[level-membership-for-basic-science-category]
CHAPTER 20 Endoplasmic Reticulum*
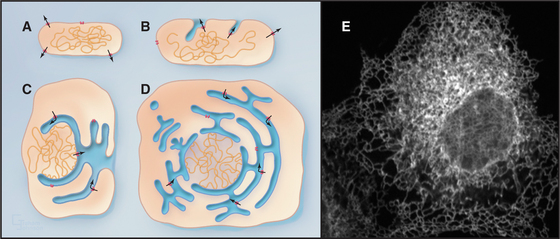
One of the key distinguishing features of eukaryotic cells is the presence of the endoplasmic reticulum (ER), the largest of numerous membrane-delineated intracellular compartments. The ER is thought to have evolved from the prokaryotic plasma membrane by expansion, internalization, and subdivision (Fig. 20-1A-D; see also Fig. 2-6). It provides both an expanded membrane surface (up to 30 times that of the plasma membrane) for carrying out vital cellular functions, including protein and lipid biosynthesis, and an internal compartment (or lumen) that collects proteins synthesized in the cytoplasm for modification and delivery into the secretory pathway. About one third of all cellular proteins are imported into the lumen of the ER or integrated into its membranes. Consisting of an extensive array of tubules or flat saccules called cisternae (cisterna means “reservoir”), the ER forms a continuous three-dimensional network (a reticulum) stretching from the nuclear envelope to the cell surface (Fig. 20-1E). Microtubules and their associated motors generate this extended network in animal cells by pulling ER membranes out toward the periphery of the cell. This allows the ER to coordinate diverse processes over large regions of the cytoplasm. The ER’s size and shape are maintained over time despite a continuous flow of proteins and lipids into and out of this compartment. The flow results from the large rate of synthesis of lipids and proteins (between 2 million and 13 million new proteins per minute) occurring at the ER membrane, as well as from the continuous export of these molecules into the secretory pathway and their selective retrieval back to the ER from the Golgi apparatus.
Endoplasmic Reticulum Functions and Organization
The membrane surface of the ER performs several functions for cells. Foremost is the production of the proteins and lipids that will make up the membranes of the other organelles, including the Golgi apparatus, nucleus, endosomes, lysosomes, and plasma membrane, as well as nearly all proteins that will be secreted from the cell. The membranes of mitochondria and peroxisomes also depend on the ER to supply much of their lipid. Another key function of the ER membrane is to form the nuclear envelope, which encloses the nucleus (see Fig. 14-5). The surface of the ER forming the outer nuclear envelope (which faces the cytoplasm) is indistinguishable from the rest of the ER except for the presence of nuclear pores that span both inner and outer nuclear envelope to allow passage of molecules between the nucleus and cytoplasm. By contrast, the ER surface forming the inner nuclear envelope, which faces the nucleus, contains specialized proteins that interact with the nuclear lamina and chromatin. ER membranes also detoxify endogenous steroids, carcinogenic compounds, and lipid-soluble drugs (xenobiotics) from the environment. This occurs by an electron transfer process carried out by ER membrane proteins such as the cytochrome P450 family of enzymes.
The lumen of the ER also performs numerous essential functions. Specialized for receiving proteins transported from the cytoplasm across ER membranes, the ER lumen is enriched in a dense meshwork of chaperones and other modifying enzymes (estimated to be 200 mg/mL in concentration) that catalyze the folding and assembly of newly synthesized proteins. These transported proteins, including both soluble and transmembrane forms, are exported from the ER for secretion or for delivery to the lumen or membrane of the Golgi apparatus, lysosome, or endosome. Proteins that are incorrectly folded or misfolded can be exported back into the cytoplasm, where they are degraded. Misfolded proteins, when accumulated in the ER at high levels, can trigger an unfolded protein response, which activates specific genes in the nucleus whose products help to modify or destroy the misfolded proteins and compensate for the decreased capacity of ER folding. The ER lumen is one of the major Ca2+ storage sites in cells, owing to ER membranes being rich in calcium pumps (see Fig. 8-7) and many Ca2+-binding proteins in the lumen. Such Ca2+ stores can be released by calcium channels in response to cellular signals such as IP3 (see Fig. 26-13). Carefully regulated release and uptake of Ca2+ by the ER control muscle contraction (see Fig. 39-15) and many other cellular processes. The lumen of the ER is also an oxidizing environment that favors disulfide bond formation, which helps to stabilize proteins after they are exported from the ER to the outside of the cell.
The diverse functions carried out by the membrane and in the lumen of the ER (Table 20-1) occur in distinctive ER regions (Fig. 20-2A). The rough ER is studded with ribosomes on its cytoplasmic surface, defining areas that are specialized for protein synthesis, folding, and degradation (Fig. 20-2B). The smooth ER, composed of tubular elements lacking ribosomes, is dedicated to enzyme pathways involved in drug metabolism (hepatocytes), steroid synthesis (endocrine cells), or calcium uptake and release (see Fig. 26-12). The cytochrome P-450 family of heme-containing membrane proteins is found in the smooth ER. Other regions of the ER that lack ribosomes, called ER export domains consist of tubulovesicular membranes that bud during export of secretory cargo to the Golgi apparatus (see Chapter 21). Regions of the ER surrounding the nucleus compose the nuclear envelope.
| ER Domain | Function | Associated Proteins |
|---|---|---|
| Rough ER | Protein translocation | Sec61 complex, TRAP, TRAM, BiP |
| Protein folding and oligomerization | PDI, Calnexin, Calreticulin, BiP | |
| Carbohydrate addition | Oligosaccharide transferase | |
| ER degradation | EDEM, Derlin1 | |
| Smooth ER | Detoxification | Cytochrome P450 enzymes |
| Lipid metabolism | HMG-CoA reductase | |
| Heme metabolism | Cytochrome b(5) | |
| Calcium release | IP3 receptors | |
| Nuclear envelope | Nuclear pores | POM121, GP210 (see Fig. 14-8) |
| Chromatin anchoring | Lamin B receptor | |
| ER export sites | Export of proteins and lipids into secretory pathway | Sar1p, Sec12p, Sec16p |
| ER contact zones | Transport of lipids | LTPs |
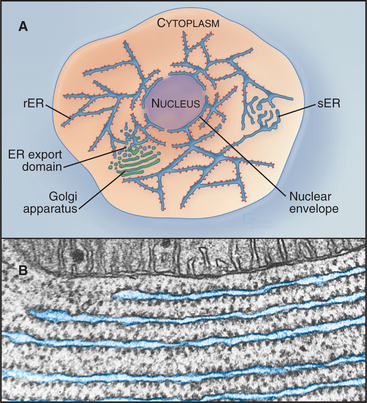
Figure 20-2 endoplasmic reticulum subdomain organization within the cell. A, ER membranes containing ribosomes, called rough ER (rER), extend from the nuclear envelope to the cell periphery. ER membranes lacking ribosomes, called smooth ER (sER), include membranes specialized for drug metabolism and steroid synthesis, as well as tubulovesicular elements composing ER exit sites. The nuclear envelope consists of ER membrane that has wrapped around DNA and other nuclear elements to compartmentalize them. Its double membrane is studded with nuclear pores, which provide a passageway for nuclear components to move between the nucleus and cytosol. (See Fig. 14-17 for more details.) B, Electron micrograph of a thin section of rough ER and neighboring mitochondrion from the pancreas.
(Micrograph by Keith R. Porter; courtesy of Don W. Fawcett, Harvard Medical School, Boston, Massachusetts.)
Overview of Protein Translocation into the Endoplasmic Reticulum
All proteins are synthesized in the cytoplasm and must be specifically targeted to the ER, where they are either fully translocated across the ER membrane and released into the ER lumen (soluble proteins) or only partly translocated across the ER membrane and embedded in the lipid bilayer of the ER membrane (transmembrane proteins). This transport to the ER can happen either as the protein is being made (cotranslational translocation) or after synthesis is complete (posttranslational translocation). The orientation of a protein in the lipid bilayer or its localization to the lumen is established during protein translocation and maintained as the protein is transferred by membrane-bound carriers through the secretory pathway (see Fig. 21-2). Thus, domains of transmembrane proteins to be exposed on the cell surface must be inserted into the ER membrane, facing the lumen. Similarly, secreted soluble proteins must be fully translocated into the lumen of the ER.
