[level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 4
Endocrine Rhythms, The Sleep-Wake Cycle, and Biological Clocks
Major Mechanisms Controlling 24-Hour Endocrine Rhythms
As is illustrated schematically in Figure 4-1, the temporal variability and organization of hormonal concentrations during the 24-hour cycle ultimately result from the activity of two interacting time-keeping mechanisms in the central nervous system: endogenous circadian rhythmicity and sleep-wake homeostasis, a mechanism that relates the timing and intensity of sleep to the duration of prior wakefulness. Although this dual control was first demonstrated for hormones of the hypothalamic-pituitary axis, a similar regulation appears to apply to other endocrine subsystems. In mammals, endogenous circadian rhythmicity is generated by a master circadian clock located in the paired suprachiasmatic nucleus (SCN) of the hypothalamus.1 The SCN controls the timing of most, if not all, circadian rhythms and partially regulates the sleep-wake cycle. The sleep-wake cycle in turn regulates the timing of many rhythms that depend on the presence or absence of sleep and wakefulness. Indeed, the timing and expression of many endocrine rhythms appear to depend upon direct control from the SCN, as well as on the presence and quality of sleep, with some 24-hour endocrine rhythms influenced more by the SCN (e.g., melatonin and cortisol), and others more regulated by the sleep-wake state (e.g., growth hormone). Thus, combined inputs of the master circadian clock and the sleep-wake state2 control the overall temporal organization of the endocrine system, as well as many other behavioral and physiologic systems, across the 24-hour cycle. The two major pathways by which circadian rhythmicity and sleep-wake homeostasis affect peripheral endocrine function are the hypothalamic-pituitary axis and the autonomous nervous system.
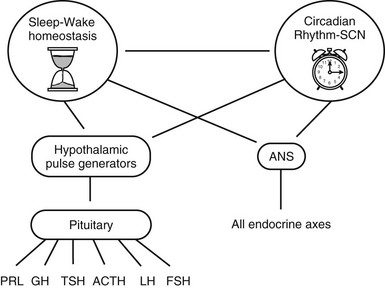
FIGURE 4-1 Schematic representation of the central mechanisms involved in the control of temporal variations in pituitary hormone secretions over the 24-hour cycle. ACTH, Adrenocorticotropic hormone; ANS, autonomous nervous system; FSH, follicle-stimulating hormone; GH, growth hormone; LH, luteinizing hormone; PRL, prolactin; SCN, suprachiasmatic nucleus of the hypothalamus; TSH, thyrotropin.
Circadian Rhythmicity
One of the most obvious characteristics of life on earth is the ability of almost all species to change their behavior on a daily or 24-hour basis. A remarkable feature of these daily or diurnal rhythms is that they are not simply a response to 24-hour changes in the physical environment imposed by the principles of celestial mechanics, but instead arise from an internal time-keeping system1,3 that has the intrinsic capability to continuously generate rhythmic activity over a nearly 24-hour period. Thus, under laboratory conditions devoid of any external time-giving cues, it has been found that nearly all 24-hour rhythms continue to be expressed. However, under such constant conditions, the period of the rhythm rarely remains exactly 24 hours but instead is “about” 24 hours, and this is why these rhythms are referred to as circadian, from the Latin “circa diem,” meaning “around a day.” When a circadian rhythm is expressed in the absence of any 24 hour signals in the external environment, it is said to be free-running. Under free-running conditions, the endogenous period generally is close to, but is nearly never exactly, 24 hours. Strictly speaking, a diurnal rhythm should not be referred to as circadian until it has been demonstrated that such a rhythm persists under constant environmental conditions. The purpose of this distinction is to separate out those rhythms that are simply a response to 24-hour changes in the environment from those that are endogenous. However, for practical purposes, there is little reason to make a distinction between diurnal and circadian rhythms, since an endogenous timing device underlies the generation of almost all diurnal rhythms. In this chapter, we therefore extend the use of the term circadian rhythm to include all diurnal variations that recur regularly at a time interval of approximately 24 hours.
The endogenous nature of human circadian rhythms has been established by experiments in which subjects were isolated with no access to the natural light-dark cycle and no time cues. Such experiments were first performed in natural caves, then in underground bunkers, and finally in specially designed windowless soundproof apartments. The results of one such early experiment, conducted in an artificial underground unit in Germany, are shown in Figure 4-2.4 The rest-activity cycle of the subject is plotted horizontally, day by day, and the times of occurrence of the daily maximum of the body temperature cycle are indicated by closed triangles. During the first 7 days of the experiment, the door of the isolation unit was left open, and the subjects knew the time of day. The average duration (t) of the rest-activity cycle and of the rhythm of body temperature was 24 hours. When, thereafter, the subject lived in complete isolation, both rhythms free-ran but with a mean period of about 26 hours. The free-running period varies from one individual to another. In humans, free-running periods of around 25 hours have been observed under conditions of prolonged temporal isolation. However, assessments of the human free-running period based on the so-called forced desynchrony protocol have provided estimations of between 24.1 and 24.2 hours.5
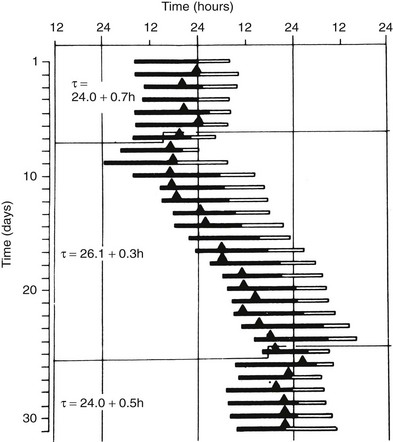
FIGURE 4-2 Circadian rhythms of wakefulness (black bars), sleep (white bars), and maximal rectal temperature (triangles) in a human subject who was exposed to external synchronizing agents for the first and last 7 days and was isolated from all time cues in an underground bunker between days 8 and 24.
The Suprachiasmatic Nucleus: A Master Circadian Pacemaker
In mammals, the suprachiasmatic nucleus (SCN), which consists of two small, bilaterally paired nuclei in the anterior hypothalamus, each of which contains about 10,000 cells in rodents, functions as the master circadian clock. Under both free-running and entrained conditions, destruction of the SCN in a variety of species leads to abolishment or severe disruption of many endocrine, behavioral, and physiologic rhythms.1,6 The role of the SCN as the control center for the circadian system, first suggested by lesion studies, was confirmed by studies involving transplantation of the SCN from one animal to another. Indeed, circadian rhythmicity can be restored in adult arrhythmic SCN-lesioned rodents by transplanting fetal SCN tissue into the region of the SCN.7,8 A number of SCN rhythms, including those of neural firing, vasopressin release, glucose metabolism, and gene expression, persist in vitro.9–12 The ability of SCN cells to generate a circadian signal does not rely on some inherent network property of many cells acting together: Single SCN cells in culture can generate circadian neural signals.13 Neurons within the SCN appear to be organized into two groups: a core group of light responsive but nonrhythmic cells, and a shell of rhythmic cells.14,15
The generation and maintenance of circadian oscillations in the SCN involve a series of clock genes (including per1, per 2, per3, cry1, cry2, tim, clock, B-mal1, and CKIε/δ), often referred to as canonical, which interact in a complex feedback loop of transcription/translation.16,17 Indeed, improved tissue culture techniques and real-time monitoring of the expression of circadian clock genes have revealed that circadian oscillations in gene expression can persist in the SCN in vitro (as well as in other tissues, see later) for many cycles.16 It is important to note that mutations or deletions of canonical circadian clock genes have been found to profoundly affect endocrine rhythms and normal endocrine function in a variety of central and peripheral tissues.18–20 For reviews on the molecular and genetic control of circadian rhythmicity, the reader is referred to references 16, 17, and 21.
In recent years, it has been recognized that circadian oscillations can be generated in areas of the brain other than the SCN, as well as in many peripheral tissues.9,22 These local oscillators appear to be under the control of the SCN and of the canonical circadian clock genes in the SCN, through the synchronization or entrainment of the same circadian clock machinery at the local tissue level. The SCN controls or synchronizes these autonomous circadian clocks in non-SCN tissues directly via neural and/or endocrine signals, or indirectly via its control of behavioral rhythms such as the sleep-wake cycle and the rhythm of feeding. However, the precise mechanisms are not well understood.
Photic Entrainment of Circadian Rhythms
The light-dark (LD) cycle is the primary agent that synchronizes most circadian rhythms. Thus, in the presence of a 24-hour LD cycle, the period of circadian rhythms exactly matches the period of the LD cycle. In addition to establishing period control, an entraining LD cycle establishes phase control, such that specific phases of the circadian rhythm occur at the same time in each cycle. Entrainment is restricted to cycles with periods that are close to 24 hours in duration and, in general, is not possible for LD cycles that are more than a few hours shorter or longer than the endogenous circadian period. If the period of the LD cycle is too short or too long for entrainment to occur, the circadian rhythm free-runs. This rigidity of the circadian pacemaker has been used in so-called forced desynchrony studies, where subjects are maintained on a light-dark and sleep-wake cycle with a period outside the range of entrainment, such as 20 hours or 28 hours.5,23 As mentioned earlier, such protocols have provided estimations of the endogenous period of the human circadian system that are closer to 24 hours (i.e., averaging 24.1 hours) than those obtained in prolonged studies of temporal isolation.5 The fact that the human endogenous circadian period probably is very close to 24 hours is consistent with the findings of a study showing that a schedule of sleep-wake and dark-light cycles with very low light intensity during wakefulness is able to maintain entrainment to the 24-hour day but not to a 23.5- or 24.6-hour day.24
The eyes are involved in relaying entraining information from the LD cycle to the circadian timing system in mammals via a unique pathway, separate from the visual system, and referred to as the retinohypothalamic tract.25 At the level of the optic chiasm, retinal projections first enter the brain in the region of the SCN and surrounding hypothalamic areas.25 Thus, the integrity of the primary visual centers of the brain and/or of the perception of light is not necessary for entrainment of circadian rhythms by the LD cycle. The independence of photic input to the circadian clock from the visual system in mammals actually is not surprising from an evolutionary point of view. In all nonmammalian vertebrates, entrainment of circadian rhythms can occur in the absence of the eyes and relies on nonretinal photoreceptors in the brain.26 The independence of the circadian light sensing system from the visual system in mammals was suggested in early studies showing that rod-less/cone-less mice could still entrain to the light-dark cycle, even though the eyes were necessary.27 Early in this century, several laboratories discovered almost simultaneously that a special subset of retinal ganglion cells containing melanopsin could act as photoreceptors for relaying light information to the circadian clock in the SCN.28–31 This breakthrough not only demonstrated a new light sensing system in the retina, but also opened up new avenues of research regarding the impact of light on brain function independent of the visual system, including possible neuroendocrine effects.32,33 The finding that visual light perception is not necessary for circadian light perception can explain why in some totally blind humans, light exposure is capable of suppressing melatonin levels, indicating that visual blindness should not be equated with circadian blindness.34 In addition to the retinohypothalamic tract, the SCN receives retinal information indirectly from the lateral geniculate nucleus (LGN), which receives a direct projection from the retina.25
To examine how a zeitgeber such as light influences the circadian system, the organism is maintained in constant conditions (e.g., constant darkness) and then is exposed to the zeitgeber for a brief period (e.g., 1 hour) before it is returned to constant conditions.35 The effects of zeitgeber exposure on a phase reference point of an overt circadian rhythm (e.g., onset of locomotor activity, minimum of body temperature) in subsequent cycles then are determined. A plot of the direction and magnitude of the phase-shift as a function of the circadian time of zeitgeber exposure is called a phase response curve (PRC).36 In the human, light exposure during the late evening and the first half of the usual sleep period results in phase delays, but light exposure at the end of the usual sleep period and in the early morning results in phase advances. The transition from phase delays to phase advances occurs around the time of the minimum of body temperature, that is, at between 04.00 and 06.00 hours in most subjects. The magnitude of the phase-shifts is also wavelength dependent, with exposure to short-wavelength monochromatic light (shifted to the blue) resulting in much larger shifts than exposure to longer-wavelength light of equal photon density,37 because the retinal ganglion cells that contain melanopsin are most sensitive to blue light.
Although differences in amplitude may exist, the general shape and characteristics of the PRC to light pulses are similar for all species. Based on this PRC, appropriate exposure to bright light can accelerate adaptation to shifts such as those occurring in jet lag and shift work. The amplitude of the phase-shifts depends on light intensity and duration, and on the number of consecutive exposures.38 For example, exposure to single 3- to 7-hour light pulses can induce phase-shifts on the order of 1 to 3 hours39–41; repeated exposure for 2 to 3 consecutive days may cause much larger 6- to 12-hour shifts.5,42 Intervening periods of dark/sleep exposure could play a role in enhancing the phase-shifting effects of repeated exposure to light.
Exposure to dark also can cause a phase-shift of mammalian circadian rhythms. Because under most circumstances exposure to dark is associated with changes in activity levels (i.e., increases in activity in nocturnal animals and decreases in activity and/or sleep induction in diurnal animals), it has been unclear whether the phase-shifts occurred in response to dark exposure per se, or in response to associated changes in activity levels.43 A study in hamsters, however, has demonstrated that resetting of the rhythm of locomotor activity and concomitant downregulation of circadian clock gene expression following exposure to dark pulses could occur independently of wheel running activity.44 In humans, abrupt 8-hour advances or 8-hour delays of the sleep-wake cycle result in immediate 2-hour phase-shifts in the same direction.45,46 Daytime naps of 6 hours’ duration in total darkness presented over a background of very dim light were found to cause delay shifts when initiated in the morning and advance shifts when initiated in the evening.47 Naps initiated in the afternoon cause no significant phase-shifts.
Nonphotic Zeitgebers
Nonphotic cues, for example, social and/or behavioral cues, also may alter the rest-activity cycle by eliciting activity during the normal rest period or by preventing activity during the normal active period, resulting in phase-shifts in circadian rhythms of activity, as well as other behavioral, physiologic, and endocrine markers.48–51 In nocturnal rodents, the PRC to activity-inducing stimuli is about 12 hours out of phase with the PRC to light pulses.52
How nonphotic information reaches the SCN still is not known, although evidence from lesion studies suggests that a distinct subdivision of the LGN, the intergeniculate leaflet (IGL), may be involved in mediating the effects of activity on the clock.53 Furthermore, the IGL is the source of neuropeptide Y (NPY) innervation of the SCN, and the administration of NPY into the SCN area, as well as electrical stimulation of the geniculohypothalamic tract, induces phase-shifts in hamster locomotor activity rhythm that are similar to those induced by activity-inducing stimuli.53 The LGN/IGL may be a common pathway by which information about the lighting environment and the activity-rest state reaches the circadian clock, and it may be involved in integrating information from the external and the internal environment. Both the LGN/IGL and the SCN receive a dense serotonergic projection from the midbrain raphe nuclei, and substantial evidence now indicates that these projections play a role in both photic and nonphotic regulation of the mammalian circadian clock.54–56
Nonphotic stimuli may affect human circadian rhythms. Exposure to a single session of 3 hours of moderate-intensity exercise during the usual nighttime period was found to result in phase-shifts of markers of circadian phase on the next day, with the direction and magnitude of the phase-shifts being dependent on the timing of exercise.57 Similar findings were obtained with nocturnal exposure to high-intensity 1 hour exercise sessions.58 Supporting evidence for a zeitgeber effect of exercise was obtained in a field study, which found that adaptation to night work could be facilitated by nocturnal exercise.59 These findings were confirmed by a study demonstrating that daily exposure to nightttime exercise facilitated phase delays in circadian melatonin rhythm, even when subjects were maintained in very dim light.60 Nocturnal exercise of low intensity also causes phase-delays in circadian rhythms in older adults, suggesting that it could be a useful treatment for the adjustment of circadian rhythmicity in older populations.61
Another nonphotic agent that has been shown to induce phase-shifts in human circadian rhythms is melatonin.62 This action of melatonin is often referred to as chronobiotic. Specific neural connections are present between the SCN and the pineal gland, and diurnal variation in plasma melatonin levels is driven by the circadian clock.63 Phase-shifts of the central circadian signal induced by changes in the LD cycle will be faithfully reflected in the synchronization of the onset of nocturnal melatonin secretion.40,64,65 Evidence suggests that, in turn, the melatonin rhythm feeds back on the clock (where melatonin receptors have been identified) and exerts synchronizing effects.63
The timing of feeding, when restricted to a narrow temporal window, can affect the entrainment pattern of behavioral rhythms through what has been referred to as a food-entrainable oscillator that is independent of the SCN. Recent findings in rodents have led to a renewed interest in the role of feeding in overall circadian organization in mammals, including the following: (1) alterations in circadian clock genes can lead to obesity and other metabolic abnormalities,2,20 (2) metabolic transcription factor and nuclear receptors involved in metabolism can alter the expression of circadian clock genes,66,67 (3) the timing of feeding can alter rhythmicity in several peripheral circadian oscillators,20,68 and (4) a high-fat diet can alter behavioral and molecular circadian rhythms in central and peripheral tissues involved in the regulation of energy balance.69 Although controversy exists as to the location of an SCN-independent food entrainable oscillator,70,71 now overwhelming evidence suggesting that the circadian and metabolic systems are linked together at molecular, cellular, and behavioral levels has fueled great interest in the possible role of circadian disorganization in obesity, diabetes, and other cardiometabolic disorders.20,72,73
Sleep-Wake Regulation
General Characteristics: The sleep-wake cycle may be viewed as a 24-hour rhythm driven partially by the circadian pacemaker and partially by the homeostatic regulation of sleep pressure. Sleep itself is an ultradian rhythm in that it involves two states of distinct brain activity, each of which is generated in specific brain regions. The ultradian rhythm of normal sleep is an approximate 90-minute oscillation between non-REM (rapid eye movement) and REM stages. In healthy subjects, this pattern usually is repeated four to six times per night. REM sleep and non-REM sleep are characterized by distinct patterns of cerebral and peripheral activity.
The all-night recording of EEG, muscle tone, and eye movements, called the polysomnogram, is scored visually over 20 or 30 second periods in stages I, II, III, IV, REM, and Wake with the use of standardized criteria.74 This procedure allows determination of the duration of each sleep stage but does not quantify the intensity of non-REM sleep. In contrast, the quantification of EEG recordings by power spectral analysis provides useful information regarding sleep depth or sleep intensity, because spectral analysis is sensitive to the amplitude of the delta waves. Higher-amplitude delta waves reflect more intense, deeper sleep, with less sensitivity to arousal stimuli. Slow-wave activity (SWA), i.e., spectral EEG power in the low-frequency range (also called delta range; 0.5 to 4.0 Hz), is a marker of the intensity of non-REM sleep.
Neuroanatomic Basis of Sleep Regulation: Normal waking is associated with neuronal activity in regions of the so-called ascending arousal system, which includes monoaminergic neurons in the brain stem and posterior hypothalamus, cholinergic neurons in the brain stem and basal forebrain, and orexin (hypocretin) neurons in the lateral hypothalamus.75,76 Initiation of sleep therefore requires the inhibition of these multiple arousal systems. In recent years, the ventrolateral preoptic area (VLPO) of the hypothalamus has been identified as involved in the inhibition of arousal. The VLPO contains sleep-active neurons that use the inhibitory neurotransmitter GABA (gamma-aminobutyric acid) and have much higher firing rates during deep sleep than during wakefulness.77–79 Lesions of the central cell cluster of the VLPO drastically reduce SW activity. Neurons of the VLPO provide GABAergic inhibitory innervation of the major monoamine arousal systems in the brain stem. Reciprocally, inhibitory pathways result from the monoamine arousal nuclei to the VLPO.80 The orexin neurons in the lateral hypothalamus project to all components of the ascending arousal system and stimulate the cortex.81 REM sleep is regulated primarily by cholinergic nuclei in the pons.
Interactions Between Circadian Rhythmicity and Sleep-Wake Homeostasis: Several features of the interaction between sleep and circadian rhythmicity appear to be fairly unique to the human species. First, human sleep generally is consolidated in a single 6- to 9-hour period, whereas fragmentation of the sleep period in several bouts is the rule in most other mammals. Possibly as a result of this consolidation of the sleep period, the wake-sleep transition in man is associated with physiologic changes that usually are more marked than those observed in animals. For example, the secretion of growth hormone (GH) in normal adults is tightly associated with the beginning of the sleep period, whereas the relationship between GH secretory pulses and sleep stages is much less evident in rodents, primates, and dogs. Second, man is unique in his capacity to ignore circadian signals and to maintain wakefulness despite increased pressure to go to sleep. Finally, approximately 25% of human subjects maintained for prolonged periods in temporal isolation have shown behavioral modifications that have not been observed in laboratory animals under constant conditions. These modifications consist of a desynchronization between the sleep-wake cycle and other rhythms, such as those of body temperature and cortisol secretion, which continue to free-run with a circadian period. Under conditions of so-called internal desynchronization, the sleep-wake cycle may be lengthened suddenly to 30 hours or more, while the rhythm of body temperature continues to free-run with a circadian period.4 Wakefulness may last longer than 30 hours. Remarkably, the subjects are not aware of these drastic changes in their way of living. Instead, most of them believe that they are living on a more or less regular 24-hour schedule. This can be explained by the observation that time perception is altered profoundly: Subjective estimations of 1 hour intervals are positively correlated with the duration of wakefulness.82 Of particular interest is that subjects continue to have three meals per “day,” irrespective of the actual number of hours they are awake.83 The intervals between meals as well as those between wake-up and breakfast, or between dinner and bedtime, are stretched or compressed in strong proportionality to the duration of wakefulness.84 The mechanisms that cause spontaneous internal desynchronization are not completely understood.
Detailed analyses of data obtained during temporal isolation and forced desynchrony protocols show that the timing, duration, and architecture of sleep are regulated in part by circadian rhythmicity.23,85 Thus, the duration of sleep episodes is correlated with the phase of the circadian rhythm of body temperature and not with the duration of prior wakefulness. Short (i.e., 7 to 8 hours) sleep episodes occur in free-running conditions when the subject goes to sleep around the minimum of body temperature, whereas long (i.e., 12 to 14 hours) sleep episodes occur when sleep starts at around the maximum of body temperature. Moreover, the distribution of REM sleep is markedly modulated by circadian timing. In contrast, the hourglass-like mechanism of sleep-wake homeostasis originally was thought to be largely independent of the circadian system and to involve one or several putative neural sleep factors, which rise during waking and decay exponentially during sleep.86 This homeostatic mechanism regulates the timing, amount, and intensity of SWS and SWA. The VLPO has been proposed as a neuroanatomic locus for the interaction of the homeostatic process and central circadian rhythmicity, as it receives dense projections from the dorsomedial hypothalamic nucleus, which itself receives direct and indirect projections from the SCN.87 Based on human studies described later, it is thought that the SCN generates a waking signal that promotes alertness during the active period. In support of this theory, studies have shown that rodents and monkeys with SCN lesions have increased sleep duration.88,89 Furthermore, studies in rats have described an indirect neuronal circuit from the SCN to the locus coeruleus, an area of the midbrain involved in the control of arousal.90 A role for the SCN in promoting sleep at other circadian times is suggested by the finding of decreased sleep in a mouse with a mutation of the Clock gene.91 The recent finding that the mutation or deletion of a number of circadian clock genes affects not only the timing of sleep but also many other sleep-wake traits, including traits linked to the homeostatic drive to sleep,15,91–94 indicates that circadian and homeostatic processes underlying the regulation of the sleep-wake cycle may be linked at molecular and anatomic levels of organization.
The dual control of sleep by circadian and homeostatic mechanisms extends to the control of objective and subjective measures of sleep tendency, mood, and vigilance.95–98 When wakefulness is extended beyond the usual 16 to 18 hours, maximum subjective sleepiness coincides with the minimum of body temperature, mood, and performance. Remarkably, despite continued sleep deprivation, subjective fatigue then decreases, and mood and performance partially recover during the daytime hours, reflecting an interaction of circadian timing with the accumulation of waking time.96–102 Currently, it is believed that the circadian clock generates a waking signal that increases from morning to evening and is expressed maximally in the early evening hours, 1 to 2 hours before the onset of nocturnal melatonin secretion.97 This circadian waking signal counteracts the buildup of the putative factor “S” underlying the homeostatic process, allowing the individual to maintain a high level of alertness throughout the usual waking period. Current data from human studies are compatible with the hypothesis that the SCN generates a sleep signal in the early evening hours.103
Circadian rhythmicity and sleep-wake homeostasis also interact to regulate hormonal secretion. These modulatory effects were long thought to be present only in hormones directly dependent on the hypothalamic-pituitary axis. However, it is now clear that modulation by circadian rhythmicity and sleep is also present in other endocrine systems, such as glucose regulation and the renin-angiotensin system.104,105 The multiple pathways by which circadian rhythmicity, sleep-wake homeostasis, and their interaction modulate hormonal release are incompletely understood. As is illustrated in Fig. 4-1, humoral and/or neural signals originating from the hypothalamic circadian pacemaker and from brain regions involved in sleep regulation affect the pulsatile release of hypothalamic neuroendocrine factors, which stimulate or inhibit intermittent secretion of pituitary hormones. The autonomic nervous system is another pathway linking the central control of sleep-wake homeostasis and circadian rhythmicity with peripheral endocrine organs. It appears that stimulatory or inhibitory effects of sleep on endocrine release are primarily associated with SW rather than REM sleep.106–110 Pituitary hormones that influence endocrine systems not directly controlled by hypothalamic factors probably mediate, together with the autonomous nervous system, the modulatory effects of sleep and circadian rhythmicity on these systems (e.g., counterregulatory effects of GH and cortisol on glucose regulation105).
To delineate the relative roles of circadian and sleep effects in the temporal organization of hormonal secretion, strategies based on the fact that circadian rhythmicity needs several days to adapt to abrupt shifts in the sleep-wake cycle have been used. Thus, by shifting sleep times by 8 to 12 hours, masking effects of sleep on circadian inputs are removed, and the effects of sleep at an abnormal circadian time are revealed. Fig. 4-3 illustrates the mean profiles of plasma cortisol, GH, prolactin, and thyrotropin (TSH) as observed in normal subjects who were studied before and during an abrupt 12-hour shift in the sleep-wake and dark-light cycles. The study period extended over a 53-hour span and included an 8-hour period of nocturnal sleep, a 28-hour period of continuous wakefulness, and a daytime period of recovery sleep. To eliminate the effects of feeding, fasting, and postural changes, subjects remained recumbent throughout the study, and the normal meal schedule was replaced by intravenous glucose infusion at a constant rate. As shown in Figure 4-3, this drastic manipulation of sleep had only modest effects on the wave shape of the cortisol profile, in sharp contrast with the immediate shift of GH and prolactin rhythms, which followed the shift in the sleep-wake cycle. As will be reviewed in subsequent sections, numerous studies have indicated that control of diurnal rhythms of corticotropic activity is primarily dependent on circadian timing, whereas sleep-wake homeostasis appears to be an important factor in control of the 24-hour profiles of GH and prolactin.111 Nevertheless, small modulatory effects of sleep-wake homeostasis on cortisol secretion and, conversely, influences of circadian timing on somatotropic function have been clearly demonstrated.112 The diurnal variation in TSH levels includes an evening elevation that is thought to be under circadian control and nocturnal inhibition by sleep-dependent processes that is clearly demonstrated during sleep deprivation, when a large increase in nocturnal TSH levels is apparent, as is shown in the lower panel of Fig. 4-3.111
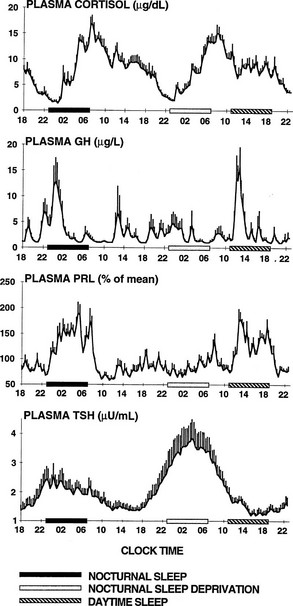
FIGURE 4-3 Mean (+standard error of the mean [SEM]) 24-hour profiles of plasma cortisol, growth hormone (GH), prolactin (PRL), and thyrotropin (TSH) in a group of eight normal young men (20 to 27 years old) studied during a 53-hour period that include 8 hours of nocturnal sleep, 28 hours of sleep deprivation, and 8 hours of daytime sleep. The black bars represent the sleep periods. The open bars represent the period of nocturnal sleep deprivation. The dashed bars represent the period of daytime sleep. Data were sampled at 20-minute intervals. (From Van Cauter E, Spiegel K: Circadian and sleep control of endocrine secretions. In Turek FW, Zee PC [eds]: Neurobiology of Sleep and Circadian Rhythms, vol 133. New York, Marcel Dekker, 1999, pp 397–426.)
