CHAPTER 8 Erythroenzyme disorders
Introduction
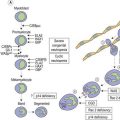
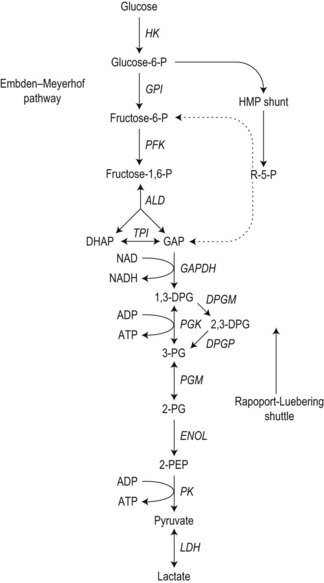
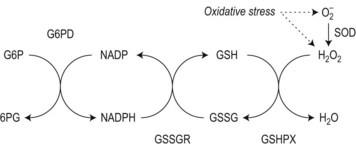
To achieve optimal performance as an oxygen transporter the mature red cell has sacrificed metabolic versatility.1 Red cell structural and functional integrity depend on catabolism of glucose via the anerobic Embden–Meyerhof pathway to replenish adenosine triphosphate (ATP) required for cation homeostasis and other energy-dependent processes in conjunction with the oxidative pentose phosphate pathway (hexose monophosphate shunt) to maintain redox capacity. In the resting state 90% of glucose is catabolized anerobically through the Embden–Meyerhof pathway, which also serves to generate nicotinamide adenine dinucleotide in its reduced form (NADH) required as a cofactor for cytochrome b5 reductase for the conversion of methemoglobin to hemoglobin (Fig. 8.1). The pentose phosphate pathway serves mainly to supply the reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) necessary to regenerate reduced glutathione (GSH), which acts as a sacrificial reductant to protect the membrane and contents of the red cell against oxidative damage (Fig. 8.2). Glucose-6-phosphate dehydrogenase (G6PD) catalyzes the first and rate limiting step, the conversion of glucose-6-phosphate (G6P) to 6-phosphogluconate (6PG), in this pathway. Synthesis of the tripeptide glutathione from its constituent amino acids and nucleotide salvage complete the essential metabolic repertoire active in the red cell cytosol. Conservation of adenine nucleotides to maintain intracellular ATP and elimination of pyrimidine nucleotides is facilitated through the action of pyrimidine 5′-nucleotidase (P5N) which specifically dephosphorylates pyrimidine nucleoside-5′-monophosphates formed by RNA breakdown. This permits removal of toxic pyrimidines by passive diffusion and prevents their accumulation within the red cell. Defects in each of these key pathways produces hemolytic anemia.
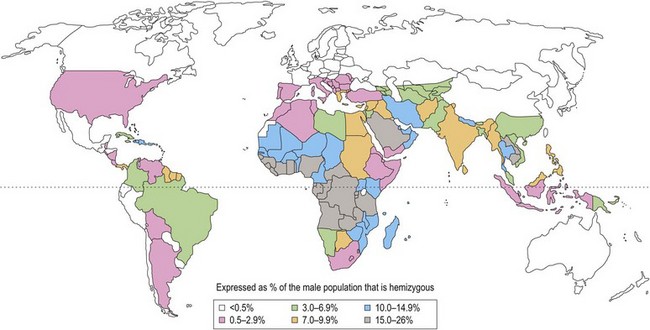
With the exception of polymorphic G6PD variants (Fig. 8.3) estimated to affect up to 400 million people worldwide, most inherited disorders of red cell metabolism are uncommon. Pyruvate kinase (PK) deficiency is the most commonly encountered defect of the Embden–Meyerhof pathway with around 500 cases reported to date. Heterozygote frequencies based on biochemical population studies vary from 0.14% in the US2 to 6% in Saudi Arabia.3 There is some evidence that reduced red cell PK activity affords protection against malaria.4 Mice deficient in PK are protected from malaria and the growth of Plasmodium falciparum is impaired in PK-deficient red cells. The impact of this on populations in areas of malarial endemicity is, however, likely to be small compared with the selective advantage conferred by the more common genetic red cell variants, G6PD deficiency and the hemoglobinopathies.
