[level-membership-for-opthalmology-category]
Chapter 33 Epigenetic Mechanisms of Retinal Disease
Brief history
The term “epigenetics” was coined by C. H. Waddington in the 1940s, fusing the word “genetics” with “epigenesis,” the latter indicating the theory by which the adult form develops from the embryo through gradual steps, as opposed to being fully preformed as a zygote.1,2 With the discovery of inheritable patterns of DNA methylation, the idea that epigenetic traits were inherited as regulatory signals in addition to genetic information quickly took hold, and the definition of epigenetics became “the study of mitotically and/or meiotically heritable changes in gene function that cannot be explained by changes in DNA sequence.” Since the 1990s, the term “epigenetics” has become commonly used to refer to heritable changes that do not involve changes in DNA sequence.3 The term “epimutation” is used to indicate a heritable change in gene expression that does not affect the actual basepair sequence of DNA. In 2003, the Human Epigenome Consortium established the Human Epigenome Project (HEP) to identify, catalog, and interpret the genomewide DNA methylation patterns of all human genes in all major tissues.4 The field of epigenetics has garnered increasing attention over recent years.
Concept
DNA methylation
DNA methylation involves the addition of a methyl group to the 5’ position of the cytosine pyrimidine ring or the number 6 nitrogen of the adenine purine ring, and typically occurs in a CpG dinucleotide context. This modification can be inherited through cell division. In mammals, 60–90% of all CpGs are methylated in the nonpromoter area.5 DNA methylation may affect the transcription of genes in two ways. First, the methylation of DNA may itself physically impede the binding of transcriptional factors to the gene, and second, and likely more important, methylated DNA may be bound by proteins known as methyl-CpG-binding domain (MBD) proteins. MBD proteins then recruit additional proteins to the locus, such as histone deacetylases (HDACs) and other chromatin remodeling proteins that can modify histones, thereby forming compact, inactive chromatin. This process has been termed silent gene expression.6
Histone methylation
Histone methylation is the modification of certain amino acids in a histone protein by the addition of one, two, or three methyl groups. Histone methylation is generally associated with transcriptional repression. However, methylation of some lysine and arginine residues of histones results in transcriptional activation.7 Histone lysine methylation has been well studied at the K4, K9, and K27 residues. These lysine residues can be monomethylated, dimethylated, or trimethylated. Generally, trimethylation of lysine 4 on histone H3 (H3K4me3) is associated with a fully activated promoter, which correlates with gene transcription, whereas dimethylation (H3K4me2) occurs at both inactive and active euchromatic genes. H3K9 is a major negative regulator of the H3K4 mark. Dimethylation at lysine 9 (H3K9me2) marks the silence of gene expression in euchromatin, important in proliferating cells, whereas H3K9me3 is enriched in regions of “gene-poor” pericentric heterochromatin. Methylation at lysine 27 on histone H3 (H3K27me) is associated with transcriptional repression in many developmental processes.8
Histone acetylation and deacetylation
In histone acetylation, the histones are acetylated on lysine residues in the N-terminal tail as an important regulatory factor of gene expression. Steady-state levels of histone acetylation are maintained by a balance between the opposing activities of histone acetyltransferases (HATs) and HDACs. Acetylation brings in a negative charge, which acts to neutralize the positive charge on the histones and decreases the interaction of the N termini of histones with the negatively charged phosphate groups of DNA. As a consequence, the condensed chromatin is transformed to a more relaxed structure, which allows transcription factors access for DNA binding and gene transcription. Aberrant histone acetylation/deacetylation and other epigenetic modulations have been implicated in many pathological conditions, including cancer and multiple sclerosis.9
Histone acetylation is catalyzed by HATs, which are broadly classified in two different classes, types A and B, based on their functional localization.9 Type A HATs are nuclear HATs that catalyze transcription-related acetylation events. Type B HATs are cytoplasmic HATs that catalyze acetylation events linked to the transport of newly synthesized histones from the cytoplasm to the nucleus for deposition on to newly replicated DNA.10 There are five main classes of HATs (GNAT, MYST, p300/CBP, transcription factor, and nuclear hormone-related), which are characterized by specific functions controlled by its specific structural folding.
The process of histone acetylation is reversed by the HDACs, which catalyze acetyl group removal.10 There are four main classes (I–IV) of HDACs. HDAC1–3, and 8 are members of class I, and are located in the nucleus and involved in epigenetic regulation. HDAC4–6, 7, 9, and 10 are members of class II, and are characterized by nucleocytoplasmic shuttling. Class III HDACs are characterized by their NAD dependence. HDAC 11 has been categorized into its own group, a class IV deacetylase. These molecules have been implicated in aging and calorie restriction as well as disease.11
Noncoding RNA
miRNAs are a class of small (on average 22 nucleotides long), noncoding RNA molecules that regulate gene expression and are implicated in many cellular processes, including development, tissue morphogenesis, apoptosis, and tumor growth.12–14 Genes encoding primary miRNAs are scattered throughout the genomes of eukaryotes and are transcribed to generate RNA species that are cleaved to 60–70-nucleotide stem–loop miRNA precursors (pre-miRNAs) by a microprocessor complex containing the nuclear RNaseIII Drosha and DiGeorge syndrome critical region protein. After export from the nucleus, the pre-miRNAs are cleaved by a cytoplasmic RNase III (Dicer). After strand selection and separation, mature 22-basepair miRNAs are incorporated into an RNA-induced silencing complex to act as target recognition sequences. miRNAs may contain target sequences and can undergo cleavage or translational repression.15
Epigenetic factors in the retina
DNA methylation in mammalian retina
DNA methyltransferases (DNMT) are expressed in mammalian retina. Three active DNMTs have been identified in mammals: DNMT1, DNMT3a, and DNMT3b. DNMT3a methylates CpG sites at a rate much slower than that of DNMT1 but greater than that of DNMT3b. DMNT1 is expressed in human retinal pigment epithelium (RPE) cells. DNMT3a is weakly expressed in some inner nuclear layer cells in the adult mouse retina.16
DNA methylation regulates the expression of genes in mammalian retina. Interphotoreceptor retinoid-binding protein (IRBP) is the major soluble component of the interphotoreceptor matrix. This important extracellular matrix (ECM) surrounds the outer and inner segments of the photoreceptors and separates the neural retina from the RPE. IRBP is a large glycolipoprotein, which is secreted into this matrix by both rods and cones. The IRBP gene is hypomethylated in retina, and DNA methylation regulates IRBP gene expression.17 Exogenous methylation of specific CCGG sites in the murine IRBP 5’ flanking region suppressed promoter activity in transient transfection assays of cultured retinal cells. Methylation of the IRBP promoter suppresses transcription in nonphotoreceptor cells by precluding specific DNA:protein-binding events, whereas the lack of methylation in photoreceptors allows for transcription.17 Another example is EphA5, a member of the ephrin receptor subfamily of the protein-tyrosine kinase family. It regulates multiple aspects of development and disease, including vascular development. In the retina of the adult mouse, EphA5 mediates 1.2 ± 0.3% of CpG methylation.18 This represents a relatively low level of promoter methylation. Although methylation of all CpG sites resulted in the silencing of EphA5 promoter activity, lower levels of methylation resulted in differential activation or repression of EphA5 promoter activity, depending on the sites methylated.18 We found that methyl CpG-binding protein 2 (MeCP2) and DNMT1 are expressed in human RPE cells.
