitors) |
| Advanced breast cancer (aromatase inhibitors, SERMs) |
| Advanced endometrial cancer (progestins) |
| Advanced prostate cancer (estrogens) |
| Breast cancer prevention (SERMs) |
| Others |
| Endometriosis (estrogens plus progestins, progestins, progesterone analog, progestin plus GnRH analog) |
| Dysfunctional uterine bleeding (progestins, estrogens plus progestins) |
| Luteal phase dysfunction (progestins) |
Mechanisms of Action
Biosynthesis of Estrogens and Progestins
Estrogens and progestins are produced by steroidogenesis in various tissues (see Fig. 38-1). The ovary is the predominant source of these steroids in nonpregnant, premenopausal women. A significant amount of estrogenic activity is also produced by skeletal muscle, liver, and adipose tissue through the conversion of circulating androgens to estrone. Certain brain areas in males and females may also produce estrogens through the action of aromatase on circulating androgens. Small amounts of estradiol can be produced in the male testes.
During the menstrual cycle, the pituitary gonadotropins FSH and LH regulate the synthesis and release of estrogen and progesterone from the ovary. The pulsatile release of hypothalamic gonadotropin-releasing hormone (GnRH), in turn, regulates FSH and LH synthesis and release. GnRH concentrations are regulated through negative and positive feedback by the steroid hormones. Estrogens and progestins also act directly on the pituitary gonadotrophs to decrease FSH and LH concentrations. In addition, an ovarian protein, inhibin, negatively affects FSH synthesis. The pathways for the integrated control of hormone regulation are shown in Figure 40-1, A.
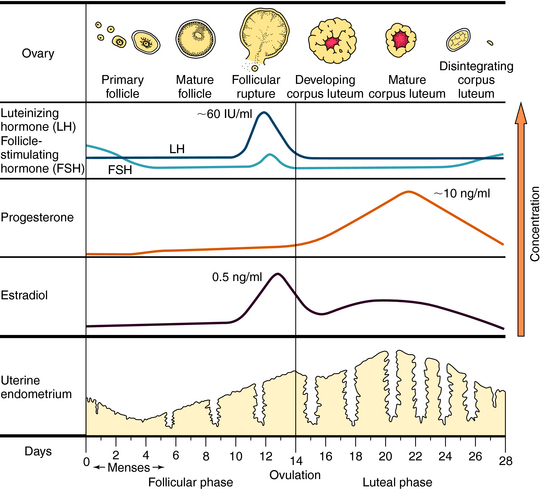
The ovulatory-menstrual cycle normally spans 25 to 35 days. The steps in the ovarian and endometrial cycles are shown in Figure 40-2. The ovarian cycle is divided into the follicular (preovulatory) phase, when ova maturation and estrogen release occurs, ovulation, when follicular rupture leads to ova release, and the postovulatory phase, when the corpus luteum maximally releases progesterone and stimulates growth of the endometrial lining. The follicle is the basic reproductive unit of the ovary and consists of an oocyte surrounded by granulosa cells. At the onset of a menstrual cycle, FSH accelerates maturation of several follicles. Through interactions with its receptor, FSH increases aromatase activity, which stimulates conversion of androgens to estradiol. By days 8 to 10, FSH decreases, and the dominant follicle becomes more sensitive to circulating gonadotropin because of an increased number of FSH receptors. In the late follicular phase, estradiol levels increase rapidly and initiate a mid-cycle LH surge (16 to 24 hours before ovulation). Increased LH levels promote follicular production of progesterone, prostaglandin F2α, and proteolytic enzymes, and ultimately, follicular rupture and ovulation occur. After ovulation, the granulosa and theca cells become the corpus luteum, which produces and releases progesterone throughout the first half of the luteal phase (10 to 20 ng/mL). The suppression of FSH and LH release promotes the decline of progesterone and estrogen, luteolysis, initiation of menses, and ultimately a new cycle.
Compounds with estrogenic activity can be classified as either steroidal or nonsteroidal. Steroidal estrogens can be subdivided into natural and synthetic forms. The structures of progesterone, the three endogenous human estrogens (β-estradiol, estrone, and estriol), and their biosynthetic pathways are shown in Figures 38-1 and 38-2. Estradiol is the most potent of the three estrogens. Synthetic hormones that are used therapeutically generally have a heterocyclic structure resembling endogenous steroids.
