[level-membership-for-critical-care-medicine-category]Chapter 33
Endocrine Disorders and Therapeutic Management
Neuroendocrinology of Stress and Critical Illness
Major neurologic and endocrine changes occur when an individual is confronted with physiologic stress caused by any critical illness, sepsis,1 trauma, major surgery, or underlying cardiovascular disease.2 The normal “fight or flight” response that is initiated in times of physiologic or psychologic stress is exacerbated in critical illness through activation of the neuroendocrine system, specifically the hypothalamic-pituitary-adrenal axis (HPA).3 All endocrine organs are affected by acute critical illness, as shown in Table 33-1.
TABLE 33-1
| GLAND OR ORGAN | HORMONE | RESPONSE OR PHYSICAL EXAMINATION |
| Adrenal cortex | Cortisol | ↑ Insulin resistance → ↑ glycogenolysis → ↑ glucose circulation |
| ↑ Hepatic gluconeogenesis → ↑ glucose available | ||
| ↑ Lipolysis | ||
| ↑ Protein catabolism | ||
| ↑ Sodium → ↑ water retention to maintain plasma osmolality by movement of extravascular fluid into the intravascular space | ||
| ↓ Connective tissue fibroblasts → poor wound healing | ||
| Glucocorticoid | ↓ Histamine release → suppression of immune system | |
| ↓ Lymphocytes, monocytes, eosinophils, basophils | ||
| ↑ Polymorphonuclear leukocytes → ↑ infection risk | ||
| ↑ Glucose | ||
| ↓ Gastric acid secretion | ||
| Mineralocorticoids | ↑ Aldosterone → ↓ sodium excretion → ↓ water excretion → ↑ intravascular volume | |
| ↑ Potassium excretion → hypokalemia | ||
| ↑ Hydrogen ion excretion → metabolic acidosis | ||
| Adrenal medulla | Epinephrine | ↑ Endorphins → ↓ pain |
| Norepinephrine, epinephrine | ↑ Metabolic rate to accommodate stress response | |
| ↑ Live glycogenolysis → ↑ glucose | ||
| ↑ Insulin (cells are insulin resistant) | ||
| ↑ Cardiac contractility | ||
| ↑ Cardiac output | ||
| ↑ Dilation of coronary arteries | ||
| ↑ Blood pressure | ||
| ↑ Heart rate | ||
| ↑ Bronchodilation → ↑ respirations | ||
| ↑ Perfusion to heart, brain, lungs, liver, and muscle | ||
| ↓ Perfusion to periphery of body | ||
| ↓ Peristalsis | ||
| Norepinephrine | ↑ Peripheral vasoconstriction | |
| ↑ Blood pressure | ||
| ↑ Sodium retention | ||
| ↑ Potassium excretion | ||
| Pituitary | All hormones | ↑ Endogenous opioids → ↓ pain |
| Anterior pituitary | Corticotropin | ↑ Aldosterone → ↓ sodium excretion → ↓ water excretion → ↑ intravascular volume |
| ↑ Cortisol → ↑ blood volume | ||
| Growth hormones | ↑ Protein anabolism of amino acids to protein | |
| ↑ Lipolysis → ↑ gluconeogenesis | ||
| Posterior pituitary | Antidiuretic hormone | ↑ Vasoconstriction |
| ↑ Water retention → restoration of circulating blood volume | ||
| ↓ Urine output | ||
| ↑ Hypo-osmolality | ||
| Pancreas | Insulin | ↑ Insulin resistance → hyperglycemia |
| Glucagon | ↑ Glycolysis (directly opposes action of insulin) | |
| ↑ Glucose for fuel | ||
| ↑ Glycogenolysis | ||
| ↑ Gluconeogenesis | ||
| ↑ Lipolysis | ||
| Thyroid | Thyroxine | ↓ Routine metabolic demands during stress |
| Gonads | Sex hormones | Energy and oxygen supply diverted to brain, heart, muscles, and liver |
Acute Neuroendocrine Response to Critical Illness
The fight or flight acute response to physiologic threat is a rapid discharge of the catecholamines norepinephrine and epinephrine into the bloodstream.3 Norepinephrine is released from the nerve endings of the sympathetic nervous system (SNS).