Ultradian Rhythmicity
The term ultradian is used primarily to designate rhythms with periods ranging from fractions of hours to several hours. Ultradian oscillations often are less regular and less reproducible than circadian rhythms. In most cases, they appear to represent an optimal functional status within the system where they occur rather than serving the primary function of a clock, that is, an accurate time measuring device. A wide variety of ultradian rhythms have been noted. The most prominent include pulsatile hormonal release and the alternation of REM and non-REM stages in sleep. In the human, the approximately 90-minute REM–non-REM cycle is accompanied by similar periodicity of dreaming, penile erections, sympathovagal balance, and breathing. It has been suggested that this ultradian rhythm during sleep may be a reflection of a basic rest-activity cycle (BRAC), which would occur during wakefulness as well.113 This concept has received some experimental support from a study demonstrating the existence of an ultradian rhythm of brain electrical activity in the frequency range of 13 to 35 Hz, an index of central alertness, during waking.114 It is interesting to note that pulses of cortisol release were significantly associated with ultradian oscillations in alertness.114
Oscillations at frequencies higher than the hourly (i.e., circhoral) range characterizing pulsatile release have been observed for a variety of hormones and metabolic variables. In particular, rapid oscillations of insulin secretion with periods in the 10- to 15-minute range have been well characterized in humans.115,116 Oscillations with a similar period also characterize lipolysis from omental fat, resulting in pulsatile free fatty acid release that appears driven by bursts of sympathetic drive to the adipocytes.117
Properties and Clinical Implications of Pulsatile Hormonal Release
In the endocrine system, ultradian variations have been observed for anterior and posterior pituitary hormones, for hormones under direct pituitary control, and for other endocrine variables such as parathyroid hormone, norepinephrine, plasma renin activity, and leptin and insulin secretion. The interval of recurrence of pulses varies from hormone to hormone and from species to species. The relative importance of pulsatile or oscillatory secretory activity versus tonic release also varies from one axis to another. For some hormones, secretory activity appears to be entirely pulsatile, with no detectable secretion between pulses. In normal men, evidence suggestive of intermittent secretion without tonic release has been obtained for luteinizing hormone (LH), follicle-stimulating hormone (FSH), GH, and adrenocorticotropic hormone (ACTH).118–120 For some hormones, pulsatile release is superimposed on a tonic level of secretion, or secretion occurs continuously but is increased and decreased in an oscillatory fashion. Pancreatic insulin secretion is a well-established example of this type of ultradian oscillation.121,122 Evidence for the existence of tonic secretion also has been obtained for pituitary prolactin and TSH release.118,119
The physiologic significance of pulsatile hormone secretion was first proved when the essential role of the episodic nature of gonadotropin-releasing hormone (GnRH) release for normal functioning of the pituitary-ovarian axis was demonstrated.123 Landmark studies showed that continuous infusions of exogenous GnRH in Rhesus monkeys with lesions of the arcuate nucleus, which abolished endogenous GnRH production, inhibited the secretion of LH and FSH. In contrast, the pulsatile administration of the synthetic hypothalamic hormone at a rate of one 6-minute pulse per hour restored normal LH and FSH levels.123 Furthermore, if the rate of pulse delivery was increased to three pulses per hour, or if it was decreased to one pulse every 2 hours, serum LH and FSH levels were partially inhibited. These findings were rapidly applied to treatment for a variety of disorders of the pituitary-gonadal axis124 and led the way to the discovery of the functional significance of pulsatility in other endocrine systems. For example, it was found that oscillatory administration of insulin with a period matching that of the normal pulsatility of endogenous insulin secretion is more effective in lowering glucose levels than is constant infusion.125
Impact of Aging On Mechanisms Controlling Endocrine Rhythms
Age-related changes in endocrine, metabolic, and behavioral circadian rhythms have been reported in a variety of species, including humans.126–128 One of the most prominent changes is a reduction in rhythm amplitude. The overall findings of a study that examined age-related differences in 24-hour endocrine rhythms in healthy subjects are shown in Fig. 4-4.126 A marked decrease in nocturnal release of TSH, melatonin, prolactin, GH, and melatonin was observed among older volunteers. The amplitude of the cortisol rhythm was decreased among elderly men, primarily because of an elevation of the nocturnal nadir. A retrospective analysis of polygraphic sleep recordings and concomitant profiles of plasma GH and cortisol from 149 normal healthy men, ages 16 to 83 years, showed a different rate of aging of SW sleep and REM sleep129 (Fig. 4-5). SW sleep decreased markedly from early adulthood to midlife and was replaced by lighter sleep (stages I and II) without significant increases in sleep fragmentation or decreases in REM sleep. The transition from midlife to late life involved an increase in wake at the expense of both non-REM and REM sleep. The chronology of aging of GH secretion paralleled that of SW sleep. In contrast, the elevation in evening cortisol levels became significant only after 50 years, when sleep became more fragmented and REM sleep declined. These results were confirmed by a meta-analysis of 65 studies, which found that most of the impact of age on sleep occurs before midlife.130 Despite the fact that older women have more subjective sleep complaints than men,131–133 it was generally assumed that SWS and SWA are less affected by aging in women than in men.134 However, when SWA in non-REM sleep is expressed relative to SWA in REM sleep (i.e., accounting for background SWA), SWA is actually lower in older women than in older men, consistent with the higher frequency of subjective complaints.135
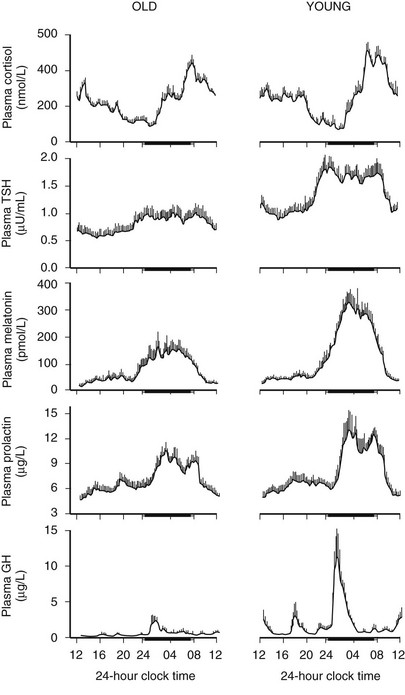
FIGURE 4-4 Mean (+standard error of the mean [SEM]) 24-hour profiles of plasma cortisol, thyrotropin (TSH), melatonin, prolactin (PRL), and growth hormone (GH) levels in eight old (67 to 84 years) and eight young (20 to 27 years) subjects. Data were sampled at 15-minute intervals. The black bars represent the mean sleep period. (From van Coevorden A, Mockel J, Laurent E, et al: Neuroendocrine rhythms and sleep in aging men. Am J Physiol 260:E651–E661, 1991.)
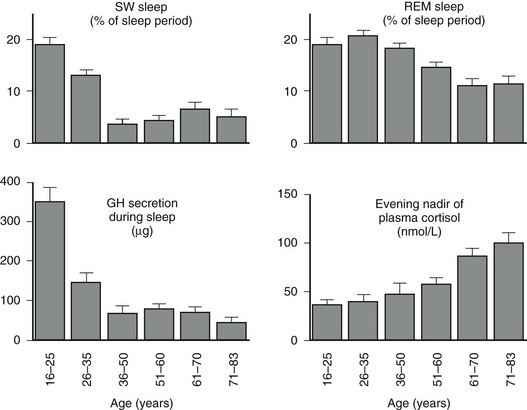
FIGURE 4-5 Left, Slow-wave (SW) sleep and growth hormone (GH) secretion during sleep as a function of age. Note the temporal concomitance between the decrease in SW sleep and in GH secretion. Right, Rapid eye movement (REM) sleep and level of evening nadir of plasma cortisol as a function of age. Note the temporal concomitance between the decrease in REM sleep and the increase in evening cortisol levels. Values shown are means (+standard error of the mean [SEM]) for each age group. Data were obtained in 149 healthy men, ages 16 to 83 years. (Data from Van Cauter E, Leproult R, Plat L: Age-related changes in slow-wave sleep and relationship with growth hormone and cortisol levels in healthy men. JAMA 284:861–868, 2000.)
Deficits in the maintenance and quality of nocturnal sleep in older adults are paralleled by decreased alertness, attention and memory deficits, and decreased performance during the daytime.136 In both rodents and humans, many circadian rhythms are advanced under entrained conditions such that specific phase points of these rhythms occur earlier than in young subjects.126,137 Both amplitude reduction and phase advance of the rhythm of body temperature have been observed in elderly subjects, and these alterations in circadian regulation were closely associated with changes in sleep-wake habits (i.e., earlier bedtimes and wake times).127,138
Age-related changes in the amplitude and/or the phase of circadian rhythms could be due to changes in the inner workings of the master clock, to alterations in the input pathways to the clock, or to factors downstream between the circadian clock and the system that is expressing the rhythm. Some early studies139,140 have reported that the free-running period of various rhythms in rodents is systematically shortened with age, suggesting that the circadian clock itself is altered in advanced age. This concept has been confirmed in rats via measurement of Per1-luc expression in transgenic animals.141 Age-related phase advances of a number of different behavioral and endocrine rhythms are consistent with the hypothesis that the period of the human circadian clock is shorter in the elderly. However, a study that measured the free-running period in healthy young and older adults using the forced desynchrony protocol has found no age difference.5 Nevertheless, the very healthy older individuals who participated in this demanding protocol exhibited marked decreases in sleep consolidation and had greater difficulties sleeping at adverse circadian phases than did young subjects. These alterations and the clear advance of the propensity to awaken from sleep are thought to be related to both a reduction in the homeostatic drive to sleep and a reduction in the strength of the circadian signal.142 In older adults, in contrast to young subjects, no significant correlation has been noted between the length of the circadian period and the phase angle of entrainment.143
Studies in rodents indicate that aging is associated with decreased responsiveness to the phase-shifting effects of both photic and nonphotic stimuli. Old hamsters show a decreased response to the phase-shifting effects of low-intensity light pulses.144 This observation raises the possibility that in old age, signal transmission of light information to the SCN itself may be decreased, or that SCN responsiveness to photic stimulation may be lessened. Similarly, although induction of locomotor activity during a time of normal inactivity can induce pronounced phase-shifts in the circadian rhythm of locomotor activity in young animals, in old animals the response is greatly diminished or is completely abolished.145,146 It is interesting to note that transplantation of fetal SCN tissue into the SCN region of old hamsters with an intact SCN can restore the response to the phase-shifting effects of triazolam on the activity rhythm.147
Behavioral changes in the elderly also may lead to changes in environmental inputs to the clock. In older adults, exposure to bright light and social cues, both potential entraining agents, is markedly diminished when compared with that seen in young adults.148–150 Furthermore, age-related lens pigmentation decreases the transmission of blue light (i.e., the wavelength to which the circadian system is most sensitive) to the retina.151 Absence of professional constraints, decreased mobility due to illness, and reduced socialization and outdoor activities are all hallmarks of old age. Thus, decreased exposure to environmental stimuli that entrain circadian rhythms could contribute to disruptions in circadian rhythmicity. The use of exposure to bright light and enriched social schedules to reinforce circadian rhythms in older adults and improve nighttime sleep and daytime alertness has proved beneficial in several studies.152 In elderly insomniacs living in a nursing home with limited exposure to environmental light, supplementary light exposure during the middle of the day significantly increased nocturnal melatonin secretion without circadian phase-shifting.153 Evidence indicates that older adults are capable of phase-shifting to the same extent as younger subjects in response to appropriately timed light exposure.154
The ultradian rhythmicity of pulsatile hormonal release is also affected by aging. For a number of individual hormones, including GH, insulin, LH, and ACTH, analyses of 24-hour profiles have shown increased irregularity, or disorderliness, of pulsatile release. Furthermore, the synchrony of pulsatile release between the pituitary hormone and the peripheral hormone (e.g., LH and testosterone, ACTH and cortisol) is partially disrupted in healthy older adults.155
Methodologic Aspects of the Study of Endocrine Rhythms
Procedures to Quantify Circadian Variations
Among the methods proposed for and applied to the analysis of 24-hour profiles of blood components, the oldest is the Cosinor test.156 The major disadvantage of this test and of its derivatives is its assumption that the observed profile may be described adequately by a single sinusoidal curve. This assumption is practically never met for biological rhythms that are asymmetric in nature (e.g., the sleep-wake cycle is a 08:16 alternation, not a 12:12). Therefore, the Cosinor test generally provides unreliable estimations of rhythm parameters. Unfortunately, in recent years, the Cosinor analysis has regained popularity and has been applied indiscriminately to asymmetric profiles.
Other procedures for the detection and estimation of circadian variation have been based on periodogram calculations or on nonlinear regression procedures.157,158 These methods provide an adequate description of asymmetric wave shapes. The times of occurrence of the maximum and the minimum of the best-fit curve often are referred to as the acrophase and the nadir, respectively. The amplitude of the rhythm may be estimated as 50% of the difference between the maximum and the minimum of the best-fit curve. With the periodogram procedure, confidence intervals for the amplitude, acrophase, and nadir may be calculated.
Procedures to Quantify Pulsatile Hormonal Secretion
Analysis of pulsatile variations may be considered at two levels.159 One may wish to define and characterize significant variations in peripheral levels, based on estimations of the size of the measurement error (i.e., primarily assay error). However, under certain circumstances, it is possible to mathematically derive secretory rates from the peripheral concentrations.160–162 This procedure, often referred to as deconvolution, often reveals more pulses of secretion than does analysis of peripheral concentrations. It also more accurately defines the temporal limits of each pulse. However, deconvolution involves amplification of measurement error, with increased risk for false-positive error. Whether peripheral concentrations or secretory rates are examined, two major approaches are used to analyze the episodic fluctuations. The first and most commonly used is the time domain analysis, wherein the data are plotted against time, and pulses are detected and identified. The second is the frequency domain analysis, in which amplitude is plotted against frequency or period. The time domain analysis provides an estimation of pulse frequency, calculated as the total number of pulses detected divided by the duration of the study period. The regularity of pulsatile behavior may be quantified by examining the distribution of interpulse intervals. Alternatively, the issue of regularity of pulsatile behavior may be approached by examining the distribution of spectral power in a frequency domain analysis.163 Finally, another analytic tool, the approximate entropy (ApEn), has been introduced to quantify the regularity of oscillatory behavior in endocrine and other physiologic time series.164,165
A number of computer algorithms for identification of pulses of hormonal concentration have been proposed. A detailed presentation of the operating principles of each of these procedures is beyond the scope of this chapter. Review articles166,167 have provided comparisons of performance of several pulse detection algorithms. These comparisons have indicated that ULTRA and CLUSTER perform similarly when used with appropriate choices of parameters.167
Endocrine Rhythms in Health and Disease
Pituitary Axes
Normal Rhythms of Adrenocorticotropic Hormone and Adrenal Secretions: Outputs from the SCN activate rhythmic release of corticotropin-releasing hormone (CRH) that stimulates circadian ACTH release. The 24-hour rhythm of adrenal secretion is primarily dependent on the diurnal pattern of ACTH release. In addition, neuronal signals generated by the SCN are transmitted by a multisynaptic neural pathway to the adrenal cortex.168 The presence in the adrenal cortex of an intrinsic circadian oscillator consisting of interacting positive and negative feedback loops in circadian gene expression has been demonstrated in various animals, including monkeys,169–171 and it has been shown that this adrenal circadian pacemaker gates the physiologic adrenal response to ACTH (i.e., defines a time window during which the adrenal most effectively responds to ACTH).172
The 24-hour profiles of ACTH and cortisol show an early morning maximum, declining levels throughout daytime, a quiescent period of minimal secretory activity centered around midnight, and an abrupt elevation during late sleep, resulting in an early morning maximum. Mathematical derivations of secretory rates from plasma concentrations have suggested that the 24-hour profile of plasma cortisol reflects a succession of secretory pulses of magnitude modulated by a circadian rhythm with no evidence of tonic secretion.120,173 In normal conditions, the acrophase of the pituitary-adrenal periodicity occurs between 06.00 and 10.00 hours. With a 15-minute sampling interval, 12 to 18 significant pulses of plasma ACTH and cortisol can be detected per 24-hour span.174 Circadian and pulsatile variations parallel to those of cortisol have been demonstrated for the plasma levels of several other adrenal steroids, in particular dehydroepiandrosterone (DHEA).175 The temporal concomitance of 24-hour profiles of ACTH, cortisol, and DHEA is illustrated in Fig. 4-6.
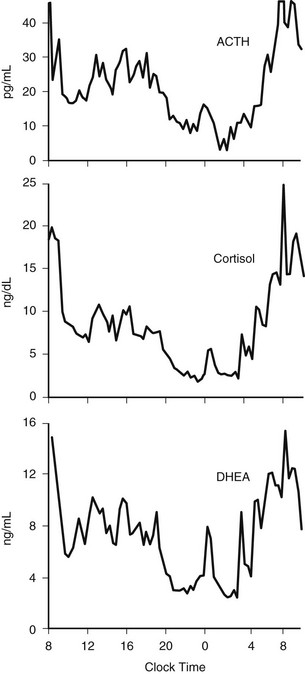
FIGURE 4-6 Twenty-four-hour profiles of plasma adrenocorticotropic hormone (ACTH), cortisol, and dehydroepiandrosterone (DHEA) levels sampled at 20-minute intervals in a healthy young man. Note the temporal concomitance of circadian and pulsatile variations of the three hormones. (Unpublished data kindly provided by Dr. K. Spiegel.)
The profile shown in the upper panel of Fig. 4-3 illustrates the remarkable persistence of the cortisol and, by inference, ACTH secretory rhythm when sleep is manipulated. Indeed, the overall wave shape of the profile was not markedly affected by the absence of sleep or the presence of sleep at an abnormal time of day. Thus, this rhythm is controlled primarily by the circadian pacemaker.
However, modulatory effects of sleep-wake homeostasis have been clearly demonstrated. As illustrated in Fig. 4-7, sleep onset is consistently associated with short-term inhibition of cortisol secretion, which may not be detectable when sleep is initiated in the morning (i.e., at the peak of corticotropic activity).176–179 This inhibitory effect of sleep appears to be related to SW sleep.109,180 Conversely, final awakenings from sleep, as well as transient awakenings that interrupt the sleep period, consistently trigger pulses of cortisol secretion (see Fig. 4-7),120,178,180–183 and the number of nocturnal microarousals predicts morning plasma and saliva cortisol levels.184 In an analysis of cortisol profiles during nocturnal sleep, it was observed that all transient awakenings that interrupt sleep and last at least 10 minutes were followed within the next 20 minutes by significant bursts of cortisol secretion.183 In addition, a temporal coupling between pulses of cortisol secretion and ultradian variations in an EEG marker of alertness has been reported.114 Total nocturnal wake time is associated with increased 24-hour plasma cortisol concentrations,185 and chronic insomnia with reduced total sleep time is associated with higher cortisol levels across the night.186
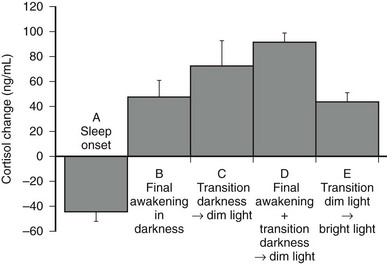
FIGURE 4-7 Mean (and standard error of the mean [SEM]) changes in plasma cortisol levels: A, Within 120 minutes following sleep onset at 15.00 hours (n = 32). B, Within 20 minutes following final spontaneous awakening in darkness (scheduled sleep period 23.00 to 07.00 hours or 15.00 to 23.00 hours; n = 10). C, Within 20 minutes following transition from darkness to dim light at 07.00 or 23.00 hours in subjects awake at bed rest (n = 10). D, Within 20 minutes following final awakening concomitant with transition from darkness to dim light at 07.00 or 23.00 hours (n = 38). E, Within 15 minutes following transition from dim to bright light at 05.00 hours in subjects awake at bed rest (n = 8). (Panels A to D, Data from Caufriez A, Moreno-Reyes R, Leproult R, et al: Immediate effects of an 8-h advance shift of the rest-activity cycle on 24-h profiles of cortisol. Am J Physiol 282:E1147–E1153, 2002; panel E, Data from Leproult R, Colecchia EF, L’Hermite-Balériaux M, et al: Transition from dim to bright light in the morning induces an immediate elevation of cortisol levels. J Clin Endocrinol Metab 86:151–157, 2001.)
Modulatory effects of dark-light transitions also have been noted. Cortisol secretory pulses associated with morning awakening are enhanced by increasing light intensity.187 Moreover, the transition from darkness to dim light and from dim to bright light may stimulate cortisol secretion in subjects who are awake at bed rest.183,188 The stimulatory effects of bright light exposure on nocturnal cortisol levels appear to be dependent on the timing of exposure.189 When dark-light and sleep-wake transitions occur concomitantly, associated cortisol elevations are nearly two times as high as when the final awakening occurs in continuous darkness183 (see Fig. 4-7). Thus, under usual bedtime schedules, both sleep-wake and dark-light transitions amplify the effects of circadian rhythmicity.
Studies of the 24-hour cortisol profile in the course of adaptation to shifts of the sleep-wake cycle have demonstrated that the end of the quiescent period, which coincides with the onset of the early morning rise, takes longer to adjust and appears to be a robust marker of circadian timing. Twin studies have demonstrated that the timing of the nadir is influenced by genetic factors,190 providing evidence for genetic control of the human circadian phase. In contrast, the timing of the morning acrophase is more labile and may be influenced by the timing of sleep offset,182 the transition from dark to bright light,187 and breakfast intake.191 Finally, anticipation of the expected time of waking has been reported to be associated with a rise in levels of ACTH, but not cortisol, during the end of the sleep period.192
In addition to the immediate modulatory effects of sleep-wake transitions on ACTH and cortisol levels, acute total or partial (4 hours in bed) nocturnal sleep deprivation results in elevated cortisol concentrations in late afternoon and evening on the following day,193 and the amplitude of the cortisol circadian variations is reduced by approximately 15%. Similarly, as illustrated in Fig. 4-8, recurrent partial sleep deprivation (4 hours in bed per night for 6 nights) also results in an elevation of cortisol levels in the late afternoon and evening.194 These disturbances, which were observed in young healthy subjects, are strikingly similar to those noted in older healthy subjects with normal sleep schedules.126,195,196 In any case, sleep loss appears to delay the return to quiescence of the hypothalamic-pituitary-adrenal axis (HPA) that normally occurs in the evening. This suggests that sleep loss, similar to aging, may slow down the rate of recovery of the HPA axis response following a challenge and therefore could facilitate the development of central and peripheral disturbances associated with glucocorticoid excess, in particular when cortisol concentrations are elevated at the time of the normal daily nadir, such as memory deficits, insulin resistance, and osteoporosis.197–201 Conversely, decreased HPA resiliency results in HPA hyperactivity that inhibits SW sleep and promotes nocturnal awakenings, initiating a feed-forward cascade of negative events generated by both HPA and sleep disruptions.
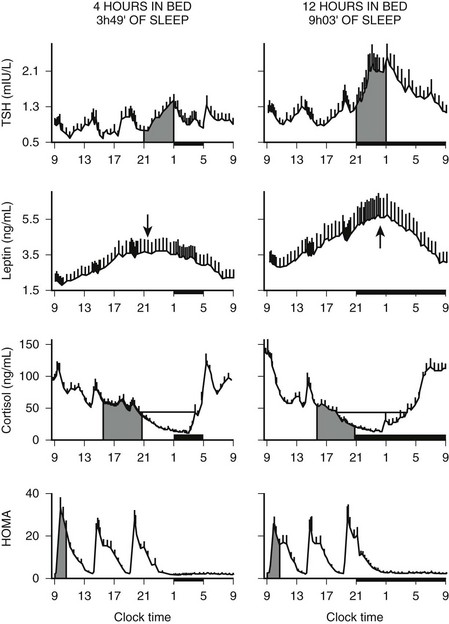
FIGURE 4-8 Impact of recurrent sleep curtailment (4 hours in bed for 6 nights) and of sleep recovery (12 hours in bed for 6 nights) on 24-hour plasma profiles of thyrotropin (TSH), leptin, and cortisol, and on homeostatic model assessment (HOMA) in healthy young men. HOMA was calculated as glucose concentration (mg/dL) × insulin concentration (µU/mL). Values shown are means ± SEM. (Adapted from Spiegel K, Leproult R, Van Cauter E: Impact of sleep debt on metabolic and endocrine function. Lancet 354:1435–1439, 1999, and from Spiegel K, Leproult R, L’Hermite-Balériaux M, et al: Leptin levels are dependent on sleep duration: relationships with sympathovagal balance, carbohydrate regulation, cortisol and thyrotropin. J Clin Endocrinol Metab 89:5762–5771, 2004.)
The circadian rhythm of cortisol persists throughout adulthood and has been observed through the ninth decade.126,196 Among young adults, 24-hour cortisol levels are slightly lower in women than in men, primarily because of lower morning maxima. With aging, evening cortisol levels increase progressively in both men and women, so that the cortisol nadir is markedly higher in healthy subjects over 70 years of age than in young adults (see Fig. 4-4). It is interesting to note that this elevation of evening cortisol levels occurs with a chronology similar to that observed for a progressive decrease in the duration of REM sleep129 (see Fig. 4-5). As a result, older subjects have elevated 24-hour mean cortisol levels and reduced amplitude of cortisol variations. In addition, the timing of the nadir is advanced by 1 to 2 hours, indicating that aging is associated with an advance in the circadian phase.126,196
In pregnancy, placental CRH is secreted into the maternal circulation in a pulsatile but not in a circadian fashion, and no correlation has been noted between maternal levels of CRH and ACTH. However, ACTH and cortisol concentrations remain strongly correlated with each other over time, suggesting that diurnal variation in maternal ACTH probably is driven by another ACTH secretagogue.202
Alterations in Disease States: The 24-hour profile of pituitary-adrenal secretion remains largely unaltered in a wide variety of pathologic states. Disease states in which pronounced alterations of the cortisol rhythm have been observed include primarily (1) disorders involving abnormalities in binding and/or metabolism of cortisol; (2) the various forms of Cushing’s syndrome; and (3) depression and posttraumatic stress disorder (PTSD).
The relative amplitude of the circadian rhythm and of the episodic fluctuations in cortisol is blunted in patients with liver disease203 and in those with anorexia nervosa,204 primarily because of the decreased metabolic clearance of cortisol. Pulsatile secretion of ACTH, but not cortisol, was reportedly enhanced in obese premenopausal women.205 In hypothyroid patients, the mean level is markedly elevated and the relative amplitude of the rhythm is dampened.206 These alterations are thought to be due to both diminished clearance and decreased efficiency of feedback control. In contrast, in hyperthyroidism, where cortisol production and peripheral metabolism are increased, episodic pulses are enhanced.207
A low-amplitude circadian variation may persist in pituitary-dependent Cushing’s disease. Cortisol pulsatility is blunted in about 70% of patients with Cushing’s disease, suggesting autonomous tonic secretion of ACTH by a pituitary tumor. However, in about 30% of these patients, the magnitude of the pulses is enhanced instead.208 These hyperpulsatile patterns could be caused by enhanced hypothalamic release of CRH or persistent pituitary responsiveness to CRH. It also has been shown that patients with Cushing’s disease secrete ACTH and cortisol jointly more asynchronously than healthy subjects.209 The left and middle panels of Fig. 4-9 compare representative and mean 24-hour cortisol profiles in normal subjects and in patients with Cushing’s disease.
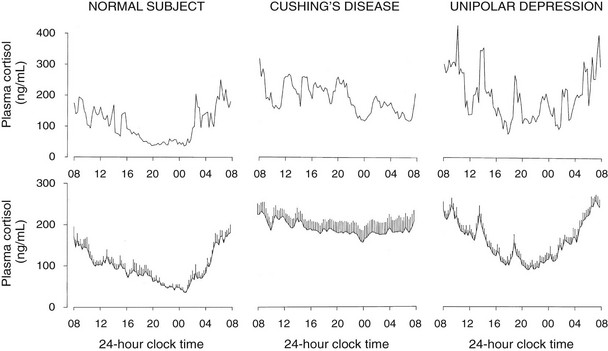
FIGURE 4-9 Twenty-four-hour profiles of plasma cortisol in normal subjects, patients with pituitary Cushing’s disease, and patients with major endogenous depression of the unipolar subtype. For each condition, a representative example is shown in the top panel, and mean (+standard error of the mean [SEM]) profiles from 8 to 10 subjects are shown in the lower panel. (From Van Cauter E: Physiology and pathology of circadian rhythms. In Edwards CW, Lincoln DW [eds]: Recent Advances in Endocrinology and Metabolism. Edinburgh, Churchill Livingstone, 1989, vol 3, pp 109–134.)
In patients with primary adrenal Cushing’s syndrome, increased cortisol secretion appears to result from both increased basal secretion and increased pulse frequency.210 Although the persistence of a low-amplitude circadian cortisol variation has been claimed,210 examination of the individual profiles does not support this notion. Nevertheless, the partial persistence of cortisol rhythmicity in the absence of ACTH could reflect the newly described neural pathway between the SCN and the adrenal cortex.168
Hypercortisolism with persistent circadian rhythmicity and increased pulsatility is found in a majority of severely depressed patients.211–213 This is illustrated in the right panels of Fig. 4-9. In these patients, who do not develop the clinical signs of Cushing’s syndrome despite high circulating cortisol levels, the quiescent period of cortisol secretion is shorter and more fragmented, and it often starts later and ends earlier than in normal subjects of comparable age. These alterations could reflect the impact of sleep disturbances, as well as an advance in circadian phase. When clinical remission of the depressed state is obtained, hypercortisolism and alterations in the quiescent period disappear, indicating that these disturbances are “state” rather than “trait” dependent.214
In PTSD, some authors have reported that plasma cortisol levels are decreased in the afternoon and/or in the evening, and that the amplitude of circadian variations is enhanced.215–217 In fibromyalgia and in the chronic fatigue syndrome, cortisol levels have been reported to be low, normal, or elevated, depending on the study, but ACTH and cortisol pulsatility were found to be normal.218 In a well-documented study performed in constant routine conditions, normal circadian variations of plasma cortisol levels were evidenced in women with fibromyalgia.219
The Somatotropic Axis
The 24 Hour Profile of Growth Hormone in Normal Subjects: Pituitary secretion of GH is stimulated by hypothalamic GH-releasing hormone (GHRH) and is inhibited by somatostatin. In addition, the acylated form of ghrelin (acyl-ghrelin), a peptide produced predominantly by the stomach, binds to the GH-secretagogue receptor and therefore is another potent endogenous stimulus of GH secretion.220,221 In normal adult subjects, the 24-hour profile of plasma GH levels consists of stable low levels abruptly interrupted by bursts of secretion. The most reproducible pulse occurs shortly after sleep onset, in association with the first phase of SW sleep.222 Other secretory pulses may occur in later sleep and during wakefulness, in the absence of any identifiable stimulus. Studies in young male twins have evidenced a major genetic effect on GH secretion during waking, but not during sleep.223 In adult men, the sleep-onset GH pulse generally is the largest pulse observed over the 24-hour span. In normally cycling women, 24-hour GH levels are higher than in age-matched men, daytime pulses are more frequent, and the sleep-associated pulse, although still present in most cases, does not generally account for most of the 24-hour GH release.224 Typical profiles of young men and women are shown in Fig. 4-10. Well-documented studies have demonstrated that in women, the amplitude of GH secretory pulses is correlated with the circulating level of estradiol.224,225 In normally cycling young women, it also was observed that daytime GH secretion was increased during the luteal phase as compared with the follicular phase, and that this elevation correlated positively with plasma levels of progesterone but not estradiol.226
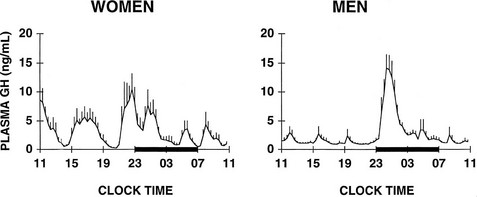
FIGURE 4-10 Mean (+standard error of the mean [SEM]) 24-hour plasma growth hormone profiles in nine men, age 18 to 30 years, and in seven women, age 21 to 33 years, during the follicular phase. The black bars represent the sleep periods. (Adapted from Van Cauter E, Plat L, Copinschi G: Interrelations between sleep and the somatotropic axis. Sleep 21:533–566, 1998.)
Sleep onset will elicit a GH secretory pulse, whether sleep is advanced, delayed, interrupted, or fragmented.222 Thus, as illustrated in Fig. 4-3, shifts in the sleep-wake cycle are followed immediately by parallel shifts in GH rhythm.222 In night workers, the main GH secretory episode occurs during the first half of the shifted sleep period.227 The release of GH in early sleep is temporally and quantitatively associated with the amount of SW sleep.228,229 Both SW sleep and GH levels are increased during the recovery night following nocturnal total sleep deprivation as compared with the baseline predeprivation night, especially during the first 4 hours of sleep.230 Good evidence suggests that the mechanisms underlying the relationship between SW sleep and GH release involve synchronous activity of at least two different populations of hypothalamic GHRH neurons.231 Indeed, inhibition of endogenous GHRH by administration of a specific antagonist or by immunoneutralization inhibits both sleep and GH secretion.232 Additional evidence for the existence of a robust relationship between SW activity and GH release has been provided by studies using pharmacologic stimulation of SW sleep. Indeed, enhancement of SW sleep by oral administration of low doses of gamma-hydroxybutyrate (GHB), a natural metabolite of GABA used in treatment for narcolepsy, or of ritanserin, a selective serotonin (5-HT)2 antagonist, results in simultaneous and highly correlated increases in nocturnal GH secretion.108,233 Conversely, transient awakenings during sleep inhibit GH secretion.234 Thus, sleep fragmentation generally will decrease nocturnal GH release.