The synthetic estrogens, ethinyl estradiol and mestranol, are used predominantly in combination oral contraceptives. These compounds have an ethinyl group at C17, which retards hepatic inactivation. Mestranol requires activation by hepatic conversion to ethinyl estradiol. More recently, attention has focused on the nonsteroidal synthetic compounds, selective estrogen receptor modulators (SERMs), that interact with estrogen receptors (ER). The first available SERM, tamoxifen, is a triphenylethylene derivative that acts as an estrogen antagonist in breast; clinical applications include treatment, and recently prevention, of breast cancer (see Chapter 55). A clinically important benefit of tamoxifen is its agonist activity in bone, which antagonizes osteopenia. A concern with tamoxifen is the significantly increased risk of endometrial cancer and venous thrombosis related to its estrogenic activity. Raloxifene is a SERM developed to delay osteoporosis in postmenopausal women who are not candidates for estrogen treatment. It has agonist activity in bone but displays little estrogen-like activity in breast or uterus. Clomiphene citrate is a racemic mixture of two stereoisomers that has both agonist and antagonist properties and is used to treat infertility by inducing ovulation. Considerable research is directed at identifying new SERMS with tissue-specific agonist and antagonist properties for each therapeutic goal.
Two additional important ligands of the PGR include danazol and mifepristone (formerly called RU486). Danazol is a steroid derivative that has significant agonist activity at both progesterone and androgen receptors and is used in treating endometriosis. Mifepristone is a steroid derivative that binds to both progesterone and glucocorticoid receptors and displays progestin antagonist activity in most target tissues.
The molecular basis for hormone action, including estrogen and progesterone, is reviewed in Chapter 1. Free steroid passively diffuses into any cell but accumulates only in cells expressing the specific cytoplasmic steroid-binding proteins. Both estrogen and progesterone receptors are members of the nuclear receptor superfamily. The two distinct ER subtypes, ERα and ERβ, are products of different genes, and estrogen binds with high affinity to both receptor subtypes. ERα is expressed in the reproductive tract and breast and mediates many of the effects of estrogen on sexual development and reproductive function. ERβ is highly expressed in ovary and brain. All currently available drugs that target estrogen receptors can bind to both receptor dimers, suggesting that selectivity of action is not simply dependent on the presence of receptor subtypes. Because the ligand-binding domains of these two ER subtypes are different, specific ligands for each receptor are likely to be developed. Only one gene encodes the PGR, but two protein isoforms are produced, PGR-A and PGR-B. These proteins display some functional differences in experimental systems, and their ratio shows some variability. However, they have the same ligand-binding domain, and pharmacological effects result from activation of both isoforms.
The classical mechanism of action of nuclear steroid hormones is that the steroid-hormone complex can act as a steroid-activated transcription factor (see Chapter 1). The response of a tissue to a specific ligand is highly regulated at multiple levels:
Pharmacokinetics
The pharmacokinetic parameters of these agents are summarized in Table 40-1.
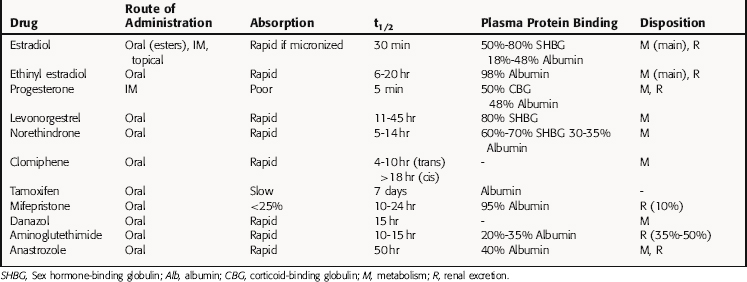
Oral progesterone is almost completely inactivated in the liver; thus synthetic modifications are necessary to produce orally active progestins. Progesterone can be given parenterally but has an elimination half-life of only a few minutes. It is converted in the liver to pregnanediol and conjugated with glucuronic acid at C3, and the conjugate is excreted mainly in urine. The 19-nortestosterone derivatives are all orally active. Medroxyprogesterone acetate can be administered orally or intramuscularly (IM), whereas megestrol acetate is administered orally only. The plasma half-lives of the C21 derivatives and the 19-nortestosterone compounds are longer than those of progesterone. Most of them are metabolized in the liver and conjugated to glucuronides and excreted in the urine.