Hypothalamic-Pituitary-Adrenal Axis in Critical Illness
Epinephrine (adrenalin) is released from the medulla of the adrenal glands. Epinephrine increases cerebral blood flow and cerebral oxygen consumption and may be the trigger for recruitment of the hypothalamic-pituitary axis.3
The pituitary gland has two parts (anterior and posterior) that function under control of the hypothalamus, as described in Chapter 31.
The posterior pituitary gland releases antidiuretic hormone (ADH), also known as vasopressin (pitressin), as a component of the physiologic stress response. This hormone is an antidiuretic with a powerful vasoconstrictive effect on blood vessels.3 The combination of epinephrine and vasopressin quickly raises blood pressure; it also decreases gastric motility.3 Epinephrine increases heart rate, causes ventricular dysrhythmias in susceptible patients, and provides some analgesia or lack of pain awareness during acute physical stress.3
The anterior pituitary gland produces several hormones including corticotropin (also called ACTH), which stimulates release of cortisol from the adrenal cortex.4 Cortisol release is an important protective response to stress. Increased cortisol levels alter carbohydrate, fat, and protein metabolism so that energy is immediately and selectively available to vital organs such as the brain. However, if critical illness is prolonged, the HPA may not be able to respond adequately to prolonged physiologic stress.
• Primary hypoadrenalism describes an intrinsic failure of the adrenal gland to produce normal endogenous glucocorticosteroid hormones (e.g., cortisol) and mineralocorticosteroid hormones (e.g., aldosterone). Primary adrenal failure is rare.
• Secondary adrenal dysfunction, or Cushing syndrome, occurs as a result of the administration of therapeutic steroids. In response to exogenous glucocorticosteroids, the adrenal glands stop production of intrinsic hormones. Patients who have taken steroids before their admission to the hospital need their dosage increased during illness.
• Critical illness—related corticosteroid insufficiency (CIRCI) describes a situation in which the adrenal gland produces glucocorticosteroids but the quantity is insufficient for the disease process. Peripheral cortisol resistance occurs as inflammatory cytokines induce cellular resistance to cortisol.5
Cosyntropin Stimulation Test
Further confirmation of adrenal dysfunction may be obtained by performance of a corticotropin stimulation test (cosyntropin test). Cosyntropin is a medication made from the first 24 amino acids of corticotropin. In the test, 250 mcg cosyntropin is administered by the intravenous (IV) route, and serum blood levels are measured 30 minutes later. A serum cortisol rise from baseline of less than 9 mcg/dL after 30 minutes denotes inability of the adrenal gland to respond to a stress stimulus (nonresponder).5 If the cortisol rise is greater than 9 mcg/dL in response to corticotropin stimulation, the adrenal glands are functioning normally (responder).5 Corticosteroids are given only to nonresponders. The combination of a low baseline cortisol value (less than 10 mcg/dL) with minimal or no rise (less than 9 mcg/dL) after cosyntropin stimulation is evidence of adrenal gland dysfunction with corticosteroid deficiency.5
Corticosteroid Replacement
Clinical practice guidelines5 recommend short-term provision of low-dose hydrocortisone for patients who have a diagnosis of septic shock with refractory vasopressor-dependent hypotension. Hydrocortisone is the recommended replacement because it is the pharmacologic steroid that most resembles endogenous cortisol.