After PK the most common enzyme deficiencies implicated in hemolytic anemia are in approximate order of frequency: glucosephosphate isomerase (GPI); class I G6PD variants (associated with chronic hemolytic anemia); phosphofructokinase (PFK), triosephosphate isomerase (TPI), phosphoglycerate kinase (PGK) and hexokinase (HK). Deficiencies of P5N,5 which has been described in populations of diverse geographic origin, and glutathione synthetase6 occur at a comparable frequency. Other erythroenzyme disorders are rare.
Clinical features
The presence of neurological, myopathic or other non-hematologic manifestations is of considerable diagnostic value. Neurodevelopmental abnormalities must be interpreted with caution since severe neonatal hyperbilirubinemia sufficient to cause kernicterus has been described in deficiency of several red cell enzymes including G6PD and PK. Distinctive features of erythroenzymopathies are summarized in Table 8.1.
The pattern of somatic abnormalities manifest in individual enzyme disorders is determined by several factors. Deficiency of an isoenzyme, expression of which is restricted (e.g. pyruvate kinase-L/R), generally causes isolated hemolysis. Conversely, defects of ubiquitously expressed enzymes (e.g. triosephosphate isomerase) often result in a more generalized phenotype. The physiochemical properties of a mutant enzyme also influence clinical expression. Enzyme variants associated with impaired catalytic efficiency generally produce greater metabolic perturbance than those resulting in structural instability, the consequences of which are offset in tissues which retain the capacity for protein synthesis. This is reflected in a correspondingly more severe clinical phenotype. Examples include stable GPI mutants associated with multisystem7 disease and high Km class I G6PD variants which in addition to hemolysis manifest impaired leukocyte function or cataract due to reduction of enzyme activity in non-erythroid tissues.8
Blood cell morphology
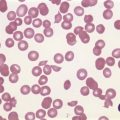
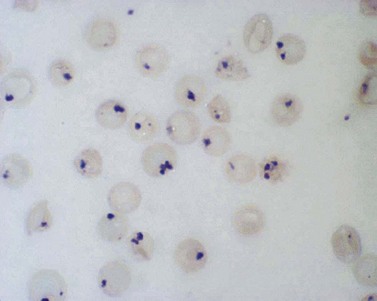
Red cell enzyme defects have traditionally been grouped under the heading congenital non-spherocytic hemolytic anemia. While morphological atypia in the red cells are usually apparent, these overlap with other causes of hemolysis and are seldom exclusive to a specific enzyme defect. A notable exception is the striking basophilic stippling associated with P5N deficiency which may be seen in up to 5% of red cells on a freshly stained blood film prepared from an ethylenediaminetetraacetic acid (EDTA) sample (Fig. 8.4). Stippling may disappear if the stain is delayed by more than a few hours, presumably because EDTA chelates metal ions required for ribonucleoprotein aggregation. This problem may be circumvented by examination of blood taken into lithium heparin. Conspicuous punctate basophilia akin to that in P5N deficiency accompanies unstable hemoglobin variants and CDP-choline phosphotransferase deficiency,9 a putative defect of nucleotide metabolism described in only a few families. Lead is a potent inhibitor of P5N activity. This explains the finding of punctate basophilia in plumbism. The presence of other clinical and morphologic stigmata, the latter including red cell hypochromia and microcytosis, reticulocytosis or sideroblastic erythropoiesis, usually render distinction from primary P5N deficiency straightforward. Basophilic stippling may also be found in a wide range of congenital and acquired dyserythropoietic states including thalassemia and occasionally in some glycolytic defects (e.g. pyruvate kinase or phosphofructokinase deficiency).
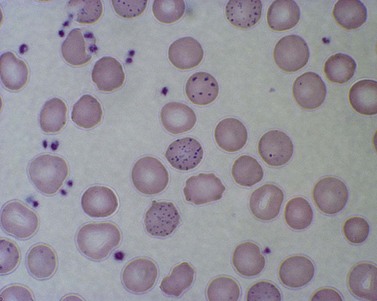
Fig. 8.4 Coarse basophilic stippling in pyrimidine 5′-nucleotidase deficiency. May–Grünwald–Giemsa. × 1000.