Relationship of Mechanisms of Action to Clinical Response
Combination Oral Contraception
The most common use for administered combination estrogens and progestins is oral contraception. Oral contraceptives are one of the most effective, reversible ways to prevent pregnancy. The failure rate in users of combination oral contraceptives is less than 1 per 100 women-years, and serious risks are rare. Combination oral contraceptives currently available in the United States contain one of two synthetic estrogens with one of several synthetic progestins. The estrogen component is usually ethinyl estradiol or, less commonly, mestranol. The progestins include norethindrone, norgestrel, and its active isomer levonorgestrel, desogestrel, ethynodiol diacetate, and drospirenone. The present low-dose (50 µg or less) estrogen contraceptives are associated with a decreased incidence of adverse side effects and have prevented pregnancy at rates equal to those of earlier higher-dose formulations. The most commonly used oral contraceptives consist of a combination preparation taken for 21 days followed by 7 days without any steroids to induce withdrawal bleeding, but other dosing regimens are available. The dose and type of progestin are available in different formulations. If the dosage of progestin is fixed, altered two times, or altered three times, these are referred to as monophasic, biphasic, or triphasic, respectively. In addition, there have been four “generations” of progestins created for use in oral contraceptives. The major differences are in their side effects (usually androgenic).
Progestin-only contraception has a variety of delivery methods. A progestin-only oral contraceptive (the mini-pill) is taken daily and contains one of the synthetic progestins, norgestrel or norethindrone. Failure rates of 1 to 3 per 100 woman-years have been observed. To avoid the inconvenience of taking a pill daily and to obtain lower continuous doses of steroid to minimize side effects, alternate methods of progestin-only administration have been developed such as silastic capsules containing levonorgestrel that are placed subdermally, usually in the arm. The major benefits of this form of contraception are that it is effective for up to 5 years, and much lower doses of steroid are released. Failure rates are less than 1 per 100 woman-years. Problems include irregular uterine bleeding and the need for surgical insertion and removal. Medroxyprogesterone acetate in microcrystals is given as an IM injection at a dose of 150 mg every 3 months. Failure rates are 0.3 to 1 per 100 woman-years. Another benefit is that normal menstrual cycles return very quickly after removal. Progestin-releasing intrauterine devices (IUD) deliver low, continuous doses of the steroid locally instead of systemically. The progestin-induced endometrial atrophy decreases bleeding, which is a significant problem of the nonsteroid-containing IUD. Failure rates of 0.1 to 2 per 100 woman-years have been reported for these devices.
ERT in postmenopausal women remains the most effective treatment for the acute symptoms of menopause. It is used for treatment of moderate to severe symptoms of vulvar and vaginal atrophy and moderate to severe vasomotor symptoms, such as hot flushes (also termed “hot flashes”) and night sweats. It is currently recommended that ERT for postmenopausal women be used for the shortest time and at the lowest dose possible to relieve acute symptoms. A progestin is added to the treatment for women with an intact uterus to oppose the proliferative actions of estrogen on the endometrium. The most commonly used preparation is a mixture of conjugated estrogens taken orally. Transdermal and vaginal delivery preparations are also available. Vaginal delivery is recommended when treatment is only for symptoms of genitourinary atrophy. ERT is also very effective at reducing the risk of osteoporosis. However, if the only clinical goal is prevention of postmenopausal osteoporosis, alternative treatments are available (see Chapter 44). Because raloxifene has been shown to be an agonist in bone but have little estrogenic effects at the uterus or breast, it has been approved for prevention of postmenopausal osteoporosis. There are also other drugs for prevention and treatment of osteoporosis (see Chapter 44).
A reduction in estrogen production in postmenopausal women as a means of preventing breast cancer recurrence can be achieved by inhibition of adrenal steroidogenesis or peripheral aromatization of adrenal androgens. Aminoglutethimide acts by inhibiting two enzymes. One is the cholesterol side-chain-cleaving enzyme, which converts cholesterol to pregnenolone, and the other is the aromatase enzyme, which converts adrenal androstenedione to estrone, and testosterone to estradiol (see Fig. 38-1). Glucocorticoid replacement therapy is needed in such patients, mainly to inhibit the compensatory rise in adrenocorticotropic hormone, which can antagonize the action of aminoglutethimide. Several drugs are now available that specifically inhibit the aromatase enzyme and not the cholesterol side-chain-cleaving enzyme. Adrenal steroidogenesis is not inhibited, avoiding the need for glucocorticoid replacement. Circulating estrogen concentrations are effectively suppressed. These compounds include anastrozole, letrozole, and exemestane. These agents are used in the adjuvant treatment of breast cancer but also as first-line treatment in advanced disease (see Chapter 55). Clinical studies have shown that the sequential use of tamoxifen for 5 years followed by an aromatase inhibitor reduced the risk of breast cancer recurrence compared with tamoxifen use alone.
