[level-membership-for-hematology-oncology-and-palliative-medicine-category]
Chapter 29 Essential Thrombocythemia
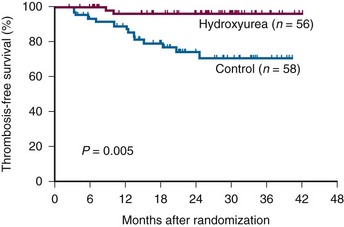
Figure 29-1 PROBABILITY OF THROMBOSIS-FREE SURVIVAL IN 114 PATIENTS WITH ESSENTIAL THROMBOCYTHEMIA TREATED WITH HYDROXYUREA OR LEFT UNTREATED.
(From Cortelazzo S, Finazzi G, Ruggeri M, et al: Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med 332:1132, 1995.)
Table 29-1 Frequency of Neurologic Complaints Associated With Essential Thrombocythemia

Data from Jabaily J, Iland HJ, Laszlo J, et al: Neurologic manifestations of essential thrombocythemia. Ann Intern Med 99:513, 1983.
Table 29-2 Risk Stratification in Essential Thrombocythemia Based on Thrombotic Risk*
| Risk Category | Age >60 Years or History of Thrombosis | Cardiovascular Risk Factors |
|---|---|---|
| Low | No | No |
| Intermediate | No | Yes |
| High | Yes | Yes |
*Cardiovascular risk factors: hypertension, hypercholesterolemia, diabetes, smoking, and congestive heart failure. Extreme thrombocytosis (platelet count >1500 × 109/L) is a risk factor for bleeding. Its role as a risk factor for thrombosis in essential thrombocythemia is uncertain.
Data from Finazzi G, Barbui T: Risk-adapted therapy in essential thrombocythemia and polycythemia vera. Blood Rev 19:243, 2005.
Table 29-3 Proposed Revised World Health Organization Criteria for the Diagnosis of Primary Myelofibrosis and Essential Thrombocythemia and British Committee for Standards in Hematology Criteria for Diagnosis of Essential Thrombocythemia
| Proposed Revised WHO Criteria for Primary Myelofibrosis |
|---|
| Major Criteria |
|
1. Presence of megakaryocyte proliferation and atypia,* usually accompanied by either reticulin or collagen fibrosis or in the absence of significant reticulin fibrosis, the megakaryocyte changes must be accompanied by an increased BM cellularity characterized by granulocytic proliferation and often decreased erythropoiesis (i.e., prefibrotic cellular phase disease) 2. Not meeting WHO criteria for PV,† CML,‡ MDS,§ or other myeloid neoplasm 3. Demonstration of JAK2617V->F or other clonal marker (e.g., MPL515W->L/K) or in the absence of a clonal marker, no evidence of BM fibrosis caused by underlying inflammatory or other neoplastic diseases‖ |
| Minor Criteria |
| Diagnosis requires meeting all three major criteria and two minor criteria. |
| BM, Bone marrow; CML, chronic myeloid leukemia; LDH, lactate dehydrogenase; MDS, myelodysplastic syndrome; PV, polycythemia vera; WHO, World Health Organization. *Small to large megakaryocytes with an aberrant nuclear-to-cytoplasmic ratio and hyperchromatic, bulbous, or irregularly folded nuclei and sense clustering. †Requires the failure of iron replacement therapy to increase hemoglobin level to the PV range in the presence of decreased serum ferritin. Exclusion of PV is based on hemoglobin and hematocrit levels, and RBC mass measurement is not required. ‡Requires the absence of BCR-ABL. §Requires absence of dyserythropoiesis and dysgranulopoiesis. ‖Secondary to infection, autoimmune disorder or other chronic inflammatory condition, hairy cell leukemia or other lymphoid neoplasm, metastatic malignancy, or toxic (chronic) myelopathies. It should be noted that patients with conditions associated with reactive myelofibrosis are not immune to primary myelofibrosis, and the diagnosis should be considered in such cases if other criteria are met. ¶Degree of abnormality could be borderline or marked. |
| Proposed Revised World Health Organization Criteria for Essential Thrombocythemia |
|
1. Sustained platelet count ≥450 × 109/L* 2. BM biopsy specimen showing proliferation mainly of the megakaryocytic lineage with increased numbers of enlarged, mature megakaryocytes; no significant increase or left shift of neutrophil granulopoiesis or erythropoiesis 3. Not meeting WHO criteria for PV,† PMF,‡ CML,§ MDS,‖ or other myeloid neoplasm 4. Demonstration of JAK2V617F or other clonal marker or in the absence of a clonal marker, no evidence for reactive thrombocytosis¶ |
