Published on 19/03/2015 by admin
Filed under Dermatology
Last modified 22/04/2025
This article have been viewed 3087 times
Anna F. Falabella, Ysabel M. Bello and Lawrence A. Schachner
Evidence Levels: A Double-blind study B Clinical trial ≥ 20 subjects C Clinical trial < 20 subjects D Series ≥ 5 subjects E Anecdotal case reports
Epidermolysis bullosa (EB) is a complex group of mechanobullous disorders characterized by painful blister formation as a result of minor trauma to the skin. With the exception of the acquisita type, EB is an inherited disorder. The classification system for hereditary EB was revised in 2007 and now includes four major types according to the level of blister formation: simplex or intraepidermal (’epidermolytic’), junctional or intralamina lucida (‘lamina lucidolytic’), dystrophic or sublamina densa (‘dermolytic’), and Kindler syndrome or mixed. The disease can involve the skin, mucosae, and internal organs. The severity of EB ranges from mild to severe, and skin involvement can be localized or generalized.
In addition to the four major types of EB, there are several dozen subtypes, resulting from more than 1000 documented mutations on at least 14 structural genes. The genetic variability of EB results in a wide variety of clinical presentations, extra-cutaneous manifestations, degree of morbidity, and risk for early mortality.
The management of inherited EB has classically been supportive: avoidance of trauma, blister management, wound management, treatment of infections, and nutritional support. More recent efforts have been directed at identifying and treating the underlying cause of disease with the goal of improving wound healing and preventing new wound formation.
Avoidance of trauma to intact skin is important but difficult. Sewing foam pads into the lining of clothing is helpful, especially over the elbows, knees, and other pressure points. Blister management involves puncturing new blisters with a sterile needle to avoid extension of the blister. The punctured area should be covered with a topical antibiotic and a non-adherent dressing.
Wound management comprises assessing the location and characteristics of wound; cleansing with low toxicity solutions (e.g., saline, water); gentle debridement of eschar/slough if present; and covering the wound with an appropriate non-adherent dressing. Wounds need to be evaluated and treated on a daily basis. In general, it is beneficial to soak wounds in a bath tub for 5 to 10 minutes. This facilitates cleansing and non-traumatic removal of dressings. Use of low concentration acetic acid or bleach in the bath water may also help control bacterial load in wounds.
Minimizing trauma to wounds is vital. Mepilex is a non-adherent, absorbent polyurethane foam pad that can be applied, removed, and reapplied to wounds with little discomfort, no trauma to the wound bed or surrounding skin, and no disruption of wound healing. Other non-adherent dressings, such as white petrolatum-impregnated gauzes, hydrogels, and foams, can be used and held in place with soft, roller gauze bandages or elastic tube dressings.
A variety of skin grafts have been used to treat the wounds of EB, including split-thickness skin grafts, allogeneic and autogeneic cultured keratinocytes, and cryopreserved acellular human dermis. A study using intradermal injections of allogeneic fibroblasts demonstrated therapeutic potential in patients with recessive dystrophic epidermolysis bullosa (RDEB). Several trials have reported impressive results with Apligraf, a bilayered, tissue-engineered skin derived from neonatal foreskin that contains living keratinocytes and fibroblasts. An allogeneic composite cultured skin (OrCel) has been approved by the US Food and Drug Administration to treat hands and donor sites in patients with RDEB. More recently, amniotic membranes have been used to treat patients with EB with chronic, non-healing wounds.
Avoidance of wound infection is also critical to promote more rapid healing and to avoid overwhelming infections and sepsis, which are associated with an increased mortality rate. Topical antibiotics are routinely used, but should be rotated monthly to avoid the development of resistant organisms. Cutaneous infections unresponsive to topical measures need to be treated with systemic antibiotics, but the chronic use of systemic antibiotics is not recommended as a preventive measure.
Common nutritional problems in patients with EB include chewing and swallowing difficulties, malnutrition, constipation, and vitamin and mineral deficiencies. Avoidance of malnutrition depends on active and continuous nutritional support. Early nutritional supplementation can promote better childhood growth rates and promote healing of skin lesions. In patients who develop esophageal strictures, balloon dilatation, surgery is sometimes needed. Daily multivitamin trace elements and zinc supplementation is recommended. Anemia may be profound in EB, and oral iron replacement is mandatory for patients with iron deficiency. Erythropoietin has been recommended if the iron level is <500 mU/mL. Some have recommended intravenous iron therapy for patients resistant to oral replacement. Albumin should be monitored to assess the patient’s nutritional status. If necessary, protein supplements can be added to the diet. Severe malnutrition can be treated with enteral feeding via gastrostomy tube if necessary.
Systemic therapies, such as psoralens in combination with UVA irradiation, corticosteroids, vitamin E, cyclosporine, antimalarials, retinoids, phenytoin, tetracycline, trimethoprim–sulfamethoxazole, and cyproheptadine, have been used to treat EB, but their efficacy is unproven.
