[level-membership-for-pathology-category]
CHAPTER 22 Erythrocytosis and polycythemia
Introduction
The term ‘erythrocytosis’ is derived from Greek words meaning ‘too many red cells’ and should be distinguished from ‘polycythemia’, meaning ‘too many cells in the blood’.1 Erythrocytosis has been defined as a greater than two standard deviation-increase from the age-, sex- and race-adjusted norm in hematocrit or hemoglobin level.2 It is clear, however, that these laboratory parameters may be affected by decreases in plasma volume. Therefore, a clinical diagnosis of ‘erythrocytosis’ might represent true erythrocytosis, indicating a true increase in red cell mass (RCM), or in fact apparent polycythemia, resulting from either reduced plasma volume (relative polycythemia) or failure to recognize otherwise normal values for hematocrit (Hct) or hemoglobin (Hgb) level that lie in the extreme right tail of the Gaussian distribution.3 Hence, for an individual patient, interpretation of laboratory results without knowledge of the personal baseline value remains inaccurate because of the inevitable statistical overlap of extreme values between subjects with and without disease.3
The recent development of molecular tests for the JAK2V617F and MPL mutations (see Chapter 21 for details) has allowed the reliable distinction of clonal from non-clonal erythrocytosis and has radically altered the investigation of erythrocytosis. Furthermore the value of aggressive phlebotomy to lower Hct levels below 45% in men and 42% in women has not been substantiated.3 The therapeutic relevance of distinguishing polycythemia vera (PV) from so-called ‘essential thrombocythemia (ET) with borderline increased Hct’ has diminished and has further undermined the value of RCM measurement, which is no longer fundamental to the diagnosis of many patients.3
In this chapter, the recent revision of the WHO criteria,4 which reflects developments in molecular pathogenesis and international consensus on clinico-pathological classification, is used for definition purposes although it is recognized that dissenting views have been published.5 Erythrocytosis is therefore practically defined according to the thresholds used in the WHO classification,3,4 i.e.:
The classification of erythrocytosis has recently been the subject of two excellent reviews.3,6 The term ‘idiopathic erythrocytosis’ was used by McMullin6 but has been criticized by Patnaik and Tefferi3 as an entity, due to misuse of the term for patients who have an inappropriate diagnosis of erythrocytosis or who have been inadequately investigated. Nevertheless, there remain patients who have been fully investigated and who are likely to have abnormalities that have not as yet been defined. These may include defects of the erythropoietin (Epo) signaling pathway or oxygen sensing pathway.3,6 Therefore, the category ‘unclassifiable’ is introduced here in accordance with the WHO classification and strictly defined for patients who have been fully investigated and for whom no currently defined cause of the erythrocytosis has been found. Patients who are partially investigated should not be placed in this category. While there are several potential divisions of a classification of patients with proven erythrocytosis, the classification used here (Box 22.1) is adapted from Patnaik and Tefferi.3
The pathology of erythrocytosis
The discovery of the JAK2V617F mutation in 20057–10 (see Chapter 21) has profoundly changed both understanding and investigation of the erythrocytoses. Accordingly, modern diagnostic evaluation of a patient with proven erythrocytosis, and for whom no obvious cause is apparent, may begin with screening of the peripheral blood for the JAK2V617F mutation and serum Epo. A working classification of erythrocytosis into primary, or clonal, i.e. PV, versus secondary has therefore emerged,6 but the more classical pathogenetic approach is used here.
Congenital erythrocytosis
Associated with reduced P50
High-oxygen-affinity hemoglobinopathy
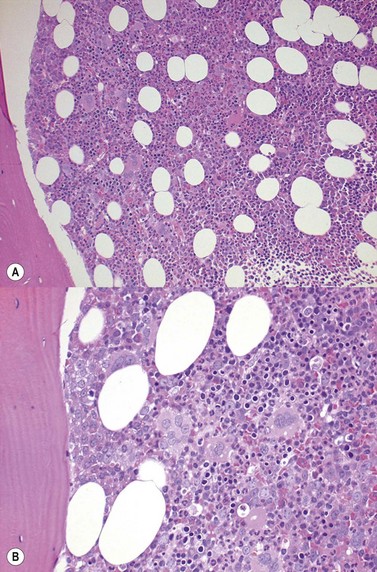
High-oxygen-affinity hemoglobins release oxygen at a lower rate than normal and thus create relative tissue hypoxia, which might result in compensatory erythrocytosis in approximately one third of affected patients. Affected patients often present with isolated erythrocytosis, in the absence of signs and symptoms of systemic disease.3 Erythrocytosis is accompanied by chronic hemolysis where the Hgb variant is unstable.6 Bone marrow trephine biopsy (BMTB) investigation shows erythroid hyperplasia but normal megakaryocytes (Fig. 22.1A, B).These patients may have family members who are similarly affected and a family history is therefore essential in the investigation of patients with erythrocytosis. Transmission in affected patients is usually autosomal dominant.
More than 90 mutations have been described and are covered in an exemplary fashion in a web resource: http://globin.bx.psu.edu/hbvar/. Most of the high-oxygen-affinity mutations involve the β-globin chain and α1β2 contact zones.11 Serum Epo levels are either normal or elevated and P50 (partial pressure of oxygen at which 50% of Hgb is saturated with oxygen) is decreased.12 Structurally abnormal high-affinity Hgb should be suspected3 if the P50 level is <20 mmHg.
