Chapter 61 Epilepsy Surgery in the Pediatric Population
Epilepsy is one of the most common chronic disorders facing children and adolescents. The overall prevalence of epilepsy has been estimated to be 5–8 per 1000 [Hauser et al., 1975, Hauser, 1994, 1996, 1998; Olafsson et al., 1996; Osuntokun et al., 1987]. Extrapolating the Hauser data from Rochester, Minnesota, of 6.66 per 1000 to the total population in 2004, approximately 2.3 million persons in the United States have epilepsy [Hauser et al., 1991]. In children, there are approximately 10.5 million worldwide with epilepsy. The annual incidence of epilepsy in children is reported to be 61–124 per 100,000 in developing countries and 41–50 per 100,000 in developed countries [Guerrini, 2006]. The cumulative risk of developing epilepsy from birth through adolescence is 1 percent [Hauser et al., 1991, 1994]. Unfortunately, only 60–70 percent of patients will achieve seizure freedom with antiepileptic medications [Kwan and Brodie, 2000; Mohanraj and Brodie, 2006]. The introduction of several new antiepileptic drugs over the past 15 years has not changed the fact that approximately 30–40 percent of patients with epilepsy will be medically refractory [Perucca et al., 2007; Mohanraj and Brodie, 2006].
In addition, the longer epilepsy persists without control, the less likely is the chance of remission. Specifically, if seizures remain inadequately controlled for longer than 4 years, the chance of remission decreases to approximately 10 percent [Annegers et al., 1979]. Seizure duration of over 10 years also decreases the likelihood of achieving control in patients who undergo surgery. The presence of multiple seizure types and frequent generalized tonic-clonic seizures also lessens the chance for complete remission. As stated above, 30–40 percent [Mohanraj and Brodie, 2006] of all persons with epilepsy will be intractable. There are approximately 20,000 cases of new-onset epilepsy annually. Therefore, 6000–8000 cases of new-onset epilepsy are intractable each year. In the United States, the number of patients with drug-resistant epilepsy – adult and pediatric – is estimated to be 700,000, a higher number than the number of individuals affected with Parkinson’s disease and multiple sclerosis combined [Hiritiz et al., 2007]. Approximately 60 percent of these patients will have partial seizures. Estimates by several investigators suggest that many of these patients are epilepsy surgery candidates [Hauser, 1993; Unnwongse et al., 2010]. This figure may increase as new technologies enable more precise identification of an underlying epileptogenic focus. Additionally, as medical intractability for children with epilepsy is further defined, the number of pediatric epilepsy surgical procedures will probably increase. However, currently, epilepsy surgery is underutilized in the treatment of intractable epilepsy. In fact, over the past 15 years, the mean duration of epilepsy before referral to a tertiary care epilepsy center for evaluation for epilepsy surgery has been over 20 years [Engel et al., 2003; Unnwongse et al., 2010; Choi et al., 2009].
The developing brain is highly susceptible to recurrent seizures. Until recently, the brain was believed to be relatively resistant to injury. A growing body of evidence in animal models, however, suggests that early seizures, even if brief and recurrent, can result in demonstrable structural and physiologic changes in the developing brain’s circuitry, resulting in aberrant excitation and inhibition. Clinically, these defects produces spontaneous seizures (epilepsy) and cognitive impairments [Holmes and Ben-Ari, 1998; Holmes et al., 1998; Stafstrom et al., 2000; Galanopoulou and Moshé, 2009], with the possibility of missed windows of developmental opportunity. Thus, plasticity of the brain in a young infant and child is a “double-edged sword”. It protects the brain from the neurologic consequences of destructive lesions and status epilepticus; however, recurrent seizures, or even frequent interictal epileptiform discharges, in this age group can produce permanent abnormal neuronal circuitry, resulting in long-term developmental delays and continued, intractable seizures [Holmes and Lenck-Santini, 2006]. In addition, chronic uncontrolled epilepsy in infants and children poses a significant risk for emotional, behavioral, social, cognitive, and family dysfunction. Population studies have demonstrated that epilepsy reduces life expectancy, and poorly controlled seizures further increase the risk of death in children and adults [Nashef et al., 1995; Hitiris et al., 2007]. In population-based studies, the estimated risk of sudden unexpected death in epilepsy (SUDEP) is estimated to be between 1:500 and 1:1000 per year. For those with uncontrolled epilepsy, the rate of SUDEP is approximately 1:200 per year. [Harvey et al., 1993c; Hauser et al., 1980; Meyer et al., 2010; So et al., 2009]. It should be noted that patients being evaluated for epilepsy surgery have the highest risk of SUDEP, estimated at between 2.2 and 9.3 per 1000 patient-years [Hiritiz et al., 2007].
Several studies have examined outcomes after epilepsy surgery. A majority of these studies has been performed in older adolescents and adults. The focus of these studies has been primarily on the improvement of seizure control, with 64–69 percent of patients having seizure-free outcomes [Engel, 1996; Wyllie, 1998; Wyllie et al., 1998]. These studies have placed less emphasis on improvement in quality of life – for example, enhancement of self-image, improvement in academic performance and psychosocial functioning, and increased independence in activities of daily living [Spencer, 1996; Taylor et al., 1997]. An increasing number of long-term follow-up studies have concentrated on infants and young children. Malformations of cortical development are the most frequently cited pathologic abnormalities in pediatric surgical patients [Duchowny et al., 1996; Wyllie et al., 1998; Harvey et al., 2008; Zupanc et al., 2010]. The overall outcome of pediatric epilepsy surgery in young infants and children is roughly comparable to that in the older adolescent and adult population. In infants, 61–65 percent have seizure-free outcomes [Chugani et al., 1993; Duchowny et al., 1998; Sinclair et al., 2003; Wyllie et al., 1996, 1998; Zupanc et al., 2010]. In young children, the rate of seizure-free outcomes varies, ranging from 58 to 74 percent [Paolicchi et al., 2000; Sinclair et al., 2003; Wyllie et al., 1998; Cossu et al., 2008; Kan et al., 2008; Kim et al., 2008; Zupanc et al., 2010]. The etiology of the epilepsy appears to play the major role in determining prognosis, regardless of location (temporal versus extratemporal). In one study, children with malformations of cortical development had a seizure-free outcome of 58 percent, whereas patients with other pathologies had a 77 percent seizure-free outcome. Temporal lobectomies were more commonly performed in our older adolescent patients, consistent with other studies, and demonstrated a seizure-free outcome of 84 percent. Patients who had modified lateral hemispherectomies also demonstrated a high seizure-free outcome (i.e., approximately 100 percent), even those with cortical dysplasias [Zupanc et al., 2010].
Several studies have reported on cognitive function after surgery in children who have undergone temporal lobectomy (predominantly older children and adolescents) [Gillam et al., 1997; Gleissner et al., 2002; Mabbott and Smith, 2004; Meyer et al., 1986; Szabo et al., 1998; Westerveld et al., 2000; Lah, 2004]. These studies have generally found that memory and intelligence are unchanged. Some reports note a decline in verbal memory and improvements in language, attention, and memory, while other studies have reported that good seizure outcomes have been associated with an increase in IQ [Lah, 2004]. Reports on the cognitive effects of extratemporal resections have been relatively few, in part because of the young age of those having surgery. In a follow-up study of 24 children operated on before 3 years of age, younger age at surgery was correlated with an improvement in developmental quotients [Loddenkemper et al., 2007]. In patients who have undergone successful frontal lobe resection, outcomes include improvements in attention and concentration but no change in executive functions, manual coordination, and language [Blanchette and Smith, 2002; Lendt et al., 2002]. Studies on postoperative psychosocial functioning in children have been relatively rare, almost exclusively retrospective, and based on subjective measures. In children who have undergone temporal lobectomies, studies indicate improvement in behavior, mood, and self-esteem, with the changes linked to improvement in seizure control [Danielsson et al., 2002; Davidson and Falconer, 1975; Duchowny et al., 1992; Meyer et al., 1986; Zupanc et al., 2010]. Of children who have undergone extratemporal resections, improved social behavior was found in about 50 percent [Adler et al., 1991]. Other retrospective studies that combine temporal and extratemporal cases suggest that reduction of seizure frequency, although not necessarily complete elimination of seizures, resulted in improved family life, socialization and behavior, and quality of life [Adler et al., 1991; Keene et al., 1998; Lendt et al., 2002; Mihara et al., 1994; Smith et al., 2004; Whittle et al., 1981; Zupanc et al., 1996, 2010]. Other reports indicate that a reduction in seizures may result in a favorable and significant improvement in the quality of life, with behavioral and developmental “catch-up” progress [Asarnow et al., 1997; Bourgeois et al., 1999; Chugani et al., 1990b; Duchowny et al., 1990, 1992; Wyllie et al., 1996; Jonas et al., 2004; Zupanc et al., 2010]. More recent studies indicate that shorter seizure durations and earlier surgical intervention result in better seizure outcome and quality of life [Jonas et al., 2004; Loddenkemper et al., 2007; Zupanc et al., 2010].
Historical Background
Epilepsy has always been a part of human existence. A generalized tonic-clonic seizure was first described in Akkadian, the oldest written language, more than 3000 years ago [Goldensohn et al., 1997]. Since that time, many descriptions of epilepsy appear in literature, including the Bible. In “On the Sacred Disease”, written in the 5th century, Hippocrates stated that epilepsy was a brain disease caused by an excess of phlegm that resulted in an abnormal brain consistency. He proposed diet and drugs as therapy [Scott, 1993]. Until the late 19th century, the treatment of epilepsy was surrounded by superstition, exorcism, magic, and alchemy. Caton’s (1842–1926) discovery in 1875 of spontaneous electrical activity of the brain and evoked potentials suggested that seizures might be the result of aberrant electrical activity in the brain [Caton, 1875].
The first effective treatment for epilepsy was potassium bromide, introduced in 1857 [Locock, 1857]. In 1886, Horsley performed the first epilepsy surgery on a patient with intractable post-traumatic epilepsy. Several decades later, the antiepileptic drug phenobarbital was introduced in 1912, followed by phenytoin in 1937. Epilepsy surgery did not advance until 1950, when Penfield published his article on 70 cases of temporal lobectomy [Flanigin et al., 1991; Penfield and Flanigin, 1950]. His neurosurgical career was devoted to the study of seizure semiology (i.e., clinical description) and its correlation with the brain cortex. He used cortical mapping and stimulation in much the same way in which it is used today. He also recognized the substrates of epilepsy, particularly trauma and infection. His seminal clinical research has been instrumental in guiding the hands of contemporary epileptologists and neurosurgeons interested in the surgical approach to epilepsy.
Epilepsy surgery was not regarded as a conventional treatment for intractable epilepsy until recently. In the past 30 years, dramatic improvements in brain imaging that identify specific anatomic substrates of epilepsy have sparked renewed interest in epilepsy surgery. Temporal lobectomy with amygdalohippocampectomy has become the standard of care in adult patients with intractable epilepsy emanating from the temporal lobe. The surgical success rate for a seizure-free outcome in these carefully selected patients approaches 80–90 percent [Duchowny et al., 1992; King et al., 1986; Penfield and Flanigin, 1950]. In a randomized, controlled trial of surgery for temporal lobe epilepsy compared to treatment with antiepileptic drugs, 58 percent of patients demonstrated seizure freedom after 1 year compared to only 8 percent of patients treated with medications [Wiebe et al., 2001]. Unfortunately, surgical success does not necessarily translate to an improved quality of life. The accumulation of years of low self-esteem, loss of independence, poor peer relations, and academic failure, coupled with high financial costs, often without benefit of full insurance coverage, translates to continued lack of employment and depression [Reeves et al., 1997]. The lifetime cost of epilepsy for an estimated 181,000 people with onset in 1995 is projected at $11.1 billion, and the annual cost for the estimated 2.3 million people with epilepsy is estimated at $12.5 billion [Bazil, 2004; Begley et al., 1994, 2000; Hathaway et al., 1995]. In one recent study, the calculated total aggregated annual economic impact of epilepsy on the U.S. economy was $9.6 billion in direct medical costs. This analysis did not consider the indirect costs of loss of productivity, quality of life, and comorbidities [Yoon et al., 2009]. With respect to children, it is estimated that the annual cost of medical care for a child with epilepsy is $6379, compared to $1032 for peers without epilepsy [Yoon et al., 2009]. Indirect costs probably account for 80–85 percent of the total costs, and include delayed or missed educational opportunities, psychiatric and social service needs, and lost employment. The direct costs of epilepsy are concentrated in the patients with intractable epilepsy. The growing recognition of the real costs of epilepsy – medical, psychological, educational – has led to increased interest in the early identification of children who might benefit from epilepsy surgery [Jalava et al., 1997]. In older children and adolescents, temporal lobectomies are common epilepsy surgical procedures [Harvey et al., 2008]. In younger children and infants, however, extratemporal resections, including multilobar resections and hemispherectomies, are the more typical procedures [Harvey et al., 2008].
In recent studies, it has been shown that the costs of epilepsy surgery are offset by a decline in health-care costs after successful surgery. One study documented that the total costs for adult patients who were seizure-free following epilepsy surgery declined by 32 percent by 2 years following surgery. In the 24 months after surgery, epilepsy-related costs were $2068–2094 in patients with persisting seizures, as opposed to $582 in patients who were seizure-free following surgery [Langfitt et al., 2007]. In addition, in adult patients, it appears that epilepsy surgery increases quality-adjusted life expectancy by 7.5 years [Choi et al., 2008]. In the pediatric population, additional benefits of epilepsy surgery appear to be improved long-term developmental outcomes and quality of life [Loddenkemper et al., 2007; Zupanc et al., 2010].
Indications for Epilepsy Surgery
Criteria have been proposed for referral and evaluation of children for epilepsy surgery, although there is currently insufficient class I evidence to produce a practice guideline [Cross et al., 2006]. Practice guidelines for temporal lobe and localized neocortical resections for epilepsy have been proposed for adults [Engel et al., 2003]. In determining whether a child is a candidate for epilepsy surgery, several key issues must be considered. The decision-making task must take into account the following:
 Failure of two or three antiepileptic medications in achieving complete seizure control in a child or adolescent.
Failure of two or three antiepileptic medications in achieving complete seizure control in a child or adolescent.


The proper classification of seizure type and epilepsy syndrome is crucial in the determination of whether or not a patient is an appropriate epilepsy surgery candidate [Aicardi, 1994; Holmes, 1993]. The benign seizure disorders, such as benign rolandic epilepsy or benign epilepsy with centrotemporal spikes, must be recognized. With rare exceptions, these syndromes usually are easily treated, and affected patients do not present to tertiary epilepsy centers.
Children with Sturge–Weber syndrome who have frequent, medically refractory seizures accompanied by progressive hemiparesis and cognitive impairment should be evaluated promptly for hemispherectomy [Thomas-Sohl et al., 2004; Vining et al., 1997]. Clinical outcome studies indicate that early surgical resection can result in the elimination of seizures, improvement in cognitive abilities, and overall improvement in quality of life, despite hemiparesis and visual field defect as residual neurologic deficits [Erba and Cavazzuti, 1990; Hoffman et al., 1979; Ogunmekan et al., 1991; van Empelen et al., 2004].
Children with hemimegalencephaly, a unilateral or focal malformation of cortical development, can present in infancy with multiple daily seizures, developmental stagnation or decline, and hemiparesis. Hemispherectomy provides relief from seizures (especially in those patients with unilateral epileptiform abnormalities) and improved developmental outcome [Andermann et al., 1993; Vigevano and DiRocco, 1990; Vigevano et al., 1989; Jonas et al., 2004]. The patients with symptomatic infantile spasms who have underlying focal cortical dysplasias, usually temporal-parietal-occipital, should be considered for early focal cortical resection. University of California at Los Angeles investigators have provided the seminal clinical research in this area and have documented a significant improvement in seizure control and enhanced developmental gains following epilepsy surgery, greater than would have been predicted using the natural history of infantile spasms as a comparison [Asarnow et al., 1997; Chugani et al., 1990a, 1993; Duchowny et al., 1990].