Histone methylation and acetylation in mammalian retina
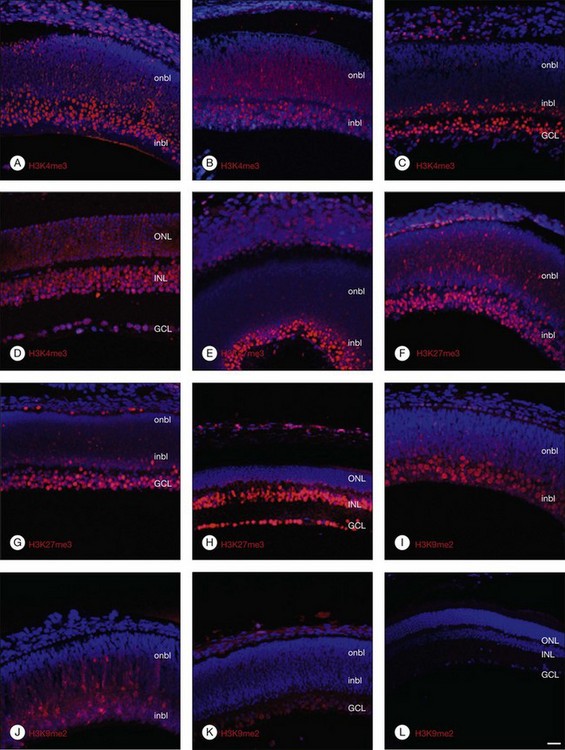
Histone lysine methylation is catalyzed by histone methyltransferases (HMTases). The HMTases constitute a family of enzymes that catalyze the methylation of histones on specific residues. The HMTases have been observed in mammalian retina. In the adult murine retina, H3K4me3, a mark associated with active transcription, is observed in all layers of the neural retina, including rhodopsin-positive photoreceptors, Müller glia, and retinal ganglion cells (RGCs). H3K27me3, a mark associated with transcriptional repression, is enriched in the inner nuclear layers and in a subset of outer nuclear layer in the adult murine retina. However, H3K9me2, another repressive mark, is not observed in the inner layers of the adult retina.8 The Ezh2 and G9a HMTases are the two best-characterized HMTases, which catalyze H3K27me3 and H3K9me2 modifications, respectively. Both can be found in the fetal murine retina. The results suggest that epigenetic regulation of gene transcription by Ezh2- and G9a-mediated histone lysine methylation plays a crucial role in retinal neuron differentiation and survival.8
HDAC is expressed in mammalian retina.19 Acetylated histone 3 and 4 are found in fetal and adult retina; however, acetylated histone 3 and 4 is reduced in dry age-related macular degeneration (AMD) specimens. In contrast to the decrease of acetylated histones, HDAC1 is highly upregulated in human AMD retinal sections. In addition, exposure of adult mice retina to trichostatin A (TSA) induces upregulation of apoptotic genes. Activation of HDAC may decrease the retina’s resistance to ischemic injury.20
miRNA in mammalian retina
The expression of miRNAs in the mammalian retina has been analyzed in several studies. The most highly expressed adult mouse retinal miRNAs were miR-181a, 182, 183, 204, 125b, 26a, and 124a. miR-181 is expressed in the ganglion cell, inner plexiform and inner nuclear layers; miR182 is expressed throughout all layers of the retina; and miR-183 is expressed only in the outer nuclear layer.21 Another study found strong expression of miR-204 in the RPE and the ciliary body, and miR-224 was detected in the RPE. In contrast, miR-124a was expressed in all layers of the neural retina except the RPE. Most recently, miRNA-132 was found to be expressed in human RPE cells. miR-204 and 211 were also found to be highly expressed in the RPE. Transforming growth factor-β (TGF-β) receptor-2 and SNAIL2 are direct targets of miR-204. Notably, anti-miR-204/211 decreased transepithelial resistance and reduced cell membrane voltage and conductance, indicating a critical role of miR-204/211 in maintaining epithelial barrier function and cell physiology.22
Epigenetic mechanisms in retinal development
Although epigenetic mechanisms have been shown to regulate neural stem cell renewal and differentiation, it was not until recent years that their involvement in retinal development has been realized. The retina is composed of specialized glia and neuronal cells which are generated from multipotent retinal progenitor cells in a highly conserved temporal sequence with overlapping phases during development.23,24 RGCs, horizontal cells, and cone photoreceptors are born in early phases, while rod photoreceptors, bipolar cells, and Müller glial cells are born in late phases. The cell fate decision of the progenitor cells depends on both intrinsic and environmental cues, which are regulated by specific networks of transcription factors.23,24 Through covalent modifications of DNA and histones, chromatin remodeling regulates the interactions of these transcription factors and their effector genes, and is an important epigenetic mechanism in retinal development.
DNA and histone methylation in retinal development
DNA and histone methylation are mediated by DNMT and HMTases. In zebrafish retina, antisense-based morpholino knockdown of DNMT3 and H3K9 HMTases Suv39h1 and G9a lead to defects in retinal cell differentiation, supporting a role of both DNA and histone methylation in zebrafish retinal development.25 In mice, immunohistochemical analysis shows changing patterns of histone methylation markers in the developing retina. Specifically, the transcriptionally activating H3K4me3 and repressive H3K27me3 histone marks are found in differentiated neurons from embryo to adulthood, corresponding to the expression of the HMTase Ezh2 that catalyzes the H3K27me3 mark. In contrast, the repressive H3K9me2 mark and the corresponding HMTase G9a were found primarily in early differentiating RGCs, but decreased after birth (Fig. 33.1).8 These changing patterns of histone methylation may at least in part account for the temporal sequence of retinal progenitor cell differentiation during development. Evidence also implicates DNA methylation in regulating the expression of genes involved in the topographic patterning of RGC axons, such as EphA5 receptor.18 These studies suggest that DNA and histone modifications may regulate both temporal and spatial expression of these genes in different populations of retinal progenitor cells to orchestrate the precise timing of cell proliferation and differentiation during development.
Histone acetylation in retinal development
During photoreceptor differentiation, HAT-containing coactivators, such as general control nondepressible 5 (Gcn5) and CREB-binding protein (Cbp), interact with cone–rod homeobox (Crx) transcription factor to promote the transcription of opsin genes.26 Crx-deficient mutant mice have decreased histone acetylation at the promoter/enhancer region of the opsin gene, resulting in photoreceptor dysfunction.27 Such results indicate that HATs are required for differentiation of retinal photoreceptors during development.
In addition to HATs, HDACs have been studied more extensively and are also implicated in retinal development. HDAC1 is recruited by the retinoblastoma protein (Rb) and related family members to promoters with E2F-binding sites, resulting in transcriptional repression of cell cycle genes and suppression of cell growth. In zebrafish, hdac1-deficient mutants exhibit retinal cell proliferation and differentiation defects, resulting in severe reduction in the inner and outer plexiform layers and absence of RGC, rod and cone photoreceptors, and Müller glia.27 These defects result from failure in downregulation of cyclin D and E transcription and the canonical Wnt and Notch signaling pathways, which are necessary for retinal progenitor cells to exit the cell cycle. Thus, histone deacetylation in zebrafish may be a regulatory mechanism to switch off stem cell proliferation and initiate a program for retinal differentiation. In mouse retinal explants, pharmacological inhibition of HDAC with TSA also results in defects in rod differentiation, but unlike in zebrafish, causes a reduction in retinal progenitor cell proliferation. In vivo knockdown HDAC4 by RNA interference leads to apoptosis of rod photoreceptors and bipolar cells, while overexpression of HDAC4 reduced naturally occurring cell death of bipolar cells, supporting a role of HDAC4 in promoting the survival of developing retinal neurons as well.28 Further work will help clarify what specific downstream effectors are regulated by HATs and HDACs during the various stages of retinal development.
Chromatin remodeling complexes in retinal development
While covalent DNA and histone modifications decondense chromatin and promoter regions, ATP-dependent displacement from promoters and enhancer regions requires the action of chromatin-remodeling complexes. These include the SWItch/Sucrose NonFermentable (SWI/SNF) and the Imitation SWItch (ISWI) families. Zebrafish mutants lacking the brahma-related gene 1 (BRG1), the catalytic ATPase subunit of SWI/SNF chromatin remodeling complex, show defects in retinal differentiation, suggesting that BRG1 is involved in triggering retinal cell differentiation.29 In mice, however, loss of BRG1 in neural stem cells results in precocious neuronal differentiation, suggesting that it represses differentiation and maintains neural stem and progenitor cells. These differences may be related to changes in the composition of accessory units known as BRG1-associated factors (BAFs) in the SWI/SNF complexes in different cell states. Nevertheless, chromatin-remodeling complexes are likely to be critically involved in regulating retinal cell differentiation during development.
microRNAs in retinal development
Recently, miRNAs have also been implicated in retinal development. Early studies using a conditional knockout mouse lacking the RNA endonuclease Dicer in the retina showed no defects in the retina before the second postnatal week, suggesting that miRNAs are not required for mouse retinal development.30 However, subsequent research using a different strain of Dicer conditional knockout mouse showed increased production of early generated cell types such as RGC and horizontal cells, and failure to generate late-born cell types such as rod photoreceptors and Müller glia.31 This is also supported by data from Xenopus morpholinos, where Dicer inactivation resulted in defects in cell cycle, lamination, and timing of retinal differentiation. These observations suggest that Dicer and miRNAs may provide a common regulatory mechanism to signal changes in retinal progenitor cell competence.
