[level-membership-for-pathology-category]
CHAPTER 23 Essential thrombocythemia and primary myelofibrosis
Introduction
Among the classical Philadelphia-chromosome-negative myeloproliferative neoplasms (MPN), essential thrombocythemia (ET) and primary myelofibrosis (PMF) represent two subtypes with considerable overlap in clinical and hematological presentation in particular at early stages of disease.1 Differentiation between those two MPN subtypes is of clinical relevance, since PMF is characterized by a considerably higher risk of progression and leukemic transformation. In contrast, ET generally represents a stable disease with only minimal risk of myelofibrotic progression.2 There is also a significant difference in long-term survival between both groups. The 15-year age-adjusted relative survival rate for ET is nearly 84%. In contrast, PMF is characterized by a significant loss in life expectancy. Even when diagnosed in very early stages of disease, PMF patients have a 15-year survival of only 55–67%.2 Although the discovery of the JAK2V617F mutation (see Chapter 21) led to important progress in the understanding of the molecular pathogenesis of ET and PMF,3 the finding of this mutation in several MPN subtypes and the absence of the JAK2V617F allele in many MPN patients preclude the use of JAK2V617F testing alone to establish a diagnosis of ET or PMF. This is reflected in the revised WHO diagnostic criteria for ET and PMF (Box 23.1), which include mutation screening in the diagnostic work-up, but explicitly require a bone marrow (BM) morphological examination for making the diagnosis of both ET and PMF. Although a significantly lower JAK2V617F allele burden has been found in ET when compared to early stages of PMF,4 such variability in the allele burden does not represent a sufficient criterion for distinguishing among different clinical entities. Furthermore, JAK2V617F-negative cases reveal a similar clinical profile and outcome5 and increase in JAK2V617F allele frequencies is not linked to myelofibrotic transformation or progression to acute myeloid leukemia (AML).6
Box 23.1 WHO criteria for the diagnosis of essential thrombocythemia (ET) and primary myelofibrosis (PMF)
| Essential thrombocythemia (ET) | Primary myelofibrosis (PMF) |
|---|---|
|
1. Presence of megakaryocyte proliferation and atypia, usually accompanied by either reticulin and/or collagen fibrosis, or, in the absence of significant reticulin fibrosis, the megakaryocyte changes must be accompanied by an increased bone marrow cellularity characterized by granulocytic proliferation and often decreased erythropoiesis (i.e. prefibrotic/early phase disease).
|
|
| Diagnosis requires meeting all four criteria | Diagnosis requires meeting all three major criteria and two minor criteria* |
* In prefibrotic/early stages these features are generally only borderline expressed.
a) Causes of reactive thrombocytosis include iron deficiency, splenectomy, surgery, infection, inflammation, connective tissue disease, metastatic cancer, and lymphoproliferative disorders. The presence of a condition associated with reactive thrombocytosis does not exclude the possibility of ET if the first three criteria are met.
b) Causes of bone marrow fibrosis include infection, autoimmune disorders or other chronic inflammatory conditions, hairy cell leukemia or other lymphoid neoplasm, metastatic malignancy, or toxic (chronic) myelopathies.
ET and PMF usually affect the elderly population, but they can occasionally be found in children, and in this instance, they raise age-related diagnostic and management issues.7 Familial clustering of ET and also PMF is known, and this observation led to a suggestion of predisposition alleles even before the discovery of the JAK2V617F mutation.8 Relatives of patients with ET have a more than sevenfold increase in the relative risk of developing a similar MPN.9 However, the coexistence of different clinical entities and of JAK2V617F-positive and JAK2V617F-negative diseases in the same family is noteworthy.8,10
When addressing the current WHO criteria (Box 23.1) for diagnosing the different MPN entities,11,12 it is essential to emphasize that these guidelines do not claim that a single histological parameter defines a subgroup, but that the different subtypes of MPN are characterized by specific morphological BM patterns.13 These patterns are composed of distinctive features and should always be reviewed in close relation to clinical, hematological and molecular-genetic findings to achieve a consensus-based working diagnosis.1,3,14,15 In contrast to the determination of age-dependent cellularity and to the semiquantitative grading of myelofibrosis,16,17 characterization of megakaryopoiesis may cause significant difficulties concerning definition and easy recognition of disease-related patterns among different observers.18 Assessment of megakaryocytic histotopography, i.e. their arrangement within the BM space, and detection of certain nuclear abnormalities (besides maturation defects) are the keys to the diagnosis.19 In contrast to the normal BM, in which megakaryocytes show a central distribution of single isolated cells, the overall increase of megakaryocytes in MPN is often associated with the uneven distribution such as group formation: from small clusters (at least three cells) to extensive groups (more than seven cells).20 These megakaryocyte clusters may display either a loose (intermingled with other hematopoietic cells) or dense arrangement.19 An abnormal dislocation of megakaryocytes towards the endosteal (paratrabecular) border is a highly conspicuous finding that is usually not seen in reactive disorders. Other features indicating a neoplastic process are peculiar nuclear aberrations and maturation defects that imply disturbances of the normal development of megakaryopoiesis.1,21–23 These include:
Furthermore, maturation defects include a conspicuous deviation of the nuclear-cytoplasmic ratio or maturation with appearance of bizarre megakaryocytes. It is noteworthy that all these changes may be present in megakaryocytes of different sizes and ploidy status. Finally, so-called naked (denuded, bare) nuclei with condensed chromatin pattern may frequently be found implicating a stimulated thrombocyte shedding and cell turnover.20
In other hematopoietic lineages, an increase and left-shifting of neutrophil granulopoiesis or erythropoiesis may be a prominent feature or a reduction in the amount of nucleated red cell precursors may be found, depending on disease entity and phase.24
Essential thrombocythemia (ET)
Essential thrombocythemia (ET) is a MPN that involves primarily the megakaryocytic lineage. It is characterized by sustained thrombocytosis >450 × 109/l in the peripheral blood (PB), increased numbers of large, mature megakaryocytes in the BM, and clinically by episodes of thrombosis and/or hemorrhage. Because there is no known genetic or biological marker specific for ET, other causes for thrombocytosis such as other MPN, inflammatory and infectious disorders, hemorrhage and other types of hematopoietic and non-hematopoietic neoplasms must be excluded.12 The presence of the BCR-ABL1 fusion gene excludes the diagnosis of ET (Box 23.1). The reported annual incidence of ET ranges from 0.59 to 2.53/100 000 inhabitants and its prevalence is around 30/100 000, which is similar to that of polycythemia vera (PV).25 The median age at diagnosis is 65–70 years,26 but ET is occasionally found in children, and it is relatively common in female patients in their third or fourth decade of life.27,28 In women of childbearing age, ET may be a risk factor for complications during pregnancy.29 Venous thrombosis in unusual locations is typical in younger patients, and the thrombotic risk increases in patients over 60 years old.26