2,3-Bisphosphoglycerate mutase (BPGM) deficiency
BPGM deficiency is a rare cause of erythrocytosis.13,14 Deficiency of the enzyme results in a high affinity Hgb with a left shifted oxygen dissociation curve. This results in a compensatory erythrocytosis. Patients with both autosomal dominant15 and autosomal recessive inheritance16 have been described. In a fully penetrant autosomal recessive case, there is an isolated erythrocytosis with normal serum Epo level.16 Diagnosis is established by showing a low P50, a normal Hgb structure and decreased BPGM activity.3
Methemoglobinemia
Congenital methemoglobinemias are of three main types:17,18
Methemoglobin causes both an impaired O2 binding and increased oxygen affinity of the Hgb; sometimes resulting in compensatory erythrocytosis.18
Methemoglobinemia is clinically suspected when cyanosis is accompanied by normal PaO2 levels but low saturation per pulse oximeter. Both carboxyhemoglobin and methemoglobin may be measured by modern oximeters.3
Associated with normal P50
The involvement of oxygen-sensing pathways in physiologic and pathologic erythropoiesis has recently been reviewed.20 Red blood cells deliver O2 from the lungs to the tissues. Better understanding of the physiological regulation of the process has allowed a rational classification of rare cases of congenital erythrocytosis to be developed (Fig. 22.2).3,6
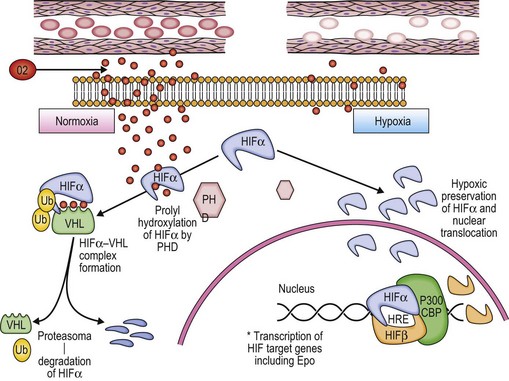
A classic physiologic response to hypoxia in humans is the up-regulation of the Erythropoietin (Epo) gene, which is the central regulator of red blood cell mass. Reduction of tissue oxygenation triggers increased production of Epo by hypoxia-inducible factor 1 (HIF-1), which is a transcriptional activator composed of an O2-regulated α subunit and a constitutively expressed β subunit. Hydroxylation of HIF-1α or HIF-2α by the asparaginyl hydroxylase FIH-1 blocks coactivator binding and transactivation. Hydroxylation of HIF-1α or HIF-2α by the prolyl hydroxylase PHD2 is required for binding of the von Hippel–Lindau (VHL) protein, leading to ubiquitination and proteasomal degradation. Mutations in the genes encoding VHL, PHD2, and HIF-2α have all been identified in patients with familial erythrocytosis.20
VHL mutations including Chuvash erythrocytosis (frequently termed Chuvash polycythemia)
Chuvash erythrocytosis is a rare, autosomal recessive, congenital erythrocytosis, first described in the Chuvash autonomous region in Russia21 but also occurring in other racial and ethnic groups.22,23 Affected patients are homozygous for a germline mutation affecting the VHL tumor suppressor gene, producing an abnormal VHL protein.24,25 The mutation in the VHL gene disrupts the normal mechanism of hypoxia sensing, ultimately resulting in increased Epo production and erythrocytosis.6
In contrast to patients with the VHL disease, an autosomal dominant familial syndrome, patients with Chuvash erythrocytosis do not display an increased predilection for tumors.3 However, other abnormalities are found.26 Plasma concentrations of endothelin-1, Epo, plasminogen activator inhibitor-1, transferrin, transferrin receptor, and vascular endothelial growth factor are elevated. Clinical manifestations include increased cardiac valvular abnormalities, hemangiomas, pulmonary arterial hypertension, thrombotic and hemorrhagic events, varicose veins, and shortened life span. In parallel, peripheral blood concentrations of CD4 positive T-helper cells and CD4/CD8 ratio have been found to be lower in the VHL598C>T homozygotes.26
Prolyl hydroxlase domain 2 (PHD2) mutations
Under normoxic conditions, PHD2 hydroxylates the α-subunits of HIF proteins, facilitating VHL binding and subsequent ubiquitin-mediated proteasomal degradation of HIF.3,6 Rare cases of PHD2 mutations have been described, with the loss of PHD2 function, associated erythrocytosis and normal serum Epo.3,6 Recently, a novel PHD2 mutation has been described, associated with both erythrocytosis and recurrent paraganglioma.27
Hypoxia inducible factor alpha (HIF2α) mutations
Several erythrocytosis-associated HIF2α mutations have been characterized. All result in impaired degradation and thus aberrant stabilization of HIF2α. However, each exhibits a distinct profile with respect to their effects on PHD2 binding and VHL interaction.28 Epo levels are usually elevated.3,6
Epo receptor (Epo-R) mutations
Epo-R signaling is regulated by the binding of the protein tyrosine phosphatase SHP-1 (or other JAK/STAT regulators) to the distal cytoplasmic region of Epo-R. This interaction results in the down-regulation of the Epo-mediated activation of the JAK2/STAT5 pathway and ultimately production of more red cells.3,6 Epo-R mutations (all in exon 8) that cause erythrocytosis have been reviewed.29–31 These mutations often result in cytoplasmic truncation of Epo-R, resulting in failure of attachment of SHP-1, ongoing production of red cells and thus erythrocytosis.
Transmission is usually autosomal dominant. Affected patients are usually asymptomatic and display subnormal (or normal) serum Epo level and hypersensitivity of erythroid progenitors to exogenous Epo.3,6
Acquired erythrocytosis
Erythrocytosis secondary to hypoxia
Chronic hypoxia leads to physiological secondary erythrocytosis, in order to compensate for the low oxygen concentrations at the pulmonary and/or tissue level. Clinically, interpretation of arterial blood gases, pulmonary function tests and chest radiography is an integral part of the investigation of erythrocytosis.3 Chronic lung disease, right-to-left cardiopulmonary shunts, high-altitude habitat, tobacco use/carbon monoxide poisoning, sleep apnea/hypoventilation syndrome and renal artery stenosis are all associated with secondary erythrocytosis.3,6 The bone marrow (BM) morphology is similar to that of patients with congenital erythrocytosis, showing hyperplasia of erythropoiesis and normal megakaryocytes (Fig. 22.1).
Erythrocytosis independent of hypoxia
Erythropoietin and related agents
Epo-stimulating agents (ESAs) were originally designed to replace endogenous Epo in patients with anemia secondary to renal failure. Their use has subsequently been expanded to include patients with anemia of other causes, including cancer patients.32 Recombinant human Epo and its hyperglycosylated analog darbepoetin alfa and an array of novel ESAs are known to be misused by athletes.33 Erythrocytosis has also been associated with supraphysiologic doses of anabolic steroids.34
Post-renal transplant erythrocytosis
Post-renal transplant erythrocytosis (PTE) affects approximately 10–15% of patients and typically manifests within the first or 2 years following transplantation.35 The persistent secretion of Epo by the retained diseased native kidney, which is unaffected by feedback inhibition, is believed to play a central role. However, many patients with PTE have a low or normal Epo level and other erythroid growth factors contribute to driving erythropoiesis. The renal angiotensin system is thought to play an important role in PTE. Insulin-like growth factor has been demonstrated to enhance Epo-induced erythropoiesis in vitro and circulates in significant amounts in anephric dialysis patients. Adenosine receptors are also present on erythroid cells and stimulate Epo release.35
BMTB is most commonly performed in renal failure patients36 due to cytopenias, pyrexia and suspected malignancies and rarely in cases of PTE, in which erythroid hyperplasia is the expected finding possibly in conjunction with the effects of treatment and chronic disease.
Erythrocytosis secondary to tumors
Several benign and malignant tumors and other lesions have been shown to produce inappropriate Epo. A high expression of Epo mRNA in the tumor tissue and the correction of erythrocytosis, together with a fall in the serum Epo levels after the tumor has been removed, is required to establish the association between the tumor and the secondary erythrocytosis.3
Uterine leiomyoma are associated with the so-called ‘myomatous erythrocytosis syndrome’, which is defined by the combination of erythrocytosis, uterine leiomyoma and persistent restoration of normal hematological values after hysterectomy.37 Vlasveld et al.37 were able to demonstrate a large gradient between the Epo levels in the uterine vein and artery during hysterectomy, providing direct evidence for in vivo Epo production by the uterine leiomyoma.