The guidelines recommend use of the cosyntropin stimulation test as described previously. However, the test is not recommended as a stand-alone method to identify patients who might receive low-dose steroids. This apparent contradiction is explained by the fact that several clinical trials of low-dose steroid replacement in sepsis have demonstrated a faster resolution of the shock symptoms but no difference in overall mortality compared with placebo.5 High-dose steroid replacement is never recommended in the management of sepsis. Corticosteroids are never discontinued abruptly and must be tapered gradually over several days.5
Liver and Pancreas in Critical Illness
The liver releases the hormone glucagon to stimulate the liver to pour additional glucose into the bloodstream in response to physiologic stress. Glucagon rapidly raises blood glucose levels. Peripheral tissues may become insulin resistant, meaning the tissues are unable to use the available insulin to transport glucose inside the cells. This further raises blood glucose levels, causing persistent stress-induced hyperglycemia.6 There is a second metabolic system that enables insulin to enter the cell (see Chapter 31). The insulin-independent glucose transporters (GLUT 1, GLUT 2, and GLUT 3) are active during physiologic stress, but may be unable to keep up with the massive increase in glucose production by the liver. Continuous infusion of insulin to return and maintain blood glucose levels within a safe, near-normal range reduces morbidity and mortality.2
Hyperglycemia in Critical Illness
Normal fasting blood glucose levels range between 70 and 100 mg/dL in a healthy person. Critically ill patients frequently have much higher blood glucose levels, and several retrospective analyses have reported that hyperglycemic patients have a higher mortality rate than patients with normal blood glucose values. In 2001, a landmark prospective, randomized study showed a significant reduction in morbidity and mortality among critically ill surgical patients whose blood glucose concentration was maintained between 80 and 110 mg/dL with a continuous insulin infusion, compared with those whose blood glucose was only treated if it was greater than 180 mg/dL.7 A study of medical critical care patients by the same group with the same protocol demonstrated a survival benefit after 3 days of tight glucose control with a continuous insulin infusion.8 These initial studies were greeted with tremendous enthusiasm and many critical care units adopted stringent glucose control standards to reduce hyperglycemia-associated morbidity and mortality. However, achievement of such tight glucose control outside of a research trial can be challenging as shown by the results of more recent clinical trials.
The NICE-SUGAR trial was a prospective randomized trial of 6014 critically ill patients. It compared continuous insulin infusion to achieve tight glucose control (target 81 to 108 mg/dL) with a conventional glucose control range (target below 180 mg/dL).9 In the tight glucose control group 6.8% had episodes of severe hypoglycemia (below 40 mg/dL); in the conventional control group only 0.5% experienced severe hypoglycemia.9 There was a 2.6% higher risk of death in the intensive glucose control group (27.5% died) compared with the conventional control group (24.9% died).9
Clinical Practice Guidelines Related to Blood Glucose Management in Critically Ill Patients
As a result of the studies just described, clinical practice guidelines were developed by the American Association of Clinical Endocrinologists (AACE) and the American Diabetes Association (ADA) that recommend the use of continuous insulin infusions to maintain blood glucose in critical care patients between 140 and 180 mg/dL, with frequent monitoring of blood glucose.2 The 140 to 180 mg/dL level was selected to minimize the risk of hypoglycemia.
Other glucose-control guidelines relevant to critical illness have also been published. The Society of Critical Care Medicine (SCCM) recommends initiating glycemic control when the blood glucose rises above 150 mg/dL.1,10 Insulin management must be initiated if the blood glucose level is above 180 mg/dL.2,10 More liberal glucose control represents the current trend of targeted values.
Insulin Management in the Critically Ill
As a result of the research that has highlighted the deleterious effects of hyperglycemia in critical illness, most hospitals have developed an institution-specific glucose–insulin algorithm to lower blood glucose into the targeted range.11 The vigilance of the critical care nurse is pivotal to the success of any intervention to lower blood glucose using a continuous insulin infusion. As discussed earlier, many glucose control protocols are using less restrictive ranges due to concerns about iatrogenic hypoglycemia.
Frequent Blood Glucose Monitoring
Monitoring the blood glucose with a point-of-care glucometer is the basis of targeted glucose control. As part of the comprehensive initial assessment, the blood sugar is measured by a standard laboratory sample or by a finger-stick capillary blood sample. In many institutions, if the blood sugar is greater than 180 mg/dL, the patient is started on a continuous IV insulin infusion. While the glucose is elevated, blood sample measurements are usually obtained hourly, to allow titration of the insulin drip to lower blood glucose.11 After the patient is stable, blood glucose measurements can be spaced approximately every 2 hours, based on individual hospital protocols.