Rasmussen’s encephalitis is characterized by intractable focal motor seizures, often evolving into epilepsia partialis continua, cognitive decline, and progressive hemiparesis. Recent findings of glutamate receptor antibodies in some patients with Rasmussen’s encephalitis implicate a possible autoimmune pathophysiology [Antel and Rasmussen, 1996; Pardo et al., 2004; Rogers et al., 1994]. Although initial trials of intravenous immunoglobulin and plasmapheresis have been encouraging, long-term studies have not confirmed efficacy [Andrews et al., 1996; Hart et al., 1994; Krauss et al., 1996]. Therefore, the only definitive treatment for Rasmussen’s encephalitis remains hemispherectomy [van Empelen et al., 2004; Jonas et al., 2004].
In addition to those with the catastrophic epilepsies of infancy and childhood, all children with tumors and concomitant localization-related epilepsy should be considered for early surgical intervention. Compelling reasons for such intervention exist. Most tumors need to be biopsied or excised. Additionally, although the tumors associated with epilepsy usually are slow-growing, cortical, and well circumscribed, some tumors, especially astrocytomas, are not necessarily benign and can undergo malignant change [Jack, 1995]. Without resection, the natural history of these tumor-associated epilepsy syndromes is one of continued seizures with little hope of remission. Antiepileptic drugs produce side effects that can affect cognitive function and behaviors, with concomitant impact on psychosocial development [Meador, 2002] Examples of tumors that usually are easily resectable are the gangliogliomas and dysembryonic neuroectodermal tumors, which have a predilection for the temporal lobe (Figure 61-1) [Duchowny et al., 1992; Tice et al., 1993; Vali et al., 1993].
Children with other types of lesional symptomatic localization-related epilepsy also should be considered as epilepsy surgery candidates. Common substrates of epilepsy include encephalomalacias, vascular malformations, tubers, and malformations of cortical development [Harvey et al., 2008; Zupanc et al., 2010].
Patients who have generalized epilepsy syndromes may also be candidates for epilepsy surgery. The presence of generalized or multifocal epileptiform discharges on surface EEG monitoring should not necessarily exclude someone from epilepsy surgery. Children with generalized or multifocal epilepsy should be considered for epilepsy surgery, if data suggest an underlying focal generator for the epileptic condition [Wyllie, 1995; Wyllie et al., 2007; Zupanc et al., 2010]. Specifically, in the presence of a lesion on magnetic resonance imaging (MRI), the epilepsy syndrome is most likely to be due to a symptomatic localization-related epilepsy with rapid secondary bisynchrony. The mechanisms of underlying generalized epileptiform discharges in focal cerebral lesions can be seen as a form of “maladaptive plasticity” of the immature brain, whereby the lesions in the immature neural network of the young brain permanently alter the neural circuitry, producing spontaneous hypersynchrony and generalized discharges [Sutula, 2004]. These generalized discharges may also involve the thalamocortical network, resulting in generalized rhythmic discharges [Van Hirtum-Das et al., 2006]. Approximately 10–15 percent of children with Lennox–Gastaut syndrome, one of the most common symptomatic “generalized” epilepsy syndromes, have underlying focal malformations of cortical development and should be evaluated carefully for epilepsy surgery. In one study, children who had generalized discharges and focal lesions had identical seizure-free outcomes (72 percent seizure-free) to children with similar lesions and ipsilateral focal epileptiform discharges [Wyllie et al., 2007]. In addition, children with intractable epilepsy who have tonic-atonic seizures associated with generalized spikes/polyspikes (usually Lennox–Gastaut syndrome) and no identifiable lesion on neuroimaging may respond to a complete corpus callosotomy, a palliative treatment that can have a significant impact on quality of life [Wyllie et al., 1993; Zupanc et al., 2010]. An alternative approach is that of a multistaged epilepsy surgery, initially performing a complete corpus callosotomy, then placing lateralizing strip electrodes with the hope of identifying epileptogenic cortex that can be resected. In one study of 14 patients undergoing this approach, 9 went on to have a focal cortical resection and 5 out of 9 (56 percent) were seizure-free [Zupanc et al., 2011).
Patients with tuberous sclerosis and medically refractory, symptomatic, localization-related epilepsy should also be considered for epilepsy surgery. In patients with multiple tubers, emerging neuroimaging techniques, particularly interictal α-[11C]methyl-l-tryptophan (AMT) positron emission tomography (PET) scans, offer promise in identifying the most highly epileptogenic tuber [Asano et al., 2000; Chugani et al., 1998; Juhasz et al., 2003]. If the presurgical evaluation points to a specific tuber, studies have shown that it can be successfully removed, with a significant improvement in seizure control [Bebin et al., 1993; Koh et al., 2000; Romanelli et al., 2004]. There is also precedent for removing multiple tubers in the brain, as several tubers may be producing medically refractory seizures [Weiner et al., 2006].
In summary, factors that favor early intervention with epilepsy surgery include the following:
Even if the lesion is outside the temporal lobe, in carefully selected patients, epilepsy surgery can generally be performed with little risk of neurologic sequelae and a high rate of surgical success [Britton et al., 1994; Cascino et al., 1990, 1992, 1993, 1994; Montes et al., 1995; Paolicchi et al., 2000; Zupanc et al., 2010].
The suspected central nervous system pathology, based on MRI, may also have an impact on whether or not a patient is an epilepsy surgery candidate. For example, patients with low-grade tumors, infarctions, and mesial temporal sclerosis are likely to undergo remission of their epilepsy and typically have an excellent seizure-free outcome. If the epilepsy is temporal in onset and pathologic features include associated hippocampal formation atrophy or mesial temporal sclerosis, the surgical success rate approaches 80–90 percent [Cascino et al., 1993; Sinclair et al., 2001] (Figure 61-2). Data from our own retrospective study and from the work of Palmini and co-workers suggest that malformations of cortical development, no matter where the cortical location, carry a significant risk for status epilepticus and intractability [Laoprasert et al., 1997; Palmini et al., 1997]. Epilepsy surgery in patients with underlying malformations of cortical development have a lower seizure-free outcome, but it is still close to 60 percent, which is significantly better than additional trials of antiepileptic medication. Patients with mild malformations of cortical development, such as focal cortical dysplasia (FCD) type 1a, fared better than patients with more severe malformations of cortical development, such as FCD type 2a [Fauser et al., 2004]. In addition, the patients with known central nervous system lesions can be identified early, with detailed MR imaging.
Preoperative Evaluation
Techniques and Technologies
Seizure Semiology
Seizure semiology can provide insightful clues to the lateralization and localization of the underlying epileptogenic focus. The presence of versive head movements, unilateral motor clonic activity, and eye deviation may constitute critical lateralizing information. Specifically, versive head movements indicate that the epileptogenic zone resides in the contralateral hemisphere. In similar fashion, seizures consisting of olfactory or gustatory hallucinations, followed by complex motor automatisms and staring unresponsively, are virtually diagnostic of involvement of the temporal lobe. These seizures generally are seen in older children or adolescents, but also may occur in younger children. It should be noted, however, that seizures emanating from the temporal lobe in infants and young children commonly are associated with behavioral arrest, motor dystonic posturing, and fewer automatisms [Brockhaus and Elger, 1995; Jayakar and Duchowny, 1990; Wyllie et al., 1993]. Additionally, young children are typically incapable of describing the premonitory symptoms before the onset of the more overt clinical seizure. Video EEG monitoring has been helpful in fully elucidating the seizure semiology in these patients. Children with infantile spasms may have partial seizures before, during, or after the onset of the infantile spasms [Kobayashi et al., 2001; Watanabe et al., 2001]. Partial seizures can be a helpful clue that prompts the epileptologist to screen carefully for an underlying focal abnormality, such as a tuber or focal cortical dysplasia.
Electroencephalography
In the adult preoperative evaluation, prolonged video EEG monitoring provides the baseline data with which all of the other data are compared. In children, however, it is becoming increasingly clear that the surface EEG data may be poorly localized and at times misleading in the preoperative evaluation [Wyllie, 1995]. Therefore, this modality may be less important in localizing the epileptogenic focus. In these children, other modalities, particularly neuroimaging studies (MRI, MR spectroscopy, PET, and SPECT), may provide the pivotal information that determines the location of the epileptogenic zone and may reduce the need for invasive subdural EEG monitoring. Indeed, in the near future, the improvement of noninvasive functional brain imaging techniques may obviate the need for invasive EEG monitoring. Even now, invasive EEG monitoring may be deferred if surface EEG monitoring and MRI data are congruent [Wyllie et al., 1998; Zupanc et al., 2010].
Magnetic Resonance Imaging
MRI scans have greatly enhanced the ability to visualize intraparenchymal brain structures. The linkage between intracranial abnormalities and epilepsy is well accepted [Zupanc, 1997a]. The mechanism(s) involved in the production of epilepsy is an area of intense research and involves structural changes, synaptic reorganization, stimulation of mossy fibers, astroglial proliferation with neuronal cell loss, and neurotransmitter or corresponding receptor changes. With the recent advances in MRI technology, the ability to identify the substrates of epilepsy has been greatly enhanced. This modality provides some of the most sensitive and specific neuroimaging data for localization of the epileptogenic zone [Brooks et al., 1990; Cascino, 1994; Cascino et al., 1989, 1991; Kuzniecky et al., 1993a, 1993c; Madan and Grant, 2009]. The following new technologies are exciting and innovative [Jack, 1995; Madan and Grant, 2009]:
 Use of thin contiguous cuts of 1.5–1.6 mm in multiple sections of the cortex, in combination with a three-dimensional volumetric pulse sequence, provides the necessary resolution to detect small lesions that would be missed with conventional MRI scans. Specific areas can be targeted, and images can be reformatted to correct for head rotation and other perturbations in data collection. This technique has allowed detection of even small amounts of unilateral hippocampal atrophy (see Figure 61-2), as well as identification of small areas of focal cortical dysplasia.
Use of thin contiguous cuts of 1.5–1.6 mm in multiple sections of the cortex, in combination with a three-dimensional volumetric pulse sequence, provides the necessary resolution to detect small lesions that would be missed with conventional MRI scans. Specific areas can be targeted, and images can be reformatted to correct for head rotation and other perturbations in data collection. This technique has allowed detection of even small amounts of unilateral hippocampal atrophy (see Figure 61-2), as well as identification of small areas of focal cortical dysplasia.
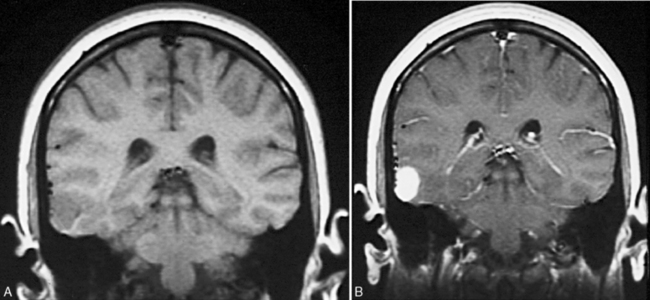
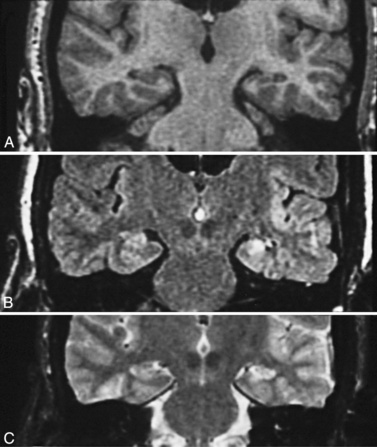
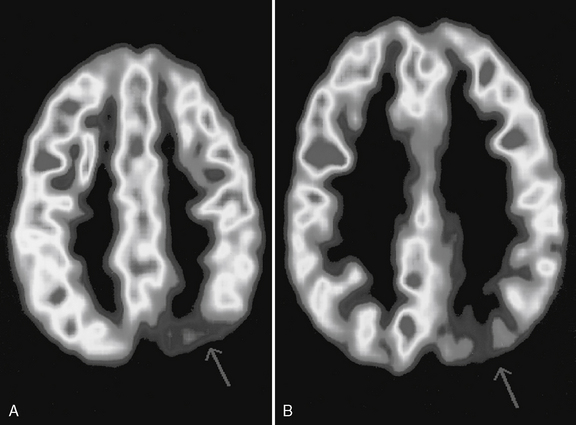
 The fluid-attenuated inversion recovery imaging (FLAIR) technique highlights lesions such as mesial temporal sclerosis and malformations of cortical development and allows detection of previously unidentifiable small lesions. This sequence produces a T2-weighted image that subtracts the cerebrospinal fluid signal (white and bright on T2), but keeps the T2 signal from intraparenchymal structures (Figure 61-3 and Figure 61-4).
The fluid-attenuated inversion recovery imaging (FLAIR) technique highlights lesions such as mesial temporal sclerosis and malformations of cortical development and allows detection of previously unidentifiable small lesions. This sequence produces a T2-weighted image that subtracts the cerebrospinal fluid signal (white and bright on T2), but keeps the T2 signal from intraparenchymal structures (Figure 61-3 and Figure 61-4). Diffusion tensor imaging is an MRI imaging technique that can identify white-matter tracts [Rugg-Gunn et al., 2001] that may be disrupted in areas of cortical dysplasia.
Diffusion tensor imaging is an MRI imaging technique that can identify white-matter tracts [Rugg-Gunn et al., 2001] that may be disrupted in areas of cortical dysplasia. Multichannel coils (32 phased array and beyond and higher field strengths (3 Tesla, 7 Tesla, and greater), coupled with newer imaging sequences, including arterial spin labeling (ASL) and susceptibility weighted imaging (SWI), as well as diffusion tensor imaging (DTI/DSI), are likely to increase our detection of focal cortical dysplasias [Madan and Grant, 2009].
Multichannel coils (32 phased array and beyond and higher field strengths (3 Tesla, 7 Tesla, and greater), coupled with newer imaging sequences, including arterial spin labeling (ASL) and susceptibility weighted imaging (SWI), as well as diffusion tensor imaging (DTI/DSI), are likely to increase our detection of focal cortical dysplasias [Madan and Grant, 2009].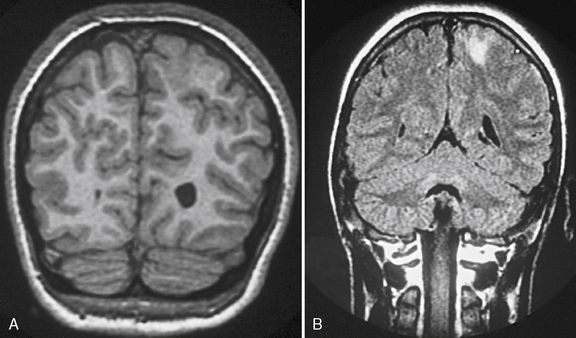
Fig. 61-3 MRI scans from a patient with focal cortical dysplasia of the posterior left parasagittal region.
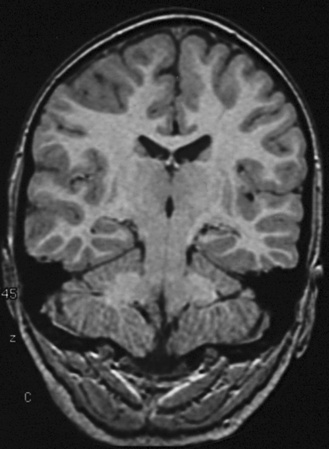
With use of these techniques, MRI scans can identify many substrates of epilepsy, including malformations of cortical development, tumors, vascular malformations, and encephalomalacias secondary to trauma, infection, and infarction. Malformations of cortical development are increasingly recognized as being highly epileptogenic [Kuzniecky and Ruben, 1995; Kuzniecky and Barkovich, 2001; Palmini et al., 1995; Raymond et al., 1995]. Advances in MRI technology have greatly improved the ability to identify these abnormalities. They may account for more than 60 percent of the intractable localization-related epilepsies of childhood [Kuzniecky et al., 1993c]. These malformations can be small and difficult to detect, even with sophisticated MRI scans of the brain, or can be widespread and diffuse, as with lissencephaly [Dobyns and Truwit, 1995; Dobyns et al., 1996] (Figure 61-5 and Figure 61-6). The unilateral and focal malformations of cortical development are most often targeted for surgical excision. Clinically, information on the natural history of the malformations of cortical development is emerging. Status epilepticus is a common initial presentation, usually in the latter half of the first decade of life [Laoprasert and Zupanc, 1997]. Many of the malformations of cortical development produce an epilepsy syndrome that is intractable to medical management [Palmini et al., 1997].