Microarray analysis of the miRNA transcriptome in mouse retina reveals at least 78 miRNAs, 21 of which are potentially retina-specific.32 The miRNA transcriptome of the mouse retina is similar to that of humans, and shows dozens of miRNAs that are differentially expressed during different stages of development. In Xenopus, miRNAs have been found to inhibit the translation of homeodomain transcription factors (Xotx2 and Xvsx1) involved in bipolar cell differentiation by binding the 3’ untranslated region of mRNAs. Inactivation of these miRNAs in vivo releases the inhibition and supports the generation of additional bipolar cells.33 Another miRNA, miR-24a, negatively regulates proapoptotic factors caspase-9 and apoptotic protease-activating factor 1 (apaf1) in Xenopus retina. Inhibition of miR-24a leads to increased apoptosis during retina development and reduction in eye size.34 These studies suggest that specific miRNAs can play distinct roles in signaling differentiation or promoting survival of retinal neurons. In mice, the miRNAs most enriched in the retina include miR-96, miR-182, and miR-183, which are transcribed as a single precursor pri-miRNA.32 Target prediction and in vitro functional studies showed that the microphthalmia-associated transcription factor (MITF), a transcription factor necessary for the development and function of RPE, is directly inhibited by miR-96 and miR-182, supporting a role of miRNAs in RPE maintenance also.32 MiR-124a, which is expressed in all neuronal subtypes in the adult retina, has also been shown to repress retinol dehydrogenase 10 (Rdh10), which is selectively expressed in Müller glia and RPE.35 Hence, some miRNAs may impact retinal development not by affecting neuronal proliferation and differentiation, but via effects on supporting glial and RPE cells. MicroRNA research is still in its infancy, and more work is necessary to determine the landscape of noncoding RNAs in different retinal cell populations.
Epigenetic mechanisms in retinal diseases
Epigenetic factors in retinal fibrosis
It is known that RPE cells are key players in the pathogenesis of proliferative vitreoretinopathy (PVR).36 In PVR, RPE cells undergo myofibroblastic transdifferentiation (MTD) characterized by an increase in mesenchymal cell components and the manifestation of a migratory and proliferative phenotype. Transdifferentiation of RPE cells into myofibroblast-like cells has been exclusively demonstrated in tissue repair during several retinal pathologic conditions, including choroidal neovascularization, diabetic retinopathy, and PVR. A major feature of RPE transdifferentiation is the increased expression of alpha smooth-muscle actin (α-SMA), and α-SMA-positive RPE cells have been shown to be the major cells that promote contraction and induce retinal detachment in PVR.37 Although a variety of inflammatory cytokines and growth factors (in particular, TGF-β) are involved in the regulation of RPE transdifferentiation, little is known about the basic mechanisms of the RPE transdifferentiation from an epithelial cell to a myofibroblast-like cell. TGF-β is the major inducer of α-SMA expression in transdifferentiated cells.37 In addition, increased TGF-β activity is associated with the downregulation of the genes of transdifferentiation inhibitors such as Smad7, IκBα, and peroxisome proliferator-activated receptor-γ (PPAR-γ).38
DNA methylation
Recent studies suggest that the wound-healing process is also regulated by epigenetic factors, including DNA methylation and histone acetylation.39 In terms of DNA methylation, MeCP2 has been demonstrated to be an orchestrator of epithelial myofibroblastic transdifferentiation and play a pivotal role in MTD and/or fibrosis.37–39 We have recently shown that RPE MTD and the pathogenesis of PVR are tightly regulated by MeCP2. Phosphorylated MeCP2 is highly and extensively expressed in human and rabbit PVR membranes and MeCP2 co-localized with cytokeratin and α-SMA in human PVR membranes. Knockdown MeCP2 by specific siRNA inhibits Smad2/3 activation and α-SMA, fibronectin expression induced by TGF-β. More importantly, PPAR-γ expression is almost absent in human PVR membranes; the upregulation of PPAR-γ is associated with the decreased expression of MeCP2 after methylation inhibitor (5-aza-2’deoxycytidine, 5-AZA) exposure. The involvement of methylation in the regulation of MTD also is demonstrated in liver cells in which liver cell transdifferentiation is blocked by treatment with 5-AZA and MeCP2 is selectively expressed in the fibrotic liver area.39
Histone acetylation/deacetylation
Besides DNA methylation, the other epigenetic factor in the regulation of MTD or fibrosis is histone acetylation/deacetylation. Although there are few reports of the effect of histone acetylation on RPE MTD and retinal fibrosis, the involvement of histone acetylation has been extensively studied in a number of systemic diseases.40,41 Recent reports reveal that histone acetylation and HDAC activity are also correlated with the development and progression of some fibrotic diseases, such as cardiac hypertrophy, kidney fibrosis, idiopathic pulmonary fibrosis, and liver fibrosis. Hyunjin et al. found that HDAC inhibition suppressed both diabetes- and TGF-β1-induced renal fibrosis.40 Inhibition of HDAC activity by TSA decreased platelet-derived growth factor (PDGF)-induced fibroblast proliferation; importantly, it was found that activation of STAT3 is required for the induction of HDAC activity; renal fibroblast activation also is blocked by the inhibition of STAT3 pathway.41 Interestingly, TSA is able to inhibit the TGF-β-mediated transdifferentiation of corneal stromal cell.42 Furthermore, research by Qin and Han43 suggests that the silencing of matrix metalloproteinase genes is under epigenetic regulation, especially by HDAC-4 during liver cell fibrogenesis. More specifically, Hyunjin et al. demonstrated that HDAC-2 plays an important role in the development of MTD and ECM accumulation in diabetic kidney induced by TGF-β and reactive oxygen species.40
Epigenetic factors in retinitis pigmentosa and other retinal degenerations
Retinitis pigmentosa (RP) is an inherited retinal degeneration that is characterized by selective cell death of photoreceptors. At least 40 gene mutations involved in human RP have been identified so far,44 although the metabolic pathways leading to photoreceptor cell death remain unknown, and no treatment is as yet available. In the well-studied retinal degeneration 1 (rd1) mouse model for RP, where a rod photoreceptor cGMP phosphodiesterase-6 mutation leads to cGMP accumulation and photoreceptor cell death, increased HDAC activity was found to precede photoreceptor degeneration.42 More importantly, pharmacological inhibition of HDAC activity reduces photoreceptor cell death, an effect that may be mediated by transcriptional regulation through the poly-ADP-ribose-polymerase family of proteins.45 Interestingly, overexpression of HDAC4, which regulates retinal neuronal survival, can prolong photoreceptor survival in rd1 mouse retina,28 suggesting different roles of HDAC family members in the pathogenesis of retinal degeneration.
Studies in recent years are beginning to unveil a role for miRNAs in retinal degenerative diseases. Conditional knockout mice lacking Dicer in the retina show no response to light stimuli, with progressive cellular disorganization and degeneration of retinal cell types as well as decreased electroretinogram responses.30 miRNAs typically enriched in the mouse retina, such as miR-96, miR-182, and miR-183, are reduced severalfold in rd1 mice where rod photoreceptors have degenerated.32 A mouse model of RP carrying a mutant Pro347Ser rhodopsin transgene demonstrates altered expression of miR-96, miR-183, miR-1, and miR-133 in the retina when compared with wild-type animals.46 A similar miRNA signature was confirmed in three other mouse models of RP. Predicted targets of these miRNAs include antiapoptotic factors such as Fas apoptotic inhibitor molecule, which offers a possible mechanism whereby defects in miRNAs expression may lead to photoreceptor degeneration through apoptosis.
Epigenetic factors in age-related macular degeneration
The detailed pathogenesis of AMD has been difficult to study due to the lack of a model that adequately recapitulates each of the pathologic features of the disease in humans. Recent work has shown that clusterin (also known as apolipoprotein J), a major component of drusen that accumulates with age, has a promoter with a CpG-rich methylation domain that may be epigenetically regulated.47 In human RPE cell culture, pharmacological induction of DNA hypomethylation or histone hyperacetylation led to increased expression of clusterin.48 If similar findings can be demonstrated in vivo, epigenetic mechanisms may potentially be exploited as targets for treating this chronic eye disease.
Another recent study showed that Dicer may also be involved in the pathogenesis of geographic atrophy in AMD. Dicer is reduced in the RPE of human patients with geographic atrophy, and conditional knockout of Dicer reproduces the RPE degeneration phenotype in mice. Surprisingly, the role of Dicer in the pathogenesis of macular degeneration does not appear to involve miRNAs, but instead the degradation of Alu elements, common noncoding, repetitive DNA sequences in the human genome which may be toxic to the RPE.49 In this way, Dicer may serve a protective role in RPE to maintain a low level of toxic long Alu RNAs in the human retina. Future studies may help determine why Dicer is reduced in geographic atrophy, and whether accumulation of noncoding RNAs with age may participate in the pathogenesis of this disease.