Renal cell carcinoma has also been associated with Epo production.38 Burk at al.39 described a 55-year-old man with clear cell renal carcinoma, pulmonary metastases and erythrocytosis. The increase in RCM was associated with an elevation in erythropoietic stimulatory activity in serum, pleural fluid and tumor-cyst fluid supporting the presence of autonomous tumor secretion of Epo or an Epo-like substance. Erythrocytosis has also been described in cases of tumors arising in polycystic kidneys40 and renal cysts,41 as well as in parathyroid adenoma,42 pheochromocytoma,43 hepatocellular adenoma44 and carcinoma,45 cerebellar hemangioblastoma46 and meningiomas.47
Bone marrow biopsy in erythrocytosis
The histological features of PV are described in detail in the following section. Biopsies of the bone marrow in cases of erythrocytosis lack the findings of panmyelosis and typical megakaryocyte morphology seen in PV. The appearance is of a hyperplastic BM with erythroid hyperplasia. Megakaryocyte morphology is typically normal or reactive (Fig. 21.1). In addition, reactive stromal changes such as perivascular plasmacytosis, eosinophils, cell debris and iron deposits may be seen in cases of erythrocytosis,48 particularly secondary to smoking, in which bronchopulmonary infection may be a concomitant finding.
Thiele et al.49 studied BMTB together with clinical data and follow-up of 208 PV patients and 113 secondary polycythemia patients. In only 13 patients (4%) of this cohort, histopathology failed to differentiate clearly between the two diagnoses. Recently, testing for the JAK2V617F mutation has been shown to have a very high sensitivity, specificity and utility for the differential diagnosis between PV and erythrocytosis50 but BMTB continues to have a role in the diagnosis of erythrocytosis, although it is now more likely to be used in atypical cases.3
Polycythemia vera (PV)
Polycythemia vera is a clonal stem cell disease with trilineage myeloid involvement,4 which in classical cases manifests as panmyeloid proliferation (panmyelosis) in the BM and peripheral erythrocytosis, thrombocytosis and neutrophil leukocytosis.3,4,6
Clinical features
The predominant symptoms of PV are related to hypertension or vascular abnormalities associated with increased RCM or to arterial or venous thrombotic episodes such as deep venous thrombosis, myocardial ischemia or cerebrovascular occlusion, portal or splenic vein thrombosis. Headaches, dizziness, visual disturbances, paresthesias, pruritus, erythromyalgia and gout are common. Patients may be stratified into low, intermediate (if generic cardiovascular risk factors are present) or high risk categories if age is greater than 60 years or a history of thrombosis is present.4,51
Laboratory features
Variations in the classical picture occur and competing classification systems have attempted to separate PV from other forms of MPN for therapeutic purposes. The WHO classification is used here as the basis for classification for the sake of reproducibility. The discovery of the JAK2 mutations (see Chapter 21) and obsolescence of some laboratory tests has radically changed the approach to diagnosis (see Fig. 21.1 for details). Various other laboratory abnormalities are associated with PV (Box 22.2).
In vitro erythroid colony formation in patients with PV does not require the addition of exogenous Epo, a phenomenon termed ‘endogenous erythroid colony growth’. This does not occur in either healthy persons or patients with non-clonal erythrocytosis3 but the test is not widely available and is not routinely used.6 The independent clonal proliferation of eythropoiesis in PV is associated with a correspondingly low serum Epo.6 However, some cases of JAK2 mutation positive ET may also have lower Epo levels, limiting the utility of the test in borderline cases.52
Hemoglobin and hematocrit: the Hct or packed cell volume is a measure of the total number of cells present, of which most are red cells. It is the most accurate way of measuring blood viscosity.6 In PV viscosity is fundamental to the pathological effects: the greater the viscosity, the greater the sluggishness of the blood flow and thus the greater the severity of complications. A raised Hct is therefore the most important criterion for a diagnosis of PV. The Hb is used as a substitute measure of viscosity but there is not always a direct correlation between the two measurements. Using the Hb as a measure of viscosity may underestimate it, particularly in iron deficient patients.6
Red cell mass (RCM): the measurement of RCM is not required in patients who have a raised Hct and a JAK2 mutation as they satisfy the diagnostic criteria for PV.4,6 Measuring RCM is an expensive test and its use is now mainly confined to documenting absolute erythrocytosis versus apparent erythrocytosis in investigating rare JAK2 mutation negative patients who are being investigated for possible PV.3,6
World Health Organization 2008 criteria for the diagnosis of PV
Significant changes in the approach to the diagnosis of PV have been brought about by the ability to define clonal erythrocytosis by the detection of JAK2 mutations, a relatively simple test using polymerase chain reaction (PCR) based techniques.6 It is also possible to determine whether an individual has a totally mutated JAK2 or a mixture of wild type and mutated, described as homozygous or heterozygous (see Chapter 21). The WHO criteria for diagnosis of PV are summarized in Box 22.3. In addition to the classical polycythemic phase of PV, the WHO classification recognizes a pre- and a post-polycythemic phase of PV.
Box 22.3 WHO 2008 criteria for the diagnosis of polycythemia vera
Major criteria
Pre-polycythemic PV
Also termed prodromal, latent or evolving PV, this condition, by definition, does not fulfill the criteria for polycythemic PV and may be diagnosed in retrospect or be only suggested in a BMTB report. The presence of marked thrombocytosis and normal or borderline high hemoglobin in the pre-polycythemic phase may lead to confusion with ET.53 However, BMTB may show the classical features of PV. Recently, Gianelli et al.54 compared the clinicopathologic and molecular features of 17 patients with evolving PV (e-PV) with those of 14 patients with ET and 19 with classical PV. The results for e-PV were more similar to those for PV than for ET. Patients with e-PV were characterized by an increase in the red cell parameters, splenomegaly and hepatomegaly, together with a hypercellular BM due to increased erythropoiesis and granulopoiesis, associated with megakaryocytic hyperplasia, with pleomorphic aggregates. The frequency of the JAK2V617F mutation was nearly 100% in both e-PV and PV but it was significantly lower in ET (54%).
ePV cases may be placed in the myeloproliferative neoplasm, unclassifiable (MPN, U) category, which should only be used for patients who have had full clinical, laboratory and morphological assessment and who still do not fulfill all the criteria for one of the other MPN diagnoses.4 Subsequent evolution of clinical, laboratory or morphological findings may allow more precise classification. The concept of a prodromal phase in chronic myeloproliferative neoplasms has recently been reviewed.55
Post-polycythemic PV
With progress of PV, erythropoiesis decreases, RCM normalizes and then decreases, and splenomegaly increases.4 Peripheral blood changes occur and a leukoerythroblastic blood film with poikilocytosis appears. Splenomegaly due to extramedullary hemopoiesis increases. Terminally, blasts may appear and more than 10% blasts in the peripheral blood or BM or the presence of significant myelodysplasia signals progression to an accelerated phase or the development of a myelodysplastic syndrome. The appearance of more than 20% blasts is considered to represent the development of an acute phase, usually acute myeloid leukemia (AML).4
The International Working Group for Myelofibrosis Research and Treatment56 recently proposed standardization criteria for post-polycythemic myelofibrosis (PPMF) and these have been adapted by the WHO4 (Box 22.4).