Signal Sequence Recognition
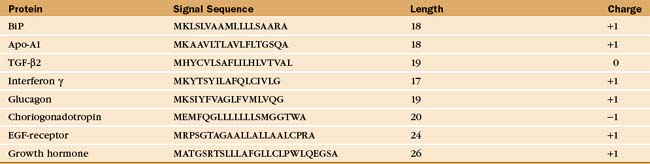
Soluble and membrane proteins destined for ER translocation contain a hydrophobic sequence either at their N-terminus or in transmembrane segments that serve as recognition signals for direction to the ER membrane. N-terminal leader sequences (termed signal sequences) are typically 15 to 35 amino acids long and contain a hydrophobic core of at least 6 residues, while transmembrane signal segments have a hydrophobic stretch of 16 to 25 residues. Aside from hydrophobicity, these signal sequences have no other features in common (Table 20-2). Nevertheless, when attached to proteins that are not normally targeted to the ER, these signal sequences direct the protein to the ER and not to other organelles such as to mitochondria or peroxisomes, which use unique targeting signals (see Fig. 18-1).
Cotranslational Translocation
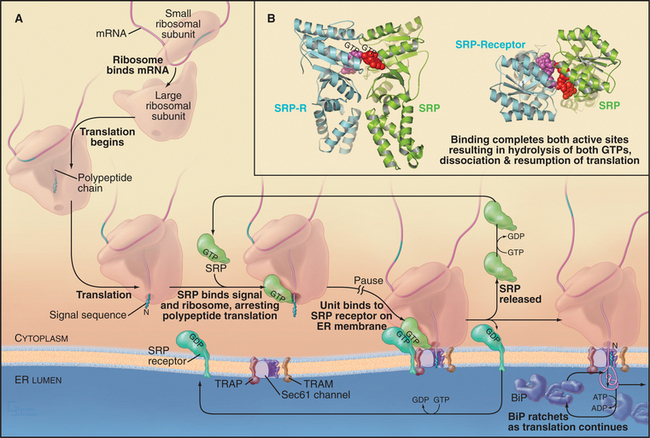
In the cotranslational pathway, the signal recognition particle (SRP) recognizes and binds the first hydrophobic domain of either the signal sequence or the transmembrane signal segment as it emerges from the ribosome slowing translation of the polypeptide (Fig. 20-3A). Then the complex consisting of ribosome, nascent chain, and SRP associates with the ER membrane by binding the SRP receptor (SR), a heterodimer consisting of one subunit that binds SRP and another that spans the ER membrane. Both the SRP and the SR have guanosine triphosphatase (GTPase) domains (Fig. 20-3B) similar to Ras (see Fig. 4-6). GTP binding and hydrolysis by SRP and SR provide directionality and order to the sequence of reactions that bring the nascent chain to the translocation channel. Once the ribosome–nascent chain–SRP complex is bound to SR at the ER membrane, SRP releases the signal sequence, allowing the ribosome–nascent chain complex to be transferred to the protein-conducting channel across the ER membrane. SRP and SR then dissociate after hydrolyzing their bound guanosine triphosphate (GTP), releasing SRP into the cytoplasm and allowing SR to diffuse away in the membrane. The targeting cycle delivers the ribosome–nascent chain complex to the protein-conducting channel (called the translocon) and recycles the targeting machinery (i.e., SRP and SR).
Posttranslational Translocation
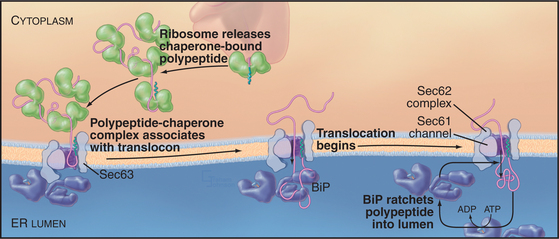
Posttranslational translocation (prevalent in fungi such as yeast) differs because neither cotranslational targeting machinery (SRP and SR) nor ribosomes participate. Instead, other components fulfill the functions provided by SRP, SR, and ribosomes (with the Sec61 complex serving as channel, as it does in cotranslational translocation). Signal-containing polypeptides destined for posttranslational insertion into the ER are held in a largely unfolded state by cytoplasmic chaperones until they can be delivered to the tetrameric Sec62/63 protein complex at the ER membrane (Fig. 20-4). A similar strategy is used to import proteins into the mitochondria (see Fig. 18-4). During posttranslational translocation, the signal sequence engages and opens the Sec61 channel in a fashion similar to cotranslational translocation. Since protein synthesis is already complete, another energy source must be exploited to move the substrate through the channel into the ER. BiP, a luminal ER chaperone belonging to the Hsp70 family, binds the substrate in the ER lumen, thereby preventing it from sliding back into the cytoplasm. Repeated rounds of substrate binding and release, catalyzed by ATP hydrolysis, allow BiP to act as a molecular ratchet to drive substrate transport into the lumen. The transmembrane protein Sec63 regulates the ATPase activity of BiP and helps to recruit BiP to the translocation channel. Thus, the peptide is “pulled” across the membrane from the luminal side instead of being “pushed” from the cytoplasmic side, as during cotranslational translocation. Hsp70 family members perform a similar function during import of proteins into mitochondria and chloroplasts (see Fig. 18-4).
Universality of Protein Translocation
In both cotranslational and posttranslational translocation, the Sec61 complex translocation channel is closed until opened by interacting with binding partners, including the translocated polypeptide. This ensures that only specific types of proteins pass through the ER membrane and that the permeability barrier of these membranes is maintained at all times. In cotranslational translocation, interactions with a ribosome and a signal sequence open the channel, and protein synthesis provides the energy for translocation. In posttranslational translocation, the Sec62/63 complex operates on both sides of the membrane. On the cytoplasmic side, it contributes to the interactions of the signal sequence that open the translocon. On the lumenal side, it recruits BiP to provide the driving force for translocation. This theme applies to protein translocation across the plasma membrane by prokaryotes. There, a homolog of the Sec61 complex (the SecYE complex) interacts with the cytoplasmic SecA adenosine triphosphatase (ATPase), a partner that drives translocation across the plasma membrane (see Fig. 18-9).
Molecular Machinery for Protein Translocation into the Endoplasmic Reticulum
Signal Recognition Particle and Signal Recognition Particle–Receptor
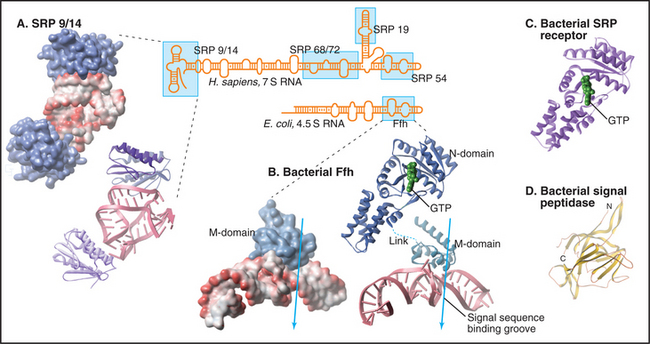
Human SRP is a ribonucleoprotein composed of six proteins (named by their apparent molecular weights and a 300-nucleotide RNA (Fig. 20-5). The SRP54 protein subunit and a portion of the RNA compose the minimal hardware for targeting to the translocon and are found in both prokaryotic and eukaryotic cells. SRP54 binds signal sequences in a deep, hydrophobic groove lined by the flexible side chains of several methionines. Like bristles of a brush, the methionines accommodate the various shapes of the hydrophobic side chains of the residues of different signal sequences. Phosphates of the SRP RNA near one end of this groove may interact with basic residues that are often (but not always) adjacent to the hydrophobic core of signal sequences and transmembrane signal segments.
SRP binding to a signal sequence slows translation, a phenomenon that is termed elongation arrest (Fig. 20-3A). The mechanism appears to involve occlusion of the elongation factor binding site on the ribosome by the SRP9 and SRP14 subunits of SRP, which structurally resembles a portion of eEF2. Slowing translation provides time to target the ribosome to the translocation channel before excessive polypeptide synthesis precludes cotranslational transport.
Interaction of SRP with SR directs the SRP–ribosome–nascent chain complex to the translocation channel (Fig. 20-3A). This interaction is regulated by GTP binding and hydrolysis by SRP and SR, whose GTPase cycles are a notable exception to the GTPase switch paradigm of Ras-like GTPases that involve GTP exchange factors (GEFs) and GTPase-activating proteins (GAPs; see Fig. 25-8). No external GEFs or GAPs are known for SRP and SR GTPases. Instead, these proteins readily exchange GDP for GTP and are in the GTP-bound state as they enter the targeting cycle. On formation of a complex, SRP and SR reciprocally activate each other’s GTPase activity, thereby obviating the need for an external GAP to drive the conversion of GTP to GDP on these proteins. Upon GTP hydrolysis, the conformations of SR and SRP change in a way that reduces their affinity. They dissociate from each other as well as from the ribosome–nascent chain and translocation channel. This frees SRP and SR for further rounds of ribosome–nascent chain targeting. Release of SRP and SR occurs only after the ribosome has become properly engaged with the translocation channel in the ER membrane, thereby ensuring that the channel is closed until the ribosome has bound.