Epigenetic factors in glaucoma
In glaucoma, a group of eye conditions that lead to optic nerve damage, RGCs undergo a complex apoptotic program in response to injury, resulting in a change in expression patterns of various genes. After acute injury to the optic nerve in experimental models of glaucoma, many genes involved in apoptosis or cell stress response are upregulated, including the B-cell lymphoma 2 (Bcl-2) family members, as well as heat shock proteins (Hsps) and caspases. Various other genes, including Thy1, Brn3b, Fem1c, Sncg, BclXI, TrkB, and members of the neurofilament family, have been shown to decrease in expression.50 Interestingly, recent evidence shows that several RGC-specific genes whose expression is downregulated after optic nerve crush injury exhibit a decrease in promoter histone acetylation, and are accompanied by an increase in HDAC-2 and 3 activity, as well as translocation of HDAC3 to the nuclei of dying RGCs.51 Moreover, inhibitors of HDACs such as TSA and valproic acid reduce RGC loss and may even enhance axonal regeneration after optic nerve damage.51 These reports implicate a potential role of histone deacetylation in RGC death in optic nerve diseases such as glaucoma.
Epigenetic factors in retinoblastoma
Retinoblastoma is the most common intraocular tumor of childhood. Although the disease is initiated by the loss of both alleles of the prototypic tumor suppressor gene, Rb1, subsequent changes in other tumor suppressor and DNA repair genes have been implicated in the pathogenesis of the disease. The retinoblastoma protein Rb is the founding member of the pocket protein family of tumor suppressors, including p107 and p130, which have been implicated in numerous cellular processes, including cell cycle regulation, DNA repair, DNA replication, differentiation, and apoptosis.52 Rb regulates cell cycle progression by binding to the E2F family of transcription factors and inducing repression of E2F-regulated cell cycle genes. The action of this repression involves two mechanisms – direct binding and blockage of the transactivation domain of E2F, and recruitment of chromatin-modifying molecules. All three pocket protein family members have been shown to associate with HDAC1 through the “pocket” domain to repress cell cycle genes by E2F-regulated promoters.53 In addition, Rb associates with the HMTase Suv39H1, DNMT1, and heterochromatin protein 1 (HP1) via its pocket domain.54 Finally, Rb also influences the accessibility of chromatin through interactions with the ATP-dependent helicase BRG1, the catalytic subunit of the SNF/SWI chromatin remodeling complex. Together, DNMT1, HMTases, HDACs, HP1, and BRG1 are all recruited by Rb to form a multiprotein chromatin remodeling complex that can heterochromatinize promoters regulated by the E2F family, and epigenetically silence transcriptional activation of cell cycle genes (Fig. 33.2).
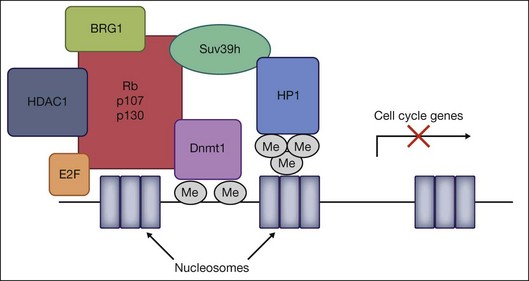
There is increasing evidence that, in addition to mutations in the Rb1 gene, epigenetic changes involving aberrant DNA methylation of other tumor suppressor gene promoters may be involved. Aberrant DNA methylation is commonly found in human cancers. Regional hypermethylation of CpG islands in promoter regions of tumor suppressor genes results in transcriptional silencing, while global DNA hypomethylation is associated with activation of proto-oncogenes and generation of genomic instability. In retinoblastoma tissues, there is promoter hypermethylation of O6-methylguanine-DNA methyltransferase,55 which encodes the DNA repair enzyme O6-methylguanine DNMT and is also hypermethylated in breast and prostate cancer, lymphoma, and gliomas. RASSF1A (the RAS-association domain family 1, isoform A, tumor suppressor gene involved in microtubule stability, apoptosis, and cell cycle regulation), is also methylated and inactivated in multiple pediatric tumors, including retinoblastoma.56 Finally, promoter methylation of caspase 8, which is involved in Fas-mediated apoptosis, and MLH1, a DNA mismatch repair gene, may provide mechanistic explanations for loss of cell cycle regulation in retinoblastoma tumors.57 It remains unclear to what degree aberrant methylation contributes to the pathogenesis of tumor growth in patients with retinoblastoma. For example, does hypermethylation of tumor suppressor genes represent the second or third “hit” required for tumor genesis? Or are they merely compensatory changes that occur in response to tumor growth? Further work will be necessary to delineate the complex epigenetic factors involved in this childhood cancer.
Epigenetic factors in uveal melanomas
Uveal melanoma is the most common primary intraocular tumor in adults, with a high mortality rate and frequent metastases. Similar to retinoblastoma, aberrant DNA methylation of the RASSF1A promoter has also been implicated in uveal melanoma.58 However, a recent investigation of promoter methylation in several tumor suppressor genes in uveal melanoma tissues identified only the human telomerase reverse transcriptase (hTERT) gene, but not RASSF1A, to be abnormally methylated.59 Such discrepancies may be related to the genetic heterogeneity of uveal melanomas in humans, and additional research is needed to identify these different patterns of DNA methylation.
The involvement of histone acetylation in uveal melanoma remains unclear. In vitro, inhibition of HDAC activity in both primary and metastatic uveal melanoma cell lines results in inhibition of cell growth and induction of apoptosis, an effect likely mediated by Fas-dependent pathways.60 It is not known, however, whether HDACs are involved in the pathogenesis of the disease in humans. Nevertheless, these findings implicate a potential role of HDAC inhibitors in the treatment of this devastating cancer.
Recent evidences also point to a role of microRNAs in uveal melanoma. miRNAs such as miR-34a and miR-137, which have been implicated in tumor suppression, are expressed in melanocytes, but not in uveal melanoma cells.61,62 Transfection of miR-34a or miR-137 into uveal melanoma cells led to decreased cell growth and migration.61,62 miR-34a is a potential key effector of the p53 tumor suppressor gene, while miR-137 is involved in downregulation of MITF, a master regulator of melanocyte cell growth, maturation, apoptosis, and pigmentation.63 Identifying other miRNAs involved in tumor suppression may provide novel targets for uveal melanoma therapy.
Diabetic retinopathy
Diabetes is a chronic metabolic disease associated with both genetic and environmental factors. Diabetic retinopathy, a microvascular complication, is a leading cause of acquired blindness. Many biochemical and molecular sequelae of hyperglycemia have been implicated in its pathogenesis, but the exact mechanism has remained elusive. Recent studies have shown that exposure of aortic endothelial cells to high levels of glucose induces epigenetic changes in the promoter of NF-κB subunit p65.64 The modification of histone H3 at lysine 9 (H3K9) at the proximal Cox2 promoter bearing the NF-κB-binding site has been shown to modulate hyperglycemia-induced thioredoxin-interacting protein-mediated inflammation in retinal capillary endothelial cells.65 However, much less is known about epigenetic regulation in diabetic retinopathy.
A recent study examined the role of histone modification in the development of diabetic retinopathy and in the resistance of retinopathy to arrest after the termination of hyperglycemia.66,67 The authors showed that HDAC activity and related gene transcripts are increased in the retina in diabetes, whereas HAT activity is decreased, resulting in the reduced acetylation of histone H3. The reversal of hyperglycemia fails to provide any benefit in terms of retinal histone acetylation or the enzymes responsible for maintaining acetylation homeostasis, suggesting that histone deacetylation is an important contributor in the progression of diabetic retinopathy after hyperglycemia is terminated.
The mechanism of histone deacetylation in diabetes may include increased oxidative stress and hypoxia. In diabetes, the retina and its capillary cells experience increased oxidative stress, and hyperglycemia-induced superoxide overproduction activates the major pathways in the development of diabetic retinopathy.68 Reactive oxygen species are shown to increase HDAC activity and decrease HAT activity, which inhibits histone acetylation. Therefore, oxidative stress may play a role in regulating histone deacetylation in diabetes.