High doses of estrogens can be used as an adjuvant and palliative treatment for advanced prostate cancer by antagonizing gonadotropin formation. High-dose estrogen therapy is associated with a high incidence of adverse cardiovascular events, predominantly thromboembolism. The use of the synthetic nonsteroidal estrogen diethylstilbestrol has been replaced with newer therapies using a combination of steroidal estrogens that have a somewhat lower incidence of side effects. The benefit of high-dose estrogen is presumed to be due to its suppression of testosterone production.
Pharmacovigilance: Clinical Problems, Side Effects, and Toxicity
Combination Oral Contraceptives
A number of clinical studies show an association between combined oral contraceptive use and thromboembolic disease in the absence of other predisposing factors. The risk of venous thromboembolism is twofold to six fold greater in those who use combined oral contraceptives than in nonusers. The increased risk is dependent on the type of progestin used. The increased risk of thromboembolic events is greater in women who smoke, in older women (over 35), and in women with higher doses of estrogen. Women who use oral contraceptives should not smoke. The risk of thromboembolic disease rapidly returns to normal after oral contraceptive use is discontinued and should be stopped at least 2 to 4 weeks before elective surgery and not restarted until at least 2 weeks after surgery. Combination oral contraceptives can cause a small increase in both systolic and diastolic blood pressure in some patients, and their use is contraindicated in patients with moderate to severe hypertension.
Antiestrogens, SERMs, and Progesterone Receptor Ligands
The serious side effects of raloxifene include an increase in the risk of pulmonary thromboembolism and deep vein thrombosis. The most common, less serious side effect seen
Antiestrogens/SERMs/Progesterone Receptor Ligands
in some women is vasomotor symptoms. Raloxifene is contraindicated in women who are lactating, pregnant, or have a history of venous thromboembolic events.
Anonymous. Choice of contraceptives. Treat Guidel Med Lett. 2007;5:101-108.
Anonymous. Drugs for prevention and treatment of postmenopausal osteoporosis. Treat Guidel Med Lett. 2005;3:69-74.
Anonymous. Low-dose transdermal estrogens. Med Lett. 2007;49:71-72.
Bray et al. 2007 Bray PF, Howard TD, Vittinghoff E, et al. Effect of genetic variations in platelet glycoproteins Ib and VI on the risk for coronary heart disease events in postmenopausal women taking hormone therapy. Blood. 2007;109:1862-1869.
Petitti DB. Combination estrogen-progestin oral contraceptives. N Engl J Med. 2003;349:1443-1450.
Rossouw et al. 2002 Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: Principal results from the Women’s Health Initiative randomized controlled trial. JAMA. 2002;288:321-333.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Chapter 40 Estrogens and Progestins
| Abbreviations | |
|---|---|
| CBG | Corticosteroid-binding globulin |
| ER | Estrogen receptor |
| ERT | Estrogen replacement therapy |
| FSH | Follicle-stimulating hormone |
| GI | Gastrointestinal |
| GnRH | Gonadotropin-releasing hormone |
| HDL | High-density lipoprotein |
| HRE | Hormone responsive element |
| IM | Intramuscular |
| IUD | Intrauterine devices |
| LDL | Low-density lipoprotein |
| LH | Luteinizing hormone |
| PGR | Progesterone receptor |
| SERM | Selective estrogen receptor modulator |
| SHBG | Sex hormone-binding globulin |
Therapeutic Overview
There are three endogenous nineteen-carbon steroids in humans that have estrogenic activity. The principal ovarian estrogens are 17β-estradiol, which is the primary circulating form, and its metabolite, estrone, which the primary postmenopausal estrogen. During pregnancy the placenta synthesizes estriol. Estrogens coordinate systemic responses during the ovulatory cycle, including regulation of the reproductive tract, pituitary, breasts, and other tissues. Also, some forms of cancer are estrogen-dependent for growth. The hypothalamic-pituitary-ovarian axis and target organs for the actions of estrogens are shown in Figure 40-1. Estrogens are also responsible for mediating development of secondary sex characteristics when females enter puberty, including progressive maturation of the fallopian tubes, uterus, vagina, and external genitalia. Upon estrogenic stimulation, more fat is deposited in the breast, buttocks, and thighs, leading to the normal adult female habitus. The following are characteristics promoted by estrogens.
