Chapter 97 Endocrine Disorders of the Hypothalamus and Pituitary in Childhood and Adolescence
Anatomic and Physiologic Aspects
The hypothalamus, the ventral part of the diencephalon, is an evolutionarily conserved region of the mammalian brain. The hypothalamus is separated by the third ventricle and connected to the pituitary by the hypophysial stalk. The hypothalamus functions as the primary control center for a variety of physiologic processes, integrating neural and hormonal signaling. Hypothalamic nuclei are not well-demarcated regions; however, these cell groups in the walls of the third ventricle possess specific physiologic functions [Pansky et al., 1988]. The supraoptic and paraventricular nuclei produce arginine vasopressin (AVP, also known as antidiuretic hormone [ADH]) and oxytocin. Paraventricular nuclei and arcuate nuclei release thyrotropin-releasing hormone (TRH), corticotropin-releasing hormone (CRH), somatostatin (SST), growth hormone-releasing hormone (GHRH), gonadotropin-releasing hormone (GnRH), and dopamine into the hypophysial portal circulation to regulate the synthesis and release of anterior pituitary hormones. In addition, hypothalamic neurons that regulate appetite and energy balance are also located in the paraventricular and arcuate nuclei of the hypothalamus, such as pro-opiomelanocortin (POMC) and neuropeptide Y (NPY) and agouti-related protein (AgRP)-expressing neurons [Cone et al., 2003].
The pituitary (hypophysis) is housed in the sella turcica, and, as noted above, is attached to the hypothalamus by the hypophysial stalk. The stalk, also called the infundibulum, is a collective term for the median eminence (the most inferior extension of the tuber cinereum) and the infundibular stalk (a hollow process extending from the tuber cinereum to the posterior hypophysis). The stalk serves as an anatomic and functional link between the hypothalamus and the pituitary. The optic chiasm is situated directly anterior to the pituitary stalk (Figure 97-1). The pituitary itself is small, weighing an average of about half a gram, and is divided into the anterior lobe (adenohypophysis), the posterior lobe (neurohypophysis), and a vestigial intermediate lobe. Six hormones, i.e., growth hormone (GH), thyroid stimulating hormone (TSH), adrenocorticotropic hormone (ACTH), follicle stimulating hormone (FSH), luteinizing hormone (LH), and prolactin (PRL), are synthesized and stored in the anterior lobe from well-differentiated distinct cell types (somatotrophs, thyrotrophs, corticotrophs, gonadotrophs, and lactotrophs). However, some cell types, such as mammosomatotrophs, can express multiple hormones (PRL and GH). The neurohypophysis synthesizes, stores, and secretes AVP and oxytocin [Cone et al., 2003].
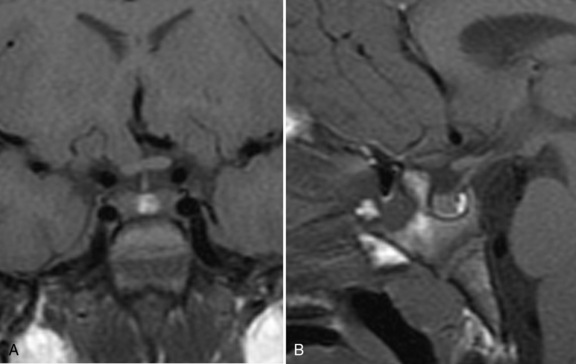
Fig. 97-1 T1-weighted magnetic resonance images of the normal hypothalamus/pituitary in an 8-year-old girl.
The blood supply in the hypothalamic-pituitary-portal system allows bidirectional hypothalamic-pituitary hormonal interaction. The superior hypophysial arteries from the internal carotid arteries form a primary plexus in the median eminence. These vessels travel down to the anterior pituitary as the major blood supply. The hypothalamic-pituitary-portal circulation carries the hypothalamic releasing and inhibiting hormones to the adenohypophysis. Retrograde blood flow within the internal capillary plexus (gomitoli), derived from the stalk branches of the superior hypophysial arteries, provides local hormonal feedback to the hypothalamus. The blood supply for the posterior pituitary gland is derived from the inferior hypophysial arteries, which are, in turn, derived from the internal carotid arteries [Stanfield, 1960; Bergland and Page, 1979].
Organogenesis of the hypothalamus and the pituitary is a complex process. In the early embryo, prosencephalon (neuroectoderm of the forebrain) forms telencephalon (endbrain, cortex) and diencephalon. The alar plates from the myelencephalon form the lateral walls of the diencephalon. The hypothalamic sulcus divides the alar plates into dorsal and ventral regions. The hypothalamus, as part of the lower and ventral portion of the alar plates, differentiates into hypothalamic nuclei that serve as control centers for life-sustaining physiologic processes [Sadler, 1990; Hill, 2009]. The pituitary, however, develops from two distinct anlagen. The adenohypophysis derives from the oral ectoderm (Rathke’s pouch), and the neurohypophysis is a downward extension of the diencephalon [Sadler, 1990]. Development of the neuroendocrine system is detectable by the third embryonic week. Rathke’s pouch forms by the fourth embryonic week, and by week 16, the adenohypophysis is fully differentiated.
Normal development of the hypothalamus and the pituitary relies on sequential expression of multiple homeodomain transcriptions factors, morphogenic proteins, and growth factors. Bone morphogenic protein (BMP)-4 and fibroblast growth factor (FGF)-8 are critical for anterior pituitary gland development. In addition, differentiation of anterior pituitary cell types requires a spatiotemporally regulated cascade of homeodomain transcription factors. Several pituitary-specific transcription factors, such as Lhx3/Lhx4 (LIM-homeobox-3 and 4), Rpx (Rathke’s pouch homeobox, also known as Hesx1), Pitx (pituitary homeobox), Prop-1, Pit-1, POU1F1, Sox2, and Sox3, are important determinants of pituitary cell lineages. Mutations in these transcription factors can lead to either isolated or combined pituitary hormone deficiencies, with or without detectable anatomic abnormalities [Dattani et al., 1998, 2000; Cohen and Radovick, 2002; Rizzoti et al., 2004; Kelberman and Dattani, 2009].
Hypothalamic/Pituitary Disorders of Pubertal Development
Normal Physiology of Puberty and Adrenarche
Puberty is the transitional period between the juvenile state and adulthood, characterized by attainment of secondary sex characteristics and reproductive capability. The control center of puberty is comprised of hypothalamic GnRH neurosecretory neurons (pulse generator) located in the medial basal hypothalamus [King et al., 1985]. Kisspeptin (product of the KISS 1 gene) and the kisspeptin receptor (KISS1R), also known as GPR54 (a member of the rhodopsin family of G protein coupled receptors), have emerged as key regulators of the hypothalamic-pituitary-gonadal (HPG) axis [de Roux et al., 2003; Seminara et al., 2003]. Kisspeptin neurons found in the hypothalamus directly innervate and stimulate GnRH neurons. Kisspeptin neurons also express the estrogen receptor and the androgen receptor, which enable negative feedback action of sex steroids. Kisspeptin signaling in the brain is critical for triggering and guiding the tempo of sexual maturation at puberty [Oakley et al., 2009]. In addition, the tachykinin neurokinin B (encoded by TAC3) and its receptor NK3R (encoded by TACR3) also serve as central regulators for normal gonadatropin secretion and pubertal development [Topaloglu et al., 2009].
Puberty is not a sudden event, but rather reflects a milestone on a continuum that begins before birth. The GnRH pulse generator becomes pulsatile by midgestation, and remains active in early infancy until about 6 months of age in boys and 12–24 months in girls [Grumbach, 2002]. Between late infancy and the onset of puberty, the GnRH pulse generator becomes relatively quiescent as a consequence of an as yet poorly defined central nervous system (CNS) inhibitory mechanism. After a quiescent period of approximately 10 years, the GnRH pulse generator is disinhibited, leading to increased amplitude and frequency of LH and FSH secretion, and resulting in increased sex steroid production (principally, estradiol and testosterone) and attainment of physical puberty [Grumbach, 2002].
Adrenarche refers to the prepubertal rise of adrenal androgen precursors – in particular, dehydroepiandrosterone (DHEA) and its sulfated form (DHEAS) – as a consequence of maturation of the zona reticularis of the adrenal cortex. DHEA can be converted peripherally to testosterone; thus, adrenarche is typified clinically by mild androgenic effects, such as a change in body odor and the appearance of axillary and pubic hair and acne. The mechanisms that regulate adrenarche are not well understood. Adrenache is independent of the maturation of the hypothalamic-pituitary-gonadal axis, preceding the onset of puberty by about 2 years [Miller, 1999].
The normal age of onset of secondary sexual characteristics (defined as 2.5 SD on either side of the mean, or where approximately 99 percent of the population falls) is 6–13 years in girls and 9–14 years in boys. The appearance of secondary sexual characteristics, acceleration of growth, and the capacity of reproduction are hallmarks of puberty. The development of puberty is characterized by sequential events that are specific for each gender. In females, secondary sexual characteristics include breast development, the appearance of pubic and axillary hair, maturation of the labia, and estrogenization of the vaginal mucosa. The development of pubic and axillary hair is influenced by androgens produced in both the adrenal cortex (adrenarche) and the ovary. Menarche usually occurs 2–3 years after initiation of breast development. The pubertal growth spurt, which normally occurs in the early stages of puberty for girls, can result in a gain in height of 25 cm or more. In males, puberty begins with testicular enlargement (≥ 2.5 cm in the longest dimension) followed by the appearance of sexual hair and phallic enlargement. The growth spurt occurs during midpuberty, which results in an average 28 cm gain in height [Grumbach and Styne, 2003]. Abnormalities of puberty include sexual precocity and delayed or arrested puberty, each with a broad differential diagnosis. While pubertal disorders can arise from defects at the level of the hypothalamus, pituitary, or gonads, the focus of this discussion is on hypothalamic and pituitary causes of sexual precocity and delayed puberty.