However, although sleep is clearly the major determinant of GH secretion in man, evidence for the existence of a circadian modulation of the occurrence and amplitude of GH pulses also has been found. This may reflect decreased somatostatin inhibitory activity in the evening and during the night,235 or it could result from the nocturnal rise in ghrelin, which occurs even in the absence of sleep.236 Thus, the major sleep-onset associated GH pulse is caused by a surge of hypothalamic GHRH coincident with a circadian period of relative somatostatin disinhibition.222,232 In normal-weight subjects, fasting, even for only 1 day, enhances GH secretion via an increase in pulse amplitude.237 Presleep GH pulses, reported by some investigators in normal men,238 may reflect the presence of a sleep debt, thus unmasking the circadian component of GH secretion.239 A recent study provides evidence for modulation of GH secretion by endogenous acyl-ghrelin, leading to higher GH peaks before times of food intake in subjects given regular meals.237
After a night of total sleep deprivation, a compensatory increase in GH release is observed during the daytime, so that the overall 24-hour secretion is not altered significantly.240 The mechanisms underlying this compensatory increase might involve decreased somatostatinergic tone and/or elevated ghrelin levels. Recurrent partial sleep restriction is associated consistently with the appearance of a presleep GH pulse.239
Aging is associated with dramatic decreases in circulating levels of GH (see Figs. 4-4 and 4-5).126,224 This reduction is achieved by a decrease in amplitude, rather than in frequency, of GH pulses.126,241,242 It also has been reported that the orderliness of GH secretion is decreased in the elderly.243 As illustrated in Fig. 4-5, this age-related GH decrease occurs in an exponential fashion between young adulthood and midlife and follows the same chronology as the decrease in SW sleep. Despite the persistence of high levels of sex steroids, plasma concentrations and pulsatile secretion rates of GH fall in midlife to less than half the values achieved in young adulthood. Thereafter, smaller and more progressive decrements occur from midlife to old age.129 Among the elderly, GH secretory profiles are similar in men and in women.224 The age-related reduction of GH secretion appears to result from increased somatostatin secretion and diminished GHRH responsiveness.244
It is interesting to note that during pregnancy, a placental GH variant, which substitutes for pituitary GH to regulate maternal insulin-like growth factor-1 (IGF-1) levels,245 is released in a tonic rather than a pulsatile fashion.246
Alterations in Disease States: Abnormalities in the 24-hour profile of plasma GH have been reported in a variety of metabolic, endocrine, neurologic, and psychiatric conditions.
An inverse relationship between adiposity and GH release has been noted, which results in marked suppression of GH levels throughout the 24-hour span in obese subjects. A normal pattern can be restored after prolonged fasting.247 In anorexia nervosa, GH pulse amplitude and frequency are increased, and the orderliness of GH release is disrupted.248 Nonobese juvenile or maturity-onset diabetic patients hypersecrete GH during wakefulness as well as during sleep, primarily because of an increase in the amplitude of pulses.249 This abnormality may disappear when glycemia is strictly controlled.
In functional hypothalamic amenorrhea, 24-hour mean GH levels are normal, but the pattern of pulsatile GH release is distinctly altered, with a decrease in pulse amplitude, a 40% increase in pulse frequency, and a twofold increase in interpulse GH concentrations.250 In lean women with the polycystic ovary syndrome (PCOS), the amplitude, but not the frequency, of GH pulses is increased as compared with body mass index (BMI)-matched normal cycling controls. In contrast, pulse amplitude is similarly reduced in obese patients with PCOS and obese controls.251
Diurnal and nocturnal episodes of GH secretion are more frequent and of higher amplitude in adult subjects with hyperthyroidism, who have an overall daily GH production rate fourfold greater than normal.252 Patients with major endogenous depression often have the major nocturnal GH pulse before, rather than after, sleep onset.253
In Cushing’s disease, an inverse relationship has been noted between the degree of cortisol hypersecretion and total GH secretion, and the frequency and the disorderliness of GH pulses are increased.254 In acromegaly, GH is hypersecreted throughout the 24-hour span, with a highly irregular pulsatile pattern superimposed on elevated basal levels, indicating the presence of tonic secretion.255,256 After pituitary surgery, a normal 24-hour pattern of GH release can be restored in most, but not all, patients.255,257
The Lactotropic Axis
The 24-Hour Profile of Prolactin in Normal Subjects: Under normal conditions, the 24-hour profile of prolactin levels exhibits minimal levels around noon, a modest increase in the afternoon, and a major nocturnal elevation starting shortly after sleep onset and culminating around midsleep at levels corresponding to an average increase of more than 200% above minimum levels (see Fig. 4-3).258,259 Episodic pulses occur throughout the 24-hour span, but their amplitude and their frequency are higher during the night than during the day. Decreased dopaminergic inhibition of prolactin secretion during sleep is likely to be the primary mechanism underlying this nocturnal elevation. Mean prolactin levels, pulse amplitude, and pulse frequency are higher in normally cycling women than in postmenopausal women or in normal young men.260 In normally cycling young women, it has also been observed that daytime prolactin pulsatility was enhanced during the luteal phase as compared with the follicular phase, leading to increased late afternoon and evening levels.226 In the luteal phase, the magnitude of the evening prolactin rise correlated positively with both estradiol and progesterone levels.226 These data indicate that endogenous estrogens and progesterone play a critical role in the differential regulation of prolactin secretion associated with sex and age. Deconvolution analysis has shown that the prolactin profile reflects both tonic and intermittent release.119 Twin studies have revealed that genetic factors determine partially the temporal organization of prolactin secretion.261
Diurnal prolactin variations are regulated primarily by sleep-wake homeostasis. Sleep onset is invariably associated with an increase in prolactin secretion, irrespective of the time of the day. Thus, as illustrated in Fig. 4-3, shifts in the sleep-wake cycle are followed immediately by parallel shifts in the prolactin rhythm,222 but the amplitude of the prolactin rise may be dampened when associated with daytime sleep as compared with nocturnal sleep.262 Conversely, modest elevations in prolactin levels may occur during waking around the time of the usual sleep onset, particularly in women.259 Thus, prolactin secretion appears to be modulated in part by circadian rhythmicity, and maximal secretion occurs when sleep and circadian effects are superimposed (i.e., at the usual bedtime).258,259,263 Benzodiazepine (e.g., triazolam) and imidazopyridine (e.g., zolpidem) hypnotics taken at bedtime generally enhance the nocturnal prolactin elevation.264,265
A close temporal relationship has been evidenced between increased prolactin secretion and SW activity when sleep structure was characterized by power spectral analysis of the EEG.110 Conversely, prolonged awakenings that interrupt sleep are consistently associated with decreasing prolactin concentrations.110 Thus, SW sleep is associated with elevated prolactin secretion, and shallow and fragmented sleep generally is associated with dampening of the nocturnal prolactin rise. This is indeed observed in elderly subjects, who have a nearly 50% dampening of the nocturnal prolactin elevation (see Fig. 4-4).126,266
During pregnancy, serum prolactin levels rise but the 24-hour pattern of secretion is maintained, albeit at a higher level. During the postpartum period, prolactin secretory pulses follow suckling episodes, and the nocturnal rise, independent of suckling, is evident only after breastfeeding has ceased.267
Alterations in Disease States: Absence or blunting of the nocturnal increase in plasma prolactin has been reported in a variety of pathologic states, including uremia and breast cancer in postmenopausal women. In Cushing’s disease, prolactin levels are elevated throughout the 24-hour cycle, and the relative amplitude of the nighttime rise is reduced.268 In subjects with insulin-dependent diabetes, the circadian and sleep modulation of prolactin secretion is preserved, but overall levels are markedly diminished.269 Obesity is associated with an increase in overall prolactin secretion, in proportion to excess visceral fat, without alteration of the diurnal variation. This enhancement is achieved by an increase in the amplitude and duration of secretory pulses.270
In hyperprolactinemia associated with prolactinomas, or secondary to functional pituitary stalk disconnection, the number of prolactin pulses is increased, and the regularity of the pulsatile pattern is decreased.271,272 The nocturnal elevation in prolactin may be preserved.271,273 Selective removal of prolactin-secreting adenomas generally results in normalization of the prolactin pattern.
The Gonadotropic Axis
Normal Diurnal Profiles of Gonadotropins and Sex Steroids: Rhythms in the gonadotropic axis cover a wide range of frequencies, from episodic release in the ultradian range, to diurnal rhythmicity and menstrual cycles. These various rhythms interact to provide a coordinated temporal program that governs the development of the reproductive axis and its operation at every stage of maturation. The following description of the current state of knowledge in this area will be limited to 24-hour rhythms and their interaction with pulsatile release during adulthood.
Patterns of LH release in adult men exhibit episodic pulses with large interindividual variability.274 LH secretory episodes mainly reflect GnRH pulsatility. A recent study indicates that LH pulsatility is inhibited by acyl-ghrelin, suggesting that the ghrelin system may play a centrally mediated inhibitory role in the gonadal axis.275 The diurnal variation is of low amplitude or is even undetectable. During the sleep period, LH pulses appear to be temporally related to the REM–non-REM cycle.276 FSH profiles may show some occasional pulses, with no diurnal variation.
In contrast, a marked diurnal rhythm in circulating testosterone levels is present in young normal men, with minimal levels in the late evening and maximal levels in the early morning.175,277 In young adult men, the amplitude of the testosterone rhythm averages 25%.175 With a 15-minute sampling interval, 17 to 18 testosterone pulses per 24-hour span can be detected.175 Growing evidence suggests that diurnal testosterone variations are controlled primarily by the sleep-wake cycle. Experimental sleep fragmentation (schedule allowing 7 minutes of sleep every 20 minutes) results in dampening of the nocturnal testosterone rise, particularly in subjects who do not achieve REM sleep.278 Daytime sleep, as well as nocturnal sleep, is associated with a robust rise in testosterone levels.279 However, a progressive elevation in testosterone levels persists during nocturnal wakefulness, albeit blunted as compared with nocturnal sleep,279 indicating the existence of a circadian component that could reflect adrenal androgen secretion. Diurnal profiles of testosterone are paralleled by inhibin B variations, with peak values in the early morning and nadirs in the late afternoon, and significant cross-correlations between inhibin B and testosterone or estradiol have been detected.280
A progressive decline in testosterone levels, together with an increase in sex hormone–binding globulin (SHBG) levels, is observed in normal men from 30 years of age onward, so that the decrease in bioavailable testosterone is more important than the decline in total testosterone.281 In elderly men, the diurnal variation in testosterone is still present but may be markedly dampened.277,282 A strong positive correlation has been evidenced between total sleep time and morning testosterone levels.283 Pulsatile testosterone secretion is attenuated, suggesting possible partial desensitization of Leydig cells to LH.284 Mean LH levels are increased, but the amplitude of LH pulses is decreased,285,286 their frequency is increased,284 and no significant diurnal pattern can be detected.282 In contrast, pulsatile FSH secretion is increased in older men.287 In addition, older males secrete LH and testosterone more irregularly, and jointly more asynchronously, than do younger men.288
In adult women, diurnal profiles of LH, FSH, estradiol, and progesterone exhibit episodic pulses throughout the 24-hour span.289 The 24-hour variations in plasma LH are markedly modulated by the menstrual cycle.290 Nocturnal slowing of pulsatile LH secretion is observed during the early follicular phase.290 This nocturnal slowing is related specifically to sleep rather than to time of day, since it is also observed during daytime sleep but not during nighttime wake.291 During periods of sleep, LH pulses were found to occur preferentially in association with brief awakenings, suggesting an inhibitory effect of sleep on pulsatile LH secretion.291 Since night and shift work is consistently associated with shorter and more fragmented sleep, these results indicate that altered menstrual function, which frequently is observed in night and shift workers, could result directly from altered sleep patterns. Evening elevation of LH levels and LH pulse amplitude is observed in the absence of sleep, suggesting the existence of a circadian modulation of LH secretion.291 Circulating levels of LH and FSH and LH pulse frequency increase with aging and are higher in normal women older than 40 years of age with regular menstrual cycles than in women younger than age 40.292 Gonadotropin levels remain elevated after menopause.
Alterations in Disease States: Early studies on the 24-hour profile of plasma LH in anorexia nervosa have established the importance of an adequate temporal secretory program in the maintenance of normal reproductive function. In women with amenorrhea secondary to anorexia nervosa, the secretory pattern of LH regresses to the pubertal or prepubertal pattern, with low daytime pulsatility and increased secretion at night.293 Secretory profiles of LH usually return to normal following weight gain and clinical remission. Short-term fasting was shown to suppress pulsatile LH secretion while enhancing its regularity in young, but not older, men.294
In most men with idiopathic hypogonadotropic hypogonadism, LH pulses are undetectable.295 In a small number of patients, an early pubertal pattern, with enhanced pulse amplitude during the nighttime, may be observed.295 Attenuation of pulsatile LH secretion has been reported in men, but not in women, during both hypocortisolism and hypercortisolism. The authors have speculated that this sex difference could be due to a higher level of hypothalamic opioid activity in men.296 Poorly controlled type 1 diabetes mellitus has been found to be associated with decreased amplitude of pulsatile LH secretion.297
The Thyrotropic Axis
The 24-Hour Profile of Thyrotropin in Normal Subjects: In normal adult men and women, TSH levels are low and relatively stable throughout the daytime and begin to increase in the late afternoon or early evening. Maximal levels occur around the beginning of the sleep period.298 TSH levels progressively decline during the latter part of sleep, and daytime values resume shortly after morning awakening. Onset of the nocturnal rise in TSH well before sleep onset is believed to reflect a circadian effect. This 24-hour pattern of TSH levels appears to be generated by frequency, as well as amplitude modulation, of thyrotropin-releasing hormone (TRH)-driven secretory pulses.118 Studies involving sleep deprivation and shifts in the sleep-wake cycle have consistently indicated that sleep exerts an inhibitory influence on TSH secretion; sleep deprivation relieves this inhibition.298,299 It is interesting to note that when sleep occurs during the daytime, TSH secretion is not suppressed significantly below normal daytime levels.300 Profiles of plasma TSH during normal nocturnal sleep, nocturnal sleep deprivation, and daytime sleep are illustrated in the lower panel of Fig. 4-3. When depth of sleep at the habitual time is enhanced by previous sleep deprivation, inhibition of the nocturnal TSH rise is more pronounced than in basal conditions. Descending slopes of TSH concentrations during sleep are consistently associated with SW stages, and negative cross-correlations have been found between TSH fluctuations and SW activity,107,301 suggesting that SW sleep probably is the primary determinant of the sleep-associated TSH decrease. Conversely, awakenings are frequently associated with TSH increments.300 The timing of the TSH evening rise seems to be controlled by circadian rhythmicity and shifts in concordance with the melatonin rhythm following exposure to light or nocturnal exercise.58 Free triiodothyronine shows a diurnal rhythm that parallels TSH variations.302
Under conditions of sleep deprivation, the increased amplitude of the TSH rhythm may result in a detectable increase in plasma triiodothyronine levels, paralleling the nocturnal TSH rise,300 although negative findings also have been reported.303 If sleep deprivation is prolonged for a second night, the nocturnal rise in TSH is markedly diminished as compared with that occurring during the first night.303 It is likely that, following the first night of sleep deprivation, elevated thyroid hormone levels, which persist during the daytime period because of the prolonged half-life of these hormones, limit the subsequent TSH rise. A study involving 64 hours of sleep deprivation demonstrated, during the second night of sleep deprivation, a nocturnal increase in both triiodothyronine and thyroxine levels, contrasting with the decreases seen during normal sleep.304 These data suggest that prolonged sleep loss may be associated with upregulation of the thyroid axis. Consistent findings have been reported in a study of 6 days of partial sleep loss (4 hours in bed per night) wherein the nocturnal TSH rise was strikingly decreased and overall mean TSH levels were reduced by more than 30%, probably as the result of increased levels of thyroid hormones caused by TSH elevation at the beginning of sleep curtailment (see Fig. 4-8).194
Because inhibitory effects of sleep on TSH secretion are time dependent, elevations in plasma TSH levels may occur in conditions of misalignment of sleep and circadian timing. This is illustrated in Fig. 4-11, which shows the mean profiles of plasma TSH observed in a group of normal young men in the course of adaptation to a simulated jet lag involving an abrupt 8-hour advance of the sleep-wake cycle and the dark period, following a 24-hour baseline period.300 In the course of adaptation, TSH levels increased progressively because nighttime wakefulness was associated with large circadian-dependent TSH elevations, and daytime sleep failed to inhibit TSH. As a result, mean TSH levels were more than twofold higher following awakening from the second shifted period than during the same time interval after normal nocturnal sleep. This study indicates that the subjective discomfort and fatigue associated with jet lag may involve prolonged elevation of a hormonal concentration in the peripheral circulation.
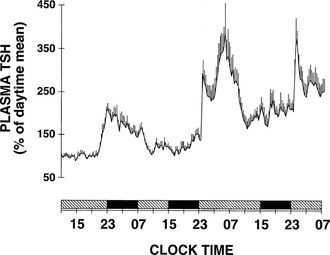
FIGURE 4-11 Mean (+standard error of the mean [SEM]) profile of plasma thyrotropin (TSH) from eight normal young men submitted to an 8-hour advance of the sleep-wake and light-dark cycles. Black bars indicate bedtime periods. (Data from Hirschfeld U, Moreno-Reyes R, Akseki E, et al: Progressive elevation of plasma thyrotropin during adaptation to simulated jet lag: effects of treatment with bright light or zolpidem. J Clin Endocrinol Metab 81:3270–3277, 1996.)
Aging is associated with a progressive decrease in overall TSH secretion (which is achieved by a decrease in amplitude, rather than in frequency, of secretory pulses) and in circulating TSH levels, and with dampening of the amplitude of the circadian variation.126 In subjects in the seventh and eighth decades, TSH levels are lower than in young adults throughout the 24-hour span, although the difference is more marked during sleep than during the daytime period (see Fig. 4-4). In middle-aged subjects, age-related decreases in TSH levels may be evidenced only in response to nocturnal sleep deprivation. Thus, it appears that TSH secretory capacity declines progressively with aging.
Alterations in Disease States: A decreased or absent nocturnal rise in TSH has been observed in a wide variety of nonthyroidal illnesses,305 which suggests that hypothalamic dysregulation generally will affect the circadian TSH surge. This contrasts with the circadian variation of plasma cortisol, which persists in a wide variety of disease states. The nocturnal TSH surge is diminished or absent in various conditions of hypercortisolism,306 as well as in hyperthyroidism and in primary and secondary hypothyroidism.307,308 In poorly controlled diabetic states, whether insulin-dependent or non–insulin-dependent, the surge also disappears.309 Correction of hyperglycemia is associated with a reappearance of the nocturnal elevation.309 It is interesting to note that morning TSH values in hyperglycemic patients do not differ from those of control subjects, and the TSH response to TRH is only marginally reduced.309 Obesity is associated with an increase in overall TSH secretion (which is achieved by an increase in amplitude, rather than in frequency, of secretory pulses) without alteration of the diurnal variation. TSH profiles tend to return toward normal levels after body weight loss induced by caloric restriction.310
Parathyroid Hormone
The 24-hour profile of circulating parathyroid hormone (PTH) levels shows a major nocturnal peak occurring at around 01.00 to 03.00 hours and morning minimal values at around 10.00 to 11.00 hours. This diurnal rhythm persists, albeit dampened, in constant routine conditions.311 Thus, it appears to be regulated primarily by the circadian pacemaker but modulated by other factors. Although serum calcium, the major modulator of PTH secretion, also may exhibit diurnal variations, the timing of the maximum was found to be highly variable among individuals and did not have any apparent relation to PTH.311 An early study suggested that the nocturnal rise in PTH was related to SW sleep,312 but in more recent studies, shifts in the sleep-wake cycle did not alter the timing of the nocturnal PTH peak.313 In contrast, this nocturnal rise was completely suppressed following a 4 day fast.314 Diurnal profiles of circulating PTH levels are temporally related to diurnal variations in urinary calcium and phosphate and are likely to play an important role in the optimization of calcium balance.311 The nocturnal rise in PTH has been reportedly abolished in osteoporotic women315 and in subjects with primary hyperparathyroidism.316
Hydromineral Hormones
Hormones of the renin-angiotensin-aldosterone system exhibit diurnal variations with higher nocturnal levels. These diurnal variations are regulated primarily by sleep-wake homeostasis, since shifts in the sleep-wake cycle are followed immediately by parallel shifts in hormonal profiles.104 During nocturnal and shifted sleep periods, a close temporal relationship has been evidenced between increases in SW activity and parallel increases in plasma renin activity (PRA) and aldosterone levels.104,317,318 Sleep-associated PRA pulses are blunted by awakenings,319 and nocturnal increases in PRA and aldosterone levels are dampened markedly by acute total sleep deprivation320 (Fig. 4-12).
FIGURE 4-12 Mean (+standard error of the mean [SEM]) 24-hour profiles of plasma aldosterone and plasma renin activity (PRA) in eight normal young men (21 to 28 years old) during a period of normal nocturnal sleep and a period of total acute sleep deprivation. (Adapted from Charloux A, Gronfier C, Chapotot F, et al: Sleep deprivation blunts the nighttime increase in aldosterone release in humans. J Sleep Res 10:27–33, 2001.)
In conditions of abnormal sleep architecture (e.g., narcolepsy, sleeping sickness), disturbances in the REM–non-REM cycle are faithfully reflected in the PRA temporal pattern.321
Vasopressin release is pulsatile but shows no apparent relationship to sleep stages.321 Diurnal variations in plasma levels of atrial natriuretic peptide (ANP) have been evidenced in some, but not all, studies, and the existence of a circadian rhythm of ANP is still a matter of controversy.322
Glucose Tolerance and Insulin Secretion
Diurnal and Ultradian Variations in Normal Subjects
In normal man, glucose tolerance varies with the time of day. Fig. 4-13 shows circadian variations in glucose tolerance to oral glucose, identical meals, constant glucose infusion, and continuous enteral nutrition. In all four conditions, plasma glucose levels are markedly higher in the evening than in the morning.105 Studies of fasting during nocturnal sleep have consistently observed that, despite the prolonged fasting condition, glucose levels remain stable or decrease only minimally during the night, contrasting with a clear decrease during daytime fasting. Thus, a number of mechanisms operative during nocturnal sleep are likely to maintain stable glucose levels during the overnight fast. Experimental protocols involving intravenous glucose infusion or enteral nutrition while allowing for normal nocturnal sleep have shown that glucose tolerance deteriorates further as the evening progresses, reaches a minimum around midsleep, and then improves to return to morning levels.178,323 During the first half of sleep, SWS is the dominant sleep stage. During SWS, cerebral glucose utilization is lower than during either wake or REM sleep.324,325 A strong correlation between SWA and regional blood flow in the prefrontal regions was demonstrated by positron emission tomography (PET) scans of subjects who were undergoing continuous polygraphic recordings.324,326 Brain glucose metabolism represents 30% to 50% of total body glucose utilization327,328; therefore, a robust link between SWS and glucose tolerance should be expected. Evidence also indicates that the diurnal variation in glucose tolerance is driven in part by the wide and highly reproducible diurnal rhythm of plasma cortisol, an important counterregulatory hormone.105,200,329 Indeed, the diurnal variation in insulin secretion was found to be inversely related to the cortisol rhythm, with significant correlation of the magnitudes of morning to evening excursions. Rises in plasma levels of glucose and insulin following short-term elevations in plasma cortisol are more pronounced in the evening than in the morning.200 Diminished insulin sensitivity and decreased β cell responsiveness are involved in reduced glucose tolerance later in the day. Under conditions of constant glucose infusion, sleep-associated rises in glucose were found to correlate with the amount of concomitant GH secreted. Thus, during the first part of the night, decreased glucose tolerance is due to decreased glucose utilization both by peripheral tissues—resulting from muscle relaxation and rapid insulin-like effects of sleep-onset GH secretion—and by the brain.325,330 During the second part of the night, these effects subside as sleep becomes shallow and more fragmented and GH is no longer secreted. Thus, complex interactions of circadian and sleep effects, possibly mediated in part by cortisol and GH, result in a consistent pattern of changes in set point of glucose regulation over the 24-hour period. A recent study has demonstrated a continuous decline in plasma glucagon levels during nighttime sleep in healthy nondiabetic subjects.331 Because this nocturnal decline is preserved in patients with type 1 diabetes, it has been suggested that nocturnal regulation of spontaneous glucagon release occurs independent of circulating glucose and insulin levels.331
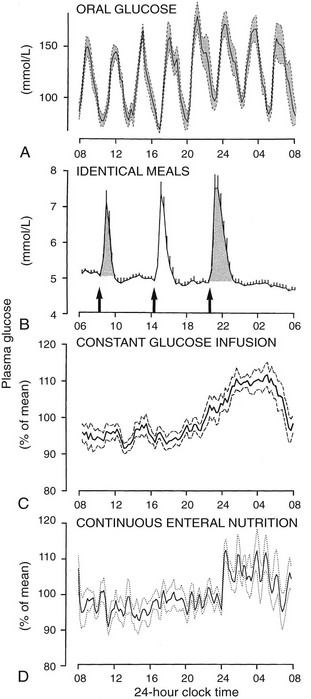
FIGURE 4-13 Mean (and standard error of the mean [SEM]) 24-hour pattern of plasma glucose changes in response to oral glucose 50 g every 3 hours, identical meals, constant glucose infusion, and continuous enteral nutrition in normal young adults. (From Van Cauter E, Polonsky KS, Scheen AJ: Roles of circadian rhythmicity and sleep in human glucose regulation. Endocr Rev 18:716–738, 1997.)
Consistent with the important modulatory effects of sleep on glucose regulation, recurrent sleep loss is associated with marked alterations in parameters of glucose tolerance. In a study of sleep curtailment (4 hours in bed per night for 6 nights) performed in young healthy subjects,194,332 the overall glucose response to breakfast was increased and subjects were more insulin resistant, as assessed by an elevation in the homeostatic model assessment (HOMA) index (see Fig. 4-8). Moreover, following an intravenous glucose tolerance test, insulin release was reduced by 30%, and glucose tolerance was found to fall in the range observed in older adults with impaired glucose tolerance. The deleterious impact of sleep restriction on glucose metabolism was subsequently confirmed in a follow-up study that used a randomized crossover design.333 Recently, a laboratory study in young healthy adults demonstrated that reduced sleep quality, without change in sleep duration, also has a clear negative impact on glucose tolerance. Indeed, this study indicated that all-night selective suppression of SWS, without any change in total sleep time, resulted in marked decreases in insulin sensitivity without adequate compensatory increases in insulin release, leading to reduced glucose tolerance and increased diabetes risk.334 To date, seven prospective epidemiologic studies have found an association between short and/or poor sleep and diabetes risk, even after controlling for many covariates such as BMI, shift work, hypertension, exercise, and depression (reviewed in reference 335).
Human insulin secretion is a complex oscillatory process involving rapid pulses of small amplitude that recur every 10 to 15 minutes superimposed on slower ultradian oscillations with periods in the 90- to 120-minute range.121,336 The ultradian oscillations are tightly coupled to glucose, with a tendency for glucose pulses to lead insulin pulses by 10 minutes, and they have been shown to promote more efficient glucose utilization.125 They are best seen in conditions in which insulin secretion is stimulated, including ingestion of a meal, continuous enteral nutrition, and constant intravenous glucose infusion.121,323 Under these conditions, their relative amplitude is about 50% to 70% for insulin secretory pulses and 20% for plasma glucose. Their amplitude is maximal immediately after a meal, then decreases progressively. Moreover, the periodicity of the insulin secretory oscillations can be entrained to the period of an oscillatory glucose infusion,337 thus supporting the concept that these ultradian oscillations are generated by the glucose-insulin feedback mechanism.338 However, ultradian oscillations, but less regular and of smaller amplitude, are still present in fasting conditions. Stimulatory effects of sleep on insulin secretion are mediated by an increase in the amplitude of the oscillation.323 During constant glucose infusion, REM sleep and wake episodes coincide significantly with decreasing levels of glucose and insulin, and increasing glucose levels occur during the deeper stages of non-REM sleep.330
Rapid 10- to 15-minute pulsations seem to have a different origin than ultradian oscillations. Indeed, they may appear independently of glucose, since they have been observed in the isolated perfused pancreas and in perifused islets.121 Rapid insulin pulsations also have been observed in perfused human islets.339 Administration of insulin by pulsatile infusion improves insulin-mediated glucose uptake.340 The frequency, amplitude, and regularity of rapid insulin pulses are decreased by aging.341 Omental lipolysis is also pulsatile and has a rapid frequency, similar to that of insulin.342 However, the oscillation of free fatty acids appears to be driven by the central nervous system, rather than by insulin.342
Alterations in Disease States
In obese and diabetic subjects, diurnal and ultradian variations in glucose regulation are abnormal. In obesity, the morning versus evening difference in glucose tolerance is abolished. Obese adult subjects show no diurnal variation in glucose tolerance, no decline in insulin sensitivity in the afternoon, and only a marginally significant decline in β cell responsiveness to glucose in the latter part of the day.343
In patients with insulin-dependent diabetes, an increase in glucose levels and/or insulin requirements occurs during a prebreakfast period ranging from 05:00 to 09:00 hours and has been called the dawn phenomenon.344 A role for nocturnal GH secretion in the pathogenesis of the dawn phenomenon has been demonstrated.345,346 The observation of a dawn phenomenon in patients with non–insulin-dependent diabetes mellitus (NIDDM) under normal dietary conditions has been less consistent. However, prominent late night and early morning elevations in glucose levels and insulin secretion in both normal subjects and diabetic patients become apparent during prolonged fasting.347
Counterregulatory mechanisms that are already deficient in type 1 diabetes are further impaired during sleep as compared with wakefulness, as autonomic responses to hypoglycemia are reduced during sleep in diabetic patients. As a result, patients with type 1 diabetes are substantially less likely to be awakened by hypoglycemia than are nondiabetic subjects.348 Patients with type 1 diabetes present distinct alterations in sleep architecture and nocturnal neuroendocrine release.349 Levels of GH, epinephrine, ACTH, and cortisol are elevated, and patients have less deep SWS and more shallow stage II sleep.349
The rapid and ultradian oscillations of insulin secretion are perturbed in NIDDM and in impaired glucose tolerance without hyperglycemia.121,350,351 Ultradian oscillations, which have an exaggerated amplitude in obese subjects without apparent changes in frequency or pattern of recurrence, are also more irregular and of lower amplitude in subjects with established NIDDM.121
Hormones Involved In Appetite Regulation
Leptin is an anorexigenic hormone, mainly released by the adipocytes, that provides information about energy status to hypothalamic regulatory centers.352,353 As illustrated in Fig. 4-14, plasma leptin levels in normal lean men and women show a robust diurnal rhythm, with minimal values during the daytime and a nocturnal rise with maximal values during early sleep to midsleep.354 The amplitude of the diurnal variation averages 25% to 30% of the mean level.355
FIGURE 4-14 Mean (±standard error of the mean [SEM]) 24-hour profiles of plasma ghrelin, insulin, and leptin levels in 10 healthy subjects 29 to 64 years of age (body mass index [BMI], 22 to 30 kg/m2) receiving breakfast, lunch, and dinner at 08.00, 12.00, and 17.30 hours, respectively. (Adapted from Cummings DE, Purnell JQ, Frayo RS, et al: A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes 50:1714–1719, 2001.)
The timing of the daily maximum of plasma leptin levels is markedly dependent on the timing of meals, as shifts in meal timings induce immediate shifts in leptin profiles: Fasting and eating are associated with a decrease and an increase in leptin levels, respectively.356 However, the diurnal rhythm was found to persist, albeit with a smaller amplitude, in subjects who received continuous enteral nutrition,323 and in subjects who received identical snacks at 2-hour intervals.357 The rhythm also persisted in subjects who were submitted to 38 or 88 consecutive hours of wakefulness.357,358 Following an abrupt shift in the sleep period, nocturnal leptin levels rose despite the absence of sleep, and a second rise was observed following the onset of daytime recovery sleep.323 Thus, leptin diurnal variations reflect the combined effects of the circadian pacemaker, the sleep-wake cycle, and the schedule of food intake.