Features of oxidative damage to red cells are most commonly associated with, though not confined to, G6PD deficiency. These are most remarkable during hemolytic crises following exposure to oxidant drugs (Table 8.2) or fava bean (broad bean) consumption and include irregularly contracted hyperchromic erythrocytes, some of which display a characteristic ‘bite’ or ‘hemighost’ appearance (Fig. 8.5). ‘Bite’ cells in which the surface of the erythrocyte is breached producing an irregular gap are thought to be generated by removal of Heinz bodies during transit through the spleen. Erythrocyte ‘hemighosts’ are forms in which the hemoglobin appears condensed and is retracted to one side leaving an empty space in the cell. In the common polymorphic G6PD variants (e.g. G6PD A− or Med) these morphologic abnormalities are visible only during hemolytic episodes. Following acute hemolysis in G6PD deficiency rapid clearance of damaged cells by the spleen ensues and during the recovery phase polychromasia and macrocytosis predominate. Similar though usually less marked features of oxidative damage may be seen in defects which impair glutathione biosynthesis (Fig. 8.6) or regeneration due to deficiency of γ-glutamylcysteine synthetase,10 glutathione synthetase6,11 or glutathione reductase12 as well as neonatal hemolysis due to deficiency of glutathione peroxidase (Fig. 8.7).13 The latter condition which does not conform to the laws of Mendelian inheritance may reflect a transient reduction in enzyme activity due to impaired selenium homeostasis in the mother or neonate. Selenium is an essential co-factor for glutathione peroxidase which serves to detoxify harmful peroxides in the red cell. Glutathione peroxidase deficiency produces an acute and usually self-limiting hemolytic anemia in the newborn period. The diagnosis may be confirmed by assay of neonatal and maternal selenium levels and red cell glutathione peroxidase. Defects of other enzymes in the pentose phosphate pathway, 6-phosphogluconolactonase14 and phosphogluconate dehydrogenase15 have been described, albeit rarely, and should be considered if changes suggestive of oxidative damage are evident and more common causes excluded.
Table 8.2 Drugs and chemicals associated with hemolysis in glucose-6-phosphate dehydrogenase (G6PD) deficiency
| Class of drug | Examples |
|---|---|
| Antimalarials | Primaquine, pentaquine, pamaquine, chloroquine* |
| Sulfonamides and sulfones | Sulfanilamide, sulfacetamide, sulfapyridine, sulfamethoxazole (including co-trimoxazole), dapsone |
| Other antibacterial agents | Nitrofurantoin, nalidixic acid, chloramphenicol, ciprofloxacin* |
| Analgesic/antipyretic | Acetanilid, acetylsalicylic acid (aspirin)†, paracetamol (acetaminophen)† |
| Miscellaneous | Probenecid |
| Dimercaprol | |
| Vitamin K analogs | |
| Naphthalene (moth balls) | |
| Methylene blue | |
| Ascorbic acid | |
| Trinitrotoluene |
† Only after high doses or overdose.
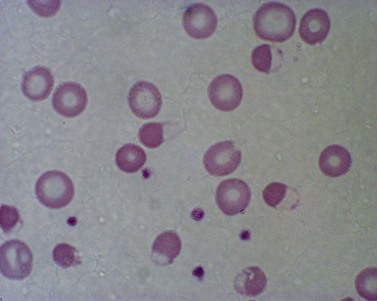
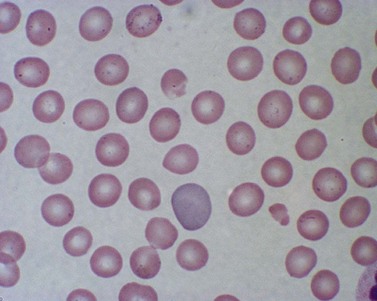
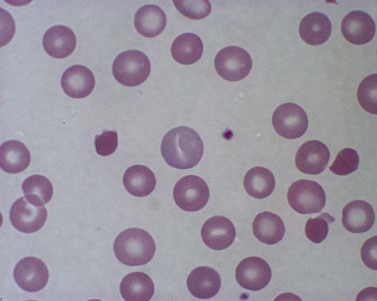
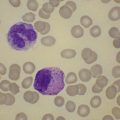
Examination of a blood film for Heinz bodies should be performed in cases of suspected oxidative hemolysis. These intraerythrocytic inclusions, first characterized in experimental studies of acetylphenylhydrazine toxicity, are visualized after staining supravitally with the basic dyes methyl violet or brilliant cresyl blue (Fig. 8.8). Heinz bodies, formed from denatured globin which attaches to the inner surface of the erythrocyte membrane, develop either spontaneously in the case of unstable hemoglobin variants or after oxidative challenge in susceptible (e.g. G6PD-deficient) red cells. The number of Heinz bodies increases dramatically after splenectomy. Similar inclusions due to precipitation of surplus α-globin chains may be visible in some thalassemia syndromes. It should be noted that unstable hemoglobin variants which produce a Heinz-body hemolytic anemia often escape detection by conventional separation techniques either because they result from structural alteration within the interior of the hemoglobin molecule and therefore do not alter surface charge or are so unstable as to undergo rapid degradation ex vivo. Stability tests in combination with mass spectrometry16 provide a reliable approach to the detection of unstable hemoglobins which should be undertaken in cases where oxidative changes or Heinz bodies are present before detailed studies of red cell metabolism are embarked upon. Similarly, if drug or toxin ingestion is suspected, screening for sulphemoglobin and methemoglobin by absorbance at 620 and 630 nm respectively is indicated to exclude drug- or chemical-induced hemolysis which may follow severe oxidant stress in the absence of any intrinsic red cell defect.