Sexual Precocity
Sexual precocity is usually defined as the development of secondary sexual characteristics before 6–7 years of age in girls [Herman-Giddens et al., 1997], and before 9 years of age in boys. In all cases thus far described, sexual precocity results from an increase in circulating sex steroids. An endocrine approach to the differential diagnosis of sexual precocity would include consideration of exogenous and endogenous sources of these steroids. Endogenous sources include the gonads and adrenal cortex, either of which may secrete sex steroids inappropriately as the result of a primary process intrinsic to these tissues, or secondary to a circulating stimulatory factor. As previously noted, this chapter will focus only on hypothalamic/pituitary causes of sexual precocity.
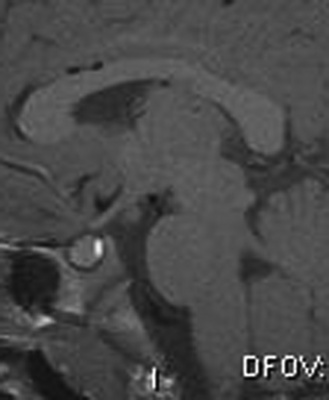
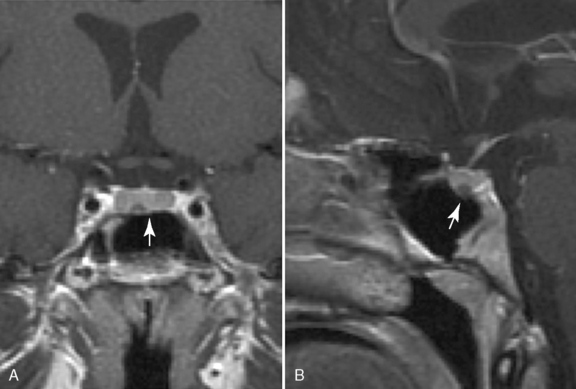
Precocious puberty (sometimes referred to as “true” or “central” precocious puberty) is defined as early puberty specifically resulting from premature reactivation of the GnRH pulse generator. Precocious puberty most commonly is idiopathic, but can result from a broad range of abnormalities, including CNS tumors, hamartoma of the tuber cinereum, congenital malformations, subarachnoid cysts, CNS infection, irradiation, and trauma. The incidence of precocious puberty is significantly higher in females. In studies of girls with precocious puberty, idiopathic precocious puberty ranges from 63 to 74 percent [Pescovitz et al., 1986; Cisternino et al., 2000]. In contrast, only 6 percent of boys with precocious puberty are idiopathic [Pescovitz et al., 1986]. Idiopathic precocious puberty is a diagnosis of exclusion and it is, thus, essential to search for underlying neurological causes. Tumors involving in the posterior hypothalamus, such as glioma, germinoma, and teratoma, can cause precocious puberty. Most of these tumors are thought to trigger early puberty by interfering with mechanisms that normally inhibit the GnRH pulse generator. Few LH/FSH-secreting adenomas have been reported [Demura et al., 1977]. Hamartoma of the tuber cinereum, a congenital malformation, can cause central precocious puberty. Hamartomas are small lesions (4–25 mm), may be sessile or pedunculated, and usually do not enlarge with time (Figure 97-2). Histologically, they appear to be composed of normal brain tissue and contain GnRH secretory neurons, which may serve as an “ectopic pulse generator” [Mahachoklertwattana et al., 1993]. In some hypothalamic harmatomas, the production of transforming growth factor (TGF)-α can initiate early puberty by activating the normal GnRH pulse generator [Jung et al., 1999]. Hamartomas of the tuber cinereum are often associated with gelastic (laughing) seizures. Recently, an autosomal-dominant activating mutation in GPR54 was found in a girl with central precocious puberty [Teles et al., 2008].
Ectopic human chorionic gonadotropin (hCG)-secreting tumors in the CNS (and elsewhere), e.g., hypothalamic germinomas, can cause sexual precocity in boys [Sklar et al., 1981]. Such patients do not have central precocious puberty, in that they do not have premature reactivation of the hypothalamic GnRH pulse generator. Rather, the ectopic hCG interacts with the LH/hCG receptor on testicular Leydig cells, resulting in increased testosterone secretion and virilization. In girls, ovarian estrogen secretion requires both LH and FSH. Thus, ectopic secretion of hCG alone causes sexual precocity only in boys [Grumbach and Styne, 2003]. The accurate diagnosis of precocious puberty and its cause requires a detailed history, physical examination, hormonal testing, and imaging studies of the CNS. Idiopathic precocious puberty is often familial, exhibiting autosomal-dominant inheritance [de Vries et al., 2004]. The occurrence of early breast and pubic hair development is often seen in girls with precocious puberty, while testicular enlargement and other signs of virilization are seen in boys with precocious puberty. Hormonal analysis demonstrates increased amplitude and frequency of LH and FSH pulsatile secretion, resulting from increased pulsatile secretion of GnRH. This finding can be demonstrated either by serial sampling of LH and FSH or by single LH/FSH measurements using highly sensitive immunochemiluminescent assays. Dynamic testing using GnRH or a GnRH agonist is also used routinely to diagnose precocious puberty. Magnetic resonance imaging (MRI), with particular attention to the hypothalamic-pituitary area, should be carried out in any child diagnosed with precocious puberty.
Management
If a CNS lesion that causes precocious puberty is identified, an appropriate treatment plan for that lesion should be developed. Previously, treatment of hamartomas of the tuber cinereum was principally surgical. However, such treatment carried a significant risk of morbidity and mortality [Valdueza et al., 1994; Rosenfeld et al., 2001]. A long-term follow-up study demonstrated an excellent response to GnRH agonist without surgical resection [Mahachoklertwattana et al., 1993]. Rarely, after treatment of CNS disorders causing precocious puberty, the rapid progression of pubertal development is reversed or arrested. More commonly, once puberty has been initiated, it will often continue, despite intervention to address a primary CNS disorder. Such patients require treatment to suppress the HPG axis. State-of-the-art treatment consists of administration of a GnRH agonist, which desensitizes pituitary GnRH receptors, leading to suppression of the HPG axis [Breyer et al., 1993]. This treatment is commonly given by a monthly depot intramuscular injection. Recent studies indicate that the gonadotropin-releasing hormone analog, histrelin (a subcutaneous implant), results in gonadotropin suppression for 12 months [Kaplowitz, 2009]. Long-term studies demonstrate resumption of normal puberty after discontinuation of a GnRH agonist [Feuillan et al., 2001].
Delayed or Arrested Puberty
Isolated Congenital Hypogonadotropic Hypogonadism
Isolated hypogonadotropic hypogonadism (IHH) may or may not be associated with olfactory abnormalities. IHH associated with anosmia or hyposmia is referred to as Kallmann’s syndrome, and is thought to be a consequence of defective embryonic migration of GnRH neurons from the olfactory placode to the hypothalamus. Kallmann’s syndrome is the most common form of IHH, occurring in 1 in 10,000 males and 1 in 50,000 females [Rugarli and Ballabio, 1993]. Considerable genetic heterogeneity exists for Kallmann’s syndrome. Classic Kallmann’s syndrome is transmitted in an X-linked or autosomal-dominant fashion with variable penetrance, and is characterized by hypogonadotropic hypogonadism and anosmia/hyposmia. Some patients may also have unilateral renal agenesis, synkinesia (mirror movements), and pes cavus. Mirror movements occur in 85 percent of patients with classic Kallmann’s syndrome, associated with bilateral hypertrophy of the corticospinal tract [Krams et al., 1999]. Mutations in the KAL-1 gene account for half of males with X-linked hypogonadotropic hypogonadism [Hardelin and Dode, 2008] and 5 percent of sporadic cases [Georgopoulos et al., 1997]. The KAL-1 gene resides in the pseudoautosomal region of the X chromosome and encodes the extracellular glycoprotein, anosmin-1. Lack of correlation between genotype and phenotype has been described in Kallmann’s syndrome. For instance, within the same family, one patient may have normal gonadal function and anosmia, while another may have hypogonadism and a normal sense of smell. However, isolated gonadotropin deficiency with normal sense of smell is rare in Kallmann’s syndrome. Autosomal forms of Kallman’s syndrome are associated with loss-of-function mutations of the fibroblast growth factor receptor 1 (FGFR1) (also known as KAL2), and with mutations in a variety of other genes, including FGF8 (ligand for FGFR1), prokineticin-2 (PROK2), and prokineticin receptor-2 (PROKR2) [Trarbach et al., 2006; Hardelin and Dode, 2008]. Another form of X-linked hypogonadotropic hypogonadism is associated with adrenal hypoplasia congenita due to defects of DAX1 (dose-sensitive sex reversal AHC (Adrenal Hypoplasia Congenita)-associated gene on the X chromosome). DAX1 encodes a transcription factor that appears to play key developmental roles in the hypothalamus, pituitary, gonad, and adrenal cortex. Boys with DAX1 mutations, who survive adrenal failure in infancy and early childhood, can present with hypogonadotropic hypogonadism. Rarely, a mild mutation may present in adulthood with mild adrenal insufficiency and incomplete pubertal development. Females homozygous for a DAX1 nonsense mutation can present with isolated hypogonadotropic hypogonadism [Kalantaridou and Chrousos, 2002].
IHH without olfactory abnormalities (normosmic IHH) is associated with mutations in a variety of genes, including the GnRH receptor, the GNRH1 gene, GPR54, TAC3, and TACR3 [Kalantaridou and Chrousos, 2002; Bouligand et al., 2009; de Roux et al., 2003; Seminara et al., 2003; Semple et al., 2005; Topaloglu et al., 2009]. In addition, mutations in the beta subunits of FSH and LH have been reported in association with primary amenorrhea and hypogonadism [Kalantaridou and Chrousos, 2002]. IHH also occurs in association with Prader–Willi syndrome and Laurence–Moon–Biedl syndrome [Hashimoto and Kumahara, 1979; Crino et al., 2003].
Hypogonadotropic Hypogonadism Associated with Multiple Hypothalamic/Pituitary Hormone Deficiencies
Hypogonadotropic hypogonadism can also present in combination with other hypothalamic/pituitary hormone deficiencies. Human mutations of pituitary transcription factors known to cause delayed puberty include Prop-1 and Lhx3. Prop-1 (prophet of Pit-1) is a paired-like homeodomain transcription factor expressed only in the anterior pituitary. Combined pituitary hormone deficiencies caused by mutations in Prop-1 occur with an incidence of about 1 in 8000 births. Patients with mutations in Prop-1 have combined pituitary hormone deficiencies, which may include deficiencies of GH, PRL, TSH, and gonadotropins. Such patients have hypogonadotropic hypogonadism in association with short stature and hypothyroidism [Wu et al., 1998]. Lhx3 is a LIM-type homeodomain protein. Mutations in Lhx3 have been associated with anterior pituitary hypoplasia and complete deficits of GH, PRL, TSH, and gonadotropins. Lhx3 mutations are also associated with decreased range of motion in the cervical spine [Netchine et al., 2000].