Several studies have reported that human leptin levels, including those in subjects receiving continuous enteral nutrition rather than separate meals, are pulsatile.323,355,359–361
Leptin levels reflect cumulative energy balance, with a decline or an increase in response to underfeeding or overfeeding, respectively.362,363 These changes have been found to be associated with reciprocal changes in hunger.363 Circulating leptin concentrations are higher and the relative amplitude of their diurnal variation is lower in obese subjects than in normal-weight controls.355 Marked sex differences have been reported in 24-hour mean leptin levels, which are twofold to tenfold higher in women than in men, regardless of fat mass.355,361 In anorexia nervosa, as in amenorrheic female athletes, leptin levels are low and diurnal variations are abolished.364,365 Aging is associated with dampening of the amplitude of the 24-hour rhythm of plasma leptin and an advance in the nocturnal acrophase.366
Sleep restriction (2 to 6 nights of 4-hour bedtimes) under controlled conditions of caloric intake and physical activity is associated with a 20% to 30% reduction in mean leptin levels, acrophase, and amplitude of the diurnal variation.332,367 The magnitude of this impact of sleep restriction on leptin levels is comparable with that observed in young adults under normal sleep conditions after 3 days of dietary restriction by approximately 900 kcal per day.363 Consistent findings have been obtained in two epidemiologic studies that found an association between short sleep and lower morning leptin levels after controlling for BMI368 or the degree of adiposity.369 Diurnal variations in leptin and cortisol levels form an approximate mirror image,360,361,370,371 and in fully rested subjects, maximal leptin levels coincide with minimal cortisol levels,332 consistent with the well-documented action of leptin on HPA activity.353 Recurrent partial sleep deprivation results in an advance in the leptin acrophase such that high levels of leptin occur when cortisol concentrations are still elevated relative to the nocturnal nadir, possibly acting in concert to increase appetite at the end of the day.332
Normal 24-hour leptin levels and diurnal leptin variations were found in patients with primary adrenal failure.371 Increased leptin levels,372,373 caused by equal amplification of basal and pulsatile secretion,374 with preservation of the diurnal pattern372 have been reported in patients with Cushing’s syndrome. A normal diurnal leptin pattern also has been reported in patients with GH deficiency375 or with perinatal stalk-transection syndrome.376
Another secretory product of differentiated adipocytes is adiponectin, a hormone that enhances insulin sensitivity.377,378 In normal-weight men, adiponectin levels exhibit ultradian pulsatility, as well as diurnal variation, with a significant nocturnal decline, reaching minimal values in the early morning.379 So far, possible relations between adiponectin rhythm and sleep have not been reported. Adiponectin levels are decreased, adiponectin pulsatility is blunted, and diurnal variations are abolished in obese subjects.380–382 In severely obese subjects, massive weight loss coupled with the reversibility of insulin resistance is associated with a restoration of adiponectin pulsatility.382
Ghrelin and Peptide YY
Ghrelin is an orexigenic hormone that is secreted primarily by the stomach and the duodenum.221,383,384 Ghrelin also stimulates GH secretion and displays ACTH- and prolactin-releasing activities.221 One report claimed that ghrelin was found to promote SW sleep in man,385 but others found suppressive effects on sleep in rats.386 Ghrelin secretion is pulsatile.381,387 Daytime profiles are regulated primarily by the schedule of food intake: Levels rise sharply before each designated mealtime and fall to trough levels within 1 hour after eating. A study examining spontaneous meal initiation in the absence of time- and food-related cues provided good evidence of a role for ghrelin in meal initiation.388 This pattern seems to be exaggerated after the dinner meal, as ghrelin levels peak at around 01:00 hours and remain elevated until the latter part of the night, when they tend to decrease spontaneously389–391 (see Fig. 4-14). Ghrelin levels are increased in anorexia nervosa and decreased in young obese subjects.390,392 In obese subjects, the diurnal pattern was found to remain largely unaltered390 or to be markedly blunted.381 A diet-induced weight loss was associated with increased 24-hour ghrelin levels.390
Diurnal variations in ghrelin levels persist after 3 days of total fasting. However, the timings of acrophase and nadir are not consistent across studies.393 Studies concur in indicating that ghrelin levels are not increased by prolonged fasting, and that women have higher levels than men.393 Diurnal variations in ghrelin likely reflect the combined effects of the circadian pacemaker, the schedule of food intake, and possibly the sleep-wake cycle.
Acute total sleep deprivation was found to be associated with a blunted but prolonged nocturnal ghrelin elevation.236 A 2-day sleep restriction (4 hours bedtime per night) under controlled conditions of caloric intake and physical activity was reportedly associated with a nearly 30% elevation in daytime levels (Fig. 4-15).367 Consistent with these laboratory findings is the recent observation of a 22% increase in plasma ghrelin levels after a single night of total sleep deprivation.394 In a large epidemiologic study, short sleep was associated with higher ghrelin levels after controlling for sex, age, and BMI.368
FIGURE 4-15 Mean (and standard error of the mean [SEM]) daytime profiles of plasma leptin and ghrelin and of hunger ratings in healthy young men after 2 days of 10-hour bedtimes and 2 days of 4-hour bedtimes. (Data from Spiegel K, Tasali E, Penev P, Van Cauter E: Brief communication: sleep curtailment in healthy young men is associated with decreased leptin levels, elevated ghrelin levels, and increased hunger and appetite. Ann Intern Med 141:846–850, 2004.)
Another gut hormone that is thought to play an important role in the regulation of hunger and body weight is peptide YY (PYY), which is synthesized by gut-endocrine L cells.395 Exogenous administration of PYY3-36 reduces food intake in obese humans and rodents. The diurnal variation of plasma PYY levels in normal adults mirrors that of ghrelin, both after meal intake and during the overnight fast.
Conditions of Altered Sleep and Circadian Rhythmicity
• Elevated evening total and free cortisol concentrations,194,332 strikingly similar to those observed in the elderly.126,195,196 This disturbance may reflect decreased efficacy of the negative feedback regulation of the hypothalamic-pituitary-adrenal axis and could promote the development of insulin resistance and memory impairments.200,396
• A clinically significant impairment of carbohydrate tolerance,194,332,333 consistent with a state of impaired glucose tolerance as observed in older adults.
• A decrease in circulating levels of leptin,332,367 an anorexigenic hormone, and a concomitant increase in circulating levels of ghrelin,367 an orexigenic hormone (see Fig. 4-15). Moreover, sleep curtailment was associated with an increase in hunger, and this increase in hunger was strongly correlated with the increase in the ghrelin-to-leptin ratio.367
These data suggest that recurrent partial sleep curtailment may increase the risk for obesity and diabetes and may accelerate the senescence of endocrine and metabolic function. An obvious limitation of these studies is that the investigation period was not extended beyond 6 days. Thus, a progressive adaptation to chronic partial sleep deprivation cannot be excluded. However, the findings from these laboratory studies are consistent with the conclusions of epidemiologic studies. Several prospective cross-sectional epidemiologic studies, which varied considerably in geographic location and subject population, were remarkably consistent in indicating that short sleep may increase the risk of developing type 2 diabetes and/or obesity (reviewed in references 335 and 397). One major limitation of all of these epidemiologic studies is that they used only subjective reporting of sleep. Laboratory and epidemiologic studies now need to be complemented by large field studies incorporating objective measures of sleep duration and interventional methods to enhance our understanding of the mechanisms linking sleep loss to endocrine and metabolic alterations. Given the morbidity and mortality associated with obesity and diabetes, the identification of novel risk factors that are potentially modifiable, such as sleep curtailment, is particularly important.
Sleep Disorders
A recent laboratory study has shown that decreased sleep quality without a change in sleep duration results in a marked decrease in insulin sensitivity, without appropriate compensation of insulin release.334 Thus, diabetes risk was markedly increased. This study suppressed SWS, thus replacing deep non-REM sleep with shallow non-REM sleep, as occurs in the course of normal aging and in a variety of sleep disorders, including obstructive sleep apnea (OSA). It is important to note that this study demonstrated that a sleep disruption can cause hormonal and metabolic alterations.
Obstructive Sleep Apnea
OSA is the most common sleep disorder, and its incidence is rising rapidly in parallel with the current epidemic of obesity. A few studies have examined pituitary hormonal release in patients with OSA before and after treatment.398–400 Nocturnal release of the two pituitary hormones that are markedly dependent on sleep (i.e., GH and prolactin) is decreased in untreated apneic subjects. As illustrated in Fig. 4-16, treatment with continuous positive airway pressure (CPAP) results in a clear increase in the amount of GH secreted during the first few hours of sleep.398,399 The total amount of prolactin secreted during the sleep period is not modified by CPAP treatment, but the frequency of prolactin pulses is restored to values similar to those observed in normal subjects.400 Nocturnal LH and testosterone secretions are decreased in men with untreated OSA. These alterations are partially corrected during long-term CPAP treatment.401
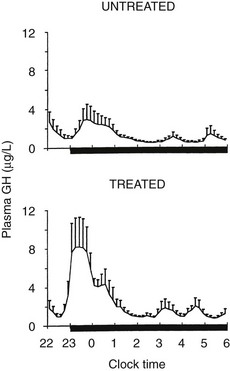
FIGURE 4-16 Mean (+standard error of the mean [SEM]) profiles of plasma growth hormone (GH) in patients with sleep apnea studied before and after treatment with continuous positive airway pressure (CPAP). (Data from Saini et al: Adapted from Van Cauter E, Spiegel K: Circadian and sleep control of endocrine secretions. In Turek FW, Zee PC [eds]: Neurobiology of Sleep and Circadian Rhythms. In Lenfant C: Lung Biology in Health and Disease, vol 133. New York, Marcel Dekker, 1999, pp 397–426.)
Morning leptin levels are elevated in patients with OSA when compared with weight-matched nonapneic subjects, and CPAP treatment decreases morning leptin levels.402–409
Whereas obesity is a major risk for OSA, OSA is now recognized as a risk for insulin resistance, independently of BMI, as supported by a large set of nine cross-sectional studies that have assessed OSA by polysomnography (reviewed in reference 410). All but the earliest study (which also involved the smallest sample size) found an association between increased severity of OSA and alterations in glucose metabolism consistent with an increased risk for diabetes. The only prospective study that used polysomnography to assess OSA did not find an independent relationship between severity of OSA at baseline and incident diabetes, but the duration of follow-up was only 4 years. Findings from clinic-based studies are largely consistent with those of epidemiologic studies. Indeed, despite differences in sample size, study design, measurement techniques, cut points, and control for possible confounders, most clinic-based studies (10 out of 13) were consistent in finding an independent association between OSA and abnormal glucose metabolism.410
Accumulating evidence suggests that metabolic abnormalities can be partially corrected by CPAP treatment, which supports the concept of a causal link between OSA and altered glucose control.410,410a When hyperinsulinemic euglycemic clamp evaluations were performed in nondiabetic patients with OSA, it was found that CPAP significantly improves insulin sensitivity after 2 days of treatment, and this improvement persists after nearly 3 years of treatment. In obese patients with type 2 diabetes, insulin sensitivity was improved after 3 months, but not after 2 days, of CPAP treatment. This finding suggests that the time course of improvement may be longer in obese patients who have diabetes. Two studies found that postprandial interstitial glucose levels and elevated hemoglobin A1C levels were reduced by CPAP use in type 2 diabetes. A population-based study showed reductions in fasting insulin levels and insulin resistance (estimated by HOMA) after 3 weeks of CPAP treatment in men with OSA compared with nonapneic controls followed over the same time period without CPAP. However, several studies did not show a beneficial effect of CPAP treatment on glucose metabolism.410 Conflicting results could be due to differences in sample sizes and populations, variable durations of therapy, variable compliance, and changes in body composition during the study period.
The overall prevalence of OSA in diabetic men has been estimated at 23%, compared with 6% in a community-based sample.411 However, preliminary analysis of cross-sectional data from a multicenter study recently revealed an exceptionally high prevalence of undiagnosed OSA in obese patients with type 2 diabetes, with more than 75% of patients having moderate to severe OSA diagnosed by polysomnography.412 These remarkable associations raise the possibility that OSA may be a novel risk factor for type 2 diabetes and/or, conversely, that chronic hyperglycemia may promote OSA. Whether treatment for OSA may delay the development or reduce the severity of type 2 diabetes is another important question.
Other Sleep Disorders
Narcolepsy is a sleep disorder that is characterized by excessive daytime sleepiness, reduced quality of nocturnal sleep with sleep-onset REM episodes, and cataleptic attacks. Narcolepsy is caused by impaired orexin (hypocretin) neurotransmission. Consistent with the role of orexin in the control of energy balance, narcoleptic patients have increased BMI and lower basal metabolism. The 24-hour rhythm of ACTH and cortisol persists in narcolepsy, suggesting that the circadian clock is not affected.413,414 In contrast, the 24-hour profiles of hormones known to be dependent on sleep-wake homeostasis, such as GH and prolactin, are markedly disrupted, with dampened or absent nocturnal GH and prolactin release.413,415 Leptin levels are decreased, and the nocturnal rise is abolished.416
Despite the high prevalence of insomnia in modern society, very little is known regarding the neuroendocrine and metabolic consequences of poor or insufficient sleep in this condition. The 24-hour profiles of ACTH and cortisol have been assessed in patients with insomnia who were monitored in a sleep laboratory for four consecutive nights. An increase in ACTH and cortisol secretion was observed in the evening and the early part of the night in patients who had objectively documented short total sleep time and poor sleep efficiency. However, patients with insomnia who had a normal sleep time did not show alterations in ACTH and cortisol profiles.186 Certain, much less prevalent, forms of sleep disorders seem to originate from a disturbance in the circadian system. Delayed sleep phase insomnia is characterized by a chronic inability to fall asleep at a normal bedtime and to awake in the morning. Nonpharmacologic chronotherapy involving repeated scheduled exposure to bright light is the treatment of choice for this disorder.417 In contrast, in the advanced sleep phase syndrome, the timing of the major sleep episode is advanced in relation to normal bedtime, resulting in symptoms of extreme evening sleepiness and early morning awakening. Familial forms of this syndrome may reflect an autosomal dominant mutation.418
Circadian Misalignment: Circadian rhythms provide synchronization with pronounced periodic fluctuations in the external environment and organize the internal milieu so that coordination and synchronization of internal processes are evident. External synchronization is of obvious importance for the survival of the species and ensures that the organism does the “right thing” at the right time of the day. Of equal importance is the fact that the circadian clock system provides internal temporal organization (i.e., internal synchrony) between the myriad of biochemical and physiologic systems in the body. The concept of internal synchrony had to be reevaluated in recent years in view of the discovery that circadian clock genes, which are part of the transcription-translation feedback loop that generates self-sustained oscillations in the central master circadian clock in the SCN, also are expressed rhythmically in other regions of the brain and in a variety of peripheral tissues, including adipocytes, hepatocytes, pancreatic β cells, cardiomyocytes, and vascular smooth muscle cells.20,419 Under normal conditions, the central SCN pacemaker maintains synchrony between central and peripheral oscillators. In conditions of circadian misalignment, such as those that occur in jet lag and shift work, the alignment of central and peripheral oscillators is disrupted. Thus, the concept of internal desynchrony has to include desynchrony between different centrally controlled rhythms (e.g., cortisol vs. GH) and desynchrony between central and peripheral rhythms. Lack of synchrony within the internal environment may lead to chronic difficulties with serious consequences for the health and well-being of the organism. The physical and mental malaise that occurs following rapid travel across time zones (i.e., the jet lag syndrome) and the pathologies associated with long-term shift work are assumed to be due in part to alterations in the normal phase relationships between various internal rhythms. In addition, it has been speculated that alterations in internal phase relationships between rhythms underlie certain forms of affective illness.
Jet Lag: Subjects who travel rapidly across time zones are confronted with a desynchronization between their internal circadian rhythms and the periodicity of the new external environment. Upon arrival, the timings of the light-dark cycle, social schedule, and meals are abnormally matched to the phase of physiologic rhythm of the traveler. Associated with this lack of synchronization are symptoms of fatigue, subjective discomfort, sleep disturbances, reduced mental and psychomotor performance, and gastrointestinal disorders.
The rate of adaptation is generally slower for overt rhythms that are strongly dependent on the circadian system, such as those of cortisol and melatonin secretions, than for those that are markedly modulated by sleep-wake homeostasis, such as prolactin and GH secretions. As a result, during the period of adaptation, abnormal phase relationships between overt rhythms occur. Thus, the jet lag syndrome involves not only desynchronization between internal and external rhythms but also perturbation of internal temporal organization of physiologic functions. Depending on the strength of the zeitgebers, the rate of adaptation can be as low as half an hour a day or as high as 3 hours a day. The rate of adaptation is not constant: Adaptation to a large shift occurs at a faster rate during the first few days and progresses at a slower pace thereafter.420 The rate of adaptation is also dependent on the direction of the shift, with adaptation generally occurring faster after a delay (i.e., westward) shift than after an advance (i.e., eastward) shift.420 This eastward-westward difference in rate of adaptation is believed to be due to the fact that the endogenous circadian period of the human is slightly longer than 24 hours, and thus adjustment by delays is achieved more easily than adjustment by advances. Strong evidence suggests that reentrainment after a transmeridian flight is facilitated by exposure to bright light at appropriate circadian phases. It is widely believed that adherence to the local social and meal schedule upon arrival will accelerate adaptation to jet lag, but this has not been rigorously demonstrated. Laboratory studies suggest that physical exercise scheduled during the period corresponding to the nighttime before travel will facilitate adaptation to a delay (i.e., westward) shift.57,421
Shift Work: Shift work, which is—voluntarily or not—accepted by millions of workers, is a major health hazard, involving increased risks for cardiometabolic illness, gastrointestinal disorders, infertility, and insomnia.422–424 Epidemiologic studies have indicated that shift work is a risk factor for weight gain,425 dyslipidemia,426,427 and insulin resistance.427,428 The medical consequences of shift work are associated with chronic misalignment of physiologic circadian rhythms and the activity-rest cycle. In addition, shift work almost invariably results in substantial sleep loss because daytime sleep generally is shorter and more fragmented than nocturnal sleep. Shift work usually creates conditions in which some zeitgebers (e.g., an artificial light-dark cycle) and additional phase-setting factors such as the rest-activity cycle are shifted, while others (e.g., the natural light-dark cycle, the routines of family life) remain unaltered. Shift workers thus live in a situation of conflicting zeitgebers that almost never allow a complete shift of the circadian system. Indeed, several studies have shown that workers on permanent or rotating night shifts do not adapt to these schedules, even after several years.227,429,430
Besides its health implications, this misalignment with the circadian system has important social and economic implications, because night work is associated with substantial decrements in performance and vigilance, resulting in diminished productivity and increased accident rates. Scheduled exposure to bright light during night work and complete darkness during daytime sleep following night work can accelerate adjustment to the new schedule and improve nighttime alertness and performance; exogenous melatonin may facilitate sleep at abnormal circadian phases.431
References
1. Rosenwasser, AM, Turek, FW. Physiology of the Mammalian Circadian System. Section 4—Chronobiology. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practices of Sleep Medicine. ed 4. New York: WB Saunders; 2005:351–362.
2. Turek, FW, Dugovic, C, Laposky, A. Master Circadian Clock, Master Circadian Rhythm. Section 4—Chronobiology. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practices of Sleep Medicine. ed 4. New York: W B Saunders; 2005:318–320.
3. Turek, FW. Circadian rhythms. Horm Res. 1998;49:103–113.
4. Aschoff, J. Circadian rhythms: general features and endocrinological aspects. In: Krieger DT, ed. Endocrine Rhythms. New York: Raven Press; 1979:1–61.
5. Czeisler, CA, Duffy, JF, Shanahan, TL, et al. Stability, precision, and near-24-hour period of the human circadian pacemaker. Science. 1999;284:2177–2181.
6. Saper, CB, Scammell, TE, Lu, J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437:1257–1263.
7. Lehman, MN, Silver, R, Bittman, EL. Anatomy of suprachiasmatic nucleus grafts. In: Klein DC, Moore RY, Reppert SM, eds. Suprachiasmatic Nucleus: The Mind’s Clock. New York: Oxford University Press; 1991:349–374.
8. Ralph, M, Foster, RG, Davis, FC, et al. Transplanted suprachiasmatic nucleus determines circadian period. Science. 1990;247:975–978.
9. Abe, M, Herzog, ED, Yamazaki, S, et al. Circadian rhythms in isolated brain regions. J Neurosci. 2002;22:350–356.
10. Earnest, DJ, Sladek, CD. Circadian vasopressin release from perifused rat suprachiasmatic explants in vitro: effects of acute stimulation. Brain Res. 1987;422:398–402.
11. Gillette, MU. SCN electrophysiology in vitro: rhythmic activity and endogenous clock properties. In: Klein DC, Moore RY, Reppert SM, eds. Suprachiasmatic Nucleus—The Mind’s Clock. New York: Oxford University Press, 1991.
12. Panda, S, Antoch, MP, Miller, BH, et al. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell. 2002;109:307–320.
13. Welsh, DK, Logothetis, DE, Meister, M, et al. Individual neurons dissociated from rat suprachiasmatic nucleus express independently phased circadian firing rhythms. Neuron. 1995;14:697–706.
14. Lee, HS, Billings, HJ, Lehman, MN. The suprachiasmatic nucleus: a clock of multiple components. J Biol Rhythms. 2003;18:435–449.
15. Laposky, A, Easton, A, Dugovic, C, et al. Deletion of the mammalian circadian clock gene BMAL1/Mop3 alters baseline sleep architecture and the response to sleep deprivation. Sleep. 2005;28:395–409.
16. Lowrey, PL, Takahashi, JS. Mammalian circadian biology: elucidating genome-wide levels of temporal organization. Annu Rev Genomics Hum Genet. 2004;5:407–441.
17. Vitaterna, MH, Pinto, LH, Turek, FW. Molecular Genetic Basis for Mammalian Circadian Rhythms. Section 4—Chronobiology. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practices of Sleep Medicine. ed 4. New York: WB Saunders; 2005:363–374.
18. Miller, BH, Olson, SL, Turek, FW, et al. Circadian clock mutation disrupts estrous cyclicity and maintenance of pregnancy. Curr Biol. 2004;14:1367–1373.
19. Turek, FW, Joshu, C, Kohsaka, A, et al. Obesity and metabolic syndrome in circadian clock mutant mice. Science. 2005;308:1043–1045.
20. Laposky, AD, Bass, J, Kohsaka, A, et al. Sleep and circadian rhythms: key components in the regulation of energy metabolism. FEBS Lett. 2008;582:142–151.
21. Reppert, SM, Weaver, DR. Coordination of circadian timing in mammals. Nature. 2002;418:935–941.
22. Yoo, SH, Yamazaki, S, Lowrey, PL, et al. PERIOD2::LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc Natl Acad Sci U S A. 2004;101:5339–5346.
23. Dijk, DJ, Czeisler, CA. Contribution of the circadian pacemaker and the sleep homeostat to sleep propensity, sleep structure, electroencephalographic slow waves, and sleep spindle activity in humans. J Neurosci. 1995;15:3526–3538.
24. Wright, KP, Jr., Hughes, RJ, Kronauer, RE, et al. Intrinsic near-24-h pacemaker period determines limits of circadian entrainment to a weak synchronizer in humans. Proc Natl Acad Sci U S A. 2001;98:14027–14032.
25. Card, JP, Moore, RY. The organization of visual circuits influencing the circadian activity of suprachiasmatic nucleus. In: Klein DC, Moore RY, Reppert SM, eds. Suprachiasmatic Nucleus: The Mind’s Clock. New York: Oxford University Press; 1991:51–76.
26. Underwood, H. Circadian Organization in Nonmammalian Vertebrates. In: Takahashi JS, Turek FW, Moore RY, eds. Handbook of Behavioral Neurobiology. New York: Plenum Press, 2001.
27. Foster, RG, Provencio, I, Hudson, D, et al. Circadian photoreception in the retinally degenerate mouse (rd/rd). J Comp Physiol A. 1991;169:39–50.
28. Panda, S, Sato, TK, Castrucci, AM, et al. Melanopsin (Opn4) requirement for normal light-induced circadian phase shifting. Science. 2002;298:2213–2216.
29. Panda, S, Provencio, I, Tu, DC, et al. Melanopsin is required for non-image-forming photic responses in blind mice. Science. 2003;301:525–527.
30. Lupi, D, Oster, H, Thompson, S, et al. The acute light-induction of sleep is mediated by OPN4-based photoreception. Nat Neurosci. 2008;11:1068–1073.
31. Guler, AD, Ecker, JL, Lall, GS, et al. Melanopsin cells are the principal conduits for rod-cone input to non-image-forming vision. Nature. 2008;453:102–105.
32. Turek, FW. Lighting up the brain. Journal of Biological Rhythms. 1999;14:171.
33. Foster, RG. Seeing the light … in a new way. J Neuroendocrinol. 2004;16:179–180.
34. Klein, T, Martens, H, Dijk, DJ, et al. Circadian sleep regulation in the absence of light perception: chronic non-24-hour circadian rhythm sleep disorder in a blind man with a regular 24-hour sleep-wake schedule. Sleep. 1993;16:333–343.
35. Turek, FW. Pharmacological probes of the mammalian circadian clock: use of the phase response curve approach. Trends Pharmacol Sciences. 1987;8:212–217.
36. Turek, FW. Pharmacological probes of the mammalian circadian clock: use of the phase response curve approach. Trends Pharmacol Sci. 1987;8:212–217.
37. Lockley, SW, Brainard, GC, Czeisler, CA. High sensitivity of the human circadian melatonin rhythm to resetting by short wavelength light. J Clin Endocrinol Metab. 2003;88:4502–4505.
38. Boivin, DB, Duffy, JF, Kronauer, RE, et al. Dose-response relationships for resetting of human circadian clock by light. Nature. 1996;379:540–542.
39. Minors, DS, Waterhouse, JM, Wirz-Justice, A. A human phase-response curve to light. Neurosci Lett. 1991;13:36–40.
40. Van Cauter, E, Sturis, J, Byrne, MM, et al. Demonstration of rapid light-induced advances and delays of the human circadian clock using hormonal phase markers. Am J Physiol. 1994;266:E953–E963.
41. Khalsa, SB, Jewett, ME, Cajochen, C, et al. A phase response curve to single bright light pulses in human subjects. J Physiol. 2003;549:945–952.
42. Jewett, M, Kronauer, RE, Czeisler, CA. Light-induced suppression of endogenous circadian amplitude in humans. Nature. 1991;350:59–62.
43. Van Reeth, O, Turek, FW. Stimulated activity mediates phase shifts in the hamster circadian clock induced by dark pulses or benzodiazepines. Nature. 1989;339:49–51.
44. Mendoza, JY, Dardente, H, Escobar, C, et al. Dark pulse resetting of the suprachiasmatic clock in Syrian hamsters: behavioral phase-shifts and clock gene expression. Neuroscience. 2004;127:529–537.
45. Van Cauter, E, Moreno-Reyes, R, Akseki, E, et al. Rapid phase advance of the 24-h melatonin profile in response to afternoon dark exposure. Am J Physiol. 1998;275:E48–E54.
46. Goichot, B, Weibel, L, Chapotot, F, et al. Effect of the shift of the sleep-wake cycle on three robust endocrine markers of the circadian clock. Am J Physiol. 1998;275:E243–E248.
47. Buxton, OM, L’Hermite-Baleriaux, M, Turek, FW, et al. Daytime naps in darkness phase shift the human circadian rhythms of melatonin and thyrotropin secretion. Am J Physiol Regul Integr Comp Physiol. 2000;278:R373–R382.
48. Mrosovsky, N. Locomotor activity and non-photic influences on the circadian clock. Biol Rev. 1996;71:343–372.
49. Mrosovsky, N. Phase response curves for social entrainment. J Comp Physiol A. 1988;162:35–46.
50. Turek, FW. Effects of stimulated physical activity on the circadian pacemaker of vertebrates. J Biol Rhythms. 1989;4:135–148.
51. Turek, FW, Smith, R, Van Reeth, O, et al. Disturbances of the activity rest cycle alter the circadian clock of mammals. In: Inouye S, Krieger JM, eds. Endogenous sleep factors, The Hague. SPB Academic Publishing; 1990:277–283.
52. Smith, R, Turek, FW, Takahashi, JS. Two families of phase-response curves characterize the resetting of the hamster circadian clock. Am J Physiol. 1992;262:R1149–R1153.
53. Zlomanczuk, P, Schwartz, W. Cellular and molecular mechanisms of circadian rhythms in mammals. In: Turek FW, Zee PC, eds. Regulation of sleep and circadian rhythms. New York: Marcel Dekker; 1999:309–342.
54. Penev, PD, Zee, PC, Wallen, EP, et al. Aging alters the phase-resetting properties of a serotonin agonist on hamster circadian rhythmicity. Am J Physiol. 1995;268:R293–R298.
55. Pickard, GE, Rea, MA. Serotonergic innervation of the hypothalamic suprachiasmatic nucleus and photic regulation of circadian rhythms. Biol Cell. 1997;89:513–523.
56. Mistleberger, RE, Rusak, B. Circadian rhythms in mammals: formal properties and environmental Influences. In: MH Kryger MH, Roth T, Dement WC, eds. Practices of Sleep Medicine. ed 4. New York: WB Saunders; 2005:363–374.
57. Van Reeth, O, Sturis, J, Byrne, MM, et al. Nocturnal exercise phase delays circadian rhythms of melatonin and thyrotropin in normal men. Am J Physiol. 1994;266:E964–E974.
58. Buxton, OM, Frank, SA, L’Hermite-Balériaux, M, et al. Roles of intensity and duration of nocturnal exercise in causing phase-shifts of human circadian rhythms. Am J Physiol (Endocrinol & Metab). 1997;273:E536–E542.
59. Eastman, CI, Hoese, EK, Youngstedt, SD, et al. Phase-shifting human circadian rhythms with exercise during the night shift. Physiol Behav. 1995;58:1287–1291.
60. Barger, LK, Wright, KP, Jr., Hughes, RJ, et al. Daily exercise facilitates phase delays of circadian melatonin rhythm in very dim light. Am J Physiol Regul Integr Comp Physiol. 2004;286:R1077–R1084.
61. Baehr, EK, Eastman, CI, Revelle, W, et al. Circadian phase-shifting effects of nocturnal exercise in older compared with young adults. Am J Physiol Regul Integr Comp Physiol. 2003;284:R1542–R1550.
62. Scheer, FA, Cajochen, C, Turek, FW, et al. Melatonin in the Regulation of Sleep and Circadian Rhythms. In: Kryger MH, Roth T, Dement WC, eds. Principles and Practices of Sleep Medicine. ed 4. New York: WB Saunders; 2005:395–404.
63. Turek, FW, Gillette, MU. Melatonin, sleep, and circadian rhythms: rationale for development of specific melatonin agonists. Sleep Med. 2004;5:523–532.
64. Rosenthal, NE. Plasma melatonin as a measure of the human clock. J Clin Endocrinol Metab. 1991;73:225–226.
65. Shanahan, TL, Czeisler, CA. Light exposure induces equivalent phase shifts of the endogenous circadian rhythms of circulating plasma melatonin and core body temperature in men. J Clin Endocrinol Metab. 1991;73:227–235.
66. Duez, H, Staels, B. The nuclear receptors Rev-erbs and RORs integrate circadian rhythms and metabolism. Diab Vasc Dis Res. 2008;5:82–88.
67. Yin, L, Wu, N, Curtin, JC, et al. Rev-erbalpha, a heme sensor that coordinates metabolic and circadian pathways. Science. 2007;318:1786–1789.
68. Stokkan, KA, Yamazaki, S, Tei, H, et al. Entrainment of the circadian clock in the liver by feeding. Science. 2001;291:490–493.
69. Kohsaka, A, Laposky, AD, Ramsey, KM, et al. High-fat diet disrupts behavioral and molecular circadian rhythms in mice. Cell Metab. 2007;6:414–421.
70. Fuller, PM, Lu, J, Saper, CB. Differential rescue of light- and food-entrainable circadian rhythms. Science. 2008;320:1074–1077.
71. Landry, GJ, Yamakawa, GR, Webb, IC, et al. The dorsomedial hypothalamic nucleus is not necessary for the expression of circadian food-anticipatory activity in rats. J Biol Rhythms. 2007;22:467–478.
72. Turek, FW. Staying off the dance floor: when no rhythm is better than bad rhythm. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1672–R1674.
73. Martino, TA, Oudit, GY, Herzenberg, AM, et al. Circadian rhythm disorganization produces profound cardiovascular and renal disease in hamsters. Am J Physiol Regul Integr Comp Physiol. 2008;294:R1675–R1683.
74. Rechtschaffen, A, Kales, A. A manual of standardized terminology, techniques and scoring system for sleep stages of human subjects. Los Angeles: UCLA Brain Information Service/Brain Research Institute; 1968.