Box 22.4 WHO4 diagnostic criteria for post-polycythemic myelofibrosis
Additional criteria (two required)
Genetics in PV
Cases of PV are by definition Philadelphia chromosome/ BCR-ABL1 negative.4 Abnormalities of the thrombopoietin receptor (MPL) (such as MPLW515K/L, MPLW515S and MPLS505N) have been described in 4% of ET and 11% of PMF but not in PV.62
The most important genetic abnormalities in PV are the JAK2V617F mutation and the functionally similar mutation in exon 12 of JAK2 (see Chapter 21 for details), which are present in more than 95% of PV cases. However, the JAK2V617F mutation is not specific for PV and is present in other forms of MPN. This subject has recently been reviewed in depth59 and is covered in detail in Chapter 21. At diagnosis, some 20% of patients with PV have cytogenetic abnormalities.4,60–62 The commonest recurring abnormalities include +8, +9, del(20q), del(13q), del(9p) and a combination of +8 and +9.
Evolution to post-PPMF and acceleration are characterized by increasing cytogenetic abnormalities. Moreover, the karyotype differs from PPMF, where +1q is the main anomaly, compared with PMF where +1q is rare and del(13q) or del(20q) are far more common.60,62 Almost 100% of PV cases that develop dysplasia or progress to an acute phase have cytogenetic abnormalities, including those associated with therapy-related myelodysplasia or AML4 and, interestingly,the leukemic blasts may loose JAK2–V617F expression.63
The bone marrow biopsy in PV
The BMTB is less often performed since the advent of detection of JAK2V617F and related mutations as a reliable diagnostic test.6 However, the BMTB is very useful in the elucidation of atypical or pre-polycythemic cases and in the assessment of cytopenias due to the effects of therapy or the development of PPMF.
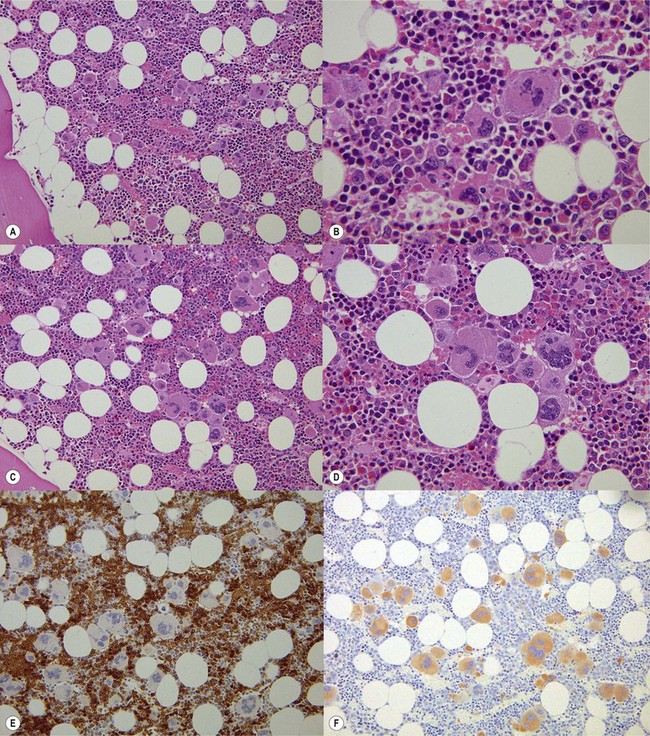
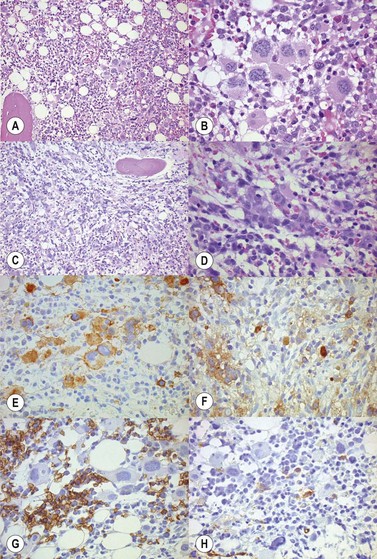
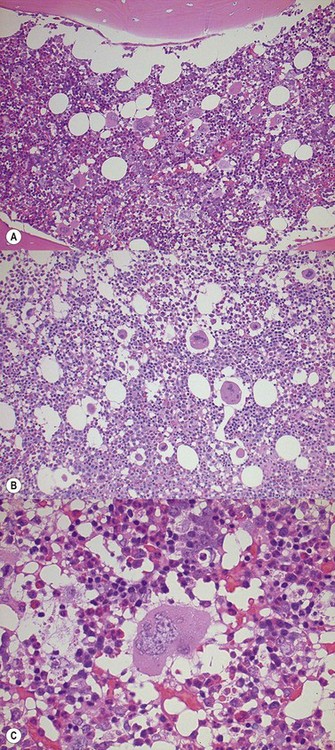
In classical cases,53 the BM is hypercellular due to panmyelosis: proliferation of generally morphologically normal erythropoiesis and granulopoiesis accompanied by abnormal megakaryopoiesis (Fig. 22.3A–F). Megakaryocytes are increased in numbers, sometimes quite markedly in cases with pronounced thrombocytosis, and have hyperlobated nuclei. They are typically pleomorphic, with a mixture of cell sizes, and loosely clustered. In the polycythemic phase megakaryocytes lack significant dysplastic features (Fig. 22.3) but become hyperchromatic and morphologically atypical as PPMF develops (Fig. 22.4A–D). Proliferation of left-shifted neutrophil granulopoiesis and reduced nucleated erythroid precursors is also a feature of PPMF (Fig. 22.5A–B).
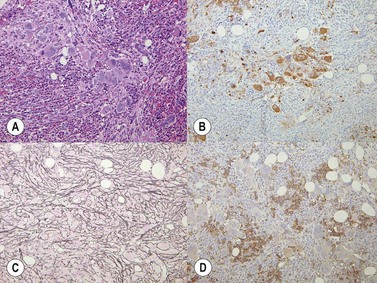
Reticulin is initially normal in 80% of cases, the rest showing varying degrees of increase.4,53 There is a marked increase in the PPMF stage with increasing collagen fibrosis and finally osteomyelofibrosis (Fig. 22.6A, B). Sinusoidal dilatation and other feature of myelofibrosis are also seen (Fig. 22.7). Reticulin is typically not increased in cases of erythrocytosis.53
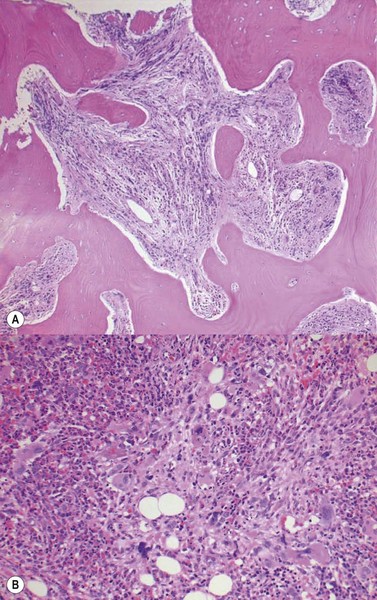
Fig. 22.6 (A, B) Post-polycythemic myelofibrosis, osteomyelofibrosis. (A) Bone changes are readily apparent. (B) More cellular areas may persist.