Other genetic factors that lead to delayed puberty include mutations of prohormone convertase (PC)1, leptin, and leptin receptor genes. A defect in PC1 has been shown to disrupt GnRH processing, and results in hypogonadotropic hypogonadism and obesity associated with impaired processing of insulin and POMC. Mutations in leptin and the leptin receptor are associated with a similar clinical picture of hypogonadotropic hypogonadism, hyperinsulinemia, and obesity [Kalantaridou and Chrousos, 2002; Beier and Dluhy, 2003]. Congenital midline defects of the CNS, such as septo-optic dysplasia (SOD), empty sella syndrome, and Rathke’s cyst, are often associated with hypothalamic/pituitary dysfunction, which may include hypogonadotropic hypogonadism. The genetics of SOD and its related pituitary hormone deficiencies will be reviewed in detail in the section on hypothalamic/pituitary disorders of statural growth.
Numerous CNS lesions can lead to hypogonadotropic hypogonadism, including CNS tumors (primarily, third ventricular), CNS infection, invasive diseases, cranial irradiation, and trauma. These CNS disorders often present with combined anterior and posterior pituitary hormone deficiencies. CNS tumors of the sella and parasellar region associated with multiple hypothalamic/pituitary hormone deficiencies include craniopharyngioma, pituitary adenomas, optic and hypothalamic gliomas, and germ-cell tumors. Delayed or arrested puberty is the second most common presenting symptom of CNS tumors following headache. Craniopharyngiomas, the most common tumor of the sella and parasellar region in children, are slow-growing, space-occupying tumors (Figure 97-3). Most patients present before their teenage years with headache, visual loss, and multiple hypothalamic/pituitary hormone deficits. Pituitary adenomas in children usually present as microadenomas with pituitary hormone hypersecretion. Nonsecreting adenomas in children are rare, and are usually macroadenomas. Prolactinoma is the most common pituitary adenoma in the pediatric population and frequently is associated with delayed or arrested puberty. Nonsecreting macroadenomas can also cause delayed puberty, at least in part, through elevation of prolactin as a consequence of stalk compression [Kunwar and Wilson, 2001]. Cranial irradiation to the third ventricular area also may be associated with hypogonadotropic hypogonadism.
Disorders of Prolactin Secretion
Clinical Features and Management of Hyperprolactinemia
The principal clinical feature of hyperprolactinemia, regardless of the cause, is galactorrhea, which may be unilateral or bilateral. Other clinical features may include delayed or arrested puberty, amenorrhea in females, and gynecomastia in males. Prolactinoma is the most common tumor of the pituitary, comprising about 50 percent of anterior pituitary adenomas. Most prolactinomas are less than 1 cm in diameter, i.e., microprolactinomas, and normally do not cause significant mass effects. However, with macroprolactinoma, headache and visual disturbance can be the first presenting symptoms. Rarely, patients with macroprolactinoma present with hydrocephalus, cranial nerve palsies, and seizures [Colao et al., 1998].
If a prolactinoma is diagnosed, therapeutic options include medical management with dopamine agonists, surgery, and adjunctive radiotherapy. While medical management is often considered the principal intervention, numerous reports indicate that microprolactinomas can be removed surgically, though with variable recurrence rates. Macroprolactinomas are less likely to be cured surgically and often require chronic dopamine agonist treatment. Dopamine agonists are effective and safe pharmacologic interventions for both micro- and macroprolactinoma [Pivonello et al., 2004; Verhelst et al., 1999]. Cabergoline is better tolerated and more effective than bromocriptine [Di Sarno et al., 2001]. Withdrawal of cabergoline after long-term therapy has been safe, although careful monitoring of tumor progression is necessary [Colao et al., 2003]. Nearly all prolactinomas have been reported to respond to cabergoline treatment, irrespective of tumor size and preceding treatment with higher doses (>3 mg/week) [Ono et al., 2008]. Adjunctive radiotherapy has been considered beneficial in some patients with macroprolactinoma. The skill and experience of the surgeon clearly play a role in determining the optimal treatment and outcome.
Hypothalamic/Pituitary Disorders of Glucocorticoid Production
Adrenocorticotropic Excess
Excessive production of ACTH from the pituitary arises either from CRH overproduction or from a primary ACTH-producing adenoma (Cushing’s disease). The first sign of Cushing’s disease in a growing child is often impaired linear growth [Mindermann and Wilson, 1995]. Other classic signs and symptoms of Cushing’s disease include excessive weight gain (central obesity), buffalo hump, plethora, “moon facies,” acne, striae, hypertension, hirsutism, fatigue, pubertal delay or arrest, bruising, and headache [Magiakou et al., 1994; Devoe et al., 1997; Magiakou and Chrousos, 2002]. Usually, at the time of diagnosis, ACTH-producing adenomas are significantly smaller than other pituitary adenomas (Figure 97-4). These tumors are usually not well demarcated, and are often less than 10 mm in diameter (microadenoma).
The diagnosis of Cushing’s disease can pose significant challenges. Endogenous hypercortisolism is either ACTH-dependent or ACTH-independent. Of the ACTH-dependent causes, a pituitary adenoma (Cushing’s disease) is most common. A less common ACTH-dependent cause is ectopic ACTH production associated with a variety of extrapituitary neoplasms. ACTH-independent causes of hypercortisolism are rare in the pediatric population. Biochemical evidence of hypercortisolism can be demonstrated by 24-hour urine collections for free cortisol. Patients with hypercortisolism will demonstrate loss of normal diurnal variation in plasma cortisol and ACTH. A single midnight serum cortisol value has been reported to distinguish Cushing’s syndrome effectively from a pseudo-Cushing condition (e.g., exogenous obesity, depression, stress) [Papanicolaou et al., 1998]. The principal challenge in the differential diagnosis of Cushing’s syndrome is distinguishing Cushing’s disease from ectopic ACTH production. In general, patients with Cushing’s disease show suppression of cortisol and ACTH with dexamethasone administration (20 μg/kg every 6 hours for 2 days). However, up to 20 percent of patients with Cushing’s disease do not suppress under these conditions. An MRI of the hypothalamic/pituitary area should be obtained if Cushing’s disease is suspected, though small adenomas may not be visualized [Magiakou et al., 1994; Devoe et al., 1997].
Trans-sphenoidal resection of pituitary adenomas has emerged as the treatment of choice for Cushing’s disease [Devoe et al., 1997]. Due to small size, some of these tumors are difficult to identify intraoperatively. The recurrence rate after surgery is about 15–25 percent, even in the hands of skilled neurosurgeons. Radiotherapy has been shown to be effective for unsuccessful trans-sphenoidal surgery [Estrada et al., 1997]. Other therapeutic options for recurrent Cushing’s disease include repeated pituitary exploration, bilateral adrenalectomy, and the use of pharmacologic agents that directly impair cortisol synthesis and secretion.
Adrenocorticotropic Hormone Deficiency
Isolated ACTH deficiency is a rare cause of secondary adrenal insufficiency, due to either CRH deficiency or a primary decrease in ACTH production by corticotrophs. Clinical presentation is highly variable. Neonatal onset of isolated ACTH deficiency usually presents with sudden, severe episodes of hypoglycemia, sometimes with seizure and coma. Neonates may also present with prolonged cholestatic jaundice. Death may occur if treatment is not initiated promptly. Over 70 percent of these patients have a loss-of-function mutation in the TPIT gene, which encodes a transcription factor that is expressed specifically in corticotrophs and is required for expression of POMC, the precursor of ACTH [Pulichino et al., 2003; Vallette-Kasic et al., 2003, 2007]. Other genetic factors leading to isolated ACTH deficiency remain to be elucidated, since later-onset, isolated ACTH deficiency can occur peripubertally [Bremer et al., 2008]. As noted, ACTH deficiency can be associated with multiple hypothalamic/pituitary hormone deficiencies. The genetic basis of some forms of combined pituitary hormone deficiencies has been elucidated. As previously noted, mutations in Prop-1 can present with GH, PRL, TSH, gonadotropin, and ACTH deficiencies [Agarwal et al., 2000]. ACTH deficiency also is found in patients who have mutations in the Lhx4 gene, which is closely related to Lhx3, as described in the section on delayed puberty. Mutations in either Lhx3 or Lhx4 also can lead to deficiencies in both GH and TSH [Machinis et al., 2001]. CNS tumors involving the third ventricular area, such as craniopharyngioma, germinoma, and astrocytoma, can cause multiple pituitary deficiencies, including ACTH deficiency. In addition, ACTH deficiency with other pituitary hormone deficiencies can be seen in a variety of CNS disorders, including infection, invasive disease, irradiation, congenital malformation, and trauma. Long-term glucocorticoid therapy, e.g., in patients with chronic inflammatory and autoimmune diseases, will often suppress the HPA axis.
Cortisol (hydrocortisone) replacement is based on studies of the cortisol secretory rate in the pediatric population [Linder et al., 1990; Kerrigan et al., 1993; Metzger et al., 1993]. During periods of significant stress, such as febrile illness and surgery, such patients are managed routinely with a temporary increase in glucocorticoids (given orally or parenterally) at doses up to 50 mg/m2/day of hydrocortisone.
Hypothalamic/Pituitary Disorders of Statural Growth
Growth Hormone Deficiency
The incidence of GH deficiency is estimated to be as high as 1:3480 in the United States [Lindsay et al., 1994]. Children with congenital GH deficiency are of normal size at birth. However, after approximately 6 months of life, growth failure becomes apparent when linear growth becomes GH-dependent. Usually, children with GH deficiency are short and cherubic, with a doll-like appearance. Approximately 10–20 percent of G- deficient patients present with severe hypoglycemia in addition to short stature. In the neonatal period, hypoglycemia and microphallus strongly suggest GH deficiency.