| Diagnosis requires meeting all four criteria. From Tefferi A, Thiele J, Orazi A, et al: Proposals and rationale for revision of the World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia, and primary myelofibrosis: Recommendations from an ad hoc international expert panel. Blood 110:1092, 2007. BM, Bone marrow; CML, chronic myeloid leukemia; MDS, myelodysplastic syndrome; PV, polycythemia vera; WHO, World Health Organization. *During the workup period. †Requires the failure of iron replacement therapy to increase hemoglobin level to the PV range in the presence of decreased serum ferritin. Exclusion of PV is based on hemoglobin and hematocrit levels, and red blood cell mass measurement is not required. ‡Requires the absence of relevant reticulin fibrosis, collagen fibrosis, peripheral blood leukoerythroblastosis, or markedly hypercellular BM for age accompanied by megakaryocyte morphology that is typical for PMF—small to large with an aberrant nuclear-to-cytoplasmic ratio and hyperchromatic, bulbous, or irregularly folded nuclei and dense clustering. §Requires the absence of BCR-ABL. ||‖Requires absence of dyserythropoiesis and dysgranulopoiesis. ¶Causes of reactive thrombocytosis include iron deficiency, splenectomy, surgery, infection, inflammation, connective tissue disease, metastatic cancer, and lymphoproliferative disorders. However, the presence of a condition associated with reactive thrombocytosis does not exclude the possibility of ET if the first three criteria are met. |
| British Committee for Standards in Hematology Criteria for Diagnosis of Essential Thrombocythemia |
| Requires A1-A3 or A1+A3-A5
A1: Sustained platelet count >450 × 109/L A2: Presence of an acquired pathogenetic mutation (e.g., in JAK2 or MPL) A3: No other myeloid malignancy, especially PV, PMF, CML, or MDS A4: No reactive cause for thrombocytosis and normal iron stores A5: BM aspirate and trephine biopsy showing increased megakaryocyte numbers displaying a spectrum of morphology with predominant large megakaryocytes with hyperlobated nuclei and abundant cytoplasm |
BM, Bone marrow; CML, chronic myeloid leukemia; JAK2, Janus kinase 2; MDS, myelodysplastic syndrome; MPL, thrombopoietin receptor; PMF, primary myelofibrosis; PV, polycythemia vera.
From Harrison CN, Bareford D, Butt N, et al: Guideline for investigation and management of adults and children presenting with a thrombocytosis. Br J Haematol 149:352, 2010.
Table 29-4 Clinical and Laboratory Features Helpful in Distinguishing Essential Thrombocythemia from Reactive Thrombocytosis* (Revised)
| Feature | ET | RT |
|---|---|---|
| Chronic platelet increase | + | − |
| Known causes of RT | − | + |
| Thrombosis or hemorrhage | + | − |
| Splenomegaly | + | − |
| BM reticulin fibrosis | + | − |
| BM megakaryocyte clusters | + | − |
| Abnormal cytogenetics | + | − |
| Increased acute phase reactants | − | + |
| Spontaneous colony formation† | + | − |
| JAK2V617F mutation | + | − |
BM, bone marrow; ET, essential thrombocythemia; RT, reactive thrombocytosis.
*Acute phase reactants include C-reactive protein and fibrinogen.
Modified from Tefferi A, Hoagland HC: Issues in the diagnosis and management of primary thrombocythemia. Mayo Clin Proc 69:651, 1994.
Table 29-5 Risk Factors for Thrombosis in 100 Patients With Essential Thrombocythemia
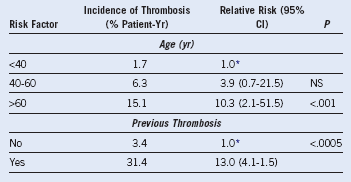
CI, Confidence interval; NS, not significant.
Data from: Finazzi G, Barbui T: Risk-adapted therapy in essential thrombocythemia and polycythemia vera. Blood Rev 19:243, 2005.
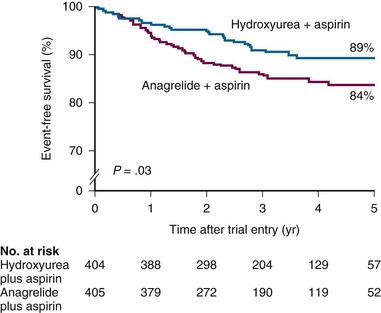
Figure 29-3 EVENT-FREE SURVIVAL IN A HYDROXYUREA-TREATED GROUP COMPARED WITH AN ANAGRELIDE-TREATED GROUP.
(From Harrison CN, Campbell PJ, Buck G, et al: Hydroxyurea compared with anagrelide in high-risk essential thrombocythemia. N Engl J Med 353:33, 2005.)

[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
Chapter 29 Essential Thrombocythemia
Figure 29-1 PROBABILITY OF THROMBOSIS-FREE SURVIVAL IN 114 PATIENTS WITH ESSENTIAL THROMBOCYTHEMIA TREATED WITH HYDROXYUREA OR LEFT UNTREATED.
(From Cortelazzo S, Finazzi G, Ruggeri M, et al: Hydroxyurea for patients with essential thrombocythemia and a high risk of thrombosis. N Engl J Med 332:1132, 1995.)
Table 29-1 Frequency of Neurologic Complaints Associated With Essential Thrombocythemia
Data from Jabaily J, Iland HJ, Laszlo J, et al: Neurologic manifestations of essential thrombocythemia. Ann Intern Med 99:513, 1983.
Table 29-2 Risk Stratification in Essential Thrombocythemia Based on Thrombotic Risk*
| Risk Category | Age >60 Years or History of Thrombosis | Cardiovascular Risk Factors |
|---|---|---|
| Low | No | No |
| Intermediate | No | Yes |
| High | Yes | Yes |
*Cardiovascular risk factors: hypertension, hypercholesterolemia, diabetes, smoking, and congestive heart failure. Extreme thrombocytosis (platelet count >1500 × 109/L) is a risk factor for bleeding. Its role as a risk factor for thrombosis in essential thrombocythemia is uncertain.
Data from Finazzi G, Barbui T: Risk-adapted therapy in essential thrombocythemia and polycythemia vera. Blood Rev 19:243, 2005.
Table 29-3 Proposed Revised World Health Organization Criteria for the Diagnosis of Primary Myelofibrosis and Essential Thrombocythemia and British Committee for Standards in Hematology Criteria for Diagnosis of Essential Thrombocythemia
| Proposed Revised WHO Criteria for Primary Myelofibrosis |
|---|
| Major Criteria |
|
1. Presence of megakaryocyte proliferation and atypia,* usually accompanied by either reticulin or collagen fibrosis or in the absence of significant reticulin fibrosis, the megakaryocyte changes must be accompanied by an increased BM cellularity characterized by granulocytic proliferation and often decreased erythropoiesis (i.e., prefibrotic cellular phase disease) 2. Not meeting WHO criteria for PV,† CML,‡ MDS,§ or other myeloid neoplasm 3. Demonstration of JAK2617V->F or other clonal marker (e.g., MPL515W->L/K) or in the absence of a clonal marker, no evidence of BM fibrosis caused by underlying inflammatory or other neoplastic diseases‖ |
| Minor Criteria |
| Diagnosis requires meeting all three major criteria and two minor criteria. |
| BM, Bone marrow; CML, chronic myeloid leukemia; LDH, lactate dehydrogenase; MDS, myelodysplastic syndrome; PV, polycythemia vera; WHO, World Health Organization. *Small to large megakaryocytes with an aberrant nuclear-to-cytoplasmic ratio and hyperchromatic, bulbous, or irregularly folded nuclei and sense clustering. †Requires the failure of iron replacement therapy to increase hemoglobin level to the PV range in the presence of decreased serum ferritin. Exclusion of PV is based on hemoglobin and hematocrit levels, and RBC mass measurement is not required. ‡Requires the absence of BCR-ABL. §Requires absence of dyserythropoiesis and dysgranulopoiesis. ‖Secondary to infection, autoimmune disorder or other chronic inflammatory condition, hairy cell leukemia or other lymphoid neoplasm, metastatic malignancy, or toxic (chronic) myelopathies. It should be noted that patients with conditions associated with reactive myelofibrosis are not immune to primary myelofibrosis, and the diagnosis should be considered in such cases if other criteria are met. ¶Degree of abnormality could be borderline or marked. |
| Proposed Revised World Health Organization Criteria for Essential Thrombocythemia |
|
1. Sustained platelet count ≥450 × 109/L* 2. BM biopsy specimen showing proliferation mainly of the megakaryocytic lineage with increased numbers of enlarged, mature megakaryocytes; no significant increase or left shift of neutrophil granulopoiesis or erythropoiesis 3. Not meeting WHO criteria for PV,† PMF,‡ CML,§ MDS,‖ or other myeloid neoplasm 4. Demonstration of JAK2V617F or other clonal marker or in the absence of a clonal marker, no evidence for reactive thrombocytosis¶ |