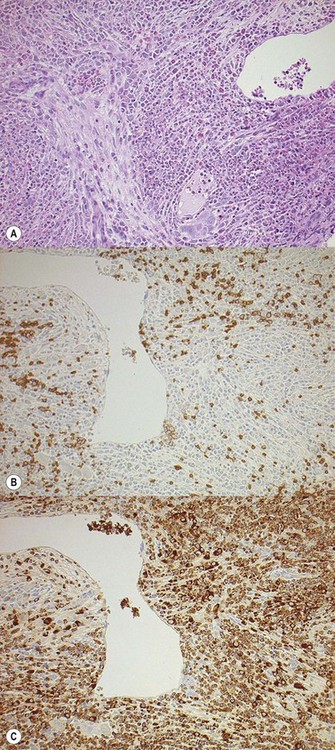
Iron deposits are absent (Perls staining) in approximately 95% of cases, in contrast to cases of erythrocytosis in which iron is readily identified.53 Immunohistochemistry has no particular pattern but is useful in defining the components of hemopoiesis. CD34, CD117 and megakaryocyte markers such as CD61 and CD42 are useful in the diagnosis of the accelerated phase although CD117 positivity of immature erythroid precursors as well as early myeloid cells must be borne in mind64 (Fig. 22.8A–C).
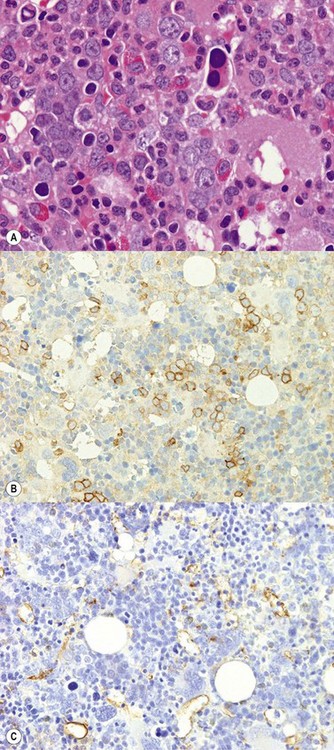
Fig. 22.8 (A–C) (A) An erythroid island showing dyserythropoiesis (B) and CD117 positivity (C) but no expression of CD34.
Frank dysplastic features develop in the accelerated phase4 (Fig. 22.9A–D). The appearance of more than 20% blasts herald transformation to acute leukemia (Fig. 22.10A,B). Atypical morphology and reduced erythropoiesis may also be seen after cytoreductive therapy4 (Fig. 22.11). This finding should not be mistaken for myelodysplasia. Thus, a complete clinical history is essential for the interpretation of a BMTB in PV.
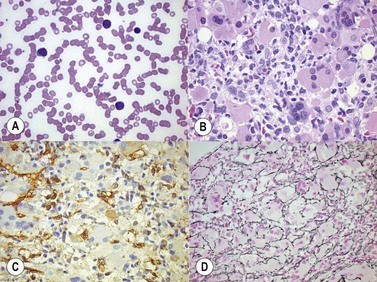
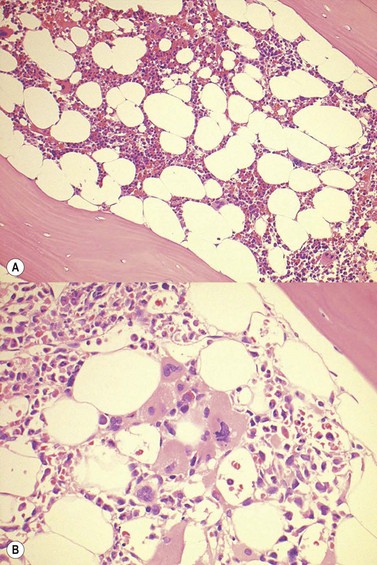
Fig. 22.10 (A, B) PV post-cytoreductive therapy. (A) Reduced cellularity and (B) dysplastic changes in megakaryocytes are apparent.
Cases harboring the exon 12 JAK2 mutation65 have been reported to show predominant erythroid proliferation with normal megakaryocyte morphology and lack of clustering. However, others66 and personal experience indicate that the megakaryocytes can be abnormal in some cases (Fig. 22.11A–C), with loose rather than tight clustering and variable nuclear morphology.
Familial PV
The familial nature of many primary erythrocytoses has already been described. Familial cases fulfilling the definition of the MPNs also occur but are rare. They differ from primary erythrocytoses because of clinical phenotype and multi-lineage proliferation in most instances.67
Only a small number of kindred have been described with four or more affected family members.67,68 Affected family members have clonal hemopoiesis and form spontaneous erythroid colonies. Most but not all affected family members carry an acquired somatic mutation in the JAK2 gene. This strongly suggests that only the predisposition to acquiring additional somatic mutations, such as JAK2V617F, is inherited in these families, thus explaining the late onset and low penetrance of the MPN phenotype and the clonal hemopoiesis.68
Unclassifiable erythrocytosis
In the decades after the first analysis of the Epo gene, regulatory elements led to the discovery of HIF, which led in turn to the discovery of VHL and the PHDs.20 Mutations in genes encoding all three of these essential components of the oxygen-sensing system have been identified in patients with familial erythrocytosis. Gain-of-function mutations in the EpoR gene also cause familial erythrocytosis.3,6,20 It is highly likely that novel mutations in loci encoding other components of the O2-sensing pathway remain to be identified.20
In this group of patients with erythrocytosis in whom no cause has been identified after full investigation, one third has Epo levels below the normal range. A recent study69 of so-called idiopathic erythrocytosis with low serum Epo levels showed that in fact 27% harbored the JAK2 exon 12 mutation. The other two thirds have normal or elevated Epo levels, and so have a secondary erythrocytosis of unknown cause.70
1 Messinezy M, van der Walt JD, Pearson TC. Polycythemia (the erythrocytoses). In: Wickramasinghe SN, McCullough J, editors. Blood and Bone Marrow Pathology. Edinburgh: Elsevier, 2002.
2 Hollowell JG, van Assendelft OW, Gunter EW, et al. Hematological and iron-related analytes – reference data for persons aged 1 year and over: United States, 1988–1994. Vital Health Stat 11. 2005;247:1-156.
3 Patnaik MM, Tefferi A. The complete evaluation of erythrocytosis: congenital and acquired. Leukemia. 2009;23:834-844.
4 Thiele J, Kvasnicka HM, Orazi A, et al. Polycythaemia vera. In: Swerlow SH, Campo E, Harris NL, et al, editors. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC, 2008.
5 Spivak JL, Silver RT. The revised World Health Organization diagnostic criteria for polycythemia vera, essential thrombocythemia and primary myelofibrosis: an alternative proposal. Blood. 2008;112:231-239.