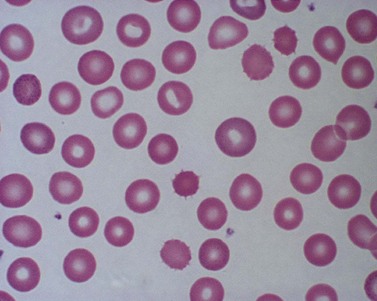
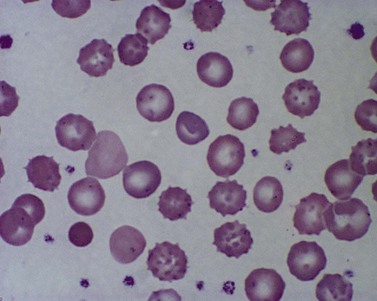
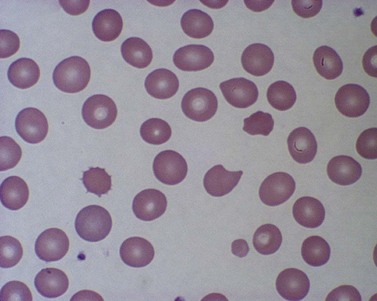
True spherocytes, such as seen in hereditary spherocytosis in which both the normal discoid shape of the erythrocyte is lost and cell volume reduced, are generally not seen in erythroenzymopathies. A possible exception is enolase 1 deficiency, a disorder hitherto described in only a single kindred with a dominant mode of inheritance and spherocytosis but normal acidified glycerol lysis test. Morphologic variants, particularly spheroechinocytes, derived from the Greek for sea urchin (Echinus), are frequently present in variable numbers in glycolytic disorders (Fig. 8.9). These crenated cells have multiple short spicules of uniform appearance and represent effete red cells in which ATP depletion has led to failure of cation homeostasis and cellular dehydration. They are most striking in, though not specific to, PK deficiency where the number of such cells often increases dramatically (up to 30% of red cells) after splenectomy (Fig. 8.10). Poikilocytosis with elliptocytic, ovalocytic and dacrocytic (tear-drop) forms may also be seen in PK deficiency (Fig. 8.11). These findings are nonspecific and may be ascribable to dyserythropoiesis. Evidence of ineffective erythropoiesis with defective utilization of 59Fe has been observed in some cases and experimental models indicate PK deficiency is associated with increased apoptosis of erythroid progenitors.17
Biochemical investigation of erythroenzyme disorders
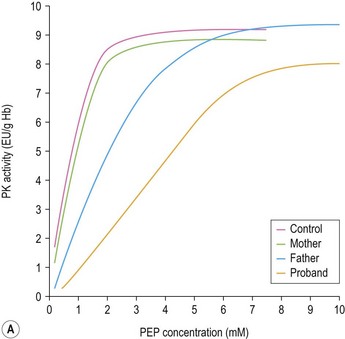
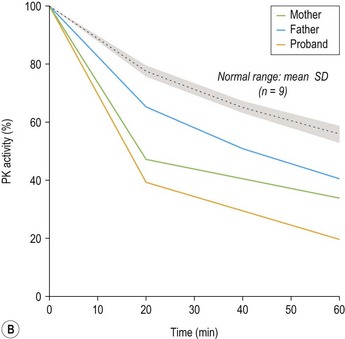
While useful screening methods18 exist for detection of some more common enzyme defects (e.g. G6PD and PK deficiency) definitive diagnosis relies on quantitation of enzyme activity in red cells in conjunction with physiochemical properties of the mutant enzyme.19 Rigorous removal of leukocytes in which residual enzyme activity is substantially higher or reflects expression of a different isoenzyme from that in red cells potentially masking deficiency and correction for the higher activity of some enzymes (HK, PK, aldolase, G6PD and P5N) in younger red cells by comparison with a control matched for a reticulocyte count or another red cell age-dependent enzyme is essential. In most clinical erythroenzymopathies residual enzyme activity in red cells is 5–40% of normal. Higher levels do not exclude an erythroenzyme disorder and particular care must be taken in interpretation of studies performed in patients who have received transfusion due to interference from donor red cells and neonates. Significant differences in erythrocyte metabolism have been observed between neonatal and adult red cells. These include a higher activity for some enzymes (PK, GPI, G6PD) and lower activity for others (PFK, glutathione peroxidase, adenylate kinase) in erythrocytes from cord blood. Under the saturating substrate conditions employed for quantitation of enzyme activity in vitro relatively stable mutants with impaired catalytic efficiency in vivo may elude detection. If a strong suspicion of enzymopathy remains, measurement of enzyme activity at low substrate concentration or studies of enzyme kinetics and response to physiologic modulators may be necessary (Fig. 8.12A, B).
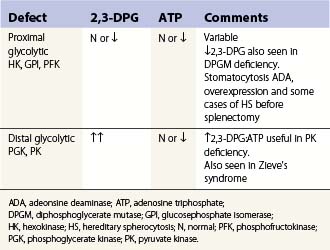
