[level-membership-for-neonatal-and-perinatal-medicine-category]Chapter 8
Endocrinology and Metabolism
Hypocalcemia
1. What perinatal factors are associated with hypocalcemia in the immediate newborn period?






2. How are calcium levels expected to change in premature infants during the first few days of life?
In newborn infants there is a physiologic decline in serum total and ionized calcium during the first 48 hours of life. This decline is exaggerated in preterm infants compared with term infants, with a direct correlation between serum calcium and gestational age ( Fig. 8-1). Because no symptoms are specific for early hypocalcemia in preterm infants, the diagnosis is made by demonstrating a serum calcium level below 7 mg/dL (1.75 mmol/L).
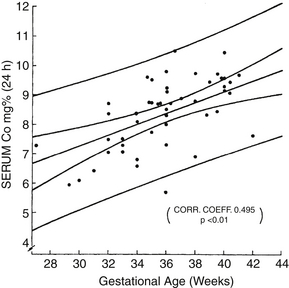
Figure 8-1 Serum calcium in relation to gestational age at 24 hours of age. (From Tsang RC, Light IJ, Sutherland JM, et al. Possible pathogenetic factors in neonatal hypocalcemia of prematurity. J Pediatr 1973;82:423–9.)
 Hypocalcemia of prematurity is usually asymptomatic.
Hypocalcemia of prematurity is usually asymptomatic.

 Long-term follow-up studies have shown no benefit with treatment.
Long-term follow-up studies have shown no benefit with treatment.
 Total serum calcium level is a poor predictor of ionized serum calcium in premature infants.
Total serum calcium level is a poor predictor of ionized serum calcium in premature infants.
 Intravenous (IV) calcium is associated with complications such as cardiac arrhythmias and ulcerations as a result of soft-tissue infiltration of the infusate. ∗†
Intravenous (IV) calcium is associated with complications such as cardiac arrhythmias and ulcerations as a result of soft-tissue infiltration of the infusate. ∗†
 KEY POINTS: HYPOCALCEMIA
KEY POINTS: HYPOCALCEMIA
1. Neonatal hypocalcemia is associated with prematurity, asphyxia, maternal diabetes, transient hypoparathyroidism, permanent congenital hypothyroidism, and (rarely) maternal hyperparathyroidism.
2. The most common reason for hypocalcemia in the newborn period is prematurity.
3. Breast milk rickets is seen in premature infants because of the relatively low mineral (e.g., calcium and phosphorus) content of breast milk.
4. Serum calcium levels are frequently elevated in patients with Williams syndrome.
5. Normal magnesium levels are needed for optimal functioning of the parathyroid glands.
Recent studies in premature infants using stable isotopes of calcium showed a true calcium absorption rate of 50% to 90%. Thus to meet an accretion rate of 100 mg/kg/day with an absorption rate of 75% and an assumed retention rate of 75% (which may be on the high side), oral intake of calcium for growing premature infants should be about 200 mg/kg/day. This large intake in infants with very low birth weight can be achieved only with special formulas for low-birth-weight infants or mineral fortifiers for breast milk–fed preterm infants. ∗
Hypercalcemia



 23–27 weeks: 10.0 +/−1.0 (8.1–11.9)
23–27 weeks: 10.0 +/−1.0 (8.1–11.9)
 28–31 weeks: 10.2 +/−1.2 (8–12.5)
28–31 weeks: 10.2 +/−1.2 (8–12.5)
 32–34 weeks: 10.5 +/−1 (8.6–12.4)
32–34 weeks: 10.5 +/−1 (8.6–12.4)








Total serum calcium above 10.8 mg/dL or ionized serum calcium above 5.4 mg/dL.
15. What are some of the causes of hypercalcemia in neonates?


 Idiopathic infantile hypercalcemia
Idiopathic infantile hypercalcemia

 Hyperparathyroidism (primary and secondary)
Hyperparathyroidism (primary and secondary)













17. A 3-day-old infant born small for gestational age at term has a total serum calcium level of 13.2 mg/dL. She was delivered by emergency cesarean section and was diagnosed with supravalvular aortic stenosis. What is the likely diagnosis?
Williams syndrome is the likely diagnosis in an infant with hypercalcemia and supravalvular stenosis who was born small for gestational age. It results from a deletion of the elastin gene on 7q11.23. Affected infants are often described as having “elfin” faces. ∗
18. A term infant is incidentally noted to have a calcium level of 12.2 mg/dL at 4 days of age. Family history reveals that the father has also been evaluated for elevated calcium levels. What would you expect to find on measurement of the infant’s urinary calcium level? What is the most likely diagnosis? What is the appropriate therapy?
The most likely diagnosis is an autosomal dominant mutation of the calcium-sensing receptor, or “hypocalciuric hypercalcemia.” The infant’s urinary calcium level will be inappropriately low for the serum calcium. In the heterozygous state this is generally thought to be a benign condition, and treatment is not indicated. Rare cases of homozygous mutations result in severe neonatal hyperparathyroidism, which is a life-threatening disorder. ∗
Hypomagnesemia and Hypermagnesemia
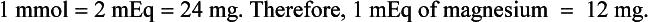
Intracellular and extracellular types of magnesium reactions are important.
24. What causes magnesium depletion in neonates?


 Renal losses of magnesium in acidotic states
Renal losses of magnesium in acidotic states
 Use of nutrient solutions containing insufficient amounts of magnesium
Use of nutrient solutions containing insufficient amounts of magnesium

 Intestinal wasting of magnesium (rare X-linked condition)
Intestinal wasting of magnesium (rare X-linked condition)
 Gastrointestinal losses (through emesis, nasogastric suctioning, and diarrhea)
Gastrointestinal losses (through emesis, nasogastric suctioning, and diarrhea)
 Prematurity, which increases the risk for magnesium deficiency
Prematurity, which increases the risk for magnesium deficiency

Most infants are asymptomatic. On rare occasions the following signs and symptoms may be seen:
 Color: pallor, cyanosis, or duskiness
Color: pallor, cyanosis, or duskiness
 Affect: out of touch with surroundings, apathetic, irritable when disturbed, restless
Affect: out of touch with surroundings, apathetic, irritable when disturbed, restless
 Eyes: staring with infrequent blinking, oculogyric crises
Eyes: staring with infrequent blinking, oculogyric crises
 Heart: tachycardia (bradycardia during apneic episodes)
Heart: tachycardia (bradycardia during apneic episodes)


Hypomagnesemia usually increases the secretion of PTH, thereby increasing calcium levels. In chronic magnesium-deficient states, however, secretion of PTH is reduced. In such circumstances hypomagnesemia may induce hypocalcemia. ∗
27. What causes hypermagnesemia in neonates?