Sec61 Complex: The Protein-Conducting Channel
The protein-conducting channel consists of three or four copies of the Sec61 complex, appearing as a donut-like structure (Fig. 20-6A). Each Sec61 complex is a heterotrimer of three transmembrane polypeptides: an a subunit with 10 transmembrane segments, and smaller b and g-subunits, each with single transmembrane sequences. The homologous bacterial proteins are called SecY, SecE, and SecG. They form the SecY complex (Fig. 20-6C), the channel for translocating proteins across the plasma membrane of bacteria.
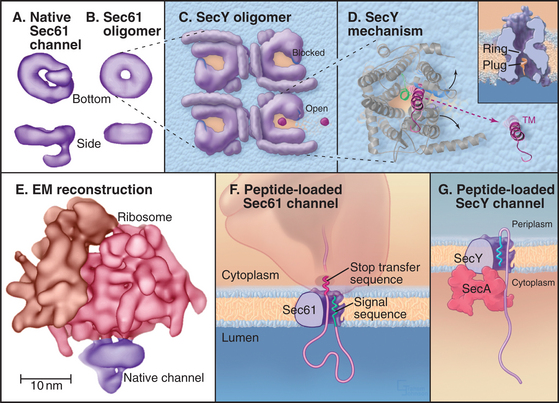
Crystal structures of the archaeal SecY complex revealed a small, hourglass-shaped pore within a single SecY complex (Fig. 20-6D). This pore is flanked on its luminal and cytoplasmic sides by funnels. The narrow constriction between the two funnels is only about 5 to 8 Å in diameter and lined by several hydrophobic side chains that together form the pore ring. Because of their small size and flexibility, the side chains forming the pore ring could fit snugly around a translocating peptide, preventing passage of ions or other small molecules. Another segment of SecY protein (termed the plug domain) occludes the pore in its inactive state and is proposed to shift away from the pore in its active state. The arrangement of the polypeptide allows the pore to open sideways, providing a lateral gate from the aqueous interior of the channel into the hydrophobic environment of the lipid bilayer that transmembrane domains of proteins use to access the lipid bilayer.
The structures strongly suggest that a single Sec61 complex functions as the pore for proteins to translocate across the ER. Several identical Sec61p complexes compose the translocon, but owing to its large size, only one ribosome can bind a translocon. Thus, only one Sec61p complex could be active for translocation. Why then is the translocon an oligomer of several Sec61 complexes? One possibility is that this creates binding sites for components associated with the translocon, such as the ER signal peptidase, the oligosaccharide transferase, TRAP, or TRAM. Oligomerization might also stabilize ribosome binding to the ER by increasing the number of binding sites that are made between Sec61 complex and the ribosome.
Signal Sequence
Binding of a signal sequence to the Sec61p complex opens or “gates” the translocon in preparation for protein translocation. Current evidence suggests that the signal sequence forms a loop in the channel with its N-terminus exposed to the cytoplasm (Fig. 20-6F). This occurs by signal sequence binding to transmembrane helices 2 and 7 on Sec61p.
Protein Insertion into the Endoplasmic Reticulum Bilayer or Lumen
Translocation of Soluble Proteins into the Lumen of the Endoplasmic Reticulum
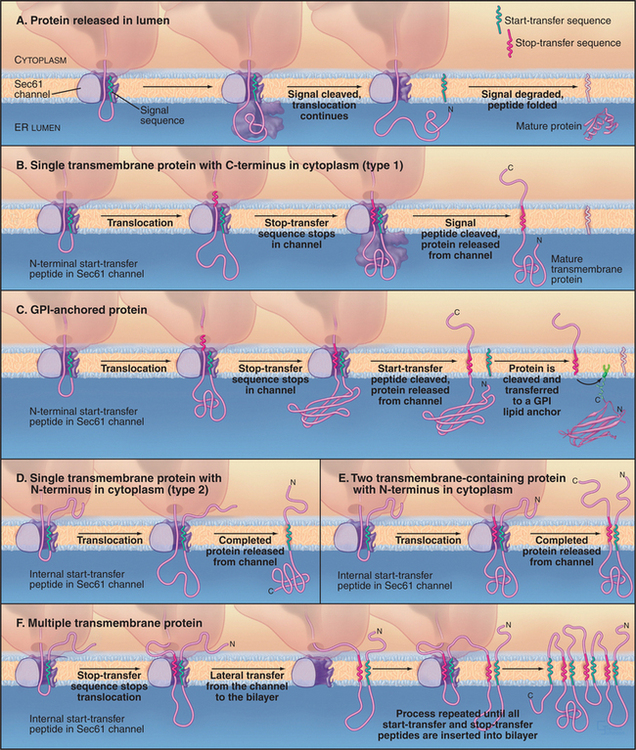
Soluble proteins destined for secretion or retention in the lumen of the ER are translocated fully across the Sec61 protein-conducting channel into the lumen of the ER (Fig. 20-7A). Once the polypeptide has grown to about 150 residues during this process, a signal peptidase in the ER lumen removes the signal sequence from the translocating polypeptide. The signal peptidase consists of five subunits ranging in size from 12 kD to 25 kD and associates with the translocon. After signal peptide cleavage, the new N-terminus of the growing polypeptide continues to pass through the translocon until it is released into the ER lumen. The remaining cleaved signal peptide either is degraded or has other functions elsewhere in the cell.
Insertion of Membrane Proteins in the Endoplasmic Reticulum Bilayer
Most proteins destined for insertion into the bilayer of the ER membrane use the Sec61 protein-conducting channel. During translocation of transmembrane proteins, some parts of the polypeptide chain are translocated across the lipid bilayer, whereas others are not. Depending on how the transmembrane protein is translocated and oriented across the bilayer, the protein is categorized as type 1, type 2, or polytopic (Fig. 20-7B-F). All copies of a particular polypeptide have the same orientation after translocation (i.e., type 1, type 2, or polytopic), and this orientation is usually maintained as the protein is carried to different membrane destinations in the cell through membrane budding and fusion events (see Chapters 21 and 22).
In type 1 transmembrane proteins, the N-terminal signal sequence initiates translocation, similar to soluble proteins (Fig. 20-7B). In the crystal structure of the archaeal SecY complex, the signal sequence binds to two adjacent helices that are proposed to open laterally to let hydrophobic segments of transmembrane proteins exit from the pore into the bilayer (see Fig. 20-6D). A stop-transfer signal (usually the future transmembrane domain) stops the transfer process before the type 1 polypeptide chain is completely translocated. The signal sequence (also called a start-transfer signal) is then cleaved off by the ER signal peptidase, and the polypeptide slides out of the translocon through the lateral exit site. The protein is now oriented with its N-terminus facing the ER lumen, its C-terminus facing the cytoplasm, and its transmembrane segment spanning the ER membrane.
Some type 1 proteins, called GPI-anchored proteins, exchange their carboxyl terminal transmembrane segment for an oligosaccharide anchored to the lipid phosphatidylinositol (Fig. 20-7C; also see Fig. 7-9). An enzyme in the ER lumen cleaves off the transmembrane segment and transfers the new C-terminus to a preassembled glycosylphosphatidylinositol (GPI) membrane anchor. Many cell surface proteins are attached to the plasma membrane in this manner. This allows them to be readily released from the cell when specific phospholipases in the plasma membrane are activated. For example, during sperm capacitation, many GPI-anchored proteins are cleaved and released from the sperm plasma membrane. This reorganization of the sperm’s cell surface is essential for a sperm to fertilize an egg.
Type 2 transmembrane proteins use a transmembrane domain as an internal signal sequence (Fig. 20-7D). Once such an internal signal sequence emerges from a ribosome, it is recognized by SRP and brought to the ER membrane, where it serves as a start-transfer signal to initiate protein translocation. When the protein is fully synthesized, the start-transfer signal, which is not cleaved off, slides out of the translocation channel into the surrounding lipid bilayer, where it serves as a transmembrane anchor.
Polytopic proteins that span the membrane multiple times (such as ion channels and carriers) utilize multiple stop-transfer signals, none of which are cleaved by signal peptides (Fig. 20-7E-F). SRP is probably required to tar-get the first signal sequence to the ER membrane. Thereafter, the dynamics of the channel must accommodate sequences that specify translocation of loops in the cytoplasm or lumen alternating with the transfer of transmembrane segments to the lipid bilayer.