Current therapies are focused on gene-, protein-, and cell-based techniques. There have been improvements in genetic manipulation of keratinocytes ex vivo and of graft techniques in vivo. Gene transfer of epidermal stem cells in combination with tissue engineering procedures is promising.
Skin biopsy for transmission electron microscopy
Skin biopsy for immunofluorescence antigenic mapping
Mutational analysis
Fine J-D, Eady RAJ, Bauer EA, Bauer JW, Heagerty A, Bruckner-Tuderman L, et al. J Am Acad Dermatol 2008; 58: 931–50.
Both transmission electron microscopy (EM) and immunofluorescence have been successfully employed to diagnose EB. Electron microscopy is likely to play a decreasing role in the diagnosis because there are only a few highly proficient EM laboratories, although it has an important place in research because it permits visualization and assessment of keratin filaments, hemidesmosomes, and anchoring fibrils. Immunofluorescence antigen mapping is relatively inexpensive and simple to perform, requiring immunofluorescence transport media. It can reveal the level of the split by defining its location relative to proteins expressed at various levels of the basement membrane zone. Mutational analysis remains a superb research tool that lets us determine the mode of inheritance, the precise site and the type of molecular mutation. However, it is not considered to be the first-line diagnostic test.
Pope E, Lara-Corrales I, Mellerio J, Martinez A, Schultz G, Burrell R, et al. J Am Acad Dermatol 2012; 67: 904–17.
Wound care is the cornerstone of treatment for patients with EB; however, there are currently no guidelines to help practitioners care for these patients. An expert panel generated a list of recommendations based on the best evidence available. The recommendations were translated into a survey, and sent to other EB experts to generate a consensus. The list was refined and grouped into four themes: treat cause; patient-centered concerns; local wound care; and develop individualized goals and plan of care. Within these four themes, 17 specific recommendations were made. Respondents also provided preventative and therapeutic pain management strategies, as well as dressing choices and topical therapy.
Bello YM, Falabella AF, Schachner LA. Clin Dermatol 2003; 21: 278–82.
Open or only partially healed erosions are best covered with polymyxin, bacitracin, or silver sulfadiazine and then covered with either petrolatum-impregnated gauze or non-adherent synthetic dressing. Such dressings are usually changed daily. Mupirocin may be substituted for those infected sites unresponsive to milder antibiotics, but is best avoided for routine use because of the potential for development of resistance.
Hubbard L, Haynes L, Sklar M, Martinez AE, Mellerio JE. Clin Exp Dermatol 2011; 36: 579–83.
This is a report of a study day held in London on 3 March 2010 to discuss measures with which to meet the nutritional requirements of patients with EB. Members of national and international multidisciplinary teams (MDTs) caring for patients with EB attended this event. The study day focused on four challenging aspects of management intimately associated with nutritional status in EB, necessitating close cooperation between MDT members: iron-deficiency anemia, gastrostomy placement and feeding, muscle mass and mobility, and dental health. The study day provided a unique forum for dietitians, doctors, nurses, physiotherapists, psychologists, psychotherapists, dentists, dental hygienists, and occupational therapists to share knowledge and debate problems common to all who strive to promote best practice in this rare and complex group of conditions.
Falabella AF, Valencia IC, Eaglstein WH, Schachner LA. Arch Dermatol 2000; 135: 1219–22.
An open-label uncontrolled study of 15 patients with 69 acute wounds and nine chronic wounds who were treated with tissue-engineered skin; a week later, 79% of the wounds were healed. The patients and their families considered that healing with the tissue-engineered skin was faster and less painful, and that quality of life was improved, compared to healing with conventional dressings.
Fivenson DP, Scherschum L, Cohen LV. Plast Reconstruct Surg 2003; 112: 584–8.
Five patients with RDEB were treated for mitten deformity with a bioengineered skin equivalent. These patients exhibited increased range of motion and have maintained web space separation for more than 12 months, improving the quality of their life.
Lo V, Lara-Corrales I, Stuparich A, Pope E. J Am Acad Dermatol 2010; 62: 1038–44.
A retrospective chart review of two patients with EB who were treated with amniotic membranes revealed the potential usefulness of amniotic membrane grafting in promoting healing of chronic wounds in patients with EB.
Wagner JE, Ishida-Yamamoto A, McGrath JA, Hordinsky M, Keene DR, Woodley DT, et al. N Engl J Med 2010; 363: 629–39. Erratum in: N Engl J Med 2010; 363(14): 1383. Woodley DT [added]; Chen M [added].