In extreme cases cardiorespiratory function ceases, and death ensues.





Thyroid Disorders
Congenital hypothyroidism occurs in 1 in 4000 liveborn infants.
32. What are the embryonic stages of development of the fetal hypothalamic–pituitary–thyroid axis?
 Thyroid tissue is first identified at the base of the tongue 16 to 17 days after conception.
Thyroid tissue is first identified at the base of the tongue 16 to 17 days after conception.

 By 10 weeks’ gestation the fetal thyroid gland is trapping iodine and synthesizing thyroxine (T4).
By 10 weeks’ gestation the fetal thyroid gland is trapping iodine and synthesizing thyroxine (T4).

 By 10 to 12 weeks’ gestation, the fetal pituitary gland is synthesizing thyroid-stimulating hormone (TSH). ∗
By 10 to 12 weeks’ gestation, the fetal pituitary gland is synthesizing thyroid-stimulating hormone (TSH). ∗
The hypothalamic–pituitary–thyroid axis is in place by the end of the first trimester. The thyroid and pituitary glands reach mature secretory capacity by 30 to 35 weeks of gestation. The feedback interrelationship among the units is fully established when hypothalamic TRH maturation is completed by 1 to 2 months after birth. ∗
The amount of T4 secreted by the fetal thyroid gland increases slowly until midgestation (20 to 24 weeks) when, stimulated by increasing amounts of TSH from the fetal pituitary, T4 levels begin to increase more rapidly, reaching a normal adult level by approximately 30 weeks’ gestation. Thereafter T4 increases slowly to high normal levels at term gestation. ∗
The placenta is a barrier to the passages of thyroid hormones and contains enzymes that break down maternal T4 and T3 into inactive metabolites. Only a small percentage of circulating maternal T4 and very little (if any) T3 reaches the fetus. However, the amount of maternal T4 that does cross the placenta is significant. During the first 10 to 12 weeks of gestation, all of the circulating T4 in the fetus is from maternal sources; thus early brain development depends on maternal hormone. Even after the fetus synthesizes its own T4 in the second and third trimesters, maternal T4 is essential for normal neurologic development, including neuronal proliferation and maturation, dendritic arborization, and synapse formation. It accounts for approximately 30% of fetal T4 levels at term. ∗†
The levels of TRH, TSH, T4, free T4, and T3 are lower in premature infants than in term infants, and the postnatal surges of TSH and T4, although qualitatively similar, are blunted. These differences are related directly to gestational age: the lower the gestational age, the lower the levels and responses of thyroid-related hormones ( Table 8-1).
TABLE 8-1
SERUM THYROXINE (µg/dL) AT DIFFERENT GESTATIONAL AGES ∗
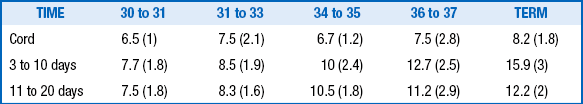
Adapted from Cuestas RA. Thyroid function in healthy premature infants. J Pediatr 1978;92:963–7.
The term refers to infants with low birth weight (30 to 35 weeks’ gestation) or very low birth weight (<30 weeks’ gestation), who have an even more attenuated rise in T4, after which T4 levels drop below cord levels in the first week of life. Then they rise gradually over 3 to 6 weeks to approach levels of term infants ( Table 8-2).
TABLE 8-2
THYROID FUNCTION IN PRETERM AND TERM INFANTS ∗
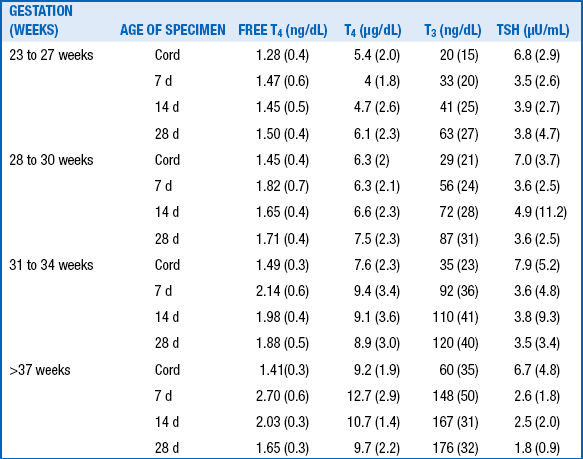
T4, Thyroxine; T3, triiodothyronine; TSH, thyroid-stimulating hormone; free T4, free thyroxine.
http://www.uptodate.com/contents/image?imageKey=PEDS%2F72215&topicKey=PEDS%2F5840&rank=4~150&source=see_link&search=thyroid+function&utdPopup=true
Adapted from Williams FL, Simpson J, Delahunty C, et al. Developmental trends in cord and postpartum serum thyroid hormones in preterm infants. J Clin Endocrinol Metab 2004;89:5314.
The premature infant with low T4 and persistently elevated TSH has either transient or permanent hypothyroidism and should be treated with T4 until the nature of the condition becomes clear. However, whether premature infants with low T4 and normal TSH levels should be treated remains controversial. ∗†
43. Does breastfeeding provide needed T4 to premature infants with an immature hypothalamic–pituitary–thyroid axis?
This question has not yet been answered. There are some case reports in the literature suggesting that breastfeeding delays the onset of hypothyroidism, but others argue against that finding.
A heel-stick blood sample is taken at discharge or 3 days of life, whichever is earlier. In most parts of the United States, T4 is measured first, then TSH is measured in samples with the lowest 10% to 29% of T4 results. ∗†
TABLE 8-3
CAUSES OF CONGENITAL HYPOTHYROIDISM AND INCIDENCE OF EACH

From Fisher FA. Disorders of the thyroid in the newborn and infant. In: Sperling MA, editor. Pediatric endocrinology. Philadelphia: Saunders; 1996. p. 57.
Only approximately 1 in 70 neonates born to thyrotoxic mothers exhibit clinical thyrotoxicosis. Such infants may show a phase of transient hypothyroidism caused by antithyroid drugs (half-life, 2 to 3 days), then thyrotoxicosis resulting from maternal TSIs. Transient congenital hypothyroidism can result from transplacental transfer of maternal thyrotopin-blocking antibodies. ∗
48. How may treatment of maternal Graves disease affect the fetus and neonate?
 Antithyroid drugs (e.g., propylthiouracil [PTU], methimazole) cross the placenta and may block the fetal thyroid, leading to fetal hypothyroidism.
Antithyroid drugs (e.g., propylthiouracil [PTU], methimazole) cross the placenta and may block the fetal thyroid, leading to fetal hypothyroidism.
 Radioactive iodine crosses the placenta and ablates the fetal thyroid.
Radioactive iodine crosses the placenta and ablates the fetal thyroid.
 Beta-adrenergic agents (e.g., propranolol) cross the placenta and have been associated with intrauterine growth retardation, bradycardia, respiratory distress, and hypoglycemia. ∗
Beta-adrenergic agents (e.g., propranolol) cross the placenta and have been associated with intrauterine growth retardation, bradycardia, respiratory distress, and hypoglycemia. ∗