Continuous Insulin Infusion
Many hospitals use insulin infusion protocols for management of stress-induced hyperglycemia that are implemented by the critical care nurse.2,11 Effective glucose protocols gauge the insulin infusion rate based on two parameters: 1) the immediate blood glucose result and 2) the rate of change in the blood glucose level since the last hourly measurement. The following three examples illustrate this concept:
• Patient A receives 3 units of continuous IV regular insulin per hour and has a blood glucose measurement of 110 mg/dL, but 1 hour ago it was 190 mg/dL; the insulin rate must be decreased to avoid sudden hypoglycemia.
• Patient B receives 3 units of continuous IV regular insulin per hour and has a blood glucose measurement of 110 mg/dL, but 1 hour ago it was 112 mg/dL; in this situation, no change is made in the insulin infusion rate.
• Patient C receives 3 units of continuous IV regular insulin per hour and has a blood glucose measurement of 190 mg/dL, and 1 hour ago it was 197 mg/dL; the insulin rate must be increased to more rapidly move the patient’s blood sugar toward the targeted glucose range (i.e., 140 to 180 mg/dL, although this range will vary by individual hospital protocol).
The important point to emphasize is that the rate of change of the blood glucose is as important as the most recent blood glucose measurement.11 Each of the patients described in the examples may have the same insulin infusion rate, depending on their catabolic state, but individualization among patients with different diagnoses can be safely achieved as long as the rate of change is also considered.
Transition from Continuous to Intermittent Insulin Coverage
The transition from a continuous insulin infusion to intermittent insulin coverage must be handled with care to avoid large fluctuations in blood glucose levels. Before the conversion, the regular insulin infusion should be at a stable and preferably low rate, and the patient’s blood glucose level should be maintained consistently within the target range. The transition from IV to subcutaneous administration depends on numerous factors, including whether the patient is able to eat a consistent amount of dietary carbohydrate.12
Clinicians use various methods to calculate the quantity of insulin to prescribe during the transition from IV to subcutaneous insulin to maintain stable blood glucose levels. Figure 33-1 depicts hypothetical examples of how a combination of basal and bolus insulin regimens (prandial insulin) can work in clinical practice. The following paragraphs describe the application of one calculation method for a 67-year-old patient, Alice Smith, who is recovering from critical illness and has recently been weaned from the ventilator and extubated.
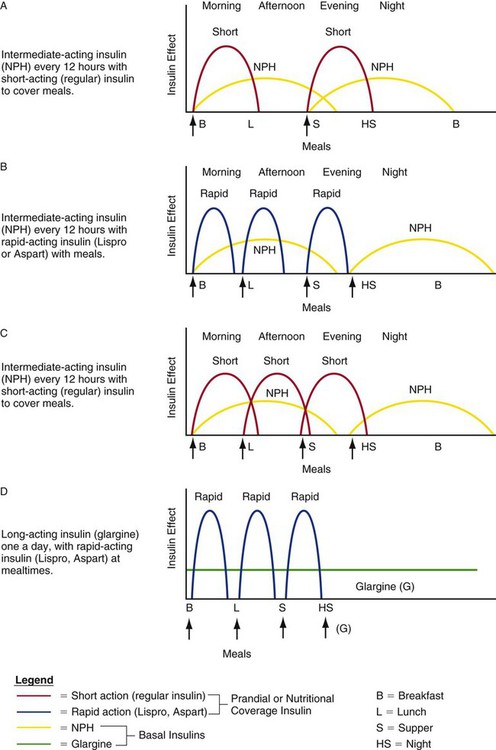
1. Ms. Smith is in stable condition on a regular insulin drip at 1 unit per hour. She is ready to be transitioned to subcutaneous insulin and will be taking food and liquids by mouth. Her total insulin requirement over the previous 24 hours was 32 units. Ms. Smith will now require basal coverage (provided by subcutaneous intermediate or long-acting insulin) and prandial coverage for mealtimes (provided by short-acting subcutaneous insulin).
2. The 30 units of insulin infused during the previous 24 hours is Ms. Smith’s required daily insulin dose. To transition to subcutaneous insulin, a proportion of this amount (i.e., 75% to 80%) will be divided between basal and prandial components.2 In this situation, 75% of the 32 units = 24 units. Half of this amount (12 units) will be administered subcutaneously as intermediate or long-acting insulin; the other half will be administered as short-acting insulin to coincide with meals (i.e., 4 units with each of three meals).