75. Jones, BE. Modulation of cortical activation and behavioral arousal by cholinergic and orexinergic systems. Ann N Y Acad Sci. 2008;1129:26–34.
76. Szymusiak, R, McGinty, D. Hypothalamic regulation of sleep and arousal. Ann N Y Acad Sci. 2008;1129:275–286.
77. Sherin, JE, Shiromani, PJ, McCarley, RW, et al. Activation of ventrolateral preoptic neurons during sleep. Science. 1996;271:216–219.
78. Gallopin, T, Fort, P, Eggermann, E, et al. Identification of sleep-promoting neurons in vitro. Nature. 2000;404:992–995.
79. Gaus, SE, Strecker, RE, Tate, BA, et al. Ventrolateral preoptic nucleus contains sleep-active, galaninergic neurons in multiple mammalian species. Neuroscience. 2002;115:285–294.
80. Saper, CB, Chou, TC, Scammell, TE. The sleep switch: hypothalamic control of sleep and wakefulness. Trends Neurosci. 2001;24:726–731.
81. Sakurai, T. Roles of orexin/hypocretin in regulation of sleep/wakefulness and energy homeostasis. Sleep Med Rev. 2005;9:231–241.
82. Aschoff, J. On the perception of time during prolonged temporal isolation. Hum Neurobiol. 1985;4:41–52.
83. Aschoff, J, von Goetz, C, Wildgruber, C, et al. Meal timing in humans during isolation without time cues. J Biol Rhythms. 1986;1:151–162.
84. Aschoff, J. On the dilatability of subjective time. Persp Biol Med. 1992;35:276–280.
85. Czeisler, CA, Weitzman, ED, Moore-Ede, MC, et al. Human sleep: its duration and organization depends on its circadian phase. Science. 1980;210:1264–1267.
86. Borbely, AA. Processes underlying sleep regulation. Horm Res. 1998;49:114–117.
87. Chou, TC, Bjorkum, AA, Gaus, SE, et al. Afferents to the ventrolateral preoptic nucleus. J Neurosci. 2002;22:977–990.
88. Edgar, DM, Dement, WC, Fuller, CA. Effect of SCN lesions on sleep in squirrel monkeys: evidence for opponent processes in sleep-wake regulation. J Neurosci. 1993;13:1065–1079.
89. Mendelson, WB, Bergmann, BM, Tung, A. Baseline and post-deprivation recovery sleep in SCN-lesioned rats. Brain Res. 2003;980:185–190.
90. Aston-Jones, G, Chen, S, Zhu, Y, et al. A neural circuit for circadian regulation of arousal. Nat Neurosci. 2001;4:732–738.
91. Naylor, E, Bergmann, BM, Krauski, K, et al. The circadian clock mutation alters sleep homeostasis in the mouse. J Neurosci. 2000;20:8138–8143.
92. Franken, P, Thomason, R, Heller, HC, et al. A non-circadian role for clock-genes in sleep homeostasis: a strain comparison. BMC Neurosci. 2007;8:87.
93. Viola, AU, Archer, SN, James, LM, et al. PER3 polymorphism predicts sleep structure and waking performance. Curr Biol. 2007;17:613–618.
94. Landolt, HP. Sleep homeostasis: a role for adenosine in humans? Biochem Pharmacol. 2008;75:2070–2079.
95. Monk, TH, Buysse, DJ, Reynolds, CF, III., et al. Rhythmic versus homeostatic influences on mood, activation and performance in the elderly. J Gerontol. 1991;47:221–227.
96. Monk, TH, Buysse, DJ, Reynolds, CF, et al. Circadian rhythms in human performance and mood under constant conditions. J Sleep Res. 1997;6:9–18.
97. Dijk, DJ, Czeisler, CA. Paradoxical timing of the circadian rhythm of sleep propensity serves to consolidate sleep and wakefulness in humans. Neurosci Lett. 1994;166:63–68.
98. Boivin, DB, Czeisler, CA, Dijk, DJ, et al. Complex interaction of the sleep-wake cycle and circadian phase modulates mood in healthy subjects. Arch Gen Psychiatry. 1997;54:145–152.
99. Folkard, S, Hume, KI, Minors, DS, et al. Independence of the circadian rhythm in alertness from the sleep-wake cycle. Nature. 1985;313:678–679.
100. Johnson, MP, Duffy, JF, Dijk, DJ, et al. Short-term memory, alertness and performance: a reappraisal of their relationship to body temperature. J Sleep Res. 1992;1:24–29.
101. Akerstedt, T, Folkard, S. Validation of the S and C components of the three-process model of alertness regulation. Sleep. 1995;18:1–6.
102. Leproult, R, Van Reeth, O, Byrne, MM, et al. Sleepiness, performance and neuroendocrine function during sleep deprivation: Effects of exposure to bright light or exercise. J Biol Rhythms. 1997;12:245–258.
103. Dijk, DJ, Duffy, JF. Circadian regulation of human sleep and age-related changes in its timing, consolidation and EEG characteristics. Ann Med. 1999;31:130–140.
104. Brandenberger, G, Follenius, M, Goichot, B, et al. Twenty-four hour profiles of plasma renin activity in relation to the sleep-wake cycle. J of Hypertension. 1994;12:277–283.
105. Van Cauter, E, Polonsky, KS, Scheen, AJ. Roles of circadian rhythmicity and sleep in human glucose regulation. Endocr Rev. 1997;18:716–738.
106. Follenius, M, Brandenberger, G, Simon, C, et al. REM sleep in humans begins during decreased secretory activity of the anterior pituitary. Sleep. 1988;11:546–555.
107. Gronfier, C, Luthringer, R, Follenius, M, et al. Temporal link between plasma thyrotropin levels and electroencephalographic activity in man. Neurosci Letters. 1995;200:97–100.
108. Gronfier, C, Luthringer, R, Follenius, M, et al. A quantitative evaluation of the relationships between growth hormone secretion and delta wave electroencephalographic activity during normal sleep and after enrichment in delta waves. Sleep. 1996;19:817–824.
109. Gronfier, C, Luthringer, R, Follenius, M, et al. Temporal relationships between pulsatile cortisol secretion and electroencephalographic activity during sleep in man. Electroencephalogr Clin Neurophysiol. 1997;103:405–408.
110. Spiegel, K, Luthringer, R, Follenius, M, et al. Temporal relationship between prolactin secretion and slow-wave electroencephalographic activity during sleep. Sleep. 1995;18:543–548.
111. Van Cauter, E, Spiegel, K. Circadian and sleep control of endocrine secretions. In: Turek FW, Zee PC, eds. Neurobiology of Sleep and Circadian Rhythms. New York: Marcel Dekker, 1999.
112. Van Cauter, E, Copinschi, G. Interactions between growth hormone secretion and sleep. In: Smith RG, Thorner MO, eds. Human growth hormone secretion: basic and clinical research. Totowa, NJ: Humana Press, Inc, 1999.
113. Kleitman, N. Basic rest activity cycle: 22 years later. Sleep. 1982;5:311–317.
114. Chapotot, F, Gronfier, C, Jouny, C, et al. Cortisol secretion is related to electroencephalographic alertness in human subjects during daytime wakefulness. J Clin Endocrinol Metab. 1998;83:4263–4268.
115. Lang, DA, Matthews, DR, Peto, J, et al. Cyclic oscillations of basal plasma glucose and insulin concentrations in human beings. N Engl J Med. 1979;301:1023–1027.
116. O’Meara, NM, Sturis, J, Blackman, JD, et al. Analytical problems in detecting rapid insulin secretory pulses in normal humans. J Clin Endocrinol Metab. 1993;27:231–238.
117. Hucking, K, Hamilton-Wessler, M, Ellmerer, M, et al. Burst-like control of lipolysis by the sympathetic nervous system in vivo. J Clin Invest. 2003;111:257–264.
118. Veldhuis, JD, Iranmanesh, A, Johnson, ML, et al. Twenty-four-hour rhythms in plasma concentrations of adenohypophyseal hormones are generated by distinct amplitude and/or frequency modulation of underlying pituitary secretory bursts. J Clin Endocrinol Metab. 1990;71:1616–1623.
119. Veldhuis, JD, Johnson, ML, Lizarralde, G, et al. Rhythmic and nonrhythmic modes of anterior pituitary gland secretion. Chronobiology International. 1992;9:371–379.
120. Van Cauter, E, van Coevorden, A, Blackman, JD. Modulation of neuroendocrine release by sleep and circadian rhythmicity. In: Yen S, Vale W, eds. Advances in neuroendocrine regulation of reproduction. Norwell: Serono Symposia USA; 1990:113–122.
121. Polonsky, KS, Sturis, J, Van Cauter, E. Temporal profiles and clinical significance of pulsatile insulin secretion. Horm Res. 1998;49:178–184.
122. Simon, C, Brandenberger, G, Saini, J, et al. Slow oscillations of plasma glucose and insulin secretion rate are amplified during sleep in humans under continuous enteral nutrition. Sleep. 1994;17:333–338.
123. Knobil, E, Hotchkiss, J. The menstrual cycle and its neuroendocrine control. In: Knobil E, Neil JD, eds. The Physiology of Reproduction. New York: Raven Press; 1988:1971–1994.
124. Conn, PM, Crowley, WF. Gonadotropin-releasing hormone and its analogues. The New England Journal of Medicine. 1991;324:93–103.
125. Sturis, J, Scheen, AJ, Leproult, R, et al. 24-hour glucose profiles during continuous or oscillatory insulin infusion. J Clin Invest. 1995;95:1464–1471.
126. van Coevorden, A, Mockel, J, Laurent, E, et al. Neuroendocrine rhythms and sleep in aging men. Am J Physiol. 1991;260:E651–E661.
127. Czeisler, CA, Dumont, M, Duffy, JF, et al. Association of sleep-wake habits in older people with changes in output of circadian pacemaker. Lancet. 1992;340:933–936.
128. Van Cauter, E, Plat, L, Leproult, R, et al. Alterations of circadian rhythmicity and sleep in aging: endocrine consequences. Horm Res. 1998;49:147–152.
129. Van Cauter, E, Leproult, R, Plat, L. Age-related changes in slow-wave sleep and REM sleep and relationship with growth hormone and cortisol levels in healthy men. JAMA. 2000;284:861–868.
130. Ohayon, MM, Carskadon, MA, Guilleminault, C, et al. Meta-analysis of quantitative sleep parameters from childhood to old age in healthy individuals: developing normative sleep values across the human lifespan. Sleep. 2004;27:1255–1273.
131. Unruh, ML, Newman, AB, Larive, B, et al. The Influence of Age on Changes in Health-Related Quality of Life over Three Years in a Cohort Undergoing Hemodialysis. J Am Geriatr Soc. 2008;56:1608–1617.
132. Vitiello, MV, Larsen, LH, Moe, KE. Age-related sleep change: gender and estrogen effects on the subjective-objective sleep quality relationships of healthy, noncomplaining older men and women. J Psychosom Res. 2004;56:503–510.
133. Groeger, JA, Zijlstra, FR, Dijk, DJ. Sleep quantity, sleep difficulties and their perceived consequences in a representative sample of some 2000 British adults. J Sleep Res. 2004;13:359–371.
134. Mourtazaev, MS, Kemp, B, Zwinderman, AH, et al. Age and gender affect different characteristics of slow waves in the sleep EEG. Sleep. 1995;18:557–564.
135. Latta, F, Leproult, R, Tasali, E, et al. Sex differences in delta and alpha EEG activities in healthy older adults. Sleep. 2005;28:1525–1534.
136. Ancoli-Israel, S. Sleep and aging: prevalence of disturbed sleep and treatment considerations in older adults. J Clin Psychiatry. 2005;66(Suppl 9):24–30.
137. Zee, PC, Rosenberg, RS, Turek, FW. Effects of aging on entrainment and rate of resynchronization of the circadian locomotor activity. Am J Physiol. 1992;263:1099–1103.
138. Monk, TH. Aging human circadian rhythms: conventional wisdom may not always be right. J Biol Rhythms. 2005;20:366–374.
139. Pittendrigh, CS, Daan, S. Circadian oscillations in rodents: a systematic increase of their frequency with age. Science. 1974;186:548–550.
140. Morin, LP. Age-related changes in hamster circadian period, entrainment and rhythm splitting. J Biol Rhythms. 1988;3:237–248.
141. Yamazaki, S, Straume, M, Tei, H, et al. Effects of aging on central and peripheral mammalian clocks. Proc Natl Acad Sci U S A. 2002;99:10801–10806.
142. Dijk, DJ, Duffy, JF, Riel, E, et al. Ageing and the circadian and homeostatic regulation of human sleep during forced desynchrony of rest, melatonin and temperature rhythms. J Physiol. 1999;516(Pt 2):611–627.
143. Duffy, JF, Czeisler, CA. Age-related change in the relationship between circadian period, circadian phase, and diurnal preference in humans. Neurosci Lett. 2002;318:117–120.
144. Zhang, Y, Kornhauser, JM, Zee, PC, et al. Effects of aging on light-induced phase-shifting of circadian behavioral rhythms, Fos expression, and Creb phosphorylation in the hamster suprachiasmatic nucleus. Neuroscience. 1996;70:951–961.
145. Van Reeth, O, Zhang, Y, Zee, PC, et al. Aging alters feedback effects of the activity-rest cycle in the circadian clock. Am J Physiol. 1992;263:R981–R986.
146. Van Reeth, O, Zhang, Y, Reddy, A, et al. Aging alters the entraining effects of an activity-inducing stimulus on the circadian clock. Brain Res. 1993;607:286–292.
147. Van Reeth, O, Zhang, Y, Zee, PC, et al. Grafting fetal suprachiasmatic nuclei in the hypothalamus of old hamsters restores responsiveness of the circadian clock to a phase shifting stimulus. Brain Res. 1994;643:338–342.
148. Campbell, SS, Kripke, DF, Gillin, JC, et al. Exposure to light in healthy elderly subjects and Alzheimer’s patients. Physiol Behav. 1988;42:141–144.
149. Ehlers, CL, Frank, E, Kupfer, DJ. Social zeitgebers and biological rhythms. Arch Gen Psychiatry. 1988;45:948–952.
150. Ancoli-Israel, S, Kripke, DF, Jones, DW, et al. 24-hour sleep and light rhythms in nursing home patients. Sleep Res. 1991;20A:410.
151. Brainard, GC, Rollag, MD, Hanifin, JP. Photic regulation of melatonin in humans: ocular and neural signal transduction. J Biol Rhythms. 1997;12:537–546.
152. Campbell, SS, Dawson, D, Anderson, MW. Alleviation of sleep maintenance insomnia with timed exposure to bright light. J Am Geriatric Soc. 1993;41:829–836.
153. Mishima, K, Okawa, M, Shimizu, T, et al. Diminished melatonin secretion in the elderly caused by insufficient environmental illumination. J Clin Endocrinol Metab. 2001;86:129–134.
154. Benloucif, S, Green, K, L’Hermite-Baleriaux, M, et al. Responsiveness of the aging circadian clock to light. Neurobiol Aging. 2006;27:1870–1879.
155. Veldhuis, JD. Mechanisms and biological significance of pulsatile hormone secretion. Symposium proceedings. London, United Kingdom, 2–4 March 1999. Novartis Found Symp. 2000;227:1–4.
156. Halberg, F, Tong, YL, Johnson, EA. Circadian system phase: an aspect of temporal morphology; procedures and illustrative examples. In: Cellular aspects of biorhythms. Berlin. Springer-Verlag; 1967:20–48.
157. Cleveland, WS. Robust locally weighted regression and smoothing scatterplots. J Am Stat Assoc. 1979;74:829–836.
158. Van Cauter, E. Method for characterization of 24-h temporal variation of blood constituents. Am J Physiol. 1979;237:E255–E264.
159. Van Cauter, E. Computer-assisted analysis of endocrine rhythms. In: Rodbard D, Forti G, eds. Computers in Endocrinology. New York: Raven Press; 1990:59–70.
160. Polonsky, KS, Licinio-Paixao, J, Given, BD, et al. Use of biosynthetic human C-peptide in the measurement of insulin secretion rates in normal volunteers and type I diabetic patients. J Clin Invest. 1986;77:98–105.
161. De Nicolao, G, Rocchetti, M. Stable and efficient techniques for the deconvolution of hormone time series. In: Guardabasso V, Rodbard D, Forti G, eds. Computers in Endocrinology: Recent Advances. New York: Raven Press; 1990:83–91.
162. Veldhuis, JD, Johnson, ML. A review and appraisal of deconvolution methods to evaluate in vivo neuroendocrine secretory events. J Neuroendocrinol. 1990;2:755–771.
163. Sturis, J, Polonsky, KS, Shapiro, ET, et al. Abnormalities in the ultradian oscillations of insulin secretion and glucose levels in type 2 (non-insulin-dependent) diabetic patients. Diabetologia. 1992;35:681–689.
164. Pincus, SM, Keefe, DL. Quantification of hormone pulsatility via an approximate entropy algorithm. Am J Physiol. 1992;262:E741–E754.
165. Pincus, SM. Orderliness of hormone release. Novartis Found Symp. 2000;227:82–96.
166. Urban, RJ, Evans, WS, Rogol, AD, et al. Contemporary aspects of discrete peak-detection algorithms. I. The paradigm of the luteinizing hormone pulse signal in men. Endocr Rev. 1988;9:3–37.
167. Urban, RJ, Kaiser, DL, Van Cauter, E, et al. Comparative assessment of objective pulse detection algorithms. II. Studies in men. Am J Physiol. 1988;254:E113–E119.
168. Buijs, RM, Wortel, J, Van Heerikhuize, JJ, et al. Anatomical and functional demonstration of a multisynaptic suprachiasmatic nucleus adrenal (cortex) pathway. Eur J Neurosci. 1999;11:1535–1544.
169. Lemos, DR, Downs, JL, Urbanski, HF. Twenty-four-hour rhythmic gene expression in the rhesus macaque adrenal gland. Mol Endocrinol. 2006;20:1164–1176.
170. Torres-Farfan, C, Rocco, V, Monso, C, et al. Maternal melatonin effects on clock gene expression in a nonhuman primate fetus. Endocrinology. 2006;147:4618–4626.
171. Valenzuela, FJ, Torres-Farfan, C, Richter, HG, et al. Clock gene expression in adult primate suprachiasmatic nuclei and adrenal: is the adrenal a peripheral clock responsive to melatonin? Endocrinology. 2008;149:1454–1461.
172. Oster, H, Damerow, S, Kiessling, S, et al. The circadian rhythm of glucocorticoids is regulated by a gating mechanism residing in the adrenal cortical clock. Cell Metab. 2006;4:163–173.
173. Veldhuis, JD, Iranmanesh, A, Johnson, ML, et al. Amplitude, but not frequency, modulation of adrenocorticotropin secretory bursts gives rise to the nyctohemeral rhythm of the corticotropic axis in man. J Clin Endocrinol Metab. 1989;71:452–463.
174. Van Cauter, E, Honinckx, E. Pulsatility of Pituitary Hormones. Exp Brain Res Suppl. 1985;12:41–60.
175. Lejeune-Lenain, C, Van Cauter, E, Desir, D, et al. Control of circadian and episodic variations of adrenal androgens secretion in man. J Endocrinol Invest. 1987;10:267–276.
176. Weitzman, ED, Zimmerman, JC, Czeisler, CA, et al. Cortisol secretion is inhibited during sleep in normal man. J Clin Endocrinol Metab. 1983;56:352–358.
177. Born, J, Muth, S, Fehm, HL. The significance of sleep onset and slow wave sleep for nocturnal release of growth hormone (GH) and cortisol. Psychoneuroendocrinology. 1988;13:233–243.
178. Van Cauter, E, Blackman, JD, Roland, D, et al. Modulation of glucose regulation and insulin secretion by circadian rhythmicity and sleep. J Clin Invest. 1991;88:934–942.
179. Weibel, L, Follenius, M, Spiegel, K, et al. Comparative effect of night and daytime sleep on the 24-hour cortisol secretory profile. Sleep. 1995;18:549–556.
180. Follenius, M, Brandenberger, G, Bardasept, J, et al. Nocturnal cortisol release in relation to sleep structure. Sleep. 1992;15:21–27.
181. Spath-Schwalbe, E, Gofferje, M, Kern, W, et al. Sleep disruption alters nocturnal ACTH and cortisol secretory patterns. Biol Psychiatry. 1991;29:575–584.
182. Pruessner, JC, Wolf, OT, Hellhammer, DH, et al. Free cortisol levels after awakening: a reliable biological marker for the assessment of adrenocortical activity. Life Sci. 1997;61:2539–2549.
183. Caufriez, A, Moreno-Reyes, R, Leproult, R, et al. Immediate effects of an 8-h advance shift of the rest-activity cycle on 24-h profiles of cortisol. Am J Physiol Endocrinol Metab. 2002;282:E1147–E1153.
184. Ekstedt, M, Akerstedt, T, Soderstrom, M. Microarousals during sleep are associated with increased levels of lipids, cortisol, and blood pressure. Psychosom Med. 2004;66:925–931.
185. Vgontzas, AN, Zoumakis, M, Bixler, EO, et al. Impaired nighttime sleep in healthy old versus young adults is associated with elevated plasma interleukin-6 and cortisol levels: physiologic and therapeutic implications. J Clin Endocrinol Metab. 2003;88:2087–2095.
186. Vgontzas, AN, Bixler, EO, Lin, HM, et al. Chronic insomnia is associated with nyctohemeral activation of the hypothalamic-pituitary-adrenal axis: clinical implications. J Clin Endocrinol Metab. 2001;86:3787–3794.
187. Scheer, FA, Buijs, RM. Light affects morning salivary cortisol in humans. J Clin Endocrinol Metab. 1999;84:3395–3398.
188. Leproult, R, Colecchia, EF, L’Hermite-Baleriaux, M, et al. Transition from dim to bright light in the morning induces an immediate elevation of cortisol levels. J Clin Endocrinol Metab. 2001;86:151–157.
189. Ruger, M, Gordijn, MC, Beersma, DG, et al. Time-of-day-dependent effects of bright light exposure on human psychophysiology: comparison of daytime and nighttime exposure. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1413–R1420.
190. Linkowski, P, Van Onderbergen, A, Kerkhofs, M, et al. Twin study of the 24-h cortisol profile: evidence for genetic control of the human circadian clock. Am J Physiol. 1993;264:E173–E181.
191. Van Cauter, E, Shapiro, ET, Tillil, H, et al. Circadian modulation of glucose and insulin responses to meals: relationship to cortisol rhythm. Am J Physiol. 1992;262:E467–E475.
192. Born, J, Hansen, K, Marshall, L, et al. Timing the end of nocturnal sleep. Nature. 1999;397:29–30.
193. Leproult, R, Copinschi, G, Buxton, O, et al. Sleep loss results in an elevation of cortisol levels the next evening. Sleep. 1997;20:865–870.
194. Spiegel, K, Leproult, R, Van Cauter, E. Impact of sleep debt on metabolic and endocrine function. Lancet. 1999;354:1435–1439.
195. Sherman, B, Wysham, C, Pfohl, B. Age-related changes in the circadian rhythm of plasma cortisol in man. J Clin Endocrinol Metab. 1985;61:439–443.
196. Van Cauter, E, Leproult, R, Kupfer, DJ. Effects of gender and age on the levels and circadian rhythmicity of plasma cortisol. J Clin Endocrinol Metab. 1996;81:2468–2473.
197. McEwen, BS, Stellar, E. Stress and the individual. Arch Intern Med. 1993;153:2093–2101.
198. McEwen, B. Protective and damaging effects of stress mediators. N Engl J Med. 1998;338:171–179.
199. Dallman, MF, Strack, AL, Akana, SF, et al. Feast and famine: Critical role of glucocorticoids with insulin in daily energy flow. Frontiers in Neuroendocrinology. 1993;14:303–347.
200. Plat, L, Féry, F, L’Hermite-Balériaux, M, et al. Metabolic effects of short-term physiological elevations of plasma cortisol are more pronounced in the evening than in the morning. J Clin Endocrinol Metab. 1999;84:3082–3092.
201. Dennison, E, Hindmarsh, P, Fall, C, et al. Profiles of endogenous circulating cortisol and bone mineral density in healthy elderly men. J Clin Endocrinol Metab. 1999;84:3058–3063.
202. Magiakou, MA, Mastorakos, G, Rabin, D, et al. The maternal hypothalamic-pituitary-adrenal axis in the third trimester of human pregnancy. Clin Endocrinol (Oxf). 1996;44:419–428.
203. Rosman, PM, Farag, A, Benn, R, et al. Modulation of pituitary-adrenal function: decreased secretory episodes and blunted circadian rhythmicity in patients with alcoholic liver disease. J Clin Endocrinol Metab. 1981;55:709–717.
204. Boyar, RM, Hellman, LD, Roffwarg, H, et al. Cortisol secretion and metabolism in anorexia nervosa. N Engl J Med. 1977;296:190–193.
205. Kok, P, Kok, SW, Buijs, MM, et al. Enhanced circadian ACTH release in obese premenopausal women: reversal by short-term acipimox treatment. Am J Physiol Endocrinol Metab. 2004;287:E848–E856.
206. Iranmanesh, A, Lizarralde, G, Johnson, ML, et al. Dynamics of 24-hour endogenous cortisol secretion and clearance in primary hypothyroidism assessed before and after partial thyroid hormone replacement. J Clin Endocrinol Metab. 1990;70:155–161.
207. Gallagher, TF, Hellman, L, Finkelstein, J, et al. Hyperthyroidism and cortisol secretion in man. J Clin Endocrinol Metab. 1972;34:919–927.
208. Van Cauter, E, Refetoff, S. Evidence for two subtypes of Cushing’s disease based on the analysis of episodic cortisol secretion. N Engl J Med. 1985;312:1343–1344.
209. Roelfsema, F, Pincus, SM, Veldhuis, JD. Patients with Cushing’s disease secrete adrenocorticotropin and cortisol jointly more asynchronously than healthy subjects. J Clin Endocrinol Metab. 1998;83:688–692.
210. van Aken, MO, Pereira, AM, van Thiel, SW, et al. Irregular and frequent cortisol secretory episodes with preserved diurnal rhythmicity in primary adrenal Cushing’s syndrome. J Clin Endocrinol Metab. 2005;90:1570–1577.
211. Linkowski, P, Mendlewicz, J, Leclercq, R, et al. The 24-hour profile of adrenocorticotropin and cortisol in major depressive illness. J Clin Endocrinol Metab. 1985;61:429–438.
212. Rubin, RT, Poland, RE, Lesser, IM, et al. Neuroendocrine aspects of primary endogenous depression: I. Cortisol secretory dynamics in patients and matched controls. Arch Gen Psychiatry. 1987;44:328–336.
213. Sachar, ED, Twenty-four-hour cortisol secretory patterns in depressed and manic patients. Hormones, homeostasis and the brain prog in brain research. Gispen, WH, Van Wimersma Greidanus, TB, Bohus, B, De Wied, D, eds. Hormones, homeostasis and the brain prog in brain research, vol 42. Amsterdam: Elsevier, 1975:81–91.
214. Linkowski, P, Mendlewicz, J, Kerkhofs, M, et al. 24-hour profiles of adrenocorticotropin, cortisol, and growth hormone in major depressive illness: effect of antidepressant treatment. J Clin Endocrinol Metab. 1987;65:141–152.
215. Yehuda, R, Teicher, MH, Trestman, RL, et al. Cortisol regulation in posttraumatic stress disorder and major depression: a chronobiological analysis. Biol Psychiatry. 1996;15:79–88.
216. Yehuda, R. Hypothalamic-pituitary-adrenal alterations in PTSD: are they relevant to understanding cortisol alterations in cancer? Brain Behav Immun. 2003;17(Suppl 1):S73–S83.
217. Bremner, JD, Vythilingam, M, Anderson, G, et al. Assessment of the hypothalamic-pituitary-adrenal axis over a 24-hour diurnal period and in response to neuroendocrine challenges in women with and without childhood sexual abuse and posttraumatic stress disorder. Biol Psychiatry. 2003;54:710–718.
218. Crofford, LJ, Young, EA, Engleberg, NC, et al. Basal circadian and pulsatile ACTH and cortisol secretion in patients with fibromyalgia and/or chronic fatigue syndrome. Brain Behav Immun. 2004;18:314–325.
219. Klerman, EB, Goldenberg, DL, Brown, EN, et al. Circadian rhythms of women with fibromyalgia. J Clin Endocrinol Metab. 2001;86:1034–1039.
220. Muccioli, G, Tschop, M, Papotti, M, et al. Neuroendocrine and peripheral activities of ghrelin: implications in metabolism and obesity. Eur J Pharmacol. 2002;440:235–254.
221. van der Lely, AJ, Tschop, M, Heiman, ML, et al. Biological, physiological, pathophysiological, and pharmacological aspects of ghrelin. Endocr Rev. 2004;25:426–457.
222. Van Cauter, E, Plat, L, Copinschi, G. Interrelations between sleep and the somatotropic axis. Sleep. 1998;21:553–566.
223. Mendlewicz, J, Linkowski, P, Kerkhofs, M, et al. Genetic control of 24-hour growth hormone secretion in man: a twin study. J Clin Endocrinol Metab. 1999;84:856–862.
224. Ho, KY, Evans, WS, Blizzard, RM, et al. Effects of sex and age on the 24-hour profile of growth hormone secretion in man: importance of endogenous estradiol concentrations. J Clin Endocrinol Metab. 1987;64:51–58.
225. Shah, N, Evans, WS, Veldhuis, JD. Actions of estrogen on pulsatile, nyctohemeral, and entropic modes of growth hormone secretion. Am J Physiol. 1999;276:R1351–R1358.
226. Caufriez, A, Leproult, R, L’Hermite-Balériaux, M, et al. A potential role of endogenous progesterone in modulation of GH, prolactin and thyrotropin secretion during normal menstrual cycle. Clin Endocrinol (Oxf). 2009;71:535–542.
227. Weibel, L, Spiegel, K, Gronfier, C, et al. Twenty-four-hour melatonin and core body temperature rhythms: their adaptation in night workers. Am J Physiol. 1997;272:R948–R954.
228. Van Cauter, E, Kerkhofs, M, Caufriez, A, et al. A quantitative estimation of GH secretion in normal man: reproducibility and relation to sleep and time of day. J Clin Endocrinol Metab. 1992;74:1441–1450.
229. Holl, RW, Hartmann, ML, Veldhuis, JD, et al. Thirty-second sampling of plasma growth hormone in man: correlation with sleep stages. J Clin Endocrinol Metab. 1991;72:854–861.
230. Vgontzas, AN, Mastorakos, G, Bixler, EO, et al. Sleep deprivation effects on the activity of the hypothalamic-pituitary-adrenal and growth axes: potential clinical implications. Clin Endocrinol (Oxf). 1999;51:205–215.
231. Obal, F, Jr., Krueger, JM. Biochemical regulation of non-rapid-eye-movement sleep. Front Biosci. 2003;8:d520–d550.
232. Ocampo-Lim, B, Guo, W, DeMott Friberg, R, et al. Nocturnal growth hormone (GH) secretion is eliminated by infusion of GH-releasing hormone antagonist. J Clin Endocrinol Metab. 1996;81:4396–4399.
233. Van Cauter, E, Plat, L, Scharf, M, et al. Simultaneous stimulation of slow-wave sleep and growth hormone secretion by gamma-hydroxybutyrate in normal young men. J Clin Invest. 1997;100:745–753.
234. Van Cauter, E, Caufriez, A, Kerkhofs, M, et al. Sleep, awakenings and insulin-like growth factor I modulate the growth hormone secretory response to growth hormone-releasing hormone. J Clin Endocrinol Metab. 1992;74:1451–1459.