 KEY POINTS: THYROID-RELATED DISORDERS
KEY POINTS: THYROID-RELATED DISORDERS
1. Hypothyroxinemia of prematurity is associated with a developmental immaturity of the hypothalamic–pituitary–thyroid axis and therefore should never be used as an explanation for low thyroid levels in the presence of an elevated TSH level.
2. Hypopituitarism in a neonate most often presents with hypoglycemia and may also cause hyponatremia, jaundice, micropenis, and undescended testes.
3. The most common cause of virilized external genitalia in a 46,XX infant is 21-hydroxylase deficiency.
4. The fetal hypothalamic–pituitary–thyroid axis is in place by the end of the first trimester but is not fully mature until 2 months postpartum (term).
5. Maternal T4 does cross the placenta and is essential for the normal neurologic development of the fetus.
6. TSH and T4 levels rise in both premature and term infants immediately after birth; however, the rise in premature infants is blunted.
7. Infants with neonatal thyrotoxicosis are at an increased risk for congestive heart failure and learning disorders.
Note: Premature infants are also unable to escape from the inhibitory effect of iodine and may become hypothyroid when subjected to multiple povidone-iodine washings or iodinated contrast agents, associated with an elevation of TSH. This is particularly important in infants who have required repeated procedures. ∗
Adrenal Disorders
53. Which disorders of adrenal steroidogenesis should be suspected as a possible cause for virilization of a 46,XX fetus?
 21-hydroxylase deficiency: This disorder results in virilization in females, salt wasting (aldosterone deficient, 75%), and signs of cortisol deficiency (e.g., hypoglycemia, shock).
21-hydroxylase deficiency: This disorder results in virilization in females, salt wasting (aldosterone deficient, 75%), and signs of cortisol deficiency (e.g., hypoglycemia, shock).

54. Which disorders of adrenal steroidogenesis should be suspected as a possible cause for undervirilization of a 46,XY fetus?
 Steroidogenic acute regulatory (StAR) protein deficiency (i.e., congenital adrenal lipoid hyperplasia) leads to salt wasting and ambiguous genitalia in males.
Steroidogenic acute regulatory (StAR) protein deficiency (i.e., congenital adrenal lipoid hyperplasia) leads to salt wasting and ambiguous genitalia in males.

 17-β-hydroxylase deficiency results in hypertension and ambiguous genitalia in males.
17-β-hydroxylase deficiency results in hypertension and ambiguous genitalia in males.
55. In infants with congenital adrenal hyperplasia (CAH) caused by 21-hydroxylase deficiency, which of the following is abnormal: (1) genetic sex, (2) gonadal differentiation, (3) internal genital formation and structure, or (4) external genitalia in females?
The answer is (4). In female infants with CAH, the karyotype (genetic sex) is normal (46XX). The müllerian ducts develop normally into a uterus and fallopian tubes without secretion of antimüllerian hormone. No wolffian duct derivatives are formed because no fetal testis is present. The elevated adrenal androgen levels cause virilization of the external genitalia. ∗
56. List the sources of maternal androgens that cause masculinization.


57. A male fetus is exposed to maternal progestin at 10 weeks of gestation. What is the possible manifestation?
Exposure of male fetuses to progestin at 8 to 14 weeks of gestation may result in hypospadias. ∗
61. A pregnant woman has a low urinary estriol level. At delivery, her male infant develops hyponatremia, hyperkalemia, and hypoglycemia. What diagnosis should you consider?
Congenital adrenal hypoplasia is an X-linked disorder affecting 1 in 12,500 live births. ∗



StAR protein is necessary for proper reduction of aldosterone, cortisone, and sex hormones. Its absence leads to feminization of males as part of congenital lipoid adrenal hyperplasia. In a subset of patients with congenital lipoid adrenal hyperplasia, mutations in StAR protein result in severe impairment of steroid biosynthesis in the adrenal glands and gonads. ∗
Collect the blood specimen at any time. Circadian rhythms do not affect the level of cortisol in very premature infants. Infants with extremely low birth weight may have quite low cortisol levels (9.2 ± 9.8 μg/dL) and lack the typical early-morning rise in cortisol. Whether such low corticosteroid levels in premature infants with very low birth weight indicate adrenal insufficiency is not fully known. ∗
 KEY POINTS: ADRENAL DISORDERS
KEY POINTS: ADRENAL DISORDERSPituitary Disorders
No. Placental growth hormone is secreted only into the maternal circulation.
71. What manifestations of adrenocorticotropic hormone insufficiency are seen in neonates?