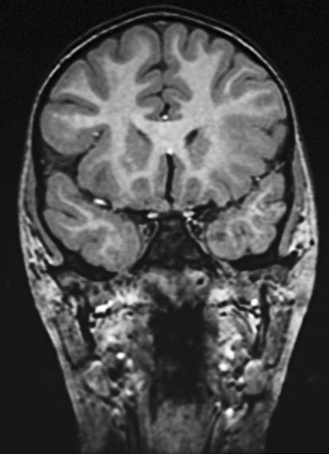
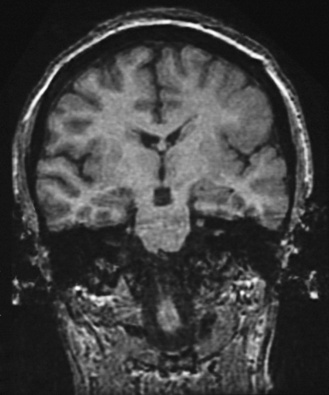
Fig. 61-6 MRI scan from a patient with unilateral perisylvian dysplasia with polymicrogyria (left hemisphere).
Finally, although mesial temporal sclerosis and hippocampal atrophy are not commonly found in children with intractable epilepsy who are younger than 10 years of age, identification of these abnormalities is a powerful indicator of the zone of epileptogenesis [Cascino, 1994; Cascino et al., 1991; Jack, 1995; Swartz et al., 1992]. The pathophysiology of mesial temporal sclerosis and hippocampal formation atrophy is poorly understood. Do the seizures themselves cause mesial temporal sclerosis and hippocampal formation atrophy? Does an underlying malformation of cortical development cause the initial seizures, ultimately resulting in mesial temporal sclerosis and hippocampal formation atrophy [Cendes et al., 1993; Kuks et al., 1993; Trenerry et al., 1993]? Prolonged febrile seizures, head injury, nonfebrile status epilepticus, encephalitis, hypertensive encephalopathy, and viruses, including human herpesvirus 6 (HHV6), have also been implicated as potential causes of mesial temporal sclerosis [Scott et al., 2001; Solinas et al., 2003; Donati et al., 2003; Theodore et al., 2008]. These questions have not yet been clearly answered. It is known, however, that the degree of volume loss correlates with the amount of cellular loss, as measured in pathologic specimens [Cascino et al., 1991]. The neuronal cell loss, coupled with the presence of aberrant mossy fibers, and synaptic reorganization and excessive glutamatergic activity, probably accounts for the recurrent, recalcitrant seizures [Fuerst et al., 2003; Holmes and Ben-Ari, 1998; Jokeit et al., 1999; Kalviainen et al., 1998; Kotloski et al., 2002; Sutula et al., 1988, 1989; Tasch et al., 1999; Eid et al., 2008].
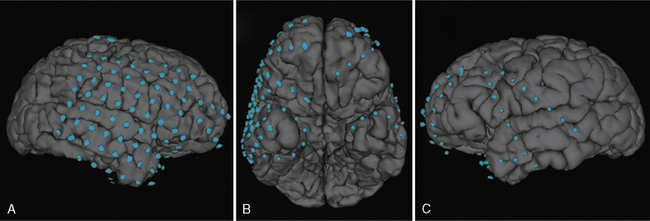
Co-registration of scalp EEG and MRI data has become an important tool in the determination of source localization relative to the patient’s brain anatomy [Lamm et al., 2001]. This technique provides a three-dimensional rendition of the topographic EEG activity on to the patient’s head, taking into consideration anatomical differences specific to the patient. More recently, co-registration of the intracranial EEG and MRI data has provided the epileptologist and neurosurgeon with a three-dimensional map of electrode placement used to define the epileptogenic zone, and hence, the boundaries of surgical resection [LaViolette et al., 2011] (Figure 61-7).
Single-Photon Emission Computed Tomography
SPECT also has enhanced the ability to identify the epileptogenic zone. Penfield and colleagues observed relative hyperperfusion in the epileptogenic zone during a seizure [Penfield, 1958]. Interictally, blood flow and metabolism decrease. SPECT scan technology enables quantification of cerebral blood flow and identification of areas of relative blood flow change. SPECT images are reconstructed from data obtained by recording photon emissions from radiotracers injected intravenously. These radiotracers rapidly cross the blood–brain barrier because of their lipophilic nature and bind within minutes to the brain, producing an instantaneous picture of cerebral blood flow [English and Brown, 1990]. Clinical research has focused on both interictal and ictal SPECT scans, with a substantial portion of the clinical research using the radioisotope 99mtechnetium-hexamethylpropyleneamine oxime (99mTc-HMPAO) [Cross et al., 1995, 1997; Harvey et al., 1993a, b]. More recently, 99mTc ethyl cysteinate dimer (ECD) (i.e., 99mTc-N,N′(1,2-ethylenediyl)bis-l-cysteine diethyl ester), prepared as technetium 99mTc bicisate (Neurolite), has been introduced [Lanceman et al., 1997]. Logistically, Neurolite provides distinct advantages for ictal SPECT scans because it is a stable isotope tracer that can be mixed well ahead of the time of injection, as opposed to 99mTc-HMPAO, which decomposes quickly in vitro and must be used less than 30 minutes after it is reconstituted. For ictal SPECT scans, a technologist or nurse trained in the delivery of these radioisotopes can sit at the bedside and deliver the Neurolite within seconds after the onset of a seizure. Spatial resolution with SPECT scans also has improved because of the development of gamma cameras with multiple detectors that provide more data points, with subsequent enhanced sensitivity.
Interictal SPECT scans have been used for longer than 10 years as a method of identifying the epileptogenic focus in patients with medically intractable localization-related epilepsy who are candidates for epilepsy surgery. With interictal SPECT scans, the epileptogenic zone can be identified by a regional area of reduced cerebral blood flow [Adams et al., 1992; Berkovic et al., 1992, 1993; Cordes et al., 1990; Coubes et al., 1993; Denays et al., 1988; Dietrich et al., 1991; Grunwald et al., 1991; Hajek et al., 1991; Kuzniecky et al., 1993b; Lamanna et al., 1989; Launes et al., 1992; Lee et al., 1988; Rowe et al., 1989, 1991; Ryding et al., 1988; Ryvlin et al., 1992; Shen et al., 1990; Verhoeff et al., 1992]. Clinical research clearly indicates that interictal studies alone have a relatively low sensitivity for identification of the epileptogenic focus in adults with temporal lobe epilepsy and even lower sensitivity with extratemporal epilepsy. Data pooled from several studies yield estimates of interictal SPECT sensitivity of 66 percent for temporal lobe epilepsy and 60 percent for extratemporal epilepsy localized by EEG [Spencer, 1994; Knowlton, 2006].
Ictal SPECT scan data, however, have proved valuable with respect to localization of the epileptogenic focus. Ictal SPECT scans typically reveal an area of regional hyperperfusion that corresponds to the underlying epileptogenic focus, as verified by surgical pathology and surface EEG localization [Bauer et al., 1989; Grunwald et al., 1991; Ho et al., 1994; Hwang et al., 1990; Katz et al., 1990; Lee et al., 1988; Marks et al., 1992; Newton et al., 1992b; Rowe et al., 1989, 1991; Shen et al., 1990; Stefan et al., 1990]. Using data pooled from several centers, the sensitivity of ictal SPECT (as judged by EEG correlation) has been estimated at 90 percent for temporal and 81 percent for extratemporal epilepsy, with specificity at 77 percent and 93 percent, respectively [Spencer, 1994; Knowlton, 2006]. Critical to the efficacy of the ictal SPECT scan is the timing of the injection. If the injection can be given within 30 seconds of the seizure onset, the isotope remains localized and can “capture” the epileptogenic focus or generator before the epileptogenic discharge spreads [Newton et al., 1995]. If the seizure propagation is rapid, ictal injections are less sensitive and unreliable.
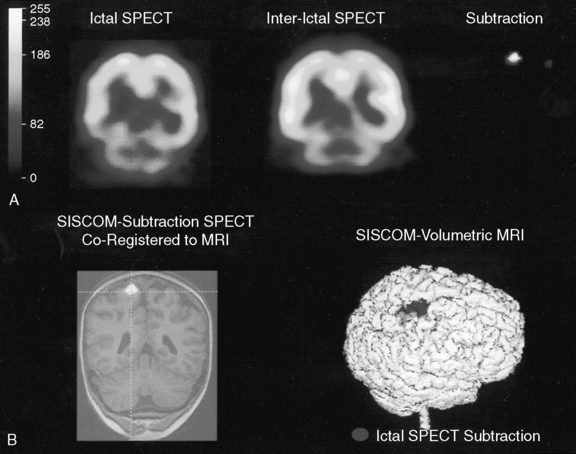
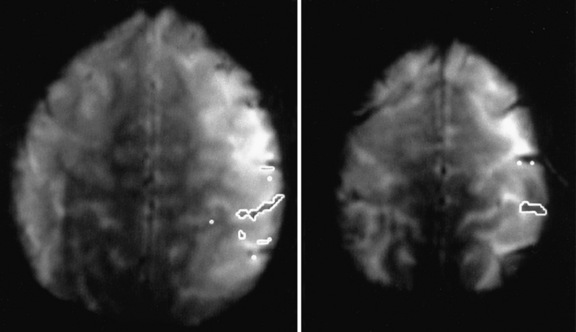
Comparison of ictal and interictal scans also is important in determining whether any abnormality in blood flow is significant. With the assistance of computerized technology and surface matching techniques, co-registration of the ictal SPECT scan to the volumetric MRI scan has demonstrated a close relationship between the region of ictal hyperperfusion and MRI structural lesions [Hogan et al., 1996; Mountz et al., 1994]. A technique has been developed whereby the ictal and interictal SPECT scan data are co-registered with one another and the interictal image is subtracted from the ictal image, producing the area of true ictal hyperperfusion [Zubal et al., 1995]. This difference image, called a subtraction SPECT scan, is then co-registered with a three-dimensional representation of the MRI scan. In nonlesional extratemporal epilepsy, this information has been proven to be especially helpful in either guiding placement for subdural invasive EEG monitoring or obviating the need for invasive monitoring altogether. Several studies have demonstrated that peri-ictal subtraction SPECT provides useful information for seizure localization in patients with focal malformations of cortical development, even when the MRI study is nonlocalizing (i.e., “nonlesional”) [O’Brien et al., 1998, 2000, 2004; Tan et al., 2008]. In a large series involving pediatric and adult epilepsy patients, if the site of the surgical resection was concordant with the subtraction SPECT localization (using SISCOM technology, a Mayo Clinic-patented computer program that performs subtraction SPECT and then co-registers the results to a volumetric MRI scan of the brain), postoperative seizure frequency scores were significantly lower and postoperative improvement was greater [O’Brien et al., 1998] (Figure 61-8). A recent multicenter study further confirmed the incremental benefit of the SISCOM technology over traditional side-by-side comparison in the presurgical evaluation, particularly in patients with extratemporal lobe epilepsy [Matsuda et al., 2009].
Positron Emission Tomography
PET is another imaging modality used for localization of the epileptogenic focus [Mohan et al., 1999]. It uses radiotracers labeled with specific positron-emitting isotopes (11C, 15O, and 18F) to measure a variety of biochemical brain functions. With the aid of computerized technology and mathematical modeling, the source and concentration of the emission are either qualitatively or quantitatively plotted on a three-dimensional representation of the brain. Cerebral glucose metabolism is the most commonly measured parameter, using 18F-fluorodeoxyglucose (FDG) (Figure 61-9). Other tracers also can be used to measure cerebral blood flow, benzodiazepine and opiate receptors, pH, serotonin metabolism, and amino acid transport [Henry et al., 1993; Mohan et al., 1999; Shah et al., 1995].
FDG PET images are averaged over a 40-minute time interval, suggesting the limited value of this technique for ictal studies. The interictal images, on the other hand, are highly sensitive in complex partial seizures emanating from the temporal lobe. In several studies in adult patients with medically refractory epilepsy of temporal lobe origin, glucose hypometabolism in the temporal lobe correlated highly with localized ictal EEGs and MRI abnormalities in this region [Abou-Khali et al., 1987; Chugani et al., 1990b; Coubes et al., 1993; Debets et al., 1990; Engel et al., 1982; Hajek et al., 1993; Henry et al., 1993; Leiderman et al., 1992; Radtke et al., 1993; Sackellares et al., 1990; Stefan et al., 1987, 1990; Swartz et al., 1992; Theodore et al., 1986, 1990; Valk et al., 1993]. Glucose hypometabolism in the temporal lobe, as obtained on interictal FDG PET scan, has been found to have an overall sensitivity of 84 percent and a specificity of 86 percent [Spencer, 1994; Knowlton, 2006].
In a study by Theodore et al. [1997], the presence of temporal lobe glucose hypometabolism in the presence of a nonlocalizing surface ictal EEG predicted successful outcome with temporal lobectomy. Localization to the temporal lobe was confirmed with invasive EEG monitoring but the authors make the point that invasive EEG monitoring may be unnecessary and may even provide false localizing information in some patients being evaluated for epilepsy surgery. As technology improves, concordance of noninvasive neuroimaging techniques may be all that is necessary before proceeding with the surgery.
Analysis of nonlesional extratemporal epilepsy in adult patients undergoing PET scans has provided data that have been less definitive [Chugani et al., 1990a; Sackellares et al., 1990; Stefan et al., 1990]. In one recent analysis, the sensitivity and specificity of FDG PET scans decreased to 40 percent in MRI-negative extratemporal cases [Yun et al., 2006]. In children with refractory epilepsy, however, with poor localization on surface EEG and negative findings on MRI scans of the brain, FDG PET scans may still provide useful information in identifying an underlying epileptogenic focus. Specifically, University of California at Los Angeles investigators were the first to recognize a small subset of children with intractable infantile spasms and underlying deficits of focal glucose metabolism [Chugani et al., 1988, 1990a; Olson et al., 1990]. These deficits usually were temporal-parietal-occipital in origin. Many of these patients had partial seizures before, during, or after the onset of their infantile spasms, often providing a clue to localization. Additionally, interictal surface EEG examined retrospectively often disclosed focal delta slowing or an asymmetry in beta activity. Large cortical resections of the underlying epileptogenic zone were performed, guided by PET scan data and electrocorticography. After surgery, the seizures (infantile spasms and/or partial seizures) disappeared. Results indicate, not only that seizure control improved, but also that these patients’ development improved at a faster rate and to a greater degree than would have been predicted without surgery [Asarnow et al., 1997; Chugani et al., 1988].
Newer ligands also have been developed. In flumazenil PET scans, the flumazenil binds to benzodiazepine receptors [Juhasz et al., 1999; Mohan et al., 1999]. In the area of the epileptogenic zone, benzodiazepine binding appears to be decreased. In one clinical study, the flumazenil PET scan demonstrated a more restricted area of decreased binding than was apparent on the FDG PET scan; the resection of this cortical region was associated with good surgical outcome [Juhasz et al., 2000]. In addition, diffuse cortical abnormalities on flumazenil PET scans predict poor seizure control following epilepsy surgery [Juhasz et al., 2001].