Clinical features
Many patients are asymptomatic when an excess in platelets is discovered by a routine blood count.26,28,29 Most investigators have argued that the use of a threshold level 600 × 109/l platelets compromises the detection of early-phase disease because the adjusted 95th percentile for normal platelet count is below 400 × 109/l.30,31 Therefore, the platelet threshold count required for ET diagnosis has been lowered to 450 × 109/l (Box 23.1). Initial presentation may include vascular occlusions, hemorrhage or microvascular complications that lead to transient ischemic attacks and digital ischemia with paresthesias.32,33 Some patients may present with a thrombosis of major arteries and veins such as splenic or hepatic vein thrombosis as in the Budd–Chiari syndrome.26,34,35 Increase in spleen size at time of diagnosis is seen only in a minority of patients.2 Generally, there is no anemia, no circulating blasts, and serum levels of lactate dehydrogenase (LDH) are within the normal range.23,36 In patients with borderline anemia, palpable spleen, slight elevation of LDH serum level or evidence of erythroid and/or granulopoietic precursors in the PB, a differential diagnosis of very early (prefibrotic) stages of PMF should be excluded by careful bone marrow trephine biopsy (BMTB) examination.1,2,21,23,24,36,37 In contrast to PV cases, the utility of JAK2V617F mutation screening for the diagnosis of ET is limited by suboptimal negative predictive value and lack of diagnostic specificity in the context of MPN.14 Therefore, a BMTB is mandatory to differentiate ET from other MPN and to help with the differential diagnosis between JAK2V617F-negative ET and reactive thrombocytosis.1,14,21,24
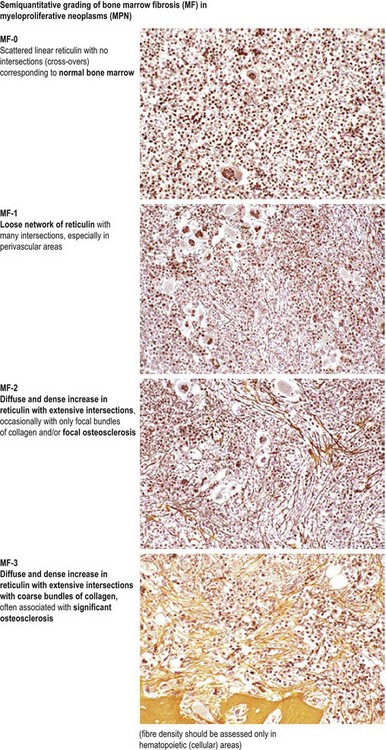
In most patients, ET is an indolent disorder characterized by long symptom-free intervals, interrupted by occasional life-threatening thromboembolic or hemorrhagic episodes.26,28,38 Although after many years a few patients with ET may develop BM fibrosis such progression is very uncommon.39–41 The exact incidence of myelofibrotic transformation in ET persists to be a controversial issue42 and the reported results are certainly influenced by the risk status of the patients and the applied diagnostic criteria.1,21,43,44 Careful morphological examination of BMTB is crucial in the diagnostic workup of post ET myelofibrosis (Post-ET MF), since appearance of BM fibrosis with a grade MF-2 or MF-3 is a prerequisite for the diagnosis (Box 23.2 and Fig. 23.1). Secondly, only cases with predefined and documented ET according to the WHO guidelines fall into this category.45 Strict adherence to the WHO criteria (Box 23.1) is necessary to prevent diagnostic confusion associated with early PMF accompanied by thrombocytosis.1,23,36 The frequency of true post-ET myelofibrosis is low; the transformation rate is 2.8% at a median follow-up of 9.1 years (or a 10-year risk of 3.9% and a 15-year risk of 6%).46 Transformation of ET to acute myeloid leukemia or MDS occurs in fewer than 5% of patients, and in chemotherapy treated patients may be related to previous cytotoxic therapy.26,28,47–49 The life expectancy in ET is near normal for most patients.2,50,51
Box 23.2 WHO criteria for the diagnosis of post-essential thrombocythemia myelofibrosis (Post-ET MF)
Molecular data
A JAK2V617F or a functionally similar mutation may be detected in approximately 40–50% of patients (see Chapter 21 for details).5,52–55 However, these findings are not specific for ET and are also seen in other MPN subtypes. It has to be emphasized that significant differences in the JAK2V617F allele burden were described between ET and early PMF.4,6,56 As an independent marker this feature validates very nicely corresponding BMTB findings and thus supports the concept of differentiating ET from early PMF.1,21,23,24,57 A gain-of-function mutation of MPL has been reported in approximately 1–3% of patients with ET.58,59 None of these mutations are found in cases of reactive thrombocytosis. Although ET is usually characterized by a normal karyotype, an isolated del(5q) has been reported in few cases with ET. However, it is more likely that these cases represent MDS associated with this abnormality.60,61
Blood and bone marrow findings
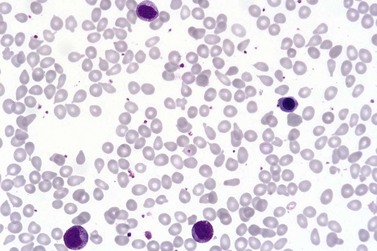
Marked and sustained thrombocytosis is the hallmark of ET. The platelets often display anisocytosis, ranging from tiny forms to atypical large, giant platelets that may reveal bizarre shapes, pseudopods and agranularity. The white blood cell count (WBC) and leukocyte differential are usually normal, although a borderline elevation in the neutrophil lineage may occur.28,62,63 In ET classified according to the histological guidelines of the WHO classification, neither a relevant increase in cellularity nor a significant left-shifted neutrophil granulopoiesis is observed.1,3,57 Any case with a mild to moderate granulocytic and erythroid growth pattern (panmyelosis) and an EPO level below the reference range is suspicious for occult (pre-polycythemic) PV mimicking ET.64,65 Regarding megakaryopoiesis, gross disturbances of the histological topography (significant abnormal localization and/or extensive dense clustering) are not seen (Figs 23.2 and 23.3). Megakaryocytes show a more or less random distribution, with scattered forms or a few loose clusters (Figs 23.4 and 23.5). A predominance of large to giant mature megakaryocytes with extensively folded (staghorn-like) nuclei19,20,22,24,66,67 surrounded by a correspondingly mature cytoplasm is found (Fig. 23.6). These features are clearly different from prefibrotic early PMF, where megakaryocytes show an extensive dense clustering and have hypolobulated (cloud-like or bulbous) and hyperchromatic nuclei with striking maturation defects (Fig. 23.7) leading to a marked anomaly of their nuclear-cytoplasmic ratio.1,19–22,24,36,65,67 Finally, there is no substantial increase in reticulin fibers at presentation and collagen fibrosis is never observed in ET.44 In a large series of ET patients, minimal to slight reticulin fibrosis was described in only 3% of cases.19,40,41,68,69 Any marked increase in reticulin fibers is not compatible with ET,1 in contrast to prefibrotic PMF, which is a differential diagnosis in patients with thrombocytosis.2,21,22,57 In PMF cases, a relevant increase in age-matched cellularity and a significantly expressed left-shifted neutrophil granulopoiesis is regularly found. At times, the clinical and morphological distinction between ET and early phase PMF with associated thrombocytosis might not be clear cut. As the therapeutic relevance in these early stages is still unclear, a strict adherence to the WHO criteria for making a working diagnosis and close monitoring of the patient to capture any substantial changes that might warrant revision of diagnosis is recommended.3,11,15
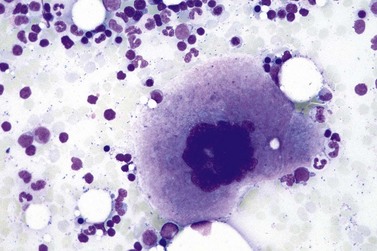
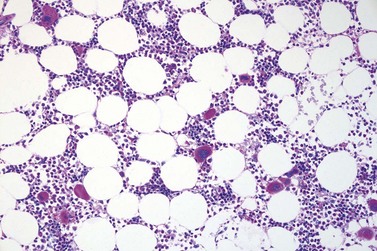
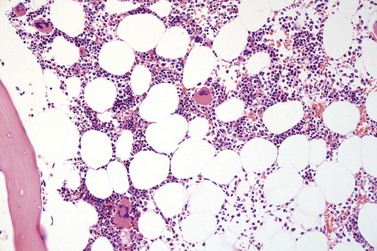
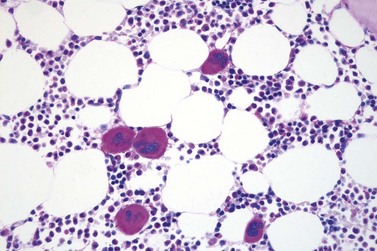
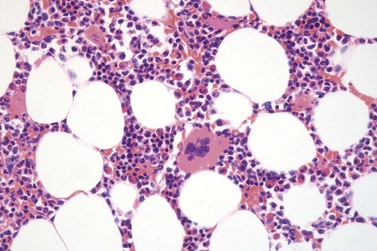
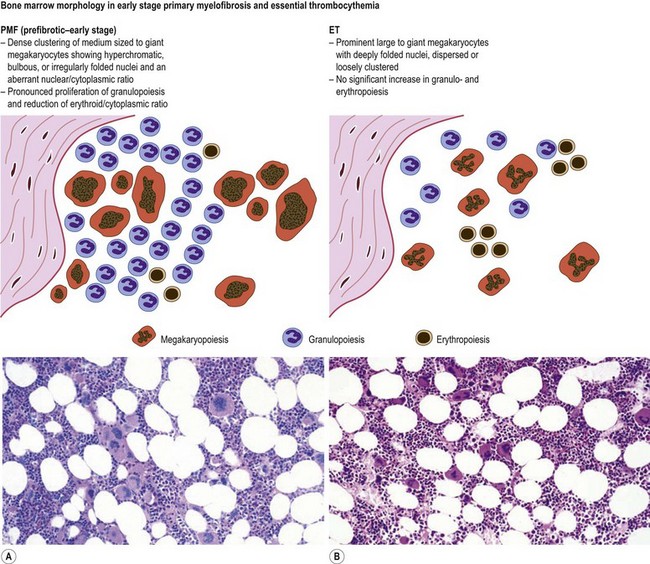
Fig. 23.7 Bone marrow morphology in early stage primary myelofibrosis and essential thrombocythemia.