72. What are the symptoms of growth hormone deficiency in neonates?
The most common presenting symptom is hypoglycemia. Micropenis is also common in male neonates. Growth hormone deficiency may result in an exagerated jaundice (direct and indirect hyperbilirubinemia). Because growth hormone is not necessary for intrauterine linear growth, intrauterine growth restriction is not a feature of growth hormone deficiency. ∗
 KEY POINTS: PITUITARY DISORDERS
KEY POINTS: PITUITARY DISORDERS74. What major malformations may be associated with disorders of the hypothalamic–pituitary axis in neonates?
Cleft lip and palate, optic nerve atrophy, septo-optic dysplasia, and holoprosencephaly have been noted. ∗
Disorders of Sexual Development
75. What is the initial gene thought to be responsible for differentiation of the bipotential gonad into the testis?
There are a number of cases of 46,XX sex reversal in the literature. Only a minority of these cases have been shown to have been caused by translocation of SRY. At least one case of SOX9 duplication (a transcription factor downstream of SRY) has been reported. The majority of cases are unexplained at this time. ∗
78. You are asked to assess a neonate with nonpalpable gonads and genital ambiguity (i.e., severe hypospadias and an intermediate-sized phallic structure). What is the most likely diagnosis? Why?
80. A neonate presents with severe penoscrotal hypospadias and a palpable gonad in the left hemiscrotum; the right hemiscrotum is empty. Amniocentesis shows a classic 46,XX karyotype, and ultrasound shows a cystic structure behind the bladder but no uterus. The genitogram shows a vagina with low insertion and a tiny atretic uterine cavity. What is the differential diagnosis?
81. A neonate presents with genital ambiguity, including significant clitoromegaly and a palpable gonad on the left side in a labioscrotal fold. The right gonad is palpable in the right inguinal canal. The infant’s family recently migrated from the Dominican Republic. What is the most likely diagnosis?
82. You are asked to evaluate a neonate in the delivery room. Amniocentesis during pregnancy revealed a 46,XY karyotype, but the infant has a perfectly normal female phenotypic appearance. What is the most likely diagnosis?
Androgen insensitivity syndrome (AIS) is the most likely diagnosis. Patients with AIS have a normal XY karyotype. The testes are fully developed but never descend, and the external genitalia are those of a normal female. Serum testosterone levels are markedly elevated, but no virilization takes place. Because of a mutation in the androgen receptor, androgen has no effect on its target tissues. AIS, in effect, is end-organ failure based on molecular mutation; it is a syndrome in the sense that several point mutations have been identified.
84. A male neonate in the intensive care unit has a right hernia and a left undescended testis. When the bulging hernia enlarges, intervention is recommended. The surgeon reports that a fallopian tube has been found in the hernia sac. What is the diagnosis?
The diagnosis is hernia uteri inguinalis.
 KEY POINTS: DISORDERS OF SEXUAL DEVELOPMENT
KEY POINTS: DISORDERS OF SEXUAL DEVELOPMENT
1. Informative findings in narrowing the differential diagnosis in an infant with ambiguous genitalia are the presence or absence of palpable gonads, the presence or absence of a uterus, or a combination thereof.
2. MIS levels may be useful in documenting the presence of testicular tissue (Sertoli cells).
3. Complete androgen insensitivity is rarely diagnosed in infancy but may present in the newborn period when a phenotypically normal female infant is born after a prenatal karyotype of 46,XY has been documented on amniocentesis.
Absence of MIS, which is produced by the testis and results in involution of müllerian ducts during the course of normal male sexual differentiation, causes hernia uteri inguinalis. A normal-appearing testis that produces testosterone may lack the capacity to synthesize or secrete MIS. The result is a normal external prominent utricle. ∗
Hypoglycemia
In adults hypoglycemia is defined as a condition involving a plasma glucose level below 40 mg/dL. A plasma glucose concentration of 70 to 100 mg/dL is considered normal, and the therapeutic target range for adults with hypoglycemia is above 60 mg/dL. The definition in neonates is controversial. Some physicians accept significantly lower plasma glucose concentrations as normal for neonates. However, in the absence of scientific evidence that neonates tolerate lower concentrations than adults, many clinicians now believe that values below 50 mg/dL are abnormal. This definition is supported by Koh and colleagues, who demonstrated electrophysiologic changes in the brains of infants when glucose reaches 50 mg/dL. ∗†‡
88a. Which infant catgories are at high risk for hypoglycemia?



89. What physical features suggest the cause of hypoglycemia in neonates?
 Macrosomia: Because insulin is a growth factor, hyperinsulinism leads to macrosomia. Infants of diabetic mothers and infants with severe forms of congenital hyperinsulinism typically are large for gestational age. In addition, neonates with Beckwith–Wiedemann syndrome are macrosomic and may have hyperinsulinism.
Macrosomia: Because insulin is a growth factor, hyperinsulinism leads to macrosomia. Infants of diabetic mothers and infants with severe forms of congenital hyperinsulinism typically are large for gestational age. In addition, neonates with Beckwith–Wiedemann syndrome are macrosomic and may have hyperinsulinism.

 Micropenis: Congenital gonadotropin deficiency can cause micropenis.
Micropenis: Congenital gonadotropin deficiency can cause micropenis.

90. Which hormonal abnormalities cause hypoglycemia in neonates?


92. Which defects in fasting metabolic systems cause hypoglycemia in neonates?
 Defects of glycogenolysis (i.e., GSDs) are associated with hepatomegaly. Examples include deficiencies of debranching enzyme (GSD type 3), liver phosphorylase (GSD type 6), and phosphorylase kinase (GSD type 9).
Defects of glycogenolysis (i.e., GSDs) are associated with hepatomegaly. Examples include deficiencies of debranching enzyme (GSD type 3), liver phosphorylase (GSD type 6), and phosphorylase kinase (GSD type 9).

 Fatty acid oxidation disorders include medium-chain acyl dehydrogenase deficiency. Unless a neonate is breastfeeding poorly or experiences an illness that limits oral intake, a fatty acid oxidation disorder is unlikely to present in infancy. This disorder, however, can cause serious problems during fasting later in life and should be tested for as part of neonatal screening.
Fatty acid oxidation disorders include medium-chain acyl dehydrogenase deficiency. Unless a neonate is breastfeeding poorly or experiences an illness that limits oral intake, a fatty acid oxidation disorder is unlikely to present in infancy. This disorder, however, can cause serious problems during fasting later in life and should be tested for as part of neonatal screening.
93. List and explain the hormonal controls necessary for fasting adaptation.
 Insulin inhibits fasting metabolic systems.
Insulin inhibits fasting metabolic systems.
 Epinephrine stimulates hepatic glycogenolysis, hepatic gluconeogenesis, and hepatic ketogenesis.
Epinephrine stimulates hepatic glycogenolysis, hepatic gluconeogenesis, and hepatic ketogenesis.
 Glucagon stimulates hepatic glycogenolysis.
Glucagon stimulates hepatic glycogenolysis.


94. What tests should be included in the “critical sample” during a hypoglycemic episode?








Congenital hyperinsulinism caused by severe sulfonylurea receptor/potassium channel mutation is often resistant to diazoxide. Octreotide (a somatostatin analog) tempers excessive insulin secretion but rarely prevents hypoglycemia completely or normalizes fasting tolerance. Continuous glucagon infusion can stabilize blood glucose until surgery is performed, but experience with long-term use is limited. If the combination of octreotide and frequent feeds fails, pancreatectomy is necessary. Surgery may be curative if a focal lesion is present and completely resected. ∗†
These defects necessitate frequent feedings.
 KEY POINTS: HYPOGLYCEMIA
KEY POINTS: HYPOGLYCEMIA
1. A work-up for hypoglycemia should be considered in any newborn who is documented to have a blood sugar level persistently lower than 50 mg/dL.
2. The three common etiologies for hypoglycemia in a neonate are decreased production owing to an inborn error of metabolism, increased utilization, and altered hormonal regulation.
3. An infant with hypoglycemia should be carefully examined for hepatomegaly, macroglossia, macrosomia, and midline abnormalities (including clefting defects).
4. The etiology of hypoglycemia is most readily identified by measuring metabolites and hormones at the time of hypoglycemia and should include measures of glucose, free fatty acids, ketones, lactate, pyruvate, ammonia, insulin, cortisol, and growth hormone. Abnormalities in any of these studies may then necessitate definitive diagnostic studies.
5. Treatment of hypoglycemia depends on the etiology but may include avoidance of fasting, a diet altered to circumvent a metabolic block, insulin suppression with diazoxide or pancreatic resection, or replacement of growth hormone or cortisol deficiency.
6. Abnormalities of both the β-cell sulfonylurea receptor and the potassium channel have been documented to cause congenital hyperinsulinism.
Neonatal Screening
Routine neonatal screening tests for the following:


 Glucose-6-phosphate dehydrogenase (G6PD) deficiency
Glucose-6-phosphate dehydrogenase (G6PD) deficiency

 Maple syrup urine disease (MSUD) (G6PD deficiency)
Maple syrup urine disease (MSUD) (G6PD deficiency)

 Medium-chain acyl-coenzyme A (CoA) dehydrogenase
Medium-chain acyl-coenzyme A (CoA) dehydrogenase
 Fatty acid oxidation disorders
Fatty acid oxidation disorders







102. Which of these groups of diseases is likely to be life-threatening in the neonatal period: (1) galactosemia, MSUD, and CAH; or (2) sickle cell disease, G6PD deficiency, and biotinidase deficiency?
103. In which of these groups of diseases is delayed or impaired development of the central nervous system expected if effective treatment is begun at 3 months of age and not shortly after birth: PKU, hypothyroidism, MSUD, galactosemia, or homocystinuria?
Effective treatment of PKU, hypothyroidism, and MSUD must begin within the first few weeks of life to prevent significant problems in development. In infants with galactosemia, learning disabilities are quite prominent if treatment is not initiated early. Developmental disabilities are found in 50% of untreated homocystinuric patients, but the age at which treatment must begin is not known. ∗
104. In which of these groups of diseases may physical signs be present at or shortly after birth: (1) sickle cell disease, G6PD deficiency, and homocystinuria; (2) galactosemia and CAH; or (3) galactosemia, CAH, and PKU?
Sickle cell disease presents at various ages and in various ways, but the major threat to life for small infants is bacterial sepsis, with Streptococcus pneumoniae high on the list of causative organisms. Preclinical detection of sickle cell disease allows prophylaxis against pneumococcal infection. ∗
106. A 5-day-old breastfed infant has a strongly positive test result for urinary-reducing substance but a negative test result for urinary glucose. What action should be taken?
It most states it is a two-tiered test in which immunoreactive trypsinogen is first measured. Babies with the highest immunoreactive trypsinogen levels are then genetically tested for the most common mutations in cystic fibrosis transmembrane conductance regulator, including the most common delta F508 mutation. ∗
 KEY POINTS: NEONATAL SCREENING
KEY POINTS: NEONATAL SCREENING
1. New technologic advances in tandem mass spectrometry have revolutionized newborn screening and allow for detection of dozens of inborn errors of metabolism from blood spots.
2. Specimens should be collected after 24 hours of life and, with the exception of sickle cell disease and G6PD deficiency, should not be affected by transfusions.
3. DNA diagnostic tests are also being added to newborn screening regimens to increase the specificity of testing and reduce the number of false-positive results.
4. The majority of positive results from newborn screening are false-positive results, and repeat or additional diagnostic testing is required to distinguish true positive results from false-positive results.
5. Treatment should be initiated as soon as the diagnosis is made; for some conditions, such as congenital hypothyroidism, PKU, and MSUD, permanent neurologic damage can result if treatment is delayed.
109. What new diagnostic method is being used in newborn screening to increase testing sensitivity and specificity, decrease cost, and increase the number of inborn errors of metabolism that can be effectively screened?
Tandem mass spectrometry (MS-MS) can be performed on dried blood spots and can measure hundreds of metabolites to facilitate screening of dozens of inborn errors of metabolism while more precisely quantitating the levels of the metabolites to improve screening sensitivity and specificity. ∗
110. What is the rationale for adding screening for Krabbe disease to one state’s newborn screening panel?
Neonatal diagnosis before the onset of symptoms would allow for use of bone marrow transplantation as treatment at an age when it is most likely to be effective and allow for more normal brain development. Treatment initiation after the onset of symptoms often leaves children in a state of severe intellectual disability. ∗
The newborn screening test for SCID based on measurement of T-cell receptor excision circles (TRECs) by real-time qPCR using DNA extracted from newborn screening of dried blood spots. RECs are by-products generated during T-cell maturation and are consistently absent or present in low numbers in newborns with SCID. Identification of infants with SCID allows for prevention of life threatening infections and early bone marrow transplantation.
Inborn Errors of Metabolism
The following clinical signs suggest metabolic disease:













The following metabolic disorders are associated with distinctive odors:







The following metabolic disorders are associated with acidosis:










116. What are the first items the neonatal transport team must address in an infant with a suspected inborn error of metabolism?
The neonatal transport team should first address the following:


The transport team may encounter the following:






The following inborn errors are common with neonatal seizures:

 Pyridoxine-responsive seizure disorders
Pyridoxine-responsive seizure disorders
 Peroxisomal disorders (e.g., neonatal adrenoleukodystrophy)
Peroxisomal disorders (e.g., neonatal adrenoleukodystrophy)

 Glucose transporter (e.g., GLUT 1) deficiency with hypoglycorrhachia
Glucose transporter (e.g., GLUT 1) deficiency with hypoglycorrhachia
 Disorders of ammonia metabolism (e.g., ornithine transcarbamylase deficiency)
Disorders of ammonia metabolism (e.g., ornithine transcarbamylase deficiency)
 Disorders causing hypoglycemia (e.g., fatty acid oxidation disorders, GSDs, hyperinsulinemia)
Disorders causing hypoglycemia (e.g., fatty acid oxidation disorders, GSDs, hyperinsulinemia)
Treatment involves institution of a ketogenic diet, which shifts the brain’s metabolism to the utilization of ketone bodies rather than carbohydrates. ∗
The following are possible causes of Fanconi syndrome:






122. What should the initial diagnostic assessment of an infant with suspected metabolic disease include?
Diagnositic assessment should include the following:


 Blood pH and partial pressure of carbon dioxide
Blood pH and partial pressure of carbon dioxide



 Urine Clinitest reaction (while the infant is ingesting a lactose-containing formula)
Urine Clinitest reaction (while the infant is ingesting a lactose-containing formula)