Isolated GH deficiency is rarely due to a GH gene deletion. About 3–30 percent of cases with isolated GH deficiency (IGHD) have a genetic etiology. Most of these genetic defects remain elusive, with 11 percent of the mutations being reported in GH1 and GHRHR genes [Alatzoglou et al., 2009]. Mutations in the homeobox gene Rpx/Hesx1 are associated with septo-optic dysplasia, also known as de Morsier’s syndrome, a common midline malformation. This syndrome is characterized by the classical triad of optic nerve hypoplasia, midline malformations (such as agenesis of the corpus callosum and absence of the septum pellucidum), and pituitary hypoplasia with consequent panhypopituitarism. Mutations in Rpx/Hesx1 also can cause isolated GH deficiency. However, more commonly, these mutations found in pituitary transcription factors cause multiple hormonal deficiencies and congenital midline defects. The phenotypes of mutations in the Rpx/Hesx1 gene are highly variable, ranging from isolated GH deficiency without any midline defects, to the complete spectrum of the disease with panhypopituitarism [Thomas et al., 2001].
Growth Hormone Excess
GH excess can lead to tall stature (gigantism) in children, who have open epiphyses, and acromegaly in adolescents, who have closed epiphyses. Gigantism is a rare condition caused by GH oversecretion, primarily due to increased GH production from the pituitary [Abe and Ludecke, 1999; Kunwar and Wilson, 1999]. GH-producing adenomas constitute up to 10 percent of pituitary adenomas [Kunwar and Wilson, 2001]. Ectopic GHRH overproduction is a rare cause of GH excess [Thorner et al., 1982]. In addition, McCune–Albright syndrome can present with gigantism. The constitutively active G proteins in somatotrophs can result in a rise in cAMP, leading to increased GH secretion in patients with McCune–Albright syndrome [Geffner et al., 1987; Cuttler et al., 1989].
The goal of treatment in GH-secreting adenomas is normalization of GH and IGF-I levels. Trans-sphenoidal surgery has resulted in a greater than 80 percent cure rate, and thus remains the first choice of treatment. If surgery is unsuccessful, somatostatin analogs, GH antagonists, and dopamine agonists can be used. Long-term studies have shown a clear advantage to using a long-acting-release somatostatin analog over a slow-release analog, which can be used as first-line treatment should the patient prefer medication over surgery [Colao et al., 2009]. Radiotherapy may be beneficial.
Hypothalamic/Pituitary Disorders of Thyroid Function
Normal Thyroid Physiology
Under regulation by the hypothalamus and pituitary, the thyroid gland produces thyroxine (T4) and triiodothyronine (T3), which have broad effects on metabolism, general growth, and CNS development [Anderson et al., 2000]. Embryonic development of the hypothalamic-pituitary-thyroid (HPT) axis begins during the first trimester, marked by significant concentrations of TRH in the hypothalamus, the median eminence, and the supraoptic tract. The thyroid gland forms as an invagination of endoderm at the base of the tongue, and descends along the midline to its final position anterior to the second to fourth tracheal cartilage rings [Fisher and Brown, 2000]. Maturation of the human HPT axis is a complex process, involving ontogenesis of hypothalamic TRH, pituitary TSH, and thyroid hormone secretory processes, as well as maturation of their respective receptors, and of pituitary iodothyronine monodeiodinase enzyme activities [Fisher and Brown, 2000]. TSH is a heterodimeric glycoprotein consisting of alpha and beta subunits. The alpha subunit is common to TSH, LH, FSH, and hCG, while the beta subunit confers specificity. TSH acts on the thyroid gland to stimulate thyroid hormone synthesis and secretion. TSH is negatively regulated by thyroid hormones and positively regulated by TRH. Thyroid hormone signaling is mediated by specific nuclear receptors that regulate the expression of target genes at the levels of transcription and protein synthesis [Anderson et al., 2000]. Over 90 percent of circulating T4 and T3 is bound to thyroid binding proteins (thyroid-binding globulin, transthyretin, and albumin) [Refetoff, 1989]. T4 serves largely as prohormone, which is converted peripherally to the more potent T3 [Anderson et al., 2000].
Central Hypothyroidism
Congenital TSH deficiency occurs in 1:50,000–150,000 newborns [Fisher and Grueters, 2008]. Underlying etiologies include isolated TSH deficiency, developmental defects of the hypothalamus and pituitary, and familial panhypopituitarism. Patients with congenital TSH deficiency are usually not detected by neonatal screening for hypothyroidism, as most programs are designed to screen for the hyperthyrotropinemia that accompanies the more primary hypothyroidism.
Isolated TSH deficiency is usually autosomal-recessive. Mutations in the TSH beta subunit prevent formation of the active TSH heterodimer [Hayashizaki et al., 1989, 1990; McDermott et al., 2002; Karges et al., 2004]. TSH deficiency in combination with other pituitary hormone deficiencies is seen frequently in patients with HESX1, PIT-1, and PROP-1 mutations. As described previously in the sections on disorders of statural growth and puberty, these genes are pituitary transcription factors that are critical for pituitary development. In addition, isolated central hypothyroidism can result from a loss-of-function mutation in the TRH receptor [Collu et al., 1997].
With respect to laboratory assessment of thyroid function, it is critical to recognize that normal ranges vary significantly with age [Nelson et al., 1993]. In particular, TSH and free T4 are relatively high in newborns in comparison to older infants, children, and adolescents. The principal goals of treating central hypothyroidism are prevention of mental retardation and growth impairment. Thus, thyroxine replacement should be instituted promptly following diagnosis of hypothyroidism, regardless of etiology.
Central Hyperthyroidism
Most cases of hyperthyroidism in the pediatric population are autoimmune and TSH-independent. Central hyperthyroidism as a consequence of a pituitary TSH-secreting adenoma is exceedingly rare [Tolis et al., 1978]. Such patients may have a prominent local mass effect of the tumor, such as loss or partial loss of vision from optic atrophy, and may have hydrocephalus. Other signs and symptoms of hyperthyroidism, such as agitation, tall stature, goiter, tremor, and palpitation, also are manifested in central hyperthyroidism. Biochemically, patients with central hyperthyroidism have elevated concentrations of TSH in the context of elevated T4 and T3. If a pituitary adenoma is not identifiable on imaging studies, rare selective pituitary T3 resistance should be considered. Surgical removal of the tumor is the treatment of choice for TSH-secreting pituitary adenoma. However, the treatment of selective pituitary T3 resistance remains a therapeutic challenge.
Hypothalamic Disorders of Appetite Regulation and Energy Balance
Appetite and energy balance are regulated by a complex network, in which the hypothalamus serves as the central processing unit for hormonal, nutritional, and neuronal input from both peripheral tissue and higher brain centers [Elmquist et al., 1999]. In humans, it is well known that hypothalamic damage (e.g., from a tumor, surgery, trauma, or radiation) can lead to excessive weight gain, i.e. hypothalamic obesity [Bray and Gallagher, 1975]. These patients present with excessive weight gain without response to caloric restriction or exercise [Bray et al., 1981]. Traditionally, the ventromedial hypothalamus has been viewed as the “satiety” center, since damage to the ventromedial hypothalamus can cause hyperphagic obesity. In contrast, the lateral hypothalamus has been viewed as the “feeding” center, since its disruption leads to hypophagia and weight loss [Flier and Maratos-Flier, 1998].
Insights into mechanisms of hypothalamic obesity derive from advances in our understanding of hypothalamic control of appetite regulation and energy balance (Figure 97-5). Energy storage and appetite are regulated at the hypothalamus through signaling by leptin, an anorexigenic hormone secreted by adipocytes, and by insulin. Both leptin and insulin stimulate anorexigenic and inhibit orexigenic pathways in the hypothalamus. In the anorexigenic pathway, leptin and insulin maintain expression of POMC in the arcuate nuclei within the ventromedial hypothalamus, and POMC-derived α-melanocyte stimulating hormone (α-MSH) activates the melanocortin-4 receptor (MC4R) in the paraventricular nuclei and the lateral hypothalamic area. This melanocortin signal is appetite-suppressing and leads to decreased food intake. In addition, leptin and insulin inhibit NPY and AgRP within the arcuate nuclei, both of which are appetite-stimulating peptides [Schwartz et al., 2003]. NPY decreases POMC expression through the neuropeptide Y1 receptor (Y1R). AgRP is an antagonist for MC4R, competing with α-MSH for binding to the MC4R and leading to increased food intake [Korner and Leibel, 2003].
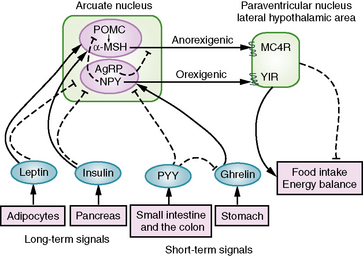
Short-term signals that affect appetite and determine meal initiation and termination are derived from gut hormones, such as ghrelin, secreted by oxyntic cells in the stomach fundus, and peptide YY3-36 (PYY), secreted by endocrine L cells in the distal small bowel and colon. Ghrelin signals hunger and increases food intake by stimulating ghrelin receptors on hypothalamic NPY- and AgRP-expressing neurons [Cummings et al., 2002]. In contrast, PYY signals satiety and decreases food intake by inhibiting NPY-/AgRP-expressing neurons via the neuropeptide Y2 receptor (Y2R) and by decreasing ghrelin levels [Batterham et al., 2002, 2003].
Both long-term and short-term hormonal signals are received in the arcuate nuclei, constituting the afferent system of the feedback loop for appetite and energy balance [Schwartz et al., 2000]. Through the paraventricular nuclei and lateral nuclei, these signals are transmitted into a complex and less well-understood efferent system that controls appetite and energy balance [Lustig, 2003]. The autonomic system plays a critical role in regulating energy expenditure. It is known that the ventromedial hypothalamus has neuronal projections to the spinal cord that regulate sympathetic outflow. Activation of the sympathetic nervous system results in increased energy expenditure, such as increased local heat production and increased lipolysis [Lowell and Spiegelman, 2000]. In addition, the ventromedial hypothalamus has projections to the dorsal motor nucleus of the vagus, which innervate the gastrointestinal system. Disinhibition of vagal firing by ventromedial hypothalamus lesions is associated with relative insulin hypersecretion and subsequent increased energy storage [Lustig, 2002].