6 McMullin MF. The classification and diagnosis of erythrocytosis. Int Jnl Lab Hem. 2008;30:447-459.
7 Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7:387-397.
8 James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434:1144-1148.
9 Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779-1790.
10 Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054-1061.
11 Gonzalez Fernandez FA, Villegas A, Ropero P, et al. Haemoglobinopathies with high oxygen affinity. Experience of Erythropathology Cooperative Spanish Group. Ann Hematol. 2009;88:235-238.
12 Rumi E, Passamonti F, Pagano L, et al. Blood p50 evaluation enhances diagnostic definition of isolated erythrocytosis. J Intern Med. 2009;265:266-274.
13 Cartier P, Labie D, Leroux JP, et al. [Familial diphosphoglycerate mutase deficiency: hematological and biochemical study]. Nouv Rev Fr Hematol. 1972;12:269-287.
14 Rosa R, Prehu MO, Beuzard Y, Rosa J. The first case of a complete deficiency of diphosphoglycerate mutase in human erythrocytes. J Clin Invest. 1978;62:907-915.
15 Galacteros F, Rosa R, Prehu MO, et al. Diphosphoglyceromutase deficiency: new cases associated with erythrocytosis. Nouv Rev Fr Hematol. 1984;26:69-74.
16 Hoyer JD, Steven LA, Beutler E, et al. Erythrocytosis due to bisphosphoglycerate mutase deficiency with concurrent glucose-6-phosphate dehydrogenase (G-6-PD) deficiency. Am J of Hematol. 2004;75:205-208.
17 Yilmaz D, Cogulu O, Ozkinay F, et al. A novel mutation in the DIA1 gene in a patient with methemoglobinemia type II. Am J Med Genet A. 2005;133A:101-102.
18 Fermo E, Bianchi P, Vercellati C, et al. Recessive hereditary methemoglobinemia: two novel mutations in the NADH-cytochrome b5 reductase gene. Blood Cells Mol Dis. 2008;41:50-55.
19 Percy MJ, Lappin TR. Recessive congenital methaemoglobinaemia: cytochrome b5 reductase deficiency. B J of Haematol. 2008;141:298-308.
20 Semenza GL. Involvement of oxygen-sensing pathways in physiologic and pathologic erythropoiesis. Blood. 2009;114:2015-2019.
21 Liu E, Percy MJ, Amos CI, et al. The worldwide distribution of the VHL 598C>T mutation indicates a single founding event. Blood. 2004;103:1937-1940.
22 Percy MJ, McMullin MF, Jowitt SN, et al. Chuvash-type congenital polycythemia in 4 families of Asian and Western European ancestry. Blood. 2003;102:1097-1099.
23 Perrotta S, Nobili B, Ferraro M, et al. Von Hippel-Lindau-dependent polycythemia is endemic on the island of Ischia: identification of a novel cluster. Blood. 2006;107:514-519.
24 Ang SO, Chen H, Gordeuk VR, et al. Endemic polycythemia in Russia: mutation in the VHL gene. Blood Cells Mol Dis. 2002;28:57-62.
25 Ang SO, Chen H, Hirota K, et al. Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat Genet. 2002;32:614-621.
26 Niu X, Miasnikova GY, Sergueeva AI, et al. Altered cytokine profiles in patients with Chuvash polycythemia. Am J Hematol. 2009;84:74-78.
27 Ladroue C, Carcenac R, Leporrier M, et al. PHD2 mutation and congenital erythrocytosis with paraganglioma. N Engl J Med. 2008;359:2685-2692.
28 Furlow PW, Percy MJ, Sutherland S, et al. Erythrocytosis-associated HIF-2alpha mutations demonstrate a critical role for residues C-terminal to the hydroxylacceptor proline. J Biol Chem. 2009;284:9050-9058.
29 Percy ML. Genetically heterogeneous origins of idiopathic erythrocytosis. Hematology. 2007;12:131-139.
30 Al-Sheikh M, Mazurier E, Gardie B, et al. A study of 36 unrelated cases with pure erythrocytosis revealed three new mutations in the erythropoietin receptor gene. Haematologica. 2008;93:1072-1075.
31 Percy MJ, Lee FS. Familial erythrocytosis: molecular links to red blood cell control. Haematologica. 2008;93:963-967.
32 Hadland BK, Longmore GD. Erythroid-stimulating agents in cancer therapy: potential dangers and biologic mechanisms. J Clin Oncol. 2009;27:4217-4226.
33 Jelkmann W. Erythropoiesis stimulating agents and techniques: a challenge for doping analysts. Curr Med Chem. 2009;16:1236-1247.
34 Stergiopoulos K, Brennan JJ, Mathews R, et al. Anabolic steroids, acute myocardial infarction and polycythemia: a case report and review of the literature. Vasc Health Risk Man. 2008;4:1475-1480.
35 Marinella MA. Hematologic abnormalities following renal transplantation. Int Urol Nephrol. 2009. [Epub ahead of print]
36 Garewal G, Ahluwalia J, Kumar V, et al. The utility of bone marrow examination in renal transplantation: nine years of experience from north India. Transplantation. 2006;81:1354-1356.
37 Vlasveld LT, de Wit CW, Verweij RA. Myomatous erythrocytosis syndrome: further proof for the pathogenic role of erythropoietin. Neth J Med. 2008;66:283-285.
38 Shiramizu M, Katsuoka Y, Grodberg J, et al. Constitutive secretion of erythropoietin by human renal adenocarcinoma cells in vivo and in vitro. Exp Cell Res. 1994;215:249-256.
39 Burk JR, Lertora JJ, Martinez IRJr, Fisher JW. Renal cell carcinoma with erythrocytosis and elevated erythropoietic stimulatory activity. South Med J. 1977;70:955-958.
40 Hama Y, Kaji T, Ito K, et al. Erythropoietin-producing renal cell carcinoma arising from autosomal dominant polycystic kidney disease. Br J Radiol. 2005;78:269-271.
41 Blake-James B, Attar KH, Rabbani S, et al. Secondary polycythaemia associated with unilateral renal cystic disease. Int Urol Nephrol. 2007;39:955-958.
42 Godeau P, Bletry O, Brochard C, Hussonois C. Polycythemia vera and primary hyperparathyroidism. Arch Intern Med. 1981;141:951-953.
43 Drenou B, Le Tulzo Y, Caulet-Maugendre S, et al. Pheochromocytoma and secondary erythrocytosis: role of tumour erythropoietin secretion. Nouv Rev Fr Hematol. 1995;37:197-199.
44 Vik A, Cui G, Isaksen V, Wik T, Hansen JB. Erythropoietin production by a hepatic adenoma in a patient with severe erythrocytosis. Acta Haematol. 2009;121:52-55.
45 Matsuyama M, Yamazaki O, Horii K, et al. Erythrocytosis caused by an erythropoietin-producing hepatocellular carcinoma. J Surg Oncol. 2000;75:197-202.
46 Trimble M, Caro J, Talalla A, Brain M. Secondary erythrocytosis due to a cerebellar hemangioblastoma: demonstration of erythropoietin mRNA in the tumor. Blood. 1991;78:599-601.