Association of Tail-Anchored Proteins with the Endoplasmic Reticulum Membrane
Although most integral membrane proteins that integrate into the ER bilayer do so by being targeted to and translocated through the Sec61 translocation channel, a group of integral membrane proteins known as C-tail-anchored proteins (tail-anchored proteins) do not. These proteins are held in the phospholipid bilayer by a single stretch of hydrophobic amino acids close to the C-terminus and have their entire functional N-terminus facing the cytoplasm (Fig. 20-8).
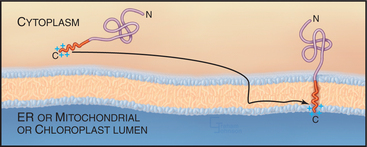
Tail-anchored proteins lack an N-terminal signal sequence and their membrane-interacting region is so close to the C-terminus that it emerges from the ribosome only on termination of translation. Because of this, tail-anchored proteins do not bind to SRP, which recognizes only signal peptides or signal anchors as long as they are part of a nascent polypeptide chain (i.e., still attached to the ribosome). Tail-anchored proteins also do not posttranslationally target to the Sec61 channel. Instead, they use a still unclear mechanism for membrane insertion that involves ATP, cytosolic factors, and membrane components, and that requires that the C-terminal anchor of the protein be hydrophobic. The C-terminus of a tail-anchored protein that crosses the bilayer doubles as a transmembrane anchor after insertion into the appropriate bilayer with only two to three hydrophilic residues translocated across the membrane. By contrast, most other transmembrane and soluble proteins translocate a much larger hydrophilic region across the membrane during their biogenesis.
Tail-anchored proteins have diverse roles in membrane biogenesis and traffic, as well as in cell metabolism (Table 20-3). They include SNARE proteins, such as syntaxins and synaptobrevins, which are responsible for membrane fusion events within cells (see Chapter 21); Sec61g and b of the translocation channel; cytochrome b5, which participates in lipid metabolism; and Bcl and Bax, which are found on the mitochondrial outer membrane and regulate apoptosis.
| Protein | Function |
|---|---|
| ER-Inserted | |
| Target SNAREs (syntaxin) | Target membrane for vesicle insertion |
| Vesicle SNAREs (synaptobrevin) | Target membrane for vesicle insertion |
| Giantin | Golgi tethering protein |
| Sec61γ, Sec61β | ER protein translocation |
| Cytochrome b(5) | Heme metabolism |
| Heme oxygenase I and II | Heme metabolism |
| UBC 6 | ER degradation |
| Mitochondrial-Inserted | |
| Bcl-2 | Apoptosis |
| Bax | Apoptosis |
| Tom5, Tom6 | Mitochondrial protein translocation |
Protein Folding and Oligomerization in the Endoplasmic Reticulum
Once proteins that translocate across the ER bilayer through the Sec61 translocation channel emerge into the ER lumen, they encounter a wealth of proteins that interact with the nascent polypeptide. The proteins remove the signal sequence, add oligosaccharides, and direct folding by catalyzing disulfide bond formation and oligomerization. One such factor, BiP, binds unfolded polypeptides by interacting with hydrophobic regions that are normally sequestered in the interior of a protein. This prevents newly synthesized proteins from aggregating and promotes their folding, and it helps to bias the movement of the polypeptide into the lumen but not back out. Another factor involved in protein folding and assembly is oligosaccharyl transferase, which adds core sugars to the growing chain when an asparagine in an appropriate sequence passes by. An additional factor is protein disulfide isomerase (PDI), which catalyses disulfide exchange between sulfhydryl (SH) groups on cysteines allowing the formation of disulfide (S-S) bonds. The oxidizing equivalents to form disulfide bonds flow from flavin adenine dinucleotide (FAD) through two pairs of cysteines of an ER membrane protein called Ero1p, which oxidizes a pair of cysteines in the active site of PDI. PDI then mediates correct formation of disulfide bonds by forming reversible mixed disulfides with polypeptide substrates until the correct disulfides are formed. Retention of these folding factors in the ER depends on the sequence lysine–aspartic acid–glutamic acid–leucine (KDEL), present at the C-termini of these proteins. If this sequence is deleted, the mutated protein is transported to the Golgi apparatus and secreted from the cell. Addition of KDEL to a normally secreted protein results in its accumulation in the ER.
N-linked Glycosylation
The majority of proteins synthesized in association with the ER are glycoproteins with covalently attached carbohydrates. One class of protein glycosylation takes place cotranslationally in the ER by the addition of a preformed oligosaccharide complex to asparagine residues (Fig. 20-9). These asparagine or N-linked oligosaccharides form flexible hydrated branches that can extend 3 nm or more out from the polypeptide. They frequently make up a sizable portion of the mass of a glycoprotein and cover a large fraction of its surface. Because the oligosaccharides are polar, glycosylation makes the protein more hydrophilic and is less likely to aggregate. By avoiding aggregation, the protein has a higher probability of folding into its correct conformation. Hence, oligosaccharides on proteins play a key role in enabling a newly synthesized protein to fold properly. Once correctly folded, the protein can leave the ER and move through the secretory pathway, where the sugars can be modified further. The great diversity of oligosaccharides found on secreted proteins is also crucial for their functions outside the cell.
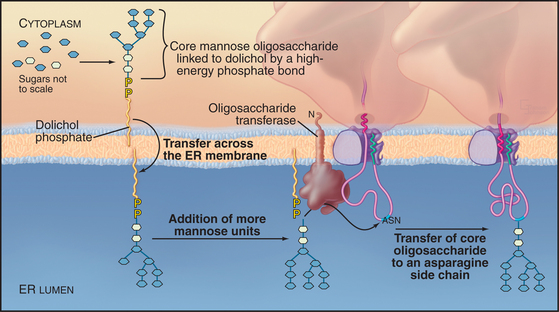
A single, preformed oligosaccharide precursor, composed of 14 sugars (three glucoses, nine mannoses, and two N-acetyl glucosamines) (Glc3Man9GlcNAc2), serves as the core for N-linked oligosaccharides. This precursor is synthesized in a stepwise fashion while attached to the ER membrane by dolichol phosphate (a long-chained, unsaturated isoprenoid alcohol with pyrophosphate at one end; Fig. 20-9). Assembly of the oligosaccharide precursor involves 14 separate transfer reactions: seven on the cytoplasmic face of the ER and seven in the lumen. The mechanism that flips the glycolipid across the bilayer is unknown. The enzyme oligosaccharyltransferase recognizes dolichol and transfers the complete oligosaccharide to asparagine side chains on the nascent polypeptide contained in the sequences Asn-X-Ser/Thr, where X is any amino acid other than proline. The sequence must have emerged a distance of 12 to 14 residues out of the translocon into the ER lumen before it can be recognized.
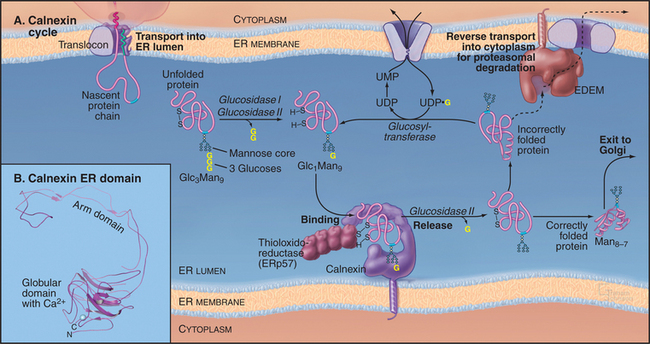
Calnexin/Calreticulin Cycle
Once the core oligosaccharide has been added to the protein, the glycoprotein begins a cycle of modifications that help it to achieve its fully folded state. This calnexin cycle (Fig. 20-10) starts when glucosidases I and II remove the first two glucose residues of a core glycan. The resulting monoglucosylated transmembrane or soluble proteins bind to calnexin, a type I transmembrane protein in the ER lumen (Fig. 20-10). Monoglucosylated soluble proteins also bind to calreticulin, a soluble protein in the ER lumen similar to calnexin. Both proteins are related to sugar-binding lectin proteins from legumes. Both are monomeric, calcium-binding proteins. Binding to calnexin or calreticulin sequesters the glycoprotein and prevents its aggregation. It also exposes the glycoprotein to ERp57, a thiol-disulfide oxidoreductase. A close homolog of protein disulfide isomerase, ERp57 forms a complex with calnexin and calreticulin and specifically interacts with glycoproteins. ERp57 catalyzes intramolecular disulfide bond interchange during the folding process.