C-alphamethyl-l-tryptophan (AMT) PET scans also have been studied. AMT is an analog of trytophan and a precursor for serotonin synthesis [Chugani et al., 1998; Juhasz et al., 2003]. Data suggest that the AMT PET scans may be useful in identifying the most epileptogenic tuber in patients with tuberous sclerosis, multiple tubers, and medically intractable epilepsy. Concordance of the epileptogenic tuber with increased AMT uptake has been observed in PET scans [Asano et al., 2000; Chugani et al., 1998, Chugani and Muzik, 2000]. In addition, AMT PET scans may also be helpful in reevaluating patients in whom epilepsy surgery has failed to improve seizure control. In the patients studied with AMT PET, the area of increased AMT binding correlated closely with the epileptogenic zone [Juhasz et al., 2004].
Magnetic Resonance Spectroscopy
MR spectroscopy has been used in the study of patients with intractable epilepsy [Kuzniecky et al., 1992; Laxer et al., 1992; Matthews et al., 1990; Novotny, 1995; Connelly et al., 1994]. Specifically, phosphorus MR spectroscopy measures phospholipid metabolism. In the region of the epileptogenic focus, investigators have found abnormal phosphocreatine to inorganic phosphate ratios. Phosphocreatine (Pcr), intracellular pH, and inorganic phosphorus (Pi) increase during a seizure. The adenosine triphosphate concentration, however, only decreases slightly [Duncan, 1997; Prichard, 1994]. Proton MR spectroscopy can also measure regional abnormalities in lactate, N-acetyl-aspartate (NAA), creatine (Cr), and choline (Cho). Lactate levels increase during a seizure and remain elevated for several hours. Data also indicate reductions in the NAA/Cho and NAA/Cr ratios in the region of the epileptogenic zone, presumed to reflect neuronal loss and reactive astrocytosis [Petroff et al., 1984, 1986; Prichard, 1994]. Therefore, abnormal NAA/Cr and NAA/Cho ratios may serve as indices of regional cellular pathology. MR spectroscopy holds promise as an important adjunctive noninvasive technique for assisting with the identification of the underlying epileptogenic zone.
Magnetoencephalography
MEG is another technology that has been developed to improve the ability to identify epileptogenic foci. It measures tiny magnetic fields in the brain that are created by the electrical activity of the brain. Most institutions are using 128-channel MEG technology to enhance resolution. MEG offers several advantages over EEG. First, the magnetic fields are not attenuated by the skull, scalp, and skin, as are electrical potentials. Therefore, the MEG signal contains fewer distortions or changes [Barth, 1993]. Second, MEG is a monopolar measure and does not require a dipolar montage, eliminating the possibility of artifact associated with an “active reference.” Third, MEG provides high temporal resolution, which can be useful in determining the functional activity of different brain areas (i.e., motor cortex) or propagation of seizure activity. Finally, and of greatest importance, MEG measures postsynaptic intracellular currents in the dendrites of neurons situated tangentially to the skull, whereas the EEG measures the extracellular postsynaptic ionic currents [Tovar-Spinoza et al., 2008; Barth, 1993; Barth et al., 1984].
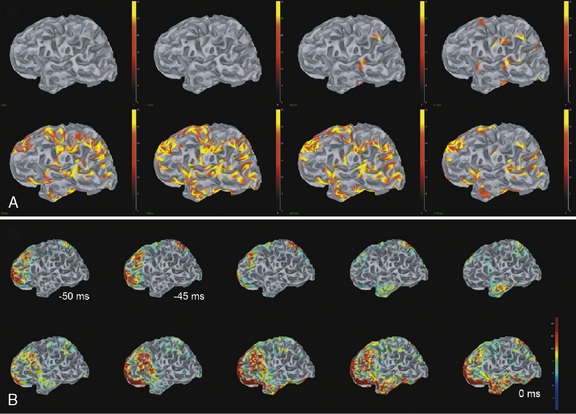
Clinical research suggests that, although surface interictal EEG spike recordings may indicate multifocal activity, MEG can more precisely localize the underlying epileptogenic focus [Barth, 1993; Stefan et al., 2003; Sutherling et al., 1988; Wheless et al., 1999]. In this regard, MEG provides complementary data to EEG and there is a growing belief that combined MEG/EEG data should be used routinely in the presurgical evaluation of patients with intractable epilepsy [Funke et al., 2009]. MEG has provided pivotal information and may become the most precise way of identifying the size, location, and dipole orientation of the epileptogenic zone [Minassian et al., 1999; Stefan et al., 2003; Wheless et al., 1999, 2004]. In particular, there is widespread agreement that MEG provides the best tool to distinguish a temporal from an extratemporal epileptogenic zone [Funke et al., 2009]. In fact, MEG has been employed in children with intractable epilepsy to provide spatial information to be used in planning the excision area [Iida et al., 2005]. MEG spike source clusters have been used to indicate the epileptogenic zone [Iida et al., 2005]. Figure 61-10 demonstrates simultaneously recorded MEG and EEG data obtained at our center and used to map the epileptogenic focus of a child with frontal lobe epilepsy. As shown in Figure 61-10A and B, a left frontal MEG spike is observed while the EEG demonstrates theta range activity without clear epileptiform activity. Figure 61-10C demonstrates the distributed source estimates for the MEG data displayed on a representation of the cortical surface reconstructed from MRI data from the same patient. Note the basomesial frontal location of the epileptogenic zone, which would have been difficult to observe on scalp EEG. In addition, MEG may be a very useful tool in children with respect to functional imaging, particularly imaging language cortex [Stufflebeam et al., 2009; Tovar-Spinoza et al., 2008; Papanicolaou et al., 2004; Simos et al., 1999]. Figure 61-11A demonstrates locations of the equivalent current dipoles (ECD) for the MEG data for a visual reading task displayed in sagittal slices of an anatomical representation of the same patient’s brain as in Figure 61-10. The high temporal resolution of MEG allows for the dissociation of functional brain activity in different areas. The patient’s language was localized to the left hemisphere, based on the Wada procedure (see below). The extraordinary temporal resolution of MEG also enables us to understand the propagation patterns of interictal activity occurring over a period of 50 msec or less (Figure 61-11B)!
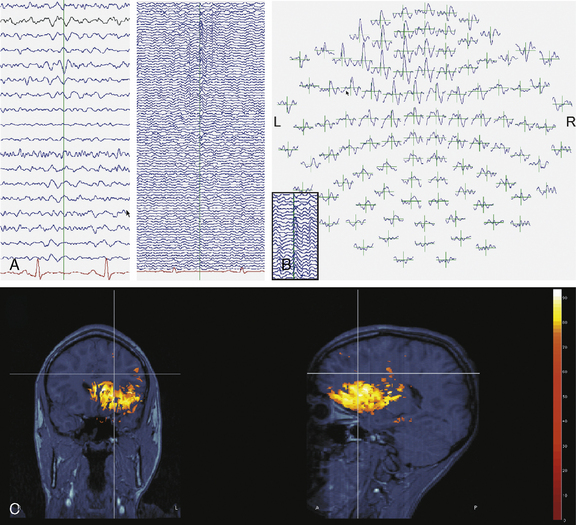
Fig. 61-10 Magnetoencephalography (MEG) complements EEG data in the presurgical evaluation.
(Courtesy of Dr. Sylvain Baillet, Medical College of Wisconsin, Milwaukee, Wisconsin.)
Functional Mapping
Classically, in the older child and adolescent, the sodium amytal test (Wada test) is used in the preoperative evaluation for the localization of speech and language and to determine whether memory can be supported in the contralateral hemisphere [Loring, 1997; Wyllie et al., 1990]. This test involves injecting sodium amytal into either the left or the right internal carotid artery, in an attempt to ameliorate ipsilateral hemispheric function chemically and to determine which hemisphere is “dominant” (i.e., responsible for speech and language function and, to a lesser extent, memory). It is a time-consuming test that is invasive and provides a broad but nonspecific overview of hemispheric function. Additionally, controversy continues over its interpretation and its ability to predict postoperative function, particularly with respect to memory [Loring et al., 1992; Perrine, 1994]. As an example, language is a complex function. Although speech arrest after sodium amytal injection usually is in the dominant hemisphere, this is not always the case [Loring et al., 1992]. Language involves spontaneous speech, repetition, comprehension, reading, and counting. These aspects of language are all interactive, but their corresponding cortical areas may be located in different areas of the brain, making it difficult to relegate language to one specific hemisphere or region. In younger children, the Wada test can be even more challenging and technically difficult. Obtaining full cooperation from a child requires preparation, time, and patience. If the child becomes frightened during the test, test validity becomes questionable. Two other techniques also used frequently to identify eloquent cortex (i.e., cortex controlling vital motor, language, or memory functions) are somatosensory-evoked potentials and stimulation mapping. The measurement of somatosensory-evoked potentials has the advantage of being able to be applied successfully, regardless of the state of the patient. This modality can be used in the operating room in the anesthetized patient, or in an awake and cooperative patient. Somatosensory-evoked potentials are used primarily to identify the sensorimotor cortex. Stimulation mapping involves the application of subdural electrodes followed by sequential electrical stimuli at various intensities and durations. Penfield pioneered this technique during the 1930s through the 1950s to localize language and motor functions intraoperatively, in order to avoid postoperative neurologic deficits. Subsequent investigators have used cortical stimulation preoperatively (using implanted grid electrodes) and intraoperatively to map out functional cortex, such as the sensorimotor cortex or expressive language cortex [Ojemann, 1978, 1979, 1993; Ojemann and Dodrill, 1987]. Although cortical stimulation mapping has yielded a tremendous amount of information about the localization of functions, several other emerging techniques are providing valid, noninvasive methods for mapping out functional areas of the brain. These techniques include functional MRI (fMRI) scans, MEG and magnetic source imaging, and transcranial magnetic stimulation [Binder, 1997; Detre, 2004; Knowlton and Shih, 2004; Peresson et al., 1998; Perrine, 1994; Powell et al., 2004; Sabsevitz et al., 2003; Wheless et al., 2004]. In view of the limitations of the previously described techniques for functional mapping, these noninvasive techniques, which might provide more specific and salient information, are being developed. fMRI scans are being used in a number of epilepsy centers as adjunctive techniques to define eloquent cortex [Kwong et al., 1992; Lee et al., 1996; Ogawa et al., 1992].
fMRI is based on the fact that performance of a specific act will activate the anatomically appropriate cortex in the brain. With activation, a concomitant increase in blood flow occurs, resulting in a change in the paramagnetic properties of the affected cortex. This produces a signal that can then be detected by the MRI scanner. fMRI is a technique that will be increasingly used to map out eloquent functions, such as sensorimotor cortex and speech and language centers [Logan et al., 1995, 1997, 1998; Sachs et al., 2003; O’Shaughnessy et al., 2008]. It is still not suitable for the young, uncooperative infant or child, except for fMRI of the sensorimotor cortex, which can be performed under sedation. Language and memory testing using fMRI can be utilized in cooperative older children and adolescents. In fact, clinical research has yielded increasing proof that fMRI can provide important localization data. With respect to language lateralization, fMRI language examinations, in combination with comprehensive neuropsychometric testing, can play a very important role in estimating the risk for postoperative cognitive changes and in selecting patients for invasive functional mapping procedures [Sachs et al., 2003; O’Shaughnessy et al., 2008; Liegeois et al., 2002; Hertz-Pannier et al., 1997; Stapleton et al., 1997; Anderson et al., 2006]. In the young infant and child, functional studies are less likely to alter the surgical plan because brain plasticity in these age groups makes localization less critical [Shields, 2000; Stafstrom et al., 2000; Wyllie, 1998, Liegeois et al., 2004; Kadis et al., 2007].
An example of a functional MRI scan is illustrated in Figure 61-12.
Concept of Congruence
Under ideal conditions, identification of the epileptogenic focus is made by the congruence of data obtained during the preoperative evaluation, with the precise localization based on seizure semiology, physical examination, surface ictal and interictal EEG monitoring (and, if necessary, invasive-depth electrodes or subdural electrode strips or grid), MRI scan of the brain, ictal and interictal SPECT scans, interictal PET scan, MEG, fMRI scan, and/or MR spectroscopy. Each case must be individualized, with some cases requiring the acquisition of data from all of these studies. Other cases may be resolved with a less complicated approach. At a minimum, seizure semiology, surface ictal EEG monitoring, and MRI scan of the brain should be congruent. With lesional localization-related epilepsy, the use of invasive EEG monitoring may not be necessary. Electrocorticography at the time of surgery usually can assist with identifying the dimensions of the epileptogenic zone. The sensorimotor cortex can be identified intraoperatively using motor-evoked potentials, somatosensory-evoked potentials, or direct electrical cortical stimulation mapping. In a cooperative patient, fMRI brain imaging or MEG may be able to identify the sensorimotor cortex accurately and display it on a three-dimensional volumetric MRI scan of the brain. If the surgical excision is near functional speech and language cortex, older children and adolescents usually can be cooperative enough to tolerate an awake surgical procedure. In addition, in older cooperative children and adolescents, fMRI has also been very helpful in identifying cortex involved with receptive and expressive language. In younger children (before the age of 5–6 years), brain plasticity is still adequate, so that removal of primary speech and language cortex will result in transition of these functions to the contralateral hemisphere [Peacock, 1995; Shields, 2000].
Types of Surgery
Several types of epilepsy surgery are performed in children and adults, depending on the identification of the epileptogenic focus and its location and extent [Zupanc, 1997b]. The most common surgical procedures are:
Implantation of a vagus nerve stimulator accounts for approximately 16 percent of the total number of operations, while multiple subpial transections are relatively uncommon procedures, accounting for only 0.6 percent [Harvey et al., 2008].
Temporal lobectomy is the most common epilepsy surgery performed in adolescents and adults. This procedure is almost exclusively a temporal lobectomy with amygdalohippocampectomy, because removal of the mesial temporal structures is correlated with a better surgical outcome. Often an associated abnormality or lesion, such as a tumor (dysembryoplastic neuroepithelial tumor [DNET] or ganglioglioma), mesial temporal sclerosis, hippocampal formation atrophy, or malformation of cortical development, is found to be present. The new MRI technologies have been helpful in identification of these substrates of epilepsy. Those patients with mesial temporal sclerosis or hippocampal atrophy concordant with ictal surface EEG abnormalities have an excellent prognosis for successful epilepsy surgery, with a 90 percent chance of becoming seizure-free [Duchowny et al., 1992; Falconer, 1970; Mizrahi et al., 1990]. Younger children do not commonly have mesial temporal sclerosis or hippocampal atrophy [Ng et al., 2004]. Many of the intractable epilepsies in childhood are extratemporal and nonlesional.
Extratemporal cortical resection is more commonly performed in children, often involving extensive lobar or multilobar resections. The extent of the resection is dictated primarily by the extent of the lesion: e.g., a tuberectomy in a patient with tuberous sclerosis versus a multilobar resection in a child with infantile spasms and an underlying focal cortical dysplasia involving the temporal-parietal-occipital lobes. As the ability to identify focal cortical dysplasias and the concomitant epileptogenic zone improves, epilepsy surgical outcomes also will improve. Focal cortical dysplasias are a common cause of intractable partial epilepsy in children, accounting for 60 percent of the cases [Kuzniecky and Barkovich, 2001; Kuzniecky et al., 1993c]. The best predictive factors in successful surgical outcome in focal cortical dysplasia are completeness of the resection and the presence of an identifiable lesion on MRI brain imaging [ILAE Pediatric Epilepsy Surgery Consortium data, submitted for publication].
Stereotactic lesionectomy is performed in highly selected cases in children and adults, with a reported 50–60 percent chance of rendering the patient seizure-free [Britton et al., 1994; Cascino et al., 1990, 1992, 1993, 1994]. Outcome is improved if intraoperative electrocorticography is used to remove not only the lesion, but also the surrounding “epileptogenic zone” [Jooma et al., 1995; Montes et al., 1995; Palmini et al., 1995; Pilcher et al., 1993]. To date, no prospective, controlled studies of statistically significant numbers of patients have critically compared the different operative strategies with respect to outcome [Shields, 2000; Tonini et al., 2004; Wyllie, 1998]. The surgical outcome in these patients may vary, depending on the age of the patient, the location of the lesion, and most important, the type of lesion. In a review of 47 published articles about epilepsy surgery outcome, the best predictors for seizure-free outcome included a history of febrile seizures as a child, mesial temporal sclerosis, tumors, EEG and MRI data concordance, and an extensive surgical resection [Tonini et al., 2004].