As previously noted, hypothalamic obesity frequently is seen in children with hypothalamic insults, such as brain tumor, surgery, irradiation, or trauma. Approximately 50 percent of children with craniopharyngioma develop hypothalamic obesity. Other tumors, such as germinoma, optic glioma, prolactinoma, and hypothalamic astrocytoma, also are associated with hypothalamic obesity. These patients usually have unrelenting weight gain after tumor removal or radiotherapy, without documented excessive food intake [Harz et al., 2003]. Reduced physical activity, rather than increased energy intake, was shown to be primarily responsible for obesity in patients with craniopharyngioma [Harz et al., 2003]. Decreased resting energy expenditure also may be present in children after brain tumor therapy [Shaikh et al., 2008]. Other risk factors associated with hypothalamic obesity include dose of hypothalamic irradiation (greater than 51 Gy), and associated hypothalamic endocrinopathy (GH deficiency, hypothyroidism, ACTH deficiency, precocious or delayed puberty, and diabetes insipidus) [Lustig et al., 2003]. In patients with hypothalamic obesity and apparent insulin hypersecretion, the somatostatin agonist, octreotide, has been shown to decrease insulin secretion associated with a decrease or stabilization of body weight [Lustig et al., 1999].
Numerous examples of monogenic obesity have been described at many levels of the leptin-melanocortin system of energy balance, including leptin, leptin receptor, POMC, prohormone convertase 1/3 (PC1/3), and MC4R [Ranadive and Vaisse, 2008; O’Rahilly and Farooqi, 2008]. Mutations in leptin (autosomal-recessive) and leptin receptor (homozygous or compound heterozygote) are extremely rare monogenic forms of morbid obesity [Montague et al., 1997; Farooqi et al., 2002; Gibson et al., 2004; Licinio et al., 2004]. The first three children reported with leptin deficiency were born to two consanguineous Pakistani families [Farooqi et al., 2002]. They had normal birth weight, but became hyperphagic in infancy and developed severe obesity with undetectable levels of leptin. Leptin-deficient patients also demonstrate pubertal delay as a consequence of hypogonadotropic hypogonadism, and may also have impaired immune function. Exogenous administration of leptin induced sustained weight loss due to loss of fat mass in these patients. Similar results were observed in three adults with congenital leptin deficiency [Licinio et al., 2004]. Leptin replacement in these adults resulted in profound weight loss, increased physical activity, and resolution of insulin resistance and hypogonadism. Homozygous and compound heterozygous mutations of the leptin receptor also are associated with severe childhood obesity. Leptin receptor-deficient patients have early onset of morbid obesity associated with absent pubertal development, reduced levels of both thyrotropin and GH, and impaired immune function [Clement et al., 1998]. Loss-of-function mutations in POMC (homozygous or compound heterozygous) result in severe hyperphagia and obesity as a consequence of lack of MC4R activation by α-MSH, and adrenal insufficiency as a consequence of defective synthesis of ACTH [Krude et al., 1998, 2003]. Patients may also have red hair from lack of MC1R activation by α-MSH in melanocytes [Krude et al., 1998]. Mutations in PC1/3 (a serine endoprotease), thought to result in defective processing of POMC to α-MSH, as well as defective ACTH synthesis, lead to hyperphagia, early-onset obesity, and partial ACTH deficiency [O’Rahilly et al., 1995]. MC4R mutations (primarily autosomal-dominant) have been found in 2.5 percent of obese children, representing the most common genetic defects in human obesity [Farooqi et al., 2000; Vaisse et al., 2000; Lubrano-Berthelier et al., 2006]. Of note, other than severe hyperphagia and early-onset obesity, MC4R mutations typically do not result in other physical, hormonal, or developmental consequences [Ranadive and Vaisse, 2008].
Childhood obesity also is associated with mutations in a number of genes thought to play a role in hypothalamic development [Ranadive and Vaisse, 2008]. Haploinsufficiency of SIM1 results in hyperphagia, severe early-onset obesity, and increased linear growth [Holder et al., 2000]. In addition, mutations of brain-derived neurotrophic factor (BDNF) and of its receptor, tropomysin-related kinase B (TRKB), have been associated with hyperphagia, early-onset obesity, and neurocognitive deficits [Gray et al., 2006; Yeo et al., 2004].
Hypothalamic/Pituitary Disorders of Water Balance
Under normal circumstances, plasma osmolality is maintained within a relatively narrow range (280–295 mOsm/kg). This homeostasis requires adequate water intake, regulated by an intact thirst mechanism, and appropriate free water excretion by the kidneys, mediated by appropriate secretion of vasopressin (AVP, i.e., antidiuretic hormone [ADH]) [Robertson, 2001]. AVP is produced in the magnocellular neurons in the paraventricular and supraoptic nuclei of the hypothalamus. Axons from these neurons project through the pituitary stalk and terminate in the posterior pituitary gland [Robertson, 2001]. A gene on chromosome 20p13 encodes both AVP and its carrier protein, neurophysin II (NPII). AVP and NPII are synthesized as a single polypeptide, cleaved within neurosecretory granules, reassembled into an AVP/NPII complex, and secreted [Rutishauser et al., 2002]. AVP exerts its antidiuretic action by binding to the X chromosome-encoded V2 vasopressin receptor (V2R), a G-protein-coupled receptor on the basolateral membrane of renal collecting duct epithelial cells. Following V2R activation, increased intracellular cAMP mediates shuttling of the water channel aquaporin 2 (AQP-2) to the apical membrane of collecting duct epithelial cells, resulting in increased water permeability and antidiuresis [Schrier and Cadnapaphornchai, 2003].
AVP secretion is regulated principally by changes in plasma osmolality and in effective circulating volume [Robertson, 2001]. Osmoreceptors in the hypothalamus stimulate secretion of AVP when plasma osmolality increases by as little as 1 percent in healthy individuals. AVP levels are normally low and do not increase until plasma osmolality exceeds 280 mOsm/kg [Robinson and Verbalis, 2003]. AVP secretion also is regulated by changes in blood volume. Baroreceptors in the systemic venous circulation, right side of the heart, and left atrium (“low-pressure” areas), as well as in the systemic arterial systems of the carotid sinus and aortic arch (“high-pressure” areas), signal the hypothalamus via the vagus and glossopharyngeal nerves, respectively. These baroreceptors become activated when stretched by increases in intravascular volume, leading to inhibition of AVP secretion. In addition, a variety of other factors affect AVP secretion. AVP is stimulated by pain, stress, and a variety of drugs, and is inhibited by multiple factors [Robinson and Verbalis, 2003]. Adequate water intake, governed by an intact thirst mechanism, is regulated by hypothalamic osmoreceptors located near vasopressinergic neurons.
Clinical disorders of water balance are common, and abnormalities in many steps involving AVP secretion and responsiveness have been described [Robertson, 2001; Ranadive and Rosenthal, 2009]. The focus of this section is on the principal hypothalamic/pituitary disorders of water balance, diabetes insipidus (DI) and the syndrome of inappropriate antidiuretic hormone secretion (SIADH).
Diabetes Insipidus
DI may result from a deficiency of AVP or from nephrogenic causes [Baylis and Cheetham, 1998; Robinson and Verbalis, 2003; Verbalis, 2003]. Nephrogenic DI may be congenital, resulting from inactivating mutations in the V2R gene, or from autosomal-recessive or dominant lesions in the AQP-2 gene; or acquired, resulting from a variety of conditions, including some forms of primary renal disease, obstructive uropathy, hypokalemia, hypercalcemia, sickle cell disease, and a variety of drugs, including lithium and demeclocycline [Baylis and Cheetham, 1998; Morello and Bichet, 2001; Robinson and Verbalis, 2003; Verbalis, 2003; Spanakis et al., 2008]. Prolonged polyuria of any cause can result in some degree of nephrogenic DI secondary to a reduction of tonicity in the renal medullary interstitium and a subsequent decrease in the gradient necessary to concentrate the urine.
Central DI is rarely congenital and more frequently acquired. Congenital central DI may be caused by hypothalamic structural malformations and through both autosomal-dominant and recessive mutations in the AVP/NPII gene. Of the latter, the autosomal-dominant causes are more common, and are thought to be a consequence of heterozygous mutations in the AVP/NPII gene, which lead to misfolding of the precursor AVP/NPII protein [Rutishauser et al., 2002]. The dominant negative effect is thought to occur as a consequence of the misfolded precursor protein, which accumulates in the endoplasmic reticulum of vasopressinergic neurons, ultimately resulting in death of these neurons and gliosis [Rutishauser et al., 2002; Robinson and Verbalis, 2003]. In such patients, clinical DI usually develops several months to years after birth. A rare autosomal-recessive form of central DI has been reported in association with a mutation in the AVP/NPII gene, resulting in a biologically inactive AVP [Willcutts et al., 1999].
Acquired forms of DI occur in association with a variety of disorders in which there is destruction or degeneration of vasopressinergic neurons. Etiologies include primary tumors (e.g., craniopharyngioma, germinoma) or metastases, infection (meningitis, encephalitis), histiocytosis, granuloma, vascular disorders, and autoimmune disorders (lymphocytic infundibuloneurohypophysitis) [Baylis and Cheetham, 1998; Robinson and Verbalis, 2003; Verbalis, 2003]. Acquired DI may occur in association with trauma and surgery. Idiopathic DI is a diagnosis of exclusion, and one that is made with decreasing frequency concurrent with improved sensitivity of MRI of the CNS, and of cerebrospinal fluid and serum tumor markers [Mootha et al., 1997; Maghnie et al., 2000].
The principal presenting sign of DI is polyuria, which, in addition to deficiency or impaired responsiveness to AVP, may result from an osmotic agent (e.g., hyperglycemia in diabetes mellitus) or from excessive water intake (primary polydipsia). Hypernatremia usually does not occur if patients have an intact thirst mechanism, adequate access to fluids, and no additional on-going fluid losses (e.g., diarrhea). Infants with DI, in addition to polyuria and polydipsia, may be irritable and have fever of unknown origin, growth failure secondary to inadequate caloric intake, and hydronephrosis. Older children may also have nocturia and enuresis. DI may not be apparent in patients with coexisting untreated anterior pituitary-mediated adrenal glucocorticoid insufficiency, as cortisol is required to generate a normal free water loss [Robinson and Verbalis, 2003].