Protein Degradation in the Endoplasmic Reticulum and the Unfolded Protein Response
Many polypeptides transferred to the ER are subunits of homo-oligomeric or hetero-oligomeric protein complexes. Oligomer assembly generally occurs prior to ER export and involves chaperones such as BiP that protect hydrophobic surfaces found at subunit interfaces. Because each polypeptide is synthesized on its own ribosome and because the synthesis of the chains composing a complex may be unbalanced, additional chaperones promote subunit interactions and prevent premature export or degradation. For example, chaperones play a critical role in antigen presentation by ensuring that only peptide-loaded major histocompatibility complex type I proteins exit the ER. These chaperones also protect cell surface receptors and other secreted proteins from binding potential ligands that are also imported into the ER and that could activate them prematurely.
Protein Degradation in Endoplasmic Reticulum
The ER has a highly regulated mechanism to prevent the export of dysfunctional proteins into the secretory pathway. Improperly folded polypeptides, excess subunits of oligomeric assemblies, or incorrectly assembled oligomers are degraded rather than being exported from the ER (Fig. 20-10). The degradation process, termed ER-associated degradation (ERAD), prevents accumulation of unsalvageable, misfolded proteins in the ER. ERAD occurs in four sequential steps: a misfolded glycoprotein is first recognized, retrotranslocated across the ER membrane to the cytoplasm, ubiquitinated (see Fig. 23-8), and then degraded by a proteasome in the cytoplasm.
For a protein to be targeted for ERAD, it must be misfolded or unassembled. The cell distinguishes a misfolded or unassembled protein subunit from a bona fide folding intermediate by linking degradation of glycoproteins to the trimming of mannoses (Fig. 20-10). The longer an unfolded glycoprotein remains in the ER, the more likely it will be exposed to mannosidases, which trim terminal mannose residues from the core oligosaccharide. When such trimming occurs, the glycoprotein becomes a substrate for ERAD and is recognized by a membrane-bound ER protein called EDEM (for “ER degradation-enhancing α-mannosidase-like protein”), which helps to direct the glycoprotein to the retrotranslocation channel. (Elimination of the EDEM homolog in yeast retards ERAD of glycoproteins but not of nonglycosylated proteins, which use a different pathway for degradation.) Because the concentration of mannosidases in the ER is low, newly synthesized proteins and nascent chains have time to fold correctly and thereby avoid having their terminal mannoses trimmed. Once mannose trimming on these molecules has occurred, EDEM begins to compete for these substrates with the calnexin/calreticulin cycle. The channel that exports ERAD substrates back into the cytoplasm for degradation (termed the retrotranslocon) remains to be identified.
Endoplasmic Reticulum Stress Responses and Endoplasmic Reticulum Folding Diseases
The folding pathway in the ER is tightly linked to the physiological state of the cell. Conditions that flood the ER with excess protein or result in accumulation of misfolded proteins trigger the unfolded protein response (UPR; Fig. 20-11). Essentially, any condition that exceeds the protein-folding capacity of the ER triggers the unfolded protein response: misfolding of mutant proteins, inhibition of ER glycosylation (by the drug tunicamycin), inhibition of disulfide formation (by reducing agents), or even overproduction of normal proteins. To compensate for these events, this stress-induced signaling pathway upregulates genes that are required to synthesize the entire ER, including folding machinery. In yeast, the unfolded protein response activates more than 300 genes involved with all aspects of ER function, including lipid synthesis, protein translocation, protein folding, glycosylation, and degradation, as well as export to and retrieval from the Golgi apparatus. Developmental programs might work through the same genetic controls to determine the abundance of ER in differentiated cells, producing, for example, extensive ER in secretory cells such as plasma, liver, and pancreatic acinar cells.
The Unfolded Protein Response
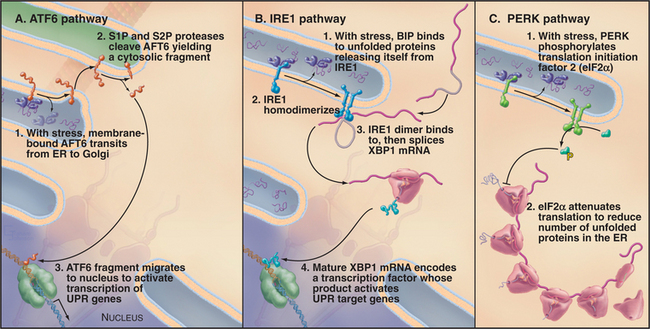
The response to ER stress (i.e., UPR) is regulated by three key ER transmembrane proteins: IRE1 (inositol requiring 1), PERK/PEK (PKR-like endoplasmic reticulum kinase/pancreatic eIF2a kinase), and ATF6 (activating transcription factor 6). These proteins serve as stress sensors to regulate the production of bZIP (basic leucine zipper) domain–containing transcription factors (see Fig. 15-17) that upregulate genes involved in ER function. This allows cells to adjust the capacity of the ER to promote ER folding depending on the demand. Whereas only IRE1 is present in yeast, all three transmembrane proteins are present and function in metazoan cells.
Accumulation of unfolded proteins in the ER lumen of metazoan cells results in activation of ATF6, IRE1, and PERK, each by a different mechanism (Fig. 20-11). Under normal conditions, free BiP is thought to inhibit the UPR pathway. BiP binds to the lumenal domains of IRE1 and PERK, preventing their dimerization. BiP also associates with ATF6, retaining it in the ER. When unfolded proteins accumulate during ER stress, BiP binds to them rather than to IRE1, ATF6, and PERK.
Aspects of the UPR pathway involving IRE1 and PERK are also important for promoting differentiation in higher eukaryotic cells. For example, IRE1 is activated during β-lymphocyte differentiation into a plasma cell (see Fig. 28-9), in which the ER expands 5-fold to accommodate immunoglobulin synthesis. Activation of the innate immune response (i.e., the inflammatory response), for example by lipopolysaccharide (LPS) treatment, also activates IRE1. Furthermore, PERK activity is required for β-cell differentiation and/or survival. These findings have led to the view that the UPR allows cells to respond to ER stress, to provide a way to sense nutrients and to promote differentiation.
Endoplasmic Reticulum Folding Diseases
Given the central role of the ER in the synthesis of proteins for the entire exocytic and endocytic pathways, it is not surprising that many inherited diseases are a direct consequence of proteins failing to pass ER quality control. Many metabolic disorders, including some lysosomal storage diseases (see Appendix 23-1), are a direct consequence of key enzymes failing to be exported from the ER. The most common form of cystic fibrosis is due to the inability of cells to export a mutant form of the cystic fibrosis transmembrane regulator (CFTR) to the cell surface, where it would normally function as a chloride channel for the respiratory system and pancreas (see Fig. 11-4). Similarly, the inability of the liver to secrete mutated forms of a1-antitrypsin predisposes to the lung disease emphysema. Normally, a1-antitrypsin protects tissues by inhibiting extracellular proteases such as elastase, which is produced by neutrophils. Mutations prevent a1-antitrypsin folding in the ER, resulting in its degradation. The resulting deficiency in a1-antitrypsin circulating in the blood allows elastase to destroy lung tissue, leading to emphysema. In severe cases, mutant forms of the protein not only fail to be exported from the ER but also elude degradation pathways, accumulating as insoluble aggregates that induce stress responses and liver failure.
In some conditions, the ER uses the unfolded protein response to compensate, in part, for mutations in cargo proteins. In congenital hypothyroidism, mutant thyroglobulin (the precursor of thyroid hormone) is not exported efficiently from the ER. Excess protein accumulates as insoluble aggregates in the ER. Feedback pathways trigger massive proliferation of ER in an attempt to produce normal levels of circulating hormone. Similarly, in mild forms of osteogenesis imperfecta (see Chapter 32), osteoblasts assemble and secrete defective procollagen chains for bone synthesis, even though the resulting bone tissue is weak. The alternative, complete loss of procollagen by retention and degradation of the mutant procollagen in the ER would be lethal. Faulty ER quality control may also contribute to diseases of the central and peripheral nervous systems, including Alzheimer’s disease.