Finally, when myelodysplastic and myeloproliferative BM features are simultaneously present and more than 15% ring sideroblasts are found in the aspirate, the case should be defined as refractory anemia with ring sideroblasts (RARS-T), an entity that is included in the MDS/MPN, unclassifiable category (see Chapter 27 for details).11 It is important to note that the diagnostic criteria for RARS-T include not only the finding of an elevated platelet count in conjunction with anemia and presence of ring sideroblasts in the BM, but also morphologically abnormal megakaryocytes.15
Primary myelofibrosis (PMF)
PMF is a chronic hematologic malignancy characterized by a stepwise evolution from an initial prefibrotic phase revealing a hypercellular BM with absent or minimal reticulin fibrosis to an overt fibrotic phase with marked reticulin or collagen fibrosis that is often accompanied by osteosclerosis.3,15,70,71 One of the hallmarks of PMF in the BM is a proliferation of predominantly megakaryocytes and granulocytes1,20,21,24,36 that in fully developed fibrotic disease stages is associated with hepatosplenomegaly, leukoerythroblastosis in the PB, cytopenias, teardrop-shaped red cells, extramedullary hematopoiesis, increased BM microvessel density71–73 and constitutive mobilization of CD34+ hematopoietic progenitors and stem cells as well as endothelial progenitor cells into the peripheral blood.74–76 However, endothelial progenitor cell mobilization predominates during the early phase of PMF, while hematopoietic stem and progenitor cell mobilization characteristically occurs in more clinically advanced phases of the disease.74 This dysregulation of stem cell trafficking most likely leads to the seeding of extramedullary sites with primitive hematopoietic and endothelial cells, resulting in extramedullary hematopoiesis in the spleen, liver and in a variety of other organs. It has been documented that several proteolytic pathways play an important role in cytokine-mediated stem cell mobilization.77 The interaction between stroma cells, endothelial cells and osteoblast-derived stroma cell derived factor-1 (SDF-1) and the CXC chemokine receptor-4 (CXCR-4) expressed by hematopoietic stem and progenitor cells is also believed to determine patterns of stem cell trafficking. Furthermore, proteases, including neutrophil elastase, soluble matrix metalloproteinase-9 (MMP-9) and cell bound MMP-9 have been shown to play a role in the constitutive mobilization of CD34+ cells.77
In the overt stages, which were formerly termed agnogeneic myeloid metaplasia (AMM) or myelofibrosis with myeloid metaplasia (MMM),45 profound BM fibrosis is a response to the clonal proliferation of hematopoietic stem cells.78 Collagen type 3, also known as reticulin, and collagen type 1 are the predominant extracellular components of BM fibrosis in PMF.79,80 These matrix components are produced by BM fibroblasts that do not belong to the malignant clone. This deposition of collagen is a result of the release of fibrogenic cytokines by abnormal megakaryocytes and monocytes derived from the malignant stem cell population.73 However, BM fibrosis is not unique to PMF and may develop in the course of many other disorders.79
Prefibrotic, early stages of PMF presenting with only borderline or minimal clinical features indicating myelofibrosis with myeloid metaplasia have been more and more acknowledged.1,3,20,21 The striking variability in the hematological findings of patients with PMF at the time of the first presentation is paralleled by corresponding BM features that may initially present only a hypercellular hematopoiesis without or with only slight increase of reticulin fibres.1,21,24,36,39 Considering the dynamics of the disease process in PMF, the former gold standard for the diagnosis of AMM/MMM72 should be avoided, because these criteria included only the advanced or overt stages of a wide spectrum of clinical and morphological disease manifestations. Therefore, overt myelofibrosis is not a necessary diagnostic feature of PMF, as outlined in Box 23.1. Furthermore, terminal stages of PV and ET presenting with findings of myeloid metaplasia45,81 should not be included in this MPN category.20,21,24,71
The reported annual incidence of PMF is significantly biased by the change in diagnostic criteria.82 When the manifest fibrotic stage is considered, the frequency is estimated at 0.5–1.5 per 100 000 individuals per year.83,84 Disease onset is most common in the sixth to seventh decade of life,71,73 but rarely even children may be affected.85
Clinical features
In the initial prefibrotic phase of PMF, the only relevant hematological finding may be sustained thrombocytosis mimicking ET and a borderline anemia and/or splenomegaly.1,36,86 Depending on the grade of BM fibrosis, hepatosplenomegaly of varying degree is detected in many patients.71,73,87 In the overt stages of the disease, extramedullary hematopoiesis in the spleen is a common finding, while liver, lymph nodes and other organs or soft tissue are other possible sites of involvement.71 Early stages of PMF with only mild increase in reticulin (fibrosis grades MF-0 or MF-1; see Fig. 23.1) may not be recognized by clinical features alone. Many of these patients are asymptomatic at the time of diagnosis and only fortuitously discovered by the detection of marginal splenomegaly during a routine physical examination and/or the presence of borderline anemia, leukocytosis and/or thrombocytosis in routine blood counts.36 Only a minority of these cases are discovered due to unexplained leukoerythroblastosis or a significantly increased LDH serum level.3,87
Although younger patients may experience longer survival,50,85 patients with symptomatic forms of PMF have a median survival of less than 5 years,87,88 while appropriately treated ET is associated with nearly normal life expectancy. However, the overall prognosis significantly depends on the stage of PMF at diagnosis.2,89 Adverse prognostic factors generally include higher age (>65 years), anemia (Hb <10 g/dl), WBC >25 × 109/l, presence of circulating blast cells, thrombocytopenia (platelet count <100 × 109/l), and an abnormal karyotype.2,50,85,88,90,91 Major causes of death are related to the sequelae of portal hypertension or hepatic-splenoportal thrombosis, thromboses in various anatomic sites, heart failure due to splenic pooling, infections, pulmonary hypertension, bleeding caused by thrombocytopenia or hemostatic defects.71–73 Terminal leukemic transformation is seen in about 5–30% of PMF patients as part of the natural history of this disease.87,92
Molecular data
Almost 50% of patients with PMF reveal a clonal hematopoiesis but lack JAK255 or MPL mutations (see Chapter 21 for details).58,59 These patients, however, have a similar clinical history and prognosis to JAK2V617F-positive patients.5 Thus, the presence of the mutations confirms the clonality of the proliferation, but it does not distinguish PMF from other MPN subtypes, in particular ET or PV.3 A remarkable finding is that progression of prodromal PMF is not triggered by the JAK2V617F mutation status,6 but by a number of relevant target genes that are involved in matrix modeling and regulation of fibrillogenesis.93 Comparative genomic hybridization studies have shown that gains of cytogenetic material occur in more than 50% of patients with PMF and most commonly involve gains of 9p, 2q, 3p, chromosome 4, 12q and 13q.94 Furthermore, an unbalanced translocation between chromosomes 1 and 6 with specific breakpoints t(1;6)(q21-23;p21.3) is strongly suggestive but not diagnostic of PMF.95 Deletions affecting the long arms of chromosomes 7 and 5 have been reported as well, but may be associated with prior cytotoxic therapy used to control the myeloproliferation.96