Hypoxia is a major stimulus for the retinal neovascularization observed in diabetes69; ischemia and hypoxia also stimulate HDAC activity. The activation of retinal HDAC by increased retinal hypoxia in diabetes remains a strong possibility.
miRNA also plays an important role in retinal neovascularization. Several miRNAs are upregulated (such as miR-92, miR-27b, and miR-15a) or downregulated (such as miR-31, miR-150, and miR-184) in the murine ischemic retina.70 The downregulation of miR-31 and miR-150 will lead to increased promoter activity at the loci that code for vascular endothelial growth factor (VEGF), PDGF-B, and HIF-1α.70 PDGF-B induces pericyte proliferation. HIF-1α may increase the expression of growth factors and ECM proteases, which will lead to retinal angiogenesis.
Choroidal neovascularization
The pathogenesis of CNV is mediated by a multitude of factors. Epigenetics can potentially participate in various pathological aspects of CNV. miR-155, which is expressed in immune cells, was demonstrated to activate macrophages. Macrophages facilitate the inflammatory response that promotes angiogenesis, for instance, through the production of tumor necrosis factor, which induces the expression of VEGF and matrix metalloproteinases.71 However, retinal production of tumor necrosis factor and its stimulatory effects on matrix metalloproteinases can be reversed by an HDAC inhibitor.20 The expression of pro- and anti-inflammatory genes, such as interleukin (IL)-2, IL-8, and IL-10, can also be regulated by HDAC activity.72 Furthermore, oxidative stress can also induce expression of the major player in angiogenesis, VEGF, as well as expression of HIF-1α, which stimulates VEGF production. HIF-1α can also be downregulated by the HDAC inhibitor,73 TSA, whereas the antiangiogenic and neuroprotective molecule pigment epithelium-derived factor is upregulated by TSA.73 TSA can also inhibit the proliferation and migration of RPE cells, implying the potential regulation of pro- and antiproliferative, ECM-modifying genes by HDACs.
Perspectives and challenges of epigenetics
Over the years, an individual may accumulate various intrinsic insults caused by chronic diseases such as diabetes mellitus or accumulate environmental alterations that could affect the epigenome, and participate in the induction of age-related diseases. However, another feature of epigenetics may reverse this phenomenon. The class III family of HDACs, Sirtuins, specifically Sirt1, has been demonstrated to inhibit cell death.74 It acts to prevent the overactivation of p53 in causing apoptosis due to DNA damage, resulting in DNA repair and cell survival.73
1. What is the detailed mechanism of epigenetic regulation of retinal development?
2. In the physiological condition, how do epigenetic factors contribute to normal retinal function?
3. How is retinal inflammation regulated by epigenetic factors?
4. How much of a role does epigenetics play in the pathogenesis of retinal angiogenesis and fibrosis?
5. How do cytokines and growth factors regulate epigenetic factor expression and vice versa?
6. How is epigenetics involved in the process of retinal degeneration, including AMD?
7. What is the role of epigenetics in mitochondrial,75 endoplasmic reticulum, and Golgi stress in retinal disease?
8. What is the role of crosstalk among epigenetics factors in retinal development and disease?
Treatment of retinal disease with epigenetic-modifying drugs
Dysregulation of DNA methylation, histone acetylation, and miRNA expression may be a common theme in many retinal diseases. Preclinical studies using epigenetic modifying drugs are in progress and some epigenetic modifying drugs such as 5-AZA and suberoylanilide hydroxamic acid are in clinical trials.76–78 However, the major concern about epigenetic drugs is lack of target specificity.79 The DNA methylation inhibitors result in global demethylation; similarly, the HDAC inhibitors can affect many isoforms of HDAC and nonhistone proteins. As well, even one miRNA can act on multiple targets, so the development of compounds with higher specificity and greater efficacy is required. Currently, in terms of treatment of certain cancers, the combination of epigenetic treatments with other therapies such as cytotoxic agents or radiation has been shown to be more effective.80 In the future, the use of epigenetic modifying drugs combined with other traditional therapies may be considered to improve therapeutic efficacy in those who suffer from retinal disease.
1 Waddington CH. The epigenotype. Endeavour. 1942;1:18–20.
2 Bonasio R, Tu S, Reinberg D. Molecular signals of epigenetic states. Science. 2010;330:612–616.
3 Haig D. The (dual) origin of epigenetics. Cold Spring Harb Symp Quant Biol. 2004;69:67–70.
4 Novik KL, Nimmrich I, Genc B, et al. Epigenomics: genome-wide study of methylation phenomena. Curr Issues Mol Biol. 2002;4:111–128.
5 Ehrlich M, Gama-Sosa MA, Huang LH, et al. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res. 1982;10:2709–2721.
6 Boyes J, Bird A. DNA methylation inhibits transcription indirectly via a methyl-CpG binding protein. Cell. 1991;64:1123–1134.
7 Bannister AJ, Schneider R, Kouzarides T. Histone methylation: dynamic or static? Cell. 2002;109:801–806.
8 Rao RC, Tchedre KT, Malik MT, et al. Dynamic patterns of histone lysine methylation in the developing retina. Invest Ophthalmol Vis Sci. 2010;51:6784–6792.
9 Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389:349–352.
10 Selvi RB, Kundu TK. Reversible acetylation of chromatin: implication in regulation of gene expression, disease and therapeutics. Biotechnol J. 2009;4:375–390.
11 de Ruijter AJ, van Gennip AH, Caron HN, et al. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749.
12 Huang KM, Dentchev T, Stambolian D. MiRNA expression in the eye. Mamm Genome. 2008;19:510–516.
13 Bartel DP, Chen CZ. Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat Rev Genet. 2004;5:396–400.
14 Stefani G, Slack FJ. Small non-coding RNAs in animal development. Nat Rev Mol Cell Biol. 2008;9:219–230.
15 Pillai RS, Bhattacharyya SN, Filipowicz W. Repression of protein synthesis by miRNAs: how many mechanisms? Trends Cell Biol. 2007;17:118–126.
16 Watanabe D, Uchiyama K, Hanaoka K. Transition of mouse de novo methyltransferases expression from Dnmt3b to Dnmt3a during neural progenitor cell development. Neuroscience. 2006;142:727–737.
17 Boatright JH, Nickerson JM, Borst DE. Site-specific DNA hypomethylation permits expression of the IRBP gene. Brain Res. 2000;887:211–221.
18 Petkova TD, Seigel GM, Otteson DC. A role for DNA methylation in regulation of EphA5 receptor expression in the mouse retina. Vision Res. 2011;51:260–268.
19 Pelzel HR, Schlamp CL, Nickells RW. Histone H4 deacetylation plays a critical role in early gene silencing during neuronal apoptosis. BMC Neurosci. 2010;11:62.
20 Crosson CE, Mani SK, Husain S, et al. Inhibition of histone deacetylase protects the retina from ischemic injury. Invest Ophthalmol Vis Sci. 2010;51:3639–3645.
21 Ryan DG, Oliveira-Fernandes M, Lavker RM. MicroRNAs of the mammalian eye display distinct and overlapping tissue specificity. Mol Vis. 2006;12:1175–1184.
22 Wang FE, Zhang C, Maminishkis A, et al. MicroRNA-204/211 alters epithelial physiology. FASEB J. 2010;24:1552–1571.
23 Rapaport DH, Wong LL, Wood ED, et al. Timing and topography of cell genesis in the rat retina. J Comp Neurol. 2004;474:304–324.
24 Kumar JP. Retinal determination at the beginning of eye development. Curr Top Dev Biol. 2010;93:1–28.
25 Rai K, Jafri IF, Chidester S, et al. Dnmt3 and G9a cooperate for tissue-specific development in zebrafish. J Biol Chem. 2010;285:4110–4121.
26 Peng GH, Chen S. Crx activates opsin transcription by recruiting HAT-containing co-activators and promoting histone acetylation. Hum Mol Genet. 2007;16:2433–2452.
27 Stadler JA, Shkumatava A, Norton WH, et al. Histone deacetylase 1 is required for cell cycle exit and differentiation in the zebrafish retina. Dev Dyn. 2005;233:883–889.
28 Chen B, Cepko CL. HDAC4 regulates neuronal survival in normal and diseased retinas. Science. 2009;323:256–259.
29 Gregg RG, Willer GB, Fadool JM, et al. Positional cloning of the young mutation identifies an essential role for the Brahma chromatin remodeling complex in mediating retinal cell differentiation. Proc Natl Acad Sci U S A. 2003;100:6535–6540.
30 Damiani D, Alexander JJ, O’Rourke JR, et al. Dicer inactivation leads to progressive functional and structural degeneration of the mouse retina. J Neurosci. 2008;28:4878–4887.