Lipid Biosynthesis, Metabolism, and Transport within the Endoplasmic Reticulum
The ER membrane synthesizes all the major classes of lipids or their precursors that are formed within cells. These include phosphoglycerides, cholesterol, and ceramide. ER enzymes participating in phosphoglyceride synthesis have their active sites facing the cytoplasm, the site of synthesis of most lipid precursors. Synthesis begins with the conjugation of two activated fatty acids to glycerol-3 phosphate to form phosphatidic acid, which can be dephosphorylated to produce diacylglycerol (DAG; see Fig. 26-4). Neither phosphatidic acid nor DAG is a bulk component of membranes; however, both are used in the synthesis of the four major phospholipids: phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS), and phosphatidylinositol (PI) (see Fig. 7-2). The most abundant phospholipids, PC and PE, are produced with the activated head groups, cytidine diphosphate (CDP)–choline, and CDP-ethanolamine. PI synthesis is by a distinct route using inositol and CDP-DAG produced from phosphatidic acid (see Fig. 26-7). PS synthesis (in mammalian cells) is an energy-independent exchange of polar head groups of PE. Head group exchange of phospholipids occurs primarily in the ER but may also occur in other organelles.
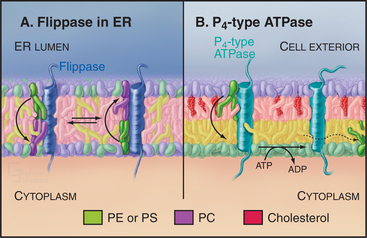
Like other types of biosynthesis carried out in the ER, phospholipid synthesis creates a topologic problem: Synthesis occurs in the cytoplasmic leaflet, restricting membrane growth to that leaflet. Phospholipids move to the luminal leaflet by individual molecules flip-flopping across the bilayer (Fig. 20-12). Flip-flopping of phospholipids across the ER bilayer is much faster than that in vesicles of pure phospholipids, owing to protein translocators called flippases. ER flippases function independently of metabolic energy and catalyze the transverse movement of most phospholipid classes in both directions promoting a symmetric lipid distribution across the ER bilayer (Fig. 20-12A). Catalysis of flip-flopping in the ER is probably not the role of one specific protein, as peptides that mimic the α-helices of transmembrane proteins can stimulate flip-flopping of phospholipids in liposomes. Cholesterol inhibits helix-induced flip-flopping. Cholesterol is a minor component of the ER but abundant in the plasma membrane. Because of this, the mechanism of transbilayer lipid movement in the plasma membrane, in contrast to that for ER membrane, involves tightly controlled translocation mechanisms, including energy-dependent inward and outward flippases (Fig. 20-12B).
Cholesterol Synthesis and Metabolism
Cholesterol is maintained in animal cells by a combination of de novo synthesis (Fig. 20-13) in the ER and receptor-mediated endocytosis of lipoprotein particles containing esterified cholesterol (see Chapter 22). Coordinated regulation of these two processes precisely maintains the physiological level of cholesterol in cellular membranes. Peroxisomes also may participate in aspects of cholesterol synthesis and metabolism.
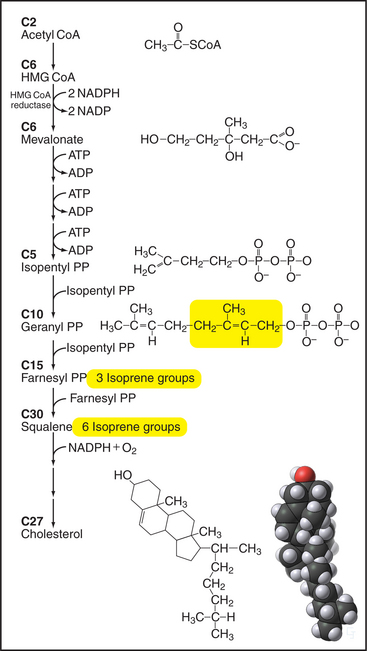
Figure 20-13 Key steps in the biosynthesis of cholesterol from acetyl CoA. Synthesis of mevalonate from 3-hydroxy-3-methylglutaryl CoA is closely regulated by controlling the concentration of the enzyme, HMG-CoA reductase, as shown in Figure 20-14. Five-carbon isoprene groups are the building blocks (yellow) for making a 10-carbon geranyl-pyrophosphate (PP), a 15-carbon farnesyl-pyrophosphate, and 30-carbon squalene intermediates. Several reactions add a hydroxyl group and join squalene into four rings to make cholesterol. ADP, adenosine diphosphate; NADP, nicotinamide adenine dinucleotide phosphate.
Enzymes in the cytoplasm and ER use 22 sequential steps to synthesize cholesterol from acetate (Fig. 20-13). Cytoplasmic enzymes catalyze the initial steps, using water-soluble molecules to produce farnesyl-pyrophosphate (farnesyl-PP) from acetyl coenzyme A (acetyl CoA). An important exception is the step going from 3-hydroxy-3methylglutaryl CoA (HMG-CoA) to mevalonate. A carefully regulated, integral membrane protein of the ER (Fig. 20-13), HMG-CoA reductase, catalyzes this key step. Enzymes in the ER bilayer catalyze the subsequent condensation of farnesyl-PP to make squalene, and cyclization to cholesterol, progressively less-polar molecules. Although the final steps leading to cholesterol take place in the ER, cholesterol is not a resident ER lipid and is rapidly exported to post-ER membranes, including the plasma membrane, where it constitutes up to 50% of the bilayer. Chapter 21 discusses the distribution of cholesterol in different organelles and current ideas regarding its transport.
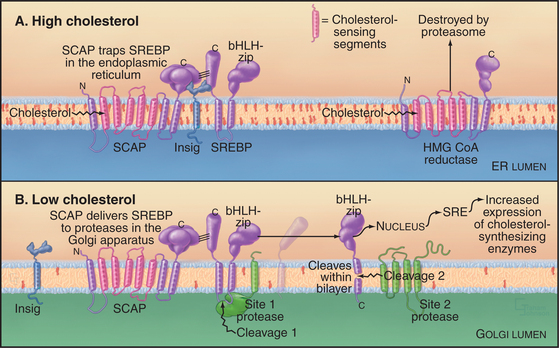
Feedback loops sense the cholesterol content of the ER membrane and regulate both the synthesis and degradation of enzymes that synthesize cholesterol (Fig. 20-14). High cholesterol levels inhibit the synthesis and stimulate the destruction of key synthetic enzymes. A novel transcription factor precursor, steroid regulator element-binding proteins (SREBP), controls the expression of these genes. When cholesterol is abundant, SREBP cleavage-activating protein (SCAP), a partner protein with cholesterol-sensing transmembrane segments, retains SREBP in the ER owing to interactions with the protein Insig. When the membrane cholesterol level is low, SCAP and Insig proteins do not interact. The SREBP-SCAP complex is then free to move to the Golgi apparatus, where two successive proteolytic cleavages release the N-terminal domain of SREBP, a basic helix-loop-helix leucine zipper transcription factor, into the cytoplasm. (The Golgi proteases that are responsible for cleaving SREBP are SP1 and SP2, the same ones used to process ATF6 during UPR.) In the nucleus, the transcription factor binds steroid regulatory elements, enhancers for a wide range of genes encoding enzymes that synthesize cholesterol and other lipids, as well as low-density lipoprotein receptors that take up cholesterol from the medium (see Fig. 23-9). Cholesterol also regulates degradation of HMG-CoA reductase, which has cholesterol-sensing transmembrane domains similar to SCAP. Abundant cholesterol targets HMG-CoA reductase for degradation by the proteolytic pathway that disposes of unfolded proteins through the proteasome (Fig. 20-10 and Chapter 23). The enzyme acyl-CoA-cholesterol transferase (ACAT) helps to lower cholesterol levels in the ER bilayer by catalyzing the formation of cholesterol esters, a storage form of cholesterol.
Ceramide Synthesis
Ceramide, the backbone of all sphingolipids (see Fig. 7-3), also begins its synthesis on the cytoplasmic face of ER membranes. It is made through sequential condensation of the amino acid serine with two fatty acids. Ceramide is transported to the Golgi apparatus by a nonvesicular pathway (see later), where enzymes on the lumenal leaflet either add oligosaccharide chains to it to form glycosphingolipids or add a choline head group to form sphingomyelin (see Chapter 21).
Lipid Movement between Organelles
While lipids can rapidly diffuse along one side of the bilayer as well as flip-flop across the bilayer, the hydrophobic nature of lipids makes free diffusion between adjacent bilayers thermodynamically unfavorable. Thus, lipids use two other mechanisms to move between organelles (Fig. 20-15). The major pathway is movement through the use of vesicle carriers (see Chapter 21). In addition, small, soluble phospholipid exchange proteins promote movements between membranes by a nonvesicular mechanism. These proteins—called lipid-transfer proteins (LTPs)—have a common overall structure and are specific for particular phospholipids. One example of an LTP is ceramide transport protein (CERT), which mediates ceramide transport to the Golgi apparatus by extracting newly synthesized ceramide from the ER and carrying it to the Golgi apparatus. To accomplish this, CERT has a domain that recog-nizes ceramide and mediates its intermembrane transfer. CERT also has a motif for targeting to the ER and a PH domain (see Fig. 25-11) for binding polyphosphoinositides in the Golgi apparatus.