47 Bruneval P, Sassy C, Mayeux P, et al. Erythropoietin synthesis by tumor cells in a case of meningioma associated with erythrocytosis. Blood. 1993;81:1593-1597.
48 Thiele J. Is it justified to perform a bone marrow biopsy examination in sustained erythrocytosis? Cur Hematol Malig Rep. 2006;1:87-92.
49 Thiele J, Kvasnicka HM, Diehl V. Bone marrow features of diagnostic impact in erythrocytosis. Ann Hematol. 2005;84:362-367.
50 Tutaeva V, Misurin AV, Michiels JJ, et al. Application of PRV-1 mRNA expression level and JAK2-V617F mutation for the differentiating between polycythemia vera and secondary erythrocytosis and assessment of treatment by interferon or hydroxyurea. Hematol. 2007;12:473-479.
51 Vannucchi AM, Guglielmelli P, Ayalew Tefferi A. Advances in understanding and management of myeloproliferative neoplasms. CA Cancer J Clin. 2009;59:171-191.
52 Campbell P, Scott LM, Buck G. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2-V617F mutation status: a prospective study. Lancet. 2005;366:1945-1953.
53 Kvasnicka HM, Thiele J. Classification of Ph-negative chronic myeloproliferative disorders: morphology as the yardstick of classification. Pathobiology. 2007;74:63-71.
54 Gianelli U, Iurlo A, Vener C, et al. Early prepolycythemic phase of PV. The significance of bone marrow biopsy and JAK2-V617F Mutation in the differential diagnosis between the ‘early’ prepolycythemic phase of polycythemia vera and essential thrombocythemia. Am J Clin Pathol. 2008;130:336-342.
55 Kvasnicka HM, Thiele J. Prodromal myeloproliferative neoplasms: The 2008 WHO classification. Am J Hematol. 2010;85:62-69.
56 Barosi G, Mesa RA, Thiele J, et al. Proposed criteria for the diagnosis of post-polycythemia vera and post-essential thrombocythemia myelofibrosis: a consensus statement from the International Working Group for Myelofibrosis Research and Treatment. Leukemia. 2008;22:437-438.
57 Thiele J, Kvasnicka HM, Facchetti F, et al. European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica. 2005;90:1128-1132.
58 Manoharan A, Horsley R, Pitney WR. The reticulin content of bone marrow in acute leukaemia in adults. Br J Haematol. 1979;43:185-190.
59 Kralovics R. Genetic complexity of myeloproliferative neoplasms. Leukemia. 2008;22:1841-1848.
60 Andrieux J, Demory JL, Caulier MT, et al. Karyotypic abnormalities in myelofibrosis following polycythemia vera. Cancer Genet Cytogenet. 2003;140:118-123.
61 Andrieux JL, Demory JL. Karyotype and molecular cytogenetic studies in polycythemia vera. Curr Hematol Rep. 2005;4:224-229.
62 Tefferi A, Skoda R, Vardiman JW. Myeloproliferative neoplasms: contemporary diagnosis using histology and genetics. Nat Rev Clin Oncol. 2009;6:627-637.
63 Theocharides A, Boissinot M, Girodon F, et al. Leukemic blasts in transformed JAK2-V617F-positive myeloproliferative disorders are frequently negative for the JAK2-V617F mutation. Blood. 2007;1(110):375-379.
64 Naresh KN, Lampert IA. CD117 Expression as an aid to identify immature myeloid cells and foci of ALIP in bone marrow trephines. Am J Hematol. 2006;81:79.
65 Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007;356(5):459-469.
66 Pardanani A, Lasho TL, Finke C, et al. Prevalence and clinicopathological correlates of JAK2 exon 12 mutations in JAK2V617F-negative polycythemia vera. Leukemia. 2007;21:1960-1963.
67 Rumi E. Familial chronic myeloproliferative disorders: the state of the art. Hematol Oncol. 2008;26:131-138.
68 Skoda R. The genetic basis of myeloproliferative disorders. Hematology Am Soc Hematol Educ Program. 2007:1-10.
69 Percy MJ, Scott LM, Erber WN, et al. The frequency of JAK2 exon 12 mutations in idiopathic erythrocytosis patients with low serum erythropoietin levels. Haematologica. 2007;92:1607-1614.
70 McMullin MF. Idiopathic erythrocytosis: a disappearing entity. Hematology Am Soc Hematol Educ Program. 2009:629-635.
[/level-membership-for-pathology-category][not-level-membership-for-pathology-category]
CHAPTER 22 Erythrocytosis and polycythemia
Introduction
The term ‘erythrocytosis’ is derived from Greek words meaning ‘too many red cells’ and should be distinguished from ‘polycythemia’, meaning ‘too many cells in the blood’.1 Erythrocytosis has been defined as a greater than two standard deviation-increase from the age-, sex- and race-adjusted norm in hematocrit or hemoglobin level.2 It is clear, however, that these laboratory parameters may be affected by decreases in plasma volume. Therefore, a clinical diagnosis of ‘erythrocytosis’ might represent true erythrocytosis, indicating a true increase in red cell mass (RCM), or in fact apparent polycythemia, resulting from either reduced plasma volume (relative polycythemia) or failure to recognize otherwise normal values for hematocrit (Hct) or hemoglobin (Hgb) level that lie in the extreme right tail of the Gaussian distribution.3 Hence, for an individual patient, interpretation of laboratory results without knowledge of the personal baseline value remains inaccurate because of the inevitable statistical overlap of extreme values between subjects with and without disease.3
The recent development of molecular tests for the JAK2V617F and MPL mutations (see Chapter 21 for details) has allowed the reliable distinction of clonal from non-clonal erythrocytosis and has radically altered the investigation of erythrocytosis. Furthermore the value of aggressive phlebotomy to lower Hct levels below 45% in men and 42% in women has not been substantiated.3 The therapeutic relevance of distinguishing polycythemia vera (PV) from so-called ‘essential thrombocythemia (ET) with borderline increased Hct’ has diminished and has further undermined the value of RCM measurement, which is no longer fundamental to the diagnosis of many patients.3
In this chapter, the recent revision of the WHO criteria,4 which reflects developments in molecular pathogenesis and international consensus on clinico-pathological classification, is used for definition purposes although it is recognized that dissenting views have been published.5 Erythrocytosis is therefore practically defined according to the thresholds used in the WHO classification,3,4 i.e.:
The classification of erythrocytosis has recently been the subject of two excellent reviews.3,6 The term ‘idiopathic erythrocytosis’ was used by McMullin6 but has been criticized by Patnaik and Tefferi3 as an entity, due to misuse of the term for patients who have an inappropriate diagnosis of erythrocytosis or who have been inadequately investigated. Nevertheless, there remain patients who have been fully investigated and who are likely to have abnormalities that have not as yet been defined. These may include defects of the erythropoietin (Epo) signaling pathway or oxygen sensing pathway.3,6 Therefore, the category ‘unclassifiable’ is introduced here in accordance with the WHO classification and strictly defined for patients who have been fully investigated and for whom no currently defined cause of the erythrocytosis has been found. Patients who are partially investigated should not be placed in this category. While there are several potential divisions of a classification of patients with proven erythrocytosis, the classification used here (Box 22.1) is adapted from Patnaik and Tefferi.3
The pathology of erythrocytosis
The discovery of the JAK2V617F mutation in 20057–10 (see Chapter 21) has profoundly changed both understanding and investigation of the erythrocytoses. Accordingly, modern diagnostic evaluation of a patient with proven erythrocytosis, and for whom no obvious cause is apparent, may begin with screening of the peripheral blood for the JAK2V617F mutation and serum Epo. A working classification of erythrocytosis into primary, or clonal, i.e. PV, versus secondary has therefore emerged,6 but the more classical pathogenetic approach is used here.