Hemispherectomy also is performed in young children [Vining et al., 1997]. The indications for this type of surgery are catastrophic epilepsies in which the substrate of epilepsy is limited to one hemisphere. Epilepsy syndromes that frequently meet these criteria include the following:
In Sturge–Weber syndrome, the pathologic features consist of unilateral leptomeningeal angiomatosis, frequently resulting in changes in the involved hemisphere with concomitant focal seizures, progressive hemiparesis, and cognitive decline [Roach et al., 1994]. Hemimegalencephaly, by definition, is a malformation of cortical development involving one hemisphere, and is characterized by general disorganization and overgrowth of the neuronal tissue. Early hemispherectomy, particularly if the EEG reveals unilateral discharges, can significantly alter the prognosis in affected children [Andermann et al., 1993; Vigevano et al., 1989; Vigevano and DiRocco, 1990]. With Rasmussen’s encephalitis, children have intractable focal seizures, often progressing to epilepsia partialis continua, accompanied by progressive hemiparesis and cognitive decline. Although debate is on-going about the pathophysiology (autoimmune versus infectious), the only long-term successful treatment has been hemispherectomy [Antel and Rasmussen, 1996].
Multiple subpial transection is a newer surgical technique that is used when the epileptogenic zone overlies an area of functional cortex. Multiple subpial transection involves the disruption of connecting horizontal fibers, rather than resection of actual tissue [Blount et al., 2004; Devinsky et al., 2003; Schramm et al., 2002; Spencer et al., 2002]. This technique has been used in the treatment of Landau–Kleffner syndrome [Morrell et al., 1995]. Children with this syndrome have an acquired epileptic aphasia, often intractable to medication. The multiple subpial transection technique has been used over the area deemed to be the epileptogenic zone on electrocorticography. Although good surgical results have been reported, the technique remains controversial. Multiple subpial transection also is being used in areas involving functional cortex. Data indicate that, although multiple subpial transection may be an appropriate adjunctive surgical technique, it will not eliminate seizures if the primary epileptogenic focus is not completely removed [Hufnagel et al., 1997; Spencer et al., 2002].
A complete corpus callosotomy is a palliative surgery that can reduce the seizure burden in carefully selected patients. It most commonly is used in children with Lennox–Gastaut syndrome, with the goal being reduction of tonic and atonic seizures. It can be highly effective [Carson, 2000; Maehara and Shimizu, 2001; McInerney et al., 1999; Sassower et al., 2001; Sorenson et al., 1997; Wyler, 1993]. It is now known that sectioning of the anterior two-thirds corpus callosotomy is rarely effective in controlling seizures long-term. The use of a complete corpus callosotomy, coupled with lateralizing strips, has been demonstrated to be very effective in identifying an epileptogenic zone as part of a multistage surgery [Zupanc et al., 2011].
Goals of Surgery
With use of innovative, noninvasive technologies, the ability of the clinician to identify the underlying epileptogenic zone has improved. In patients with medically intractable epilepsy, this ability allows one of the principal goals of epilepsy surgery to be achieved – that is, the elimination of seizures. The goals of epilepsy surgery, however, may vary, depending on the epilepsy syndrome, the underlying pathophysiology, the cognitive and developmental status of the child or adolescent, and the identification and location of an epileptogenic zone [Taylor et al., 1997]. Specifically, if a cognitively normal patient has temporal lobe epilepsy, as documented by congruence of seizure semiology and EEG and MRI data, the goals of epilepsy surgery are clear: elimination of seizures and improvement in psychosocial, behavioral, emotional, and family functioning without significant loss of cognitive abilities. On the other hand, a cognitively impaired patient with an extensive bilateral malformation of cortical development might be considered for a corpus callosotomy. In this case, the primary goals of epilepsy surgery would be palliative, with the reduction of seizures and possible improvement in cognitive and behavioral functions. Although improvement in cognition, development, and behavior usually is achieved by virtue of decreased seizure frequency and reduction of antiepileptic drug doses, such results are not always obtained, and outcomes will vary with each patient, depending on the epilepsy syndrome and the identified etiology for the epilepsy. The goals of epilepsy surgery and potential limitations of the results of surgery need to be discussed openly with the family before any decision is made.
In patients with malformations of cortical development (in children, typically extratemporal), epilepsy surgery usually has involved lobar or multilobar resections, as well as hemispherectomies. Approximately 60–65 percent of these patients are seizure-free after surgery, and a majority achieves a significant reduction in seizure burden [Shields, 2000; Wyllie, 1998; Zupanc et al., 2010]. At critical stages of development, this reduction in seizure burden is associated with an improvement in development that appears to be sustained. In patients with malformations of cortical development and medically refractory epilepsy who undergo surgical resection, withdrawal of antiepileptic medication is rarely successful. The cortical dysplasia often is very extensive, involving multiple lobes. On the other hand, children with lesions such as mesial temporal sclerosis, low-grade tumors, or middle cerebral artery infarctions can achieve seizure-free status, with eventual discontinuation of antiepileptic medication.
Deep Brain Stimulation
Deep brain stimulation has been known to be effective in the treatment of movement disorders for years. However, there is increasing interest in deep brain stimulation in the treatment of epilepsy [Kahane and Depaulis, 2010]. Several neurology and neurosurgical groups have applied this technique in the treatment of pharmacoresistant epilepsy. Recently, the SANTE study group has published their results. This was a multicenter, double-blind, randomized trial of bilateral stimulation of the anterior nuclei of the thalamus for localization-related epilepsy. The stimulation of the anterior nucleus of the thalamus was chosen because it projects to both the superior frontal and the temporal lobe structures commonly involved in epileptic seizures, produces discrete EEG changes, and inhibits chemically induced seizures in animal models. In this study, which involved adult patients only, with localization-related epilepsy, bilateral stimulation of the anterior nuclei of the thalamus significantly reduced seizure frequency. Specifically, by 2 years, there was a 56 percent median reduction in seizure frequency; 54 percent of patients had a seizure reduction of at least 50 percent. Fourteen patients were seizure-free for over 6 months [Fischer et al., 2010].
In addition to the anterior nucleus, the centromedian nucleus has also been the subject of both clinical and experimental interest. This nucleus is part of the reticulothalamocortical system, which modulates cortical excitability. There have been several studies that have documented significant improvement in seizure control, particularly in patients with generalized tonic-clonic seizures and atypical absence seizures found in Lennox–Gastaut syndrome [Velasco et al., 1997, 2006; Cukiert et al., 2009].
Research Issues: Trends for the Future
 What are the age-specific developmental mechanisms in childhood epilepsy? Can they be targeted for the development of new and more effective antiepileptic drugs or surgical therapy?
What are the age-specific developmental mechanisms in childhood epilepsy? Can they be targeted for the development of new and more effective antiepileptic drugs or surgical therapy?




Future trends for exploration will involve several avenues of research: source localization and predictive EEG patterns for identification of the epileptogenic zone [Smart et al., 2004; Worrell et al., 2004]; implantable devices that can detect predictive EEG patterns before a clinical seizure and deliver either an abortive dose of antiepileptic medication or an abortive electrical stimulus; deep brain cortical-thalamic stimulation to diminish seizure frequency in those patients with subcortical-cortical epileptogenic networks (intractable nonlesional, generalized epilepsy syndromes); and, finally, “designer” antiepileptic drugs targeting specific mechanisms of epilepsy – most likely sodium, potassium, calcium, and Gamma-aminobutyric acid (GABA) channels – to be delivered locally or systemically.
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Abou-Khalil B.W., Siegel G.J., Sackellares J.C., et al. Positron emission tomography studies of cerebral glucose metabolism in chronic partial epilepsy. Ann Neurol. 1987;22:480.
Adams C., Hwang P.A., Gilday D.L., et al. Comparison of SPECT, EEG, CT, MRI, and pathology in partial epilepsy. Pediatr Neurol. 1992;8:97.
Adler J., Erba G., Winston K.R., et al. Results of surgery for extratemporal partial epilepsy that began in childhood. Arch Neurol. 1991;48:133.
Aicardi J.. Benign partial epilepsies. Epilepsy in children. ed 2. New York: Raven Press; 1994.
Andermann F.A., Freeman J.M., Vivegano F., et al. Surgically remediable diffuse hemispheric syndromes. In Engel J.J., editor: Surgical treatment of the epilepsies, ed 2, New York: Raven Press, 1993.
Anderson D.P., Harvey A.S., Saling M.M., et al. FMRI lateralization of expressive language in children with cerebral lesions. Epilepsia. 2006;47(6):998-1008.
Andrews P.I., Dichter M.A., Berkovic S.F., et al. Plasmapheresis in Rasmussen’s encephalitis. Neurology. 1996;46:242.
Annegers J.F., Hauser W.A., Elveback L.R. Remission of seizures and relapse in patients with epilepsy. Epilepsia. 1979;20:729.
Antel J.P., Rasmussen T. Rasmussen’s encephalitis and the new hat. Neurology. 1996;46:9.
Arts W.F.M., Visser L.H., Loonen M.C.B., et al. Follow-up of 146 children with epilepsy after withdrawal of antiepileptic therapy. Epilepsia. 1988;29:244.
Asano E., Chugani D.C., Muzik O., et al. Multimodality imaging for improved detection of epileptogenic foci in tuberous sclerosis complex. Neurology. 2000;54:1976.
Asarnow R.F., LoPresti C., Elliott T. Developmental outcomes in children receiving resection surgery for medically intractable infantile spasms. Dev Med Child Neurol. 1997;39:430.
Barth D.S. Magnetoencephalography. In: Wyllie E., editor. The treatment of epilepsy: Principles and practice. Philadelphia: Lea and Febiger, 1993.
Barth D.S., Sutherling W.W., Engel J., et al. Neuromagnetic evidence of spatially distributed sources underlying epileptiform spikes in the human brain. Science. 1984;223:293.
Bauer J., Stefan H., Huk W.J., et al. CT, MRI and SPECT neuroimaging in status epilepticus with simple partial and complex partial seizures: Case report. J Neurol. 1989;236:296.
Bazil C.W. Comprehensive care of the epilepsy patient – control, comorbidity, and cost. Epilepsia. 2004;45(Suppl 6):3.
Bebin E.M., Kelly P.J., Gomez M.R. Surgical treatment for epilepsy in cerebral tuberous sclerosis. Epilepsia. 1993;34:651.
Begley C.E., Annegers J.F., Lairson D.R., et al. Cost of epilepsy in the United States: A model based on incidence and prognosis. Epilepsia. 1994;35:1230.
Begley C.E., Famulari M., Annegers J.F., et al. The cost of epilepsy in the US: An estimate from population-based clinical and survey data. Epilepsia. 2000;41:342.
Berg A.T., Shinnar S., Levy S.R., et al. Two year remission and subsequent relapse in children with newly diagnosed epilepsy. Epilepsia. 2001;42:1553.
Berkovic S.F., Newton M.R., Chiron C., et al. Single photon emission tomography. In Engel J.Jr, editor: Surgical treatment of the epilepsies, ed 2, New York: Raven Press, 1993.
Berkovic S.F., Rowe C.C. The use of SPECT in focal epilepsy. In: Luders H.O., editor. Epilepsy surgery. New York: Raven Press, 1992.
Binder J. Functional magnetic resonance imaging language mapping. Neurosurg Clin N Am. 1997;8:383.
Blanchette N., Smith M.L. Language after temporal or frontal lobe surgery in children with epilepsy. Brain Cogn. 2002;3:51.
Blount J.P., Langburt W., Otsubo H., et al. Multiple subpial transactions in the treatment of pediatric epilepsy. J Neurosurg Spine. 2004;100:118.
Bourgeois M., Sainte-Rose C., Lellouch-Tubiana A., et al. Surgery of epilepsy associated with focal lesions in childhood. J Neurosurg. 1999;90:833.
Britton J.W., Cascino G.D., Sharbrough F.W., et al. Low-grade glial neoplasms and intractable partial epilepsy: Efficacy of surgical treatment. Epilepsia. 1994;35:1130.
Brockhaus A., Elger C.E. Complex partial seizures of temporal lobe origin in children of different age groups. Epilepsia. 1995;36:1173.
Brooks B., King D., El Gammal T., et al. MRI in patients with intractable complex partial seizures. Am J Neuroradiol. 1990;11:93.
Camfield P., Camfield C. Childhood epilepsy: What is the evidence for what we think and what we do? J Child Neurol. 2003;18:272.
Carson B.S. Indications and outcomes for lobectomy, corpus callosotomy, and hemispherectomy in pediatric neurosurgical patients. Clin Neurosurg. 2000;47:385.
Cascino G.D. Commentary: How has neuroimaging improved patient care? Epilepsia. 1994;35:S103.
Cascino G.D., Hirschorn K.A., Jack C.R., et al. Gadolinium-DTPA enhanced MRI in intractable partial epilepsy. Neurology. 1989;39:1115.
Cascino G.D., Hulihan J.F., Sharbrough F.W., et al. Parietal lobe lesional epilepsy: Electroclinical correlation and operative outcome. Epilepsia. 1993;34:52.
Cascino G.D., Jack C.R.Jr, Parisi J.E., et al. MRI-based volume studies in temporal lobe epilepsy: Pathologic correlations. Ann Neurol. 1991;30:31.
Cascino G.D., Kelly P.J., Hirschorn K.A., et al. Stereotactic resection of intra-axial cerebral lesions in partial epilepsy. Mayo Clin Proc. 1990;65:1053.
Cascino G.D., Kelly P.J., Sharbrough F.W., et al. Long-term follow-up of stereotactic lesionectomy in partial epilepsy: Predictive factors and electroencephalographic results. Epilepsia. 1992;33:639.
Cascino G.D., Sharbrough F.W., Trenerry M.R., et al. Extratemporal cortical resections and lesionectomies for partial epilepsy: Complications of surgical treatment. Epilepsia. 1994;35:1085.
Caton R. The electrical currents of the brain. BMJ. 1875;2:278.
Cendes F., Andermann F., Gloor P., et al. Atrophy of mesial structures in patients with temporal lobe epilepsy: Causes or consequence of repeated seizures? Ann Neurol. 1993;34:795.
Choi H., Carlino R., Heiman G., et al. Evaluation of duration of epilepsy prior to temporal lobe epilepsy surgery during the past two decades. Epilepsy Res. 2009;86:224-227.
Chugani D.C., Chugani H.T., Muzik O., et al. Imaging epileptogenic tubers in children with tuberous sclerosis complex using alpha 11C-methyl-l-tryptophan positron emission tomography. Ann Neurol. 1998;44:858.
Chugani D.C., Muzik O. Alpha (C-11) methyl-L-tryptophan PET maps brain serotonin synthesis and kynurenine pathway metabolism. J Cereb Blood Flow Metab. 2000;20:2.
Chugani H.T., Shewmon A., Shields W.D., et al. Pediatric epilepsy surgery: Pre-and postoperative evaluation with PET. J Epilepsy. 1990;3:75.
Chugani H.T., Shewmon D.A., Peacock W.J., et al. Surgical treatment of intractable neonatal onset seizures: The role of positron emission tomography. Neurology. 1988;38:1178.
Chugani H.T., Shewmon D.A., Shields W.D., et al. Surgery for intractable infantile spasms. Neuroimaging perspectives. Epilepsia. 1993;34:764.
Chugani H.T., Shields W.D., Shewmon D.A., et al. Infantile spasms: I. PET identified focal cortical dysgenesis in cryptogenic cases for surgical treatment. Ann Neurol. 1990;24:405.
Connelly A., Jackson G.D., Duncan J.S., et al. Magnetic resonance spectroscopy in temporal lobe epilepsy. Neurology. 1994;44:1411.
Cordes M., Christie W., Henkes H., et al. Focal epilepsies: HMPAO SPECT compared with CT, MR, and EEG. J Comput Assist Tomogr. 1990;14:402.