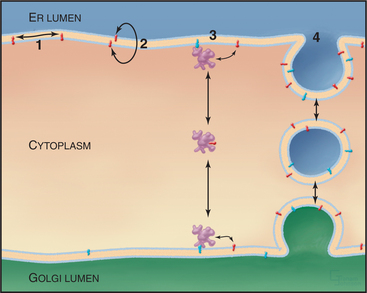
LTPs catalyze lipid exchange but not net transfer. When the protein delivers a lipid to a target membrane, the protein exchanges that lipid for another one and returns with the second lipid (Fig. 20-15). The lipid-binding pocket of LTPs is largely lined with hydrophobic residues that stabilize the internal lipid. In the open conformation, a hinged cover projects outward away from the protein, allowing the LTP to embed itself partially into the cytosolic leaflet of the bilayer. Interactions with other membrane components (either lipids or proteins) dictate what compartments the LTP targets to. Zones of close apposition between the ER and other organelles—such as mitochondria, chloroplasts, lipid droplets, TGN, endosomes, and lysosomes—are enriched in certain types of LTPs, suggesting that lipid trafficking occurs across these sites. Some proteins with lipid transfer domains might function as lipid sensors rather than as lipid carriers.
Borgese N, Colombo S, Pedrazzini E. The tale of tail-anchored proteins: Coming from the cytosol and looking for a membrane. J Cell Biol. 2003;161:1013-1019.
Choy E, Chiu VK, Silletti J, et al. Endomembrane trafficking of Ras: The CAAX motif targets proteins to the ER and Golgi. Cell. 1999;98:69-80.
Clemons WM, Menetret J-F, Akey CW, Rapoport TA. Structural insight into the protein translocation channel. Curr Opin Struct Biol. 2004;14:390-396.
Egea PF, Stroud RM, Walter P. Targeting proteins to membranes: Structure of the signal recognition particle. Curr Opin Struct Biol. 2005;15:213-220.
Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem. 2004;73:1019-1049.
Holthuis JCM, Levine TP. Lipid traffic: Floppy drives and a superhighway. Nat Rev Mol Cell Biol. 2005;6:209-220.
Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD, the long road to destruction. Nat Cell Biol. 2005;7:766-772.
Osborne AR, Rapoport TA, van den Berg B. Protein translocation by the Sec61/SecY channel. Annu Rev Cell Dev Biol. 2005;21:529-550.
Rawson RB. The SREBP pathway: Insights from insigs and insects. Mol Cell Biol. 2003;4:631-640.
Schröder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;24:739-789.
Sitia R, Braakman I. Quality control in the endoplasmic reticulum protein factory. Nature. 2003;426:891-894.
Sprong H, van der Sluijs P, van Meer G. How proteins move lipids and lipids move proteins. Nat Rev. 2001;2:804-813.
* This chapter was revised using material from the first edition written by William E. Balch, Ann L. Hubbard, J. David Castle, and Pat Shipman
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
CHAPTER 20 Endoplasmic Reticulum*
One of the key distinguishing features of eukaryotic cells is the presence of the endoplasmic reticulum (ER), the largest of numerous membrane-delineated intracellular compartments. The ER is thought to have evolved from the prokaryotic plasma membrane by expansion, internalization, and subdivision (Fig. 20-1A-D; see also Fig. 2-6). It provides both an expanded membrane surface (up to 30 times that of the plasma membrane) for carrying out vital cellular functions, including protein and lipid biosynthesis, and an internal compartment (or lumen) that collects proteins synthesized in the cytoplasm for modification and delivery into the secretory pathway. About one third of all cellular proteins are imported into the lumen of the ER or integrated into its membranes. Consisting of an extensive array of tubules or flat saccules called cisternae (cisterna means “reservoir”), the ER forms a continuous three-dimensional network (a reticulum) stretching from the nuclear envelope to the cell surface (Fig. 20-1E). Microtubules and their associated motors generate this extended network in animal cells by pulling ER membranes out toward the periphery of the cell. This allows the ER to coordinate diverse processes over large regions of the cytoplasm. The ER’s size and shape are maintained over time despite a continuous flow of proteins and lipids into and out of this compartment. The flow results from the large rate of synthesis of lipids and proteins (between 2 million and 13 million new proteins per minute) occurring at the ER membrane, as well as from the continuous export of these molecules into the secretory pathway and their selective retrieval back to the ER from the Golgi apparatus.
Endoplasmic Reticulum Functions and Organization
The membrane surface of the ER performs several functions for cells. Foremost is the production of the proteins and lipids that will make up the membranes of the other organelles, including the Golgi apparatus, nucleus, endosomes, lysosomes, and plasma membrane, as well as nearly all proteins that will be secreted from the cell. The membranes of mitochondria and peroxisomes also depend on the ER to supply much of their lipid. Another key function of the ER membrane is to form the nuclear envelope, which encloses the nucleus (see Fig. 14-5). The surface of the ER forming the outer nuclear envelope (which faces the cytoplasm) is indistinguishable from the rest of the ER except for the presence of nuclear pores that span both inner and outer nuclear envelope to allow passage of molecules between the nucleus and cytoplasm. By contrast, the ER surface forming the inner nuclear envelope, which faces the nucleus, contains specialized proteins that interact with the nuclear lamina and chromatin. ER membranes also detoxify endogenous steroids, carcinogenic compounds, and lipid-soluble drugs (xenobiotics) from the environment. This occurs by an electron transfer process carried out by ER membrane proteins such as the cytochrome P450 family of enzymes.
The lumen of the ER also performs numerous essential functions. Specialized for receiving proteins transported from the cytoplasm across ER membranes, the ER lumen is enriched in a dense meshwork of chaperones and other modifying enzymes (estimated to be 200 mg/mL in concentration) that catalyze the folding and assembly of newly synthesized proteins. These transported proteins, including both soluble and transmembrane forms, are exported from the ER for secretion or for delivery to the lumen or membrane of the Golgi apparatus, lysosome, or endosome. Proteins that are incorrectly folded or misfolded can be exported back into the cytoplasm, where they are degraded. Misfolded proteins, when accumulated in the ER at high levels, can trigger an unfolded protein response, which activates specific genes in the nucleus whose products help to modify or destroy the misfolded proteins and compensate for the decreased capacity of ER folding. The ER lumen is one of the major Ca2+ storage sites in cells, owing to ER membranes being rich in calcium pumps (see Fig. 8-7) and many Ca2+-binding proteins in the lumen. Such Ca2+ stores can be released by calcium channels in response to cellular signals such as IP3 (see Fig. 26-13). Carefully regulated release and uptake of Ca2+ by the ER control muscle contraction (see Fig. 39-15) and many other cellular processes. The lumen of the ER is also an oxidizing environment that favors disulfide bond formation, which helps to stabilize proteins after they are exported from the ER to the outside of the cell.
The diverse functions carried out by the membrane and in the lumen of the ER (Table 20-1) occur in distinctive ER regions (Fig. 20-2A). The rough ER is studded with ribosomes on its cytoplasmic surface, defining areas that are specialized for protein synthesis, folding, and degradation (Fig. 20-2B). The smooth ER, composed of tubular elements lacking ribosomes, is dedicated to enzyme pathways involved in drug metabolism (hepatocytes), steroid synthesis (endocrine cells), or calcium uptake and release (see Fig. 26-12). The cytochrome P-450 family of heme-containing membrane proteins is found in the smooth ER. Other regions of the ER that lack ribosomes, called ER export domains consist of tubulovesicular membranes that bud during export of secretory cargo to the Golgi apparatus (see Chapter 21). Regions of the ER surrounding the nucleus compose the nuclear envelope.
| ER Domain | Function | Associated Proteins |
|---|---|---|
| Rough ER | Protein translocation | Sec61 complex, TRAP, TRAM, BiP |
| Protein folding and oligomerization | PDI, Calnexin, Calreticulin, BiP | |
| Carbohydrate addition | Oligosaccharide transferase | |
| ER degradation | EDEM, Derlin1 | |
| Smooth ER | Detoxification | Cytochrome P450 enzymes |
| Lipid metabolism | HMG-CoA reductase | |
| Heme metabolism | Cytochrome b(5) | |
| Calcium release | IP3 receptors | |
| Nuclear envelope | Nuclear pores | POM121, GP210 (see Fig. 14-8) |
| Chromatin anchoring | Lamin B receptor | |
| ER export sites | Export of proteins and lipids into secretory pathway | Sar1p, Sec12p, Sec16p |
| ER contact zones | Transport of lipids | LTPs |
Figure 20-2 endoplasmic reticulum subdomain organization within the cell. A, ER membranes containing ribosomes, called rough ER (rER), extend from the nuclear envelope to the cell periphery. ER membranes lacking ribosomes, called smooth ER (sER), include membranes specialized for drug metabolism and steroid synthesis, as well as tubulovesicular elements composing ER exit sites. The nuclear envelope consists of ER membrane that has wrapped around DNA and other nuclear elements to compartmentalize them. Its double membrane is studded with nuclear pores, which provide a passageway for nuclear components to move between the nucleus and cytosol. (See Fig. 14-17 for more details.) B, Electron micrograph of a thin section of rough ER and neighboring mitochondrion from the pancreas.