Blood and bone marrow findings
It has been estimated that 30–40% of patients present with a prefibrotic or early fibrotic stage of PMF.36,39,57,69 Clinical data in these prodromal stages of PMF are usually characterized by only minimal abnormalities such as borderline to slight anemia, minimal splenomegaly, a very low or missing peripheral blast count but often a pronounced thrombocytosis.1,21,23 In the prodromal stages of PMF, the BMTB shows a marked hypercellularity with prominent granulocytic and megakaryocytic myeloproliferation (Figs 23.8, 23.9 and 23.10) and a concomitant reduction and/or maturation arrest of the nucleated erythroid precursors.1,20,21,86 Reticulin fibrosis is minimal (Fig. 23.11) or even absent at this stage (corresponding to fibrosis grades MF-0 and MF-1; Box 23.2). If present, fibrosis is usually seen focally and tends to be concentrated around vessels.17 There may be a mild left shift in granulopoiesis (see Fig. 23.10), with predominance of metamyelocytes, bands and segmented forms. Percentages of myeloblasts in BM aspirates are not increased and conspicuous clusters of blasts or CD34+ progenitors are not observed.1,36,86 Abnormal megakaryopoiesis is most conspicuous, and thus the histotopography and morphology of megakaryocytes is the key to the recognition of the prefibrotic stage of PMF.1 Characteristically, an extensive clustering of megakaryocytes with loose to dense groupings with abnormal localization towards the endosteal borders is seen (Figs 23.12 and 23.13), but there are also striking abnormalities in megakaryocyte morphology and maturation.1,19,21,24,36,86 Significant anomalies of megakaryocytes include a high degree of cellular pleomorphism with variations in size that range from small to giant forms. Abnormal nuclear folding and an aberration of the nuclear cytoplasmic ratio created by large, bulbous and hyperchromatic cloud-like nuclei are common. Apart from the disorganized nuclear lobulation of megakaryocytes, many so-called naked (bare) megakaryocytic nuclei are observed.19,21,24,69,86 Increased vascular proliferation is usual in the BM97 and lymphoid nodules are found in about 20–30% of cases.22,36
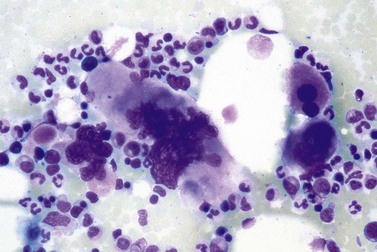
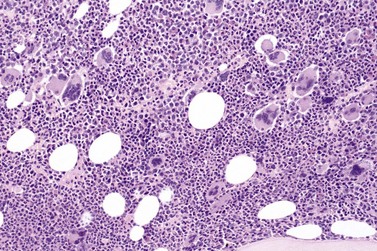
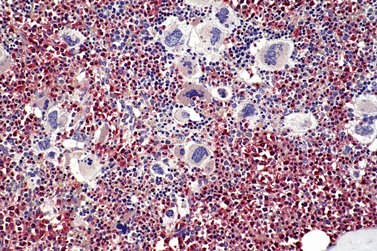
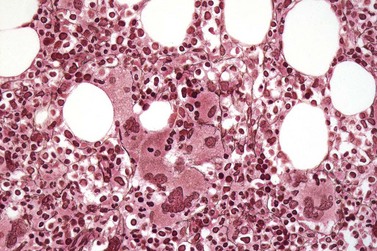
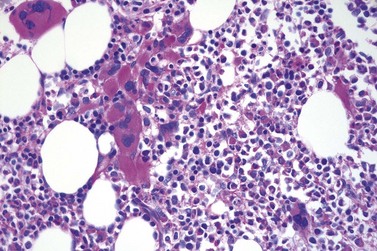
Fig. 23.12 Dense clustering of abnormal megakaryocytes in prefibrotic/early PMF. PAS, ×20 objective.
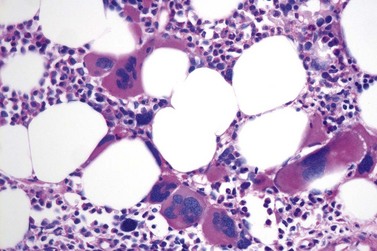
Overall, the megakaryocytes in PMF are characterized by a higher degree of cytological atypia (megakaryocytic dysplasia) than any other subtype of MPN, in particular ET1,19,21,24,66,67 Thus, megakaryocytic dysplasia is one of the most important features discriminating prefibrotic/early stage PMF from ET (Fig. 23.7). Careful morphological BMTB examination is crucial in distinguishing PMF cases with accompanying thrombocytosis from ET, because early stages of myelofibrosis with only borderline to mild increase in reticulin may not be recognized by clinical features alone.2,3,14,21,22,24 The significant difference between these two MPN subtypes with regard to both survival and rate of myelofibrotic transformation has to be kept in mind.2,21 An accurate distinction between both subtypes is not a matter of semantics, but ultimately does exert an influence on therapeutic strategies and, most importantly, is predictive of possible complications and outcome. The majority of patients with prefibrotic/early stages of PMF eventually transform into overt fibrotic/sclerotic myelofibrosis associated with extramedullary hematopoiesis.36,39,41,69,98 It has been estimated that PMF patients have a more than 65% probability of progression to a full-blown myelofibrosis with a median time of 4.2 years.2,44 However, usually this change in BM morphology is significantly preceding the clinical progression towards signs of myeloid metaplasia.1
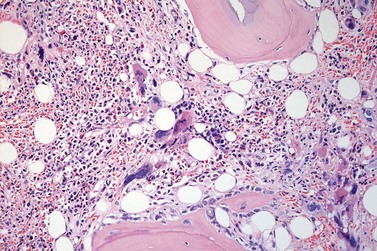
The classical picture of advanced PMF includes a leukoerythroblastic PB smear with teardrop poikilocytosis (Fig. 23.14), splenomegaly and anemia of varying degree, associated with a marked reticulin and/or collagen BM fibrosis (fibrosis grades MF-2 and MF-3; see Fig. 23.1).73,87 Progressive accumulation of fibrous tissue generally parallels disease progression; however, a striking variability in the hematological findings may be observed at the time of presentation.21,87 Besides the significant amount of reticulin deposition and the appearance of coarse bundles of collagen fibers in the BM, additional features indicating an advanced to terminal PMF stage include plaque to bud-like osteosclerosis (endophytic bone formation) that is often associated with patchy hematopoiesis replaced by adipose tissue (Figs 23.15 and 23.16), i.e. progressive hypoplasia.36,69 Foci of immature cells may be more prominent, although myeloblasts account for fewer than 10% of the BM cells.36 As in prodromal stages, atypical megakaryopoiesis remains the most prominent feature, including the presence of large clusters or sheets of megakaryocytes and numerous naked nuclei (Fig. 23.16). Dilated BM sinuses97,99 with intraluminal hematopoiesis (Fig. 23.17), especially megakaryocytes, are prominent in most cases.36,39 Rarely the BM is almost devoid of hematopoietic cells, showing mainly dense reticulin or collagen fibrosis with small islands of hematopoietic precursors situated mostly within the vascular sinuses.36 In osteosclerotic terminal phase of PMF, the marrow space is progressively replaced by broad, irregular trabeculae and appositional bud-like endophytic new bone formation.69
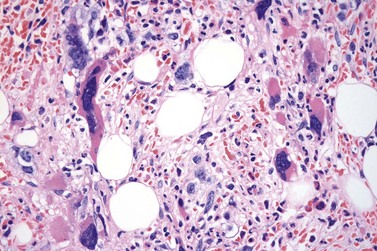
Increase in blood and/or BM blasts (<20%) as well as increased numbers of CD34+ cells with cluster formation and/or an abnormal endosteal location in the BM36,100 indicate an accelerated phase of the disease, whereas 20% or more blasts in PB or BM are considered as blastic transformation, i.e. acute leukemia.
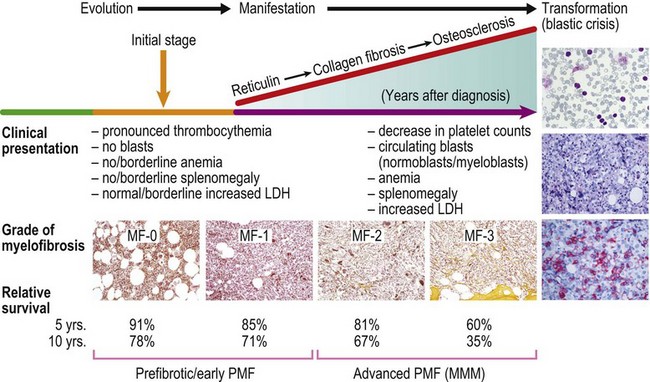
PMF usually displays an insidious onset and stepwise progression, which comprises the full spectrum of prodromal and terminal stages, the latter terminating in BM insufficiency, myelodysplastic changes and blast crisis (Fig. 23.18). Early stages may pose a diagnostic problem, because the classical clinical criteria for diagnosis may not be present at onset of the disease. Early PMF may be only recognized by careful BMTB examination in combination with clinical, hematological and molecular findings.1 Therefore, demonstration of reticulin fibrosis, although characteristic, is not a required criterion for diagnosis of PMF. Instead, the cardinal and ultimately required features of PMF include an increase in megakaryocyte growth associated with conspicuous morphological abnormalities as well as granulocyte proliferation.36 The speed of disease progression in the individual patient is unpredictable and development of myelofibrosis cannot be significantly influenced by treatment modalities with the exception of allogeneic stem cell transplantation.37
1 Kvasnicka HM, Thiele J. Prodromal myeloproliferative neoplasms: The 2008 WHO classification. Am J Hematol. 2010 Jan;85(1):62-69.