Cossu M., Lo Russo G., Francione S., et al. Epilepsy surgery in children: results and predictors of outcome on seizures. Epilepsia. 2008;49:65.
Coubes P., Awad I.A., Antar M., et al. Comparison and spatial correlation of interictal HMPAO-SPECT and FDG-PET in intractable temporal lobe epilepsy. Neurol Res. 1993;15:160.
Cross J.H., Gordon I., Connelly A., et al. Interictal 99mTc HMPAO SPECT and 1H MRS in children with temporal lobe epilepsy. Epilepsia. 1997;38:338.
Cross J.H., Gordon I., Jackson G.D., et al. Children with intractable focal epilepsy: Ictal and interictal 99mTc HMPAO single photon emission computed tomography. Dev Med Child Neurol. 1995;37:673.
Cross J.H., Jayakar P., Nordli D., et al. Proposed criteria for referral and evaluation of children for epilepsy surgery: recommendations of the subcommission for pediatric epilepsy surgery. Epilepsia. 2006;47:952.
Cukiert A., Burattini J.A., Cukiert C.M., et al. Centro-median stimulation yields additional seizure frequency and attention improvement in patients previously submitted to callosotomy. Seizure. 2009;18:588-592.
Danielsson S., Rydenhag B., Uvebrant P., et al. Temporal lobe resection in children with epilepsy: Neuropsychiatric status in relation to neuropathology and seizure outcome. Epilepsy Behav. 2002;3:76.
Davidson S., Falconer M.A. Outcome of surgery in 40 children with temporal-lobe epilepsy. Lancet. 1975;1:1260.
Debets R.M.C., van Veelen C.W.M., Maquet P., et al. Quantitative analysis of 18FDG-PET in the presurgical evaluation of patients suffering from refractory partial epilepsy: Comparison with CT, MRI, and combined subdural and depth EEG. Acta Neurochir Suppl (Wien). 1990;50:88.
Denays R., Rubinstein M., Ham H., et al. Single photon emission computed tomography in seizure disorders. Arch Dis Child. 1988;63:1184.
Detre J.A., Functional M.R.I. Applications in epilepsy. Epilepsia. 2004;45(Suppl 4):26.
Devinsky O., Romanelli P., Orbach D., et al. Surgical treatment of multifocal epilepsy involving eloquent cortex. Epilepsia. 2003;44:718.
Dietrich M.E., Bergen D., Smith M.C., et al. Correlation of abnormalities of interictal N-isopropyl-p-iodoamphetamine single-photon emission tomography with focus of seizure onset in complex partial seizure disorders. Epilepsia. 1991;32:187.
Dobyns W.B., Andermann E., Andermann F., et al. X-linked malformations of neuronal migration. Neurology. 1996;47:331.
Dobyns W.B., Truwit C.L. Lissencephaly and other malformations of cortical development: 1995 update. Neuropediatrics. 1995;26:132.
Donati D., Akhyani N., Fogdell-Hahn A., et al. Detection of human herpesvirus-6 in mesial temporal lobe epilepsy surgical brain resections. Neurology. 2003;61(10):1405-1411.
Duchowny M., Jayakar P., Resnick T., et al. Epilepsy surgery in patients under age 3 years. Ann Neurol. 1996;40:286.
Duchowny M., Jayakar P., Resnick T., et al. Epilepsy surgery in the first three years of life. Epilepsia. 1998;39:737.
Duchowny M., Lewin B., Jayakar P., et al. Temporal lobectomy in children. Epilepsia. 1992;33:298.
Duchowny M.S., Resnick T.K., Alvarez L.A., et al. Focal resection for malignant partial seizures in children. Neurology. 1990;40:980.
Duncan J.S. Magnetic resonance spectroscopy. Epilepsia. 1997;37:598.
Eid T., Williamson A., Lee T.S., et al. Glutamate and astrocytes – key players in human mesial temporal lobe epilepsy? Epilepsia. 2008;49(Suppl 2):42-52.
Emerson R., D’Souza B.J., Vining E.P., et al. Stopping medication in children and epilepsy predictors of outcome. N Engl J Med. 1981;304:1125.
Engel J.Jr. Surgery for seizures. N Engl J Med. 1996;334:647.
Engel J.Jr, Kuhl D.E., Phelps M.E., et al. Interictal cerebral glucose metabolism in partial epilepsy and its relation to EEG changes. Ann Neurol. 1982;12:510.
Engel J.Jr, Wiebe S., French J., et al. Practice parameter: temporal lobe and localized neocortical resections for epilepsy. Neurology. 2003;60:538.
English R.J., Brown S.E. Single photon emission computed tomography – a primer. New York: Society of Nuclear Medicine; 1990.
Erba G., Cavazzuti V. Sturge-Weber syndrome: Natural history and indications for surgery. J Epilepsy. 1990;3(Suppl 1):287.
Falconer M.A. Significance of surgery for temporal lobe epilepsy in childhood and adolescence. J Neurosurg. 1970;33:233.
Fauser S., Schulze-Bonhage A., Honeggar J., et al. Focal cortical dysplasias: surgical outcome in 67 patients in relation to histological subtypes and dual pathology. Brain. 2004;127:2406.
Fischer R., Salanova V., Witt T., et al. SANTE study group. Electrical stimulation of the anterior nucleus of thalamus for treatment of refractory epilepsy. Epilepsia. 2010;51(5):899-908.
Flanigin H.F., Hermann B.P., King D.W., et al. The history of surgical treatment of epilepsy in North America prior to 1975. In: Luders H., editor. Epilepsy surgery. New York: Raven Press, 1991.
Fuerst D., Shah J., Shah A., et al. Hippocampal sclerosis is a progressive disorder: A longitudinal volumetric MRI study. Ann Neurol. 2003;53:413.
Funke M., Constantino T., Van Orman C., et al. Magnetoencephalography and Magnetic Source Imaging in Epilepsy. Clin EEG Neurosci. 2009;40:271.
Galanopoulou A.S., Moshé S. The epileptic hypothesis: developmentally related arguments based on animal models. Epilepsia. 2009;50(Suppl 7):37.
Gillam F., Wyllie E., Kashden J., et al. Epilepsy surgery outcome: Comprehensive assessment in children. Neurology. 1997;48:1368.
Gleissner U., Sassen R., Lendt M., et al. Pre- and postoperative verbal memory in pediatric patients with temporal lobe epilepsy. Epilepsy Res. 2002;51:287.
Goldensohn E.S., Porter R.J., Schwartzkroin P.A. The American Epilepsy Society: An historic perspective on 50 years of advances in research. Epilepsia. 1997;38:124.
Grunwald F., Durwen H.F., Bockisch A., et al. Technetium-99m-HMPAO brain SPECT in medically intractable temporal lobe epilepsy: A postoperative evaluation. J Nucl Med. 1991;32:388.
Guerrini R. Epilepsy in chilren. Lancet. 2006;367(9509):499-524.
Hajek M., Antonini A., Leenders K.L., et al. Mesiobasal versus lateral temporal lobe epilepsy: Metabolic differences in the temporal lobe shown by interictal 18F-FDG positron emission tomography. Neurology. 1993;43:79.
Hajek M., Siegel A., Haldemann R., et al. Value of HMPAO-SPECT in selective temporal lobe surgery for epilepsy. J Epilepsy. 1991;4:43.
Hart Y.M., Cortez M., Andermann F., et al. Medical treatment of Rasmussen’s syndrome (chronic encephalitis and epilepsy): Effect of high-dose steroids or immunoglobulin in 19 patients. Neurology. 1994;44:1030.
Harvey A.S., Bowe J.M., Hopkins I.J., et al. Ictal 99Tc HMPAO single photon emission computed tomography in children with temporal lobe epilepsy. Epilepsia. 1993;34:869.
Harvey A.S., Hopkins I.J., Bowe J.M., et al. Frontal lobe epilepsy: Clinical seizure characteristics and localization with ictal 99mTc-HMPAO SPECT. Neurology. 1993;43:1966.
Harvey A.S., Nolan T., Carlin J.B. Community-based study of mortality in children with epilepsy. Epilepsia. 1993;34:597.
Harvey H., Cross J.H., Shinnar S., et al. Defining the spectrum of international practice in pediatric epilepsy surgery patients. Epilepsia. 2008;49:146.
Hathaway S., Kasnic K., Barkley G., et al. Comparison of medical and surgical costs for the treatment of intractable epilepsy. Epilepsia. 1995;36:95. (abstract)
Hauser W.A. The natural history of seizures. In: Wyllie E., editor. The treatment of epilepsy: Principles and practice. Philadelphia: Lea and Febiger, 1993.
Hauser W.A. The prevalence and incidence of convulsive disorders in children. Epilepsia. 1994;35(Suppl 2):S1.
Hauser W.A., Annegers J.F., Brodie M.J., et al. Mortality in patients with epilepsy. Epilepsia. 1980;21:399.
Hauser W.A., Annegers J.F., Kurland L.T. The prevalence of epilepsy in Rochester, Minnesota, 1940–1980. Epilepsia. 1991;32:429.
Hauser W.A., Annegers J.F., Rocca W.A. Descriptive epidemiology of epilepsy: Contributions of population-based studies from Rochester, Minnesota. Mayo Clin Proc. 1996;71:576.
Hauser W.A., Kurland L.T. The epidemiology of epilepsy in Rochester, Minnesota, 1935 through 1967. Epilepsia. 1975;16:1.
Hauser W.A., Rich S.S., Lee J.R., et al. Risk of recurrent seizures after two unprovoked seizures. N Engl J Med. 1998;338:429.
Henry T.R., Chugani H.T., Abou-Khalil B.W., et al. Positron emission tomography. In Engel J.Jr, editor: Surgical treatment of the epilepsies, ed 2, New York: Raven Press, 1993.
Hertz-Pannier L., Baillard W.D., Mott S.H., et al. Noninvasive assessment of language dominance in children and adolescents with functional MRI: a preliminary study. Neurology. 1997;48(4):1003-1012.
Hiritiz N., Mohanraj R., Norrie J., et al. Mortality in epilepsy. Epilepsy Behav. 2007;10:363.
Ho S.S., Berkovic S.F., Newton M.R., et al. Parietal lobe epilepsy: Clinical features and seizure localization by ictal SPECT. Neurology. 1994;44:2277.
Hoffman H.H., Hendrick E.G., Dennis M., et al. Hemispherectomy for Sturge-Weber syndrome. Child Brain. 1979;5:233.
Hogan R.E., Cook M.J., Kilpatrick C.J., et al. Accuracy of coregistration of single photon emission tomography with MR images using a brain surface matching technique. Am J Neuroradiol. 1996;17:793.
Holmes G.L. Benign focal epilepsies of childhood. Epilepsia. 1993;34:S49.
Holmes G.L., Ben-Ari Y. Seizures in the developing brain: Perhaps not so benign after all. Neuron. 1998;21:1231.
Holmes G.L., Gaiarsa J.-L., Chevassus-Au-Louis N., et al. Consequences of neonatal seizures in the rat: Morphological and behavioral effects. Ann Neurol. 1998;44:845.
Holmes G.L., Lenck-Santini P.-P. Role of interictal epileptiform abnormalities in cognitive impairment. Epilepsy Behav. 2006;8:504.
Hufnagel A., Zentner J., Fernandez G., et al. Multiple subpial transection for control of epileptic seizures: Effectiveness and safety. Epilepsia. 1997;38:678.
Hwang P.A., Gilday D.L., Adams C., et al. SPECT studies in epilepsy: Application to epilepsy surgery in children. J Epilepsy. 1990;3:83.
Iida K., Otsubo H., Matsumoto Y., et al. Characterizing magnetic spike sources by using magnetoencephalography-guided neuronavigation in epilepsy surgery in pediatric patients. J Neurosurg. 2005;102:187.
Jack C.R. Neuroimaging and anatomy: Magnetic resonance imaging. Neuroimag Clin North Am. 1995;5:1.
Jalava M., Sillanpaa M., Camfield C., et al. Social adjustment and competence 35 years after onset of childhood epilepsy: A prospective study. Epilepsia. 1997;38:708.
Jayakar P., Duchowny M.S. Complex partial seizures of temporal lobe origin in early childhood. J Epilepsy. 1990;3(Suppl):41.
Jokeit H., Ebner A., Arnold S., et al. Bilateral reductions of hippocampal volume, glucose metabolism, and Wada hemispheric memory performance are related to the duration of mesial temporal lobe epilepsy. J Neurol. 1999;246:926.
Jonas R., Nguyen S., Hu B., et al. Cerebral hemispherectomy: Hospital course, seizure, developmental, language, and motor outcomes. Neurology. 2004;62:1712-1721.
Jooma R., Yeh H.S., Privitera M.D., et al. Lesionectomy versus electrophysiologically guided resection for temporal lobe tumors manifesting with complex partial seizures. J Neurosurg. 1995;83:231.
Juhasz C., Chugani D.C., Muzik O., et al. Alpha-methyl-L-tryptophan PET detects epileptogenic cortex in children with intractable epilepsy. Neurology. 2003;60:960.
Juhasz C., Chugani D.C., Muzik O., et al. Electroclinical correlates of flumazenil and fluorodeoxyglucose PET abnormalities in lesional epilepsy. Neurology. 2000;55:825.
Juhasz C., Chugani D.C., Muzik O., et al. Relationship of flumazenil and glucose PET abnormalities to neocortical epilepsy surgery outcome. Neurology. 2001;56:1650.
Juhasz C., Chugani D.C., Padhye U.N., et al. Evaluation with alpha-(11C)methyl-L-tryptophan positron emission tomography for reoperation after failed epilepsy surgery. Epilepsia. 2004;45:124.
Juhasz C., Nagy F., Muzik O., et al. [11C]flumazenil PET in patients with epilepsy with dual pathology. Epilepsia. 1999;40:566.
Juul-Jensen P., Foldsprang A. Natural history of epileptic seizures. Epilepsia. 1983;24:297.
Kadis D.S., Lida K., Kerr E.N., et al. Intrahemispheric reorganization of language in children with medically intractable epilepsy of the left hemisphere. J Int Neuropsychol Soc. 2007;13(3):505-516.
Kahane P., Depaulis A. Deep brain stimulation in epilepsy : what is next? Curr Opin Neurol. 2010;23:177-183.
Kalviainen R., Salmenpera T., Partanen K., et al. Recurrent seizures may cause hippocampal damage in temporal lobe epilepsy. Neurology. 1998;50:1377.
Kan P., Van Orman C., Kestle J.R.W. Outcomes after surgery for focal epilepsy in children. Childs Nerv Syst. 2008;24(5):587-591.
Katz A., Bose A., Lind S.J., et al. SPECT in patients with epilepsia partialis continua. Neurology. 1990;40:1848.
Keene D.L., Loy-English I., Ventureya E.C. Long-term socioeconomic outcome following surgical intervention in the treatment of refractory epilepsy in childhood and adolescence. Childs Nerv Syst. 1998;14:362.
Kim S.K., Wang K.C., Hwang Y.S., et al. Epilepsy surgery in children: outcomes and complications. J Neurosurg Pediatr. 2008;1(4):277-283.
King D.W., Flanigin H.F., Gallagher B.B., et al. Temporal lobectomy for partial complex seizures: Evaluation, results and 1-year follow-up. Neurology. 1986;36:334.
Knowlton R.C. The role of FDG-PET, ictal SPECT, and MEG in the epilepsy surgery evaluation. Epilepsy Behav. 2006;8:91-101.
Knowlton R.C., Shih J. Magnetoencephalography in epilepsy. Epilepsia. 2004;45(Suppl 4):61.
Kobayashi K., Ohtsuka Y., Ohno S., et al. Clinical spectrum of epileptic spasms associated with cortical malformation. Neuropediatrics. 2001;32:236.
Koh S., Jayakar P., Dunoyer C., et al. Epilepsy surgery in children with tuberous sclerosis complex: Presurgical evaluation and outcome. Epilepsia. 2000;41:1206.