Seven children with RDEB were treated with immunomyeloablative chemotherapy and allogeneic stem-cell transplantation. One patient died of cardiomyopathy before transplantation. Of the remaining six patients, one had severe regimen-related cutaneous toxicity, with all having improved wound healing and a reduction in blister formation between 30 and 130 days after transplantation. Increased C7 deposition was observed at the dermal–epidermal junction in five of the six recipients, albeit without normalization of anchoring fibrils. Five recipients were alive 130 to 799 days after transplantation; one died at 183 days as a consequence of graft rejection and infection. The six recipients had substantial proportions of donor cells in the skin, and none had detectable anti-C7 antibodies.
Further studies are needed to assess the long-term risks and benefits of such therapy in patients with this disorder.
Yuen WY, Huizinga J, Jonkman MF. J Am Acad Dermatol 2012; 68: 93–7.
Four patients with laminin-332-deficient JEB-nH were treated with punch grafting. Over a 10-year period, 23 ulcers were treated using punch grafting without any complications or adverse effects. The ulcers had on average persisted 6 years before treatment. Healing rate after punch grafting was 70% (n = 16), with a mean healing time of 2 months. Thirty percent (n = 7) of the treated ulcers did not completely heal, but did show improvement. The recurrence rate after 3 months was 13% (n = 2), and was a result of renewed blistering. Punch grafting can be used as a first-line treatment in small persistent ulcers in patients with JEB-nH.
Veien NK, Buus SK. Arch Dermatol 2000; 136: 424–5.
A number of patients using tetracycline were observed over a 7-year period. The article reports an increase in bulla formation during the summer months, but healing was more rapid and less painful while patients took the tetracycline.
Weiner M, Stein A, Cash S, de Leoz J, Fine JD. Br J Dermatol 2004; 150: 613–14.
Six of 12 patients completed at least the first arm: four experienced a reduction in the total number of EB lesions; two experienced an increased number of lesions after 4 months of active therapy (oral tetracycline administered 1000 mg every morning and 500 mg every evening). The risk of tetracycline-induced dental discoloration in children needs to be balanced against the severity and chronicity of the symptoms.
Lara-Corrales I, Parkin PC, Stephens D, Hamilton J, Koren G, Weinstein M, et al. J Am Acad Dermatol 2012; 66: 264–70.
Ten patients with RDEB were treated with trimethoprim (TMP). The study assessed lesion counts, quality of life, and emergence of antibiotic resistance. All patients showed improved wound healing on TMP, but primary and secondary outcome measures did not achieve statistical significance. This proof-of-concept study demonstrates the potential efficacy of TMP in improving wound healing in RDEB, and provides useful information for further prospective studies.
Fine JD, Johnson LB, Weiner M, Stein A, Suchindran C. J Am Acad Dermatol 2004; 50: 563–71.
Patients with RDEB are at high risk of developing squamous cell carcinoma. A initial study on 20 patients aged 15 years or older who were treated with isotretinoin 0.5 mg/kg/day for 8 months reported that isotretinoin may be safely used. However, a chemoprevention effect has yet to be proven.
Mavillo F, Pellegrini G, Ferrari S, Di Nunzio F, Di Nunzio F, Di Iorio E, et al. Nature Med 2006; 12: 1397–402.
Ex vivo transduction of autologous epidermal stem cells with a normal copy of the defective gene, followed by reconstitution of the patient’s skin with epithelial sheets that were grown from these genetically corrected cells, kept the epidermis firmly adherent and stable for the duration of follow-up (1 year).
Yuen WY, Jonkman MF. J Am Acad Dermatol 2011; 65: 780–9.
Squamous cell carcinoma (SCC) is the most severe complication and most common cause of death in patients with RDEB. The SCCs have a high recurrence rate and follow an aggressive course that results in death in one of five patients. It is recommended that annual checks of all JEB patients for SCC start at 25 years of age.
Treatment of Skin Disease Comprehensive Therapeutic Strategies 4e
WhatsApp us
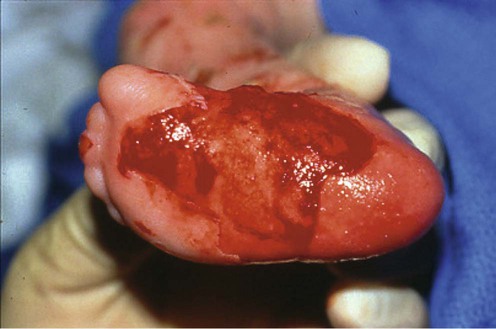




 Sterile dressing and topical antibiotics
Sterile dressing and topical antibiotics Nutritional support
Nutritional support Skin grafts
Skin grafts Cultured keratinocytes
Cultured keratinocytes Fibroblast cell therapy
Fibroblast cell therapy Amniotic cell membrane
Amniotic cell membrane Phenytoin (ineffective)
Phenytoin (ineffective) Tetracycline
Tetracycline Trimethoprim–sulfamethoxazole
Trimethoprim–sulfamethoxazole Cyproheptadine
Cyproheptadine Isotretinoin
Isotretinoin Gene therapy
Gene therapy Bone marrow transplantation
Bone marrow transplantation