2 Kvasnicka HM, Thiele J. The impact of clinicopathological studies on staging and survival in essential thrombocythemia, chronic idiopathic myelofibrosis, and polycythemia rubra vera. Semin Thromb Hemost. 2006;32:362-371.
3 Tefferi A, Thiele J, Orazi A, et al. Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: recommendations from an ad hoc international expert panel. Blood. 2007;110:1092-1097.
4 Malysz J, Crisan D. Correlation of JAK2 V617F mutant allele quantitation with clinical presentation and type of chronic myeloproliferative neoplasm. Ann Clin Lab Sci. 2009;39:345-350.
5 Vannucchi AM, Guglielmelli P, Tefferi A. Advances in understanding and management of myeloproliferative neoplasms. CA Cancer J Clin. 2009;59:171-191.
6 Hussein K, Bock O, Theophile K, et al. JAK2(V617F) allele burden discriminates essential thrombocythemia from a subset of prefibrotic-stage primary myelofibrosis. Exp Hematol. 2009;37:1186-1193.
7 Teofili L, Giona F, Martini M, et al. The revised WHO diagnostic criteria for Ph-negative myeloproliferative diseases are not appropriate for the diagnostic screening of childhood polycythemia vera and essential thrombocythemia. Blood. 2007;110:3384-3386.
8 Rumi E, Passamonti F, Della Porta MG, et al. Familial chronic myeloproliferative disorders: clinical phenotype and evidence of disease anticipation. J Clin Oncol. 2007;25:5630-5635.
9 Landgren O, Goldin LR, Kristinsson SY, et al. Increased risks of polycythemia vera, essential thrombocythemia, and myelofibrosis among 24,577 first-degree relatives of 11,039 patients with myeloproliferative neoplasms in Sweden. Blood. 2008;112:2199-2204.
10 Bellanne-Chantelot C, Chaumarel I, Labopin M, et al. Genetic and clinical implications of the Val617Phe JAK2 mutation in 72 families with myeloproliferative disorders. Blood. 2006;108:346-352.
11 Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the WHO classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114:937-951.
12 Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed. Lyon: IARC Press; 2008.
13 Thiele J, Kvasnicka HM. A critical reappraisal of the WHO classification of the chronic myeloproliferative disorders. Leuk Lymphoma. 2006;47:381-396.
14 Tefferi A, Vardiman JW. Classification and diagnosis of myeloproliferative neoplasms: the 2008 World Health Organization criteria and point-of-care diagnostic algorithms. Leukemia. 2008;22:14-22.
15 Tefferi A, Thiele J, Vardiman JW. The 2008 World Health Organization classification system for myeloproliferative neoplasms: order out of chaos. Cancer. 2009;115:3842-3847.
16 Vener C, Fracchiolla NS, Gianelli U, et al. Prognostic implications of the European consensus for grading of bone marrow fibrosis in chronic idiopathic myelofibrosis. Blood. 2008;111:1862-1865.
17 Thiele J, Kvasnicka HM, Facchetti F, et al. European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica. 2005;90:1128-1132.
18 Wilkins BS, Erber WN, Bareford D, et al. Bone marrow pathology in essential thrombocythemia: interobserver reliability and utility for identifying disease subtypes. Blood. 2008;111:60-70.
19 Thiele J, Kvasnicka HM, Diehl V. Standardization of bone marrow features – does it work in hematopathology for histological discrimination of different disease patterns? Histol Histopathol. 2005;20:633-644.
20 Thiele J, Kvasnicka HM, Orazi A. Bone marrow histopathology in myeloproliferative disorders – current diagnostic approach. Semin Hematol. 2005;42:184-195.
21 Kvasnicka HM, Thiele J. Classification of Ph-negative chronic myeloproliferative disorders – morphology as the yardstick of classification. Pathobiology. 2007;74:63-71.
22 Thiele J, Kvasnicka HM. Diagnostic differentiation of essential thrombocythaemia from thrombocythaemias associated with chronic idiopathic myelofibrosis by discriminate analysis of bone marrow features – a clinicopathological study on 272 patients. Histol Histopathol. 2003;18:93-102.
23 Thiele J, Kvasnicka HM. Clinicopathological criteria for differential diagnosis of thrombocythemias in various myeloproliferative disorders. Semin Thromb Hemost. 2006;32:219-230.
24 Thiele J, Kvasnicka HM, Vardiman J. Bone marrow histopathology in the diagnosis of chronic myeloproliferative disorders: a forgotten pearl. Best Pract Res Clin Haematol. 2006;19:413-437.
25 Johansson P. Epidemiology of the myeloproliferative disorders polycythemia vera and essential thrombocythemia. Semin Thromb Hemost. 2006;32:171-173.
26 Fabris F, Randi ML. Essential thrombocythemia: past and present. Intern Emerg Med. 2009.
27 Randi ML, Putti MC, Scapin M, et al. Pediatric patients with essential thrombocythemia are mostly polyclonal and V617FJAK2 negative. Blood. 2006;108:3600-3602.
28 Finazzi G, Harrison C. Essential thrombocythemia. Semin Hematol. 2005;42:230-238.
29 Tefferi A, Passamonti F. Essential thrombocythemia and pregnancy: observations from recent studies and management recommendations. Am J Hematol. 2009;84:629-630.
30 Lengfelder E, Hochhaus A, Kronawitter U, et al. Should a platelet limit of 600 × 10(9)/l be used as a diagnostic criterion in essential thrombocythaemia? An analysis of the natural course including early stages. Br J Haematol. 1998;100:15-23.
31 Sacchi S, Vinci G, Gugliotta L, et al. Diagnosis of essential thrombocythemia at platelet counts between 400 and 600×10(9)/L. Gruppo Italiano Malattie Mieloproliferative Croniche(GIMMC). Haematologica. 2000;85:492-495.
32 Besses C, Cervantes F, Pereira A, et al. Major vascular complications in essential thrombocythemia: a study of the predictive factors in a series of 148 patients. Leukemia. 1999;13:150-154.
33 Regev A, Stark P, Blickstein D, et al. Thrombotic complications in essential thrombocythemia with relatively low platelet counts. Am J Hematol. 1997;56:168-172.
34 Allegra A, Alonci A, Penna G, et al. JAK2 V617F-positive latent essential thrombocythemia and splanchnic vein thrombosis: the role of bone marrow biopsy for the diagnosis of myeloproliferative disease. Acta Haematol. 2009;121:218-220.
35 Kiladjian JJ, Cervantes F, Leebeek FW, et al. The impact of JAK2 and MPL mutations on diagnosis and prognosis of splanchnic vein thrombosis: a report on 241 cases. Blood. 2008;111:4922-4929.
36 Thiele J, Kvasnicka HM. Hematopathologic findings in chronic idiopathic myelofibrosis. Semin Oncol. 2005;32:380-394.
37 Thiele J, Kvasnicka HM. Grade of bone marrow fibrosis is associated with relevant hematological findings – a clinicopathological study on 865 patients with chronic idiopathic myelofibrosis. Ann Hematol. 2006;85:226-232.
38 Murphy S, Peterson P, Iland H, et al. Experience of the Polycythemia Vera Study Group with essential thrombocythemia: a final report on diagnostic criteria, survival, and leukemic transition by treatment. Semin Hematol. 1997;34:29-39.