Kotloski R., Lynch M., Lauersdorf S., et al. Repeated brief seizures induce progressive hippocampal neuron loss and memory deficits. Prog Brain Res. 2002;135:95.
Krauss G.L., Campbell M.L., Roche K.W., et al. Chronic steroid-responsive encephalitis without autoantibodies to glutamate receptor GluR3. Neurology. 1996;46:247.
Kuks J.B.M., Cook K.J., Fish D.R., et al. Hippocampal sclerosis in epilepsy and childhood febrile seizures. Lancet. 1993;342:1391.
Kuzniecky R., Cascino G., Palmini A., et al. Structural neuroimaging. In: Engel J.E., editor. Surgical treatment of the epilepsies. New York: Raven Press, 1993.
Kuzniecky R., Mountz J.M., Wheatley G., et al. Ictal single photon emission computed tomography demonstrates localized epileptogenesis in cortical dysplasia. Ann Neurol. 1993;34:627.
Kuzniecky R., Murro A., King D., et al. Magnetic resonance imaging in childhood intractable partial epilepsies: Pathologic correlations. Neurology. 1993;43:681.
Kuzniecky R., Elgavish G.A., Hetherington H.P., et al. In vivo 31P nuclear magnetic resonance spectroscopy of human temporal lobe epilepsy. Neurology. 1992;42:1586.
Kuzniecky R.I., Barkovich A.J. Malformations of cortical development and epilepsy. Brain Dev. 2001;23:2.
Kuzniecky Ruben I. MRI in cerebral developmental malformations and epilepsy. Magn Reson Imaging. 1995;13:1137.
Kwan P., Brodie M.J. Early identification of refractory epilepsy. N Engl J Med. 2000;342:314.
Kwong K.K., Belliveau J.W., Chesler D.A., et al. Dynamic magnetic resonance imaging of human brain activity during primary sensory stimulation. Proc Natl Acad Sci USA. 1992;89:5675.
Lah S. Neuropsychological outcome following focal cortical removal for intractable epilepsy in children. Epilepsy Behav. 2004;5(6):804-817.
Lamanna M.M., Sussman N.M., Harner R.N., et al. Initial experience with SPECT imaging of the brain using I-123 p-iodoamphetamine in focal epilepsy. Clin Nucl Med. 1989;14:428.
Lamm C., Windischberger C., Leodolter U., et al. Co-registration of EEG and MRI data using matching of spline interpolated and MRI-segmented reconstructions of the scalp surface. Brain Topogr. 2001;14:93.
Lanceman M.E., Morris H.H., Raja S., et al. Usefulness of ictal and interictal 99mTc ethyl cysteinate dimer single photon emission computed tomography in patients with refractory partial epilepsy. Epilepsia. 1997;38:472.
Langfitt J.T., Holloway R.G., McDermott M.D., et al. Health Care Costs Decline after successfull epilepsy surgery. Neurology. 2007;68(16):1290-1298.
Laoprasert P., Zupanc M.L. Malformations of cortical development and epilepsy surgery outcome in children and adults. Epilepsia. 1997;38:78.
Launes J., Iivanainen M., Salmi T., et al. Interictal brain 99m-Tc-HMPAO SPECT hypoperfusion in patients with unstable partial epilepsy and normal CT. Acta Neurol Scand. 1992;86:558.
LaViolette P.S., Rand S.D., Raghavan M., et al. 3D visualization of subdural electrodes for presurgical planning. Neurosurgery. 2011;68(1 Suppl operative):152-161.
Laxer K.D., Hubesch B., Sappey M.D., et al. Increased pH and inorganic phosphate in temporal seizure foci demonstrated by 31P MRS. Epilepsia. 1992;33:178.
Lee B.I., Markand O.N., Wellman H.N., et al. HIPDM-SPECT in patients with medically intractable complex partial seizures: Ictal study. Arch Neurol. 1988;46:397.
Lee C.C., Riederer S.J., Jack C.R. The specificity of sensorimotor mapping using functional MRI. Epilepsia. 1996;37:206.
Leiderman O.B., Balish M., Sato S., et al. Comparison of PET measurements of cerebral blood flow and glucose metabolism for the localization of human epileptic foci. Epilepsy Res. 1992;13:153.
Lendt M., Gleissner U., Helmstaedter C., et al. Neuropsychological outcome in children after frontal lobe epilepsy surgery. Epilepsy Behav. 2002;3:51.
Liegeois F., Connelly A., Cross J.H., et al. Language reorganization in children with early onset lesions of the left hemisphere: an fMRI study. Brain. 2004;127(pt 6):1229-1236.
Liegeois F., Connelly A., Salmond C.H., et al. A direct test for lateralization of language activation using fMRI : comparison with invasive assessments in children with epilepsy. Neuroimage. 2002;17(4):1861-1867.
Locock C. Discussion of a paper by EH Sievking: Meeting of the Royal Medical and Chirurgical Society of London. Lancet. 1857;1:527.
Loddenkemper T., Holland K.D., Stanford L.D., et al. Developmental outcome after epilepsy srgery in infancy. Pediatrics. 2007;119(5):930-935.
Logan W.J., Benson R.R., Cosgrove G.R., et al. Functional MRI (fMRI) localization of language in children. Can J Neurol Sci. 1995;22:24.
Logan W.J., Crawley A.P., Mikulis D.J., et al. An imaging technique particularly suited for children. Ann Neurol. 1997;42:530.
Logan W.J., McAndrews M.P., Crawley A., et al. Functional MRI (fMRI) language localization and lateralization in children using object naming and word generation tasks. Ann Neurol. 1998;44:551.
Loring D., Meador K., Lee G., et al. Amobarbital effects and lateralized brain function. New York: Springer-Verlag; 1992.
Loring D.W. Neuropsychological evaluation in epilepsy surgery. Epilepsia. 1997;38(Suppl 4):S18.
Mabbott D.M., Smith M.L. Material-specific memory in children with temporal and extratemporal lobectomies. Neuropsychologia. 2004;41:995.
Madan N., Grant P.E. New directions in clinical imaging of cortical dysplasias. Epilepsia. 2009;50(Suppl 9):9-18.
Maehara T., Shimizu H. Surgical outcome of corpus callosotomy in patients with drop attacks. Epilepsia. 2001;42:67.
Marks D.A., Katz A., Hoffer P., et al. Localization of extra-temporal epileptic foci during ictal single photon emission computed tomography. Ann Neurol. 1992;31:250.
Matsuda H., Matsuda K., Nakamura F., et al. Contribution of subtraction ictal SPECT coregistered to MRI to epilepsy surgery: a multicenter study. Ann Nucl Med. 2009;23:283-291.
Matthews P.M., Andermann F., Arnold D.L. A proton magnetic resonance spectroscopy study of focal epilepsy in humans. Neurology. 1990;40:985.
McInerney J., Siegel A.M., Nordgren R.E., et al. Long-term seizure outcome following corpus callosotomy in children. Stereotact Funct Neurosurg. 1999;73:79.
Meador K.J. Cognitive outcomes and predictive factors in epilepsy. Neurology. 2002;58:S21-S26.
Meyer F.B., Marsh W.R., Lawa E.R., et al. Temporal lobectomy in children with epilepsy. J Neurosurg. 1986;64:371.
Meyer S., Sharndeen M., Gottschling S., et al. Sudden unexpected death in epilepsy in children. J Paediatr Child Health. 2010:1-6.
Mihara T., Inoue Y., Watanabe Y., et al. Improvement in quality-of-life following resective surgery for temporal lobe epilepsy: Results of patient and family assessments. Jpn J Psychiatry Neurol. 1994;48:221.
Minassian B.A., Otsubo H., Weiss S., et al. Magnetoencephalographic localization in pediatric epilepsy surgery: Comparison with invasive intracranial electroencephalography. Ann Neurol. 1999;46:627.
Mizrahi E.M., Kellaway P., Grossman R.G., et al. Anterior temporal lobectomy and medically refractory temporal lobe epilepsy of childhood. Epilepsia. 1990;31:302.
Mohan K.K., Chugani D.C., Chugani H.T. Positron emission tomography in pediatric neurology. Semin Ped Neurol. 1999;6:111.
Mohanraj R., Brodie M.J. Diagnosing refractory epilepsy: response to sequential treatment schedules. Eur J Neurol. 2006;13:277-282.
Montes J.L., Rosenblatt B., Farmer J.P., et al. Lesionectomy of MRI detected lesions in children with epilepsy. Pediatr Neurosurg. 1995;22:167.
Morrell F., Whisler W.W., Smith M.C., et al. Landau Kleffner syndrome: Treatment with subpial intracortical transection. Brain. 1995;118:1529.
Mountz J.M., Zhang B., Liu H.G., et al. A reference method for correlation of anatomic and functional brain images: Validation and clinical applications. Semin Nucl Med. 1994;4:256.
Nashef L., Fish D.R., Sander J.W., et al. Incidence of sudden unexpected death in adult outpatient cohort with epilepsy at a tertiary referral centre. J Neurol Neurosurg Psychiatry. 1995;58:462.
Newton M.R., Berkovic S.F., Austin M.C., et al. A postictal switch in blood flow distribution characterizes human temporal lobe seizures. J Neurol Neurosurg Psychiatry. 1992;55:891.
Newton M.R., Berkovic S.F., Austin M.C., et al. SPECT in the localization of extratemporal and temporal epilepsy. J Neurol Neurosurg Psychiatry. 1995;59:26.
Ng Y.T., McGregor A.L., Wheless J.W. Magnetic resonance imaging detection of mesial temporal sclerosis in children. Pediatr Neurol. 2004;30:81.
Novotny E.J. Overview – the role of NMR spectroscopy. Magn Reson Imaging. 1995;13:1171.
O’Brien T.J., So E.L., Cascino G.D., et al. Subtraction SPECT coregistered to MRI in focal malformations of cortical development: Localization of epileptogenic zone in epilepsy surgery candidates. Epilepsia. 2004;45:367.
O’Brien T.J., So E.L., Mullan B.P., et al. Subtraction ictal SPECT coregistered to MRI improves clinical usefulness of SPECT in localizing the surgical seizure focus. Neurology. 1998;50:445.
O’Brien T.J., So E.L., Mullan B.P., et al. Subtraction peri-ictal SPECT is predictive of extratemporal epilepsy surgery outcome. Neurology. 2000;55:1668.
O’Shaughnessy ES, Berl M.M., Moore E.N., et al. Pediatric functional magnetic resonance imaging (fMRI): issues and applications. J Child Neurol. 2008;23(7):791-801.
Ogawa S., Tank D., Menon R., et al. Intrinsic signal changes accompanying sensory stimulation: Functional brain mapping with magnetic resonance imaging. Proc Natl Acad Sci USA. 1992;89:5951.
Ogunmekan A.O., Hwang P.A., Hoffman H.J. Sturge-Weber-Dimitri disease: Role of hemispherectomy in prognosis. Can J Neurol Sci. 1991;78:80.
Ojemann G.A. Organization of short-term verbal memory in language areas of the human cortex: Evidence from electrical stimulation. Brain Lang. 1978;5:331.
Ojemann G.A. Individual variability in cortical localization of language. J Neurosurg. 1979;50:164.
Ojemann G.A. Cortical stimulation. In Engel J.Jr, editor: Surgical treatment of the epilepsies, ed 2, New York: Raven Press, 1993.
Ojemann G., Dodrill C.B. Intraoperative techniques for reducing language and memory deficits with left temporal lobectomy. In: Wolf P., Dam M., Janz D., et al, editors. Advances in epileptology. XVIth Epilepsy International Symposium. New York: Raven Press, 1987.
Olafsson E., Hauser W.A., Ludvigsson P., et al. Incidence of epilepsy in rural Iceland: A population based study. Epilepsia. 1996;37:951.
Olson D.M., Chugani H., Shewmon A., et al. Electrocorticographic confirmation of focal positron emission tomographic abnormalities in children with intractable epilepsy. Epilepsia. 1990;31:731.
Osuntokun B.O., Adeuja A.O.G., Nottidge V.A., et al. Prevalence of the epilepsies in Nigerian Africans: A community-based study. Epilepsia. 1987;28:272.
Palmini A., da Costa J.C., Calcogrotto M.E., et al. Patients with specific histopathological types of cortical dysplasia have specific degrees of severity of the epileptic condition – a study of 78 patients. Epilepsia. 1997;38:5.
Palmini A., Gambardella A., Andermann F., et al. Intrinsic epileptogenicity of human dysplastic cortex as suggested by corticography and surgical results. Ann Neurol. 1995;37:476.
Paolicchi J.M., Jayakar P., Dean P., et al. The outcome of focal resection in preadolescent children with intractable partial epilepsy. Neurology. 2000;54:642.
Papanicolaou A.C., Simos P.G., Castillo E.M., et al. Magnetocephalography: A noninvasive alternative to the Wada procedure. J Neurosurg. 2004;100:867.
Pardo C.A., Vining E.P.G., Guo L., et al. The pathology of Rasmussen syndrome: Stages of cortical involvement and neuropathological studies in 45 hemispherectomies. Epilepsia. 2004;45:516.
Peacock W.J. Hemispherectomy for the treatment of intractable seizures in childhood. Neurosurg Clin N Am. 1995;6:549.
Penfield W. Pitfalls and success in surgical treatment of focal epilepsy. BMJ. 1958;45:669.
Penfield W., Flanigin H. Surgical therapy of temporal lobe seizures. Arch Neurol Psychiatry. 1950;64:491.
Peresson M., Lopez L., Narici L., et al. Magnetic source imaging and reactivity to rhythmical stimulation in tuberous sclerosis. Brain Dev. 1998;20:512.
Perrine K. Future directions for functional mapping. Epilepsia. 1994;35:S90.
Perucca E., French J., Bialer M. Development of new antiepileptic drugs: challenges, incentives, and recent advances. Lancet Neurol. 2007;6:793.
Petroff O.A.C., Prichard J.W., Behar K.L., et al. In vivo phosphorus nuclear magnetic resonance spectroscopy in status epilepticus. Ann Neurol. 1984;16:169.
Petroff O.A.C., Prichard J.W., Ogino T., et al. Combined 1H and 31P nuclear magnetic resonance studies of bicuculline-induced seizures in vivo. Ann Neurol. 1986;20:185.
Pilcher W.H., Silbergeld D.L., Berger M.S., et al. Intraoperative electrocorticography during tumor resection: Impact on seizure outcome in patients with gangliogliomas. J Neurosurg. 1993;78:891.
Powell N.W., Koepp M.J., Richardson M.P., et al. The application of fMRI of memory in temporal lobe epilepsy: A clinical review. Epilepsia. 2004;45:855.
Prichard J.W. Nuclear magnetic resonance spectroscopy of seizure states. Epilepsia. 1994;35:514.
Radtke R.A., Hanson M.W., Hoffman J.M., et al. Temporal lobe hypometabolism on PET: Predictor of seizure control after temporal lobectomy. Neurology. 1993;43:1088.
Raymond A.A., Fish D.R., Sisdiya S.M., et al. Abnormalities of gyration, heterotopias, tuberous sclerosis, focal cortical dysplasia, microdysgenesis, dysembryoplastic neuroepithelial tumor and dysgenesis of the archiocortex in epilepsy – clinical, EEG and neuroimaging features in 100 adult patients. Brain. 1995;118:629.
Reeves A.L., So E.L., Evans R.W., et al. Factors associated with work outcome after anterior temporal lobectomy for intractable epilepsy. Epilepsia. 1997;38:689.
Roach E.S., Ricla A.R., Chugani H.T., et al. Sturge-Weber syndrome: Recommendations for surgery. J Child Neurol. 1994;9:190.
Rogers S.W., Andrews P.I., Gahring L.C., et al. Autoantibodies to glutamate receptor 3 in Rasmussen’s encephalitis. Science. 1994;265:648.
Romanelli P., Verdecchia M., Rodas R., et al. Epilepsy surgery for tuberous sclerosis. Pediatr Neurol. 2004;31:239.