 KEY POINTS: INBORN ERRORS OF METABOLISM
KEY POINTS: INBORN ERRORS OF METABOLISM
1. Common presentations for inborn errors of metabolism include lethargy and coma, dysmorphism, recurrent vomiting, ocular abnormalities, tachypnea unrelated to pulmonary disease, visceromegaly, unusual odors, marked hypotonia, skin or hair abnormalities, seizures, unstable body temperature, bleeding, and jaundice.
2. Common strategies for treating inborn errors of metabolism include avoidance of fasting; dietary manipulation to avoid substrates that cannot be metabolized; medications to clear toxic by-product; supplementation with high doses of cofactors and vitamins used by the deficient enzyme; and, when appropriate, enzyme replacement therapy or organ transplants (e.g., liver or bone marrow transplant).
3. Infants with inborn errors of metabolism may not be symptomatic until metabolically stressed by an intercurrent illness or fasting.
4. Fetal development for inborn errors of metabolism may be normal if the metabolites are able to cross the placenta and may be metabolized by the mother for the fetus.
5. Sudden infant death syndrome can be caused by inborn errors of metabolism, and a family history of a death in infancy of unknown etiology should prompt screening for inborn errors of metabolism.
Secure the airway; if necessary, intubate preemptively. Make sure that the infant receives nothing by mouth, and maintain on IV glucose only. Give IV arginine. Hemodialyze if ammonia levels are above 300 mmol/L and increasing. Administer sodium phenylbutyrate (trade name Buphenyl) and sodium benzoate as ammonia scavenger. ∗
TABLE 8-4
SUMMARY OF MAJOR FINDINGS IN THE FIVE MAJOR KINDS OF METABOLIC DISEASE
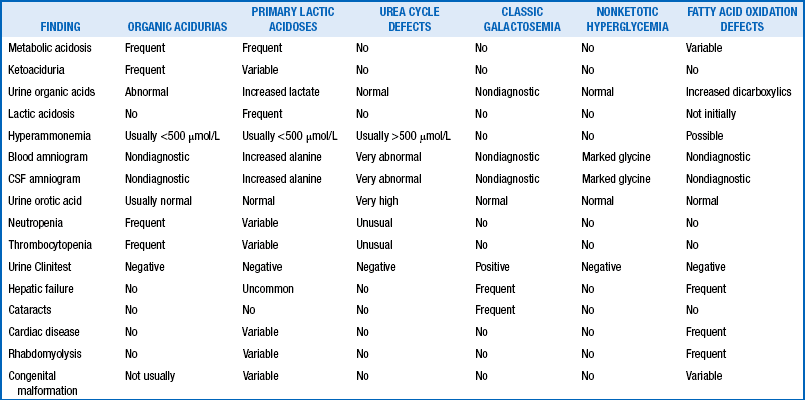
From Spitzer A. Intensive care of the neonate. St Louis: Mosby; 2005. p 1209.
128. If an inborn error of metabolism is suspected, when is the best time to obtain samples for diagnostic testing?
At the time the baby is most severely clinically affected, the diagnostic yield is highest.