39 Buhr T, Busche G, Choritz H, et al. Evolution of myelofibrosis in chronic idiopathic myelofibrosis as evidenced in sequential bone marrow biopsy specimens. Am J Clin Pathol. 2003;119:152-158.
40 Thiele J, Kvasnicka HM, Schmitt-Graeff A, et al. Follow-up examinations including sequential bone marrow biopsies in essential thrombocythemia (ET): a retrospective clinicopathological study of 120 patients. Am J Hematol. 2002;70:283-291.
41 Kreft A, Buche G, Ghalibafian M, et al. The incidence of myelofibrosis in essential thrombocythaemia, polycythaemia vera and chronic idiopathic myelofibrosis: a retrospective evaluation of sequential bone marrow biopsies. Acta Haematol. 2005;113:137-143.
42 Campbell PJ, Bareford D, Erber WN, et al. Reticulin accumulation in essential thrombocythemia: prognostic significance and relationship to therapy. J Clin Oncol. 2009;27:2991-2999.
43 Cervantes F, Alvarez-Larran A, Talarn C, et al. Myelofibrosis with myeloid metaplasia following essential thrombocythaemia: actuarial probability, presenting characteristics and evolution in a series of 195 patients. Br J Haematol. 2002;118:786-790.
44 Thiele J, Kvasnicka HM, Vardiman JW, et al. Bone marrow fibrosis and diagnosis of essential thrombocythemia. J Clin Oncol. 2009;27:e220-e221. author reply e222–3
45 Mesa RA, Verstovsek S, Cervantes F, et al. Primary myelofibrosis (PMF), post polycythemia vera myelofibrosis (post-PV MF), post essential thrombocythemia myelofibrosis (post-ET MF), blast phase PMF (PMF-BP): consensus on terminology by the international working group for myelofibrosis research and treatment (IWG-MRT). Leuk Res. 2007;31:737-740.
46 Passamonti F, Rumi E, Arcaini L, et al. Prognostic factors for thrombosis, myelofibrosis, and leukemia in essential thrombocythemia: a study of 605 patients. Haematologica. 2008;93:1645-1651.
47 Passamonti F, Rumi E, Arcaini L, et al. Blast phase of essential thrombocythemia: a single center study. Am J Hematol. 2009;84:641-644.
48 Passamonti F, Rumi E, Arcaini L, et al. Leukemic transformation of polycythemia vera: a single center study of 23 patients. Cancer. 2005;104:1032-1036.
49 Radaelli F, Mazza R, Curioni E, et al. Acute megakaryocytic leukemia in essential thrombocythemia: an unusual evolution? Eur J Haematol. 2002;69:108-111.
50 Cervantes F, Passamonti F, Barosi G. Life expectancy and prognostic factors in the classic BCR/ABL-negative myeloproliferative disorders. Leukemia. 2008;22:905-914.
51 Passamonti F, Rumi E, Pungolino E, et al. Life expectancy and prognostic factors for survival in patients with polycythemia vera and essential thrombocythemia. Am J Med. 2004;117:755-761.
52 Kralovics R. Genetic complexity of myeloproliferative neoplasms. Leukemia. 2008;22:1841-1848.
53 Lippert E, Boissinot M, Kralovics R, et al. The JAK2-V617F mutation is frequently present at diagnosis in patients with essential thrombocythemia and polycythemia vera. Blood. 2006;108:1865-1867.
54 Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779-1790.
55 Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054-1061.
56 Larsen TS, Pallisgaard N, Moller MB, et al. The JAK2 V617F allele burden in essential thrombocythemia, polycythemia vera and primary myelofibrosis – impact on disease phenotype. Eur J Haematol. 2007;79:508-515.
57 Thiele J, Kvasnicka HM. Chronic myeloproliferative disorders with thrombocythemia: a comparative study of two classification systems (PVSG, WHO) on 839 patients. Ann Hematol. 2003;82:148-152.
58 Pardanani AD, Levine RL, Lasho T, et al. MPL515 mutations in myeloproliferative and other myeloid disorders: a study of 1182 patients. Blood. 2006;108:3472-3476.
59 Vannucchi AM, Antonioli E, Guglielmelli P, et al. Characteristics and clinical correlates of MPL 515W>L/K mutation in essential thrombocythemia. Blood. 2008;112:844-847.
60 Panani AD. Cytogenetic findings in untreated patients with essential thrombocythemia. In Vivo. 2006;20:381-384.
61 Gangat N, Tefferi A, Thanarajasingam G, et al. Cytogenetic abnormalities in essential thrombocythemia: prevalence and prognostic significance. Eur J Haematol. 2009;83:17-21.
62 Tefferi A, Murphy S. Current opinion in essential thrombocythemia: pathogenesis, diagnosis, and management. Blood Rev. 2001;15:121-131.
63 Gisslinger H. Update on diagnosis and management of essential thrombocythemia. Semin Thromb Hemost. 2006;32:430-436.
64 Thiele J, Kvasnicka HM, Diehl V. Initial (latent) polycythemia vera with thrombocytosis mimicking essential thrombocythemia. Acta Haematol. 2005;113:213-219.
65 Gianelli U, Iurlo A, Vener C, et al. The significance of bone marrow biopsy and JAK2V617F mutation in the differential diagnosis between the ‘early’ prepolycythemic phase of polycythemia vera and essential thrombocythemia. Am J Clin Pathol. 2008;130:336-342.
66 Gianelli U, Vener C, Raviele PR, et al. Essential thrombocythemia or chronic idiopathic myelofibrosis? A single-center study based on hematopoietic bone marrow histology. Leuk Lymphoma. 2006;47:1774-1781.
67 Florena AM, Tripodo C, Iannitto E, et al. Value of bone marrow biopsy in the diagnosis of essential thrombocythemia. Haematologica. 2004;89:911-919.
68 Buhr T, Georgii A, Choritz H. Myelofibrosis in chronic myeloproliferative disorders. Incidence among subtypes according to the Hannover classification. Pathol Res Pract. 1993;189:121-132.
69 Georgii A, Buesche G, Kreft A. The histopathology of chronic myeloproliferative diseases. Baillière’s Clin Haematol. 1998;11:721-749.
70 Tefferi A, Skoda R, Vardiman JW. Myeloproliferative neoplasms: contemporary diagnosis using histology and genetics. Nat Rev Clin Oncol. 2009;6:627-637.
71 Tefferi A. Primary myelofibrosis. Cancer Treat Res. 2008;142:29-49.
72 Barosi G. Myelofibrosis with myeloid metaplasia: diagnostic definition and prognostic classification for clinical studies and treatment guidelines. J Clin Oncol. 1999;17:2954-2970.
73 Barosi G, Hoffman R. Idiopathic myelofibrosis. Semin Hematol. 2005;42:248-258.
74 Massa M, Rosti V, Ramajoli I, et al. Circulating CD34+, CD133+, and vascular endothelial growth factor receptor 2-positive endothelial progenitor cells in myelofibrosis with myeloid metaplasia. J Clin Oncol. 2005;23:5688-5695.
75 Oppliger Leibundgut E, Horn MP, Brunold C, et al. Hematopoietic and endothelial progenitor cell trafficking in patients with myeloproliferative diseases. Haematologica. 2006;91:1465-1472.
76 Passamonti F, Rumi E, Pietra D, et al. Relation between JAK2 (V617F) mutation status, granulocyte activation, and constitutive mobilization of CD34+ cells into peripheral blood in myeloproliferative disorders. Blood. 2006;107:3676-3682.
77 Xu M, Bruno E, Chao J, et al. Constitutive mobilization of CD34+ cells into the peripheral blood in idiopathic myelofibrosis may be due to the action of a number of proteases. Blood. 2005;105:4508-4515.
78 Le Bousse-Kerdiles MC, Martyre MC, Samson M. Cellular and molecular mechanisms underlying bone marrow and liver fibrosis: a review. Eur Cytokine Netw. 2008;19:69-80.
79 Kuter DJ, Bain B, Mufti G, et al. Bone marrow fibrosis: pathophysiology and clinical significance of increased bone marrow stromal fibres. Br J Haematol. 2007;139:351-362.
80 Bock O, Hoftmann J, Theophile K, et al. Bone morphogenetic proteins are overexpressed in the bone marrow of primary myelofibrosis and are apparently induced by fibrogenic cytokines. Am J Pathol. 2008;172:951-960.