235. Jaffe, C, Turgeon, D, DeMott Friberg, R, et al. Nocturnal augmentation of growth hormone (GH) secretion is preserved during repetitive bolus administration of GH-releasing hormone: potential involvement of endogenous somatostatin—A clinical research center study. J Clin Endocrinol Metab. 1995;80:3321–3326.
236. Dzaja, A, Dalal, MA, Himmerich, H, et al. Sleep enhances nocturnal plasma ghrelin levels in healthy subjects. Am J Physiol Endocrinol Metab. 2004;286:E963–E967.
237. Nass, R, Farhy, LS, Liu, J, et al. Evidence for acyl-ghrelin modulation of growth hormone release in the fed state. J Clin Endocrinol Metab. 2008;93:1988–1994.
238. Steiger, A, Herth, T, Holsboer, F. Sleep-electroencephalography and the secretion of cortisol and growth hormone in normal controls. Acta Endocrinol. 1987;116:36–42.
239. Spiegel, K, Leproult, R, Colecchia, EF, et al. Adaptation of the 24-h growth hormone profile to a state of sleep debt. Am J Physiol Regul Integr Comp Physiol. 2000;279:R874–R883.
240. Brandenberger, G, Gronfier, C, Chapotot, F, et al. Effect of sleep deprivation on overall 24 h growth-hormone secretion. Lancet. 2000;356:1408.
241. Vermeulen, A. Nyctohemeral growth hormone profiles in young and aged men: correlation with somatomedin-C levels. J Clin Endocrinol Metab. 1987;64:884–888.
242. Veldhuis, J, Liem, A, South, S, et al. Differential impact of age, sex steroid hormones, and obesity on basal versus pulsatile growth hormone secretion in men as assessed in an ultrasensitive chemiluminescence assay. J Clin Endocrinol Metab. 1995;80:3209–3222.
243. Veldhuis, JD, Iranmanesh, A, Weltman, A. Elements in the pathophysiology of diminished growth hormone (GH) secretion in aging humans. Endocrine. 1997;7:41–48.
244. Martin, FC, Yeo, AL, Sonksen, PH. Growth hormone secretion in the elderly: ageing and the somatopause. Baillieres Clin Endocrinol Metab. 1997;11:223–250.
245. Caufriez, A, Frankenne, F, Hennen, G, et al. Regulation of maternal IGF-I by placental GH in normal and abnormal human pregnancies. Am J Physiol. 1993;265:E572–E577.
246. Eriksson, L, Frankenne, F, Eden, S, et al. Growth hormone 24-h serum profiles during pregnancy–lack of pulsatility for the secretion of the placental variant. Br J Obstet Gynaecol. 1989;96:949–953.
247. Copinschi, G, De Laet, MH, Brion, JP, et al. Simultaneous study of cortisol, GH and PRL circadian variations of hourly integrated concentrations in normal and obese subjects. Clin Endocrinol. 1978;9:15–26.
248. Stoving, RK, Veldhuis, JD, Flyvbjerg, A, et al. Jointly amplified basal and pulsatile growth hormone (GH) secretion and increased process irregularity in women with anorexia nervosa: indirect evidence for disruption of feedback regulation within the GH-insulin-like growth factor I axis. J Clin Endocrinol Metab. 1999;84:2056–2063.
249. Edge, JA, Dunger, DB, Matthews, DR, et al. Increased overnight growth hormone concentrations in diabetic compared with normal adolescents. J Clin Endocrinol Metab. 1990;71:1356–1362.
250. Laughlin, GA, Dominguez, CE, Yen, SS. Nutritional and endocrine-metabolic aberrations in women with functional hypothalamic amenorrhea. J Clin Endocrinol Metab. 1998;83:25–32.
251. Morales, AJ, Laughlin, GA, Butzow, T, et al. Insulin, somatotropic, and luteinizing hormone axes in lean and obese women with polycystic ovary syndrome: common and distinct features. J Clin Endocrinol Metab. 1996;81:2854–2864.
252. Iranmanesh, A, Lizarralde, G, Johnson, ML, et al. Nature of altered growth hormone secretion in hyperthyroidism. J Clin Endocrinol Metab. 1991;72:108–115.
253. Mendlewicz, J, Linkowski, P, Kerkhofs, M, et al. Diurnal hypersecretion of growth hormone in depression. J Clin Endocrinol Metab. 1985;60:505–512.
254. Veldman, RG, Frolich, M, Pincus, SM, et al. Growth hormone and prolactin are secreted more irregularly in patients with Cushing’s disease. Clin Endocrinol (Oxf). 2000;52:625–632.
255. Hartman, ML, Veldhuis, JD, Vance, ML, et al. Somatotropin pulse frequency and basal concentrations are increased in acromegaly and are reduced by successful therapy. J Clin Endocrinol Metab. 1990;70:1375–1384.
256. Hartman, ML, Pincus, SM, Johnson, ML, et al. Enhanced basal and disorderly growth hormone secretion distinguish acromegalic from normal pulsatile growth hormone release. J Clin Invest. 1994;94:1277–1288.
257. van den Berg, G, Pincus, SM, Frolich, M, et al. Reduced disorderliness of growth hormone release in biochemically inactive acromegaly after pituitary surgery. Eur J Endocrinol. 1998;138:164–169.
258. Spiegel, K, Follenius, M, Simon, C, et al. Prolactin secretion and sleep. Sleep. 1994;17:20–27.
259. Waldstreicher, J, Duffy, JF, Brown, EN, et al. Gender differences in the temporal organization of prolactin (PRL) secretion: evidence fort a sleep-independent circadian rhythm of circulating PRL levels—A Clinical Research Center study. J Clin Endocrinol Metab. 1996;81:1483–1487.
260. Katznelson, L, Riskind, PN, Saxe, VC, et al. Prolactin pulsatile characteristics in postmenopausal women. J Clin Endocrinol Metab. 1998;83:761–764.
261. Linkowski, P, Spiegel, K, Kerkhofs, M, et al. Genetic and environmental influences on prolactin secretion during wake and during sleep. Am J Physiol. 1998;274:E909–E919.
262. Van Cauter, E, Refetoff, S. Multifactorial control of the 24-hour secretory profiles of pituitary hormones. J Endocrinol Invest. 1985;8:381–391.
263. Desir, D, Van Cauter, E, L’Hermite, M, et al. Effects of “jet lag” on hormonal patterns. III. Demonstration of an intrinsic circadian rhythmicity in plasma prolactin. J Clin Endocrinol Metab. 1982;55:849–857.
264. Copinschi, G, Van Onderbergen, A, L’Hermite-Balériaux, M, et al. Effects of the short-acting benzodiazepine triazolam, taken at bedtime, on circadian and sleep-related hormonal profiles in normal men. Sleep. 1990;13:232–244.
265. Copinschi, G, Akseki, E, Moreno-Reyes, R, et al. Effects of bedtime administration of zolpidem on circadian and sleep-related hormonal profiles in normal women. Sleep. 1995;18:417–424.
266. Greenspan, SL, Klibanski, A, Rowe, JW, et al. Age alters pulsatile prolactin release: influence of dopaminergic inhibition. Am J Physiol. 1990;258:E799–E804.
267. Tay, CC, Glasier, AF, McNeilly, AS. Twenty-four hour patterns of prolactin secretion during lactation and the relationship to suckling and the resumption of fertility in breast-feeding women. Hum Reprod. 1996;11:950–955.
268. Caufriez, A, Désir, D, Szyper, M, et al. Prolactin secretion in Cushing’s disease. J Clin Endocrinol Metab. 1981;53:843–846.
269. Iranmanesh, A, Veldhuis, JD, Carlsen, EC, et al. Attenuated pulsatile release of prolactin in men with insulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1990;71:73–78.
270. Kok, P, Roelfsema, F, Frolich, M, et al. Prolactin release is enhanced in proportion to excess visceral fat in obese women. J Clin Endocrinol Metab. 2004;89:4445–4449.
271. Groote Veldman, R, van den Berg, G, Pincus, SM, et al. Increased episodic release and disorderliness of prolactin secretion in both micro- and macroprolactinomas. Eur J Endocrinol. 1999;140:192–200.
272. Veldman, RG, Frolich, M, Pincus, SM, et al. Basal, pulsatile, entropic, and 24-hour rhythmic features of secondary hyperprolactinemia due to functional pituitary stalk disconnection mimic tumoral (primary) hyperprolactinemia. J Clin Endocrinol Metab. 2001;86:1562–1567.
273. Boyar, RM, Kapen, S, Finkelstein, JW, et al. Hypothalamic-pituitary function in diverse hyperprolactinemic states. J Clin Invest. 1974;53:1588–1598.
274. Spratt, DI, O’Dea, LL, Schoenfeld, D, et al. Neuroendocrine-gonadal axis in men: frequent sampling of LH, FSH and testosterone. Am J Physiol. 1988;254:E658–E666.
275. Lanfranco, F, Bonelli, L, Baldi, M, et al. Acylated ghrelin inhibits spontaneous luteinizing hormone pulsatility and responsiveness to naloxone but not that to gonadotropin-releasing hormone in young men: evidence for a central inhibitory action of ghrelin on the gonadal axis. J Clin Endocrinol Metab. 2008;93:3633–3639.
276. Fehm, HL, Clausing, J, Kern, W, et al. Sleep-associated augmentation and synchronization of luteinizing hormone pulses in adult men. Neuroendocrinology. 1991;54:192–195.
277. Bremner, WJ, Vitiello, MV, Prinz, PN. Loss of circadian rhythmicity in blood testosterone levels with aging in normal men. J Clin Endocrinol Metab. 1983;56:1278–1280.
278. Luboshitzky, R, Zabari, Z, Shen-Orr, Z, et al. Disruption of the nocturnal testosterone rhythm by sleep fragmentation in normal men. J Clin Endocrinol Metab. 2001;86:1134–1139.
279. Axelsson, J, Ingre, M, Akerstedt, T, et al. Effects of acutely displaced sleep on testosterone. J Clin Endocrinol Metab. 2005;90:4530–4535.
280. Carlsen, E, Olsson, C, Petersen, JH, et al. Diurnal rhythm in serum levels of inhibin B in normal men: relation to testicular steroids and gonadotropins. J Clin Endocrinol Metab. 1999;84:1664–1669.
281. Lejeune, H, Dechaud, H, Pugeat, M. Contribution of bioavailable testosterone assay for the diagnosis of androgen deficiency in elderly men. Ann Endocrinol (Paris). 2003;64:117–125.
282. Tenover, JS, Matsumoto, AM, Clifton, DK, et al. Age-related alterations in the circadian rhythms of pulsatile luteinizing hormone and testosterone secretion in healthy men. J of. Gerontology. 1988;43:M163–M169.
283. Penev, PD. Association between sleep and morning testosterone levels in older men. Sleep. 2007;30:427–432.
284. Mulligan, T, Iranmanesh, A, Gheorghiu, S, et al. Amplified nocturnal luteinizing hormone (LH) secretory burst frequency with selective attenuation of pulsatile (but not basal) testosterone secretion in healthy aged men: possible Leydig cell desensitization to endogenous LH signaling–a clinical research center study. J Clin Endocrinol Metab. 1995;80:3025–3031.
285. Vermeulen, A, Deslypere, JP, Kaufman, JM. Influence of antiopioids on luteinizing hormone pulsatility in aging men. J Clin Endocrinol Metab. 1989;68:68–72.
286. Veldhuis, JD, Urban, RJ, Lizarralde, G, et al. Attenuation of luteinizing hormone secretory burst amplitude as a proximate basis for the hypoandrogenism of healthy aging in men. J Clin Endocrinol Metab. 1992;75:52–58.
287. Veldhuis, JD, Iranmanesh, A, Demers, LM, et al. Joint basal and pulsatile hypersecretory mechanisms drive the monotropic follicle-stimulating hormone (FSH) elevation in healthy older men: concurrent preservation of the orderliness of the FSH release process: a general clinical research center study. J Clin Endocrinol Metab. 1999;84:3506–3514.
288. Pincus, SM, Mulligan, T, Iranmanesh, A, et al. Older males secrete luteinizing hormone and testosterone more irregularly, and jointly more asynchronously, than younger males. Proc Natl Acad Sci U S A. 1996;93:14100–14105.
289. Backstrom, CT, McNeilly, AS, Leask, RM, et al. Pulsatile secretion of LH, FSH, prolactin, oestradiol and progesterone during the human menstrual cycle. Clin Endocrinol (Oxf). 1982;17:29–42.
290. Filicori, M, Santoro, N, Merriam, GR, et al. Characterization of the physiological pattern of episodic gonadotropin secretion throughout the menstrual cycle. J Clin Endocrinol Metab. 1986;62:1136–1144.
291. Hall, JE, Sullivan, JP, Richardson, GS. Brief wake episodes modulate sleep-inhibited luteinizing hormone secretion in the early follicular phase. J Clin Endocrinol Metab. 2005;90:2050–2055.
292. Reame, NE, Kelche, RP, Beitins, IZ, et al. Age effects of follicle-stimulating hormone and pulsatile luteinizing hormone secretion across the menstrual cycle of premenopausal women. J Clin Endocrinol Metab. 1996;81:1512–1518.
293. Turek, FW, Van Cauter, E. Rhythms in Reproduction. In: Knobil E, Neil JD, eds. The Physiology of Reproduction. New York: Raven Press; 1993:487–540.
294. Bergendahl, M, Aloi, JA, Iranmanesh, A, et al. Fasting suppresses pulsatile luteinizing hormone (LH) secretion and enhances orderliness of LH release in young but not older men. J Clin Endocrinol Metab. 1998;83:1967–1975.
295. Spratt, DI, Carr, DB, Merriam, GR, et al. The spectrum of abnormal patterns of gonadotropin-releasing hormone secretion in men with idiopathic hypogonadotropic hypogonadism: clinical and laboratory correlations. J Clin Endocrinol Metab. 1987;64:283–291.
296. Hangaard, J, Andersen, M, Grodum, E, et al. Pulsatile luteinizing hormone secretion in patients with Addison’s disease. Impact of glucocorticoid substitution. J Clin Endocrinol Metab. 1998;83:736–743.
297. Lopez-Alvarenga, JC, Zarinan, T, Olivares, A, et al. Poorly controlled type I diabetes mellitus in young men selectively suppresses luteinizing hormone secretory burst mass. J Clin Endocrinol Metab. 2002;87:5507–5515.
298. Brabant, G, Prank, K, Ranft, U, et al. Physiological regulation of circadian and pulsatile thyrotropin secretion in normal man and woman. J Clin Endocrinol Metab. 1990;70:403–409.
299. Parker, DC, Rossman, LG, Pekary, AE, et al. Effect of 64-hour sleep deprivation on the circadian waveform of thyrotropin (TSH): further evidence of sleep-related inhibition of TSH release. J Clin Endocrinol Metab. 1987;64:157–161.
300. Hirschfeld, U, Moreno-Reyes, R, Akseki, E, et al. Progressive elevation of plasma thyrotropin during adaptation to simulated jet lag: effects of treatment with bright light or zolpidem. J Clin Endocrinol Metab. 1996;81:3270–3277.
301. Goichot, B, Brandenberger, G, Saini, J, et al. Nocturnal plasma thyrotropin variations are related to slow-wave sleep. J Sleep Res. 1992;1:186–190.
302. Russell, W, Harrison, RF, Smith, N, et al. Free triiodothyronine has a distinct circadian rhythm that is delayed but parallels thyrotropin levels. J Clin Endocrinol Metab. 2008;93:2300–2306.
303. Allan, JS, Czeisler, CA. Persistence of the circadian thyrotropin rhythm under constant conditions and after light-induced shifts of circadian phase. Journal of Clinical Endocrinology and. Metabolism. 1994;79:508–512.
304. Gary, KA, Winokur, A, Douglas, SD, et al. Total sleep deprivation and the thyroid axis: effects of sleep and waking activity. Aviat Space Environ Med. 1996;67:513–519.
305. Romijn, JA, Wiersinga, WM. Decreased nocturnal surge of thyrotropin in nonthyroidal illness. J Clin Endocrinol. Metabolism. 1990;70:35–42.
306. Bartalena, F, Martino, E, Petrini, L, et al. The nocturnal serum thyrotropin surge is abolished in patients with ACTH-dependent or ACTH-independent Cushing’s syndrome. J Clin Endocrinol Metab. 1991;72:1195–1199.
307. Caron, PJ, Nieman, LK, Rose, SR, et al. Deficient nocturnal surge of thyrotropin in central hypothyroidism. J Clin Endocrinol Metab. 1986;62:960–964.
308. Samuels, MH, Lillehei, K, Kleinschmidt-Demasters, BK, et al. Patterns of pulsatile pituitary glycoprotein secretion in central hypothyroidism and hypogonadism. J Clin Endocrinol Metab. 1990;70:391–395.
309. Bartalena, L, Cossu, E, Grasso, L, et al. Relationship between nocturnal serum thyrotropin peak and metabolic control in diabetic patients. J Clin Endocrinol Metab. 1993;76:983–987.
310. Kok, P, Roelfsema, F, Langendonk, JG, et al. High circulating thyrotropin levels in obese women are reduced after body weight loss induced by caloric restriction. J Clin Endocrinol Metab. 2005;90:4659–4663.
311. el-Hajj Fuleihan, G, Klerman, EB, Brown, EN, et al. The parathyroid hormone circadian rhythm is truly endogenous—a general clinical research center study. J Clin Endocrinol Metab. 1997;82:281–286.
312. Kripke, DF, Lavie, P, Parker, D, et al. Plasma parathyroid hormone and calcium are related to sleep stage cycles. J Clin Endocrinol Metab. 1978;47:1021–1027.
313. Logue, FC, Fraser, WD, O’Reilly, DS, et al. Sleep shift dissociates the nocturnal peaks of parathyroid hormone (1–84), nephrogenous cyclic adenosine monophosphate, and prolactin in normal men. J Clin Endocrinol Metab. 1992;75:25–29.
314. Fraser, WD, Logue, FC, Christie, JP, et al. Alteration of the circadian rhythm of intact parathyroid hormone following a 96-hour fast. Clin Endocrinol (Oxf). 1994;40:523–528.
315. Fraser, WD, Logue, FC, Christie, JP, et al. Alteration of the circadian rhythm of intact parathyroid hormone and serum phosphate in women with established postmenopausal osteoporosis. Osteoporos Int. 1998;8:121–126.
316. Lobaugh, B, Neelon, FA, Oyama, H, et al. Circadian rhythms for calcium, inorganic phosphorus, and parathyroid hormone in primary hyperparathyroidism: functional and practical considerations. Surgery. 1989;106:1009–1016.
317. Luthringer, R, Brandenberger, G, Schaltenbrand, N, et al. Slow wave electroencephalographic activity parallels renin oscillations during sleep in humans. Electroenceph and. Clin Neurophysiol. 1995;95:318–322.
318. Charloux, A, Gronfier, C, Lonsdorfer-Wolf, E, et al. Aldosterone release during the sleep-wake cycle in humans. Am J Physiol. 1999;276:E43–E49.
319. Brandenberger, G, Follenius, M, Simon, C, et al. Nocturnal oscillations in plasma renin activity and REM—NREM sleep cycles in man: a common regulatory mechanism? Sleep. 1988;11:242–250.
320. Charloux, A, Gronfier, C, Chapotot, F, et al. Sleep deprivation blunts the night time increase in aldosterone release in humans. J Sleep Res. 2001;10:27–33.
321. Brandenberger, G, Charloux, A, Grongier, C, et al. Ultradian rhythms in hydromineral hormones. Horm Res. 1998;49:131–135.
322. Follenius, M, Brandenberger, G, Saini, J. Lack of diurnal rhythm in plasma atrial natriuretic peptide. Life Sci. 1992;51:143–149.
323. Simon, C, Gronfier, C, Schlienger, JL, et al. Circadian and ultradian variations of leptin in normal man under continuous enteral nutrition: relationship to sleep and body temperature. J Clin Endocrinol Metab. 1998;83:1893–1899.
324. Maquet, P. Positron emission tomography studies of sleep and sleep disorders. J Neurol. 1997;244:S23–S28.
325. Maquet, P. Functional neuroimaging of normal human sleep by positron emission tomography. J Sleep Res. 2000;9:207–231.
326. Dang-Vu, TT, Desseilles, M, Laureys, S, et al. Cerebral correlates of delta waves during non-REM sleep revisited. Neuroimage. 2005;28:14–21.
327. DeFronzo, RA. Pathogenesis of type 2 diabetes mellitus. Med Clin North Am. 2004;88:787–835.
328. Magistretti, PJ. Neuron-glia metabolic coupling and plasticity. J Exp Biol. 2006;209:2304–2311.
329. Plat, L, Byrne, MM, Sturis, J, et al. Effects of morning cortisol elevation on insulin secretion and glucose regulation in humans. Am J Physiol. 1996;270:E36–E42.
330. Scheen, AJ, Byrne, MM, Plat, L, et al. Relationships between sleep quality and glucose regulation in normal humans. Am J Physiol. 1996;271:E261–E270.
331. Jauch-Chara, K, Hallschmid, M, Schmid, SM, et al. Plasma glucagon decreases during night-time sleep in Type 1 diabetic patients and healthy control subjects. Diabet Med. 2007;24:684–687.
332. Spiegel, K, Leproult, R, L’Hermite-Baleriaux, M, et al. Leptin levels are dependent on sleep duration: relationships with sympathovagal balance, carbohydrate regulation, cortisol, and thyrotropin. J Clin Endocrinol Metab. 2004;89:5762–5771.
333. Spiegel, K, Knutson, K, Leproult, R, et al. Sleep loss: a novel risk factor for insulin resistance and Type 2 diabetes. J Appl Physiol. 2005;99:2008–2019.
334. Tasali, E, Leproult, R, Ehrmann, DA, et al. Slow-wave sleep and the risk of type 2 diabetes in humans. Proc Natl Acad Sci U S A. 2008;105:1044–1049.
335. Knutson, KL, Van Cauter, E. Associations between sleep loss and increased risk of obesity and diabetes. Ann N Y Acad Sci. 2008;1129:287–304.
336. Simon, C, Brandenberger, G. Ultradian oscillations of insulin secretion in humans. Diabetes. 2002;51(Suppl 1)):S258–S261.
337. Sturis, J, Van Cauter, E, Blackman, JD, et al. Entrainment of pulsatile insulin secretion by oscillatory glucose infusion. J Clin Invest. 1991;87:439–445.
338. Sturis, J. Possible mechanisms underlying slow oscillations of human insulin secretion [PhD dissertation]. Lyngby, Denmark: The Technical University of Denmark; 1991.
339. Song, SH, Kjems, L, Ritzel, R, et al. Pulsatile insulin secretion by human pancreatic islets. J Clin Endocrinol Metab. 2002;87:213–221.
340. Porksen, N, Hollingdal, M, Juhl, C, et al. Pulsatile insulin secretion: detection, regulation, and role in diabetes. Diabetes. 2002;51(Suppl 1)):S245–S254.
341. Meneilly, GS, Veldhuis, JD, Elahi, D. Disruption of the pulsatile and entropic modes of insulin release during an unvarying glucose stimulus in elderly individuals. J Clin Endocrinol Metab. 1999;84:1938–1943.
342. Getty, L, Panteleon, AE, Mittelman, SD, et al. Rapid oscillations in omental lipolysis are independent of changing insulin levels in vivo. J Clin Invest. 2000;106:421–430.
343. Van Cauter, E, Polonsky, KS, Blackman, JD, et al. Abnormal temporal patterns of glucose tolerance in obesity: relationship to sleep-related growth hormone and circadian cortisol rhythmicity. J Clin Endocrinol Metab. 1994;79:1797–1805.
344. Bolli, GB, Gerich, JE. The “dawn phenomenon”—a common occurrence in both non-insulin-dependent and insulin-dependent diabetes mellitus. N Engl J Med. 1984;310:746–750.
345. Campbell, PJ, Bolli, GB, Cryer, PE, et al. Pathogenesis of the dawn phenomenon in patients with insulin-dependent diabetes mellitus. N Engl J Med. 1985;312:1473–1479.
346. Davidson, MB, Harris, MD, Ziel, FH, et al. Suppression of sleep-induced growth hormone secretion by anticholinergic agent abolishes dawn phenomenon. Diabetes. 1988;37:166–171.
347. Shapiro, ET, Polonsky, KS, Copinschi, G, et al. Nocturnal elevation of glucose levels during fasting in noninsulin-dependent diabetes. J Clin Endocrinol Metab. 1991;72:444–454.
348. Banarer, S, Cryer, PE. Sleep-related hypoglycemia-associated autonomic failure in type 1 diabetes: reduced awakening from sleep during hypoglycemia. Diabetes. 2003;52:1195–1203.
349. Jauch-Chara, K, Schmid, SM, Hallschmid, M, et al. Altered neuroendocrine sleep architecture in patients with type 1 diabetes. Diabetes Care. 2008;31:1183–1188.
350. O’Meara, NM, Sturis, J, Van Cauter, E, et al. Lack of control by glucose of ultradian insulin secretory oscillations in impaired glucose tolerance and in non-insulin-dependent diabetes mellitus. J Clin Invest. 1993;92:262–271.
351. Schmitz, O, Juhl, CB, Hollingdal, M, et al. Irregular circulating insulin concentrations in type 2 diabetes mellitus: an inverse relationship between circulating free fatty acid and the disorderliness of an insulin time series in diabetic and healthy individuals. Metabolism. 2001;50:41–46.
352. Kershaw, EE, Flier, JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab. 2004;89:2548–2556.
353. Flier, JS. Obesity wars: molecular progress confronts an expanding epidemic. Cell. 2004;116:337–350.
354. Sinha, MK, Ohannesian, JP, Heiman, ML, et al. Nocturnal rise of leptin in lean, obese and non-insulin-dependent diabetes mellitus subjects. J Clin Invest. 1996;97:1344–1347.
355. Saad, MF, Riad-Gabriel, MG, Khan, A, et al. Diurnal and ultradian rhythmicity of plasma leptin: effects of gender and adiposity. J Clin Endocrinol Metab. 1998;83:453–459.
356. Schoeller, DA, Cella, LK, Sinha, MK, et al. Entrainment of the diurnal rhythm of plasma leptin to meal timing. J Clin Invest. 1997;100:1882–1887.
357. Shea, SA, Hilton, MF, Orlova, C, et al. Independent circadian and sleep/wake regulation of adipokines and glucose in humans. J Clin Endocrinol Metab. 2005;90:2537–2544.
358. Mullington, JM, Chan, JL, Van Dongen, HP, et al. Sleep loss reduces diurnal rhythm amplitude of leptin in healthy men. J Neuroendocrinol. 2003;15:851–854.
359. Sinha, MK, Sturis, J, Ohannesian, J, et al. Ultradian oscillations of leptin secretion in humans. Biochem Biophys Res Commun. 1996;228:733–738.
360. Licinio, J, Mantzoros, C, Negrao, AB, et al. Human leptin levels are pulsatile and inversely related to pituitary-adrenal function. Nat Med. 1997;3:575–579.
361. Licinio, J, Negrao, AB, Mantzoros, C, et al. Sex differences in circulating human leptin pulse amplitude: clinical implications. J Clin Endocrinol Metab. 1998;83:4140–4147.
362. Kolaczynski, JW, Considine, RV, Ohannesian, J, et al. Responses of leptin to short-term fasting and refeeding in humans: a link with ketogenesis but not ketones themselves. Diabetes. 1996;45:1511–1515.
363. Chin-Chance, C, Polonsky, KS, Schoeller, DA. Twenty-four-hour leptin levels respond to cumulative short-term energy imbalance and predict subsequent intake. J Clin Endocrinol Metab. 2000;85:2685–2691.
364. Laughlin, GA, Yen, SS. Hypoleptinemia in women athletes: absence of a diurnal rhythm with amenorrhea. J Clin Endocrinol Metab. 1997;82:318–321.
365. Balligand, JL, Brichard, SM, Brichard, V, et al. Hypoleptinemia in patients with anorexia nervosa: loss of circadian rhythm and unresponsiveness to short-term refeeding. Eur J Endocrinol. 1998;138:415–420.
366. Franceschini, R, Corsini, G, Cataldi, A, et al. Twenty-four-hour variation in serum leptin in the elderly. Metabolism. 1999;48:1011–1014.
367. Spiegel, K, Tasali, E, Penev, P, et al. Brief communication: sleep curtailment in healthy young men is associated with decreased leptin levels, elevated ghrelin levels, and increased hunger and appetite. Ann Intern Med. 2004;141:846–850.
368. Taheri, S, Lin, L, Austin, D, et al. Short sleep duration is associated with reduced leptin, elevated ghrelin, and increased body mass index. PLoS Med. 2004;1:e62.
369. Chaput, JP, Despres, JP, Bouchard, C, et al. Short sleep duration is associated with reduced leptin levels and increased adiposity: results from the Quebec family study. Obesity (Silver Spring). 2007;15:253–261.
370. Elimam, A, Knutsson, U, Bronnegard, M, et al. Variations in glucocorticoid levels within the physiological range affect plasma leptin levels. Eur J Endocrinol. 1998;139:615–620.
371. Purnell, JQ, Samuels, MH. Levels of leptin during hydrocortisone infusions that mimic normal and reversed diurnal cortisol levels in subjects with adrenal insufficiency. J Clin Endocrinol Metab. 1999;84:3125–3128.
372. Leal-Cerro, A, Considine, RV, Peino, R, et al. Serum immunoreactive-leptin levels are increased in patients with Cushing’s syndrome. Horm Metab Res. 1996;28:711–713.
373. Masuzaki, H, Ogawa, Y, Hosoda, K, et al. Glucocorticoid regulation of leptin synthesis and secretion in humans: elevated plasma leptin levels in Cushing’s syndrome. J Clin Endocrinol Metab. 1997;82:2542–2547.
374. Veldman, RG, Frolich, M, Pincus, SM, et al. Hyperleptinemia in women with Cushing’s disease is driven by high-amplitude pulsatile, but orderly and eurhythmic, leptin secretion. Eur J Endocrinol. 2001;144:21–27.
375. Kousta, E, Chrisoulidou, A, Lawrence, NJ, et al. The circadian rhythm of leptin is preserved in growth hormone deficient hypopituitary adults. Clin Endocrinol (Oxf). 1998;48:685–690.
376. Pombo, M, Herrera-Justiniano, E, Considine, RV, et al. Nocturnal rise of leptin in normal prepubertal and pubertal children and in patients with perinatal stalk-transection syndrome. J Clin Endocrinol Metab. 1997;82:2751–2754.
377. Yamauchi, T, Kamon, J, Waki, H, et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med. 2001;7:941–946.
378. Kubota, N, Terauchi, Y, Yamauchi, T, et al. Disruption of adiponectin causes insulin resistance and neointimal formation. J Biol Chem. 2002;277:25863–25866.
379. Gavrila, A, Peng, CK, Chan, JL, et al. Diurnal and ultradian dynamics of serum adiponectin in healthy men: comparison with leptin, circulating soluble leptin receptor, and cortisol patterns. J Clin Endocrinol Metab. 2003;88:2838–2843.
380. Weyer, C, Funahashi, T, Tanaka, S, et al. Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab. 2001;86:1930–1935.
381. Yildiz, BO, Suchard, MA, Wong, ML, et al. Alterations in the dynamics of circulating ghrelin, adiponectin, and leptin in human obesity. Proc Natl Acad Sci U S A. 2004;101:10434–10439.
382. Calvani, M, Scarfone, A, Granato, L, et al. Restoration of adiponectin pulsatility in severely obese subjects after weight loss. Diabetes. 2004;53:939–947.
383. Date, Y, Kojima, M, Hosoda, H, et al. Ghrelin, a novel growth hormone-releasing acylated peptide, is synthesized in a distinct endocrine cell type in the gastrointestinal tracts of rats and humans. Endocrinology. 2000;141:4255–4261.
384. Wren, AM, Seal, LJ, Cohen, MA, et al. Ghrelin enhances appetite and increases food intake in humans. J Clin Endocrinol Metab. 2001;86:5992.