∗Loughead JL, Tsang RC. Neonatal calcium and phosphorus metabolism. In: Cowett RM, editor. Principles of perinatal-neonatal medicine. New York: Springer-Verlag; 1998. p. 879–908.
†Moya, FR, Laughon, M. Common problems of the newborn. In: Reece EA, Hobbins JC, editors. Clinical obstetrics: the fetus and mother. 3rd ed. Massachusetts: Blackwell; 2008. p. 1247.
∗Tsang RC, Lucas A, Uauy R, et al., editors. Nutritional needs of the preterm infant. Baltimore: Williams & Wilkins; 1993.
∗Loughead JL, Mimouni F, Tsang RC. Serum ionized calcium concentrations in normal neonates. Am J Dis Child 1988;142:516–18.
∗Fenton TR, Lyon AW, Rose MS. Cord blood calcium, phosphate, magnesium, and alkaline phosphatase gestational age-specific reference intervals for preterm infants. BMC Pediatr 2011;11:76.
∗Bachrach LK, Lum CK. Etidronate in subcutaneous fat necrosis of the newborn. J Pediatr 1999;135:530–1.
∗Pohlenz J, Van Vliet G. Developmental abnormalities of the thyroid. In: Weiss RE, Refetoff S, editors. Genetic diagnosis of endocrine disorders. Amsterdam: Academic Press/Elsevier; 2010. p.101.
∗Hsu SC, Levine MA. Perinatal calcium metabolism: physiology and pathophysiology. Semin Neonatol 2004;9:23–36.
∗Rubin LP. Neonatal disorders of serum magnesium. In: Taeusch HW, Ballard RA, editors. Avery’s diseases of the newborn. 7th ed. Philadelphia: Saunders; 1998. p. 1189–1206.
∗Spiegel, DM. Normal and abnormal magnesium metabolism. In: Schrier RW, editor. Renal and electrolyte disorders. 7th ed. Philadelphia: Lippincott Williams & Wilkins; 2010. p. 240.
∗Fisher DA, Dussault JH, Sack J, et al. Ontogenesis of hypothalamic-pituitary-thyroid function and metabolism in man, sheep and rat. Rec Prog Hormone Res 1977;33:59–116.
∗Fisher DA, Klein AH. Thyroid development and disorders of thyroid function in the newborn. N Engl J Med 1981;304:706.
∗Fisher DA. Fetal thyroid function: diagnosis and management of fetal thyroid disorders. Clin Obstet Gynecol 1997;40:16–31.
∗Lazarus JH, Bestwick JP, Channon S, et al. Antenatal thyroid screening and childhood cognitive function. N Engl J Med 2012;366:493–501.
†Gyamfi C, Wapner RJ, D’Alton ME. Thyroid dysfunction in pregnancy: the basic science and clinical evidence surrounding the controversy in management. Obstet Gynecol 2009;113(3):702–7.
∗Fisher DA. Thyroid function and dysfunction in premature infants. Pediatr Endocrinol Rev 2007;4(4):317–28.
†Rapaport R, Rose SR, Freemark M. Hypothyroxinemia in the preterm infant: the benefits and risks of thyroxine treatment. J Pediatr 2001;139(2):182–8.
∗LaFranchi SH. Approach to the diagnosis and treatment of neonatal hypothyroidism. J Clin Endocrinol Metab 2011;96(10):2959–67.
†American Academy of Pediatrics. Update of newborn screening and therapy for congenital hypothyroidism. Pediatrics. 2006;117(6):2290–303.
∗Feingold SB, Brown RS. Neonatal thyroid function. NeoReviews 2010;11;e640–e646.
∗Downing S, Halpern L, Carswell J, et al. Severe maternal hypothyroidism corrected prior to the third trimester is associated with normal cognitive outcome in the offspring thyroid. 2012;22(6):625–30.
∗Allemand D, Grüters A, Beyer P, et al. Iodine in contrast agents and skin disinfectants is the major cause for hypothyroidism in premature infants during intensive care. Hormone Res 1987;28:42–9.
∗Speiser PW, White PC. Congenital adrenal hyperplasia. N Engl J Med. 2003;349(8):776–788.
∗Carmichael SL, Shaw GM, Laurent C, et al. Maternal progestin intake and risk of hypospadias. Arch Pediatr Adolesc Med 2005;159(10):957–62.
∗McCabe ERB. Adrenal hypoplasias and aplasias. In: Scriver CR, Beaudet AL, Valle D, Sly WS, Childs B, Kinzler KW, Vogelstein B, editors. The Metabolic and Molecular Bases of Inherited Diseases. 8th ed. Vol 3. New York: McGraw-Hill; 2001. p. 4263–74.
∗Bassett JH, O’Halloran DJ, Williams GR, et al. Novel DAX1 mutations in X-linked adrenal hypoplasia congenita and hypogonadotrophic hypogonadism. Clin Endocrinol (Oxf) 1999;50:69–75.
∗Jean A, Mansukhani M, Oberfield SE, et al. Prenatal diagnosis of congenital lipoid adrenal hyperplasia (CLAH) by estriol amniotic fluid analysis and molecular genetic testing. Prenat Diagn 2008;28:11–14.
∗Metzger DL, Wright NM, Veldhuis JD, et al. Characterization of pulsatile secretion and clearance of plasma cortisol in premature and term neonates using deconvolution analysis. J Clin Endocrinol Metab 1993;77:458–63.
∗Palma Sisto PA. Endocrine disorders in the neonate. Pediatr Clin North Am 2004;1:1141–68.
∗Traggiai C, Stanhope R. Endocrinopathies associated with midline cerebral and cranial malformations. J Pediatr 2002;140:252–55.
∗Ergun-Longmire B, Vinci G, Alonso L, et al. Clinical, hormonal and cytogenetic evaluation of 46,XX males and review of the literature. J Pediatr Endocrinol Metab 2005;18(8):739–48.
∗Stanley CA. Hypoglycemia in the neonate. Pediatr Endocrinol Rev 2006;4(Suppl 1):76–81.
†Cornblath M, Hawdon JM, Williams AF, et al. Controversies regarding definition of neonatal hypoglycemia: suggested operational thresholds. Pediatrics 2000;105:1141–5.
†Koh TH, Aynsley-Green A, Tarbit M. et al. Neural dysfunction during hypoglycaemia. Arch Dis Child 1988;63:1353–8.
∗Glaser B, Thornton P, Otonkoski T, et al. Genetics of neonatal hyperinsulinism. Arch Dis Child Fetal Neonatal Ed 2000;82:79–86.
†Stanley C. Advances in diagnosis and treatment of hyperinsulinism in infants and children. J Clin Endocrinol Metab 2002;87:4857–9.
∗Walter JH, Jahnke N, Remmington T. Newborn screening for homocystinuria. Cochrane Database Syst Rev. 2011;10(8):CD008840.
∗Ellison AM, Ota KV, McGowan KL, et al. Pneumococcal bacteremia in a vaccinated pediatric sickle cell disease population. Pediatr Infect Dis J 2012;31:534–6.
∗Wagener JS, Zemanick ET, Sontag MK. Newborn screening for cystic fibrosis. Curr Opin Pediatr 2012;24:329–35.
∗Chace DH, Spitzer AR. Altered metabolism and newborn screening: lessons learned from the bench to the bedside. Curr Pharm Biotechnol 2011;12:965–75.
∗Perlman SJ, Mar S. Leukodystrophies. Adv Exp Med Biol 2012;724:154–71.
∗Pong AW, Geary BR, Engelstad KM, et al. Glucose transporter type I deficiency syndrome: epilepsy phenotypes and outcomes. Epilepsia 2012;53:1503–10.
∗Bireley WR, Van Hove JL, Gallagher RC, et al. Urea cycle disorders: brain MRI and neurological outcome. Pediatr Radiol 2012;42:455–62.
[/level-membership-for-neonatal-and-perinatal-medicine-category][not-level-membership-for-neonatal-and-perinatal-medicine-category]Chapter 8
Endocrinology and Metabolism
Hypocalcemia
1. What perinatal factors are associated with hypocalcemia in the immediate newborn period?
2. How are calcium levels expected to change in premature infants during the first few days of life?
In newborn infants there is a physiologic decline in serum total and ionized calcium during the first 48 hours of life. This decline is exaggerated in preterm infants compared with term infants, with a direct correlation between serum calcium and gestational age ( Fig. 8-1). Because no symptoms are specific for early hypocalcemia in preterm infants, the diagnosis is made by demonstrating a serum calcium level below 7 mg/dL (1.75 mmol/L).
Figure 8-1 Serum calcium in relation to gestational age at 24 hours of age. (From Tsang RC, Light IJ, Sutherland JM, et al. Possible pathogenetic factors in neonatal hypocalcemia of prematurity. J Pediatr 1973;82:423–9.)
Hypocalcemia of prematurity is usually asymptomatic.
Long-term follow-up studies have shown no benefit with treatment.
Total serum calcium level is a poor predictor of ionized serum calcium in premature infants.
Intravenous (IV) calcium is associated with complications such as cardiac arrhythmias and ulcerations as a result of soft-tissue infiltration of the infusate. ∗†
KEY POINTS: HYPOCALCEMIA
1. Neonatal hypocalcemia is associated with prematurity, asphyxia, maternal diabetes, transient hypoparathyroidism, permanent congenital hypothyroidism, and (rarely) maternal hyperparathyroidism.
2. The most common reason for hypocalcemia in the newborn period is prematurity.
3. Breast milk rickets is seen in premature infants because of the relatively low mineral (e.g., calcium and phosphorus) content of breast milk.
4. Serum calcium levels are frequently elevated in patients with Williams syndrome.