3. The options for insulin administration for Ms. Smith are as follows:
Basal insulin: 12 units of glargine once daily, or 6 units twice daily of Neutral Protamine Hagedorn (NPH) administered subcutaneously
Prandial insulin: 4 units regular insulin given subcutaneously before each meal (short-acting), or 4 units Lispro or Aspart given subcutaneously with each meal (ultra–short-acting insulin); verify current blood glucose level.
Supplemental corrective insulin: A supplemental correction scale can be used to cover any hyperglycemia above the target range, and administration can be combined with scheduled blood glucose measurements; verify current blood glucose level.
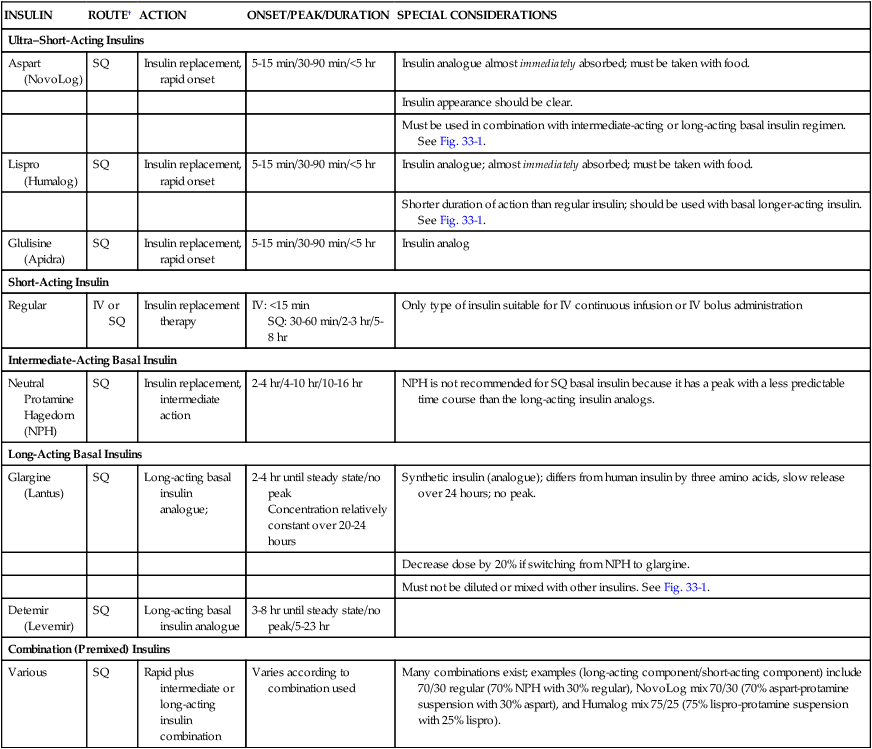
Table 33-2 describes the various types of insulin available for use. These include ultra–short-acting, short-acting, intermediate-acting, long-acting, and combination insulin replacement options.13 Even after the transition to subcutaneous insulin is completed, blood glucose is monitored frequently to maintain blood glucose within the target range and detect hyperglycemia or hypoglycemia.14
TABLE 33-2
PHARMACOLOGIC MANAGEMENT
Insulin*
| INSULIN | ROUTE† | ACTION | ONSET/PEAK/DURATION | SPECIAL CONSIDERATIONS |
| Ultra–Short-Acting Insulins | ||||
| Aspart (NovoLog) | SQ | Insulin replacement, rapid onset | 5-15 min/30-90 min/<5 hr | Insulin analogue almost immediately absorbed; must be taken with food. |
| Insulin appearance should be clear. | ||||
| Must be used in combination with intermediate-acting or long-acting basal insulin regimen. See Fig. 33-1. | ||||
| Lispro (Humalog) | SQ | Insulin replacement, rapid onset | 5-15 min/30-90 min/<5 hr | Insulin analogue; almost immediately absorbed; must be taken with food. |
| Shorter duration of action than regular insulin; should be used with basal longer-acting insulin. See Fig. 33-1. | ||||
| Glulisine (Apidra) | SQ | Insulin replacement, rapid onset | 5-15 min/30-90 min/<5 hr | Insulin analog |
| Short-Acting Insulin | ||||
| Regular | IV or SQ | Insulin replacement therapy | IV: <15 min SQ: 30-60 min/2-3 hr/5-8 hr |
Only type of insulin suitable for IV continuous infusion or IV bolus administration |
| Intermediate-Acting Basal Insulin | ||||
| Neutral Protamine Hagedorn (NPH) |
SQ | Insulin replacement, intermediate action | 2-4 hr/4-10 hr/10-16 hr | NPH is not recommended for SQ basal insulin because it has a peak with a less predictable time course than the long-acting insulin analogs. |
| Long-Acting Basal Insulins | ||||
| Glargine (Lantus) | SQ | Long-acting basal insulin analogue; | 2-4 hr until steady state/no peak Concentration relatively constant over 20-24 hours |
Synthetic insulin (analogue); differs from human insulin by three amino acids, slow release over 24 hours; no peak. |
| Decrease dose by 20% if switching from NPH to glargine. | ||||
| Must not be diluted or mixed with other insulins. See Fig. 33-1. | ||||