Quantitation of the major red cell metabolites 2,3-diphosphoglycerate (2,3-DPG) and GSH by spectrophotometry is of value in the diagnosis of glycolytic disorders and hemolytic anemias due to impaired defense against oxidative damage to the red cell. The ratio of 2,3-DPG to ATP specifically may localize a defect in glycolysis to the proximal or distal part of the Embden–Meyerhof pathway (Table 8.3). A reduced GSH concentration is found in G6PD deficiency, other pentose phosphate pathway defects and enzyme disorders directly affecting glutathione biosynthesis or regeneration. A low red cell GSH level is, however, a relatively nonspecific finding which may be seen in other causes of hemolytic anemia particularly unstable hemoglobins as well as some glycolytic (e.g. GPI deficiency) and membrane defects. Marked reduction in GSH implies a defect in glutathione biosynthesis due to γ-glutamylcysteine synthetase or glutathione synthetase deficiency.
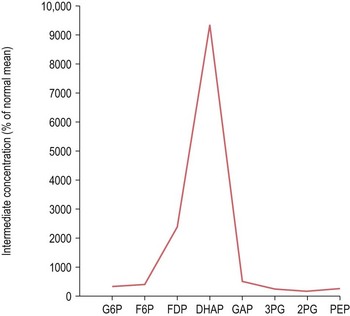
To overcome the limitation of in vitro measurement of enzyme activity under conditions which may not accurately reflect enzyme function in vivo, defects in the Embden–Meyerhof pathway may be identified by measurement of the concentration of intermediate metabolites in a deproteinized red cell extract. Typically, metabolic block is indicated by accumulation of intermediates proximal and depletion distal to the step catalyzed by the deficient enzyme. In some instances substrate accumulation may be dramatic and the resulting intermediate profile pathognomonic of a specific disorder (Fig. 8.13). Unfortunately, in many countries, the reagents required for quantitation of glycolytic intermediates are no longer readily available. Prenatal diagnosis by biochemical or molecular analysis has been undertaken for several severe erythroenzymopathies including deficiencies of TPI, GPI, PK and G6PD.20–23
Molecular basis of erythroenzyme disorders
Over the past decade the molecular defects that underlie hematologically important erythroenzyme disorders have been elucidated. This has revealed a striking bias towards missense mutations mainly affecting conserved residues in the encoded protein (Fig. 8.14).24–26 The paucity of null mutations found among patients with clinical enzyme deficiencies is consistent with evidence from murine models that complete disruption of the Embden–Meyerhof, pentose phosphate or glutathione biosynthetic pathways is lethal during embryogenesis. Exceptions to this are severe forms of PK deficiency due to mutations that abolish PK-L/R expression, for example the PK Gypsy mutation, a 1149bp deletion which results in the loss of exon 11. Surviving homozygotes may be rescued from what would otherwise be a lethal phenotype by persistence of the muscle isoenzyme PK-M2, normally expressed during early erythroid differentiation, which is encoded by a separate genetic locus.27 Relatively few examples of regulatory mutations have been implicated in erythroenzyme disorders, exceptions being the −72G and −83C mutations which disrupt conserved elements in the promoter of the PK-L/R gene. Sequence variation within the TATA box and other essential promoter elements of the TPI gene is widely distributed in human populations and has been linked to a reduction of enzyme activity in vivo.26
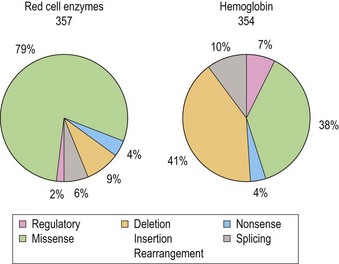
Fig. 8.14 Distribution of gene mutations in human erythroenzyme (n = 357) and hemoglobin (n = 354) disorders.