31 Georgi SA, Reh TA. Dicer is required for the transition from early to late progenitor state in the developing mouse retina. J Neurosci. 2010;30:4048–4061.
32 Xu S, Witmer PD, Lumayag S, et al. MicroRNA (miRNA) transcriptome of mouse retina and identification of a sensory organ-specific miRNA cluster. J Biol Chem. 2007;282:25053–25066.
33 Decembrini S, Bressan D, Vignali R, et al. MicroRNAs couple cell fate and developmental timing in retina. Proc Natl Acad Sci U S A. 2009;106:21179–21184.
34 Walker JC, Harland RM. microRNA-24a is required to repress apoptosis in the developing neural retina. Genes Dev. 2009;23:1046–1051.
35 Arora A, McKay GJ, Simpson DA. Prediction and verification of miRNA expression in human and rat retinas. Invest Ophthalmol Vis Sci. 2007;48:3962–3967.
36 Cardillo JA, Stout JT, La Bree L, et al. Post-traumatic proliferative vitreoretinopathy. The epidemiologic profile, onset, risk factors, and visual outcome. Opthalmol. 1997;104:1166–1173.
37 Grisanti S, Guidry C. Transdifferentiation of retinal pigment epithelial cells from epithelial to mesenchymal phenotype. Invest Ophthalmol Vis Sci. 1995;36:391–405.
38 Burgess HA, Daugherty LE, Thatcher TH, et al. PPARg agonist inhibit TGF-β induced pulmonarymyofibroblast differentiation and collagen production: implications for therapy of lung fibrosis. Am J Physiol. 2005;288:L1146–L1153.
39 Mann J, Chu DC, Maxwell A, et al. MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology. 2010;138:705–714.
40 Hyunjin Noh, Eun Young Oh, Ji Yeon Seo, et al. Histone deacetylase-2 is a key regulator of diabetes- and transforming growth factor-β1-induced renal injury. Am J Physiol Renal Physiol. 2009;297:F729–F739.
41 Pang M, Kothapally J, Mao H, et al. Inhibition of histone deacetylase activity attenuates renal fibroblast activation and interstitial fibrosis in obstructive nephropathy. Am J Physiol Renal Physiol. 2009;297:F996–F1005.
42 Zhou Q, Yang L, Wang Y, et al. TGFβ mediated transition of corneal fibroblasts from a proinflammatory state to a profibrotic state through modulation of histone acetylation. J Cell Physiol. 2010;224:135–143.
43 Qin L, Han YP. Epigenetic repression of matrix metalloproteinases in myofibroblastic hepatic stellate cells through histone. Am J Pathol. 2010;177:1915–1928.
44 Sancho-Pelluz J, Arango-Gonzalez B, Kustermann S, et al. Photoreceptor cell death mechanisms in inherited retinal degeneration. Mol Neurobiol. 2008;38:253–269.
45 Sancho-Pelluz J, Alavi MV, Sahaboglu A, et al. Excessive HDAC activation is critical for neurodegeneration in the rd1 mouse. Cell Death Dis. 2010;1:e24.
46 Loscher CJ, Hokamp K, Kenna PF, et al. Altered retinal microRNA expression profile in a mouse model of retinitis pigmentosa. Genome Biol. 2007;8:R248.
47 Rosemblit N, Chen CL. Regulators for the rat clusterin gene: DNA methylation and cis-acting regulatory elements. J Mol Endocrinol. 1994;13:69–76.
48 Suuronen T, Nuutinen T, Ryhanen T, et al. Epigenetic regulation of clusterin/apolipoprotein J expression in retinal pigment epithelial cells. Biochem Biophys Res Commun. 2007;357:397–401.
49 Kaneko H, Dridi S, Tarallo V, et al. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature. 2011;471:325–330.
50 Soto I, Oglesby E, Buckingham BP, et al. Retinal ganglion cells downregulate gene expression and lose their axons within the optic nerve head in a mouse glaucoma model. J Neurosci. 2008;28:548–561.
51 Pelzel HR, Schlamp CL, Nickells RW. Histone H4 deacetylation plays a critical role in early gene silencing during neuronal apoptosis. BMC Neurosci. 2010;11:62.
52 Gonzalo S, Blasco MA. Role of Rb family in the epigenetic definition of chromatin. Cell Cycle. 2005;4:752–755.
53 Luo RX, Postigo AA, Dean DC. Rb interacts with histone deacetylase to repress transcription. Cell. 1998;92:463–473.
54 Robertson KD, Ait-Si-Ali S, Yokochi T, et al. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat Genet. 2000;25:338–342.
55 Choy KW, Pang CP, To KF, et al. Impaired expression and promotor hypermethylation of O6-methylguanine-DNA methyltransferase in retinoblastoma tissues. Invest Ophthalmol Vis Sci. 2002;43:1344–1349.
56 Harada K, Toyooka S, Maitra A, et al. Aberrant promoter methylation and silencing of the RASSF1A gene in pediatric tumors and cell lines. Oncogene. 2002;21:4345–4349.
57 Poulaki V, Mitsiades CS, McMullan C, et al. Human retinoblastoma cells are resistant to apoptosis induced by death receptors: role of caspase-8 gene silencing. Invest Ophthalmol Vis Sci. 2005;46:358–366.
58 Maat W, van der Velden PA, Out-Luiting C, et al. Epigenetic inactivation of RASSF1a in uveal melanoma. Invest Ophthalmol Vis Sci. 2007;48:486–490.
59 Moulin AP, Clement G, Bosman FT, et al. Methylation of CpG island promoters in uveal melanoma. Br J Ophthalmol. 2008;92:281–285.
60 Klisovic DD, Katz SE, Effron D, et al. Depsipeptide (FR901228) inhibits proliferation and induces apoptosis in primary and metastatic human uveal melanoma cell lines. Invest Ophthalmol Vis Sci. 2003;44:2390–2398.
61 Yan D, Zhou X, Chen X, et al. MicroRNA-34a inhibits uveal melanoma cell proliferation and migration through downregulation of c-Met. Invest Ophthalmol Vis Sci. 2009;50:1559–1565.
62 Chen X, Wang J, Shen H, et al. Epigenetics, microRNAs, and carcinogenesis: functional role of microRNA-137 in uveal melanoma. Invest Ophthalmol Vis Sci. 2011;52:1193–1199.
63 Levy C, Khaled M, Fisher DE. MITF: master regulator of melanocyte development and melanoma oncogene. Trends Mol Med. 2006;12:406–414.
64 El-Osta A, Brasacchio D, Yao D, et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med. 2008;205:2409–2417.
65 Perrone L, Devi TS, Hosoya K, et al. Thioredoxin interacting protein (TXNIP) induces inflammation through chromatin modification in retinal capillary endothelial cells under diabetic conditions. J Cell Physiol. 2009;221:262–272.
66 Zhong Q, Kowluru RA. Role of histone acetylation in the development of diabetic retinopathy and the metabolic memory phenomenon. J Cell Biochem. 2010;110:1306–1313.
67 Zhong Q, Kowluru RA. Role of histone acetylation in the development of diabetic retinopathy and the metabolic memory phenomenon. J Cell Biochem. 2011;110:1306–1313.
68 Kowluru RA, Kanwar M. Oxidative stress and the development of diabetic retinopathy: contributory role of matrix metalloproteinase-2. Free Radic Biol Med. 2009;46:1677–1685.
69 Frank RN. Diabetic retinopathy. N Engl J Med. 2004;350:48–58.
70 Shen J, Yang X, Xie B, et al. MicroRNAs regulate ocular neovascularization. Mol Ther. 2008;16:1208–1216.
71 Campa C, Harding SP. Two-year visual results for older Asian women treated with photodynamic therapy or bevacizumab for myopic choroidal neovascularization. Am J Ophthalmol. 2010;149:1014–1015.
72 Villagra A, Sotomayor EM, Seto E. Histone deacetylases and the immunological network: implications in cancer and inflammation. Oncogene. 2010;29:157–173.
73 Histone deacetylase inhibitors: the epigenetic therapeutics that repress hypoxia-inducible factors. J Biomed Biotechnol. 2011;2011:197946.
74 Ozawa Y, Kubota S, Narimatsu T, et al. Retinal aging and sirtuins. Ophthal Res. 2010;44:199–203.
75 Wallace DC, Weiwei Fan. Energetics, epigenetics, mitochondrial genetics. Mitochondrion. 2010;10:12–31.
76 Fandy TE. Development of DNA methyltransferase inhibitors for the treatment of neoplastic diseases. Curr Med Chem. 2009;16:2075–2085.