| Detemir (Levemir) | SQ | Long-acting basal insulin analogue | 3-8 hr until steady state/no peak/5-23 hr | |
| Combination (Premixed) Insulins | ||||
| Various | SQ | Rapid plus intermediate or long-acting insulin combination | Varies according to combination used | Many combinations exist; examples (long-acting component/short-acting component) include 70/30 regular (70% NPH with 30% regular), NovoLog mix 70/30 (70% aspart-protamine suspension with 30% aspart), and Humalog mix 75/25 (75% lispro-protamine suspension with 25% lispro). |
IV, Intravenous; SQ, subcutaneous.
*Dosages are individualized according to patient’s age and size.
Corrective Insulin Coverage
A patient may be prescribed supplemental or corrective doses of insulin in addition to the basal/prandial insulin combination. The use of the trio of basal, prandial, and corrective insulin is designed to eliminate the use of the traditional sliding scale. Criticisms of the sliding scale method are that the dosages are rarely re-evaluated or adjusted once established and that the scales treat hyperglycemia only after it has occurred; they are not proactive in the manner of the basal/bolus/corrective insulin method.11
Hypoglycemia Management
It is important to have a protocol for the management of hypoglycemia. The major drawback to use of intensive insulin protocols, as described earlier, is the potential for hypoglycemia.14 Whenever hypoglycemia is detected, it is important to stop any continuous infusion of insulin. An example of one protocol to reverse hypoglycemia follows:
Nursing Management
Nursing management of the patient with neuroendocrine stress resulting from critical illness incorporates a variety of nursing diagnoses (Box 33-1). The goals of nursing management are to monitor the hyperglycemic side effects of vasopressor therapy; administer prescribed corticosteroids; monitor blood glucose and insulin effectiveness; avoid hypoglycemia; provide nutrition; and provide education to the patient’s family and supportive others (Box 33-2).
 Box 33-1 Nursing Diagnoses
Box 33-1 Nursing Diagnoses
Stress of Critical Illness
• Deficient Fluid Volume related to absolute loss, p. 1132
• Imbalanced Nutrition: Less Than Body Requirements related to exogenous nutrients and increased metabolic demand, p. 1143
• Risk for Ineffective Peripheral Tissue Perfusion, p. 1158
• Compromised Family Coping related to critically ill family member, p. 1127
Administer Prescribed Corticosteroids
Critically ill patients with below-normal cortisol levels may be prescribed IV hydrocortisone.1 Therapeutic steroids raise blood glucose levels and can make glycemic control more difficult. Frequent monitoring of the blood glucose concentration is necessary to guide treatment of hyperglycemia in the patient receiving IV corticosteroids. Ongoing monitoring for the presence of new infection is mandatory.