Congenital erythrocytosis
Associated with reduced P50
High-oxygen-affinity hemoglobinopathy
High-oxygen-affinity hemoglobins release oxygen at a lower rate than normal and thus create relative tissue hypoxia, which might result in compensatory erythrocytosis in approximately one third of affected patients. Affected patients often present with isolated erythrocytosis, in the absence of signs and symptoms of systemic disease.3 Erythrocytosis is accompanied by chronic hemolysis where the Hgb variant is unstable.6 Bone marrow trephine biopsy (BMTB) investigation shows erythroid hyperplasia but normal megakaryocytes (Fig. 22.1A, B).These patients may have family members who are similarly affected and a family history is therefore essential in the investigation of patients with erythrocytosis. Transmission in affected patients is usually autosomal dominant.
More than 90 mutations have been described and are covered in an exemplary fashion in a web resource: http://globin.bx.psu.edu/hbvar/. Most of the high-oxygen-affinity mutations involve the β-globin chain and α1β2 contact zones.11 Serum Epo levels are either normal or elevated and P50 (partial pressure of oxygen at which 50% of Hgb is saturated with oxygen) is decreased.12 Structurally abnormal high-affinity Hgb should be suspected3 if the P50 level is <20 mmHg.
2,3-Bisphosphoglycerate mutase (BPGM) deficiency
BPGM deficiency is a rare cause of erythrocytosis.13,14 Deficiency of the enzyme results in a high affinity Hgb with a left shifted oxygen dissociation curve. This results in a compensatory erythrocytosis. Patients with both autosomal dominant15 and autosomal recessive inheritance16 have been described. In a fully penetrant autosomal recessive case, there is an isolated erythrocytosis with normal serum Epo level.16 Diagnosis is established by showing a low P50, a normal Hgb structure and decreased BPGM activity.3
Methemoglobinemia
Congenital methemoglobinemias are of three main types:17,18
Methemoglobin causes both an impaired O2 binding and increased oxygen affinity of the Hgb; sometimes resulting in compensatory erythrocytosis.18
Methemoglobinemia is clinically suspected when cyanosis is accompanied by normal PaO2 levels but low saturation per pulse oximeter. Both carboxyhemoglobin and methemoglobin may be measured by modern oximeters.3
Associated with normal P50
The involvement of oxygen-sensing pathways in physiologic and pathologic erythropoiesis has recently been reviewed.20 Red blood cells deliver O2 from the lungs to the tissues. Better understanding of the physiological regulation of the process has allowed a rational classification of rare cases of congenital erythrocytosis to be developed (Fig. 22.2).3,6
A classic physiologic response to hypoxia in humans is the up-regulation of the Erythropoietin (Epo) gene, which is the central regulator of red blood cell mass. Reduction of tissue oxygenation triggers increased production of Epo by hypoxia-inducible factor 1 (HIF-1), which is a transcriptional activator composed of an O2-regulated α subunit and a constitutively expressed β subunit. Hydroxylation of HIF-1α or HIF-2α by the asparaginyl hydroxylase FIH-1 blocks coactivator binding and transactivation. Hydroxylation of HIF-1α or HIF-2α by the prolyl hydroxylase PHD2 is required for binding of the von Hippel–Lindau (VHL) protein, leading to ubiquitination and proteasomal degradation. Mutations in the genes encoding VHL, PHD2, and HIF-2α have all been identified in patients with familial erythrocytosis.20
VHL mutations including Chuvash erythrocytosis (frequently termed Chuvash polycythemia)
Chuvash erythrocytosis is a rare, autosomal recessive, congenital erythrocytosis, first described in the Chuvash autonomous region in Russia21 but also occurring in other racial and ethnic groups.22,23 Affected patients are homozygous for a germline mutation affecting the VHL tumor suppressor gene, producing an abnormal VHL protein.24,25 The mutation in the VHL gene disrupts the normal mechanism of hypoxia sensing, ultimately resulting in increased Epo production and erythrocytosis.6
In contrast to patients with the VHL disease, an autosomal dominant familial syndrome, patients with Chuvash erythrocytosis do not display an increased predilection for tumors.3 However, other abnormalities are found.26 Plasma concentrations of endothelin-1, Epo, plasminogen activator inhibitor-1, transferrin, transferrin receptor, and vascular endothelial growth factor are elevated. Clinical manifestations include increased cardiac valvular abnormalities, hemangiomas, pulmonary arterial hypertension, thrombotic and hemorrhagic events, varicose veins, and shortened life span. In parallel, peripheral blood concentrations of CD4 positive T-helper cells and CD4/CD8 ratio have been found to be lower in the VHL598C>T homozygotes.26
Prolyl hydroxlase domain 2 (PHD2) mutations
Under normoxic conditions, PHD2 hydroxylates the α-subunits of HIF proteins, facilitating VHL binding and subsequent ubiquitin-mediated proteasomal degradation of HIF.3,6 Rare cases of PHD2 mutations have been described, with the loss of PHD2 function, associated erythrocytosis and normal serum Epo.3,6 Recently, a novel PHD2 mutation has been described, associated with both erythrocytosis and recurrent paraganglioma.27
Hypoxia inducible factor alpha (HIF2α) mutations
Several erythrocytosis-associated HIF2α mutations have been characterized. All result in impaired degradation and thus aberrant stabilization of HIF2α. However, each exhibits a distinct profile with respect to their effects on PHD2 binding and VHL interaction.28 Epo levels are usually elevated.3,6
Epo receptor (Epo-R) mutations
Epo-R signaling is regulated by the binding of the protein tyrosine phosphatase SHP-1 (or other JAK/STAT regulators) to the distal cytoplasmic region of Epo-R. This interaction results in the down-regulation of the Epo-mediated activation of the JAK2/STAT5 pathway and ultimately production of more red cells.3,6 Epo-R mutations (all in exon 8) that cause erythrocytosis have been reviewed.29–31 These mutations often result in cytoplasmic truncation of Epo-R, resulting in failure of attachment of SHP-1, ongoing production of red cells and thus erythrocytosis.
Transmission is usually autosomal dominant. Affected patients are usually asymptomatic and display subnormal (or normal) serum Epo level and hypersensitivity of erythroid progenitors to exogenous Epo.3,6