77 Santini V, Gozzini A, Ferrari G. Histone deacetylase inhibitors: molecular and biological activity as a premise to clinical application. Curr Drug Metab. 2007;8:383–393.
78 Shabason JE, Tofilon PJ, Camphausen K. HDAC inhibitors in cancer care. Oncology (Williston Park). 2010;24:180–185.
79 Boumber Y, Issa JP. Epigenetics in cancer: what’s the future? Oncology (Williston Park). 2011;25:220–226. 228
80 Ellis L, Atadja PW, Johnstone RW. Epigenetics in cancer: targeting chromatin modifications. Mol Cancer Ther. 2009;8:1409–1420.
[/level-membership-for-opthalmology-category][not-level-membership-for-opthalmology-category]
Chapter 33 Epigenetic Mechanisms of Retinal Disease
Brief history
The term “epigenetics” was coined by C. H. Waddington in the 1940s, fusing the word “genetics” with “epigenesis,” the latter indicating the theory by which the adult form develops from the embryo through gradual steps, as opposed to being fully preformed as a zygote.1,2 With the discovery of inheritable patterns of DNA methylation, the idea that epigenetic traits were inherited as regulatory signals in addition to genetic information quickly took hold, and the definition of epigenetics became “the study of mitotically and/or meiotically heritable changes in gene function that cannot be explained by changes in DNA sequence.” Since the 1990s, the term “epigenetics” has become commonly used to refer to heritable changes that do not involve changes in DNA sequence.3 The term “epimutation” is used to indicate a heritable change in gene expression that does not affect the actual basepair sequence of DNA. In 2003, the Human Epigenome Consortium established the Human Epigenome Project (HEP) to identify, catalog, and interpret the genomewide DNA methylation patterns of all human genes in all major tissues.4 The field of epigenetics has garnered increasing attention over recent years.
Concept
DNA methylation
DNA methylation involves the addition of a methyl group to the 5’ position of the cytosine pyrimidine ring or the number 6 nitrogen of the adenine purine ring, and typically occurs in a CpG dinucleotide context. This modification can be inherited through cell division. In mammals, 60–90% of all CpGs are methylated in the nonpromoter area.5 DNA methylation may affect the transcription of genes in two ways. First, the methylation of DNA may itself physically impede the binding of transcriptional factors to the gene, and second, and likely more important, methylated DNA may be bound by proteins known as methyl-CpG-binding domain (MBD) proteins. MBD proteins then recruit additional proteins to the locus, such as histone deacetylases (HDACs) and other chromatin remodeling proteins that can modify histones, thereby forming compact, inactive chromatin. This process has been termed silent gene expression.6
Histone methylation
Histone methylation is the modification of certain amino acids in a histone protein by the addition of one, two, or three methyl groups. Histone methylation is generally associated with transcriptional repression. However, methylation of some lysine and arginine residues of histones results in transcriptional activation.7 Histone lysine methylation has been well studied at the K4, K9, and K27 residues. These lysine residues can be monomethylated, dimethylated, or trimethylated. Generally, trimethylation of lysine 4 on histone H3 (H3K4me3) is associated with a fully activated promoter, which correlates with gene transcription, whereas dimethylation (H3K4me2) occurs at both inactive and active euchromatic genes. H3K9 is a major negative regulator of the H3K4 mark. Dimethylation at lysine 9 (H3K9me2) marks the silence of gene expression in euchromatin, important in proliferating cells, whereas H3K9me3 is enriched in regions of “gene-poor” pericentric heterochromatin. Methylation at lysine 27 on histone H3 (H3K27me) is associated with transcriptional repression in many developmental processes.8
Histone acetylation and deacetylation
In histone acetylation, the histones are acetylated on lysine residues in the N-terminal tail as an important regulatory factor of gene expression. Steady-state levels of histone acetylation are maintained by a balance between the opposing activities of histone acetyltransferases (HATs) and HDACs. Acetylation brings in a negative charge, which acts to neutralize the positive charge on the histones and decreases the interaction of the N termini of histones with the negatively charged phosphate groups of DNA. As a consequence, the condensed chromatin is transformed to a more relaxed structure, which allows transcription factors access for DNA binding and gene transcription. Aberrant histone acetylation/deacetylation and other epigenetic modulations have been implicated in many pathological conditions, including cancer and multiple sclerosis.9
Histone acetylation is catalyzed by HATs, which are broadly classified in two different classes, types A and B, based on their functional localization.9 Type A HATs are nuclear HATs that catalyze transcription-related acetylation events. Type B HATs are cytoplasmic HATs that catalyze acetylation events linked to the transport of newly synthesized histones from the cytoplasm to the nucleus for deposition on to newly replicated DNA.10 There are five main classes of HATs (GNAT, MYST, p300/CBP, transcription factor, and nuclear hormone-related), which are characterized by specific functions controlled by its specific structural folding.
The process of histone acetylation is reversed by the HDACs, which catalyze acetyl group removal.10 There are four main classes (I–IV) of HDACs. HDAC1–3, and 8 are members of class I, and are located in the nucleus and involved in epigenetic regulation. HDAC4–6, 7, 9, and 10 are members of class II, and are characterized by nucleocytoplasmic shuttling. Class III HDACs are characterized by their NAD dependence. HDAC 11 has been categorized into its own group, a class IV deacetylase. These molecules have been implicated in aging and calorie restriction as well as disease.11
Noncoding RNA
miRNAs are a class of small (on average 22 nucleotides long), noncoding RNA molecules that regulate gene expression and are implicated in many cellular processes, including development, tissue morphogenesis, apoptosis, and tumor growth.12–14 Genes encoding primary miRNAs are scattered throughout the genomes of eukaryotes and are transcribed to generate RNA species that are cleaved to 60–70-nucleotide stem–loop miRNA precursors (pre-miRNAs) by a microprocessor complex containing the nuclear RNaseIII Drosha and DiGeorge syndrome critical region protein. After export from the nucleus, the pre-miRNAs are cleaved by a cytoplasmic RNase III (Dicer). After strand selection and separation, mature 22-basepair miRNAs are incorporated into an RNA-induced silencing complex to act as target recognition sequences. miRNAs may contain target sequences and can undergo cleavage or translational repression.15
Epigenetic factors in the retina
DNA methylation in mammalian retina
DNA methyltransferases (DNMT) are expressed in mammalian retina. Three active DNMTs have been identified in mammals: DNMT1, DNMT3a, and DNMT3b. DNMT3a methylates CpG sites at a rate much slower than that of DNMT1 but greater than that of DNMT3b. DMNT1 is expressed in human retinal pigment epithelium (RPE) cells. DNMT3a is weakly expressed in some inner nuclear layer cells in the adult mouse retina.16
DNA methylation regulates the expression of genes in mammalian retina. Interphotoreceptor retinoid-binding protein (IRBP) is the major soluble component of the interphotoreceptor matrix. This important extracellular matrix (ECM) surrounds the outer and inner segments of the photoreceptors and separates the neural retina from the RPE. IRBP is a large glycolipoprotein, which is secreted into this matrix by both rods and cones. The IRBP gene is hypomethylated in retina, and DNA methylation regulates IRBP gene expression.17 Exogenous methylation of specific CCGG sites in the murine IRBP 5’ flanking region suppressed promoter activity in transient transfection assays of cultured retinal cells. Methylation of the IRBP promoter suppresses transcription in nonphotoreceptor cells by precluding specific DNA:protein-binding events, whereas the lack of methylation in photoreceptors allows for transcription.17 Another example is EphA5, a member of the ephrin receptor subfamily of the protein-tyrosine kinase family. It regulates multiple aspects of development and disease, including vascular development. In the retina of the adult mouse, EphA5 mediates 1.2 ± 0.3% of CpG methylation.18 This represents a relatively low level of promoter methylation. Although methylation of all CpG sites resulted in the silencing of EphA5 promoter activity, lower levels of methylation resulted in differential activation or repression of EphA5 promoter activity, depending on the sites methylated.18 We found that methyl CpG-binding protein 2 (MeCP2) and DNMT1 are expressed in human RPE cells.