Monitor Blood Glucose, Insulin Effectiveness, Avoid Hypoglycemia
The critical care nurse is responsible for the hourly monitoring of blood glucose and titration of the insulin infusion according to the hospital’s protocol while the patient is hyperglycemic. The use of standardized protocols makes possible a systematic approach to the control of blood glucose. It is essential and recommended that nurses receive effective and ongoing education about the anabolic impact of insulin therapy in critical illness.2 Hospital protocols to minimize development of hypoglycemia, such as using the 140-180 mg/dL target range,2 and rapid reversal of any occurrence of severe hypoglycemia (below 40 mg/dL) by provision of IV dextrose (D50W) are essential.10
Provide Nutrition
Whenever an insulin infusion is started to lower blood glucose, enteral nutritional support should be considered. In the absence of nutrition, a 10% dextrose solution may temporarily be infused. The 10% dextrose offers the advantage of carbohydrate calories for metabolism, limits fluctuations in the blood sugar, and reduces the risk of hypoglycemia. After the patient’s metabolic condition is stable, introduction of non-glucose nutrition (protein and fat) is recommended.15
Patient Education
While the patient is acutely ill, most of the educational interventions are directed to the family at the bedside. Numerous explications are required to describe the IV medications, the nutritional needs, the purpose of insulin, the role of steroids (if applicable), the ongoing nursing care, prevention of complications, and management of the underlying disease process. Educational issues that may be discussed are listed in the Box 33-2.
Collaborative Management
It is well established that standardized protocols designed to manage the complications of critical illness result in lower morbidity and mortality for patients. Optimally, all disciplines concerned with the endocrine status of the patient will have participated in the hospital’s guidelines related to targeted glucose control. Guidelines for blood glucose monitoring in critical illness are described in Box 33-3.2,16
Box 33-3 Evidence-Based Practice
Hyperglycemia Management in Critical Illness
 Summary of evidence and evidence-based recommendations for controlling hyperglycemic symptoms related to physiologic stress of critical illness
Summary of evidence and evidence-based recommendations for controlling hyperglycemic symptoms related to physiologic stress of critical illness• Initiate insulin therapy for persistent hyperglycemia above 180 mg/dL.
• Once a continuous insulin infusion is initiated, maintain the target blood glucose level between 140 and 180 mg/dL.
• Frequent blood glucose monitoring to avoid hypoglycemia (hypoglycemia is defined as a blood glucose below 70 mg/dL; severe hypoglycemia <40 mg/dL).
• For patients who are eating, maintain preprandial blood glucose below 130 mg/dL; maintain 2-hour postprandial blood glucose below 180 mg/dL.
• A multidisciplinary team approach to implement institutional guidelines, protocols, and standardized order-sets results in fewer hypoglycemic and hyperglycemic events.
Diabetes Mellitus
Diagnosis of Diabetes
Diabetes mellitus is diagnosed by measurement of the fasting plasma glucose (FPG) or by a glycated hemoglobin above 6.5%.16 The blood glucose may also be called a fasting blood glucose (FBG). The benchmarks for a normal blood glucose value have been progressively lowered as more knowledge has been gained about the benefits of maintaining the plasma glucose level as close to normal as possible.
Blood glucose values endorsed by the ADA are as follows16:
• An FPG level 70 to 100 mg/dL (5.6 mmol/L) signifies normal fasting glucose.
• An FPG level between 100 and 125 mg/dL (5.6 and 6.9 mmol/L) denotes impaired fasting glucose (IFG).
• An FPG level greater than 126 mg/dL (7 mmol/L) provides a diagnosis of diabetes (result is verified by testing more than once).
Types of Diabetes
Two distinct types of diabetes are discussed in this chapter17:
• Type 1 diabetes results from beta-cell destruction, usually leading to absolute insulin deficiency.
• Type 2 diabetes results from a progressive insulin secretory defect in addition to insulin resistance.
The two diseases are different in nature, cause, treatment, and prognosis. A further category of prediabetes describes patients with impaired fasting glucose (FPG between 100 and 125 mg/dL) who are likely to develop diabetes in the future and who are at increased risk for coronary artery disease and stroke.16 Other conditions, such as gestational diabetes, are not discussed in this chapter.