A diagnosis of DI can be made if screening laboratory studies reveal serum hyperosmolality concurrent with urine that is inappropriately dilute. However, as most patients with DI do not have hyperosmolality and hypernatremia, as noted above, a standardized water deprivation test is useful to distinguish DI from primary polydipsia. Urine osmolality of >750 mOsm/kg after 7–8 hours of water deprivation is thought to exclude DI [Baylis and Cheetham, 1998]. Urine osmolality in the 300–750 mOsm/kg range may indicate partial DI [Baylis and Cheetham, 1998]. If DI is suspected, a plasma sample should be obtained for AVP radioimmunoassay. AVP or a synthetic analog (desmopressin) should then be administered to distinguish AVP deficiency from AVP unresponsiveness.
Once a diagnosis of central DI is made, a brain MRI, with particular attention to the third ventricular area, should be obtained. An absent posterior pituitary “bright spot” is seen in virtually all patients with central DI. Under normal circumstances, a posterior pituitary bright spot is seen on T1-weighted images due to stored AVP in neurosecretory granules (see Figure 97-1). In patients with DI as a consequence of an autosomal-dominant mutation in the AVP/NPII gene, a posterior pituitary bright spot may be seen in the early stages of the disease [Robinson and Verbalis, 2003]. In central DI patients with an absent posterior pituitary bright spot, an otherwise normal MRI warrants close follow-up with cerebrospinal fluid tumor markers and cytology, serum tumor markers, and serial contrast-enhanced brain MRIs for early detection of an evolving occult hypothalamic-stalk lesion [Mootha et al., 1997].
Syndrome of Inappropriate Antidiuretic Hormone Secretion
SIADH is characterized by the inability to excrete a free water load, with inappropriately concentrated urine, and resultant hyponatremia, hypo-osmolality, and natriuresis [Bartter and Schwartz, 1967; Baylis, 2003; Verbalis et al., 2007]. Following from the original criteria established by Bartter and Schwartz, a diagnosis of SIADH is made when the following occur [Bartter and Schwartz, 1967]:
While most patients with SIADH have inappropriately measurable or elevated levels of plasma AVP relative to plasma osmolality, 10–20 percent of patients with SIADH do not have measurable AVP levels. This may reflect issues of assay sensitivity or may indicate a syndrome resembling SIADH, such as the recently described nephrogenic syndrome of inappropriate antidiuresis (NSIAD), associated with an activating mutation in the X-linked G-protein-coupled V2R and unmeasurable circulating levels of AVP [Feldman et al., 2005].
Euvolemia in chronic SIADH is an important distinguishing factor in the evaluation of a patient with serum hypo-osmolality, and has a bearing on treatment issues, as will be discussed subsequently. Euvolemia in chronic SIADH is thought to represent an adaptation to water overload. This adaptation is mediated, in part, at the cellular level through depletion of intracellular electrolytes (potassium) and organic osmolytes [Verbalis et al., 2007]. The loss of brain solutes is thought to allow effective regulation of brain volume during chronic hyponatremia and SIADH. Natriuresis, thought to be mediated in part through secretion of atrial natriuretic peptide, also contributes to volume regulation in chronic SIADH [Cogan et al., 1988]. Cerebral salt wasting, associated with some intracranial diseases (e.g., subarachnoid hemorrhage), is often considered in the differential diagnosis of SIADH. However, the hypo-osmolality, hyponatremia, and natriuresis in cerebral salt wasting are associated with volume contraction, which distinguishes this disorder from the euvolemic condition of SIADH [Verbalis et al., 2007].
Therapy for SIADH includes treatment of the underlying disorder (or discontinuation of an offending drug) and fluid restriction. Replacement of lost body sodium also may be necessary but usually can be achieved through normal dietary salt intake. Severe hyponatremia (serum sodium <120 mEq/L) may be associated with CNS abnormalities, including seizures, and may require treatment with hypertonic (3 percent) intravenous sodium chloride solution. Concurrent use of a diuretic, such as furosemide, may be indicated when volume expansion is severe. Other therapeutic approaches include the use of agents that induce nephrogenic DI, such as demeclocycline and lithium, although both are contraindicated, particularly in younger pediatric patients, because of untoward side effects. Urea has been used as an osmotic diuretic in pediatric SIADH and NSIAD [Huang et al., 2006]. A variety of nonpeptide V2R antagonists are currently in various stages of clinical trials or have been approved by the Food and Drug Administration for use in adults [Verbalis et al., 2007].
If SIADH and hyponatremia are acute (<48 hours), it is thought that hyponatremia can be corrected quickly. However, if SIADH and hyponatremia are chronic (>48 hours), overzealous treatment can result in CNS damage, including central pontine myelinolysis [Verbalis et al., 2007]. Brain solute loss, while an important regulatory mechanism in chronic SIADH, may predispose to the development of central pontine myelinolysis with rapid correction of serum osmolality. Generally, it is recommended that plasma sodium be corrected to a “safe” level of approximately 120–125 mEq/L, at a rate of no greater than 0.5 mEq/L per hour, with an overall correction that does not exceed 12 mEq/L in the initial 24 hours and 18 mEq/L in the initial 48 hours of treatment [Verbalis et al., 2007].
References
 The complete list of references for this chapter is available online at www.expertconsult.com.
The complete list of references for this chapter is available online at www.expertconsult.com.
Abe T., Ludecke D.K. Recent primary transnasal surgical outcomes associated with intraoperative growth hormone measurement in acromegaly. Clin Endocrinol (Oxf). 1999;50:27.
Agarwal G., Bhatia V., Cook S., et al. Adrenocorticotropin deficiency in combined pituitary hormone deficiency patients homozygous for a novel PROP1 deletion. J Clin Endocrinol Metab. 2000;85:4556.
Alatzoglou K.S., Turton J.P., Kelberman D., et al. Expanding the Spectrum of Mutations in GH1 and GHRHR: Genetic Screening in a Large Cohort of Patients with Congenital Isolated Growth Hormone Deficiency. J Clin Endocrinol Metab. 2009;94:3191.
Anderson G.W., Mariash C.N., Oppenheimer J.H. Molecular Actions of Thyroid Hormone, ed 8. Philadelphia: Lippincott Williams & Wilkins; 2000.
Bartter F.C., Schwartz W.B. The syndrome of inappropriate secretion of antidiuretic hormone. Am J Med. 1967;42:790.
Batterham R.L., Cohen M.A., Ellis S.M., et al. Inhibition of food intake in obese subjects by peptide YY3-36. N Engl J Med. 2003;349:941.
Batterham R.L., Cowley M.A., Small C.J., et al. Gut hormone PYY(3-36) physiologically inhibits food intake. Nature. 2002;418:650.
Baylis P.H. The syndrome of inappropriate antidiuretic hormone secretion. Int J Biochem Cell Biol. 2003;35:1495.
Baylis P.H., Cheetham T. Diabetes insipidus. Arch Dis Child. 1998;79:84.
Beier D.R., Dluhy R.G. Bench and bedside – the G protein-coupled receptor GPR54 and puberty. N Engl J Med. 2003;349:1589.
Bergland R.M., Page R.B. Pituitary-brain vascular relations: a new paradigm. Science. 1979;204:18.
Bouligand J., Ghervan C., Tello J.A., et al. Isolated familial hypogonadotropic hypogonadism and a GNRH1 mutation. N Engl J Med. 2009;360:2742.
Bray G.A., Gallagher T.F.Jr. Manifestations of hypothalamic obesity in man: a comprehensive investigation of eight patients and a reveiw of the literature. Medicine (Baltimore). 1975;54:301.
Bray G.A., Inoue S., Nishizawa Y. Hypothalamic obesity. The autonomic hypothesis and the lateral hypothalamus. Diabetologia. 1981;20(Suppl):366.
Bremer A.A., Ranadive S., Conrad S.C., et al. Isolated adrenocorticotropic hormone deficiency presenting as an acute neurologic emergency in a peripubertal girl. J Pediatr Endocrinol Metab. 2008;21:799.
Breyer P., Haider A., Pescovitz O.H. Gonadotropin-releasing hormone agonists in the treatment of girls with central precocious puberty. Clin Obstet Gynecol. 1993;36:764.
Cisternino M., Arrigo T., Pasquino A.M., et al. Etiology and age incidence of precocious puberty in girls: a multicentric study. J Pediatr Endocrinol Metab. 2000;13(Suppl 1):695.
Clement K., Vaisse C., Lahlou N., et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. 1998;392:398.
Cogan E., Debieve M.F., Pepersack T., et al. Natriuresis and atrial natriuretic factor secretion during inappropriate antidiuresis. Am J Med. 1988;84:409.
Cohen L.E., Radovick S. Molecular basis of combined pituitary hormone deficiencies. Endocr Rev. 2002;23:431.
Colao A., Auriemma R.S., Galdiero M., et al. Effects of initial therapy for five years with somatostatin analogs for acromegaly on growth hormone and insulin-like growth factor-I levels, tumor shrinkage, and cardiovascular disease: a prospective study. J Clin Endocrinol Metab. 2009;94:3746.
Colao A., Di Sarno A., Cappabianca P., et al. Withdrawal of Long-Term Cabergoline Therapy for Tumoral and Nontumoral Hyperprolactinemia. N Engl J Med. 2003;349:2023.
Colao A., Loche S., Cappa M., et al. Prolactinomas in children and adolescents. Clinical presentation and long-term follow-up. J Clin Endocrinol Metab. 1998;83:2777.
Collu R., Tang J., Castagne J., et al. A novel mechanism for isolated central hypothyroidism: inactivating mutations in the thyrotropin-releasing hormone receptor gene. J Clin Endocrinol Metab. 1997;82:1561.
Cone R.D., Low M.J., Elmquist J.K., et al. Neuroendocrinology, ed 10. Philadelphia: Saunders; 2003.
Crino A., Schiaffini R., Ciampalini P., et al. Hypogonadism and pubertal development in Prader-Willi syndrome. Eur J Pediatr. 2003;162:327.