81 Alvarez-Larran A, Bellosillo B, Martinez-Aviles L, et al. Postpolycythaemic myelofibrosis: frequency and risk factors for this complication in 116 patients. Br J Haematol. 2009;146:504-509.
82 Rollison DE, Howlader N, Smith MT, et al. Epidemiology of myelodysplastic syndromes and chronic myeloproliferative disorders in the United States, 2001–2004, using data from the NAACCR and SEER programs. Blood. 2008;112:45-52.
83 Mesa RA, Silverstein MN, Jacobsen SJ, et al. Population-based incidence and survival figures in essential thrombocythemia and agnogenic myeloid metaplasia: an Olmsted County study, 1976–1995. Am J Hematol. 1999;61:10-15.
84 Johansson P, Kutti J, Andreasson B, et al. Trends in the incidence of chronic Philadelphia chromosome negative (Ph−) myeloproliferative disorders in the city of Goteborg, Sweden, during 1983–99. J Intern Med. 2004;256:161-165.
85 Cervantes F, Barosi G, Demory JL, et al. Myelofibrosis with myeloid metaplasia in young individuals: disease characteristics, prognostic factors and identification of risk groups. Br J Haematol. 1998;102:684-690.
86 Thiele J, Kvasnicka HM. Prefibrotic chronic idiopathic myelofibrosis – a diagnostic enigma? Acta Haematol. 2004;111:155-159.
87 Cervantes F, Barosi G. Myelofibrosis with myeloid metaplasia: diagnosis, prognostic factors, and staging. Semin Oncol. 2005;32:395-402.
88 Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113:2895-2901.
89 Kvasnicka HM, Thiele J, Werden C, et al. Prognostic factors in idiopathic (primary) osteomyelofibrosis. Cancer. 1997;80:708-719.
90 Hussein K, Huang J, Lasho T, et al. Karyotype complements the International Prognostic Scoring System for primary myelofibrosis. Eur J Haematol. 2009;82:255-259.
91 Tam CS, Abruzzo LV, Lin KI, et al. The role of cytogenetic abnormalities as a prognostic marker in primary myelofibrosis: applicability at the time of diagnosis and later during disease course. Blood. 2009;113:4171-4178.
92 Huang J, Li CY, Mesa RA, et al. Risk factors for leukemic transformation in patients with primary myelofibrosis. Cancer. 2008;112:2726-2732.
93 Bock O, Neuse J, Hussein K, et al. Aberrant collagenase expression in chronic idiopathic myelofibrosis is related to the stage of disease but not to the JAK2 mutation status. Am J Pathol. 2006;169:471-481.
94 Al-Assar O, Ul-Hassan A, Brown R, et al. Gains on 9p are common genomic aberrations in idiopathic myelofibrosis: a comparative genomic hybridization study. Br J Haematol. 2005;129:66-71.
95 Dingli D, Grand FH, Mahaffey V, et al. Der(6)t(1;6)(q21-23;p21.3): a specific cytogenetic abnormality in myelofibrosis with myeloid metaplasia. Br J Haematol. 2005;130:229-232.
96 Hussein K, Van Dyke DL, Tefferi A. Conventional cytogenetics in myelofibrosis: literature review and discussion. Eur J Haematol. 2009;82:329-338.
97 Kvasnicka HM, Thiele J. Bone marrow angiogenesis: methods of quantification and changes evolving in chronic myeloproliferative disorders. Histol Histopathol. 2004;19:1245-1260.
98 Thiele J, Kvasnicka HM, Schmitt-Graeff A, et al. Dynamics of fibrosis in chronic idiopathic (primary) myelofibrosis during therapy: a follow-up study on 309 patients. Leuk Lymphoma. 2003;44:949-953.
99 Mesa RA, Hanson CA, Rajkumar SV, et al. Evaluation and clinical correlations of bone marrow angiogenesis in myelofibrosis with myeloid metaplasia. Blood. 2000;96:3374-3380.
100 Thiele J, Kvasnicka HM. CD34+ stem cells in chronic myeloproliferative disorders. Histol Histopathol. 2002;17:507-521.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
CHAPTER 23 Essential thrombocythemia and primary myelofibrosis
Introduction
Among the classical Philadelphia-chromosome-negative myeloproliferative neoplasms (MPN), essential thrombocythemia (ET) and primary myelofibrosis (PMF) represent two subtypes with considerable overlap in clinical and hematological presentation in particular at early stages of disease.1 Differentiation between those two MPN subtypes is of clinical relevance, since PMF is characterized by a considerably higher risk of progression and leukemic transformation. In contrast, ET generally represents a stable disease with only minimal risk of myelofibrotic progression.2 There is also a significant difference in long-term survival between both groups. The 15-year age-adjusted relative survival rate for ET is nearly 84%. In contrast, PMF is characterized by a significant loss in life expectancy. Even when diagnosed in very early stages of disease, PMF patients have a 15-year survival of only 55–67%.2 Although the discovery of the JAK2V617F mutation (see Chapter 21) led to important progress in the understanding of the molecular pathogenesis of ET and PMF,3 the finding of this mutation in several MPN subtypes and the absence of the JAK2V617F allele in many MPN patients preclude the use of JAK2V617F testing alone to establish a diagnosis of ET or PMF. This is reflected in the revised WHO diagnostic criteria for ET and PMF (Box 23.1), which include mutation screening in the diagnostic work-up, but explicitly require a bone marrow (BM) morphological examination for making the diagnosis of both ET and PMF. Although a significantly lower JAK2V617F allele burden has been found in ET when compared to early stages of PMF,4 such variability in the allele burden does not represent a sufficient criterion for distinguishing among different clinical entities. Furthermore, JAK2V617F-negative cases reveal a similar clinical profile and outcome5 and increase in JAK2V617F allele frequencies is not linked to myelofibrotic transformation or progression to acute myeloid leukemia (AML).6
Box 23.1 WHO criteria for the diagnosis of essential thrombocythemia (ET) and primary myelofibrosis (PMF)
| Essential thrombocythemia (ET) | Primary myelofibrosis (PMF) |
|---|---|
|
1. Presence of megakaryocyte proliferation and atypia, usually accompanied by either reticulin and/or collagen fibrosis, or, in the absence of significant reticulin fibrosis, the megakaryocyte changes must be accompanied by an increased bone marrow cellularity characterized by granulocytic proliferation and often decreased erythropoiesis (i.e. prefibrotic/early phase disease).
|
|
| Diagnosis requires meeting all four criteria | Diagnosis requires meeting all three major criteria and two minor criteria* |
* In prefibrotic/early stages these features are generally only borderline expressed.
a) Causes of reactive thrombocytosis include iron deficiency, splenectomy, surgery, infection, inflammation, connective tissue disease, metastatic cancer, and lymphoproliferative disorders. The presence of a condition associated with reactive thrombocytosis does not exclude the possibility of ET if the first three criteria are met.
b) Causes of bone marrow fibrosis include infection, autoimmune disorders or other chronic inflammatory conditions, hairy cell leukemia or other lymphoid neoplasm, metastatic malignancy, or toxic (chronic) myelopathies.