5. Normal magnesium levels are needed for optimal functioning of the parathyroid glands.
Recent studies in premature infants using stable isotopes of calcium showed a true calcium absorption rate of 50% to 90%. Thus to meet an accretion rate of 100 mg/kg/day with an absorption rate of 75% and an assumed retention rate of 75% (which may be on the high side), oral intake of calcium for growing premature infants should be about 200 mg/kg/day. This large intake in infants with very low birth weight can be achieved only with special formulas for low-birth-weight infants or mineral fortifiers for breast milk–fed preterm infants. ∗
Hypercalcemia
23–27 weeks: 10.0 +/−1.0 (8.1–11.9)
28–31 weeks: 10.2 +/−1.2 (8–12.5)
32–34 weeks: 10.5 +/−1 (8.6–12.4)
Total serum calcium above 10.8 mg/dL or ionized serum calcium above 5.4 mg/dL.
15. What are some of the causes of hypercalcemia in neonates?
Idiopathic infantile hypercalcemia
Hyperparathyroidism (primary and secondary)
17. A 3-day-old infant born small for gestational age at term has a total serum calcium level of 13.2 mg/dL. She was delivered by emergency cesarean section and was diagnosed with supravalvular aortic stenosis. What is the likely diagnosis?
Williams syndrome is the likely diagnosis in an infant with hypercalcemia and supravalvular stenosis who was born small for gestational age. It results from a deletion of the elastin gene on 7q11.23. Affected infants are often described as having “elfin” faces. ∗
18. A term infant is incidentally noted to have a calcium level of 12.2 mg/dL at 4 days of age. Family history reveals that the father has also been evaluated for elevated calcium levels. What would you expect to find on measurement of the infant’s urinary calcium level? What is the most likely diagnosis? What is the appropriate therapy?
The most likely diagnosis is an autosomal dominant mutation of the calcium-sensing receptor, or “hypocalciuric hypercalcemia.” The infant’s urinary calcium level will be inappropriately low for the serum calcium. In the heterozygous state this is generally thought to be a benign condition, and treatment is not indicated. Rare cases of homozygous mutations result in severe neonatal hyperparathyroidism, which is a life-threatening disorder. ∗
Hypomagnesemia and Hypermagnesemia
Intracellular and extracellular types of magnesium reactions are important.
24. What causes magnesium depletion in neonates?
Renal losses of magnesium in acidotic states
Use of nutrient solutions containing insufficient amounts of magnesium
Intestinal wasting of magnesium (rare X-linked condition)
Gastrointestinal losses (through emesis, nasogastric suctioning, and diarrhea)
Prematurity, which increases the risk for magnesium deficiency
Most infants are asymptomatic. On rare occasions the following signs and symptoms may be seen:
Color: pallor, cyanosis, or duskiness
Affect: out of touch with surroundings, apathetic, irritable when disturbed, restless
Eyes: staring with infrequent blinking, oculogyric crises
Heart: tachycardia (bradycardia during apneic episodes)
Hypomagnesemia usually increases the secretion of PTH, thereby increasing calcium levels. In chronic magnesium-deficient states, however, secretion of PTH is reduced. In such circumstances hypomagnesemia may induce hypocalcemia. ∗
27. What causes hypermagnesemia in neonates?
In extreme cases cardiorespiratory function ceases, and death ensues.
Thyroid Disorders
Congenital hypothyroidism occurs in 1 in 4000 liveborn infants.
32. What are the embryonic stages of development of the fetal hypothalamic–pituitary–thyroid axis?
Thyroid tissue is first identified at the base of the tongue 16 to 17 days after conception.
By 10 weeks’ gestation the fetal thyroid gland is trapping iodine and synthesizing thyroxine (T4).
By 10 to 12 weeks’ gestation, the fetal pituitary gland is synthesizing thyroid-stimulating hormone (TSH). ∗
The hypothalamic–pituitary–thyroid axis is in place by the end of the first trimester. The thyroid and pituitary glands reach mature secretory capacity by 30 to 35 weeks of gestation. The feedback interrelationship among the units is fully established when hypothalamic TRH maturation is completed by 1 to 2 months after birth. ∗
The amount of T4 secreted by the fetal thyroid gland increases slowly until midgestation (20 to 24 weeks) when, stimulated by increasing amounts of TSH from the fetal pituitary, T4 levels begin to increase more rapidly, reaching a normal adult level by approximately 30 weeks’ gestation. Thereafter T4 increases slowly to high normal levels at term gestation. ∗
The placenta is a barrier to the passages of thyroid hormones and contains enzymes that break down maternal T4 and T3 into inactive metabolites. Only a small percentage of circulating maternal T4 and very little (if any) T3 reaches the fetus. However, the amount of maternal T4 that does cross the placenta is significant. During the first 10 to 12 weeks of gestation, all of the circulating T4 in the fetus is from maternal sources; thus early brain development depends on maternal hormone. Even after the fetus synthesizes its own T4 in the second and third trimesters, maternal T4 is essential for normal neurologic development, including neuronal proliferation and maturation, dendritic arborization, and synapse formation. It accounts for approximately 30% of fetal T4 levels at term. ∗†
The levels of TRH, TSH, T4, free T4, and T3 are lower in premature infants than in term infants, and the postnatal surges of TSH and T4, although qualitatively similar, are blunted. These differences are related directly to gestational age: the lower the gestational age, the lower the levels and responses of thyroid-related hormones ( Table 8-1).
TABLE 8-1
SERUM THYROXINE (µg/dL) AT DIFFERENT GESTATIONAL AGES ∗
Adapted from Cuestas RA. Thyroid function in healthy premature infants. J Pediatr 1978;92:963–7.
The term refers to infants with low birth weight (30 to 35 weeks’ gestation) or very low birth weight (<30 weeks’ gestation), who have an even more attenuated rise in T4, after which T4 levels drop below cord levels in the first week of life. Then they rise gradually over 3 to 6 weeks to approach levels of term infants ( Table 8-2).
TABLE 8-2
THYROID FUNCTION IN PRETERM AND TERM INFANTS ∗
T4, Thyroxine; T3, triiodothyronine; TSH, thyroid-stimulating hormone; free T4, free thyroxine.
http://www.uptodate.com/contents/image?imageKey=PEDS%2F72215&topicKey=PEDS%2F5840&rank=4~150&source=see_link&search=thyroid+function&utdPopup=true
Adapted from Williams FL, Simpson J, Delahunty C, et al. Developmental trends in cord and postpartum serum thyroid hormones in preterm infants. J Clin Endocrinol Metab 2004;89:5314.
The premature infant with low T4 and persistently elevated TSH has either transient or permanent hypothyroidism and should be treated with T4 until the nature of the condition becomes clear. However, whether premature infants with low T4 and normal TSH levels should be treated remains controversial. ∗†
43. Does breastfeeding provide needed T4 to premature infants with an immature hypothalamic–pituitary–thyroid axis?
This question has not yet been answered. There are some case reports in the literature suggesting that breastfeeding delays the onset of hypothyroidism, but others argue against that finding.
A heel-stick blood sample is taken at discharge or 3 days of life, whichever is earlier. In most parts of the United States, T4 is measured first, then TSH is measured in samples with the lowest 10% to 29% of T4 results. ∗†
TABLE 8-3
CAUSES OF CONGENITAL HYPOTHYROIDISM AND INCIDENCE OF EACH
From Fisher FA. Disorders of the thyroid in the newborn and infant. In: Sperling MA, editor. Pediatric endocrinology. Philadelphia: Saunders; 1996. p. 57.
Only approximately 1 in 70 neonates born to thyrotoxic mothers exhibit clinical thyrotoxicosis. Such infants may show a phase of transient hypothyroidism caused by antithyroid drugs (half-life, 2 to 3 days), then thyrotoxicosis resulting from maternal TSIs. Transient congenital hypothyroidism can result from transplacental transfer of maternal thyrotopin-blocking antibodies. ∗
[/not-level-membership-for-neonatal-and-perinatal-medicine-category]