Cummings D.E., Weigle D.S., Frayo R.S., et al. Plasma ghrelin levels after diet-induced weight loss or gastric bypass surgery. N Engl J Med. 2002;346:1623.
Cuttler L., Jackson J.A., Saeed uz-Zafar M., et al. Hypersecretion of growth hormone and prolactin in McCune-Albright syndrome. J Clin Endocrinol Metab. 1989;68:1148.
Dattani M.L., Martinez-Barbera J., Thomas P.Q., et al. Molecular genetics of septo-optic dysplasia. Horm Res. 2000;53(Suppl 1):26.
Dattani M.T., Martinez-Barbera J.P., Thomas P.Q., et al. Mutations in the homeobox gene HESX1/Hesx1 associated with septo-optic dysplasia in human and mouse. Nat Genet. 1998;19:125.
Demura R., Kubo O., Demura H., et al. FSH and LH secreting pituitary adenoma. J Clin Endocrinol Metab. 1977;45:653.
de Roux N., Genin E., Carel J.C., et al. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci USA. 2003;100:10972.
Devoe D.J., Miller W.L., Conte F.A., et al. Long-term outcome in children and adolescents after transsphenoidal surgery for Cushing’s disease. J Clin Endocrinol Metab. 1997;82:3196.
de Vries L., Kauschansky A., Shohat M., et al. Familial central precocious puberty suggests autosomal dominant inheritance. J Clin Endocrinol Metab. 2004;89:1794.
Di Sarno A., Landi M.L., Cappabianca P., et al. Resistance to Cabergoline as Compared with Bromocriptine in Hyperprolactinemia: Prevalence, Clinical Definition, and Therapeutic Strategy. J Clin Endocrinol Metab. 2001;86:5256.
Elmquist J.K., Elias C.F., Saper C.B. From lesions to leptin: hypothalamic control of food intake and body weight. Neuron. 1999;22:221-232.
Estrada J., Boronat M., Mielgo M., et al. The long-term outcome of pituitary irradiation after unsuccessful transsphenoidal surgery in Cushing’s disease. N Engl J Med. 1997;336:172.
Farooqi I.S., Matarese G., Lord G.M., et al. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest. 2002;110:1093.
Farooqi I.S., Yeo G.S., Keogh J.M., et al. Dominant and recessive inheritance of morbid obesity associated with melanocortin 4 receptor deficiency. J Clin Invest. 2000;106:271.
Feldman B.J., Rosenthal S.M., Vargas G.A., et al. Nephrogenic syndrome of inappropriate antidiuresis. N Engl J Med. 2005;352:1884.
Feuillan P.P., Jones J.V., Barnes K., et al. Follow-up of children and young adults after GnRH-agonist therapy or central precocious puberty. J Endocrinol Invest. 2001;24:734.
Fisher D.A., Brown R.S. Thyroid physiology in the perinatal period and during childhood, ed 8. Philadelphia: Lippincott Williams & Wilkins; 2000.
Fisher D.A., Grueters A. Disorders of the Thyroid in the Newborn and Infant. In: Sperling M.A., editor. Pediatric Endocrinology. ed 3. Philadelphia: Saunders; 2008:198-226.
Flier J.S., Maratos-Flier E. Obesity and the hypothalamus: novel peptides for new pathways. Cell. 1998;92:437.
Geffner M.E., Nagel R.A., Dietrich R.B., et al. Treatment of acromegaly with a somatostatin analog in a patient with McCune-Albright syndrome. J Pediatr. 1987;111:740.
Georgopoulos N.A., Pralong F.P., Seidman C.E., et al. Genetic heterogeneity evidenced by low incidence of KAL-1 gene mutations in sporadic cases of gonadotropin-releasing hormone deficiency. J Clin Endocrinol Metab. 1997;82:213.
Gibson W.T., Farooqi I.S., Moreau M., et al. Congenital leptin deficiency due to homozygosity for the Delta133G mutation: report of another case and evaluation of response to four years of leptin therapy. J Clin Endocrinol Metab. 2004;89:4821.
Gray J., Yeo G.S., Cox J.J., et al. Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain-derived neurotrophic factor (BDNF) gene. Diabetes. 2006;55:3366-3371.
Grumbach M.M. The neuroendocrinology of human puberty revisited. Horm Res. 2002;57(Suppl 2):2.
Grumbach M.M., Styne D.M. Puberty: Ontogeny, Neuroendocrinology, Physiology, and Discorders, ed 10. Philadelphia: Saunders; 2003.
Hardelin J.P., Dode C. The complex genetics of Kallmann syndrome: KAL1, FGFR1, PROKR2, PROK2, et al. Sex Dev. 2008;2:181-193.
Harz K.J., Muller H.L., Waldeck E., et al. Obesity in patients with craniopharyngioma: assessment of food intake and movement counts indicating physical activity. J Clin Endocrinol Metab. 2003;88:5227.
Hashimoto T., Kumahara Y. Concerning hypogonadism of Laurence-Moon-Biedl syndrome. Metabolism. 1979;28:370.
Hayashizaki Y., Hiraoka Y., Endo Y., et al. Thyroid-stimulating hormone (TSH) deficiency caused by a single base substitution in the CAGYC region of the beta-subunit. EMBO J. 1989;8:2291.
Hayashizaki Y., Hiraoka Y., Tatsumi K., et al. Deoxyribonucleic acid analyses of five families with familial inherited thyroid stimulating hormone deficiency. J Clin Endocrinol Metab. 1990;71:792.
Herman-Giddens M.E., Slora E.J., Wasserman R.C., et al. Secondary sexual characteristics and menses in young girls seen in office practice: a study from the Pediatric Research in Office Settings network. Pediatrics. 1997;99:505.
Hill M. Endocrine Development-Hypothalamus. Sydney: UNSW Embryology ebook; 2009.
Holder J.L.Jr, Butte N.F., Zinn A.R. Profound obesity associated with a balanced translocation that disrupts the SIM1 gene. Hum Mol Genet. 2000;9:101-108.
Huang E.A., Feldman B.J., Schwartz I.D., et al. Oral urea for the treatment of chronic syndrome of inappropriate antidiuresis in children. J Pediatr. 2006;148:128-131.
Jung H., Carmel P., Schwartz M.S., et al. Some hypothalamic hamartomas contain transforming growth factor alpha, a puberty-inducing growth factor, but not luteinizing hormone-releasing hormone neurons. J Clin Endocrinol Metab. 1999;84:4695.
Kalantaridou S.N., Chrousos G.P. Clinical review 148: Monogenic disorders of puberty. J Clin Endocrinol Metab. 2002;87:2481.
Kaplowitz P.B. Treatment of central precocious puberty. Curr Opin Endocrinol Diabetes Obes. 2009;16:31.
Karges B., LeHeup B., Schoenle E., et al. Compound Heterozygous and Homozygous Mutations of the TSHbeta Gene as a Cause of Congenital Central Hypothyroidism in Europe. Horm Res. 2004;62:149.
Kelberman D., Dattani M.T. Role of transcription factors in midline central nervous system and pituitary defects. Endocr Rev. 2009;14:67.
Kerrigan J.R., Veldhuis J.D., Leyo S.A., et al. Estimation of daily cortisol production and clearance rates in normal pubertal males by deconvolution analysis. J Clin Endocrinol Metab. 1993;76:1505.
King J.C., Anthony E.L., Fitzgerald D.M., et al. Luteinizing hormone-releasing hormone neurons in human preoptic/hypothalamus: differential intraneuronal localization of immunoreactive forms. J Clin Endocrinol Metab. 1985;60:88.
Korner J., Leibel R.L. To eat or not to eat – how the gut talks to the brain. N Engl J Med. 2003;349:926.
Krams M., Quinton R., Ashburner J., et al. Kallmann’s syndrome: mirror movements associated with bilateral corticospinal tract hypertrophy. Neurology. 1999;52:816.
Krude H., Biebermann H., Gruters A. Mutations in the human proopiomelanocortin gene. Ann N Y Acad Sci. 2003;994:233.
Krude H., Biebermann H., Luck W., et al. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet. 1998;19:155.
Kunwar S., Wilson C.B. Pediatric pituitary adenomas. J Clin Endocrinol Metab. 1999;84:4385.
Kunwar S., Wilson C.B. Sellar and Parasellar Tumors in Children, ed 4. Philadelphia: Saunders; 2001.
Licinio J., Caglayan S., Ozata M., et al. Phenotypic effects of leptin replacement on morbid obesity, diabetes mellitus, hypogonadism, and behavior in leptin-deficient adults. Proc Natl Acad Sci USA. 2004;101:4531.
Linder B.L., Esteban N.V., Yergey A.L., et al. Cortisol production rate in childhood and adolescence. J Pediatr. 1990;117:892.
Lindsay R., Feldkamp M., Harris D., et al. Utah Growth Study: growth standards and the prevalence of growth hormone deficiency. J Pediatr. 1994;125:29.
Lowell B.B., Spiegelman B.M. Towards a molecular understanding of adaptive thermogenesis. Nature. 2000;404:652-660.
Lubrano-Berthelier C., Dubern B., Lacorte J.M., et al. Melanocortin 4 receptor mutations in a large cohort of severely obese adults: prevalence, functional classification, genotype-phenotype relationship, and lack of association with binge eating. J Clin Endocrinol Metab. 2006;91:1811-1818.
Lustig R.H. Autonomic dysfunction of the th Cranial Endocrinopathy. The Endocrinologist. 12, 2002. 210ne and Metabolic Disease 2003;4:23–32
Lustig R.H. Hypothalamic Obesity: The Sixth Cranial Endocrinopathy. The Endocrinologist. 2002;12:210.
Lustig R.H., Post S.R., Srivannaboon K., et al. Risk factors for the development of obesity in children surviving brain tumors. J Clin Endocrinol Metab. 2003;88:611.
Lustig R.H., Rose S.R., Burghen G.A., et al. Hypothalamic obesity caused by cranial insult in children: altered glucose and insulin dynamics and reversal by a somatostatin agonist. J Pediatr. 1999;135:162.
Machinis K., Pantel J., Netchine I., et al. Syndromic short stature in patients with a germline mutation in the LIM homeobox LHX4. Am J Hum Genet. 2001;69:961.
