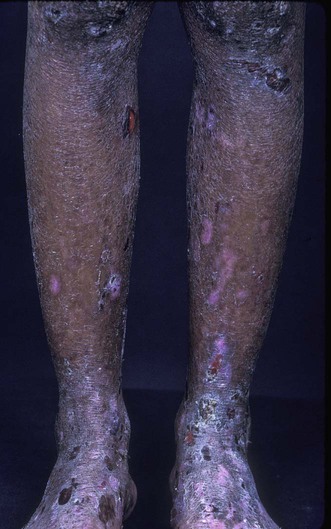
Epidermolysis bullosa acquisita
Specific investigations
 Skin biopsy and serum for direct and indirect immunofluorescence, respectively, to detect IgG or IgA class skin basement membrane-specific autoantibodies
Skin biopsy and serum for direct and indirect immunofluorescence, respectively, to detect IgG or IgA class skin basement membrane-specific autoantibodies
 Serum for ELISA to detect IgG or IgA class type VII collagen-specific autoantibodies
Serum for ELISA to detect IgG or IgA class type VII collagen-specific autoantibodies
 Gastrointestinal work-up for possible inflammatory bowel disease
Gastrointestinal work-up for possible inflammatory bowel disease
First-line therapies
 Systemic corticosteroids
Systemic corticosteroids Mycophenolate mofetil
Mycophenolate mofetil Dapsone
DapsoneSecond-line therapies
 Intravenous immunoglobulin
Intravenous immunoglobulin Colchicine
Colchicine Cyclosporine
Cyclosporine
 Anti-CD20 antibody (rituximab)
Anti-CD20 antibody (rituximab)