385. Weikel, JC, Wichniak, A, Ising, M, et al. Ghrelin promotes slow-wave sleep in humans. Am J Physiol Endocrinol Metab. 2003;284:E407–E415.
386. Szentirmai, E, Hajdu, I, Obal, F, Jr., et al. Ghrelin-induced sleep responses in ad libitum fed and food-restricted rats. Brain Res. 2006;1088:131–140.
387. Koutkia, P, Canavan, B, Breu, J, et al. Nocturnal ghrelin pulsatility and response to growth hormone secretagogues in healthy men. Am J Physiol Endocrinol Metab. 2004;287:E506–E512.
388. Cummings, DE, Frayo, RS, Marmonier, C, et al. Plasma ghrelin levels and hunger scores in humans initiating meals voluntarily without time- and food-related cues. Am J Physiol Endocrinol Metab. 2004;287:E297–E304.
389. Cummings, DE, Purnell, JQ, Frayo, RS, et al. A preprandial rise in plasma ghrelin levels suggests a role in meal initiation in humans. Diabetes. 2001;50:1714–1719.
390. Cummings, DE, Weigle, DS, Frayo, RS, et al. Plasma ghrelin levels after diet-induced weight loss or gastric bypass surgery. N Engl J Med. 2002;346:1623–1630.
391. Teff, KL, Elliott, SS, Tschop, M, et al. Dietary fructose reduces circulating insulin and leptin, attenuates postprandial suppression of ghrelin, and increases triglycerides in women. J Clin Endocrinol Metab. 2004;89:2963–2972.
392. Misra, M, Miller, KK, Kuo, K, et al. Secretory dynamics of ghrelin in adolescent girls with anorexia nervosa and healthy adolescents. Am J Physiol Endocrinol Metab. 2005;289:E347–E356.
393. Espelund, U, Hansen, TK, Hojlund, K, et al. Fasting unmasks a strong inverse association between ghrelin and cortisol in serum: studies in obese and normal-weight subjects. J Clin Endocrinol Metab. 2005;90:741–746.
394. Schmid, SM, Hallschmid, M, Jauch-Chara, K, et al. A single night of sleep deprivation increases ghrelin levels and feelings of hunger in normal-weight healthy men. J Sleep Res. 2008;17:331–334.
395. Jayasena, CN, Bloom, SR. Role of gut hormones in obesity. Endocrinol Metab Clin North Am. 2008;37:769–787.
396. McEwen, BS. Stress, adaptation, and disease. Allostasis and allostatic load. Ann N Y Acad Sci. 1998;840:33–44.
397. Van Cauter, E, Knutson, K. Sleep and the epidemic of obesity in children and adults. Eur J Endocrinol. 2008;159(suppl 1):S59–S66.
398. Cooper, BG, White, JES, Ashworth, LA, et al. Hormonal and metabolic profiles in subjects with obstructive sleep apnea syndrome and the effects of nasal continuous positive airway pressure (CPAP) treatment. Sleep. 1995;18:172–179.
399. Saini, J, Krieger, J, Brandenberger, G, et al. Continuous positive airway pressure treatment: effects on growth hormone, insulin and glucose profiles in obstructive sleep apnea patients. Hormone Metab Res. 1993;25:375–381.
400. Spiegel, K, Follenius, M, Krieger, J, et al. Prolactin secretion during sleep in obstructive sleep apnea patients. J Sleep Res. 1995;4:56–62.
401. Luboshitzky, R, Lavie, L, Shen-Orr, Z, et al. Pituitary-gonadal function in men with obstructive sleep apnea. The effect of continuous positive airways pressure treatment. Neuro Endocrinol Lett. 2003;24:463–467.
402. Ozturk, L, Unal, M, Tamer, L, et al. The association of the severity of obstructive sleep apnea with plasma leptin levels. Arch Otolaryngol Head Neck Surg. 2003;129:538–540.
403. Patel, SR, Palmer, LJ, Larkin, EK, et al. Relationship between obstructive sleep apnea and diurnal leptin rhythms. Sleep. 2004;27:235–239.
404. Phillips, BG, Kato, M, Narkiewicz, K, et al. Increases in leptin levels, sympathetic drive, and weight gain in obstructive sleep apnea. Am J Physiol Heart Circ Physiol. 2000;279:H234–H237.
405. Ip, MS, Lam, KS, Ho, C, et al. Serum leptin and vascular risk factors in obstructive sleep apnea. Chest. 2000;118:580–586.
406. Sanner, BM, Kollhosser, P, Buechner, N, et al. Influence of treatment on leptin levels in patients with obstructive sleep apnoea. Eur Respir J. 2004;23:601–604.
407. Harsch, IA, Konturek, PC, Koebnick, C, et al. Leptin and ghrelin levels in patients with obstructive sleep apnoea: effect of CPAP treatment. Eur Respir J. 2003;22:251–257.
408. Shimizu, K, Chin, K, Nakamura, T, et al. Plasma leptin levels and cardiac sympathetic function in patients with obstructive sleep apnoea-hypopnoea syndrome. Thorax. 2002;57:429–434.
409. Chin, K, Shimizu, K, Nakamura, T, et al. Changes in intra-abdominal visceral fat and serum leptin levels in patients with obstructive sleep apnea syndrome following nasal continuous positive airway pressure therapy. Circulation. 1999;100:706–712.
410. Tasali, E, Mokhlesi, B, Van Cauter, E. Obstructive sleep apnea and type 2 diabetes: interacting epidemics. Chest. 2008;133:496–506.
410a. Schahin, SP, Nechanitzky, T, Dittel, C, et al. Long-term improvement of insulin sensitivity during CPAP therapy in the obstructive sleep apnoea syndrome. Med Sci Monit. 2008;14:CR11121.
411. West, SD, Nicoll, DJ, Stradling, JR. Prevalence of obstructive sleep apnoea in men with type 2 diabetes. Thorax. 2006;61:945–950.
412. Foster, G, Kuna, S, Sanders, M, et al. Sleep Apnea In Obese Adults With Type 2 Diabetes: Baseline Results From The Sleep Ahead Study. Sleep. 25, 2008.
413. Higuchi, T, Takahashi, Y, Takahashi, K, et al. Twenty-four-hour secretory patterns of growth hormone, prolactin, and cortisol in narcolepsy. J Clin Endocrinol Metab. 1979;49:197–204.
414. Kok, SW, Roelfsema, F, Overeem, S, et al. Dynamics of the pituitary-adrenal ensemble in hypocretin-deficient narcoleptic humans: blunted basal adrenocorticotropin release and evidence for normal time-keeping by the master pacemaker. J Clin Endocrinol Metab. 2002;87:5085–5091.
415. Overeem, S, Kok, SW, Lammers, GJ, et al. Somatotropic axis in hypocretin-deficient narcoleptic humans: altered circadian distribution of GH-secretory events. Am J Physiol Endocrinol Metab. 2003;284:E641–E647.
416. Kok, SW, Meinders, AE, Overeem, S, et al. Reduction of plasma leptin levels and loss of its circadian rhythmicity in hypocretin (orexin)-deficient narcoleptic humans. J Clin Endocrinol Metab. 2002;87:805–809.
417. Rosenthal, RE, Vanderpool, JRJ, Levendosky, AA, et al. Phase-shifting effects of bright morning light as treatment for delayed sleep phase syndrome. Sleep. 1990;13:354–361.
418. Jones, CR, Campbell, SS, Zone, SE, et al. Familial advanced sleep-phase syndrome: a short-period circadian rhythm variant in humans. Nat Med. 1999;5:1062–1065.
419. Young, ME. The circadian clock within the heart: potential influence on myocardial gene expression, metabolism, and function. Am J Physiol Heart Circ Physiol. 2006;290:H1–H16.
420. Aschoff, J, Hoffmann, K, Pohl, H, et al. Re-entrainment of circadian rhythms after phase-shifts of the zeitgeber. Chronobiologia. 1975;28:119–133.
421. Buxton, OM, L’Hermite-Baleriaux, M, Hirshfeld, U, et al. Acute and delayed effects of exercise on human melatonin secretion. J Biol Rhythms. 1997;12:568–574.
422. Czeisler, CA, Johnson, MP, Duffy, JF, et al. Exposure to bright light and darkness to treat physiologic maladaptation to night work. N Engl J Med. 1990;322:1253–1259.
423. Knutsson, A, Akerstedt, T, Orth-Gomer, K, et al. Increased risk of ischaemic heart disease in shift workers. Lancet. 1986;2:89–92.
424. Rosa, RR. Extended workshifts and excessive fatigue. J Sleep Res. 1995;4:51–56.
425. van Amelsvoort, LG, Schouten, EG, Kok, FJ. Duration of shiftwork related to body mass index and waist to hip ratio. Int J Obes Relat Metab Disord. 1999;23:973–978.
426. Karlsson, B, Knutsson, A, Lindahl, B. Is there an association between shift work and having a metabolic syndrome? Results from a population based study of 27,485 people. Occup Environ Med. 2001;58:747–752.
427. Nagaya, T, Yoshida, H, Takahashi, H, et al. Markers of insulin resistance in day and shift workers aged 30–59 years. Int Arch Occup Environ Health. 2002;75:562–568.
428. Sookoian, S, Gemma, C, Fernandez Gianotti, T, et al. Effects of rotating shift work on biomarkers of metabolic syndrome and inflammation. J Intern Med. 2007;261:285–292.
429. Roden, M, Koller, M, Pirich, K, et al. The circadian melatonin and cortisol secretion pattern in permanent night shift workers. Am J Physiol. 1993;265:R261–R267.
430. Weibel, L, Spiegel, K, Follenius, M, et al. Internal dissociation of the circadian markers of the cortisol rhythm in night workers. Am J Physiol. 1996;270:E608–E613.
431. Burgess, HJ, Sharkey, KM, Eastman, CI. Bright light, dark and melatonin can promote circadian adaptation in night shift workers. Sleep Med Rev. 2002;6:407–420.
[/level-membership-for-endocrinology-diabetes-and-metabolism-category][not-level-membership-for-endocrinology-diabetes-and-metabolism-category]Chapter 4
Endocrine Rhythms, The Sleep-Wake Cycle, and Biological Clocks
Major Mechanisms Controlling 24-Hour Endocrine Rhythms
As is illustrated schematically in Figure 4-1, the temporal variability and organization of hormonal concentrations during the 24-hour cycle ultimately result from the activity of two interacting time-keeping mechanisms in the central nervous system: endogenous circadian rhythmicity and sleep-wake homeostasis, a mechanism that relates the timing and intensity of sleep to the duration of prior wakefulness. Although this dual control was first demonstrated for hormones of the hypothalamic-pituitary axis, a similar regulation appears to apply to other endocrine subsystems. In mammals, endogenous circadian rhythmicity is generated by a master circadian clock located in the paired suprachiasmatic nucleus (SCN) of the hypothalamus.1 The SCN controls the timing of most, if not all, circadian rhythms and partially regulates the sleep-wake cycle. The sleep-wake cycle in turn regulates the timing of many rhythms that depend on the presence or absence of sleep and wakefulness. Indeed, the timing and expression of many endocrine rhythms appear to depend upon direct control from the SCN, as well as on the presence and quality of sleep, with some 24-hour endocrine rhythms influenced more by the SCN (e.g., melatonin and cortisol), and others more regulated by the sleep-wake state (e.g., growth hormone). Thus, combined inputs of the master circadian clock and the sleep-wake state2 control the overall temporal organization of the endocrine system, as well as many other behavioral and physiologic systems, across the 24-hour cycle. The two major pathways by which circadian rhythmicity and sleep-wake homeostasis affect peripheral endocrine function are the hypothalamic-pituitary axis and the autonomous nervous system.
FIGURE 4-1 Schematic representation of the central mechanisms involved in the control of temporal variations in pituitary hormone secretions over the 24-hour cycle. ACTH, Adrenocorticotropic hormone; ANS, autonomous nervous system; FSH, follicle-stimulating hormone; GH, growth hormone; LH, luteinizing hormone; PRL, prolactin; SCN, suprachiasmatic nucleus of the hypothalamus; TSH, thyrotropin.
Circadian Rhythmicity
One of the most obvious characteristics of life on earth is the ability of almost all species to change their behavior on a daily or 24-hour basis. A remarkable feature of these daily or diurnal rhythms is that they are not simply a response to 24-hour changes in the physical environment imposed by the principles of celestial mechanics, but instead arise from an internal time-keeping system1,3 that has the intrinsic capability to continuously generate rhythmic activity over a nearly 24-hour period. Thus, under laboratory conditions devoid of any external time-giving cues, it has been found that nearly all 24-hour rhythms continue to be expressed. However, under such constant conditions, the period of the rhythm rarely remains exactly 24 hours but instead is “about” 24 hours, and this is why these rhythms are referred to as circadian, from the Latin “circa diem,” meaning “around a day.” When a circadian rhythm is expressed in the absence of any 24 hour signals in the external environment, it is said to be free-running. Under free-running conditions, the endogenous period generally is close to, but is nearly never exactly, 24 hours. Strictly speaking, a diurnal rhythm should not be referred to as circadian until it has been demonstrated that such a rhythm persists under constant environmental conditions. The purpose of this distinction is to separate out those rhythms that are simply a response to 24-hour changes in the environment from those that are endogenous. However, for practical purposes, there is little reason to make a distinction between diurnal and circadian rhythms, since an endogenous timing device underlies the generation of almost all diurnal rhythms. In this chapter, we therefore extend the use of the term circadian rhythm to include all diurnal variations that recur regularly at a time interval of approximately 24 hours.
The endogenous nature of human circadian rhythms has been established by experiments in which subjects were isolated with no access to the natural light-dark cycle and no time cues. Such experiments were first performed in natural caves, then in underground bunkers, and finally in specially designed windowless soundproof apartments. The results of one such early experiment, conducted in an artificial underground unit in Germany, are shown in Figure 4-2.4 The rest-activity cycle of the subject is plotted horizontally, day by day, and the times of occurrence of the daily maximum of the body temperature cycle are indicated by closed triangles. During the first 7 days of the experiment, the door of the isolation unit was left open, and the subjects knew the time of day. The average duration (t) of the rest-activity cycle and of the rhythm of body temperature was 24 hours. When, thereafter, the subject lived in complete isolation, both rhythms free-ran but with a mean period of about 26 hours. The free-running period varies from one individual to another. In humans, free-running periods of around 25 hours have been observed under conditions of prolonged temporal isolation. However, assessments of the human free-running period based on the so-called forced desynchrony protocol have provided estimations of between 24.1 and 24.2 hours.5
FIGURE 4-2 Circadian rhythms of wakefulness (black bars), sleep (white bars), and maximal rectal temperature (triangles) in a human subject who was exposed to external synchronizing agents for the first and last 7 days and was isolated from all time cues in an underground bunker between days 8 and 24.
The Suprachiasmatic Nucleus: A Master Circadian Pacemaker
In mammals, the suprachiasmatic nucleus (SCN), which consists of two small, bilaterally paired nuclei in the anterior hypothalamus, each of which contains about 10,000 cells in rodents, functions as the master circadian clock. Under both free-running and entrained conditions, destruction of the SCN in a variety of species leads to abolishment or severe disruption of many endocrine, behavioral, and physiologic rhythms.1,6 The role of the SCN as the control center for the circadian system, first suggested by lesion studies, was confirmed by studies involving transplantation of the SCN from one animal to another. Indeed, circadian rhythmicity can be restored in adult arrhythmic SCN-lesioned rodents by transplanting fetal SCN tissue into the region of the SCN.7,8 A number of SCN rhythms, including those of neural firing, vasopressin release, glucose metabolism, and gene expression, persist in vitro.9–12 The ability of SCN cells to generate a circadian signal does not rely on some inherent network property of many cells acting together: Single SCN cells in culture can generate circadian neural signals.13 Neurons within the SCN appear to be organized into two groups: a core group of light responsive but nonrhythmic cells, and a shell of rhythmic cells.14,15
The generation and maintenance of circadian oscillations in the SCN involve a series of clock genes (including per1, per 2, per3, cry1, cry2, tim, clock, B-mal1, and CKIε/δ), often referred to as canonical, which interact in a complex feedback loop of transcription/translation.16,17 Indeed, improved tissue culture techniques and real-time monitoring of the expression of circadian clock genes have revealed that circadian oscillations in gene expression can persist in the SCN in vitro (as well as in other tissues, see later) for many cycles.16 It is important to note that mutations or deletions of canonical circadian clock genes have been found to profoundly affect endocrine rhythms and normal endocrine function in a variety of central and peripheral tissues.18–20 For reviews on the molecular and genetic control of circadian rhythmicity, the reader is referred to references 16, 17, and 21.
In recent years, it has been recognized that circadian oscillations can be generated in areas of the brain other than the SCN, as well as in many peripheral tissues.9,22 These local oscillators appear to be under the control of the SCN and of the canonical circadian clock genes in the SCN, through the synchronization or entrainment of the same circadian clock machinery at the local tissue level. The SCN controls or synchronizes these autonomous circadian clocks in non-SCN tissues directly via neural and/or endocrine signals, or indirectly via its control of behavioral rhythms such as the sleep-wake cycle and the rhythm of feeding. However, the precise mechanisms are not well understood.
Photic Entrainment of Circadian Rhythms
The light-dark (LD) cycle is the primary agent that synchronizes most circadian rhythms. Thus, in the presence of a 24-hour LD cycle, the period of circadian rhythms exactly matches the period of the LD cycle. In addition to establishing period control, an entraining LD cycle establishes phase control, such that specific phases of the circadian rhythm occur at the same time in each cycle. Entrainment is restricted to cycles with periods that are close to 24 hours in duration and, in general, is not possible for LD cycles that are more than a few hours shorter or longer than the endogenous circadian period. If the period of the LD cycle is too short or too long for entrainment to occur, the circadian rhythm free-runs. This rigidity of the circadian pacemaker has been used in so-called forced desynchrony studies, where subjects are maintained on a light-dark and sleep-wake cycle with a period outside the range of entrainment, such as 20 hours or 28 hours.5,23 As mentioned earlier, such protocols have provided estimations of the endogenous period of the human circadian system that are closer to 24 hours (i.e., averaging 24.1 hours) than those obtained in prolonged studies of temporal isolation.5 The fact that the human endogenous circadian period probably is very close to 24 hours is consistent with the findings of a study showing that a schedule of sleep-wake and dark-light cycles with very low light intensity during wakefulness is able to maintain entrainment to the 24-hour day but not to a 23.5- or 24.6-hour day.24
The eyes are involved in relaying entraining information from the LD cycle to the circadian timing system in mammals via a unique pathway, separate from the visual system, and referred to as the retinohypothalamic tract.25 At the level of the optic chiasm, retinal projections first enter the brain in the region of the SCN and surrounding hypothalamic areas.25 Thus, the integrity of the primary visual centers of the brain and/or of the perception of light is not necessary for entrainment of circadian rhythms by the LD cycle. The independence of photic input to the circadian clock from the visual system in mammals actually is not surprising from an evolutionary point of view. In all nonmammalian vertebrates, entrainment of circadian rhythms can occur in the absence of the eyes and relies on nonretinal photoreceptors in the brain.26 The independence of the circadian light sensing system from the visual system in mammals was suggested in early studies showing that rod-less/cone-less mice could still entrain to the light-dark cycle, even though the eyes were necessary.27 Early in this century, several laboratories discovered almost simultaneously that a special subset of retinal ganglion cells containing melanopsin could act as photoreceptors for relaying light information to the circadian clock in the SCN.28–31 This breakthrough not only demonstrated a new light sensing system in the retina, but also opened up new avenues of research regarding the impact of light on brain function independent of the visual system, including possible neuroendocrine effects.32,33 The finding that visual light perception is not necessary for circadian light perception can explain why in some totally blind humans, light exposure is capable of suppressing melatonin levels, indicating that visual blindness should not be equated with circadian blindness.34 In addition to the retinohypothalamic tract, the SCN receives retinal information indirectly from the lateral geniculate nucleus (LGN), which receives a direct projection from the retina.25
To examine how a zeitgeber such as light influences the circadian system, the organism is maintained in constant conditions (e.g., constant darkness) and then is exposed to the zeitgeber for a brief period (e.g., 1 hour) before it is returned to constant conditions.35 The effects of zeitgeber exposure on a phase reference point of an overt circadian rhythm (e.g., onset of locomotor activity, minimum of body temperature) in subsequent cycles then are determined. A plot of the direction and magnitude of the phase-shift as a function of the circadian time of zeitgeber exposure is called a phase response curve (PRC).36 In the human, light exposure during the late evening and the first half of the usual sleep period results in phase delays, but light exposure at the end of the usual sleep period and in the early morning results in phase advances. The transition from phase delays to phase advances occurs around the time of the minimum of body temperature, that is, at between 04.00 and 06.00 hours in most subjects. The magnitude of the phase-shifts is also wavelength dependent, with exposure to short-wavelength monochromatic light (shifted to the blue) resulting in much larger shifts than exposure to longer-wavelength light of equal photon density,37 because the retinal ganglion cells that contain melanopsin are most sensitive to blue light.
Although differences in amplitude may exist, the general shape and characteristics of the PRC to light pulses are similar for all species. Based on this PRC, appropriate exposure to bright light can accelerate adaptation to shifts such as those occurring in jet lag and shift work. The amplitude of the phase-shifts depends on light intensity and duration, and on the number of consecutive exposures.38 For example, exposure to single 3- to 7-hour light pulses can induce phase-shifts on the order of 1 to 3 hours39–41; repeated exposure for 2 to 3 consecutive days may cause much larger 6- to 12-hour shifts.5,42 Intervening periods of dark/sleep exposure could play a role in enhancing the phase-shifting effects of repeated exposure to light.
Exposure to dark also can cause a phase-shift of mammalian circadian rhythms. Because under most circumstances exposure to dark is associated with changes in activity levels (i.e., increases in activity in nocturnal animals and decreases in activity and/or sleep induction in diurnal animals), it has been unclear whether the phase-shifts occurred in response to dark exposure per se, or in response to associated changes in activity levels.43 A study in hamsters, however, has demonstrated that resetting of the rhythm of locomotor activity and concomitant downregulation of circadian clock gene expression following exposure to dark pulses could occur independently of wheel running activity.44 In humans, abrupt 8-hour advances or 8-hour delays of the sleep-wake cycle result in immediate 2-hour phase-shifts in the same direction.45,46 Daytime naps of 6 hours’ duration in total darkness presented over a background of very dim light were found to cause delay shifts when initiated in the morning and advance shifts when initiated in the evening.47 Naps initiated in the afternoon cause no significant phase-shifts.
Nonphotic Zeitgebers
Nonphotic cues, for example, social and/or behavioral cues, also may alter the rest-activity cycle by eliciting activity during the normal rest period or by preventing activity during the normal active period, resulting in phase-shifts in circadian rhythms of activity, as well as other behavioral, physiologic, and endocrine markers.48–51 In nocturnal rodents, the PRC to activity-inducing stimuli is about 12 hours out of phase with the PRC to light pulses.52
How nonphotic information reaches the SCN still is not known, although evidence from lesion studies suggests that a distinct subdivision of the LGN, the intergeniculate leaflet (IGL), may be involved in mediating the effects of activity on the clock.53 Furthermore, the IGL is the source of neuropeptide Y (NPY) innervation of the SCN, and the administration of NPY into the SCN area, as well as electrical stimulation of the geniculohypothalamic tract, induces phase-shifts in hamster locomotor activity rhythm that are similar to those induced by activity-inducing stimuli.53 The LGN/IGL may be a common pathway by which information about the lighting environment and the activity-rest state reaches the circadian clock, and it may be involved in integrating information from the external and the internal environment. Both the LGN/IGL and the SCN receive a dense serotonergic projection from the midbrain raphe nuclei, and substantial evidence now indicates that these projections play a role in both photic and nonphotic regulation of the mammalian circadian clock.54–56
Nonphotic stimuli may affect human circadian rhythms. Exposure to a single session of 3 hours of moderate-intensity exercise during the usual nighttime period was found to result in phase-shifts of markers of circadian phase on the next day, with the direction and magnitude of the phase-shifts being dependent on the timing of exercise.57 Similar findings were obtained with nocturnal exposure to high-intensity 1 hour exercise sessions.58 Supporting evidence for a zeitgeber effect of exercise was obtained in a field study, which found that adaptation to night work could be facilitated by nocturnal exercise.59 These findings were confirmed by a study demonstrating that daily exposure to nightttime exercise facilitated phase delays in circadian melatonin rhythm, even when subjects were maintained in very dim light.60 Nocturnal exercise of low intensity also causes phase-delays in circadian rhythms in older adults, suggesting that it could be a useful treatment for the adjustment of circadian rhythmicity in older populations.61
Another nonphotic agent that has been shown to induce phase-shifts in human circadian rhythms is melatonin.62 This action of melatonin is often referred to as chronobiotic. Specific neural connections are present between the SCN and the pineal gland, and diurnal variation in plasma melatonin levels is driven by the circadian clock.63 Phase-shifts of the central circadian signal induced by changes in the LD cycle will be faithfully reflected in the synchronization of the onset of nocturnal melatonin secretion.40,64,65 Evidence suggests that, in turn, the melatonin rhythm feeds back on the clock (where melatonin receptors have been identified) and exerts synchronizing effects.63
The timing of feeding, when restricted to a narrow temporal window, can affect the entrainment pattern of behavioral rhythms through what has been referred to as a food-entrainable oscillator that is independent of the SCN. Recent findings in rodents have led to a renewed interest in the role of feeding in overall circadian organization in mammals, including the following: (1) alterations in circadian clock genes can lead to obesity and other metabolic abnormalities,2,20 (2) metabolic transcription factor and nuclear receptors involved in metabolism can alter the expression of circadian clock genes,66,67 (3) the timing of feeding can alter rhythmicity in several peripheral circadian oscillators,20,68 and (4) a high-fat diet can alter behavioral and molecular circadian rhythms in central and peripheral tissues involved in the regulation of energy balance.69 Although controversy exists as to the location of an SCN-independent food entrainable oscillator,70,71 now overwhelming evidence suggesting that the circadian and metabolic systems are linked together at molecular, cellular, and behavioral levels has fueled great interest in the possible role of circadian disorganization in obesity, diabetes, and other cardiometabolic disorders.20,72,73
Sleep-Wake Regulation
General Characteristics: The sleep-wake cycle may be viewed as a 24-hour rhythm driven partially by the circadian pacemaker and partially by the homeostatic regulation of sleep pressure. Sleep itself is an ultradian rhythm in that it involves two states of distinct brain activity, each of which is generated in specific brain regions. The ultradian rhythm of normal sleep is an approximate 90-minute oscillation between non-REM (rapid eye movement) and REM stages. In healthy subjects, this pattern usually is repeated four to six times per night. REM sleep and non-REM sleep are characterized by distinct patterns of cerebral and peripheral activity.
The all-night recording of EEG, muscle tone, and eye movements, called the polysomnogram, is scored visually over 20 or 30 second periods in stages I, II, III, IV, REM, and Wake with the use of standardized criteria.74 This procedure allows determination of the duration of each sleep stage but does not quantify the intensity of non-REM sleep. In contrast, the quantification of EEG recordings by power spectral analysis provides useful information regarding sleep depth or sleep intensity, because spectral analysis is sensitive to the amplitude of the delta waves. Higher-amplitude delta waves reflect more intense, deeper sleep, with less sensitivity to arousal stimuli. Slow-wave activity (SWA), i.e., spectral EEG power in the low-frequency range (also called delta range; 0.5 to 4.0 Hz), is a marker of the intensity of non-REM sleep.
Neuroanatomic Basis of Sleep Regulation: Normal waking is associated with neuronal activity in regions of the so-called ascending arousal system, which includes monoaminergic neurons in the brain stem and posterior hypothalamus, cholinergic neurons in the brain stem and basal forebrain, and orexin (hypocretin) neurons in the lateral hypothalamus.75,76 Initiation of sleep therefore requires the inhibition of these multiple arousal systems. In recent years, the ventrolateral preoptic area (VLPO) of the hypothalamus has been identified as involved in the inhibition of arousal. The VLPO contains sleep-active neurons that use the inhibitory neurotransmitter GABA (gamma-aminobutyric acid) and have much higher firing rates during deep sleep than during wakefulness.77–79 Lesions of the central cell cluster of the VLPO drastically reduce SW activity. Neurons of the VLPO provide GABAergic inhibitory innervation of the major monoamine arousal systems in the brain stem. Reciprocally, inhibitory pathways result from the monoamine arousal nuclei to the VLPO.80 The orexin neurons in the lateral hypothalamus project to all components of the ascending arousal system and stimulate the cortex.81 REM sleep is regulated primarily by cholinergic nuclei in the pons.
Interactions Between Circadian Rhythmicity and Sleep-Wake Homeostasis: Several features of the interaction between sleep and circadian rhythmicity appear to be fairly unique to the human species. First, human sleep generally is consolidated in a single 6- to 9-hour period, whereas fragmentation of the sleep period in several bouts is the rule in most other mammals. Possibly as a result of this consolidation of the sleep period, the wake-sleep transition in man is associated with physiologic changes that usually are more marked than those observed in animals. For example, the secretion of growth hormone (GH) in normal adults is tightly associated with the beginning of the sleep period, whereas the relationship between GH secretory pulses and sleep stages is much less evident in rodents, primates, and dogs. Second, man is unique in his capacity to ignore circadian signals and to maintain wakefulness despite increased pressure to go to sleep. Finally, approximately 25% of human subjects maintained for prolonged periods in temporal isolation have shown behavioral modifications that have not been observed in laboratory animals under constant conditions. These modifications consist of a desynchronization between the sleep-wake cycle and other rhythms, such as those of body temperature and cortisol secretion, which continue to free-run with a circadian period. Under conditions of so-called internal desynchronization, the sleep-wake cycle may be lengthened suddenly to 30 hours or more, while the rhythm of body temperature continues to free-run with a circadian period.4 Wakefulness may last longer than 30 hours. Remarkably, the subjects are not aware of these drastic changes in their way of living. Instead, most of them believe that they are living on a more or less regular 24-hour schedule. This can be explained by the observation that time perception is altered profoundly: Subjective estimations of 1 hour intervals are positively correlated with the duration of wakefulness.82 Of particular interest is that subjects continue to have three meals per “day,” irrespective of the actual number of hours they are awake.83 The intervals between meals as well as those between wake-up and breakfast, or between dinner and bedtime, are stretched or compressed in strong proportionality to the duration of wakefulness.84 The mechanisms that cause spontaneous internal desynchronization are not completely understood.
Detailed analyses of data obtained during temporal isolation and forced desynchrony protocols show that the timing, duration, and architecture of sleep are regulated in part by circadian rhythmicity.23,85 Thus, the duration of sleep episodes is correlated with the phase of the circadian rhythm of body temperature and not with the duration of prior wakefulness. Short (i.e., 7 to 8 hours) sleep episodes occur in free-running conditions when the subject goes to sleep around the minimum of body temperature, whereas long (i.e., 12 to 14 hours) sleep episodes occur when sleep starts at around the maximum of body temperature. Moreover, the distribution of REM sleep is markedly modulated by circadian timing. In contrast, the hourglass-like mechanism of sleep-wake homeostasis originally was thought to be largely independent of the circadian system and to involve one or several putative neural sleep factors, which rise during waking and decay exponentially during sleep.86 This homeostatic mechanism regulates the timing, amount, and intensity of SWS and SWA. The VLPO has been proposed as a neuroanatomic locus for the interaction of the homeostatic process and central circadian rhythmicity, as it receives dense projections from the dorsomedial hypothalamic nucleus, which itself receives direct and indirect projections from the SCN.87 Based on human studies described later, it is thought that the SCN generates a waking signal that promotes alertness during the active period. In support of this theory, studies have shown that rodents and monkeys with SCN lesions have increased sleep duration.88,89 Furthermore, studies in rats have described an indirect neuronal circuit from the SCN to the locus coeruleus, an area of the midbrain involved in the control of arousal.90 A role for the SCN in promoting sleep at other circadian times is suggested by the finding of decreased sleep in a mouse with a mutation of the Clock gene.91 The recent finding that the mutation or deletion of a number of circadian clock genes affects not only the timing of sleep but also many other sleep-wake traits, including traits linked to the homeostatic drive to sleep,15,91–94 indicates that circadian and homeostatic processes underlying the regulation of the sleep-wake cycle may be linked at molecular and anatomic levels of organization.