Maghnie M., Cosi G., Genovese E., et al. Central diabetes insipidus in children and young adults. N Engl J Med. 2000;343:998.
Magiakou M.A., Chrousos G.P. Cushing’s syndrome in children and adolescents: current diagnostic and therapeutic strategies. J Endocrinol Invest. 2002;25:181.
Magiakou M.A., Mastorakos G., Chrousos G.P. Final stature in patients with endogenous Cushing’s syndrome. J Clin Endocrinol Metab. 1994;79:1082.
Mahachoklertwattana P., Kaplan S.L., Grumbach M.M. The luteinizing hormone-releasing hormone-secreting hypothalamic hamartoma is a congenital malformation: natural history. J Clin Endocrinol Metab. 1993;77:118.
McDermott M.T., Haugen B.R., Black J.N., et al. Congenital isolated central hypothyroidism caused by a “hot spot” mutation in the thyrotropin-beta gene. Thyroid. 2002;12:1141.
Metzger D.L., Wright N.M., Veldhuis J.D., et al. Characterization of pulsatile secretion and clearance of plasma cortisol in premature and term neonates using deconvolution analysis. J Clin Endocrinol Metab. 1993;77:458.
Miller W.L. The molecular basis of premature adrenarche: an hypothesis. Acta Paediatr Suppl. 1999;88:60.
Mindermann T., Wilson C.B. Pituitary adenomas in childhood and adolescence. J Pediatr Endocrinol Metab. 1995;8:79.
Montague C.T., Farooqi I.S., Whitehead J.P., et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387:903.
Mootha S.L., Barkovich A.J., Grumbach M.M., et al. Idiopathic hypothalamic diabetes insipidus, pituitary stalk thickening, and the occult intracranial germinoma in children and adolescents. J Clin Endocrinol Metab. 1997;82:1362.
Morello J.P., Bichet D.G. Nephrogenic diabetes insipidus. Annu Rev Physiol. 2001;63:607.
Nelson J.C., Clark S.J., Borut D.L., et al. Age-related changes in serum free thyroxine during childhood and adolescence. J Pediatr. 1993;123:899.
Netchine I., Sobrier M.L., Krude H., et al. Mutations in LHX3 result in a new syndrome revealed by combined pituitary hormone deficiency. Nat Genet. 2000;25:182.
O’Rahilly S., Farooqi I.S. Human obesity as a heritable disorder of the central control of energy balance. Int J Obes. 2008;32:S55-S61.
O’Rahilly S., Gray H., Humphreys P.J., et al. Brief report: impaired processing of prohormones associated with abnormalities of glucose homeostasis and adrenal function. N Engl J Med. 1995;333:386-390.
Oakley A.E., Clifton D.K., Steiner R.A. Kisspetin signaling in the brain. Endocr Rev. 2009;30:713.
Ono M., Miki N., Kawamata T., et al. Prospective Study of High-Dose Cabergoline Treatment of Prolactinomas in 150 Patients. J Clin Endocrinol Metab. 2008;93:4721.
Pansky B., Allen D.J., Budd C.C. Hypothalamic Fiber Connections and Nuclear Summary, ed 2. New York: Macmillan Publishing Company; 1988.
Papanicolaou D.A., Yanovski J.A., Cutler G.B.Jr, et al. A single midnight serum cortisol measurement distinguishes Cushing’s syndrome from pseudo-Cushing states. J Clin Endocrinol Metab. 1998;83:1163.
Pescovitz O.H., Comite F., Hench K., et al. The NIH experience with precocious puberty: diagnostic subgroups and response to short-term luteinizing hormone releasing hormone analogue therapy. J Pediatr. 1986;108:47.
Pivonello R., Matrone C., Filippella M., et al. Dopamine receptor expression and function in clinically nonfunctioning pituitary tumors: comparison with the effectiveness of cabergoline treatment. J Clin Endocrinol Metab. 2004;89:1674.
Pulichino A.M., Vallette-Kasic S., Couture C., et al. Human and mouse TPIT gene mutations cause early onset pituitary ACTH deficiency. Genes Dev. 2003;17(6):711-716.
Ranadive S.A., Rosenthal S.M. Pediatric disorders of water balance. Endocrinol Metab Clin North Am. 2009;38:663-672.
Ranadive S.A., Vaisse C. Lessons from extreme human obesity: Monogenic disorders. Endocrinol Metab Clin North Am. 2008;37:733-751.
Refetoff S. Inherited thyroxine-binding globulin abnormalities in man. Endocr Rev. 1989;10:275.
Rizzoti K., Brunelli S., Carmignac D., et al. SOX3 is required during the formation of the hypothalamo-pituitary axis. Nat Genet. 2004;36:247.
Robertson G.L. Antidiuretic hormone. Normal and disordered function. Endocrinol Metab Clin North Am. 2001;30:671.
Robinson A.G., Verbalis J.G. Posterior Pituitary Gland, ed 10. Saunders; 2003.
Rosenfeld J.V., Harvey A.S., Wrennall J., et al. Transcallosal resection of hypothalamic hamartomas, with control of seizures, in children with gelastic epilepsy. Neurosurgery. 2001;48:108.
Rugarli E.I., Ballabio A. Kallmann syndrome. From genetics to neurobiology. JAMA. 1993;270:2713.
Rutishauser J., Kopp P., Gaskill M.B., et al. Clinical and molecular analysis of three families with autosomal dominant neurohypophyseal diabetes insipidus associated with a novel and recurrent mutations in the vasopressin-neurophysin II gene. Eur J Endocrinol. 2002;146:649.
Sadler T.W. Central Nervous System, ed 6. Baltimore: Williams & Wilkins; 1990.
Schrier R.W., Cadnapaphornchai M.A. Renal aquaporin water channels: from molecules to human disease. Prog Biophys Mol Biol. 2003;81:117.
Schwartz M.W., Woods S.C., Porte D., et al. Central nervous system control of food intake. Nature. 2000;404:661-671.
Schwartz M.W., Woods S.C., Seeley R.J., et al. Is the energy homeostasis system inherently biased toward weight gain? Diabetes. 2003;52:232.
Seminara S.B., Messager S., Chatzidaki E.E., et al. The GPR54 gene as a regulator of puberty. N Engl J Med. 2003;349:1614.
Semple R.K., Achermann J.C., Ellery J., et al. Two novel missense mutations in g protein-coupled receptor 54 in a patient with hypogonadotropic hypogonadism. J Clin Endocrinol Metab. 2005;90:1849.
Shaikh M.G., Grundy R.G., Kirk J.M. Reductions in basal metabolic rate and physical activity contribute to hypothalamic obesity. J Clin Endocrinol Metab. 2008;93:2588-2593.
Sklar C.A., Grumbach M.M., Kaplan S.L., et al. Hormonal and metabolic abnormalities associated with central nervous system germinoma in children and adolescents and the effect of therapy: report of 10 patients. J Clin Endocrinol Metab. 1981;52:9.
Spanakis E., Milord E., Gragnoli C. AVPR2 variants and mutations in nephrogenic diabetes insipidus: review and missense mutation significance. J Cell Physiol. 2008;217:605-617.
Stanfield J.P. The blood supply of the human pituitary gland. J Anat. 1960;94:257.
Teles M.G., Bianco S.D., Brito V.N., et al. A GPR54-activating mutation in a patient with central precocious puberty. N Engl J Med. 2008;358:709.
Thomas P.Q., Dattani M.T., Brickman J.M., et al. Heterozygous HESX1 mutations associated with isolated congenital pituitary hypoplasia and septo-optic dysplasia. Hum Mol Genet. 2001;10:39.
Thorner M.O., Perryman R.L., Cronin M.J., et al. Somatotroph hyperplasia. Successful treatment of acromegaly by removal of a pancreatic islet tumor secreting a growth hormone-releasing factor. J Clin Invest. 1982;70:965.
Tolis G., Bird C., Bertrand G., et al. Pituitary hyperthyroidism. Case report and review of the literature. Am J Med. 1978;64:177.
Topaloglu A.K., Reimann F., Guclu M., et al. TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for Neurokinin B in the central control of reproduction. Nat Genet. 2009;41:354.
Trarbach E.B., Costa E.M., Versiani B., et al. Novel fibroblast growth factor receptor 1 mutations in patients with congenital hypogonadotropic hypogonadism with and without anosmia. J Clin Endocrinol Metab. 2006;91:4006.
Vaisse C., Clement K., Durand E., et al. Melanocortin-4 receptor mutations are a frequent and heterogeneous cause of morbid obesity. J Clin Invest. 2000;106:253.
Valdueza J.M., Cristante L., Dammann O., et al. Hypothalamic hamartomas: with special reference to gelastic epilepsy and surgery. Neurosurgery. 1994;34:949.
Vallette-Kasic S., Couture C., Balsalobre A., et al. The TPIT gene mutation M86R associated with isolated adrenocorticotropin deficiency interferes with protein: protein interactions. J Clin Endocrinol Metab. 2007;92:3991.
Vallette-Kasic S., Figarella-Branger D., Grino M., et al. Differential regulation of proopiomelanocortin and pituitary-restricted transcription factor (TPIT), a new marker of normal and adenomatous human corticotrophs. J Clin Endocrinol Metab. 2003;88:3050.
Verbalis J.G. Diabetes insipidus. Rev Endocr Metab Disord. 2003;4:177.
Verbalis J.G., Goldsmith S.R., Greenberg A., et al. Hyponatremia treatment guidelines 2007: expert panel recommendations. Am J Med. 2007;120(11 Suppl 1):S1-S21.
Verhelst J., Abs R., Maiter D., et al. Cabergoline in the Treatment of Hyperprolactinemia: A Study in 455 Patients. J Clin Endocrinol Metab. 1999;84:2518.
Willcutts M.D., Felner E., White P.C. Autosomal recessive familial neurohypophyseal diabetes insipidus with continued secretion of mutant weakly active vasopressin. Hum Mol Genet. 1999;8:1303.
Wu W., Cogan J.D., Pfaffle R.W., et al. Mutations in PROP1 cause familial combined pituitary hormone deficiency. Nat Genet. 1998;18:147.
Yeo G.S., Connie Hung C.C., Rochford J., et al. A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat Neurosci. 2004;7:1187-1189.