The important endogenous progestin is progesterone, although 17α-, 20α-, and 20β-hydroxyprogesterones have weak progestational activities. Estrogen priming is necessary for progesterone receptor (PGR) expression in almost all progesterone-responsive tissues, including the uterus. Progesterone concentrations rise rapidly in the luteal phase of the menstrual cycle, resulting in a modulation of the action of estrogen on the uterus. Progesterone antagonizes estrogen-induced proliferation in the uterus and initiates secretory changes in preparation for embryo implantation. In the absence of pregnancy, plasma progesterone concentrations decrease, resulting in sloughing of the endometrial lining. Progesterone is responsible for causing the increased basal body temperature observed in the luteal phase. Progesterone is important in mammary glandular development and, unlike the uterus, probably stimulates breast cell proliferation. During pregnancy, progesterone can promote maintenance of pregnancy, inhibit uterine contraction, alter carbohydrate metabolism, decrease HDL, increase LDL, and increase Na+ and water elimination by competitive antagonism of aldosterone interaction with mineralocorticoid receptors. A variety of menstrual cycle disorders are treated with progestins, estrogens, or both.
| Therapeutic Overview |
|---|
| Fertility Control |
| Combination contraception (estrogens plus progestins) |
| Progestin-only contraception |
| Emergency contraception (estrogens plus progestins, progestins) |
| Contragestation (antiprogestin) |
| Infertility Treatment |
| Ovulation induction (SERMs, GnRH analogs, gonadotropins) |
| Replacement Therapy |
| Acute symptoms of menopause (estrogens plus progestins, estrogens) |
| Prevention of osteoporosis (SERMs, estrogens) |
| Ovarian failure (estrogens plus progestins) |
| Dysfunctional uterine bleeding (progestins, estrogens plus progestins) |
| Luteal phase dysfunction (progestins) |
| Cancer Chemotherapy |
| Breast cancer adjuvant treatment (SERMs, aromatase inhibitors, steroidogenesis inhibitors) |
| Advanced breast cancer (aromatase inhibitors, SERMs) |
| Advanced endometrial cancer (progestins) |
| Advanced prostate cancer (estrogens) |
| Breast cancer prevention (SERMs) |
| Others |
| Endometriosis (estrogens plus progestins, progestins, progesterone analog, progestin plus GnRH analog) |
| Dysfunctional uterine bleeding (progestins, estrogens plus progestins) |
| Luteal phase dysfunction (progestins) |
Mechanisms of Action
Biosynthesis of Estrogens and Progestins
Estrogens and progestins are produced by steroidogenesis in various tissues (see Fig. 38-1). The ovary is the predominant source of these steroids in nonpregnant, premenopausal women. A significant amount of estrogenic activity is also produced by skeletal muscle, liver, and adipose tissue through the conversion of circulating androgens to estrone. Certain brain areas in males and females may also produce estrogens through the action of aromatase on circulating androgens. Small amounts of estradiol can be produced in the male testes.
During the menstrual cycle, the pituitary gonadotropins FSH and LH regulate the synthesis and release of estrogen and progesterone from the ovary. The pulsatile release of hypothalamic gonadotropin-releasing hormone (GnRH), in turn, regulates FSH and LH synthesis and release. GnRH concentrations are regulated through negative and positive feedback by the steroid hormones. Estrogens and progestins also act directly on the pituitary gonadotrophs to decrease FSH and LH concentrations. In addition, an ovarian protein, inhibin, negatively affects FSH synthesis. The pathways for the integrated control of hormone regulation are shown in Figure 40-1, A.