The dual control of sleep by circadian and homeostatic mechanisms extends to the control of objective and subjective measures of sleep tendency, mood, and vigilance.95–98 When wakefulness is extended beyond the usual 16 to 18 hours, maximum subjective sleepiness coincides with the minimum of body temperature, mood, and performance. Remarkably, despite continued sleep deprivation, subjective fatigue then decreases, and mood and performance partially recover during the daytime hours, reflecting an interaction of circadian timing with the accumulation of waking time.96–102 Currently, it is believed that the circadian clock generates a waking signal that increases from morning to evening and is expressed maximally in the early evening hours, 1 to 2 hours before the onset of nocturnal melatonin secretion.97 This circadian waking signal counteracts the buildup of the putative factor “S” underlying the homeostatic process, allowing the individual to maintain a high level of alertness throughout the usual waking period. Current data from human studies are compatible with the hypothesis that the SCN generates a sleep signal in the early evening hours.103
Circadian rhythmicity and sleep-wake homeostasis also interact to regulate hormonal secretion. These modulatory effects were long thought to be present only in hormones directly dependent on the hypothalamic-pituitary axis. However, it is now clear that modulation by circadian rhythmicity and sleep is also present in other endocrine systems, such as glucose regulation and the renin-angiotensin system.104,105 The multiple pathways by which circadian rhythmicity, sleep-wake homeostasis, and their interaction modulate hormonal release are incompletely understood. As is illustrated in Fig. 4-1, humoral and/or neural signals originating from the hypothalamic circadian pacemaker and from brain regions involved in sleep regulation affect the pulsatile release of hypothalamic neuroendocrine factors, which stimulate or inhibit intermittent secretion of pituitary hormones. The autonomic nervous system is another pathway linking the central control of sleep-wake homeostasis and circadian rhythmicity with peripheral endocrine organs. It appears that stimulatory or inhibitory effects of sleep on endocrine release are primarily associated with SW rather than REM sleep.106–110 Pituitary hormones that influence endocrine systems not directly controlled by hypothalamic factors probably mediate, together with the autonomous nervous system, the modulatory effects of sleep and circadian rhythmicity on these systems (e.g., counterregulatory effects of GH and cortisol on glucose regulation105).
To delineate the relative roles of circadian and sleep effects in the temporal organization of hormonal secretion, strategies based on the fact that circadian rhythmicity needs several days to adapt to abrupt shifts in the sleep-wake cycle have been used. Thus, by shifting sleep times by 8 to 12 hours, masking effects of sleep on circadian inputs are removed, and the effects of sleep at an abnormal circadian time are revealed. Fig. 4-3 illustrates the mean profiles of plasma cortisol, GH, prolactin, and thyrotropin (TSH) as observed in normal subjects who were studied before and during an abrupt 12-hour shift in the sleep-wake and dark-light cycles. The study period extended over a 53-hour span and included an 8-hour period of nocturnal sleep, a 28-hour period of continuous wakefulness, and a daytime period of recovery sleep. To eliminate the effects of feeding, fasting, and postural changes, subjects remained recumbent throughout the study, and the normal meal schedule was replaced by intravenous glucose infusion at a constant rate. As shown in Figure 4-3, this drastic manipulation of sleep had only modest effects on the wave shape of the cortisol profile, in sharp contrast with the immediate shift of GH and prolactin rhythms, which followed the shift in the sleep-wake cycle. As will be reviewed in subsequent sections, numerous studies have indicated that control of diurnal rhythms of corticotropic activity is primarily dependent on circadian timing, whereas sleep-wake homeostasis appears to be an important factor in control of the 24-hour profiles of GH and prolactin.111 Nevertheless, small modulatory effects of sleep-wake homeostasis on cortisol secretion and, conversely, influences of circadian timing on somatotropic function have been clearly demonstrated.112 The diurnal variation in TSH levels includes an evening elevation that is thought to be under circadian control and nocturnal inhibition by sleep-dependent processes that is clearly demonstrated during sleep deprivation, when a large increase in nocturnal TSH levels is apparent, as is shown in the lower panel of Fig. 4-3.111
FIGURE 4-3 Mean (+standard error of the mean [SEM]) 24-hour profiles of plasma cortisol, growth hormone (GH), prolactin (PRL), and thyrotropin (TSH) in a group of eight normal young men (20 to 27 years old) studied during a 53-hour period that include 8 hours of nocturnal sleep, 28 hours of sleep deprivation, and 8 hours of daytime sleep. The black bars represent the sleep periods. The open bars represent the period of nocturnal sleep deprivation. The dashed bars represent the period of daytime sleep. Data were sampled at 20-minute intervals. (From Van Cauter E, Spiegel K: Circadian and sleep control of endocrine secretions. In Turek FW, Zee PC [eds]: Neurobiology of Sleep and Circadian Rhythms, vol 133. New York, Marcel Dekker, 1999, pp 397–426.)
Ultradian Rhythmicity
The term ultradian is used primarily to designate rhythms with periods ranging from fractions of hours to several hours. Ultradian oscillations often are less regular and less reproducible than circadian rhythms. In most cases, they appear to represent an optimal functional status within the system where they occur rather than serving the primary function of a clock, that is, an accurate time measuring device. A wide variety of ultradian rhythms have been noted. The most prominent include pulsatile hormonal release and the alternation of REM and non-REM stages in sleep. In the human, the approximately 90-minute REM–non-REM cycle is accompanied by similar periodicity of dreaming, penile erections, sympathovagal balance, and breathing. It has been suggested that this ultradian rhythm during sleep may be a reflection of a basic rest-activity cycle (BRAC), which would occur during wakefulness as well.113 This concept has received some experimental support from a study demonstrating the existence of an ultradian rhythm of brain electrical activity in the frequency range of 13 to 35 Hz, an index of central alertness, during waking.114 It is interesting to note that pulses of cortisol release were significantly associated with ultradian oscillations in alertness.114
Oscillations at frequencies higher than the hourly (i.e., circhoral) range characterizing pulsatile release have been observed for a variety of hormones and metabolic variables. In particular, rapid oscillations of insulin secretion with periods in the 10- to 15-minute range have been well characterized in humans.115,116 Oscillations with a similar period also characterize lipolysis from omental fat, resulting in pulsatile free fatty acid release that appears driven by bursts of sympathetic drive to the adipocytes.117
Properties and Clinical Implications of Pulsatile Hormonal Release
In the endocrine system, ultradian variations have been observed for anterior and posterior pituitary hormones, for hormones under direct pituitary control, and for other endocrine variables such as parathyroid hormone, norepinephrine, plasma renin activity, and leptin and insulin secretion. The interval of recurrence of pulses varies from hormone to hormone and from species to species. The relative importance of pulsatile or oscillatory secretory activity versus tonic release also varies from one axis to another. For some hormones, secretory activity appears to be entirely pulsatile, with no detectable secretion between pulses. In normal men, evidence suggestive of intermittent secretion without tonic release has been obtained for luteinizing hormone (LH), follicle-stimulating hormone (FSH), GH, and adrenocorticotropic hormone (ACTH).118–120 For some hormones, pulsatile release is superimposed on a tonic level of secretion, or secretion occurs continuously but is increased and decreased in an oscillatory fashion. Pancreatic insulin secretion is a well-established example of this type of ultradian oscillation.121,122 Evidence for the existence of tonic secretion also has been obtained for pituitary prolactin and TSH release.118,119
The physiologic significance of pulsatile hormone secretion was first proved when the essential role of the episodic nature of gonadotropin-releasing hormone (GnRH) release for normal functioning of the pituitary-ovarian axis was demonstrated.123 Landmark studies showed that continuous infusions of exogenous GnRH in Rhesus monkeys with lesions of the arcuate nucleus, which abolished endogenous GnRH production, inhibited the secretion of LH and FSH. In contrast, the pulsatile administration of the synthetic hypothalamic hormone at a rate of one 6-minute pulse per hour restored normal LH and FSH levels.123 Furthermore, if the rate of pulse delivery was increased to three pulses per hour, or if it was decreased to one pulse every 2 hours, serum LH and FSH levels were partially inhibited. These findings were rapidly applied to treatment for a variety of disorders of the pituitary-gonadal axis124 and led the way to the discovery of the functional significance of pulsatility in other endocrine systems. For example, it was found that oscillatory administration of insulin with a period matching that of the normal pulsatility of endogenous insulin secretion is more effective in lowering glucose levels than is constant infusion.125
Impact of Aging On Mechanisms Controlling Endocrine Rhythms
Age-related changes in endocrine, metabolic, and behavioral circadian rhythms have been reported in a variety of species, including humans.126–128 One of the most prominent changes is a reduction in rhythm amplitude. The overall findings of a study that examined age-related differences in 24-hour endocrine rhythms in healthy subjects are shown in Fig. 4-4.126 A marked decrease in nocturnal release of TSH, melatonin, prolactin, GH, and melatonin was observed among older volunteers. The amplitude of the cortisol rhythm was decreased among elderly men, primarily because of an elevation of the nocturnal nadir. A retrospective analysis of polygraphic sleep recordings and concomitant profiles of plasma GH and cortisol from 149 normal healthy men, ages 16 to 83 years, showed a different rate of aging of SW sleep and REM sleep129 (Fig. 4-5). SW sleep decreased markedly from early adulthood to midlife and was replaced by lighter sleep (stages I and II) without significant increases in sleep fragmentation or decreases in REM sleep. The transition from midlife to late life involved an increase in wake at the expense of both non-REM and REM sleep. The chronology of aging of GH secretion paralleled that of SW sleep. In contrast, the elevation in evening cortisol levels became significant only after 50 years, when sleep became more fragmented and REM sleep declined. These results were confirmed by a meta-analysis of 65 studies, which found that most of the impact of age on sleep occurs before midlife.130 Despite the fact that older women have more subjective sleep complaints than men,131–133 it was generally assumed that SWS and SWA are less affected by aging in women than in men.134 However, when SWA in non-REM sleep is expressed relative to SWA in REM sleep (i.e., accounting for background SWA), SWA is actually lower in older women than in older men, consistent with the higher frequency of subjective complaints.135
FIGURE 4-4 Mean (+standard error of the mean [SEM]) 24-hour profiles of plasma cortisol, thyrotropin (TSH), melatonin, prolactin (PRL), and growth hormone (GH) levels in eight old (67 to 84 years) and eight young (20 to 27 years) subjects. Data were sampled at 15-minute intervals. The black bars represent the mean sleep period. (From van Coevorden A, Mockel J, Laurent E, et al: Neuroendocrine rhythms and sleep in aging men. Am J Physiol 260:E651–E661, 1991.)
FIGURE 4-5 Left, Slow-wave (SW) sleep and growth hormone (GH) secretion during sleep as a function of age. Note the temporal concomitance between the decrease in SW sleep and in GH secretion. Right, Rapid eye movement (REM) sleep and level of evening nadir of plasma cortisol as a function of age. Note the temporal concomitance between the decrease in REM sleep and the increase in evening cortisol levels. Values shown are means (+standard error of the mean [SEM]) for each age group. Data were obtained in 149 healthy men, ages 16 to 83 years. (Data from Van Cauter E, Leproult R, Plat L: Age-related changes in slow-wave sleep and relationship with growth hormone and cortisol levels in healthy men. JAMA 284:861–868, 2000.)
Deficits in the maintenance and quality of nocturnal sleep in older adults are paralleled by decreased alertness, attention and memory deficits, and decreased performance during the daytime.136 In both rodents and humans, many circadian rhythms are advanced under entrained conditions such that specific phase points of these rhythms occur earlier than in young subjects.126,137 Both amplitude reduction and phase advance of the rhythm of body temperature have been observed in elderly subjects, and these alterations in circadian regulation were closely associated with changes in sleep-wake habits (i.e., earlier bedtimes and wake times).127,138
Age-related changes in the amplitude and/or the phase of circadian rhythms could be due to changes in the inner workings of the master clock, to alterations in the input pathways to the clock, or to factors downstream between the circadian clock and the system that is expressing the rhythm. Some early studies139,140 have reported that the free-running period of various rhythms in rodents is systematically shortened with age, suggesting that the circadian clock itself is altered in advanced age. This concept has been confirmed in rats via measurement of Per1-luc expression in transgenic animals.141 Age-related phase advances of a number of different behavioral and endocrine rhythms are consistent with the hypothesis that the period of the human circadian clock is shorter in the elderly. However, a study that measured the free-running period in healthy young and older adults using the forced desynchrony protocol has found no age difference.5 Nevertheless, the very healthy older individuals who participated in this demanding protocol exhibited marked decreases in sleep consolidation and had greater difficulties sleeping at adverse circadian phases than did young subjects. These alterations and the clear advance of the propensity to awaken from sleep are thought to be related to both a reduction in the homeostatic drive to sleep and a reduction in the strength of the circadian signal.142 In older adults, in contrast to young subjects, no significant correlation has been noted between the length of the circadian period and the phase angle of entrainment.143
Studies in rodents indicate that aging is associated with decreased responsiveness to the phase-shifting effects of both photic and nonphotic stimuli. Old hamsters show a decreased response to the phase-shifting effects of low-intensity light pulses.144 This observation raises the possibility that in old age, signal transmission of light information to the SCN itself may be decreased, or that SCN responsiveness to photic stimulation may be lessened. Similarly, although induction of locomotor activity during a time of normal inactivity can induce pronounced phase-shifts in the circadian rhythm of locomotor activity in young animals, in old animals the response is greatly diminished or is completely abolished.145,146 It is interesting to note that transplantation of fetal SCN tissue into the SCN region of old hamsters with an intact SCN can restore the response to the phase-shifting effects of triazolam on the activity rhythm.147
Behavioral changes in the elderly also may lead to changes in environmental inputs to the clock. In older adults, exposure to bright light and social cues, both potential entraining agents, is markedly diminished when compared with that seen in young adults.148–150 Furthermore, age-related lens pigmentation decreases the transmission of blue light (i.e., the wavelength to which the circadian system is most sensitive) to the retina.151 Absence of professional constraints, decreased mobility due to illness, and reduced socialization and outdoor activities are all hallmarks of old age. Thus, decreased exposure to environmental stimuli that entrain circadian rhythms could contribute to disruptions in circadian rhythmicity. The use of exposure to bright light and enriched social schedules to reinforce circadian rhythms in older adults and improve nighttime sleep and daytime alertness has proved beneficial in several studies.152 In elderly insomniacs living in a nursing home with limited exposure to environmental light, supplementary light exposure during the middle of the day significantly increased nocturnal melatonin secretion without circadian phase-shifting.153 Evidence indicates that older adults are capable of phase-shifting to the same extent as younger subjects in response to appropriately timed light exposure.154
The ultradian rhythmicity of pulsatile hormonal release is also affected by aging. For a number of individual hormones, including GH, insulin, LH, and ACTH, analyses of 24-hour profiles have shown increased irregularity, or disorderliness, of pulsatile release. Furthermore, the synchrony of pulsatile release between the pituitary hormone and the peripheral hormone (e.g., LH and testosterone, ACTH and cortisol) is partially disrupted in healthy older adults.155
Methodologic Aspects of the Study of Endocrine Rhythms
Procedures to Quantify Circadian Variations
Among the methods proposed for and applied to the analysis of 24-hour profiles of blood components, the oldest is the Cosinor test.156 The major disadvantage of this test and of its derivatives is its assumption that the observed profile may be described adequately by a single sinusoidal curve. This assumption is practically never met for biological rhythms that are asymmetric in nature (e.g., the sleep-wake cycle is a 08:16 alternation, not a 12:12). Therefore, the Cosinor test generally provides unreliable estimations of rhythm parameters. Unfortunately, in recent years, the Cosinor analysis has regained popularity and has been applied indiscriminately to asymmetric profiles.
Other procedures for the detection and estimation of circadian variation have been based on periodogram calculations or on nonlinear regression procedures.157,158 These methods provide an adequate description of asymmetric wave shapes. The times of occurrence of the maximum and the minimum of the best-fit curve often are referred to as the acrophase and the nadir, respectively. The amplitude of the rhythm may be estimated as 50% of the difference between the maximum and the minimum of the best-fit curve. With the periodogram procedure, confidence intervals for the amplitude, acrophase, and nadir may be calculated.
Procedures to Quantify Pulsatile Hormonal Secretion
Analysis of pulsatile variations may be considered at two levels.159 One may wish to define and characterize significant variations in peripheral levels, based on estimations of the size of the measurement error (i.e., primarily assay error). However, under certain circumstances, it is possible to mathematically derive secretory rates from the peripheral concentrations.160–162 This procedure, often referred to as deconvolution, often reveals more pulses of secretion than does analysis of peripheral concentrations. It also more accurately defines the temporal limits of each pulse. However, deconvolution involves amplification of measurement error, with increased risk for false-positive error. Whether peripheral concentrations or secretory rates are examined, two major approaches are used to analyze the episodic fluctuations. The first and most commonly used is the time domain analysis, wherein the data are plotted against time, and pulses are detected and identified. The second is the frequency domain analysis, in which amplitude is plotted against frequency or period. The time domain analysis provides an estimation of pulse frequency, calculated as the total number of pulses detected divided by the duration of the study period. The regularity of pulsatile behavior may be quantified by examining the distribution of interpulse intervals. Alternatively, the issue of regularity of pulsatile behavior may be approached by examining the distribution of spectral power in a frequency domain analysis.163 Finally, another analytic tool, the approximate entropy (ApEn), has been introduced to quantify the regularity of oscillatory behavior in endocrine and other physiologic time series.164,165
A number of computer algorithms for identification of pulses of hormonal concentration have been proposed. A detailed presentation of the operating principles of each of these procedures is beyond the scope of this chapter. Review articles166,167 have provided comparisons of performance of several pulse detection algorithms. These comparisons have indicated that ULTRA and CLUSTER perform similarly when used with appropriate choices of parameters.167
Endocrine Rhythms in Health and Disease
Pituitary Axes
Normal Rhythms of Adrenocorticotropic Hormone and Adrenal Secretions: Outputs from the SCN activate rhythmic release of corticotropin-releasing hormone (CRH) that stimulates circadian ACTH release. The 24-hour rhythm of adrenal secretion is primarily dependent on the diurnal pattern of ACTH release. In addition, neuronal signals generated by the SCN are transmitted by a multisynaptic neural pathway to the adrenal cortex.168 The presence in the adrenal cortex of an intrinsic circadian oscillator consisting of interacting positive and negative feedback loops in circadian gene expression has been demonstrated in various animals, including monkeys,169–171 and it has been shown that this adrenal circadian pacemaker gates the physiologic adrenal response to ACTH (i.e., defines a time window during which the adrenal most effectively responds to ACTH).172
The 24-hour profiles of ACTH and cortisol show an early morning maximum, declining levels throughout daytime, a quiescent period of minimal secretory activity centered around midnight, and an abrupt elevation during late sleep, resulting in an early morning maximum. Mathematical derivations of secretory rates from plasma concentrations have suggested that the 24-hour profile of plasma cortisol reflects a succession of secretory pulses of magnitude modulated by a circadian rhythm with no evidence of tonic secretion.120,173 In normal conditions, the acrophase of the pituitary-adrenal periodicity occurs between 06.00 and 10.00 hours. With a 15-minute sampling interval, 12 to 18 significant pulses of plasma ACTH and cortisol can be detected per 24-hour span.174 Circadian and pulsatile variations parallel to those of cortisol have been demonstrated for the plasma levels of several other adrenal steroids, in particular dehydroepiandrosterone (DHEA).175 The temporal concomitance of 24-hour profiles of ACTH, cortisol, and DHEA is illustrated in Fig. 4-6.
FIGURE 4-6 Twenty-four-hour profiles of plasma adrenocorticotropic hormone (ACTH), cortisol, and dehydroepiandrosterone (DHEA) levels sampled at 20-minute intervals in a healthy young man. Note the temporal concomitance of circadian and pulsatile variations of the three hormones. (Unpublished data kindly provided by Dr. K. Spiegel.)
The profile shown in the upper panel of Fig. 4-3 illustrates the remarkable persistence of the cortisol and, by inference, ACTH secretory rhythm when sleep is manipulated. Indeed, the overall wave shape of the profile was not markedly affected by the absence of sleep or the presence of sleep at an abnormal time of day. Thus, this rhythm is controlled primarily by the circadian pacemaker.
However, modulatory effects of sleep-wake homeostasis have been clearly demonstrated. As illustrated in Fig. 4-7, sleep onset is consistently associated with short-term inhibition of cortisol secretion, which may not be detectable when sleep is initiated in the morning (i.e., at the peak of corticotropic activity).176–179 This inhibitory effect of sleep appears to be related to SW sleep.109,180 Conversely, final awakenings from sleep, as well as transient awakenings that interrupt the sleep period, consistently trigger pulses of cortisol secretion (see Fig. 4-7),120,178,180–183 and the number of nocturnal microarousals predicts morning plasma and saliva cortisol levels.184 In an analysis of cortisol profiles during nocturnal sleep, it was observed that all transient awakenings that interrupt sleep and last at least 10 minutes were followed within the next 20 minutes by significant bursts of cortisol secretion.183 In addition, a temporal coupling between pulses of cortisol secretion and ultradian variations in an EEG marker of alertness has been reported.114 Total nocturnal wake time is associated with increased 24-hour plasma cortisol concentrations,185 and chronic insomnia with reduced total sleep time is associated with higher cortisol levels across the night.186
FIGURE 4-7 Mean (and standard error of the mean [SEM]) changes in plasma cortisol levels: A, Within 120 minutes following sleep onset at 15.00 hours (n = 32). B, Within 20 minutes following final spontaneous awakening in darkness (scheduled sleep period 23.00 to 07.00 hours or 15.00 to 23.00 hours; n = 10). C, Within 20 minutes following transition from darkness to dim light at 07.00 or 23.00 hours in subjects awake at bed rest (n = 10). D, Within 20 minutes following final awakening concomitant with transition from darkness to dim light at 07.00 or 23.00 hours (n = 38). E, Within 15 minutes following transition from dim to bright light at 05.00 hours in subjects awake at bed rest (n = 8). (Panels A to D, Data from Caufriez A, Moreno-Reyes R, Leproult R, et al: Immediate effects of an 8-h advance shift of the rest-activity cycle on 24-h profiles of cortisol. Am J Physiol 282:E1147–E1153, 2002; panel E, Data from Leproult R, Colecchia EF, L’Hermite-Balériaux M, et al: Transition from dim to bright light in the morning induces an immediate elevation of cortisol levels. J Clin Endocrinol Metab 86:151–157, 2001.)
Modulatory effects of dark-light transitions also have been noted. Cortisol secretory pulses associated with morning awakening are enhanced by increasing light intensity.187 Moreover, the transition from darkness to dim light and from dim to bright light may stimulate cortisol secretion in subjects who are awake at bed rest.183,188 The stimulatory effects of bright light exposure on nocturnal cortisol levels appear to be dependent on the timing of exposure.189 When dark-light and sleep-wake transitions occur concomitantly, associated cortisol elevations are nearly two times as high as when the final awakening occurs in continuous darkness183 (see Fig. 4-7). Thus, under usual bedtime schedules, both sleep-wake and dark-light transitions amplify the effects of circadian rhythmicity.
Studies of the 24-hour cortisol profile in the course of adaptation to shifts of the sleep-wake cycle have demonstrated that the end of the quiescent period, which coincides with the onset of the early morning rise, takes longer to adjust and appears to be a robust marker of circadian timing. Twin studies have demonstrated that the timing of the nadir is influenced by genetic factors,190 providing evidence for genetic control of the human circadian phase. In contrast, the timing of the morning acrophase is more labile and may be influenced by the timing of sleep offset,182 the transition from dark to bright light,187 and breakfast intake.191 Finally, anticipation of the expected time of waking has been reported to be associated with a rise in levels of ACTH, but not cortisol, during the end of the sleep period.192
In addition to the immediate modulatory effects of sleep-wake transitions on ACTH and cortisol levels, acute total or partial (4 hours in bed) nocturnal sleep deprivation results in elevated cortisol concentrations in late afternoon and evening on the following day,193 and the amplitude of the cortisol circadian variations is reduced by approximately 15%. Similarly, as illustrated in Fig. 4-8, recurrent partial sleep deprivation (4 hours in bed per night for 6 nights) also results in an elevation of cortisol levels in the late afternoon and evening.194 These disturbances, which were observed in young healthy subjects, are strikingly similar to those noted in older healthy subjects with normal sleep schedules.126,195,196 In any case, sleep loss appears to delay the return to quiescence of the hypothalamic-pituitary-adrenal axis (HPA) that normally occurs in the evening. This suggests that sleep loss, similar to aging, may slow down the rate of recovery of the HPA axis response following a challenge and therefore could facilitate the development of central and peripheral disturbances associated with glucocorticoid excess, in particular when cortisol concentrations are elevated at the time of the normal daily nadir, such as memory deficits, insulin resistance, and osteoporosis.197–201 Conversely, decreased HPA resiliency results in HPA hyperactivity that inhibits SW sleep and promotes nocturnal awakenings, initiating a feed-forward cascade of negative events generated by both HPA and sleep disruptions.
FIGURE 4-8 Impact of recurrent sleep curtailment (4 hours in bed for 6 nights) and of sleep recovery (12 hours in bed for 6 nights) on 24-hour plasma profiles of thyrotropin (TSH), leptin, and cortisol, and on homeostatic model assessment (HOMA) in healthy young men. HOMA was calculated as glucose concentration (mg/dL) × insulin concentration (µU/mL). Values shown are means ± SEM. (Adapted from Spiegel K, Leproult R, Van Cauter E: Impact of sleep debt on metabolic and endocrine function. Lancet 354:1435–1439, 1999, and from Spiegel K, Leproult R, L’Hermite-Balériaux M, et al: Leptin levels are dependent on sleep duration: relationships with sympathovagal balance, carbohydrate regulation, cortisol and thyrotropin. J Clin Endocrinol Metab 89:5762–5771, 2004.)
The circadian rhythm of cortisol persists throughout adulthood and has been observed through the ninth decade.126,196 Among young adults, 24-hour cortisol levels are slightly lower in women than in men, primarily because of lower morning maxima. With aging, evening cortisol levels increase progressively in both men and women, so that the cortisol nadir is markedly higher in healthy subjects over 70 years of age than in young adults (see Fig. 4-4). It is interesting to note that this elevation of evening cortisol levels occurs with a chronology similar to that observed for a progressive decrease in the duration of REM sleep129 (see Fig. 4-5). As a result, older subjects have elevated 24-hour mean cortisol levels and reduced amplitude of cortisol variations. In addition, the timing of the nadir is advanced by 1 to 2 hours, indicating that aging is associated with an advance in the circadian phase.126,196
In pregnancy, placental CRH is secreted into the maternal circulation in a pulsatile but not in a circadian fashion, and no correlation has been noted between maternal levels of CRH and ACTH. However, ACTH and cortisol concentrations remain strongly correlated with each other over time, suggesting that diurnal variation in maternal ACTH probably is driven by another ACTH secretagogue.202
Alterations in Disease States: The 24-hour profile of pituitary-adrenal secretion remains largely unaltered in a wide variety of pathologic states. Disease states in which pronounced alterations of the cortisol rhythm have been observed include primarily (1) disorders involving abnormalities in binding and/or metabolism of cortisol; (2) the various forms of Cushing’s syndrome; and (3) depression and posttraumatic stress disorder (PTSD).
The relative amplitude of the circadian rhythm and of the episodic fluctuations in cortisol is blunted in patients with liver disease203 and in those with anorexia nervosa,204 primarily because of the decreased metabolic clearance of cortisol. Pulsatile secretion of ACTH, but not cortisol, was reportedly enhanced in obese premenopausal women.205 In hypothyroid patients, the mean level is markedly elevated and the relative amplitude of the rhythm is dampened.206 These alterations are thought to be due to both diminished clearance and decreased efficiency of feedback control. In contrast, in hyperthyroidism, where cortisol production and peripheral metabolism are increased, episodic pulses are enhanced.207
A low-amplitude circadian variation may persist in pituitary-dependent Cushing’s disease. Cortisol pulsatility is blunted in about 70% of patients with Cushing’s disease, suggesting autonomous tonic secretion of ACTH by a pituitary tumor. However, in about 30% of these patients, the magnitude of the pulses is enhanced instead.208 These hyperpulsatile patterns could be caused by enhanced hypothalamic release of CRH or persistent pituitary responsiveness to CRH. It also has been shown that patients with Cushing’s disease secrete ACTH and cortisol jointly more asynchronously than healthy subjects.209 The left and middle panels of Fig. 4-9 compare representative and mean 24-hour cortisol profiles in normal subjects and in patients with Cushing’s disease.
FIGURE 4-9 Twenty-four-hour profiles of plasma cortisol in normal subjects, patients with pituitary Cushing’s disease, and patients with major endogenous depression of the unipolar subtype. For each condition, a representative example is shown in the top panel, and mean (+standard error of the mean [SEM]) profiles from 8 to 10 subjects are shown in the lower panel. (From Van Cauter E: Physiology and pathology of circadian rhythms. In Edwards CW, Lincoln DW [eds]: Recent Advances in Endocrinology and Metabolism. Edinburgh, Churchill Livingstone, 1989, vol 3, pp 109–134.)
In patients with primary adrenal Cushing’s syndrome, increased cortisol secretion appears to result from both increased basal secretion and increased pulse frequency.210 Although the persistence of a low-amplitude circadian cortisol variation has been claimed,210 examination of the individual profiles does not support this notion. Nevertheless, the partial persistence of cortisol rhythmicity in the absence of ACTH could reflect the newly described neural pathway between the SCN and the adrenal cortex.168
Hypercortisolism with persistent circadian rhythmicity and increased pulsatility is found in a majority of severely depressed patients.211–213 This is illustrated in the right panels of Fig. 4-9. In these patients, who do not develop the clinical signs of Cushing’s syndrome despite high circulating cortisol levels, the quiescent period of cortisol secretion is shorter and more fragmented, and it often starts later and ends earlier than in normal subjects of comparable age. These alterations could reflect the impact of sleep disturbances, as well as an advance in circadian phase. When clinical remission of the depressed state is obtained, hypercortisolism and alterations in the quiescent period disappear, indicating that these disturbances are “state” rather than “trait” dependent.214
In PTSD, some authors have reported that plasma cortisol levels are decreased in the afternoon and/or in the evening, and that the amplitude of circadian variations is enhanced.215–217 In fibromyalgia and in the chronic fatigue syndrome, cortisol levels have been reported to be low, normal, or elevated, depending on the study, but ACTH and cortisol pulsatility were found to be normal.218 In a well-documented study performed in constant routine conditions, normal circadian variations of plasma cortisol levels were evidenced in women with fibromyalgia.219
The Somatotropic Axis
The 24 Hour Profile of Growth Hormone in Normal Subjects: Pituitary secretion of GH is stimulated by hypothalamic GH-releasing hormone (GHRH) and is inhibited by somatostatin. In addition, the acylated form of ghrelin (acyl-ghrelin), a peptide produced predominantly by the stomach, binds to the GH-secretagogue receptor and therefore is another potent endogenous stimulus of GH secretion.220,221 In normal adult subjects, the 24-hour profile of plasma GH levels consists of stable low levels abruptly interrupted by bursts of secretion. The most reproducible pulse occurs shortly after sleep onset, in association with the first phase of SW sleep.222
[/not-level-membership-for-endocrinology-diabetes-and-metabolism-category]