In certain populations individual mutations account for a high proportion of deficient alleles. This applies not only to G6PD deficiency where the prevalence of individual variants reflects evolutionary selection due to protection against malaria but is also evident in PK deficiency in which 1529A(Arg510Gln) and 1456T(Arg466Trp) substitutions together account for approximately 40% and 30% of mutations in patients of northern and southern European descent respectively. Among reported Japanese PK deficient patients the most frequently found mutation is1468T(Arg490Trp).17 Even greater homogeneity is evident in TPI deficiency where a single missense mutation Glu104Asp accounts for the majority of reported cases. Haplotype studies support a single origin for these mutations. Genotype–phenotype correlations are beginning to emerge for the erythroenzyme disorders informed by the study of patients homozygous for individual mutations. Homozygotes for the 994A(Gly332Ser) mutation of the PK-L/R gene manifest a severe clinical course with transfusion-dependent anemia. At the other end of the spectrum it has been proposed, based on disparity in observed and predicted allele frequencies, that some genotypes such as homozygosity for the 1456T mutation may escape clinical detection due to their mild phenotype. Distant genetic factors may also exert an influence on the clinical course of erythroenzyme disorders. The level of unconjugated bilirubin in G6PD28 and PK27 deficiency correlates with inheritance of the (TA)7 allele of the uridinine diphosphate glucuronosyltransferase gene (UGT1A1) promoter associated with Gilbert syndrome which has been shown to potentiate gallstone formation in other hemolytic states. Coinheritance of hereditary hemochromatosis may accelerate iron loading, though the high prevalence of this complication in pyruvate kinase deficiency suggests the contribution of other mechanisms, for example ineffective erythropoiesis, may be more important.
1 Beutler E. The red cell. In: Haemolytic Anemia in Disorders of Red Cell Metabolism. New York: Plenum; 1978:1-21.
2 Mohrenweiser HW. Frequency of enzyme deficiency variants in erythrocytes of newborn infants. Proceedings of the National Academy of Sciences USA. 1981;78:5046-5050.
3 El-Hazmi MAF, Al-Swailem AR, Al-Faleh FZ, Warsy AS. Frequency of glucose-6-phosphate dehydrogenase, pyruvate kinase and hexokinase deficiency in the Saudi population. Human Hereditory. 1986;36:45-49.
4 Allison AC. Genetic control of resistance to human malaria. Current Opinion in Immunology. 2009;21:499-505.
5 Vives Corrons JL. Chronic non-spherocytic haemolytic anaemia due to congenital pyrimidine 5′ nucleotidase deficiency: 25 years later. Baillière’s Best Practice and Research in Clinical Haematology. 2000;13:103-118.
6 Ristoff E, Mayatepek E, Larsson A. Long-term clinical outcome in patients with glutathione synthetase deficiency. Journal of Pediatrics. 2001;139:79-84.
7 Schroter W, Eber SW, Bardosi A, et al. Generalized glucosephosphate isomerase (GPI) deficiency causing hemolytic anemia, neuromuscular symptoms and impairment of granulocytic function: a new syndrome due to a new stable GPI variant and diminished specific activity (GPI Homburg). European Journal of Pediatrics. 1985;144:301-305.