The ovulatory-menstrual cycle normally spans 25 to 35 days. The steps in the ovarian and endometrial cycles are shown in Figure 40-2. The ovarian cycle is divided into the follicular (preovulatory) phase, when ova maturation and estrogen release occurs, ovulation, when follicular rupture leads to ova release, and the postovulatory phase, when the corpus luteum maximally releases progesterone and stimulates growth of the endometrial lining. The follicle is the basic reproductive unit of the ovary and consists of an oocyte surrounded by granulosa cells. At the onset of a menstrual cycle, FSH accelerates maturation of several follicles. Through interactions with its receptor, FSH increases aromatase activity, which stimulates conversion of androgens to estradiol. By days 8 to 10, FSH decreases, and the dominant follicle becomes more sensitive to circulating gonadotropin because of an increased number of FSH receptors. In the late follicular phase, estradiol levels increase rapidly and initiate a mid-cycle LH surge (16 to 24 hours before ovulation). Increased LH levels promote follicular production of progesterone, prostaglandin F2α, and proteolytic enzymes, and ultimately, follicular rupture and ovulation occur. After ovulation, the granulosa and theca cells become the corpus luteum, which produces and releases progesterone throughout the first half of the luteal phase (10 to 20 ng/mL). The suppression of FSH and LH release promotes the decline of progesterone and estrogen, luteolysis, initiation of menses, and ultimately a new cycle.
Compounds with estrogenic activity can be classified as either steroidal or nonsteroidal. Steroidal estrogens can be subdivided into natural and synthetic forms. The structures of progesterone, the three endogenous human estrogens (β-estradiol, estrone, and estriol), and their biosynthetic pathways are shown in Figures 38-1 and 38-2. Estradiol is the most potent of the three estrogens. Synthetic hormones that are used therapeutically generally have a heterocyclic structure resembling endogenous steroids.
The synthetic estrogens, ethinyl estradiol and mestranol, are used predominantly in combination oral contraceptives. These compounds have an ethinyl group at C17, which retards hepatic inactivation. Mestranol requires activation by hepatic conversion to ethinyl estradiol. More recently, attention has focused on the nonsteroidal synthetic compounds, selective estrogen receptor modulators (SERMs), that interact with estrogen receptors (ER). The first available SERM, tamoxifen, is a triphenylethylene derivative that acts as an estrogen antagonist in breast; clinical applications include treatment, and recently prevention, of breast cancer (see Chapter 55). A clinically important benefit of tamoxifen is its agonist activity in bone, which antagonizes osteopenia. A concern with tamoxifen is the significantly increased risk of endometrial cancer and venous thrombosis related to its estrogenic activity. Raloxifene is a SERM developed to delay osteoporosis in postmenopausal women who are not candidates for estrogen treatment. It has agonist activity in bone but displays little estrogen-like activity in breast or uterus. Clomiphene citrate is a racemic mixture of two stereoisomers that has both agonist and antagonist properties and is used to treat infertility by inducing ovulation. Considerable research is directed at identifying new SERMS with tissue-specific agonist and antagonist properties for each therapeutic goal.
Two additional important ligands of the PGR include danazol and mifepristone (formerly called RU486). Danazol is a steroid derivative that has significant agonist activity at both progesterone and androgen receptors and is used in treating endometriosis. Mifepristone is a steroid derivative that binds to both progesterone and glucocorticoid receptors and displays progestin antagonist activity in most target tissues.
The molecular basis for hormone action, including estrogen and progesterone, is reviewed in Chapter 1. Free steroid passively diffuses into any cell but accumulates only in cells expressing the specific cytoplasmic steroid-binding proteins. Both estrogen and progesterone receptors are members of the nuclear receptor superfamily. The two distinct ER subtypes, ERα and ERβ, are products of different genes, and estrogen binds with high affinity to both receptor subtypes. ERα is expressed in the reproductive tract and breast and mediates many of the effects of estrogen on sexual development and reproductive function. ERβ is highly expressed in ovary and brain. All currently available drugs that target estrogen receptors can bind to both receptor dimers, suggesting that selectivity of action is not simply dependent on the presence of receptor subtypes. Because the ligand-binding domains of these two ER subtypes are different, specific ligands for each receptor are likely to be developed. Only one gene encodes the PGR, but two protein isoforms are produced, PGR-A and PGR-B. These proteins display some functional differences in experimental systems, and their ratio shows some variability. However, they have the same ligand-binding domain, and pharmacological effects result from activation of both isoforms.
The classical mechanism of action of nuclear steroid hormones is that the steroid-hormone complex can act as a steroid-activated transcription factor (see Chapter 1). The response of a tissue to a specific ligand is highly regulated at multiple levels:
