[level-membership-for-neurology-category]
Chapter 67 Epilepsies
Seizures and Epilepsy Definitions
Seizures are transient events that include symptoms and/or signs of abnormal excessive hypersynchronous activity in the brain (Fisher et al., 2005). In 2005, the International League Against Epilepsy (ILAE) and International Bureau for Epilepsy (IBE) proposed a definition of epilepsy as a disorder of the brain characterized by an enduring predisposition to generate epileptic seizures and by the neurobiological, cognitive, psychological, and social consequences of this condition (Fisher et al., 2005). Acute symptomatic seizures provoked by metabolic or toxic derangements or occurring acutely in the setting of head trauma or stroke do not define epilepsy.
The traditional definition of epilepsy requires at least two unprovoked seizures. The definition proposed by the ILAE in 2005 suggested that one epileptic seizure is sufficient to diagnose epilepsy if there is additional enduring alteration in the brain that increases the likelihood of future seizures, but the proposal did not specify what evidence is sufficient to define such an enduring alteration. The proposed definition has been controversial and has not been widely accepted (Beghi et al., 2010). The definition of epilepsy used in this chapter requires at least two unprovoked seizures, so a single unprovoked seizure is insufficient to define epilepsy.
In 2001, the ILAE proposed a diagnostic scheme for the classification of seizures and epilepsy (Engel, 2001). The scheme recommended axes that are helpful concepts in the evaluation of patients with epilepsy:
Ictal Phenomenology
Glossary of Seizure Terminology and Other Definitions
The terms frequently used in the description of seizures follow. Whenever possible, the definition is derived from the glossary of descriptive terminology for ictal semiology, reported by the ILAE task force on classification and terminology (Blume et al., 2001). The term ictal semiology means the signs and symptoms associated with seizures.
Automatisms can be described by the part of the body affected. Some of the most common are oroalimentary automatisms, which include lip smacking, chewing, swallowing, and other mouth movements. Ictal spitting and ictal drinking can be considered forms of oroalimentary automatisms. Automatisms affecting the distal extremities are manual or pedal. Manual or pedal automatisms can be bilateral or unilateral. Gestural automatisms include extremity movements such as those used to enhance speech. More recently introduced categories for upper extremity automatisms are manipulative and non-manipulative (Kelemen et al., 2010). Manipulative automatisms involve picking and fumbling motions, typically reflecting interaction with the environment. Non-manipulative upper extremity automatisms tend to be rhythmic and do not involve interaction with the environment. Distal non-manipulative upper-extremity automatisms have been described with the acronym RINCH (rhythmic ictal nonclonic hand) movements (Lee et al., 2006). Hyperkinetic automatisms imply an inappropriately rapid sequence of movements that predominantly involve axial and proximal limb muscles. The resulting motion can be thrashing, rocking, pelvic thrusting, kicking, or bicycling motions. Gelastic refers to abrupt laughter or giggling, while dacrystic refers to abrupt crying, both inappropriate.
Classification of Seizures
Two classifications developed by the ILAE continue to be used widely: the Clinical and Electroencephalographic Classification of Epileptic Seizures published in 1981 (Commission on Classification and Terminology, 1981) (Box 67.1) and the Classification of Epilepsies and Epileptic Syndromes introduced in 1989 (Commission on Classification and Terminology, 1989) (Box 67.2). Revisions to these classifications have been recommended based on advances made in the last 3 decades (Berg et al., 2010; Engel, 2001; Engel, 2006). The major recommended revisions are outlined later in this section, but what follows will focus on the 1981 and 1989 classifications, because these classifications are still used widely for clinical management and research and will likely continue to be used in the future.
Box 67.1
International League Against Epilepsy Classification of Epileptic Seizures
From Commission on Classification and Terminology of the International League Against Epilepsy, 1981. Proposal for revised clinical and electroencephalographic classification of epileptic seizures. Epilepsia 22, 489-501.
Box 67.2
International League Against Epilepsy Classification of Epilepsies and Epileptic Syndromes
1. Localization-related (focal, local, partial) epilepsies and syndromes
2. Generalized epilepsies and syndromes
3. Epilepsies and syndromes undetermined as to whether they are focal or generalized
From Commission on Classification and Terminology of the International League Against Epilepsy, 1989. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 30, 389-399.
One important criticism of the 1981 classification is that it requires both clinical and electroencephalographic (EEG) information, and assumptions on correlation of clinical and EEG features may be incorrect. A purely semiological classification of epileptic seizures was proposed, based solely on observed clinical features (Lüders et al., 1998). The semiological seizure classification includes somatotopic modifiers to define the somatotopic distribution of the manifestations and allows demonstration of evolution of ictal manifestations using arrows to link sequential manifestations (Lüders et al., 1998). Although this classification was not adopted by the ILAE, it is considered an optional seizure classification system that is useful for localization purposes in epilepsy surgery centers.
The latest revision of the seizure classification has maintained the division of seizures based on generalized or focal onset but has recommended replacing partial with focal. It also updated the definition of focal seizures as “originating within networks limited to one hemisphere,” with the possibility of the seizures being discretely localized or more widely distributed, and possibly originating in subcortical structures. Generalized seizures were defined as “originating at some point within, and rapidly engaging, bilaterally distributed networks,” which do not necessarily include the entire cortex (Berg et al., 2010). The revised concepts acknowledge that generalized seizures can be asymmetrical and that individual seizures may appear to have a localized onset, but the location and laterality of that onset will vary from seizure to seizure (Berg et al., 2010).
Seizure Types
Partial Seizures
Simple Partial Seizures
The most recent recommendation of the ILAE Commission on Classification and Terminology suggested eliminating the term simple partial. Instead, it recommended descriptors of focal seizures according to degree of impairment during a seizure. Seizures without impairment of consciousness or awareness are divided into those with observable motor or autonomic components and those only involving subjective sensory or psychic phenomena (Berg et al., 2010).
Partial Seizures Evolving to Generalized Tonic-Clonic Activity
These seizures may start as simple partial, complex partial, or simple partial evolving to complex partial. The transition to secondary generalization usually involves versive head turning in a direction contralateral to the hemisphere of seizure onset, and focal or lateralized tonic or clonic motor activity. The pattern of evolution may be clonic-tonic-clonic in some instances. The generalized tonic phase may be asymmetrical, with flexion on one side and extension on the other. This has been called figure-of-4 posturing (Kotagal et al., 2000). Some asymmetry and asynchrony may also occur in the clonic phase, resulting in a slight degree of side-to-side head jerking (Niaz et al., 1999). The evolution from tonic to clonic activity is gradual and not always simultaneous in all affected body parts. A phase of high-frequency tremor has been referred to as the tremulous or vibratory phase of the seizure (Theodore et al., 1994). Clonic activity typically decreases in frequency over time, with longer intervals between jerks towards the termination of the seizure. The clonic activity may end on one side of the body first, so that clonic activity may then appear lateralized to one side. In addition, there may be a late head turn ipsilateral to the hemisphere or seizure origin (Wyllie et al., 1986). After the motor activity stops, the individual is usually limp and has a loud snoring respiration often referred to as stertorous respiration. During the course of recovery, there may be variable agitation. The speed of recovery is expected to be slower with longer and more severe seizures.
Partial Seizure Semiology in Relation to Localization
Partial Seizures of Temporal Lobe Origin
Temporal lobe seizures most often are of mesial temporal amygdalohippocampal origin, in association with the pathology of hippocampal sclerosis. Patients commonly have isolated auras, and complex partial seizures tend to start with an aura. The most common aura is an epigastric sensation frequently with a rising character (French et al., 1993). Other auras occur less commonly and include fear, anxiety, and other emotions, déjà vu and jamais vu, nonspecific sensations, and autonomic experiences such as palpitation and gooseflesh. Olfactory and gustatory auras are uncommon and more likely with tumoral mesial temporal lobe epilepsy (MTLE).
Complex partial seizures may start with an aura or with altered consciousness. With nondominant temporal lobe seizures, the patient may remain responsive and verbally interactive. However, recollection of conversations is unusual. Altered consciousness is often associated with an arrest of motion and speech. Speech arrest is not synonymous with aphasia and does not distinguish dominant and nondominant temporal lobe seizures. Automatisms are one of the most prominent manifestations, and oroalimentary automatisms are the most prevalent. Extremity automatisms also occur and are most commonly manipulative, with picking or fumbling. This type of automatism is not of direct lateralizing value. However, the contralateral upper extremity is commonly involved in dystonic posturing (Kotagal et al., 1989) or milder degrees of posturing and immobility (Fakhoury and Abou-Khalil, 1995; Williamson et al., 1998). This reduces the availability of the contralateral arm for automatisms, so manipulative automatisms tend to be ipsilateral, involving the unaffected upper extremity. Nonmanipulative automatisms typically consist of rhythmic movements either distally or proximally. These tend to be contralateral, often preceding overt dystonic posturing (Lee et al., 2006). Head turning occurs commonly. Early head turning is not usually forceful. It typically occurs at the same time as dystonic posturing and is most often ipsilateral (Fakhoury and Abou-Khalil, 1995; Williamson et al., 1998). Late head turning most often occurs during evolution to generalized tonic-clonic activity. This is usually contralateral to the side of seizure origin (Williamson et al., 1998). Well-formed ictal speech may occur during seizures of nondominant temporal lobe origin (Gabr et al., 1989). Verbal output may at times be tinged with a fearful tone. Complex partial seizures of temporal lobe origin usually last between 30 seconds and 3 minutes. Postictal manifestations may be helpful in lateralizing the seizure onset. Postictal aphasia is commonly seen after dominant temporal lobe seizures (Gabr et al., 1989). In one study, such patients were unable to read a test sentence correctly in the first minute after seizure termination, but patients with nondominant right temporal lobe origin were able to read the test sentence within 1 minute of seizure termination (Privitera et al., 1991).
Seizures of lateral temporal origin or neocortical temporal origin are much less common than those of mesial temporal origin. They cannot be reliably distinguished based on their semiology, but certain features can help distinguish them. Auditory auras are the most common auras referable to the lateral temporal cortex, usually implying involvement of the Heschl gyrus. Other types of auras referable to the lateral temporal cortex are vertiginous and complex visual hallucinations (usually posterior temporal). Oroalimentary automatisms are less common, and the pattern of contralateral dystonic posturing and ipsilateral extremity automatisms is also less common (Dupont et al., 1999). Early contralateral or bilateral facial twitching may be seen as a result of propagation to the frontal operculum (Foldvary et al., 1997). Seizures of lateral temporal origin tend to be shorter in duration and have a greater tendency to evolve to generalized tonic-clonic activity than seizures of mesial temporal origin.
Partial Seizures of Frontal Lobe Origin
Many different seizure types can originate in the frontal lobe, depending on site of seizure origin and propagation. Simple partial seizures can be motor with focal clonic activity, can originate in the motor cortex, or can be the result of spread to the motor cortex. These seizures may or may not have a Jacksonian march. Asymmetrical tonic seizures or postural seizures are usually related to involvement of the supplementary motor area in the mesial frontal cortex anterior to the motor strip. The best-known posturing pattern is the fencing posture in which the contralateral arm is extended and the ipsilateral arm is flexed. Tonic posturing may involve all four extremities and is occasionally symmetrical. When these seizures originate in the supplementary motor area, consciousness is usually preserved (Morris et al., 1988). Supplementary motor seizures are an important exception to the rule that bilateral motor activity during a seizure should be associated with loss of consciousness. Supplementary motor seizures are usually short in duration and frequently arise out of sleep. They tend to occur in clusters and may be preceded by a sensory aura referable to the supplementary sensory cortex. The pattern of posturing described with supplementary motor area seizures can occur as a result of seizure spread to the supplementary motor area from other regions of the brain. In that case, consciousness is frequently impaired. Subjective simple partial seizures may also occur with frontal lobe origin, the most common being a nonspecific cephalic aura.
Complex partial seizures of frontal lobe origin tend to be very peculiar. They may be preceded by a nonspecific aura or they may start abruptly, often out of sleep. Their most characteristic features are hyperkinetic automatisms with frenzied behavior and agitation (Jobst et al., 2000; Williamson et al., 1985). There may be various vocalizations including expletives. The manifestations can be so bizarre as to suggest a psychiatric origin. The seizure duration is short, sometimes less than 30 seconds, and postictal manifestations are brief or nonexistent, further adding to the risk of misdiagnosis as psychogenic seizures. Frontal lobe complex partial seizures arise predominantly from the orbitofrontal region and from the mesial frontal cingulate region. However, they can arise from other parts of the frontal lobe and even from outside the frontal lobe, usually reflecting seizure propagation to the frontal lobe. It may be difficult to determine the region of origin in the frontal lobe based on the seizure manifestations. It has been suggested that the presence of tonic posturing on one side points to a mesial frontal origin, as does rotation along the body axis, which sometimes leads to turning prone during the seizure (Leung et al., 2008; Rheims et al., 2008).
Frontal opercular seizures originating in the frontal operculum are associated with profuse salivation, oral facial apraxia, and sometimes facial clonic activity (Williamson and Engel, 2008). Seizures originating in the dorsolateral frontal lobe may involve tonic movements of the extremities and versive deviation of the eyes and head. The head deviation preceding secondary generalization is contralateral, but earlier head turning can be in either direction (Remi et al., 2011). Seizures may begin with forced thinking. Partial seizures of frontal origin may at times resemble absence seizures (So, 1998). It is important to recognize that seizures originating in the frontal lobe can propagate to the temporal lobe and produce manifestations typical of mesial temporal lobe seizures.
Partial Seizures Originating in the Parietal Lobe
The best-recognized seizure type that originates in the parietal lobe is partial seizure with somatosensory manifestations. The somatosensory experience can be described as tingling, pins and needles, numbness, burning, or pain. The presence of a sensory march is most suggestive of involvement of the primary sensory cortex. Sensory phenomena arising from the second sensory area and the supplementary sensory area are less likely to have a march. Somatosensory auras tend to be contralateral to the hemisphere of seizure origin, but they may be bilateral or ipsilateral when arising from the second or supplementary sensory regions. Other auras of parietal lobe origin are a sensation of movement in an extremity, a feeling of the body bending forward or swaying or twisting or turning, or even a feeling of an extremity being absent (Salanova et al., 1995a; Salanova et al., 1995b). Some patients may complain of inability to move a limb. Vertigo has been reported, as well as visual illusions of objects going away or coming closer or looking larger (Siegel, 2003). Some patients may have initial auras suggesting spread to the occipital or temporal lobe. Seizures involving the dominant parietal lobe may produce aphasic manifestations. Motor manifestations tend to reflect seizure spread to the frontal lobe. These include tonic posturing of the extremities, focal motor clonic activity, and version of the head and eyes (Cascino et al., 1993; Ho et al., 1994; Williamson et al., 1992a). Negative motor manifestations may occur, with ictal paralysis (Abou-Khalil et al., 1995). Seizures may spread to the temporal lobe, producing oroalimentary or extremity automatisms (Siegel, 2003). In one study, motor manifestations were more likely with superior parietal epileptogenic foci, and oroalimentary and extremity automatisms more likely with inferior parietal epileptogenic foci (Salanova et al., 1995a). Visual manifestations seemed more likely with posterior parietal lesions.
Partial Seizures Originating in the Occipital Lobe
The best-recognized occipital lobe seizure semiology is that of simple partial seizures with visual manifestations (Salanova et al., 1992). The most common are elementary visual hallucinations that are described as flashing colored lights or geometrical figures. These are usually contralateral but may move within the visual field. Complex visual hallucinations with familiar faces or people may also occur. Negative symptoms may be reported, with loss of vision in one hemifield. Ictal blindness may involve loss of vision in the whole visual field. Objective seizure manifestations include blinking, nystagmoid eye movements, and versive eye and head deviation contralateral to the seizure focus. This version may occur while the patient is still conscious or could be a component of complex partial seizures.
Seizure manifestations that are related to seizure spread to the temporal or frontal lobe are very common. Oroalimentary automatisms are typical of seizures that spread to the temporal lobe, whereas asymmetrical tonic posturing typifies spread to the frontal lobe; both types of spread can be seen in the same patient (Williamson et al., 1992b). Spread to the temporal or frontal lobe is so common with occipital lobe seizures that it is at times reported in the majority of patients (Jobst et al., 2010b). Ictal semiology cannot distinguish seizures originating from the mesial versus lateral occipital region (Blume et al., 2005). Evolution of occipital seizures to secondary generalization is commonly reported.
Partial Seizures Originating in the Insular Cortex
Insular epilepsy is uncommon and also frequently unrecognized because of the inability to record directly from the insula with scalp electrodes. Subjective symptoms that should suggest seizure origin in the insula include laryngeal discomfort, possibly preceded or followed by a sensation in the chest or abdomen, shortness of breath, and paresthesias around the mouth or also involving other contralateral body parts (Isnard et al., 2004). Objective seizure manifestations include dysarthria/dysphonia, sometimes evolving to complete muteness. With seizure progression in some patients, tonic spasm of the face and upper limb, head and eye rotation, and at times generalized dystonia occur (Isnard et al., 2004). Hypersalivation is also very common and can be impressive. Insular-onset seizures may spread to other brain regions and can be disguised as temporal lobe, parietal lobe, or frontal lobe epilepsy (Ryvlin, 2006; Ryvlin et al., 2006).
Generalized Seizures
Generalized Absence Seizures
Typical absence seizures are characterized by a sudden blank stare with motor arrest, usually lasting less than 15 seconds (Commission on Classification and Terminology of the International League Against Epilepsy, 1981). The individual is usually unresponsive and unaware. The seizure ends as abruptly as it starts, and the patient returns immediately to a baseline level of function with no postictal confusion but may have missed conversation and seems confused as a result. If the only manifestation is altered responsiveness and awareness, with no associated motor component, the absence seizure is classified as simple absence. Most often, generalized absence seizures include mild motor components and are classified as complex absence. The most common motor components are automatisms such as licking the lips or playing with an object that was held in the hand before the seizure. Other motor components include clonic, tonic, atonic, and autonomic manifestations. Clonic activity may affect the eyelids or the mouth. An atonic component may manifest with dropping an object or slight head drop or drooping of the shoulders or trunk. Tonic components may manifest with slight increase in tone.
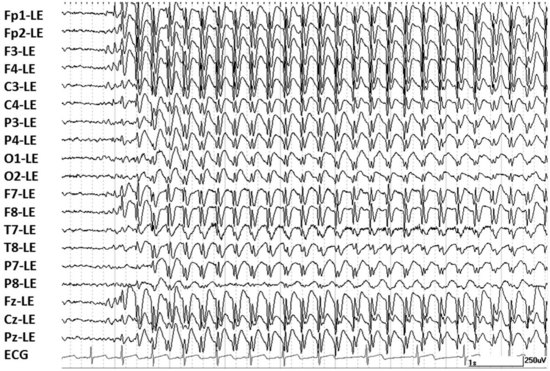
The EEG hallmark of a typical generalized absence seizure is generalized 2.5- to 4-Hz spike-and-wave activity with a normal interictal background (Fig. 67.1). Atypical absence seizures are diagnosed primarily based on a slower (<2.5 Hz) frequency of the EEG spike-and-wave activity. Less important distinctions are that the onset and termination of an atypical absence seizure may be less abrupt and the motor components a bit more pronounced than seen with typical absence seizures. Atypical absence seizures usually occur in individuals with impaired cognitive function. Affected individuals usually have associated seizure types such as generalized tonic, generalized atonic, and generalized tonic-clonic seizures.
Additional generalized absence seizure types recently recognized by the ILAE include myoclonic absences. The key manifestation of these seizures is a prominent rhythmic myoclonus predominantly affecting the limbs (Bureau and Tassinari, 2005b). Otherwise, myoclonic absences resemble typical absence seizures with respect to impairment of consciousness, although this impairment can be only partial. Another related seizure type recently recognized is eyelid myoclonia with absence. The eyelid myoclonia consists of pronounced rhythmic jerking of the eyelids, usually associated with an upward deviation of the eyes and retropulsion of the head (Caraballo et al., 2009). There may or may not be associated generalized spike-and-wave activity on EEG. Absence seizures may evolve to generalized tonic-clonic activity (Mayville et al., 2000).
Generalized Myoclonic Seizures
Myoclonic seizures are muscle contractions lasting a fraction of a second (<250 msec), in association with an ictal EEG discharge (Blume et al., 2001). The myoclonic jerk can be generalized, affecting the whole body, or could affect just the upper extremities or (rarely) the head or trunk, or even the diaphragm. The myoclonic jerks may affect one side of the body at one time, but typically the other side is affected at other times. The jerks can be single or could occur in an arrhythmic cluster. It should be noted that myoclonus is not always epileptic (Faught, 2003). Myoclonus can be generated anywhere along the central nervous system (CNS). Epileptic myoclonus is generated in the cerebral cortex and is usually associated with a single or brief serial spike-and-wave or polyspike-and-wave activity.
Negative myoclonic seizures consist of a very brief pause in muscle activity rather than a brief muscle contraction (Rubboli and Tassinari, 2006). Just as with positive myoclonus, negative myoclonus can be generalized, bilateral with limited distribution, or even focal, typically with shifting lateralization.
Myoclonic seizures may be immediately followed by a loss of tone. The seizure type is called myoclonic-atonic. Historically it was called myoclonic-astatic. The seizures are brief (1 second or less) but may be associated with falls and injuries. The EEG shows generalized spike-and-wave or polyspike-and-wave discharge. The slow wave is prolonged and associated with the electromyographic (EMG) silence characteristic of the atonic phase. Myoclonic seizures may precede a more sustained tonic contraction, and the resultant seizures may be referred to as myoclonic-tonic seizures (Berg et al., 2010). Generalized myoclonic seizures may cluster just before a generalized tonic-clonic seizure occurrence.
Generalized Clonic Seizures
Unlike myoclonic seizures, which are single jerks (but may occur in arrhythmic clusters), each generalized clonic seizure consists of a series of rhythmic jerks. Generalized clonic seizures are uncommon and particularly rare in adults (Noachtar and Arnold, 2000). They are more frequently seen in certain epileptic syndromes of infancy and childhood. For example, clonic seizures are a common seizure type of severe myoclonic epilepsy of infancy (Dravet syndrome). Clonic seizures are also noted in progressive myoclonic epilepsies.
Generalized Tonic Seizures
Generalized tonic seizures are typically brief seizures, a few seconds to 1 minute. Their onset may be gradual or abrupt. They may be initiated with a myoclonic jerk. They can vary in severity from subtle, with slight increase in neck tone with upward deviation of the eyes, to massive, with involvement of the axial muscles and extremities. Proximal muscles are the most affected. Most commonly there is neck and trunk flexion as well as abduction of the shoulders and flexion of the hips. However extension may also occur. Tonic seizures may be asymmetrical, which could result in turning to one side. The pattern of muscle involvement may change over time so that there may be a change in the position of the limbs over the course of the seizure. Autonomic changes may occur, with tachycardia, pupil dilation, and flushing. Involvement of respiratory muscles could cause apnea and cyanosis. The tonic contraction may end with one or more pauses that result in a few clonic jerks. A postictal state with confusion may occur, but recovery is usually rapid. However, tonic seizures may be followed by atypical absence, resulting in what appears to be a more prolonged postictal state. This has been referred to as tonic-absence seizure (Shih and Hirsch, 2003). Generalized tonic seizures occur most often out of sleep and drowsiness.
Epileptic Spasms
Epileptic spasms have similarities to generalized tonic seizures but a shorter duration that is intermediate between generalized myoclonic and generalized tonic seizures (Blume et al., 2001), with a typical duration of 0.5 to 2 seconds. The pattern of contraction is “diamond-shaped,” with intensity of contraction maximal in the middle of the spasm and less at the beginning and end. Epileptic spasms are also called infantile spasms and salaam attacks. Because their occurrence is not restricted to infants, the preferred current term is epileptic spasms. The classic epileptic spasm involves neck and trunk flexion and arm abduction with a jackknife pattern, but extension may be seen. Epileptic spasms typically occur in clusters recurring every 5 to 40 seconds. In a cluster, the initial spasms may be subtle or mild, increase in intensity as the cluster progresses, and decrease in intensity again toward the end of the cluster (Bleasel and Lüders, 2000).
Generalized Tonic-Clonic Seizures
Generalized tonic-clonic (GTC) seizures are dramatic and the best recognized form of seizures. They are commonly referred to as grand mal, but this term is archaic and does not distinguish seizures of focal onset from those with a generalized onset. Generalized tonic-clonic seizures do not have an aura, but they may be preceded by a prodrome—the vague sense a seizure will occur—lasting up to hours. Seizure onset is abrupt, most often with loss of consciousness and a generalized tonic contraction, but some seizures may be initiated with a series of myoclonic jerks, leading to the term clonic-tonic-clonic seizure. The tonic phase may have asymmetrical movements, and these often change from seizure to seizure. One such commonly encountered asymmetry is versive head turning, which is not evidence of a focal onset (Chin and Miller, 2004; Niaz et al., 1999). The tonic phase includes an upward eye deviation with eyes half open and the mouth open. Involvement of the respiratory muscles usually produces a forced expiration that produces a loud guttural vocalization, often referred to as the epileptic cry. Cyanosis may occur during the tonic phase in association with apnea. The tonic phase gradually evolves to clonic activity. The transition can be with initially high-frequency and low-amplitude motion, often referred to as a vibratory phase. With seizure progression, the frequency of clonic jerks decreases, and the amplitude may initially increase but later decreases just before the seizure stops. In the immediate postictal state the individual is limp and unresponsive. Respiration is loud and snoring in character (stertorous). The postictal state is often followed by sleep, although the individual may awaken briefly with postictal confusion. Tongue biting commonly occurs and most often affects the side of the tongue. Incontinence of urine is common, and incontinence of stool may also occur. After awakening, patients often have a pronounced headache and generalized muscle soreness. Generalized tonic-clonic seizures rarely last more than 2 minutes. The severity may vary. The postictal state seems to correlate with severity and duration.
Generalized Atonic Seizures
Generalized atonic are associated with very brief, sudden loss of tone and vary from extremely subtle, manifesting with only a head drop, to generalized loss of tone and falling. Atonic seizures may result in falling if the person is standing, then called a drop attack. However, drop attacks may be the result of both generalized atonic and generalized tonic seizures. There is a very brief loss of consciousness and brief postictal confusion. Seizures are usually very brief, lasting 1 second to a few seconds. They may be preceded by a brief myoclonic jerk, in which case the seizure type is called myoclonic-atonic. Very brief atonic seizures are typical of the syndrome of myoclonic-astatic epilepsy (Doose syndrome) (Oguni et al., 2001). More prolonged atonic seizures can be seen with Lennox-Gastaut syndrome or other symptomatic generalized epilepsies. Despite their brief duration, generalized atonic seizures can result in serious injury and are an important cause of morbidity in epilepsy.
Generalized-Onset Seizures with Focal Evolution
Generalized-onset seizures rarely may evolve to focal seizures (Deng et al., 2007; Williamson et al., 2009). This seems to occur with either myoclonic or absence seizures. The clinical manifestations most often are behavioral arrest and staring, with minor automatisms. However, focal motor manifestations may also occur. This type of seizure tends to be prolonged and may be associated with postictal confusion (Williamson et al., 2009).
Classification of Epilepsies and Epileptic Syndromes
The classification of seizures addresses single seizure events and not epilepsy as a condition. The 1989 classification of epilepsies and epileptic syndromes tried to organize epilepsies and epilepsy syndromes (Commission on Classification and Terminology, 1989). It defined an epileptic syndrome as “an epileptic disorder characterized by a cluster of signs and symptoms customarily occurring together; these include such items as type of seizure, etiology, anatomy, precipitating factors, age of onset, severity, chronicity, diurnal and circadian cycling, and sometimes prognosis.” A syndrome does not necessarily have a common etiology and prognosis. Two important divisions were used in the classification. The first separated epilepsies with generalized-onset seizures, called generalized epilepsies, from epilepsies with partial-onset seizures, referred to as localization-related, partial, or focal epilepsies. The other division separated epilepsies of known etiology (named symptomatic epilepsies) from those of unknown etiology. Epilepsies of unknown etiology were named idiopathic if they were pure epilepsy and “not preceded or occasioned by another condition.” These epilepsies were considered to have no underlying cause other than a possible hereditary predisposition. Thus, they were presumed genetic. The idiopathic epilepsies were also defined by an age-related onset and clinical and EEG characteristics. Epilepsies of unknown etiology were called cryptogenic if they were presumed symptomatic, but with an occult etiology. Although the term cryptogenic remains widely used in the epilepsy field, confusion exists concerning its exact meaning, which has resulted in a recommendation to replace it with the term probably symptomatic (Engel, 2001). The 1989 classification of epilepsies and epileptic syndromes also subdivided symptomatic partial epilepsies based on lobar anatomical localization of the epileptogenic zone into temporal, frontal, parietal, and occipital lobe epilepsy. Temporal lobe epilepsy was further subdivided into amygdalohippocampal and lateral temporal, and frontal lobe epilepsy into 7 subgroups: supplementary motor, cingulate, anterior frontopolar, orbitofrontal, dorsolateral, opercular, and motor cortex. The abbreviated classification is found in Box 67.2.
The 1989 classification of epilepsies and epileptic syndromes merits updating because of the recognition of new epileptic syndromes and the discovery of the genetic basis for several known epilepsies. The most recent report of the ILAE commission on classification simplified the classification of epilepsies by eliminating the division of localization-related and generalized epilepsies (Berg et al., 2010). Instead, it suggested a listing of epilepsies by age of onset, distinctive constellations, or underlying cause (Box 67.3). The list incorporated newly identified or characterized epileptic conditions. The report also recommended addressing underlying etiologies with the terms genetic, structural/metabolic, and unknown cause, thus replacing the terms idiopathic, symptomatic, and cryptogenic.
Box 67.3
Electroclinical Syndromes and Other Epilepsies
Electroclinical Syndromes Arranged by Age at Onset
Childhood
Febrile seizures plus (FS+) (can start in infancy)
Epilepsy with myoclonic atonic (previously astatic) seizures
Benign epilepsy with centrotemporal spikes (BECTS)
Autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE)
Late-onset childhood occipital epilepsy (Gastaut type)
Epilepsy with myoclonic absences
Epileptic encephalopathy with continuous spike-and-wave during sleep (CSWS)
From Berg, A.T., Berkovic, S.F., Brodie, M.J., et al., 2010. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005-2009. Epilepsia 51, 676-685; Commission on Classification and Terminology of the International League Against Epilepsy, 1981. Proposal for revised clinical and electroencephalographic classification of epileptic seizures. Epilepsia 22, 489-501; Commission on Classification and Terminology of the International League Against Epilepsy, 1989. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 30, 389-399.
Epileptic Syndromes
An epileptic syndrome was defined as a complex of signs and syndromes that define a unique epilepsy condition with different etiologies. A syndrome must involve more than just a seizure type (Engel, 2006). One important characteristic of syndromes is the characteristic age at onset. The most recent revision recommended that the term syndrome be restricted to a clinical entity that is reliably identified by a cluster of electroclinical characteristics (Berg et al., 2010). It recommended using the term constellation for epilepsies that do not quite qualify to represent a syndrome; yet there are diagnostically meaningful forms of epilepsy that may have implications for clinical treatment, particularly surgery. The classification of epilepsies in epileptic syndromes published in 1989 divides epilepsies into localization-related (focal, local, partial) and generalized. The basis for that subdivision is that most patients will have either partial-onset seizure types only or generalized-onset seizure types only. However there are syndromes in which both partial and generalized seizure types may coexist in the same individual, or the epilepsy may express itself with generalized seizure types in some individuals and partial seizure types in others. As a result, the 2010 revision recommended dropping the partial-generalized subdivision (Berg et al., 2010). In addition, it recommended using the term genetic for the “idiopathic” epilepsies that were presumed to be genetic in nature. The discussion that follows will briefly describe selected syndromes and constellations following the order suggested by the latest ILAE recommendations (see Box 67.3).
Benign Familial Neonatal Epilepsy
Benign familial neonatal epilepsy was previously referred to as benign familial neonatal convulsions (Plouin and Anderson, 2005). This rare, dominantly inherited disorder is due to mutations affecting voltage-gated potassium channel genes (KCNQ2, KCNQ3) (Biervert and Steinlein, 1999). Affected infants are usually full term and appear normal at birth. In 80% of instances, seizures start on the second or third day of life, although some infants may develop seizures later in the first month of life. The seizures are typically clonic but often preceded by a tonic component. They are more often unilateral but can also be bilateral. The seizures remit within 2 to 6 months. There is a slight increase in the risk of later epilepsy (11%-15%).
Early Myoclonic Encephalopathy and Ohtahara Syndrome
Early myoclonic encephalopathy and Ohtahara syndrome have much in common, including age at onset in the neonatal period, severe seizure manifestations, and an EEG pattern of burst-suppression, in which periods of high-voltage EEG activity are separated by periods of generalized attenuation (Aicardi and Ohtahara, 2005; Djukic et al., 2006; Ohtahara and Yamatogi, 2006).
West Syndrome
West syndrome has a later age at onset, with a peak onset between 3 and 7 months of age. It is characterized by a clinical triad of epileptic spasms, arrest or deterioration of psychomotor development, and a characteristic EEG pattern called hypsarrhythmia (Dulac and Tuxhorn, 2005). The disorder is heterogeneous in its etiology. Epileptic spasms are usually the initial manifestation. They tend to occur in clusters, sometimes multiple times a day. Approximately two-thirds of infants have brain lesions. Psychomotor development may be abnormal prior to onset, but there is a clear deterioration after onset. The spasms may have asymmetries, which are more likely when there is a focal brain lesion. The prognosis is variable, with a small portion of patients recovering quickly without sequelae. This is more likely to happen in the absence of brain pathology. Otherwise, the prognosis is unfavorable, with more than 70% developing mental retardation and other cognitive disabilities. The treatment of infantile spasms has some important differences from treatment of other seizure types. Steroids such as corticotropin (adrenocorticotropic hormone [ACTH]) and prednisone are helpful, particularly in the absence of underlying known pathology.
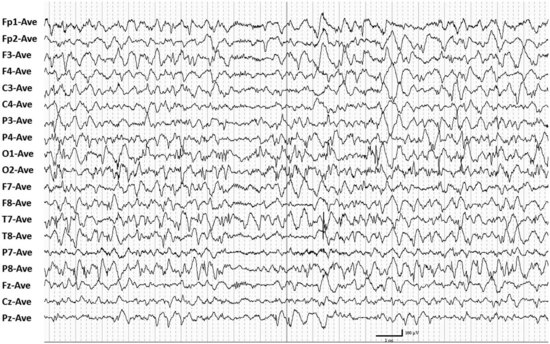
Hypsarrhythmia is characterized by high-voltage disorganized EEG activity with slow waves and multifocal spikes and sharp waves punctuated by periods of generalized attenuation (Fig. 67.2). When a spasm occurs it is usually during a period of attenuation. The attenuation may have superimposed high-frequency, low-voltage EEG activity. The periods of attenuation are typically very short in duration, lasting 1 to 2 seconds.
Dravet Syndrome
Dravet syndrome, also called severe myoclonic epilepsy of infancy, is usually due to a de novo mutation affecting the SCN1A gene encoding the α1 sodium channel subunit (Claes et al., 2001). De novo mutations account for about 95% of cases. The typical clinical presentation is that a previously normally developing infant has febrile status epilepticus at around 6 months of age, and then recurrent generalized or shifting hemiclonic seizures are seen, often triggered by fever. After 1 year of age, other seizure types appear, including myoclonic seizures, absence seizures, and complex partial seizures as well as atonic seizures at times. The seizures are drug resistant and may be exacerbated by some sodium channel blockers such as carbamazepine and lamotrigine. A delay or arrest in development may occur, and even regression may be seen, typically after episodes of prolonged seizure activity (Dravet et al., 2005; Scheffer et al., 2009). The prognosis is poor; the majority of individuals develop intellectual disability and at times ataxia and spasticity.
It has now become recognized that Dravet syndrome accounts for a large proportion of individuals previously diagnosed with vaccine encephalopathy (Berkovic et al., 2006). The fever associated with vaccination may cause an earlier age at onset of Dravet syndrome, but it does not affect the eventual course of the condition (McIntosh et al., 2010).
Epilepsy with Febrile Seizures Plus
Epilepsy with febrile seizures plus appears to be autosomal dominant in inheritance, often due to a sodium channel mutation most often in the SCN1A or SCN1B gene (Escayg et al., 2001; Wallace et al., 2002). It can also be due to a mutation in the γ2 subunit of the γ-aminobutyric acid (GABA)-A receptor (Harkin et al., 2002). No mutation has been identified in the majority of families. The condition has a heterogeneous phenotype in affected individuals (Scheffer and Berkovic, 1997; Singh et al., 1999). Some individuals have only the typical febrile seizure phenotype, with febrile seizures disappearing by 6 years of age. Other individuals have febrile seizures plus, which refers to febrile seizures persisting beyond 6 years of age or febrile seizures intermixed with afebrile generalized tonic-clonic seizures. Other individuals even have other seizure types such as generalized absence or myoclonic seizures. Less common seizure types are myoclonic-atonic and partial seizures typical of temporal lobe origin (Abou-Khalil et al., 2001; Scheffer et al., 2007).
Panayiotopoulos Syndrome
The onset of seizures in Panayiotopoulos syndrome is typically between 1 and 14 years of age, with a peak at 4 to 5 years (Covanis et al., 2005). Seizures include autonomic manifestations, particularly ictal vomiting, altered responsiveness and arrest of activity, and deviation of the eyes to one side. Autonomic manifestations are particularly pronounced (Caraballo et al., 2007). Seizures can be very prolonged, lasting longer than 30 minutes, qualifying for complex partial status epilepticus. Seizures predominate during sleep. The EEG shows multifocal spikes but with posterior predominance. Despite the alarming seizure manifestations, prognosis is generally good. Seizures are infrequent, with about a quarter of patients having only one seizure and half having two to five at most. Remission typically occurs within 1 to 3 years of onset.
Epilepsy with Myoclonic Atonic Seizures (Myoclonic Astatic Epilepsy or Doose Syndrome)
This presumed genetic epilepsy is characterized by seizure onset between 18 and 60 months of age (Guerrini et al., 2005). The characteristic seizure types are myoclonic and myoclonic-atonic seizures, present in all affected children. Tonic-clonic seizures are also seen in a majority of children. Atypical absence seizures are also common and frequently associated with reduced muscle tone. Pure atonic seizures may also occur. Tonic seizures are less frequently seen. Generalized tonic-clonic seizures are most often the seizure type that results in the diagnosis of epilepsy, with smaller seizures noticed thereafter. Seizures can be easily precipitated by inappropriate treatment with carbamazepine. The course of the condition is somewhat unpredictable. In more than half of affected children, the seizures go into remission. More than half of patients also have normal cognitive function, with less than half having mild to severe mental retardation. A worse prognosis is predicted by generalized tonic-clonic seizures in the first 2 years of life and early status epilepticus (Kelley and Kossoff, 2010).
Benign Epilepsy with Centrotemporal Spikes
Benign epilepsy with centrotemporal spikes (BECTS) is also referred to as benign rolandic epilepsy. This is the most common form of idiopathic partial epilepsy in children (Dalla Bernardina et al., 2005). Seizures begin between 3 and 13 years of age, with a peak between 5 and 8 years. Affected children will have had a normal development and normal cognitive function. Seizures typically start with paresthesias affecting one side of the face, particularly around the mouth, then contraction of that side of the face evolving into clonic activity of the face. Increased salivation and drooling occurs. Consciousness is preserved in the vast majority of children if the seizure does not secondarily generalize. Seizures are typically nocturnal and generally have a low rate of recurrence, so treatment is not always necessary. The natural history is characterized by spontaneous remission around the time of puberty. Even though long-term prognosis is excellent, patients with BECTS may have cognitive and behavioral problems while the condition is active.
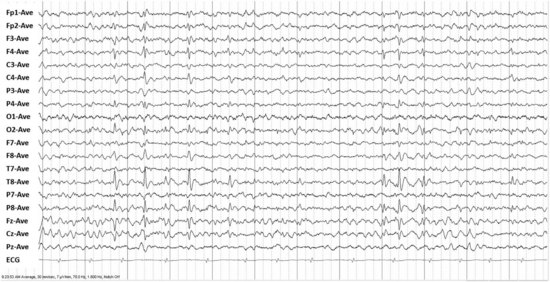
BECTS has long been thought to have a genetic basis, but the concordance in identical twins is low, suggesting that other mechanisms may play a role (Vadlamudi et al., 2004). The diagnosis of BECTS depends on the clinical presentation as well as the EEG. The typical EEG abnormality is high-voltage central-midtemporal blunt sharp waves activated in sleep (Fig. 67.3). These can become bilateral independent in deeper sleep. It is not uncommon to see atypical fields, particularly posterior temporal or parietal. The incidence of generalized spike-and-wave discharges in affected individuals is increased (Beydoun et al., 1992; Drury and Beydoun, 1991).
Autosomal Dominant Nocturnal Frontal Lobe Epilepsy
Age at seizure onset in autosomal dominant nocturnal frontal lobe epilepsy is highly variable but is most often younger than age 20, with a mean between 8 and 11 years. Seizures typically arise out of sleep. In their most pronounced expression, they may be hypermotor with vigorous frenetic movements of the extremities such as thrashing, kicking, or bicycling. The seizures may be asymmetrical tonic, sometimes with evolving posturing, or may have a mixture of hypermotor and tonic manifestations. The seizures are usually stereotyped. They are typically short in duration, lasting less than 30 seconds. They can be so short as to simply manifest with paroxysmal arousal (Provini et al., 1999). The condition is often misdiagnosed as a sleep disorder or psychogenic seizures (Scheffer et al., 1995).
This disorder is genetically heterogeneous (De Marco et al., 2007) and is typically due to mutations affecting the neuronal nicotinic acetylcholine receptor (Steinlein et al., 1995). Carbamazepine appeared particularly effective in this condition. Interestingly, the mutated nicotinic receptors were found to be more sensitive to carbamazepine than to valproate (Picard et al., 1999).
Late-Onset Childhood Occipital Epilepsy (Gastaut type)
The age at onset of seizures in late-onset childhood occipital epilepsy ranges from 3 to 16 years, with a mean age of 8 (Covanis et al., 2005). The seizures are of occipital lobe onset and manifest with visual symptoms. The ictal phenomena include elementary visual hallucinations, complex visual hallucinations and illusions, visual loss in one field or total blindness, eye deviation, and eye blinking. There may be progression of seizure manifestations with spread beyond the occipital lobe, particularly lateralized or generalized tonic-clonic activity. Consciousness is usually preserved if seizure activity does not spread beyond the occipital lobe. Postictal headache is a very common symptom, resulting in confusion with migraine. The interictal EEG is characterized by occipital spikes and sharp waves that can be extremely frequent, and typically activated with eye closure. The discharges can be so frequent as to raise concern for an ictal pattern. The activation with eye closure has been termed fixation-off photosensitivity.
Epilepsy with Myoclonic Absences
Epilepsy with myoclonic absences is a syndrome with male predominance and starts between 1 and 12 years of age, with a mean of 7 years (Bureau and Tassinari, 2005a). Its most distinctive seizure type is myoclonic absences. These seizures include impairment of consciousness of variable degree and very prominent myoclonus involving primarily the upper extremities but also the legs. The duration varies from 10 to 60 seconds, and seizures typically recur several times a day. The associated EEG usually shows 3-Hz generalized rhythmic spike-and-wave activity similar to what is seen in typical absence seizures. Approximately two-thirds of patients also have other seizure types, particularly generalized tonic-clonic seizures. Seizures tend to be resistant to monotherapy and often require dual therapy with valproate and ethosuximide or one of these agents in combination with lamotrigine.
Lennox-Gastaut Syndrome
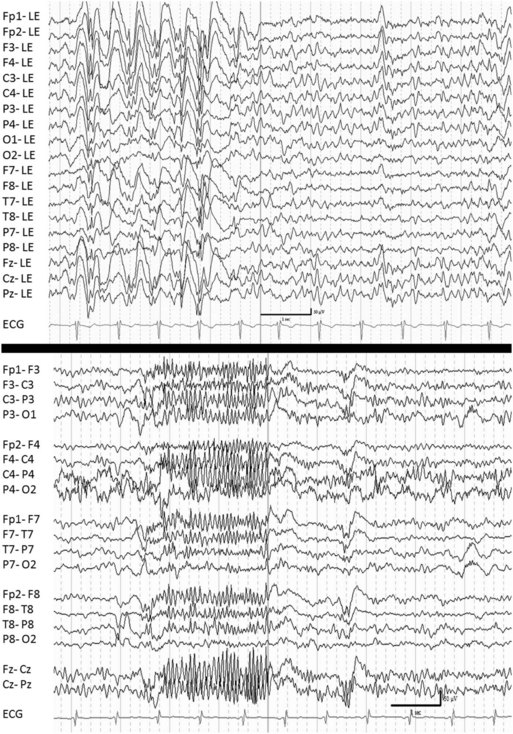
Lennox-Gastaut syndrome is defined by a triad of several seizure types including generalized tonic, generalized atonic, and atypical absence seizures, a characteristic interictal EEG abnormality of generalized slow spike-and-wave discharges (<2.5 Hz) in waking and bursts of paroxysmal fast activity (≈10 Hz) in sleep (Fig. 67.4), and cognitive dysfunction (Arzimanoglou et al., 2009; Beaumanoir and Blume, 2005). Drop attacks due to either generalized atonic or generalized tonic seizures, tend to be the most debilitating seizure type because of associated injuries. The age of onset is between 3 and 10 years with a peak between 3 and 5 years. Lennox-Gastaut syndrome may start de novo or may evolve, for example from West syndrome. Seizures tend to be drug-resistant. Lennox-Gastaut syndrome tends to be a chronic disorder even though epilepsy may become less active over time. Almost half of these patients may appear normal before onset of seizures, but deterioration occurs, and the cause is probably multifactorial.
Epileptic Encephalopathy with Continuous Spike-and-Wave During Sleep and Landau Kleffner Syndrome
The common features of the related conditions of epileptic encephalopathy with continuous spike-and-wave during sleep (CSWS) and Landau Kleffner syndrome (LKS) are a decline in cognitive function in association with an EEG pattern of continuous spike-and-wave activity during slow wave sleep (Fig. 67.5). In both conditions the associated seizures are often easy to control, and the predominant clinical manifestations are related to the EEG abnormality in sleep (Nickels and Wirrell, 2008; Tassinari et al., 2005).
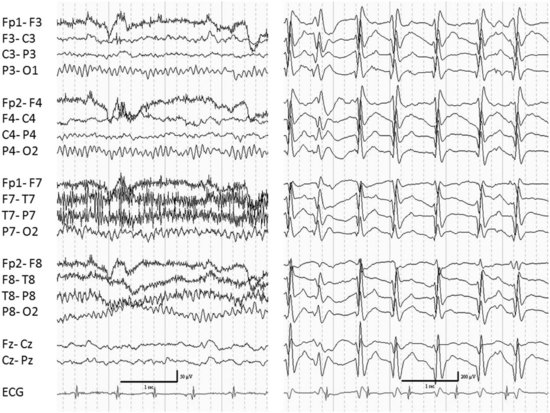
In the case of LKS, the cognitive decline is specifically in the area of speech. The condition is often called acquired epileptic aphasia. This disorder typically appears between 2 and 8 years of age, with a peak between 5 and 7 years. The most common initial manifestation is verbal auditory agnosia. The language disturbance will usually progress despite good control of clinical seizures. In fact, clinical seizures may not even occur in about a quarter of patients. The evolution is variable. Spontaneous remissions may occur within the first year. Classical antiepileptic drugs (AEDs) may be ineffective. Benefits have been reported with valproate, levetiracetam, and benzodiazepines which reduce the EEG abnormality. Steroids and immunoglobulins have been reported to be helpful. Surgical treatment with multiple subpial transections has been advocated (Morrell et al., 1995).
Childhood Absence Epilepsy
CAE syndrome, previously referred to as petit mal or pyknolepsy, typically starts between the ages of 4 and 10 years with a peak between 5 and 7 years (Hirsch and Panayiotopoulos, 2005). Affected children are normal in their development and neurological status. The key seizure type is generalized typical absence seizures occurring many times a day. In association with seizures, the EEG shows generalized synchronous and symmetrical spike-and-wave activity with a frequency around 3 Hz. It is not unusual for the spike-and-wave frequency to be initially faster (up to 4 Hz) and drop by approximately 0.5 to 1 Hz by the end of the ictal discharge. Seizure duration is brief, usually less than 15 seconds. Seizure frequency is very high, with multiple daily seizures. In 2005 the ILAE proposed strict criteria to define the syndrome, which include the absence of generalized tonic-clonic or myoclonic seizures prior to or during the active stage of absence seizures. The criteria also exclude eyelid and perioral myoclonia, high-amplitude rhythmic jerking of the limbs, and arrhythmic jerks of the head, trunk, or limbs (Loiseau and Panayiotopoulos, 2005). With this strict definition of the syndrome, many patients with a predominance of absence seizures are excluded and cannot be classified as having CAE (Ma et al., 2010; Valentin et al., 2007), but a very favorable prognosis is expected. Only 8% of patients fulfilling the strict criteria had generalized tonic-clonic seizures, compared to 30% of those who did not (Grosso et al., 2005), and 65% of those satisfying the stricter criteria had a complete seizure remission, compared to 23% of those who did not. Persistence or relapse of seizures tends to be predominately related to generalized tonic-clonic seizures. In some instances, CAE evolves into JME in the second decade. This will be discussed later under that heading.
CAE is thought to be genetically determined, with high concordance for monozygotic twins (Berkovic et al., 1998). However, the exact mode of inheritance is unknown. Although some families had a single gene mutation (including calcium channel mutations), most are thought to have polygenic inheritance (Hughes, 2009).
For children with pure absence seizures, ethosuximide is the treatment of choice (Glauser et al., 2010). Coexistence of other seizure types requires a wider-spectrum AED.
Juvenile Absence Epilepsy
Juvenile absence epilepsy is very similar to CAE except that the age at onset is in the second decade, with a peak between age 10 and 12 (Wolf and Inoue, 2005). The absence seizures are not as frequent as in CAE. In addition, the majority of patients also have generalized tonic-clonic seizures. This condition has a greater tendency for persistence of seizures into adulthood than is the case with CAE.
Juvenile Myoclonic Epilepsy
Juvenile myoclonic epilepsy (JME), also known as juvenile myoclonic epilepsy of Janz or impulsive petit mal, is common (Thomas et al., 2005) and accounts for up to 10% of all cases of epilepsy. The age at onset is typically between 12 and 18, but epilepsy may start in the first decade in a subgroup of patients who appear to have CAE early on. The defining seizure type is generalized myoclonic seizures, which occur in all patients by definition. Generalized myoclonic seizures typically occur after awakening, particularly with sleep deprivation. They are typically mild, predominately affecting the upper extremities. Although they are the first seizure type to appear, they are often not recognized as seizures and not brought to medical attention. Patients typically come to medical attention after a generalized tonic-clonic seizure, which is most likely to occur after sleep deprivation or binge drinking of alcohol. The physician has to ask about myoclonus in order to make the diagnosis. Approximately one-third of patients also have generalized absence seizures. JME is clinically and genetically heterogeneous (Martinez-Juarez et al., 2006). The most important group is classic JME, and the second largest is CAE evolving to JME. The latter tends to be more treatment resistant.
The diagnosis of the condition is based on the clinical history and EEG, which shows generalized irregular 4- to 6-Hz spike-and-wave activity occurring in bursts. The EEG is most likely to record discharges after awakening (Labate et al., 2007). JME is a lifelong condition. Even though the majority of patients can have seizure remission with medication therapy, more than 90% have a recurrence of seizures upon stopping AEDs. The prognosis for seizure freedom is lowest in individuals with CAE leading to JME and in individuals who have all three seizure types: generalized myoclonic, generalized tonic-clonic, and generalized absence seizures (Gelisse et al., 2001; Martinez-Juarez et al., 2006). Valproate appears to be the most effective medication for all three seizure types, but its teratogenicity and some adverse effects limit its use in women of childbearing age (Montouris and Abou-Khalil, 2009). Seizures may be aggravated by several AEDs that are specific for partial epilepsy (Gelisse et al., 2004; Genton et al., 2000).
JME is thought to have predominantly polygenic inheritance, but there have also been families with autosomal dominant inheritance and several identified mutations (Delgado-Escueta, 2007), including a mutation of the GABAA receptor (Cossette et al., 2002).
Autosomal Dominant Epilepsy with Auditory Features
Autosomal dominant epilepsy with auditory features (ADEAF) is related to a mutation in the leucine-rich glioma-inactivated-1 (LGI1) gene (Ottman et al., 2004). Inheritance, as the name indicates, is autosomal dominant. Seizures typically begin in adolescence or adulthood, with a mean age at onset of 24. Affected subjects commonly report an elementary auditory aura such as buzzing, ringing, humming, or even loss of hearing. Seizures may start with aphasic manifestations when the onset is in the dominant lateral temporal lobe (Gu et al., 2002).
Familial Mesial Temporal Lobe Epilepsy
Familial MTLE is a heterogeneous condition. There is a benign syndrome, first identified in twins, in which the most prominent aura is déjà vu with frequent simple partial seizures, infrequent complex partial seizures, and rare secondarily generalized tonic-clonic seizures (Berkovic et al., 1996). Prior febrile seizures are uncommon, and magnetic resonance imaging (MRI) is normal with no hippocampal sclerosis. The epilepsy is frequently not recognized when the only seizure type is subjective simple partial seizures, but when recognized is very responsive to medical therapy.
Other familial MTLE may be associated with prior febrile convulsions, hippocampal sclerosis on MRI, and less responsiveness to medical therapy, at times requiring surgical treatment (Cendes et al., 1998).
Familial MTLE is most probably polygenic in inheritance, even though there are reports of autosomal dominant inheritance (Crompton et al., 2010). No gene mutation has yet been identified.
Mesial Temporal Lobe Epilepsy with Hippocampal Sclerosis
MTLE with hippocampal sclerosis is classified as a distinctive constellation rather than a syndrome (Berg et al., 2010). Mesial temporal or hippocampal sclerosis is the most common pathology noted in surgical specimens from patients undergoing temporal lobectomy for drug-resistant temporal lobe seizures. It is characterized by neuronal loss and gliosis predominately affecting CA1 and CA3 sectors of the hippocampus, with relative sparing of CA2. Patients with MTLE and hippocampal sclerosis frequently have a history of antecedent febrile seizures (up to 80%) (French et al., 1993). The febrile seizures are usually complex, in particular prolonged. Even though febrile status epilepticus is known to injure the hippocampus in some instances, it is not clear that this is the only factor at play (VanLandingham et al., 1998). Some studies have shown evidence of prior hippocampal malformation that may predispose to injury (Fernandez et al., 1998; Park et al., 2010a). In addition, hippocampal sclerosis has been reported in familial MTLE without prior febrile seizures (Kobayashi et al., 2003). The age at onset of habitual afebrile seizures is variable but most commonly is in late childhood or adolescence. The presence of hippocampal sclerosis predicts poor response to medical therapy (Semah et al., 1998). However, the exact percentage of individuals who are drug resistant has varied between studies. It is not unusual for seizures to be drug responsive initially, with long remissions but later evolution to drug resistance (Berg et al., 2006).
The seizure pattern has already been described. The clinical seizure characteristics cannot reliably distinguish MTLE due to hippocampal sclerosis from that due to lesions (Wieser, 2004).
The hippocampal sclerosis is usually identified on MRI showing decreased volume and increased signal in the affected hippocampus (Fig. 67.6). Positron emission tomography (PET) usually shows temporal hypometabolism that is predominant in the mesial temporal region on the affected side (Fig. 67.7).
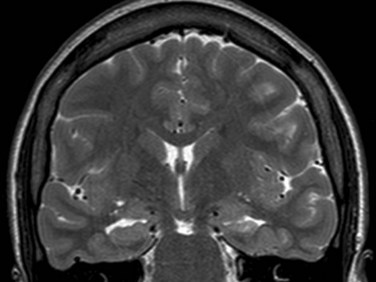
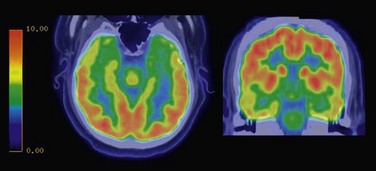
While drug resistance is common, the response rate for surgical therapy is excellent. After temporal lobectomy or selective amgydalohippocampectomy, 60% to 80% of individuals are seizure free (Wieser, 2004).
Rasmussen Syndrome
Rasmussen syndrome is a chronic progressive disorder of unknown etiology, and probably heterogeneous (Hart and Andermann, 2005). Seizures most commonly start between 1 and 14 years of age with focal-onset motor seizures. The seizures can remain simple partial or evolve to complex partial or secondary generalized tonic-clonic seizures. Seizures usually start in the same hemisphere. They become progressively more frequent with episodes of status epilepticus. Progressive hemiparesis and other deficits occur, depending on the affected hemisphere. General intellectual decline occurs at the time of hemiparesis. Imaging shows progressive hemiatrophy, with lesser atrophy on the other side. An abnormal increased T2 signal is initially most pronounced in the perisylvian region. PET reveals marked decreased metabolism in the affected hemisphere.
An autoimmune etiology is suspected. In some patients, antibodies to the GluR3 subunit of the glutamate receptor have been identified (Rogers et al., 1994). Some benefit may occur with intravenous immunoglobulin (IVIG), plasmapheresis, and corticosteroids, but hemispherectomy is generally required to achieve seizure control.
Progressive Myoclonus Epilepsies
Progressive myoclonus epilepsies (PME) are a heterogeneous group of genetic disorders characterized by myoclonus, generalized tonic-clonic seizures, and progressive neurological dysfunction, predominately with cerebellar ataxia and often with dementia (Genton et al., 2005). Included in the group are Unverricht-Lundborg disease, Lafora body disease, mitochondrial encephalopathy with ragged red fibers, and ceroid lipofuscinosis, among others. Unverricht-Lundborg disease was also called Baltic myoclonus, but it is recognized now as a worldwide condition. It is due to a mutation in the cystatin B gene (Genton, 2010). The onset is typically between 7 and 16 years of age, initially with action myoclonus then later development of tonic-clonic or clonic-tonic-clonic seizures. The myoclonus worsens progressively and greatly limits motor function. Ataxia occurs and is generally mild, but it can be very aggravated by the use of phenytoin. Phenytoin can also cause mild dementia.
Gelastic Seizures with Hypothalamic Hamartoma
Gelastic seizures with hypothalamic hamartoma typically starts with gelastic seizures in early life, with other seizures also becoming associated later on (Berkovic et al., 2003). There may be cognitive and behavioral disturbances. Some individuals have precocious puberty and a short stature. MRI reveals a hypothalamic hamartoma that can vary in size and appearance. Seizures originate within the hamartoma.
Febrile Seizures
Febrile seizures are not traditionally diagnosed as a form of epilepsy per se, even though the condition is characterized by epileptic seizures (Berg et al., 2010). The condition affects 2% to 5% of children, mostly between 3 months and 6 years of age. It is the most common form of seizures in children (Knudsen, 2000) and a benign disorder in the vast majority of those affected. Most febrile seizures are generalized tonic-clonic in semiology. They are typically symmetrical, last less than 15 minutes, and usually only one seizure occurs in association with a particular illness. Febrile seizures that satisfy the above criteria are called simple febrile seizures. Complex febrile seizures are defined by one or more of the following three criteria: prolonged duration of greater than 15 minutes, focal features (either focal ictal features or lateralized postictal weakness), or the occurrence of more than one seizure in 24 hours or with the same febrile illness. Most affected children will not have a recurrence of a febrile seizure in their lifetime. Approximately 30% to 40% will have at least one recurrence, but multiple recurrences are infrequent. Predictors of recurrence are early age at onset (<1 year), the presence of epilepsy or febrile seizures in first-degree relatives, and attendance at daycare, which increases the risk of febrile infectious illnesses.
Even though febrile seizures are a benign condition and the vast majority of affected children never develop afebrile seizures, they do increase the risk of later epilepsy. In one important study, the risk of later epilepsy was 7% by age 25 years (Annegers et al., 1987). In another study of children seen in the emergency room for their first febrile seizure, the risk of afebrile seizures was 6% at 2 years (Berg and Shinnar, 1996). The factors that predict later epilepsy include preexisting neurodevelopmental abnormalities, complex features (prolonged duration, focal features, and multiple occurrences per day), a family history of epilepsy, and recurrent febrile seizures. The presence of one complex feature increases the risk to 6% to 8%, two complex features, 17% to 22%, and all three complex features, 49% (Annegers et al., 1987). Complex features tend to predict an increased risk of partial epilepsy, while a large number of febrile seizures and a positive family history of epilepsy increase the risk of later generalized epilepsy.
Single febrile seizures are more likely to be polygenic, whereas families with single-gene inheritance are more likely to include recurrent febrile seizures. Digenic inheritance has been described (Baulac et al., 2001b). Affected individuals had two mutations, and those unaffected had either one or no mutation.
Since most febrile seizures are benign, there is usually no need to treat affected patients with prophylactic daily medication. Intermittent medication may be given at the time of fever. For example, diazepam may be given orally or rectally in patients with frequent recurrent febrile seizures. Rectal diazepam can be administered for prolonged febrile seizures (Knudsen, 2000).
Causes and Risk Factors
Infections are an important risk factor for epilepsy. The risk of later epilepsy is higher for both meningitis and encephalitis if seizures occur during the acute illness. The relative risk of later epilepsy was increased 16-fold after encephalitis and 4-fold after bacterial meningitis (Annegers et al., 1988). The risk of later epilepsy was greatest with infection prior to age 5. Early occurrence of meningitis or encephalitis prior to age 4 predicted mesial temporal localization with hippocampal sclerosis, and better outcome with temporal lobectomy (O’Brien et al., 2002).
Head Trauma
Head trauma is an important risk factor for epilepsy, with the greatest risk seen in association with penetrating head injury, head injury with depressed skull fracture, and severe head trauma with prolonged loss of consciousness. In a landmark study, mild traumatic brain injury (characterized by absence of fracture and a loss of consciousness or posttraumatic amnesia for less than 30 minutes) was associated with only a 1.5-fold increase in risk of epilepsy, which was not statistically significant (Annegers et al., 1998). Patients with moderate head injury, defined as loss of consciousness or posttraumatic amnesia for 30 minutes to 24 hours or a skull fracture, had a 2.9-fold increase in risk, while those with severe head injury, including brain contusion or intracranial hematoma or loss of consciousness or posttraumatic amnesia for more than 24 hours, had a 17-fold increased risk. The risk was highest in the first year after the injury but remained increased thereafter for a duration that varied with severity of the injury. For those with moderate brain injuries, the risk was markedly increased for up to 10 years only; for those with severe traumatic brain injury, the risk did not decrease. The Vietnam Head Injury Study (VHIS), in which 92% of subjects had penetrating head injuries, found a 53% prevalence of posttraumatic epilepsy approximately 15 years after the injury. The risk was 580 times higher than that of the general age-matched population in the first year after injury, and it was still 25 times higher after 10 years (Salazar et al., 1985). A more recent follow-up study in a subgroup of the original patients found that 12.6% of individuals who had posttraumatic epilepsy developed epilepsy more than 15 years after the injury (Raymont et al., 2010). Early seizures appeared to be a strong risk factor for late seizures, but early seizures were usually related to the severity of the head injury and intracranial lesions.
Changes in the brain reflecting the process of epileptogenesis are likely in the latent period between the head injury and onset of chronic epilepsy. Among several therapeutic measures tested for efficacy in preventing epilepsy after head injury, none have proven effective (Temkin, 2009). However, phenytoin was effective in preventing seizures in the first week (Temkin et al., 1990). The new AEDs have not been tested for the prevention of posttraumatic epilepsy.
Vascular Malformations
The two vascular malformations most commonly associated with epilepsy are arteriovenous malformations and cavernous malformations. Venous malformations, also called venous anomalies, may be accidental findings not directly related to epilepsy unless associated with a cavernous malformation. Arteriovenous malformations (AVMs) are high-pressure vascular malformations with arteriovenous shunting. They are a tangle of feeding arteries and draining veins without intervening capillary bed. They may come to attention during evaluation for seizures or after they bleed; they may also be incidental when imaging is performed for unrelated reasons. Because of the high pressure, they are susceptible to bleed at a rate of 1% to 3% per year, which is the main reason they require therapy. Surgical treatment is effective, with one series reporting 94% of patients seizure free, most on no AEDs (Piepgras et al., 1993). Patients with small AVMs were more likely to present with hemorrhage, whereas those with large AVMs were more likely to present with seizures. The best seizure outcome was seen with resection of small AVMs. Endovascular treatment with embolization and radiosurgery can also improve seizure control, though to a lesser extent. Stereotactic radiosurgery rendered more than 50% of patients seizure free and was more likely to be successful when the preoperative seizure frequency was low and the AVM was small (Schauble et al., 2004).
Cavernous malformations consist of blood-filled epithelium-lined caverns with no discrete arteries or veins (Kraemer and Awad, 1994). On MRI they have a characteristic “popcorn” appearance with mixed signal within the lesion and a rim of decreased signal, reflecting hemosiderin (Fig. 67.8). They may be multiple in approximately a third of cases. Cavernous malformations are low-pressure lesions with a much smaller risk of bleeding than AVMs. They are strongly associated with epilepsy. If seizures are controlled with medical therapy, there is no clear indication for surgical resection. However, when epilepsy is drug resistant, resection of the cavernous malformation is associated with excellent results, provided the hemosiderin-stained brain tissue surrounding it is removed (Awad and Jabbour, 2006). Intraoperative monitoring with electrocorticography can also improve surgical outcome (Van Gompel et al., 2009).
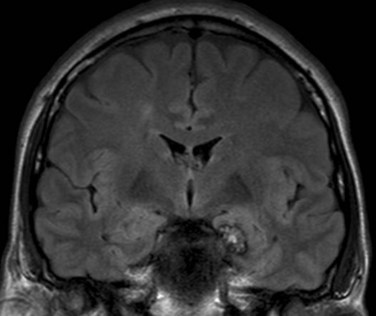
Brain Tumors
Brain tumors are a common cause of epilepsy, particularly drug-resistant epilepsy. Most drug-resistant epilepsy occurs with low-grade tumors, particularly those in the temporal lobe (Rajneesh and Binder, 2009). Benign tumors associated with epilepsy are gangliogliomas, dysembryoblastic neuroepithelial tumors (DNET), and low grade gliomas. Excellent seizure control most often occurs after removal of gangliogliomas and DNET tumors. As expected, incomplete resection is associated with less likelihood of seizure control.
Seizures contribute to the morbidity of malignant brain tumors in approximately a quarter of patients. Grade 3 anaplastic astrocytomas are more likely to present with seizures at onset than glioblastoma multiforme (Moots et al., 1995).
Parasitic Infections
Neurocysticercosis is thought to be the leading cause of acquired epilepsy in adulthood in the developing world, but it is an uncommon cause of epilepsy in developed countries. Seizures are thought to occur in 70% to 90% of patients (Pal et al., 2000). Seizures in most patients can be controlled with AEDs. When epilepsy is drug resistant, patients with living cysticerci in the brain can benefit from albendazole, an antiparasitic treatment, in combination with dexamethasone (Garcia et al., 2004).
Stroke
Stroke increases the risk of seizures and epilepsy at any age, but it is the most common cause of seizures in the elderly (Menon and Shorvon, 2009). As in posttraumatic seizures, early seizures that occur within 2 weeks of the stroke most often do not progress to chronic epilepsy, but they do increase the risk of chronic epilepsy. As with head trauma, the risk of chronic epilepsy is highest in the first year after stroke, with a 17-fold increase in the risk compared to population in the community. Compared to individuals who did not have early seizures, approximately 30% of individuals who have early post-stroke seizures develop later epilepsy. This is a 16-fold increase in risk. Seizures and even status epilepticus can be a presenting symptom of acute stroke. Nonconvulsive status epilepticus is difficult to detect.
Inflammatory and Autoimmune Disorders
Immune disorders increase the risk of epilepsy and seizures. In systemic lupus erythematosus, the risk of seizures is 12% to 20% and is more likely with anticardiolipin and anti-Smith antibodies (Najjar et al., 2008). The risk of epilepsy is also increased in primary CNS inflammatory conditions such as multiple sclerosis. Between 2% and 6% of patients with multiple sclerosis have seizures. Those who do tend to have more extensive cortical involvement with inflammatory disease. Seizures are more likely to occur in the context of acute relapse, but some patients develop chronic epilepsy. Seizures are a more common acute manifestation of acute disseminated encephalomyelitis, noted in approximately 50% of patients. However, chronic epilepsy is much less likely, with only about 5% affected.
Hashimoto encephalopathy is a steroid-responsive encephalopathy usually presenting with behavioral-cognitive abnormalities. Seizures occur in 60% of individuals. Patients have elevated antithyroid antibodies, but it is not clear that these antibodies are responsible for the clinical manifestations (Castillo et al., 2006).
Limbic encephalitis is an increasingly recognized cause of epilepsy. Suggested diagnostic criteria include one of disturbance of episodic memory, temporal lobe seizures, or affective disturbance plus the presence of either well-characterized antibodies or unexplained increased signal in the mesial temporal structures, or histopathology of mesial temporal encephalitis (Bien et al., 2007). It can be paraneoplastic or non-paraneoplastic. Neoplastic cases are associated with small cell lung cancer, testicular cancer, thymoma, breast cancer, or teratoma. The most commonly noted antibodies seen in non-paraneoplastic limbic encephalitis include anti–N-methyl-d-aspartate (NMDA) receptor antibodies, anti–potassium channel antibodies, and anti–glutamic acid decarboxylase (GAD) antibodies (Dalmau et al., 2008; Malter et al., 2010). Immunosuppressive therapies may be effective, particularly in anti–potassium channel antibody limbic encephalitis (Malter et al., 2010). An immune basis of epilepsy should be suspected in individuals who have other autoimmune disease, abrupt or recent onset of seizures (particularly if resistant to AEDs and progressive in frequency and severity), associated manifestations such as behavioral changes and psychosis, severe memory disturbances, and abnormal signal on MRI in the hippocampi.
Other Risk Factors
Other risk factors have been specifically investigated in older adults. Several risk factors for stroke are independently associated with increased risk of epilepsy, without evidence of any stroke. For example, hypertension is associated with a 1.57-fold increase in the risk (Ng et al., 1993). Left ventricular hypertrophy without diuretic treatment was associated with an 11-fold increased risk (Hesdorffer et al., 1996). Diuretics were protective, as left ventricular hypertrophy treated with diuretics carried no increase in risk (Hesdorffer et al., 1996).
Dementia, particularly Alzheimer disease, has been associated with epilepsy, with up to a 10-fold increase in risk compared to a reference population (Hauser et al., 1986). Patients who developed seizures had a younger age at onset of dementia, with seizures tending to be a late feature first seen an average of 6.8 years after onset (Mendez et al., 1994). Using very conservative criteria, the incidence of seizures was found to be 1.5%, but still with an 8-fold increase in risk compared to an age-matched population (Scarmeas et al., 2009).
Major depression seemed to increase the risk of epilepsy 6-fold in patients 55 years or older. This was the case without any prior neurological insult (Hesdorffer et al., 2000). Attempted suicide also increased the risk of epilepsy (Hesdorffer et al., 2006).
Alcohol abuse is known to be associated with acute withdrawal seizures within 48 hours of stopping alcohol. However, chronic alcohol intake was also associated with increased risk of chronic epilepsy, related to the amount of alcohol use. The increased risk was almost 20-fold for the heaviest drinkers (Ng et al., 1988).
Heroin increased the risk of unprovoked seizures threefold, while marijuana seemed to confer protection (Ng et al., 1990). Cocaine may reduce seizure threshold in individuals with epilepsy, but it is not clear that it is an important risk factor for chronic epilepsy (Koppel et al., 1996).
Causes of Acute Symptomatic Seizures
Acute symptomatic seizures occur in close temporal relationship with an acute CNS insult—metabolic, toxic, structural, infectious, or inflammatory (Beghi et al., 2010). Many of the causes of epilepsy already discussed can also cause acute symptomatic seizures. Metabolic and toxic causes of acute symptomatic seizures do not usually cause chronic epilepsy.
Metabolic disturbances known to cause seizures include hypoglycemia, hyperglycemia, hyponatremia, hypocalcemia, hypomagnesemia, and uremia (Beghi et al., 2010). Withdrawal from chronic alcohol abuse may be associated with generalized tonic-clonic seizures as well as other manifestations of withdrawal, typically within 48 hours of the last alcohol intake. Acute withdrawal from benzodiazepines and barbiturates can also be associated with withdrawal seizures, more commonly when short-acting agents are involved.
Seizure Precipitants
Some patients with epilepsy report seizure precipitants that will not trigger seizures in unaffected individuals. More than 50% of subjects with epilepsy reported at least one precipitant (Frucht et al., 2000; Nakken et al., 2005). Emotional stress and sleep deprivation were most commonly reported precipitants. Other precipitants were fatigue, fever or illness, flickering light, and menstruation. In some epileptic syndromes and epilepsies (e.g., benign epilepsy with centrotemporal spikes, frontal lobe epilepsy), seizures occur preferentially in sleep.
Several medications can reduce the seizure threshold, including tricyclic antidepressants, bupropion, various antipsychotics, CNS stimulants, fluoroquinolone antibiotics, older antihistaminics, meperidine, and tramadol. Fever can reduce the threshold for seizures in patients with known epilepsy. Hyperventilation is well known to precipitate generalized absence seizures. It can also precipitate other seizure types to a much lesser extent (Arain et al., 2009; Guaranha et al., 2005).
One likely common reason for precipitation of seizures is missing antiepileptic medication doses. Breakthrough seizures can occur with missing any of the AEDs. However, carbamazepine and oxcarbazepine are associated with more severe seizures upon abrupt withdrawal (Azar et al., 2008b; Marciani et al., 1985).
A prominent precipitation of seizures in association with the menstrual cycle has been termed catamenial epilepsy. This is reported in approximately 55% of women (Herzog, 2008). The most common pattern of clustering of seizures is perimenstrual, typically in the 3 days before and 3 days after onset of the period. Less common patterns are periovulatory (occurring around ovulation) and luteal, in association with inadequate luteal phase cycles. The mechanism of catamenial epilepsy is thought to be related to the opposite effects of estradiol and progesterone on seizure threshold. Estradiol is a proconvulsant, whereas progesterone has an anticonvulsant effect. Progesterone therapy has been suggested as a treatment when catamenial epilepsy is not responsive to standard AEDs.
Epidemiology of Epilepsy and Seizures
Descriptive Epidemiology
The incidence of epilepsy is defined as number of new cases in a unit of time divided by the total population at risk. The overall incidence in North America, adjusted for age, varied from 16 to 51 per 100,000 persons per year (Banerjee et al., 2009). The incidence is similar in European studies, but figures were at times higher in developing countries, up to 111 per 100,000 in a study in rural Chile. Incidence is high in the first decade of life, lowest in adult years until the fifth decade of life, then higher thereafter. In U.S. studies, the highest incidence was after age 75. The increased incidence in older age was not seen in developing countries. Incidence is slightly higher in males and higher in groups with lower socioeconomic status. The finding was not accounted for by known risk factors such as head injury and stroke.
Cumulative incidence is the estimate of the proportion of individuals who will have developed epilepsy by a certain age. In Rochester, Minnesota, at age 80 the cumulative risk of epilepsy was 4%, and the cumulative risk for all unprovoked seizures was more than 5% (Hauser et al., 1993). The cumulative incidence of all epilepsy was 1.2% through age 24 (Hauser et al., 1993).
Epidemiology of the First Unprovoked Seizure
Studies of recurrence after the first unprovoked seizure have predominantly focused on patients who present with a generalized tonic-clonic seizure, with or without focal onset. Milder seizures are much less likely to present to medical attention after their first occurrence. A meta-analysis of observational studies estimated a 2-year recurrence risk of approximately 40% (Berg and Shinnar, 1991). Two large randomized trials gave slightly different estimates for recurrence in patients randomized to treatment (Berg, 2008). One predicted a recurrence risk of 51% at 2 years after the initial seizure (First Seizure Trial Group, 1993), and the other estimated the risk at 39% at 2 years, 51% at 5 years, and 52% at 8 years (Marson et al., 2005). The risk of recurrence is highest in the first 2 years. Treatment reduced the risk by 60% in one study (First Seizure Trial Group, 1993) and 30% in the second study (Marson et al., 2005).
The two factors that most influenced recurrence rate were an abnormal EEG and an abnormal neurological status. Individuals with a normal EEG and normal neurological status had a risk of recurrence of 25% at 2 years and 30% at 4 years (Kim et al., 2006).
The risk of recurrence increases remarkably after a second seizure. In one prospective study in adults the 2- and 5-year risks of recurrence after the first seizure were 27% and 33%, and after the second seizure were 61% and 73% (Hauser et al., 1998).
Morbidity and Mortality
Morbidity and Comorbidity
Compared to the general population, individuals with epilepsy are at risk for increased morbidity and mortality. A large European cohort study of predominantly young patients diagnosed within the past 5 years showed a significantly greater cumulative probability of illness than controls (49% by 12 months and 86% by 24 months in patients versus 39% and 75% in controls) (Beghi and Cornaggia, 2002). The chance of illness tended to increase with number of seizures, and there was also a significant correlation between the number of illnesses and seizure frequency. However, most of the illnesses reported were trivial. Patients with epilepsy differed from controls predominantly in ear, nose, and throat complaints. Accidents were also considerably more common in patients than controls (17% and 27% by 12 and 24 months compared to 12% and 17% for controls). The number of wounds increased with the number of AEDs, which may reflect impairment of attention as a result of seizure medications. Nervous system disorders that were more common in patients than in controls included headache and vertigo/dizziness (van den Broek and Beghi, 2004).
Psychiatric disorders were not significantly different between patients and controls in this study but may have been underestimated, since the study depended on patient self-report. Several studies support an increased incidence of psychiatric disorders in epilepsy. A cross-sectional study that included 5834 individuals with epilepsy suggested that persons with epilepsy have double the risk of psychiatric disorders and an increased risk for neurodegenerative conditions, particularly dementias and Parkinson disease, cardiovascular disorders, cerebrovascular disorders, upper-gastrointestinal hemorrhages, fractures, pneumonia, chronic lung diseases, diabetes, and brain tumors (Gaitatzis et al., 2004). A U.S. survey also found an almost threefold greater frequency of serious mental illness and an increase in physical comorbid conditions including cancer, arthritis, heart disease, stroke, asthma, severe headaches, lower back pain, and neck pain (Strine et al., 2005). It is likely that individuals with longer duration of epilepsy and drug-resistant epilepsy contribute to the higher morbidity in the cross-sectional surveys. The reason for the increased morbidity is not known. One speculation is that it could be the result of stress and its adverse effects on various systems (Yuen et al., 2007).
A more recent survey designed to measure a wide spectrum of neuropsychiatric and pain comorbidities found several neuropsychiatric disorders to be more than twice as prevalent in patients with self-reported epilepsy than in those without epilepsy (Ottman et al., 2011). These disorders included bipolar disorder, attention-deficit/hyperactivity disorder (ADHD), and movement disorders/tremor. The last two had not been reported in previous national surveys. The most prevalent comorbidity was depression, reported by 32.5% of cases. The cause of depression in epilepsy may include both psychosocial and biological factors. Depression appears more prevalent in patients with medically intractable complex partial seizures (Victoroff et al., 1994). A survey of frequency and severity of depressive symptoms in community-based patients with epilepsy or asthma found evidence of depression in 36.5% of epilepsy subjects compared to 27.8% of asthma subjects and 11.8% of healthy controls. Patients with epilepsy are more likely to be depressed than patients with asthma, suggesting that depression is not solely related to a chronic illness (Ettinger et al., 2004). Epilepsy subjects were more likely to have sought consultation for depression and to have taken medication for depression. Being female, having a lower income, and being younger correlated with depression among patients with epilepsy. Epilepsy subjects with depression were less likely to be employed and reported working fewer days in the past month than those without depression. The presence of depression was associated with worse quality-of-life scores. As a result, clinicians evaluating patients with epilepsy are advised to discuss mood and quality of life as part of the evaluation (Ettinger et al., 2004).
Psychiatric symptoms need to be detected early and treated effectively, avoiding the erroneous supposition that depression is a normal reaction to the epilepsy (Garcia-Morales et al., 2008). The presence of depression should influence the selection of AEDs, avoiding drugs that are known to be associated with depression and favoring medications such as lamotrigine with a known positive mood effect. In addition, there should be a low threshold for prescribing specific antidepressant or mood-stabilizing medications, as well as consulting specialists in psychiatry for treatment. Compared to controls, the risk of suicidal behavior and suicide is increased in persons with epilepsy. The relationship between depression/suicidality and epilepsy is bidirectional, with a higher risk of developing epilepsy in those with a past history of suicidality, and a higher risk of suicidality in individuals with epilepsy (Hesdorffer et al., 2006).
Other comorbidities associated with epilepsy include migraine. In a large series of children with headache, children with epilepsy had a 4.5-fold increased risk of developing migraine over a tension-type headache (Toldo et al., 2010). The rate ratio for migraine was 2.4 for individuals with epilepsy in comparison with controls (Ottman and Lipton, 1994). In addition, specific epileptic syndromes have a greater association with migraine (Kossoff and Andermann, 2010). The comorbidity of migraine may influence the choice of AED. Some AEDs such as topiramate and valproate have proven effective in migraine prophylaxis and are approved for this indication by the U.S. Food and Drug Administration (FDA).
Sleep disorders are also a known comorbidity of epilepsy. Epilepsy patients frequently have excessive daytime sleepiness, which is multifactorial (Manni and Tartara, 2000). Several studies have suggested that the prevalence of obstructive sleep apnea may be relatively high in individuals with epilepsy as compared with controls (Kaleyias et al., 2008; Manni and Terzaghi, 2010; Manni et al., 2003). Sleep apnea is even more prevalent in individuals with drug-resistant epilepsy (Malow et al., 2000b) and may play a role in drug resistance. Sleep apnea may worsen seizure control by fragmenting nocturnal sleep and mimicking sleep deprivation. In one study in older epilepsy patients, obstructive sleep apnea was more common in those with new-onset or newly worsened epilepsy than in individuals with stable or well-controlled epilepsy (Chihorek et al., 2007). In those patients with coexistent epilepsy and sleep apnea, there is evidence that treatment of the sleep apnea may have a beneficial effect on seizure control (Malow et al., 2008). Other sleep disorders associated with epilepsy include insomnia. In particular, sleep-maintenance insomnia was reported more frequently than in controls (Khatami et al., 2006). Polysomnography in patients with epilepsy demonstrates reduced percentage of sleep time spent in rapid eye movement (REM) sleep, increased waking after sleep onset, prolonged REM latency, and increased number of stage shifts (Touchon et al., 1991). The findings are more common with temporal lobe epilepsy. The decrease in REM is more pronounced if seizures occurred during the early part of the night (Bazil et al., 2000).
Mortality in Epilepsy
Epilepsy carries a two to three times higher mortality rate than that of the general population (Tomson, 2000). The standardized mortality ratio in an early study was 2.3 through 29 years of follow-up (Hauser et al., 1980). The most significant increase in standardized mortality ratio was noted in the first 10 years after diagnosis; the increase was present even when epilepsy was in remission, suggesting that the increased mortality was not solely related to seizures. However, standardized mortality ratio remained elevated even after 30 years of follow-up (Shackleton et al., 1999). The mortality ratio does depend on the seizure type. For example, generalized tonic-clonic and myoclonic seizures were associated with increased mortality, while absence seizures were not (Hauser et al., 1980). Patients who become seizure free after epilepsy surgery have the same mortality rates as the general population (Sperling et al., 1999). The highest mortality is found in epilepsy patients with mental retardation or cerebral palsy (Forsgren et al., 2005). The increased mortality is due to a variety of factors including cerebrovascular disease, pneumonia, and neoplastic disorders, even after exclusion of primary brain tumors (Forsgren et al., 2005). One study also found increased mortality from accidents (Olafsson et al., 1998). However, deaths in epilepsy may also be related to the underlying cause of epilepsy or to seizures themselves. Seizure-related deaths include deaths from status epilepticus, accidents and drowning as a result of seizures, and sudden unexplained death in epilepsy (SUDEP) (Forsgren et al., 2005).
SUDEP accounts for about 1.7% of all deaths in epilepsy and 8.6% of deaths in the 15- to 44-year-old age group (Ficker et al., 1998). The rate of SUDEP in the 20- to 40-year-old age group was nearly 24 times that expected for the general population (Forsgren et al., 2005). The mechanisms of SUDEP are not fully understood, but there is strong evidence that SUDEP is related to seizures (So, 2008). When SUDEP is witnessed, it is most often associated with a generalized tonic-clonic seizure near the time of death (Tomson et al., 2005), and the vast majority of unwitnessed SUDEP is associated with tongue biting or incontinence, suggesting recent generalized tonic-clonic seizures (Langan, 2000).
Two main hypotheses have been put forth to explain SUDEP, one suggesting cardiac mechanisms and the other, respiratory mechanisms. In support of a cardiac mechanism is the rarely documented phenomenon of ictal bradycardia or ictal asystole, particularly in patients with temporal lobe epilepsy. In one study, the maximal ictal heart rate was significantly higher in SUDEP patients than epilepsy controls, especially with seizures arising from sleep (Nei et al., 2004). The observation that some seizures increase corrected QT interval beyond normal limits may also be relevant. This was most likely to occur with tonic-clonic seizures and right temporal seizures (Brotherstone et al., 2010).
The respiratory mechanism was proposed because of evidence of pulmonary congestion or even edema on autopsy in SUDEP patients, but the degree of pulmonary congestion is not sufficient to cause death. On the other hand, ictal apnea and hypoxia are common (Bateman et al., 2008; Nashef et al., 1996). Pronounced oxygen desaturation occurred even in partial seizures that did not progress to generalized convulsions, most commonly with seizures of temporal lobe onset. The duration of the seizure and electrographic evidence of contralateral spread influenced the degree of desaturation. The idea of hypoventilation as an initiating factor in SUDEP is supported by the clinical finding that most patients are found dead in a prone position (Kloster and Engelskjon, 1999). A recent analysis suggested that prolonged generalized EEG suppression was more commonly seen with seizures recorded in individuals who later died of SUDEP. Beyond 80 seconds of postictal generalized EEG suppression, the odds of SUDEP were quadrupled. It is thought that the generalized EEG suppression reflects profound postictal cerebral dysfunction that may be a mechanism for central apnea (Lhatoo et al., 2010).
It is now increasingly recognized that patients deemed at high risk for SUDEP need to be informed about this potential complication. Education about SUDEP is not necessary in patients with well-controlled seizures, but it is important for noncompliant individuals who could reduce the risk of SUDEP with improved compliance, and for patients with drug-resistant epilepsy who are excellent candidates for epilepsy surgery, which reduces the risk of SUDEP when successful (So et al., 2009).
Pathophysiology and Mechanisms
Epileptic Networks
On scalp and intracranial EEG, MTLE and NPE are partial epilepsy syndromes that often demonstrate discrete regions of ictal onset, frequently guiding successful surgical resection of the ictal onset zone. Nonetheless, recent imaging data demonstrate that regions distant from the ictal onset zone also participate in MTLE and NPE seizures and may serve to propagate or enhance the discharges (Kobayashi et al., 2006; Luat and Chugani, 2008; Morgan et al., 2010). The clinical and scientific implications of these epileptic networks in focal epilepsy will undoubtedly be better elucidated in the next decade. The networks involved in IGE have—and continue to be—subjects of intense study. How can the electrical discharge in a primary generalized seizure appear simultaneously throughout the entire cortex? It is generally accepted that both the cortex and thalamus are involved in this phenomenon. However, whether the thalamus is required for initiation of spike-and-wave discharges (Meeren et al., 2002), or whether the discharges arise in the cortex alone and the thalamus serves only to amplify these rhythmic cortical discharges (Berman et al., 2010; Meeren et al., 2002; Moeller et al., 2010; Polack et al., 2007; Steriade and Contreras, 1998) are still matters of debate.
Electrophysiological Abnormalities
A seizure is an abnormal rhythmic and hypersynchronous neuronal discharge. Because synchronous oscillating rhythms are necessary for normal cognition, memory, and sleep (Buzsaki and Draguhn, 2004; Jutras and Buffalo, 2010; Varela et al., 2001), the nonepileptic brain contains mechanisms to synchronize neurons that only have to be tweaked to produce a hypersynchronous seizure. Ictal hypersynchronous rhythms can oscillate at a variety of frequencies from 1 to 600 Hz (de Curtis and Gnatkovsky, 2009; Engel et al., 2009; Englot et al., 2010; Le Van Quyen et al., 2000; Schiller et al., 1998; Toczek et al., 1997; Westmoreland, 1998). Therefore, as one would expect, different mechanisms are required to synchronize neurons discharging at these very different frequencies.
The fast ripple (200-600 Hz) is a periictal rhythm that has been the subject of intensive research. Oscillations at these high frequencies can be found in normal neocortex during sensory processing (Barth, 2003). However, because fast ripples are only observed in epileptic hippocampi, and because they are present at the time of seizure onset, fast ripples are thought to contribute to the pathogenesis of hippocampal seizures in MTLE (Bragin et al., 2005). Elucidating the mechanisms by which neurons synchronize to oscillate at such high frequencies has been challenging. Some have postulated that instead of reflecting ultrafast synchronous firing of individual neurons, fast ripples arise from the out-of-phase interactions of lower-frequency ripple (80-200 Hz) oscillations (Foffani et al., 2007; Staley, 2007). Some evidence suggests that neurons in ripple oscillations are synchronized by fast-spiking interneurons that form inhibitory synaptic connections with multiple pyramidal cells (Klausberger et al., 2003, 2004; Le Van Quyen et al., 2008). In this model, all pyramidal cells innervated by the same interneuron are hyperpolarized synchronously, which thereby causes them to fire synchronously after the hyperpolarization decays.
Another putative mechanism holds that gap junctions mediate synchrony among pyramidal neurons during ripple oscillations (Roopun et al., 2010). In this model, action potentials from one neuron would depolarize nearby pyramidal neurons coupled by gap junctions. Both interneuron-mediated and gap junction–mediated mechanisms of synchrony may be altered in epileptic brains to produce pathological hypersynchrony.
The electrophysiological mechanisms that produce the rhythmic 3- to 4-Hz ictal pattern of absence seizures have been reviewed by Huguenard and McCormick (2007). As described earlier, both the cortex and thalamus are activated in absence seizures. Two regions of the thalamus, the thalamocortical relay nuclei (TC) and the thalamic reticular nuclei (RT) participate in a network that promotes rhythmic synchronization. In addition to providing excitatory output to the cortex, the TC neurons also provide excitatory output to the RT neurons which, in turn, provide feedback inhibition back to the TC. Therefore, activation of the highly interconnected RT neurons produces a synchronous postsynaptic inhibitory potential in the TC neurons. At the end of the inhibitory potential, the TC neurons engage in burst firing that is projected to both the cortex and the RT. It is thought that during an absence seizure, the inhibitory potentials are predominately mediated by GABAB receptors and persist for approximately 300 ms, approximately the inter-waveform duration in a 3- to 4-Hz absence seizure.
Histopathology
MTLE often presents with distinctive cellular changes in the hippocampus (reviewed in Sutula and Dudek, 2007). Histological studies of hippocampi from experimental animal models of chronic epilepsy, as well as those resected from human patients, demonstrate neuronal loss in the dentate gyrus and CA1 and CA3 subfields. Instead of synapsing with inhibitory interneurons as they do in normal hippocampi, dentate granule cell axons (mossy fibers) sprout and provide excitatory synapses with principal neurons in epileptic hippocampi. These histological changes most likely contribute to the development of epilepsy. Quantitative MRI studies and neuronal cell counts in surgically resected hippocampi revealed that hippocampal atrophy and reduced neuronal densities correlated with increased ratio of the occurrence of pathological fast ripples to normal ripples (Staba et al., 2007). The mechanisms by which these histological changes contribute to seizure development is unknown, but computational models suggest that processes such as neuronal loss and circuit reorganization alter the in-phase firing of neuronal clusters, which may contribute to the formation of pathological fast ripples (Ibarz et al., 2010; Stacey et al., 2009).
Histological changes in NPE are less well defined than those in MTLE. Analysis of surgical specimens revealed reduction in the number of interneurons in type II but not type I focal cortical dysplasia (Andre et al., 2010; Zamecnik et al., 2006) and alteration in the number of inhibitory versus excitatory synapses (Alonso-Nanclares et al., 2005). Histological analyses of neocortical posttraumatic epilepsy specimens have been performed primarily on experimental animal models and have revealed alterations of pyramidal neuron axonal length, budding and sprouting, as well as dendritic arborization of specific types of interneurons (Blizzard et al., 2011; Salin et al., 1995).
A controlled histological study failed to reveal any cellular differences between postmortem brain specimens from IGE patients and those of normal controls (Opeskin et al., 2000). However, this study did not examine for ultrastructural changes. An analysis of a genetic rat model of absence epilepsy revealed morphological differences in dendrite length and arborization in the cortices of epileptic animals (Karpova et al., 2005).
Molecular Pathogenesis
Pharmacological agents can provoke acute hippocampal, neocortical, and primary generalized seizures in normal subjects (Durand, 1993; Fisher, 1989; von Krosigk et al., 1993). Therefore, molecular modifications of cellular physiology are sufficient to cause seizures even in the absence of histological changes. Most likely, these molecular changes alter cellular excitability and cause the normal synchronous oscillations to develop into pathological ictal discharges.
Molecular analyses of resected surgical specimens from human mesial temporal and neocortical epilepsy patients and from experimental animal models of epilepsy revealed changes in the expression of multiple proteins including GABAA, GABAB, NMDA, AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazoleproionic acid), and metabotropic glutamate receptors, as well as voltage-gated ion channels and aquaporin proteins (Brooks-Kayal et al., 1998; Eid et al., 2002; Lee et al., 2004; Oliveira et al., 2010; Peng et al., 2004; Tang et al., 2004). Because these proteins help maintain neuronal excitability and connectivity, one would expect that their altered expression could contribute to hypersynchrony. However, thus far, there is no unifying hypothesis that predicts how all these molecular changes in toto cause seizures.
Although rare, monogenic forms of epilepsy syndromes provide invaluable tools to investigate the contributions of single molecules to pathogenesis. Geneticists have identified several families with autosomal dominant forms of MTLE (Hedera et al., 2007; Striano et al., 2008). Identifying the genes responsible for these monogenic MTLE syndromes will enable the creation of genetically modified mice possessing the mutations and help elucidate their pathogenesis.
Autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) represents an archetypal monogenic form of an NPE syndrome. Several mutations in the α2, α4, and β2 subunits of the neuronal nicotinic acetylcholine receptor are associated with this condition. Two genetically modified mice containing two ADNFLE mutations of the α4 subunit have been created (Klaassen et al., 2006). Both mouse genotypes exhibited frequent electrographic and behavioral seizures that were exacerbated by nicotine. Interestingly, the mutations augmented the inhibitory currents produced by nicotine. From these surprising findings, the authors concluded that these ADNFLE mutations promote seizures by strengthening interneuron-driven synchronization of cortical neurons.
Generalized epilepsy syndromes have been associated with mutations in GABAA receptor subunits, voltage-gated ion channels, and the EF-hand calcium binding protein, EFHC1 (Berkovic et al., 2004; Biervert et al., 1998; Chioza et al., 2001; Kleefuss-Lie et al., 2009; Kurahashi et al., 2009; Macdonald et al., 2010; Saint-Martin et al., 2009; Singh et al., 1998; Suzuki et al., 2004). These genetic mutations cause different molecular consequences in the proteins they encode, and elucidation of these consequences has identified new potential targets for novel anticonvulsant therapies. For example, mutations in GABAA receptor subunits can cause multiple different consequences including elimination of the mutant messenger (m)RNA through nonsense-mediated decay, misfolding, and degradation of the nascent polypeptides, dominant negative reductions in wild-type GABAA receptor expression, impaired receptor oligomerization, formation of insoluble protein aggregates, and altered electrophysiological properties. Specific potential therapies for each of these molecular consequences have been proposed (Table 67.1).
Table 67.1 Molecular Consequences of GABAA Receptor Subunit Mutations Associated with Generalized Epilepsy Syndromes
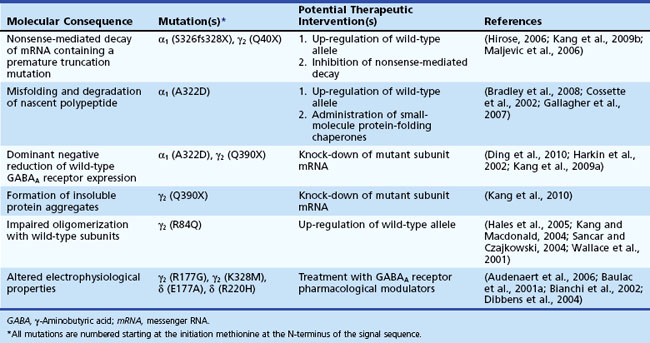
A mouse model of a human genetic generalized epilepsy syndrome has been developed. Tan et al. (2007) engineered mice containing a missense mutation in the GABAA receptor γ2 subunit that is associated with absence epilepsy and febrile seizures plus. The engineered mice exhibited absence seizures, and electrophysiological recordings demonstrated reduced GABAA currents in the cortex, but not in the thalamus. This result emphasized the importance of region-specific molecular changes within the epileptic network.
Although monogenic epilepsy syndromes allow epileptologists to probe the effect of a single molecule on the development of epilepsy, one must be mindful that the phenotype may not be directly attributable to the mutated gene. Rather, the mutation may cause downstream effects or compensatory changes that are directly linked to the production of seizures. For example, Cope et al. (2009) found that several genetic models of generalized epilepsy all caused a similar downstream effect: the augmentation of tonic GABA-ergic currents in the thalamus.
Differential Diagnosis
Seizures and epilepsy have many imitators, some of which are age-specific. In infants, the list includes apnea, either central or obstructive, which can be a seizure manifestation but can also be nonepileptic. Jitteriness associated with a variety of metabolic disturbances can imitate clonic or myoclonic seizures. Exaggerated startle can also be mistaken for tonic seizures. After the neonatal period, early childhood imitators of seizures include shuddering attacks and stereotypies or repetitive behaviors that can be mistaken for seizures. Gastroesophageal reflux may be associated with posturing (Sandifer syndrome); a greater occurrence of spells following feeding may be a clue to the diagnosis. Breath-holding spells represent a form of syncope that may be associated with tonic posturing and a few jerks (Laux and Nordli, 2005). Either cyanosis or pallor can accompany these breath-holding spells, and both are precipitated by injury or frustration, but cyanotic spells are preceded by crying.
Imitators of epilepsy that are more common in old age include transient ischemic attacks (TIAs) and transient global amnesia. Transient ischemia generally causes negative symptoms with loss of function such as weakness or numbness, whereas seizures involving sensory or motor cortex are more likely to produce positive symptoms such as twitching or paresthesias. However, seizures may occasionally present with only negative symptoms, and TIAs rarely present with limb shaking. Limb shaking as a feature of TIAs has been associated with high-grade stenosis or occlusion of the internal carotid artery (Persoon et al., 2010). Although TIAs tend to be longer in duration than seizures (most seizures last <2 minutes and most TIAs last >2 minutes), limb-shaking TIAs are short, usually shorter than 5 minutes and even shorter than 1 minute. One feature that could distinguish them from seizures is precipitation by rising or exercise and association with weakness of the affected limb (Persoon et al., 2010). TIAs can be misdiagnosed as seizures, but the more common scenario is to misdiagnose negative motor seizures or sensory seizures as TIAs. The presence of a sensory march should be suggestive of an epileptic nature.
Psychogenic Nonepileptic Seizures
Psychogenic nonepileptic seizures (PNES), also called psychogenic nonepileptic events, pseudoseizures, or pseudoepileptic seizures, are the most common imitators of seizures and epilepsy in referral centers. These are emotionally triggered attacks not associated with any paroxysmal epileptic activity in the brain. Most are the result of somatoform disorder, with a variety of reported traumatic antecedents, particularly sexual or physical abuse in women (Duncan and Oto, 2008). Antecedents or historical precipitants of PNES include head injury, which is also a common and important risk factor for epilepsy. In one study, 33% of patients with head injury and seizures had PNES on video-EEG monitoring (Hudak et al., 2004). PNES may also appear after surgery and can be one explanation for apparent failure of epilepsy surgery (Ney et al., 1998; Parra et al., 1998; Reuber et al., 2002). This emphasizes the need for video-EEG evaluation of patients who have had recurrence of seizures after epilepsy surgery. A diagnosis of fibromyalgia or a history of chronic pain were found to be predictors of PNES (Benbadis, 2005).
PNES are diagnosed in a considerable proportion of patients referred for drug-resistant seizures. The population-based incidence has been estimated at between 1.4 and 4.6 per 100,000 person-years of observation. In one study, the incidence was highest between 15 and 24 years of age (Sigurdardottir and Olafsson, 1998) and in another between 25 and 45 years of age (Szaflarski et al., 2000). All studies agree that there is a higher prevalence in women (70%-80%). The various patterns of clinical manifestations of PNES can be classified in three broad categories: psychogenic motor seizures with prominent motor activity, psychogenic minor motor or trembling seizures with tremor of the extremities, and attacks with motionless unresponsiveness or collapse (Groppel et al., 2000; Meierkord et al., 1991; Selwa et al., 2000). In children, prolonged staring and unresponsiveness was the most common pattern, while motor activity was more common in adolescents (Kramer et al., 1995).
The diagnosis of PNES depends on prolonged video-EEG monitoring with recording of typical attacks. The use of suggestion may facilitate the precipitation and recording of attacks. Hyperventilation and photic stimulation are usually adequate suggestion techniques; suggestion methods should not involve patient deception. In some individuals, suggestion may precipitate atypical attacks; to verify that recorded events are typical of what occurs at home, it is crucial to seek the input of family members who have witnessed attacks. PNES may coexist with epilepsy. Early studies suggested that more than 50% of patients with PNES also have epilepsy, but most studies now agree on a much smaller proportion, probably not more than 10% or 15% (Benbadis et al., 2001; Lesser et al., 1983; Martin et al., 2003).
A number of studies have tried to identify features that suggest psychogenic nonepileptic seizure origin (Avbersek and Sisodiya, 2010). In a comparison of epileptic generalized tonic-clonic seizures and PNES with motor activity, features that predicted PNES were out-of-phase upper and lower extremity movements, absence of vocalization or vocalization at the very onset of seizures, forward pelvic thrusting, absence of whole-body rigidity, and side-to-side head movements. No single feature was totally predictive by itself, and these features were particularly strong predictors when combined (Gates et al., 1985), but such clinical features can also be seen in frontal lobe complex partial seizures and other hypermotor seizures (Saygi et al., 1992). Other features that may help discriminate PNES from epileptic seizures include pseudosleep at onset (Benbadis et al., 1996b); preictal behavioral changes (Moore et al., 1998); discontinuous seizure activity; prolonged seizure duration; eye closure during unresponsiveness (Chung et al., 2006; DeToledo and Ramsay, 1996); resistance to eye opening; eye fluttering; certain vocalizations such as stuttering, gagging, gasping, screaming, weeping, or moaning; emotional display during events; emotional triggers; precipitation of typical events by suggestion; and attacks occurring in the clinic waiting room or admitting office (Benbadis, 2005). Tongue biting and incontinence occur more commonly with epileptic seizures but are also frequently reported by patients with PNES (Peguero et al., 1995). Injuries to the tongue during epileptic seizures tend to affect the side of the tongue. Biting the tip of the tongue or the lip was suggestive of PNES (DeToledo and Ramsay, 1996). Self-injury was also reported by patients with PNES, but one study suggested that burn injuries were specific for epileptic seizures (Peguero et al., 1995). The presence of postictal stertorous respiration is very helpful to diagnose epileptic convulsive seizures (Sen et al., 2007), whereas shallow rapid respiration was more likely after PNES (Azar et al., 2008a).
While the diagnosis of PNES depends in large part on the absence of EEG changes with typical attacks, the neurologist has to be aware that some epileptic seizures have no EEG correlate. For example, frontal lobe complex partial seizures of orbitofrontal or cingulate origin commonly have no associated EEG changes, nor do supplementary motor seizures. For definitive diagnosis, it is often necessary to record multiple attacks and observe changes in conjunction with AED withdrawal. Epileptic seizures may secondarily generalize, which provides a definitive diagnosis. In one study comparing patients with frontal lobe complex partial seizures and patients with PNES, there was no significant difference in the history of psychiatric disease, ictal pelvic thrusting, rocking of body, side-to-side head movements, or rapid postictal recovery (Saygi et al., 1992). Of interest, turning to a prone position occurred only in frontal lobe complex partial seizures. Nocturnal occurrence, short ictal duration, younger age at onset, stereotyped movements, and abnormal MRI or EEG favored frontal lobe complex partial seizures. Others have also suggested that while epileptic seizures are very stereotyped, PNES tend to have a lot of variability. However, this notion has been disputed (Seneviratne et al., 2010).
Pseudo–status epilepticus is a common occurrence in PNES, reported in a greater proportion of patients than status epilepticus in patients with epilepsy (Dworetzky et al., 2006). As expected, it tends to be resistant to treatment with AEDs, until the development of stupor or coma, which may lead to intubation. Its early recognition is essential to prevent potentially harmful interventions.
Syncope
Syncope is an abrupt transient loss of consciousness caused by decreased cerebral perfusion. It is an important condition in the differential diagnosis of epilepsy in the elderly as well as teenagers and young adults. Although syncope is mostly recognized by loss of posture and limp unresponsiveness, the majority of closely observed individuals will have brief transient motor manifestations early after loss of consciousness. The most common motor manifestation is myoclonus that is most often multifocal and arrhythmic (Lempert et al., 1994). Other motor manifestations may also occur, including posturing, head turning, upward eye movement, oral automatisms, and righting movements (Lempert et al., 1994). The motor manifestations, particularly myoclonus, are an important factor in the misdiagnosis of syncope as seizures. Although syncope with myoclonus has been referred to as “convulsive syncope,” there is no associated EEG discharge, and the origin is thought to be in the brainstem. Syncope can have a variety of mechanisms, some benign and others serious. In younger individuals, the most common is neurally mediated syncope. This can be triggered by a variety of factors including intense pain, emotion, and standing for prolonged periods of time in hot or crowded places. In addition, syncope can be precipitated in some individuals by micturition, defecation, or cough. Neurally mediated syncope tends to have a prodrome of lightheadedness, nausea, pallor, cold sweating, graying of vision, and hearing becoming distant, as well as other visual or auditory hallucinations (Carreño, 2008; Crompton and Berkovic, 2009; Lempert et al., 1994). Syncope may also be due to orthostatic hypotension, cardiac arrhythmias, and structural cardiopulmonary disease (Carreño, 2008; Crompton and Berkovic, 2009). Syncope due to cardiac arrhythmia is usually more abrupt with no preceding symptoms. In the differentiation of syncope from seizures, features that favor syncope include known heart disease, prior confirmed syncope, precipitation by prolonged standing or rising to an upright position, presence of dehydration, the typical neurocardiogenic syncope prodrome described earlier, description of pronounced pallor by witnesses, absence of tonic or clonic activity, description of multifocal myoclonus lasting less than 15 seconds, and recollection of loss of consciousness. Features that would favor seizures include previous seizures, known cortical brain lesion, presence of tongue biting, incontinence, cyanosis, postictal confusion, postictal headache, and lack of recollection of loss of consciousness (Crompton and Berkovic, 2009). Syncope may rarely trigger an epileptic seizure (Stephenson et al., 2004). These seizures, referred to as anoxic-epileptic seizures, are to be distinguished from the much more common nonepileptic “convulsive” syncope.
Migraine
Epilepsy and migraine both present with paroxysmal manifestations as a result of cerebral cortex involvement (Kossoff and Andermann, 2010). Types of migraine that are most likely to be confused with seizures are classical migraine with visual or somatosensory aura, basilar migraine, and acute confusional migraine (Carreño, 2008; Kossoff and Andermann, 2010). Occipital lobe seizures may be followed by a migraine-like headache that makes it hard to distinguish them from a classical migraine with a visual aura. Helpful distinguishing features include duration of the aura. The aura in migraine typically lasts 5 to 60 minutes (Carreño, 2008), reflecting that cortical spreading depression (the main pathophysiology of migraine) results from a slow depolarization that spreads at about 3 mm/min (Crompton and Berkovic, 2009). In contrast, epileptic discharges propagate at a much higher speed; epileptic auras are usually less than 30 seconds in duration. Another helpful distinction is that the visual aura in migraine is most commonly a fortification spectrum or scintillating scotoma, whereas colored circles are the most common aura in occipital lobe seizures (Carreño, 2008; Kossoff and Andermann, 2010). Migraine without headache, also called migraine equivalent, can be an even more difficult diagnostic challenge. Basilar migraine starts with brainstem manifestations that include dysarthria, vertigo, changes in hearing, diplopia, and ataxia, and then involves loss of consciousness.
Migraine and epilepsy have a greater overlap than would be expected by chance. The prevalence of each is increased in the presence of the other, and some epileptic syndromes (e.g., benign epilepsy with occipital paroxysms, benign epilepsy with centrotemporal spikes) have a particularly higher incidence of migraine (Kossoff and Andermann, 2010). In addition, a rare condition referred to as “migralepsy” (or migraine-triggered epilepsy or migraine-triggered seizures) is characterized by seizures that occur during or shortly after the migraine aura. Certain antiepileptic medications such as valproate and topiramate are used successfully in migraine prophylaxis, but others such as oxcarbazepine or carbamazepine do not seem to be effective. However, acute abortive therapy for the two conditions is totally different.
Sleep Disorders
Parasomnias are the most important imitators of seizures in the category of sleep disorders (see Chapter 68). These include sleep walking (somnambulism), sleep talking, night terrors, confusional arousals, and REM behavior disorder. They may imitate frontal lobe seizures that occur preferentially or even exclusively in sleep. Somnambulism, sleep talking, and night terrors typically start in childhood and tend to disappear in adolescence. They are most likely to arise out of slow-wave sleep in the first half of the night, usually after a latency of 90 minutes from sleep onset. Frontal lobe seizures are more likely to arise out of stage 1 or 2 sleep (Carreño, 2008). These events are more likely to be seizures if they occur in the first hour of sleep or in the transition between waking and sleep. REM behavior disorder is characterized by loss of muscle atonia during REM sleep, which results in acting out dreams. The behavior includes verbalization and vocalization, as well as motor activity that may be violent (e.g., kicking, punching) and getting out of bed. Affected individuals will be aware that they have been dreaming and may report the content of their dreams. REM behavior disorder is more likely in the second half of the night when REM sleep is most likely to occur (Carreño, 2008). REM behavior disorder rarely starts before age 50; it is most often a chronic disorder associated with a synucleinopathy, particularly Lewy body dementia (Crompton and Berkovic, 2009).
Paroxysmal Movement Disorders
Paroxysmal movement disorders that can be confused with epilepsy include nonepileptic myoclonus, paroxysmal dyskinesia, and hyperekplexia (Crompton and Berkovic, 2009). Myoclonus may be generated at any level of the CNS; epileptic myoclonus is generated at the level of the cortex and is usually associated with a scalp EEG discharge. In the case of focal cortical myoclonus, the distinction can be harder to establish (Crompton and Berkovic, 2009). Paroxysmal dyskinesia can be classified into two broad categories: kinesigenic and non-kinesigenic, both usually familial (Fahn and Frucht, 2008). In paroxysmal kinesigenic dyskinesia, the attacks are often brought on by a sudden movement or startle, usually after a period of inactivity. The movements are any combination of chorea, athetosis, ballism, and a dystonic posture (Fahn and Frucht, 2008). They can be bilateral or alternate sides, which is helpful in distinguishing them from epileptic seizures that generally produce consistently unilateral posturing. Other helpful distinguishing features are that consciousness is always preserved, and there is no postictal change. However, the condition responds very well to AEDs. The attacks in paroxysmal nonkinesigenic dyskinesia are longer (minutes to hours as compared to seconds), less frequent, and precipitated not by movement but rather by alcohol, caffeine, stress, excitement, or fatigue. The attacks do not usually respond to AEDs (Fahn and Frucht, 2008). Hyperekplexia is an inherited disorder in which there is an exaggeration of startle reflexes (Crompton and Berkovic, 2009). The exaggerated startle has to be distinguished from startle-evoked seizures, which are most often of supplementary motor origin.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 67 Epilepsies
Seizures and Epilepsy Definitions
Seizures are transient events that include symptoms and/or signs of abnormal excessive hypersynchronous activity in the brain (Fisher et al., 2005). In 2005, the International League Against Epilepsy (ILAE) and International Bureau for Epilepsy (IBE) proposed a definition of epilepsy as a disorder of the brain characterized by an enduring predisposition to generate epileptic seizures and by the neurobiological, cognitive, psychological, and social consequences of this condition (Fisher et al., 2005). Acute symptomatic seizures provoked by metabolic or toxic derangements or occurring acutely in the setting of head trauma or stroke do not define epilepsy.
The traditional definition of epilepsy requires at least two unprovoked seizures. The definition proposed by the ILAE in 2005 suggested that one epileptic seizure is sufficient to diagnose epilepsy if there is additional enduring alteration in the brain that increases the likelihood of future seizures, but the proposal did not specify what evidence is sufficient to define such an enduring alteration. The proposed definition has been controversial and has not been widely accepted (Beghi et al., 2010). The definition of epilepsy used in this chapter requires at least two unprovoked seizures, so a single unprovoked seizure is insufficient to define epilepsy.
In 2001, the ILAE proposed a diagnostic scheme for the classification of seizures and epilepsy (Engel, 2001). The scheme recommended axes that are helpful concepts in the evaluation of patients with epilepsy:
Ictal Phenomenology
Glossary of Seizure Terminology and Other Definitions
The terms frequently used in the description of seizures follow. Whenever possible, the definition is derived from the glossary of descriptive terminology for ictal semiology, reported by the ILAE task force on classification and terminology (Blume et al., 2001). The term ictal semiology means the signs and symptoms associated with seizures.
Automatisms can be described by the part of the body affected. Some of the most common are oroalimentary automatisms, which include lip smacking, chewing, swallowing, and other mouth movements. Ictal spitting and ictal drinking can be considered forms of oroalimentary automatisms. Automatisms affecting the distal extremities are manual or pedal. Manual or pedal automatisms can be bilateral or unilateral. Gestural automatisms include extremity movements such as those used to enhance speech. More recently introduced categories for upper extremity automatisms are manipulative and non-manipulative (Kelemen et al., 2010). Manipulative automatisms involve picking and fumbling motions, typically reflecting interaction with the environment. Non-manipulative upper extremity automatisms tend to be rhythmic and do not involve interaction with the environment. Distal non-manipulative upper-extremity automatisms have been described with the acronym RINCH (rhythmic ictal nonclonic hand) movements (Lee et al., 2006). Hyperkinetic automatisms imply an inappropriately rapid sequence of movements that predominantly involve axial and proximal limb muscles. The resulting motion can be thrashing, rocking, pelvic thrusting, kicking, or bicycling motions. Gelastic refers to abrupt laughter or giggling, while dacrystic refers to abrupt crying, both inappropriate.
Classification of Seizures
Two classifications developed by the ILAE continue to be used widely: the Clinical and Electroencephalographic Classification of Epileptic Seizures published in 1981 (Commission on Classification and Terminology, 1981) (Box 67.1) and the Classification of Epilepsies and Epileptic Syndromes introduced in 1989 (Commission on Classification and Terminology, 1989) (Box 67.2). Revisions to these classifications have been recommended based on advances made in the last 3 decades (Berg et al., 2010; Engel, 2001; Engel, 2006). The major recommended revisions are outlined later in this section, but what follows will focus on the 1981 and 1989 classifications, because these classifications are still used widely for clinical management and research and will likely continue to be used in the future.
Box 67.1
International League Against Epilepsy Classification of Epileptic Seizures
From Commission on Classification and Terminology of the International League Against Epilepsy, 1981. Proposal for revised clinical and electroencephalographic classification of epileptic seizures. Epilepsia 22, 489-501.
Box 67.2
International League Against Epilepsy Classification of Epilepsies and Epileptic Syndromes
1. Localization-related (focal, local, partial) epilepsies and syndromes
2. Generalized epilepsies and syndromes
3. Epilepsies and syndromes undetermined as to whether they are focal or generalized
From Commission on Classification and Terminology of the International League Against Epilepsy, 1989. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 30, 389-399.
One important criticism of the 1981 classification is that it requires both clinical and electroencephalographic (EEG) information, and assumptions on correlation of clinical and EEG features may be incorrect. A purely semiological classification of epileptic seizures was proposed, based solely on observed clinical features (Lüders et al., 1998). The semiological seizure classification includes somatotopic modifiers to define the somatotopic distribution of the manifestations and allows demonstration of evolution of ictal manifestations using arrows to link sequential manifestations (Lüders et al., 1998). Although this classification was not adopted by the ILAE, it is considered an optional seizure classification system that is useful for localization purposes in epilepsy surgery centers.
The latest revision of the seizure classification has maintained the division of seizures based on generalized or focal onset but has recommended replacing partial with focal. It also updated the definition of focal seizures as “originating within networks limited to one hemisphere,” with the possibility of the seizures being discretely localized or more widely distributed, and possibly originating in subcortical structures. Generalized seizures were defined as “originating at some point within, and rapidly engaging, bilaterally distributed networks,” which do not necessarily include the entire cortex (Berg et al., 2010). The revised concepts acknowledge that generalized seizures can be asymmetrical and that individual seizures may appear to have a localized onset, but the location and laterality of that onset will vary from seizure to seizure (Berg et al., 2010).
Seizure Types
Partial Seizures
Simple Partial Seizures
The most recent recommendation of the ILAE Commission on Classification and Terminology suggested eliminating the term simple partial. Instead, it recommended descriptors of focal seizures according to degree of impairment during a seizure. Seizures without impairment of consciousness or awareness are divided into those with observable motor or autonomic components and those only involving subjective sensory or psychic phenomena (Berg et al., 2010).
Partial Seizures Evolving to Generalized Tonic-Clonic Activity
These seizures may start as simple partial, complex partial, or simple partial evolving to complex partial. The transition to secondary generalization usually involves versive head turning in a direction contralateral to the hemisphere of seizure onset, and focal or lateralized tonic or clonic motor activity. The pattern of evolution may be clonic-tonic-clonic in some instances. The generalized tonic phase may be asymmetrical, with flexion on one side and extension on the other. This has been called figure-of-4 posturing (Kotagal et al., 2000). Some asymmetry and asynchrony may also occur in the clonic phase, resulting in a slight degree of side-to-side head jerking (Niaz et al., 1999). The evolution from tonic to clonic activity is gradual and not always simultaneous in all affected body parts. A phase of high-frequency tremor has been referred to as the tremulous or vibratory phase of the seizure (Theodore et al., 1994). Clonic activity typically decreases in frequency over time, with longer intervals between jerks towards the termination of the seizure. The clonic activity may end on one side of the body first, so that clonic activity may then appear lateralized to one side. In addition, there may be a late head turn ipsilateral to the hemisphere or seizure origin (Wyllie et al., 1986). After the motor activity stops, the individual is usually limp and has a loud snoring respiration often referred to as stertorous respiration. During the course of recovery, there may be variable agitation. The speed of recovery is expected to be slower with longer and more severe seizures.
Partial Seizure Semiology in Relation to Localization
Partial Seizures of Temporal Lobe Origin
Temporal lobe seizures most often are of mesial temporal amygdalohippocampal origin, in association with the pathology of hippocampal sclerosis. Patients commonly have isolated auras, and complex partial seizures tend to start with an aura. The most common aura is an epigastric sensation frequently with a rising character (French et al., 1993). Other auras occur less commonly and include fear, anxiety, and other emotions, déjà vu and jamais vu, nonspecific sensations, and autonomic experiences such as palpitation and gooseflesh. Olfactory and gustatory auras are uncommon and more likely with tumoral mesial temporal lobe epilepsy (MTLE).
Complex partial seizures may start with an aura or with altered consciousness. With nondominant temporal lobe seizures, the patient may remain responsive and verbally interactive. However, recollection of conversations is unusual. Altered consciousness is often associated with an arrest of motion and speech. Speech arrest is not synonymous with aphasia and does not distinguish dominant and nondominant temporal lobe seizures. Automatisms are one of the most prominent manifestations, and oroalimentary automatisms are the most prevalent. Extremity automatisms also occur and are most commonly manipulative, with picking or fumbling. This type of automatism is not of direct lateralizing value. However, the contralateral upper extremity is commonly involved in dystonic posturing (Kotagal et al., 1989) or milder degrees of posturing and immobility (Fakhoury and Abou-Khalil, 1995; Williamson et al., 1998). This reduces the availability of the contralateral arm for automatisms, so manipulative automatisms tend to be ipsilateral, involving the unaffected upper extremity. Nonmanipulative automatisms typically consist of rhythmic movements either distally or proximally. These tend to be contralateral, often preceding overt dystonic posturing (Lee et al., 2006). Head turning occurs commonly. Early head turning is not usually forceful. It typically occurs at the same time as dystonic posturing and is most often ipsilateral (Fakhoury and Abou-Khalil, 1995; Williamson et al., 1998). Late head turning most often occurs during evolution to generalized tonic-clonic activity. This is usually contralateral to the side of seizure origin (Williamson et al., 1998). Well-formed ictal speech may occur during seizures of nondominant temporal lobe origin (Gabr et al., 1989). Verbal output may at times be tinged with a fearful tone. Complex partial seizures of temporal lobe origin usually last between 30 seconds and 3 minutes. Postictal manifestations may be helpful in lateralizing the seizure onset. Postictal aphasia is commonly seen after dominant temporal lobe seizures (Gabr et al., 1989). In one study, such patients were unable to read a test sentence correctly in the first minute after seizure termination, but patients with nondominant right temporal lobe origin were able to read the test sentence within 1 minute of seizure termination (Privitera et al., 1991).
Seizures of lateral temporal origin or neocortical temporal origin are much less common than those of mesial temporal origin. They cannot be reliably distinguished based on their semiology, but certain features can help distinguish them. Auditory auras are the most common auras referable to the lateral temporal cortex, usually implying involvement of the Heschl gyrus. Other types of auras referable to the lateral temporal cortex are vertiginous and complex visual hallucinations (usually posterior temporal). Oroalimentary automatisms are less common, and the pattern of contralateral dystonic posturing and ipsilateral extremity automatisms is also less common (Dupont et al., 1999). Early contralateral or bilateral facial twitching may be seen as a result of propagation to the frontal operculum (Foldvary et al., 1997). Seizures of lateral temporal origin tend to be shorter in duration and have a greater tendency to evolve to generalized tonic-clonic activity than seizures of mesial temporal origin.
Partial Seizures of Frontal Lobe Origin
Many different seizure types can originate in the frontal lobe, depending on site of seizure origin and propagation. Simple partial seizures can be motor with focal clonic activity, can originate in the motor cortex, or can be the result of spread to the motor cortex. These seizures may or may not have a Jacksonian march. Asymmetrical tonic seizures or postural seizures are usually related to involvement of the supplementary motor area in the mesial frontal cortex anterior to the motor strip. The best-known posturing pattern is the fencing posture in which the contralateral arm is extended and the ipsilateral arm is flexed. Tonic posturing may involve all four extremities and is occasionally symmetrical. When these seizures originate in the supplementary motor area, consciousness is usually preserved (Morris et al., 1988). Supplementary motor seizures are an important exception to the rule that bilateral motor activity during a seizure should be associated with loss of consciousness. Supplementary motor seizures are usually short in duration and frequently arise out of sleep. They tend to occur in clusters and may be preceded by a sensory aura referable to the supplementary sensory cortex. The pattern of posturing described with supplementary motor area seizures can occur as a result of seizure spread to the supplementary motor area from other regions of the brain. In that case, consciousness is frequently impaired. Subjective simple partial seizures may also occur with frontal lobe origin, the most common being a nonspecific cephalic aura.
Complex partial seizures of frontal lobe origin tend to be very peculiar. They may be preceded by a nonspecific aura or they may start abruptly, often out of sleep. Their most characteristic features are hyperkinetic automatisms with frenzied behavior and agitation (Jobst et al., 2000; Williamson et al., 1985). There may be various vocalizations including expletives. The manifestations can be so bizarre as to suggest a psychiatric origin. The seizure duration is short, sometimes less than 30 seconds, and postictal manifestations are brief or nonexistent, further adding to the risk of misdiagnosis as psychogenic seizures. Frontal lobe complex partial seizures arise predominantly from the orbitofrontal region and from the mesial frontal cingulate region. However, they can arise from other parts of the frontal lobe and even from outside the frontal lobe, usually reflecting seizure propagation to the frontal lobe. It may be difficult to determine the region of origin in the frontal lobe based on the seizure manifestations. It has been suggested that the presence of tonic posturing on one side points to a mesial frontal origin, as does rotation along the body axis, which sometimes leads to turning prone during the seizure (Leung et al., 2008; Rheims et al., 2008).
Frontal opercular seizures originating in the frontal operculum are associated with profuse salivation, oral facial apraxia, and sometimes facial clonic activity (Williamson and Engel, 2008). Seizures originating in the dorsolateral frontal lobe may involve tonic movements of the extremities and versive deviation of the eyes and head. The head deviation preceding secondary generalization is contralateral, but earlier head turning can be in either direction (Remi et al., 2011). Seizures may begin with forced thinking. Partial seizures of frontal origin may at times resemble absence seizures (So, 1998). It is important to recognize that seizures originating in the frontal lobe can propagate to the temporal lobe and produce manifestations typical of mesial temporal lobe seizures.
Partial Seizures Originating in the Parietal Lobe
The best-recognized seizure type that originates in the parietal lobe is partial seizure with somatosensory manifestations. The somatosensory experience can be described as tingling, pins and needles, numbness, burning, or pain. The presence of a sensory march is most suggestive of involvement of the primary sensory cortex. Sensory phenomena arising from the second sensory area and the supplementary sensory area are less likely to have a march. Somatosensory auras tend to be contralateral to the hemisphere of seizure origin, but they may be bilateral or ipsilateral when arising from the second or supplementary sensory regions. Other auras of parietal lobe origin are a sensation of movement in an extremity, a feeling of the body bending forward or swaying or twisting or turning, or even a feeling of an extremity being absent (Salanova et al., 1995a; Salanova et al., 1995b). Some patients may complain of inability to move a limb. Vertigo has been reported, as well as visual illusions of objects going away or coming closer or looking larger (Siegel, 2003). Some patients may have initial auras suggesting spread to the occipital or temporal lobe. Seizures involving the dominant parietal lobe may produce aphasic manifestations. Motor manifestations tend to reflect seizure spread to the frontal lobe. These include tonic posturing of the extremities, focal motor clonic activity, and version of the head and eyes (Cascino et al., 1993; Ho et al., 1994; Williamson et al., 1992a). Negative motor manifestations may occur, with ictal paralysis (Abou-Khalil et al., 1995). Seizures may spread to the temporal lobe, producing oroalimentary or extremity automatisms (Siegel, 2003). In one study, motor manifestations were more likely with superior parietal epileptogenic foci, and oroalimentary and extremity automatisms more likely with inferior parietal epileptogenic foci (Salanova et al., 1995a). Visual manifestations seemed more likely with posterior parietal lesions.
Partial Seizures Originating in the Occipital Lobe
The best-recognized occipital lobe seizure semiology is that of simple partial seizures with visual manifestations (Salanova et al., 1992). The most common are elementary visual hallucinations that are described as flashing colored lights or geometrical figures. These are usually contralateral but may move within the visual field. Complex visual hallucinations with familiar faces or people may also occur. Negative symptoms may be reported, with loss of vision in one hemifield. Ictal blindness may involve loss of vision in the whole visual field. Objective seizure manifestations include blinking, nystagmoid eye movements, and versive eye and head deviation contralateral to the seizure focus. This version may occur while the patient is still conscious or could be a component of complex partial seizures.
Seizure manifestations that are related to seizure spread to the temporal or frontal lobe are very common. Oroalimentary automatisms are typical of seizures that spread to the temporal lobe, whereas asymmetrical tonic posturing typifies spread to the frontal lobe; both types of spread can be seen in the same patient (Williamson et al., 1992b). Spread to the temporal or frontal lobe is so common with occipital lobe seizures that it is at times reported in the majority of patients (Jobst et al., 2010b). Ictal semiology cannot distinguish seizures originating from the mesial versus lateral occipital region (Blume et al., 2005). Evolution of occipital seizures to secondary generalization is commonly reported.
Partial Seizures Originating in the Insular Cortex
Insular epilepsy is uncommon and also frequently unrecognized because of the inability to record directly from the insula with scalp electrodes. Subjective symptoms that should suggest seizure origin in the insula include laryngeal discomfort, possibly preceded or followed by a sensation in the chest or abdomen, shortness of breath, and paresthesias around the mouth or also involving other contralateral body parts (Isnard et al., 2004). Objective seizure manifestations include dysarthria/dysphonia, sometimes evolving to complete muteness. With seizure progression in some patients, tonic spasm of the face and upper limb, head and eye rotation, and at times generalized dystonia occur (Isnard et al., 2004). Hypersalivation is also very common and can be impressive. Insular-onset seizures may spread to other brain regions and can be disguised as temporal lobe, parietal lobe, or frontal lobe epilepsy (Ryvlin, 2006; Ryvlin et al., 2006).
Generalized Seizures
Generalized Absence Seizures
Typical absence seizures are characterized by a sudden blank stare with motor arrest, usually lasting less than 15 seconds (Commission on Classification and Terminology of the International League Against Epilepsy, 1981). The individual is usually unresponsive and unaware. The seizure ends as abruptly as it starts, and the patient returns immediately to a baseline level of function with no postictal confusion but may have missed conversation and seems confused as a result. If the only manifestation is altered responsiveness and awareness, with no associated motor component, the absence seizure is classified as simple absence. Most often, generalized absence seizures include mild motor components and are classified as complex absence. The most common motor components are automatisms such as licking the lips or playing with an object that was held in the hand before the seizure. Other motor components include clonic, tonic, atonic, and autonomic manifestations. Clonic activity may affect the eyelids or the mouth. An atonic component may manifest with dropping an object or slight head drop or drooping of the shoulders or trunk. Tonic components may manifest with slight increase in tone.
The EEG hallmark of a typical generalized absence seizure is generalized 2.5- to 4-Hz spike-and-wave activity with a normal interictal background (Fig. 67.1). Atypical absence seizures are diagnosed primarily based on a slower (<2.5 Hz) frequency of the EEG spike-and-wave activity. Less important distinctions are that the onset and termination of an atypical absence seizure may be less abrupt and the motor components a bit more pronounced than seen with typical absence seizures. Atypical absence seizures usually occur in individuals with impaired cognitive function. Affected individuals usually have associated seizure types such as generalized tonic, generalized atonic, and generalized tonic-clonic seizures.
Additional generalized absence seizure types recently recognized by the ILAE include myoclonic absences. The key manifestation of these seizures is a prominent rhythmic myoclonus predominantly affecting the limbs (Bureau and Tassinari, 2005b). Otherwise, myoclonic absences resemble typical absence seizures with respect to impairment of consciousness, although this impairment can be only partial. Another related seizure type recently recognized is eyelid myoclonia with absence. The eyelid myoclonia consists of pronounced rhythmic jerking of the eyelids, usually associated with an upward deviation of the eyes and retropulsion of the head (Caraballo et al., 2009). There may or may not be associated generalized spike-and-wave activity on EEG. Absence seizures may evolve to generalized tonic-clonic activity (Mayville et al., 2000).
Generalized Myoclonic Seizures
Myoclonic seizures are muscle contractions lasting a fraction of a second (<250 msec), in association with an ictal EEG discharge (Blume et al., 2001). The myoclonic jerk can be generalized, affecting the whole body, or could affect just the upper extremities or (rarely) the head or trunk, or even the diaphragm. The myoclonic jerks may affect one side of the body at one time, but typically the other side is affected at other times. The jerks can be single or could occur in an arrhythmic cluster. It should be noted that myoclonus is not always epileptic (Faught, 2003). Myoclonus can be generated anywhere along the central nervous system (CNS). Epileptic myoclonus is generated in the cerebral cortex and is usually associated with a single or brief serial spike-and-wave or polyspike-and-wave activity.
Negative myoclonic seizures consist of a very brief pause in muscle activity rather than a brief muscle contraction (Rubboli and Tassinari, 2006). Just as with positive myoclonus, negative myoclonus can be generalized, bilateral with limited distribution, or even focal, typically with shifting lateralization.
Myoclonic seizures may be immediately followed by a loss of tone. The seizure type is called myoclonic-atonic. Historically it was called myoclonic-astatic. The seizures are brief (1 second or less) but may be associated with falls and injuries. The EEG shows generalized spike-and-wave or polyspike-and-wave discharge. The slow wave is prolonged and associated with the electromyographic (EMG) silence characteristic of the atonic phase. Myoclonic seizures may precede a more sustained tonic contraction, and the resultant seizures may be referred to as myoclonic-tonic seizures (Berg et al., 2010). Generalized myoclonic seizures may cluster just before a generalized tonic-clonic seizure occurrence.
Generalized Clonic Seizures
Unlike myoclonic seizures, which are single jerks (but may occur in arrhythmic clusters), each generalized clonic seizure consists of a series of rhythmic jerks. Generalized clonic seizures are uncommon and particularly rare in adults (Noachtar and Arnold, 2000). They are more frequently seen in certain epileptic syndromes of infancy and childhood. For example, clonic seizures are a common seizure type of severe myoclonic epilepsy of infancy (Dravet syndrome). Clonic seizures are also noted in progressive myoclonic epilepsies.
Generalized Tonic Seizures
Generalized tonic seizures are typically brief seizures, a few seconds to 1 minute. Their onset may be gradual or abrupt. They may be initiated with a myoclonic jerk. They can vary in severity from subtle, with slight increase in neck tone with upward deviation of the eyes, to massive, with involvement of the axial muscles and extremities. Proximal muscles are the most affected. Most commonly there is neck and trunk flexion as well as abduction of the shoulders and flexion of the hips. However extension may also occur. Tonic seizures may be asymmetrical, which could result in turning to one side. The pattern of muscle involvement may change over time so that there may be a change in the position of the limbs over the course of the seizure. Autonomic changes may occur, with tachycardia, pupil dilation, and flushing. Involvement of respiratory muscles could cause apnea and cyanosis. The tonic contraction may end with one or more pauses that result in a few clonic jerks. A postictal state with confusion may occur, but recovery is usually rapid. However, tonic seizures may be followed by atypical absence, resulting in what appears to be a more prolonged postictal state. This has been referred to as tonic-absence seizure (Shih and Hirsch, 2003). Generalized tonic seizures occur most often out of sleep and drowsiness.
Epileptic Spasms
Epileptic spasms have similarities to generalized tonic seizures but a shorter duration that is intermediate between generalized myoclonic and generalized tonic seizures (Blume et al., 2001), with a typical duration of 0.5 to 2 seconds. The pattern of contraction is “diamond-shaped,” with intensity of contraction maximal in the middle of the spasm and less at the beginning and end. Epileptic spasms are also called infantile spasms and salaam attacks. Because their occurrence is not restricted to infants, the preferred current term is epileptic spasms. The classic epileptic spasm involves neck and trunk flexion and arm abduction with a jackknife pattern, but extension may be seen. Epileptic spasms typically occur in clusters recurring every 5 to 40 seconds. In a cluster, the initial spasms may be subtle or mild, increase in intensity as the cluster progresses, and decrease in intensity again toward the end of the cluster (Bleasel and Lüders, 2000).
Generalized Tonic-Clonic Seizures
Generalized tonic-clonic (GTC) seizures are dramatic and the best recognized form of seizures. They are commonly referred to as grand mal, but this term is archaic and does not distinguish seizures of focal onset from those with a generalized onset. Generalized tonic-clonic seizures do not have an aura, but they may be preceded by a prodrome—the vague sense a seizure will occur—lasting up to hours. Seizure onset is abrupt, most often with loss of consciousness and a generalized tonic contraction, but some seizures may be initiated with a series of myoclonic jerks, leading to the term clonic-tonic-clonic seizure. The tonic phase may have asymmetrical movements, and these often change from seizure to seizure. One such commonly encountered asymmetry is versive head turning, which is not evidence of a focal onset (Chin and Miller, 2004; Niaz et al., 1999). The tonic phase includes an upward eye deviation with eyes half open and the mouth open. Involvement of the respiratory muscles usually produces a forced expiration that produces a loud guttural vocalization, often referred to as the epileptic cry. Cyanosis may occur during the tonic phase in association with apnea. The tonic phase gradually evolves to clonic activity. The transition can be with initially high-frequency and low-amplitude motion, often referred to as a vibratory phase. With seizure progression, the frequency of clonic jerks decreases, and the amplitude may initially increase but later decreases just before the seizure stops. In the immediate postictal state the individual is limp and unresponsive. Respiration is loud and snoring in character (stertorous). The postictal state is often followed by sleep, although the individual may awaken briefly with postictal confusion. Tongue biting commonly occurs and most often affects the side of the tongue. Incontinence of urine is common, and incontinence of stool may also occur. After awakening, patients often have a pronounced headache and generalized muscle soreness. Generalized tonic-clonic seizures rarely last more than 2 minutes. The severity may vary. The postictal state seems to correlate with severity and duration.
Generalized Atonic Seizures
Generalized atonic are associated with very brief, sudden loss of tone and vary from extremely subtle, manifesting with only a head drop, to generalized loss of tone and falling. Atonic seizures may result in falling if the person is standing, then called a drop attack. However, drop attacks may be the result of both generalized atonic and generalized tonic seizures. There is a very brief loss of consciousness and brief postictal confusion. Seizures are usually very brief, lasting 1 second to a few seconds. They may be preceded by a brief myoclonic jerk, in which case the seizure type is called myoclonic-atonic. Very brief atonic seizures are typical of the syndrome of myoclonic-astatic epilepsy (Doose syndrome) (Oguni et al., 2001). More prolonged atonic seizures can be seen with Lennox-Gastaut syndrome or other symptomatic generalized epilepsies. Despite their brief duration, generalized atonic seizures can result in serious injury and are an important cause of morbidity in epilepsy.
Generalized-Onset Seizures with Focal Evolution
Generalized-onset seizures rarely may evolve to focal seizures (Deng et al., 2007; Williamson et al., 2009). This seems to occur with either myoclonic or absence seizures. The clinical manifestations most often are behavioral arrest and staring, with minor automatisms. However, focal motor manifestations may also occur. This type of seizure tends to be prolonged and may be associated with postictal confusion (Williamson et al., 2009).
Classification of Epilepsies and Epileptic Syndromes
The classification of seizures addresses single seizure events and not epilepsy as a condition. The 1989 classification of epilepsies and epileptic syndromes tried to organize epilepsies and epilepsy syndromes (Commission on Classification and Terminology, 1989). It defined an epileptic syndrome as “an epileptic disorder characterized by a cluster of signs and symptoms customarily occurring together; these include such items as type of seizure, etiology, anatomy, precipitating factors, age of onset, severity, chronicity, diurnal and circadian cycling, and sometimes prognosis.” A syndrome does not necessarily have a common etiology and prognosis. Two important divisions were used in the classification. The first separated epilepsies with generalized-onset seizures, called generalized epilepsies, from epilepsies with partial-onset seizures, referred to as localization-related, partial, or focal epilepsies. The other division separated epilepsies of known etiology (named symptomatic epilepsies) from those of unknown etiology. Epilepsies of unknown etiology were named idiopathic if they were pure epilepsy and “not preceded or occasioned by another condition.” These epilepsies were considered to have no underlying cause other than a possible hereditary predisposition. Thus, they were presumed genetic. The idiopathic epilepsies were also defined by an age-related onset and clinical and EEG characteristics. Epilepsies of unknown etiology were called cryptogenic if they were presumed symptomatic, but with an occult etiology. Although the term cryptogenic remains widely used in the epilepsy field, confusion exists concerning its exact meaning, which has resulted in a recommendation to replace it with the term probably symptomatic (Engel, 2001). The 1989 classification of epilepsies and epileptic syndromes also subdivided symptomatic partial epilepsies based on lobar anatomical localization of the epileptogenic zone into temporal, frontal, parietal, and occipital lobe epilepsy. Temporal lobe epilepsy was further subdivided into amygdalohippocampal and lateral temporal, and frontal lobe epilepsy into 7 subgroups: supplementary motor, cingulate, anterior frontopolar, orbitofrontal, dorsolateral, opercular, and motor cortex. The abbreviated classification is found in Box 67.2.
The 1989 classification of epilepsies and epileptic syndromes merits updating because of the recognition of new epileptic syndromes and the discovery of the genetic basis for several known epilepsies. The most recent report of the ILAE commission on classification simplified the classification of epilepsies by eliminating the division of localization-related and generalized epilepsies (Berg et al., 2010). Instead, it suggested a listing of epilepsies by age of onset, distinctive constellations, or underlying cause (Box 67.3
[/not-level-membership-for-neurology-category]