8 Luzzatto L, Mehta A, Vulliamy T. Glucose-6-phosphate dehydrogenase deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular basis of inherited disease. McGraw-Hill; 2001:4517-4553.
9 Paglia DE, Valentine WN, Nakatani M, et al. Selective accumulation of cytosol CDP-choline as an isolated erythrocyte defect in chronic haemolysis. Proceedings of the National Academy of Sciences USA. 1983;80:3081-3085.
10 Konrad PN, Richards FII, Valentine WN, Paglia DE. Gammaglutamyl-cysteine synthetase deficiency. New England Journal of Medicine. 1972;286:557-561.
11 Hirono A, Iyori H, Skine I, et al. Three cases of hereditary non-spherocytic haemolytic anemia associated with red blood cell glutathione deficiency. Blood. 1996;87:2071-2074.
12 Loos H, Roos D, Weening R, Houwerzijl J. Familial deficiency of glutathione reductase in human blood cells. Blood. 1976;48:53-62.
13 Necheles TF, Steinberg MH, Cameron D. Erythrocyte glutathione-peroxidase deficiency. British Journal of Haematology. 1970;19:605-612.
14 Beutler E, Kuhl W, Gelbert T. 6-Phosphogluconolactonase deficiency, a hereditary erythrocyte enzyme deficiency: possible interaction with glucose-6-phosphate dehydrogenase deficiency. Proceedings of the National Academy of Sciences USA. 1985;82:3876-3878.
15 Vives Corrons JL, Colomer D, Pujades A, et al. Congenital 6-phosphogluconate dehydrogenase (6PGD) deficiency associated with chronic hemolytic anemia in a Spanish family. American Journal of Hematology. 1996;53:221-227.
16 Wild BJ, Green BN, Cooper EK, et al. Rapid identification of hemoglobin variants by electrospray ionization mass spectrometry. Blood Cells, Molecules and Diseases. 2001;27:691-704.
17 Zanella A, Fermo E, Bianchi P, et al. Pyruvate kinase deficiency: the genotype-phenotype association. Blood Reviews. 2007;21:217-231.
18 Beutler E, Blume KG, Kaplan JC, et al. International Committee for Standardization in Haematology: recommended screening test for glucose-6-phosphate dehydrogenase (G-6-PD) deficiency. British Journal of Haematology. 1979;43:469-477.
19 Beutler E. Red Cell Metabolism. A Manual of Biochemical Methods, 2nd ed. Orlando, FL: Grune and Stratton; 1984.
20 Ayra R, Lalloz MRA, Nicolaides KH, et al. Prenatal diagnosis of triosephosphate isomerase deficiency. Blood. 1996;87:4507-4509.
21 Whitelaw AGL, Rogers PA, Hopkinson DA, et al. Congenital haemolytic anaemia resulting from glucose phosphate isomerase deficiency: genetics, clinical picture and prenatal diagnosis. Journal of Medical Genetics. 1979;16:189-196.
22 Baronciani L, Beutler E. Prenatal diagnosis of pyruvate kinase deficiency. Blood. 1994;84:2354-2356.
23 Beutler E, Kuhl W, Fox M, et al. Prenatal diagnosis of glucose-6-phosphate dehydrogenase deficiency. Acta Haematologica. 1992;87:103-110.
24 Stenson PD, Ball E, Howells K, et al. Human Gene Mutation Database: towards a comprehensive central mutation database. Journal of Medical Genetics. 2008;45:124-126.
25 Mehta A, Mason PJ, Vulliamy TJ. Glucose-6-phosphate dehydrogenase deficiency. Baillière’s Best Practice and Research in Clinical Haematology. 2000;13:21-38.
26 Marinaki AM, Escuredo E, Duley JA, et al. Genetic basis of hemolytic anemia caused by pyrimidine 5′-nucleotidase deficiency. Blood. 2001;97:3327-3332.
27 Zanella A, Bianchi P. Red cell pyruvate kinase deficiency: from genetics to clinical manifestations. Baillière’s Best Practice and Research in Clinical Haematology. 2000;13:57-81.
28 Samipietro M, Lupica L, Perrero L, et al. The expression of uridine diphosphate glucuronsyltransferase gene is a major determinant of bilirubin level in heterozygous thalassaemia and in glucose-6-phosphate dehydrogenase deficiency. British Journal of Haematology. 1997;99:437-439.