ET and PMF usually affect the elderly population, but they can occasionally be found in children, and in this instance, they raise age-related diagnostic and management issues.7 Familial clustering of ET and also PMF is known, and this observation led to a suggestion of predisposition alleles even before the discovery of the JAK2V617F mutation.8 Relatives of patients with ET have a more than sevenfold increase in the relative risk of developing a similar MPN.9 However, the coexistence of different clinical entities and of JAK2V617F-positive and JAK2V617F-negative diseases in the same family is noteworthy.8,10
When addressing the current WHO criteria (Box 23.1) for diagnosing the different MPN entities,11,12 it is essential to emphasize that these guidelines do not claim that a single histological parameter defines a subgroup, but that the different subtypes of MPN are characterized by specific morphological BM patterns.13 These patterns are composed of distinctive features and should always be reviewed in close relation to clinical, hematological and molecular-genetic findings to achieve a consensus-based working diagnosis.1,3,14,15 In contrast to the determination of age-dependent cellularity and to the semiquantitative grading of myelofibrosis,16,17 characterization of megakaryopoiesis may cause significant difficulties concerning definition and easy recognition of disease-related patterns among different observers.18 Assessment of megakaryocytic histotopography, i.e. their arrangement within the BM space, and detection of certain nuclear abnormalities (besides maturation defects) are the keys to the diagnosis.19 In contrast to the normal BM, in which megakaryocytes show a central distribution of single isolated cells, the overall increase of megakaryocytes in MPN is often associated with the uneven distribution such as group formation: from small clusters (at least three cells) to extensive groups (more than seven cells).20 These megakaryocyte clusters may display either a loose (intermingled with other hematopoietic cells) or dense arrangement.19 An abnormal dislocation of megakaryocytes towards the endosteal (paratrabecular) border is a highly conspicuous finding that is usually not seen in reactive disorders. Other features indicating a neoplastic process are peculiar nuclear aberrations and maturation defects that imply disturbances of the normal development of megakaryopoiesis.1,21–23 These include:
Furthermore, maturation defects include a conspicuous deviation of the nuclear-cytoplasmic ratio or maturation with appearance of bizarre megakaryocytes. It is noteworthy that all these changes may be present in megakaryocytes of different sizes and ploidy status. Finally, so-called naked (denuded, bare) nuclei with condensed chromatin pattern may frequently be found implicating a stimulated thrombocyte shedding and cell turnover.20
In other hematopoietic lineages, an increase and left-shifting of neutrophil granulopoiesis or erythropoiesis may be a prominent feature or a reduction in the amount of nucleated red cell precursors may be found, depending on disease entity and phase.24
Essential thrombocythemia (ET)
Essential thrombocythemia (ET) is a MPN that involves primarily the megakaryocytic lineage. It is characterized by sustained thrombocytosis >450 × 109/l in the peripheral blood (PB), increased numbers of large, mature megakaryocytes in the BM, and clinically by episodes of thrombosis and/or hemorrhage. Because there is no known genetic or biological marker specific for ET, other causes for thrombocytosis such as other MPN, inflammatory and infectious disorders, hemorrhage and other types of hematopoietic and non-hematopoietic neoplasms must be excluded.12 The presence of the BCR-ABL1 fusion gene excludes the diagnosis of ET (Box 23.1). The reported annual incidence of ET ranges from 0.59 to 2.53/100 000 inhabitants and its prevalence is around 30/100 000, which is similar to that of polycythemia vera (PV).25 The median age at diagnosis is 65–70 years,26 but ET is occasionally found in children, and it is relatively common in female patients in their third or fourth decade of life.27,28 In women of childbearing age, ET may be a risk factor for complications during pregnancy.29 Venous thrombosis in unusual locations is typical in younger patients, and the thrombotic risk increases in patients over 60 years old.26
Clinical features
Many patients are asymptomatic when an excess in platelets is discovered by a routine blood count.26,28,29 Most investigators have argued that the use of a threshold level 600 × 109/l platelets compromises the detection of early-phase disease because the adjusted 95th percentile for normal platelet count is below 400 × 109/l.30,31 Therefore, the platelet threshold count required for ET diagnosis has been lowered to 450 × 109/l (Box 23.1). Initial presentation may include vascular occlusions, hemorrhage or microvascular complications that lead to transient ischemic attacks and digital ischemia with paresthesias.32,33 Some patients may present with a thrombosis of major arteries and veins such as splenic or hepatic vein thrombosis as in the Budd–Chiari syndrome.26,34,35 Increase in spleen size at time of diagnosis is seen only in a minority of patients.2 Generally, there is no anemia, no circulating blasts, and serum levels of lactate dehydrogenase (LDH) are within the normal range.23,36 In patients with borderline anemia, palpable spleen, slight elevation of LDH serum level or evidence of erythroid and/or granulopoietic precursors in the PB, a differential diagnosis of very early (prefibrotic) stages of PMF should be excluded by careful bone marrow trephine biopsy (BMTB) examination.1,2,21,23,24,36,37 In contrast to PV cases, the utility of JAK2V617F mutation screening for the diagnosis of ET is limited by suboptimal negative predictive value and lack of diagnostic specificity in the context of MPN.14 Therefore, a BMTB is mandatory to differentiate ET from other MPN and to help with the differential diagnosis between JAK2V617F-negative ET and reactive thrombocytosis.1,14,21,24
In most patients, ET is an indolent disorder characterized by long symptom-free intervals, interrupted by occasional life-threatening thromboembolic or hemorrhagic episodes.26,28,38 Although after many years a few patients with ET may develop BM fibrosis such progression is very uncommon.39–41 The exact incidence of myelofibrotic transformation in ET persists to be a controversial issue42 and the reported results are certainly influenced by the risk status of the patients and the applied diagnostic criteria.1,21,43,44 Careful morphological examination of BMTB is crucial in the diagnostic workup of post ET myelofibrosis (Post-ET MF), since appearance of BM fibrosis with a grade MF-2 or MF-3 is a prerequisite for the diagnosis (Box 23.2 and Fig. 23.1). Secondly, only cases with predefined and documented ET according to the WHO guidelines fall into this category.45 Strict adherence to the WHO criteria (Box 23.1) is necessary to prevent diagnostic confusion associated with early PMF accompanied by thrombocytosis.1,23,36 The frequency of true post-ET myelofibrosis is low; the transformation rate is 2.8% at a median follow-up of 9.1 years (or a 10-year risk of 3.9% and a 15-year risk of 6%).46 Transformation of ET to acute myeloid leukemia or MDS occurs in fewer than 5% of patients, and in chemotherapy treated patients may be related to previous cytotoxic therapy.26,28,47–49 The life expectancy in ET is near normal for most patients.2,50,51
Box 23.2 WHO criteria for the diagnosis of post-essential thrombocythemia myelofibrosis (Post-ET MF)
Molecular data
A JAK2V617F or a functionally similar mutation may be detected in approximately 40–50% of patients (see Chapter 21 for details).5,52–55 However, these findings are not specific for ET and are also seen in other MPN subtypes. It has to be emphasized that significant differences in the JAK2V617F allele burden were described between ET and early PMF.4,6,56 As an independent marker this feature validates very nicely corresponding BMTB findings and thus supports the concept of differentiating ET from early PMF.1,21,23,24,57 A gain-of-function mutation of MPL has been reported in approximately 1–3% of patients with ET.58,59 None of these mutations are found in cases of reactive thrombocytosis. Although ET is usually characterized by a normal karyotype, an isolated del(5q) has been reported in few cases with ET. However, it is more likely that these cases represent MDS associated with this abnormality.60,61
Blood and bone marrow findings
Marked and sustained thrombocytosis is the hallmark of ET. The platelets often display anisocytosis, ranging from tiny forms to atypical large, giant platelets that may reveal bizarre shapes, pseudopods and agranularity. The white blood cell count (WBC) and leukocyte differential are usually normal, although a borderline elevation in the neutrophil lineage may occur.28,62,63 In ET classified according to the histological guidelines of the WHO classification, neither a relevant increase in cellularity nor a significant left-shifted neutrophil granulopoiesis is observed.1,3,57 Any case with a mild to moderate granulocytic and erythroid growth pattern (panmyelosis) and an EPO level below the reference range is suspicious for occult (pre-polycythemic) PV mimicking ET.64,65 Regarding megakaryopoiesis, gross disturbances of the histological topography (significant abnormal localization and/or extensive dense clustering) are not seen (Figs 23.2 and 23.3). Megakaryocytes show a more or less random distribution, with scattered forms or a few loose clusters (Figs 23.4 and 23.5). A predominance of large to giant mature megakaryocytes with extensively folded (staghorn-like) nuclei19,20,22,24,66,67 surrounded by a correspondingly mature cytoplasm is found (Fig. 23.6). These features are clearly different from prefibrotic early PMF, where megakaryocytes show an extensive dense clustering and have hypolobulated (cloud-like or bulbous) and hyperchromatic nuclei with striking maturation defects (Fig. 23.7) leading to a marked anomaly of their nuclear-cytoplasmic ratio.1,19–22,24,36,65,67 Finally, there is no substantial increase in reticulin fibers at presentation and collagen fibrosis is never observed in ET.44 In a large series of ET patients, minimal to slight reticulin fibrosis was described in only 3% of cases.19,40,41,68,69 Any marked increase in reticulin fibers is not compatible with ET,1 in contrast to prefibrotic PMF, which is a differential diagnosis in patients with thrombocytosis.2,21,22,57
[/not-level-membership-for-pathology-category]