(Micrograph by Keith R. Porter; courtesy of Don W. Fawcett, Harvard Medical School, Boston, Massachusetts.)
Overview of Protein Translocation into the Endoplasmic Reticulum
All proteins are synthesized in the cytoplasm and must be specifically targeted to the ER, where they are either fully translocated across the ER membrane and released into the ER lumen (soluble proteins) or only partly translocated across the ER membrane and embedded in the lipid bilayer of the ER membrane (transmembrane proteins). This transport to the ER can happen either as the protein is being made (cotranslational translocation) or after synthesis is complete (posttranslational translocation). The orientation of a protein in the lipid bilayer or its localization to the lumen is established during protein translocation and maintained as the protein is transferred by membrane-bound carriers through the secretory pathway (see Fig. 21-2). Thus, domains of transmembrane proteins to be exposed on the cell surface must be inserted into the ER membrane, facing the lumen. Similarly, secreted soluble proteins must be fully translocated into the lumen of the ER.
Signal Sequence Recognition
Soluble and membrane proteins destined for ER translocation contain a hydrophobic sequence either at their N-terminus or in transmembrane segments that serve as recognition signals for direction to the ER membrane. N-terminal leader sequences (termed signal sequences) are typically 15 to 35 amino acids long and contain a hydrophobic core of at least 6 residues, while transmembrane signal segments have a hydrophobic stretch of 16 to 25 residues. Aside from hydrophobicity, these signal sequences have no other features in common (Table 20-2). Nevertheless, when attached to proteins that are not normally targeted to the ER, these signal sequences direct the protein to the ER and not to other organelles such as to mitochondria or peroxisomes, which use unique targeting signals (see Fig. 18-1).
Cotranslational Translocation
In the cotranslational pathway, the signal recognition particle (SRP) recognizes and binds the first hydrophobic domain of either the signal sequence or the transmembrane signal segment as it emerges from the ribosome slowing translation of the polypeptide (Fig. 20-3A). Then the complex consisting of ribosome, nascent chain, and SRP associates with the ER membrane by binding the SRP receptor (SR), a heterodimer consisting of one subunit that binds SRP and another that spans the ER membrane. Both the SRP and the SR have guanosine triphosphatase (GTPase) domains (Fig. 20-3B) similar to Ras (see Fig. 4-6). GTP binding and hydrolysis by SRP and SR provide directionality and order to the sequence of reactions that bring the nascent chain to the translocation channel. Once the ribosome–nascent chain–SRP complex is bound to SR at the ER membrane, SRP releases the signal sequence, allowing the ribosome–nascent chain complex to be transferred to the protein-conducting channel across the ER membrane. SRP and SR then dissociate after hydrolyzing their bound guanosine triphosphate (GTP), releasing SRP into the cytoplasm and allowing SR to diffuse away in the membrane. The targeting cycle delivers the ribosome–nascent chain complex to the protein-conducting channel (called the translocon) and recycles the targeting machinery (i.e., SRP and SR).
Posttranslational Translocation
Posttranslational translocation (prevalent in fungi such as yeast) differs because neither cotranslational targeting machinery (SRP and SR) nor ribosomes participate. Instead, other components fulfill the functions provided by SRP, SR, and ribosomes (with the Sec61 complex serving as channel, as it does in cotranslational translocation). Signal-containing polypeptides destined for posttranslational insertion into the ER are held in a largely unfolded state by cytoplasmic chaperones until they can be delivered to the tetrameric Sec62/63 protein complex at the ER membrane (Fig. 20-4). A similar strategy is used to import proteins into the mitochondria (see Fig. 18-4). During posttranslational translocation, the signal sequence engages and opens the Sec61 channel in a fashion similar to cotranslational translocation. Since protein synthesis is already complete, another energy source must be exploited to move the substrate through the channel into the ER. BiP, a luminal ER chaperone belonging to the Hsp70 family, binds the substrate in the ER lumen, thereby preventing it from sliding back into the cytoplasm. Repeated rounds of substrate binding and release, catalyzed by ATP hydrolysis, allow BiP to act as a molecular ratchet to drive substrate transport into the lumen. The transmembrane protein Sec63 regulates the ATPase activity of BiP and helps to recruit BiP to the translocation channel. Thus, the peptide is “pulled” across the membrane from the luminal side instead of being “pushed” from the cytoplasmic side, as during cotranslational translocation. Hsp70 family members perform a similar function during import of proteins into mitochondria and chloroplasts (see Fig. 18-4).
Universality of Protein Translocation
In both cotranslational and posttranslational translocation, the Sec61 complex translocation channel is closed until opened by interacting with binding partners, including the translocated polypeptide. This ensures that only specific types of proteins pass through the ER membrane and that the permeability barrier of these membranes is maintained at all times. In cotranslational translocation, interactions with a ribosome and a signal sequence open the channel, and protein synthesis provides the energy for translocation. In posttranslational translocation, the Sec62/63 complex operates on both sides of the membrane. On the cytoplasmic side, it contributes to the interactions of the signal sequence that open the translocon. On the lumenal side, it recruits BiP to provide the driving force for translocation. This theme applies to protein translocation across the plasma membrane by prokaryotes. There, a homolog of the Sec61 complex (the SecYE complex) interacts with the cytoplasmic SecA adenosine triphosphatase (ATPase), a partner that drives translocation across the plasma membrane (see Fig. 18-9).
Molecular Machinery for Protein Translocation into the Endoplasmic Reticulum
Signal Recognition Particle and Signal Recognition Particle–Receptor
Human SRP is a ribonucleoprotein composed of six proteins (named by their apparent molecular weights and a 300-nucleotide RNA (Fig. 20-5). The SRP54 protein subunit and a portion of the RNA compose the minimal hardware for targeting to the translocon and are found in both prokaryotic and eukaryotic cells. SRP54 binds signal sequences in a deep, hydrophobic groove lined by the flexible side chains of several methionines. Like bristles of a brush, the methionines accommodate the various shapes of the hydrophobic side chains of the residues of different signal sequences. Phosphates of the SRP RNA near one end of this groove may interact with basic residues that are often (but not always) adjacent to the hydrophobic core of signal sequences and transmembrane signal segments.
SRP binding to a signal sequence slows translation, a phenomenon that is termed elongation arrest (Fig. 20-3A). The mechanism appears to involve occlusion of the elongation factor binding site on the ribosome by the SRP9 and SRP14 subunits of SRP, which structurally resembles a portion of eEF2. Slowing translation provides time to target the ribosome to the translocation channel before excessive polypeptide synthesis precludes cotranslational transport.
Interaction of SRP with SR directs the SRP–ribosome–nascent chain complex to the translocation channel (Fig. 20-3A). This interaction is regulated by GTP binding and hydrolysis by SRP and SR, whose GTPase cycles are a notable exception to the GTPase switch paradigm of Ras-like GTPases that involve GTP exchange factors (GEFs) and GTPase-activating proteins (GAPs; see Fig. 25-8). No external GEFs or GAPs are known for SRP and SR GTPases. Instead, these proteins readily exchange GDP for GTP and are in the GTP-bound state as they enter the targeting cycle. On formation of a complex, SRP and SR reciprocally activate each other’s GTPase activity, thereby obviating the need for an external GAP to drive the conversion of GTP to GDP on these proteins. Upon GTP hydrolysis, the conformations of SR and SRP change in a way that reduces their affinity. They dissociate from each other as well as from the ribosome–nascent chain and translocation channel. This frees SRP and SR for further rounds of ribosome–nascent chain targeting. Release of SRP and SR occurs only after the ribosome has become properly engaged with the translocation channel in the ER membrane, thereby ensuring that the channel is closed until the ribosome has bound.
[/not-level-membership-for-basic-science-category]