Rowe C.C., Berkovic S.F., Austin M., et al. Patterns of postictal cerebral blood flow in temporal lobe epilepsy: Qualitative and quantitative analysis. Neurology. 1991;41:1096.
Rowe C.C., Berkovic S.F., Sia S.T.B., et al. Localization of epileptic foci with postictal single photon emission computed tomography. Ann Neurol. 1989;26:660.
Rugg-Gunn F.J., Eriksson S.H., Symms M.R., et al. Diffusion tensor imaging of cryptogenic and acquired partial epilepsies. Brain. 2001;124:627.
Ryding E., Rosen I., Elmqvist D., et al. SPECT measurements with 99mTc-HM-PAO in focal epilepsy. J Cereb Blood Flow Metab. 1988;8(Suppl 1):S95.
Ryvlin P., Garcia-Larrea L., Philippon B., et al. High signal intensity on T2-weighted MRI correlates with hypoperfusion in temporal lobe epilepsy. Epilepsia. 1992;33:28.
Sabsevitz D.S., Swanson S.J., Hammeke T.A., et al. Use of pre-operative functional neuroimaging to predict language deficits from epilepsy surgery. Neurology. 2003;60:1788.
Sachs B.C., Gaillard W.D. Organization of language networks in children: functional magnetic resonance imaging studies. Curr Neurol Neurosci Rep. 2003;3(2):157-162.
Sackellares J.C., Siegel G.J., Abou-Khalil B.W., et al. Differences between lateral and mesial temporal metabolism interictally in epilepsy of mesial temporal origin. Neurology. 1990;40:1420.
Sassower K.C., Rollinson D.C., Duchowny M. Outcome of corpus callosotomy and other pediatric epilepsy surgery: Parental perceptions. Epileptic Disord. 2001;3:197.
Schmidt D., Tsai J.J., Janz D. Generalized tonic-clonic seizures in patients with complex partial seizures: Natural history and prognostic relevance. Epilepsia. 1983;24:43.
Schramm J., Aliashkevich A.F., Grunwald T. Multiple subpial transactions: Outcome and complications in 20 patients who did not undergo resection. J Neurosurg. 2002;97:39.
Scott D.F. The history of epileptic therapy: Account of how medication was published. Pearl River, NY: Parthenon Publishing; 1993.
Scott R.C., Gadian D.G., Cross J.H., et al. Quantitative magnetic resonance characterization of mesial temporal sclerosis in childhood. Neurology. 2001;56(12):1659-1665.
Shafer S.Q., Hauser W.A., Annegers J.F., et al. EEG and other early predictors of epilepsy remission: A community study. Epilepsia. 1988;29:590.
Shah A., Watson C., Chugani H.T., et al. Comparison between (C-11) flumazenil (FMZ) and (F-18) fluorodeoxyglucose (FDG PET) scanning in temporal lobe epilepsy. American Epilepsy Society Meeting, December 1995. Epilepsia. 1995;36:165. (abstract)
Shen W., Lee B.I., Park H.M., et al. HIPDM-SPECT brain imaging in the presurgical evaluation of patients with intractable seizures. J Nucl Med. 1990;31:1280.
Shields W.D. Catastrophic epilepsy of childhood. Epilepsia. 2000;41(Suppl 2):S2.
Sillanpaa M., Jalva M., Kaleva O., et al. Long-term prognosis of seizures with onset in childhood. N Engl J Med. 1998;338:1916.
Simos P.G., Papanicolaou A.C., Breier J.I., et al. Localization of language-specific cortex by using magnetic source imaging and electrical stimulation mapping. J Neurosurg. 1999;91:787.
Sinclair B.D., Aronyk K.E., Snyder T.J., et al. Pediatric epilepsy surgery at the University of Alberta: 1988–2000. Pediatr Neurol. 2003;29:302.
Sinclair D.B., Wheatley M., Aronyk K., et al. Pathology and neuroimaging in pediatric temporal lobectomy in intractable epilepsy. Pediatr Neurosurg. 2001;35:239.
Smart O., Worrell G., Litt B., et al. Automatic detection of HFEO from focal intracranial EEG using fuzzy c-means clustering [Abstract]. British Mechanical Engineering Science. 2004.
Smith M.L., Elliott I.M., Lach L. Cognitive, psychosocial, and family function one year after pediatric epilepsy surgery. Epilepsia. 2004;45:650.
So E., Bainbridge J., Buchhalter J.R., et al. Report of the American Epilepsy Society and the Epilepsy Foundation joint task force on sudden unexplained death in epilepsy. Epilepsia. 2009;50:917-922.
Sofijanov N.G. Clinical evolution and prognosis of childhood epilepsies. Epilepsia. 1982;21:61.
Solinas C., Briellmann R.S., Harvery A.S., et al. Hypertensive encephalopathy: antecedent to hippocampal sclerosis and temporal lobe epilepsy? Neurology. 2003;60(9):1534-1536.
Sorenson J.M., Wheless J.W., Baumgartner J.E., et al. Corpus callosotomy for medically intractable seizures. Pediatr Neurosurg. 1997;27:260.
Spencer S.S. The relative contributions of MRI, SPECT, and PET imaging in epilepsy. Epilepsia. 1994;35:572.
Spencer S.S. Long-term outcome after epilepsy surgery. Epilepsia. 1996;37:807.
Spencer S.S., Schramm J., Wyler A., et al. Multiple subpial transaction for intractable partial epilepsy: An international meta-analysis. Epilepsia. 2002;43:141.
Stafstrom C., Lynch M., Sutula T. Consequences of epilepsy in the developing brain: Implications for surgical management. Semin Pediatr Neurol. 2000;7:147.
Stapleton S.R., Kiriakopoulos E., Mikulis D., et al. Combined utility of functional MRI, cortical mapping, and frameless stereotaxy in the resection of lesions in eloquent areas of brain in children. Pediatr Neurosurg. 1997;26(2):68-82.
Stefan H., Bauer J., Feistel H., et al. Regional cerebral blood flow during focal seizures of temporal and frontocentral onset. Ann Neurol. 1990;27:162.
Stefan H., Hummel C., Scheler G., et al. Magnetic brain source imaging of focal epileptic activity: A synopsis of 455 cases. Brain. 2003;126:2396.
Stefan H., Pawlik G., Bocher-Schwarz H.G., et al. Functional and morphological abnormalities in temporal lobe epilepsy: A comparison of interictal and ictal EEG, CT, SPECT, and PET. J Neurol. 1987;234:377.
Stufflebeam S.M., Tanaka N., Ahlfors S.P. Clinical applications of magnetoencephalography. Hum Brain Mapp. 2009;30:1813.
Sutherling W.W., Crandall P., Cahan L.D., et al. The magnetic field of epileptic spikes agrees with intracranial localizations in complex partial epilepsy. Neurology. 1988;38:778.
Sutula T.P. Mechanisms of epilepsy progression: current theories and perspectives from neuroplasticity in adulthood and development. Epilespy Res. 2004;60(2–3):161-171.
Sutula T.P., Cascino G., Cavazos F., et al. Mossy fiber synaptic reorganization in the epileptic human temporal lobe. Ann Neurol. 1989;26:321.
Sutula T.P., He X.X., Davazoa J., et al. Synaptic reorganization in the hippocampus induced by abnormal functional activity. Science. 1988;26:321.
Swartz B.E., Tomiyasu U., Delgado-Escueta A.V., et al. Neuroimaging in temporal lobe epilepsy: Test sensitivity and relationship in pathology and post-surgical outcome. Epilepsia. 1992;33:624.
Szabo C.A., Wyllie E., Stanford L.D., et al. Neuropsychological effect of temporal lobe resection in preadolescent children with epilepsy. Epilepsia. 1998;39:814.
Tan K.M., Britton J.W., Buchhalter J.R., et al. Influence of subtraction ictal SPECT on surgical management in focal epilepsy of indeterminate localization: A prospective study. Epilepsy Res. 2008;82:190-193.
Tasch E., Cendes F., Li L.M., et al. Neuroimaging evidence of progressive neuronal loss and dysfunction in temporal lobe epilepsy. Ann Neurol. 1999;45:568.
Taylor D.C., Neville B.G.R., Cross J.H. New measures of outcome needed for surgical treatment of epilepsy. Epilepsia. 1997;38:625.
Theodore W.H., Dorwart R., Holmes M., et al. Neuroimaging in refractory partial seizures: Comparison of PET, CT, and MRI. Neurology. 1986;36:750.
Theodore W.H., Epstein L., Gaillard W.D., et al. Human herpes virus 6B: a possible role in epilepsy? Eplepsia. 2008;49(11):1828-1837.
Theodore W.H., Katz D., Kufta C., et al. Pathology of temporal lobe foci: Correlation with CT, MRI and PET. Neurology. 1990;40:797.
Theodore W.H., Sato S., Kufta C.V., et al. FDG positron emission tomography and invasive EEG: Seizure focus detection and surgical outcome. Epilepsia. 1997;38:81.
Thomas-Sohl K.A., Vaslow D.F., Maria B.L. Sturge-Weber syndrome: A review. Pediatr Neurol. 2004;30:303.
Tice H., Barnes P.D., Goumnerova L., et al. Pediatric and adolescent oligodendrogliomas. Am J Neuroradiol. 1993;14:1293.
Todt H. The late prognosis of epilepsy in childhood: Results of a prospective follow-up study. Epilepsia. 1984;25:137.
Tonini C., Beghi E., Berg A.T., et al. Predictors of epilepsy surgery outcome: A meta-analysis. Epilepsy Res. 2004;62:75.
Tovar-Spinoza Z.S., Ochi A., Rutka J.T., et al. The role of magnetoencephalography in epilepsy surgery. Neurosurg Focus. 2008;25:1.
Trenerry M.R., Jack C.R., Sharbrough F.W., et al. Quantitative MRI hippocampal volumes: Association with onset and duration of epilepsy and febrile convulsions in temporal lobectomy patients. Epilepsy Res. 1993;15:247.
Unnwongse K., Wehner T., Foldvary-Schaefer N. Selecting Patients for Epilepsy Surgery. Curr Neurol Neurosci Rep. 2010;10:299-307.
Vali A.M., Clarke M.A., Kelsey A. Dysembryoplastic neuroepithelial tumors as a potentially treatable cause of intractable epilepsy in children. Clin Radiol. 1993;47:255.
Valk P.E., Laxer K.D., Barbaro N.M., et al. High-resolution (2.6 mm) PET in partial complex epilepsy associated with mesial temporal sclerosis. Radiology. 1993;186:55.
Van Empelen R., Jennekens-Schinkel A., Buskens E., et al. Functional consequences of hemispherectomy. Brain. 2004;127:2071.
Van Hirtum-Das M., Licht E.A., Koh S., et al. Children with ESES: variability in the syndrome. Epilepsy Res. 2006;70(Suppl 1):S248-S258.
Velasco A.L., Velasco F., Jimenex F., et al. Neuromodulation of the centromedian thalamic nuclei in the treatment of generalized seizures and the improvement of the quality of life in patients with Lennox-Gastaut syndrome. Epilepsia. 2006;47:1203-1212.
Velasco F., Velasco M., Ogarrio C., et al. Electrical stimulation of the centromedian thalamic nucleus in the treatment of convulsive seizures: a preliminary report. Epilepsia. 1997;28:421-430.
Verhoeff N.P.L.G., Weinstein H.C., Aldenkamp A.P., et al. Focus localization in patients with partial epilepsy with 99mTc HMPAO SPECT under continuous surface EEG monitoring. Nucl Med Commun. 1992;13:127.
Vigevano F., Bertini E., Boldrini R., et al. Hemimegalencephaly and intractable epilepsy: Benefits of hemispherectomy. Epilepsia. 1989;30:833.
Vigevano F., DiRocco C. Effectiveness of hemispherectomy in hemimegalencephaly with intractable seizures. Neuropediatrics. 1990;21:222.
Vining E.P.G., Freeman J.M., Pillas D.J., et al. Why would you remove half a brain? The outcome of 58 children after hemispherectomy – the Johns Hopkins experience: 1968 to 1996. Pediatrics. 1997;100:163.
Watanabe K., Negoro T., Okumura A. Symptomatology of infantile spasms. Brain Dev. 2001;23:453.
Weiner H.L., Carlson C., Ridgway E.G., et al. Epilepsy surgery in young children with tuberous sclerosis: results of a novel approach. Pediatrics. 2006;117:1494.
Westerveld M., Sass K.J., Chelune G.L., et al. Temporal lobectomy in children: Cognitive outcome. J Neurosurg. 2000;92:24.
Wheless J.W., Castillo E., Maggio V., et al. Magnetoencephalography (MEG) and magnetic source imaging (MSI). Neurologist. 2004;10:138.
Wheless J.W., Willmore L.J., Breier J.I., et al. A comparison of magnetoencephalography, MRI and V-EEG in patients evaluated for epilepsy surgery. Epilepsia. 1999;40:931.
Whittle I.R., Ellis H.J., Simpson D.A. The surgical treatment of intractable childhood and adolescent epilepsy. Aust N Z J Surg. 1981;51:190.
Wiebe S., Blume W.T., Girvin J.P., et al. A randomized, controlled trial of surgery for temporal-lobe epilepsy. N Engl J Med. 2001;345:311.
Worrell G.A., Parish L., Craunstoun S.D., et al. High frequency oscillations and seizure generalization in neocortical epilepsy. Brain. 2004;127:1496.
Wyler A.R. Corpus callosotomy: The treatment of epilepsy. In: Wyllie E., editor. Principles and practice. Philadelphia: Lea and Febiger, 1993.
Wyllie E. Developmental aspects of seizure semiology: Problems in identifying localized onset seizures in infants and children. Epilepsia. 1995;36:1170.
Wyllie E. Surgical treatment of epilepsy in children. Pediatr Neurol. 1998;19:179.
Wyllie E., Chee M., Granström M.L., et al. Temporal lobe epilepsy in early childhood. Epilepsia. 1993;34:859.
Wyllie E., Comair Y.G., Kotagal P., et al. Epilepsy surgery in infants. Epilepsia. 1996;37:625.
Wyllie E., Comair Y.G., Kotagal P., et al. Seizure outcome after epilepsy surgery in children and adolescents. Ann Neurol. 1998;44:740.
Wyllie E., Lachhwani D.K., Gupta A., et al. Successful surgery for epilepsy due to early brain lesions despite generalized EEG findings. Neurology. 2007;69(4):389-397.
Wyllie E., Luders H., Murphy D., et al. Intracranial amobarbital (Wada) test for language dominance: Correlation with results of cortical stimulation. Epilepsia. 1990;31:156.
Yoon D., Frick K., Carr D., et al. Economic impact of epilepsy in the United States. Epilepsia. 2009;50:2186.
Yun C.H., Lee S.K., Lee S.Y., et al. Prognostic factors in neocortical epilepsy surgery: multivariate analysis. Epilepsia. 2006;47:574-579.
Zubal I.G., Spencer S.S., Imam K., et al. Difference images calculated from ictal and interictal technetium-99m-HMPAO SPECT scans of epilepsy. J Nucl Med. 1995;36:684.
Zupanc M.L. Neuroimaging in the evaluation of children and adolescents with intractable epilepsy: I. Magnetic resonance imaging and the substrates of epilepsy. Pediatr Neurol. 1997;17:19.
Zupanc M.L. Neuroimaging in the evaluation of children and adolescents with intractable epilepsy: II. Neuroimaging and pediatric epilepsy surgery. Pediatr Neurol. 1997;17:111.
Zupanc M.L., Abrahamson K.D., Cascino G.D., et al. Efficacy of surgery on seizure control and resultant quality of life in pediatric patients – Mayo Clinic experience, 1988–1994. Epilepsia. 1996;37:105.
Zupanc M.L., dos Santos Rubio E., Werner R., et al. Epilepsy Surgery Outcomes: Quality of Life and Seizure Control. Pediatr Neurol. 2010;42:12. in press
Zupanc M.L., Lin J., Lew S., et al. Corpus callosotomy and multistage resection in medically refractory pediatric epilepsy patients. 2011.