Histone methylation and acetylation in mammalian retina
Histone lysine methylation is catalyzed by histone methyltransferases (HMTases). The HMTases constitute a family of enzymes that catalyze the methylation of histones on specific residues. The HMTases have been observed in mammalian retina. In the adult murine retina, H3K4me3, a mark associated with active transcription, is observed in all layers of the neural retina, including rhodopsin-positive photoreceptors, Müller glia, and retinal ganglion cells (RGCs). H3K27me3, a mark associated with transcriptional repression, is enriched in the inner nuclear layers and in a subset of outer nuclear layer in the adult murine retina. However, H3K9me2, another repressive mark, is not observed in the inner layers of the adult retina.8 The Ezh2 and G9a HMTases are the two best-characterized HMTases, which catalyze H3K27me3 and H3K9me2 modifications, respectively. Both can be found in the fetal murine retina. The results suggest that epigenetic regulation of gene transcription by Ezh2- and G9a-mediated histone lysine methylation plays a crucial role in retinal neuron differentiation and survival.8
HDAC is expressed in mammalian retina.19 Acetylated histone 3 and 4 are found in fetal and adult retina; however, acetylated histone 3 and 4 is reduced in dry age-related macular degeneration (AMD) specimens. In contrast to the decrease of acetylated histones, HDAC1 is highly upregulated in human AMD retinal sections. In addition, exposure of adult mice retina to trichostatin A (TSA) induces upregulation of apoptotic genes. Activation of HDAC may decrease the retina’s resistance to ischemic injury.20
miRNA in mammalian retina
The expression of miRNAs in the mammalian retina has been analyzed in several studies. The most highly expressed adult mouse retinal miRNAs were miR-181a, 182, 183, 204, 125b, 26a, and 124a. miR-181 is expressed in the ganglion cell, inner plexiform and inner nuclear layers; miR182 is expressed throughout all layers of the retina; and miR-183 is expressed only in the outer nuclear layer.21 Another study found strong expression of miR-204 in the RPE and the ciliary body, and miR-224 was detected in the RPE. In contrast, miR-124a was expressed in all layers of the neural retina except the RPE. Most recently, miRNA-132 was found to be expressed in human RPE cells. miR-204 and 211 were also found to be highly expressed in the RPE. Transforming growth factor-β (TGF-β) receptor-2 and SNAIL2 are direct targets of miR-204. Notably, anti-miR-204/211 decreased transepithelial resistance and reduced cell membrane voltage and conductance, indicating a critical role of miR-204/211 in maintaining epithelial barrier function and cell physiology.22
Epigenetic mechanisms in retinal development
Although epigenetic mechanisms have been shown to regulate neural stem cell renewal and differentiation, it was not until recent years that their involvement in retinal development has been realized. The retina is composed of specialized glia and neuronal cells which are generated from multipotent retinal progenitor cells in a highly conserved temporal sequence with overlapping phases during development.23,24 RGCs, horizontal cells, and cone photoreceptors are born in early phases, while rod photoreceptors, bipolar cells, and Müller glial cells are born in late phases. The cell fate decision of the progenitor cells depends on both intrinsic and environmental cues, which are regulated by specific networks of transcription factors.23,24 Through covalent modifications of DNA and histones, chromatin remodeling regulates the interactions of these transcription factors and their effector genes, and is an important epigenetic mechanism in retinal development.
DNA and histone methylation in retinal development
DNA and histone methylation are mediated by DNMT and HMTases. In zebrafish retina, antisense-based morpholino knockdown of DNMT3 and H3K9 HMTases Suv39h1 and G9a lead to defects in retinal cell differentiation, supporting a role of both DNA and histone methylation in zebrafish retinal development.25 In mice, immunohistochemical analysis shows changing patterns of histone methylation markers in the developing retina. Specifically, the transcriptionally activating H3K4me3 and repressive H3K27me3 histone marks are found in differentiated neurons from embryo to adulthood, corresponding to the expression of the HMTase Ezh2 that catalyzes the H3K27me3 mark. In contrast, the repressive H3K9me2 mark and the corresponding HMTase G9a were found primarily in early differentiating RGCs, but decreased after birth (Fig. 33.1).8 These changing patterns of histone methylation may at least in part account for the temporal sequence of retinal progenitor cell differentiation during development. Evidence also implicates DNA methylation in regulating the expression of genes involved in the topographic patterning of RGC axons, such as EphA5 receptor.18 These studies suggest that DNA and histone modifications may regulate both temporal and spatial expression of these genes in different populations of retinal progenitor cells to orchestrate the precise timing of cell proliferation and differentiation during development.
Histone acetylation in retinal development
During photoreceptor differentiation, HAT-containing coactivators, such as general control nondepressible 5 (Gcn5) and CREB-binding protein (Cbp), interact with cone–rod homeobox (Crx) transcription factor to promote the transcription of opsin genes.26 Crx-deficient mutant mice have decreased histone acetylation at the promoter/enhancer region of the opsin gene, resulting in photoreceptor dysfunction.27 Such results indicate that HATs are required for differentiation of retinal photoreceptors during development.
In addition to HATs, HDACs have been studied more extensively and are also implicated in retinal development. HDAC1 is recruited by the retinoblastoma protein (Rb) and related family members to promoters with E2F-binding sites, resulting in transcriptional repression of cell cycle genes and suppression of cell growth. In zebrafish, hdac1-deficient mutants exhibit retinal cell proliferation and differentiation defects, resulting in severe reduction in the inner and outer plexiform layers and absence of RGC, rod and cone photoreceptors, and Müller glia.27 These defects result from failure in downregulation of cyclin D and E transcription and the canonical Wnt and Notch signaling pathways, which are necessary for retinal progenitor cells to exit the cell cycle. Thus, histone deacetylation in zebrafish may be a regulatory mechanism to switch off stem cell proliferation and initiate a program for retinal differentiation. In mouse retinal explants, pharmacological inhibition of HDAC with TSA also results in defects in rod differentiation, but unlike in zebrafish, causes a reduction in retinal progenitor cell proliferation. In vivo knockdown HDAC4 by RNA interference leads to apoptosis of rod photoreceptors and bipolar cells, while overexpression of HDAC4 reduced naturally occurring cell death of bipolar cells, supporting a role of HDAC4 in promoting the survival of developing retinal neurons as well.28 Further work will help clarify what specific downstream effectors are regulated by HATs and HDACs during the various stages of retinal development.
Chromatin remodeling complexes in retinal development
While covalent DNA and histone modifications decondense chromatin and promoter regions, ATP-dependent displacement from promoters and enhancer regions requires the action of chromatin-remodeling complexes. These include the SWItch/Sucrose NonFermentable (SWI/SNF) and the Imitation SWItch (ISWI) families. Zebrafish mutants lacking the brahma-related gene 1 (BRG1), the catalytic ATPase subunit of SWI/SNF chromatin remodeling complex, show defects in retinal differentiation, suggesting that BRG1 is involved in triggering retinal cell differentiation.29 In mice, however, loss of BRG1 in neural stem cells results in precocious neuronal differentiation, suggesting that it represses differentiation and maintains neural stem and progenitor cells. These differences may be related to changes in the composition of accessory units known as BRG1-associated factors (BAFs) in the SWI/SNF complexes in different cell states. Nevertheless, chromatin-remodeling complexes are likely to be critically involved in regulating retinal cell differentiation during development.
microRNAs in retinal development
Recently, miRNAs have also been implicated in retinal development. Early studies using a conditional knockout mouse lacking the RNA endonuclease Dicer in the retina showed no defects in the retina before the second postnatal week, suggesting that miRNAs are not required for mouse retinal development.30 However, subsequent research using a different strain of Dicer conditional knockout mouse showed increased production of early generated cell types such as RGC and horizontal cells, and failure to generate late-born cell types such as rod photoreceptors and Müller glia.31 This is also supported by data from Xenopus morpholinos, where Dicer inactivation resulted in defects in cell cycle, lamination, and timing of retinal differentiation. These observations suggest that Dicer and miRNAs may provide a common regulatory mechanism to signal changes in retinal progenitor cell competence.
Microarray analysis of the miRNA transcriptome in mouse retina reveals at least 78 miRNAs, 21 of which are potentially retina-specific.32 The miRNA transcriptome of the mouse retina is similar to that of humans, and shows dozens of miRNAs that are differentially expressed during different stages of development. In Xenopus, miRNAs have been found to inhibit the translation of homeodomain transcription factors (Xotx2 and Xvsx1) involved in bipolar cell differentiation by binding the 3’ untranslated region of mRNAs. Inactivation of these miRNAs in vivo releases the inhibition and supports the generation of additional bipolar cells.33 Another miRNA, miR-24a, negatively regulates proapoptotic factors caspase-9 and apoptotic protease-activating factor 1 (apaf1) in Xenopus retina. Inhibition of miR-24a leads to increased apoptosis during retina development and reduction in eye size.34
[/not-level-membership-for-opthalmology-category]