Glycated Hemoglobin
For individuals with diabetes, maintenance of blood glucose within a tight normal range is fundamental to avoid the development of microvascular and neuropathic secondary conditions. Although the plasma glucose produces a snapshot of the blood glucose concentration at a single point in time, the glycated hemoglobin (HbA1C), also known as a glycosylated hemoglobin, measures the percentage of glucose the red cells have absorbed from the plasma over the previous 3-month period. The optimal target for patients with diabetes is an A1C value below 6.5%.16
Type 1 Diabetes
Type 1 diabetes mellitus accounts for only about 5% to 10% of the diabetic population.17 Older names for this condition included insulin-dependent diabetes (IDDM) and juvenile diabetes. Type 1 diabetes is an autoimmune disease that causes progressive destruction of the beta cells of the islets of Langerhans in the pancreas. Autoantibodies that falsely identify self as a foreign invader to be destroyed can now be identified by laboratory analysis. Autoantibodies that contribute to pancreatic destruction include autoantibodies to the islet cell, to insulin, to glutamic acid decarboxylase (GAD65), and to the tyrosine phosphatases IA-2 and IA-2β (beta).17 One or more of these autoantibodies are present in 85% to 90% of individuals with type 1 diabetes when fasting hyperglycemia is initially detected.17 Over time, the autoantibodies render the pancreatic beta cells incapable of secreting insulin and regulating intracellular glucose. In type 1 diabetes, the rate of beta-cell destruction is highly variable. It occurs rapidly in some individuals (mainly children) and slowly in others (mainly adults). Some patients, particularly children and adolescents, may have ketoacidosis as the first manifestation of their disease.
Genetic predisposition and unknown environmental factors are also believed to play an important role.17 Patients with type 1 diabetes are prone to development of other autoimmune disorders such as Graves disease (hyperthyroidism), Hashimoto thyroiditis, Addison disease, autoimmune hepatitis, myasthenia gravis, and pernicious anemia.17 Lack of insulin impairs carbohydrate, protein, and fat metabolism.
Management of Type 1 Diabetes
Patients with type 1 diabetes must receive IV or subcutaneous insulin therapy. Treatment with exogenous insulin replacement restores normal entry of glucose into the cells. The range of insulin replacements available is expanding, and it is essential that critical care nurses be knowledgeable about this class of medications (see Table 33-2). Without insulin, the rapid breakdown of noncarbohydrate substrate, particularly fat, leads to ketonemia, ketonuria, and diabetic ketoacidosis (DKA), a life-threatening complication associated with type 1 diabetes (see later discussion).
Type 2 Diabetes
Type 2 diabetes accounts for 90% to 95% of those with diabetes.17 Previously used names for this condition were non–insulin-dependent diabetes, type II diabetes, or adult-onset diabetes.17 Type 2 diabetes is identified by insulin resistance with a relative, versus absolute, insulin deficiency. Most patients with type 2 diabetes are obese, with excess adipose tissue concentrated in the abdominal area. The onset of hyperglycemia occurs gradually and many people are unaware that they have diabetes. Initially, type 2 diabetes is managed by oral medications (noninsulin therapies) because the pancreatic beta cells remain functional. Although, as progressive beta cell dysfunction occurs, a basal long-acting insulin is added to the oral noninsulin medications.18,19
Lifestyle Management for Type 2 Diabetes
For most patients with type 2 diabetes, a program of weight reduction, increased physical exercise, and a change in diet pattern is the first step.18 The diet should contain less than 30% of calories from fat, with an increased quantity of whole grains, vegetables, and fruits. Crash diets are discouraged, and a gradual program of weight loss, if needed, is recommended. The exercise program is tailored to the individual but might start with 30 minutes of brisk walking each day if the person was previously sedentary.
Patients who have type 2 diabetes are prone to a wide range of other complications that increase morbidity and mortality. In addition to medications to control blood glucose, patients often require medications to lower their blood pressure, lower their cholesterol and triglyceride levels, treat ischemic heart disease, and manage symptoms of heart failure.16,19
Pharmacologic Management of Type 2 Diabetes
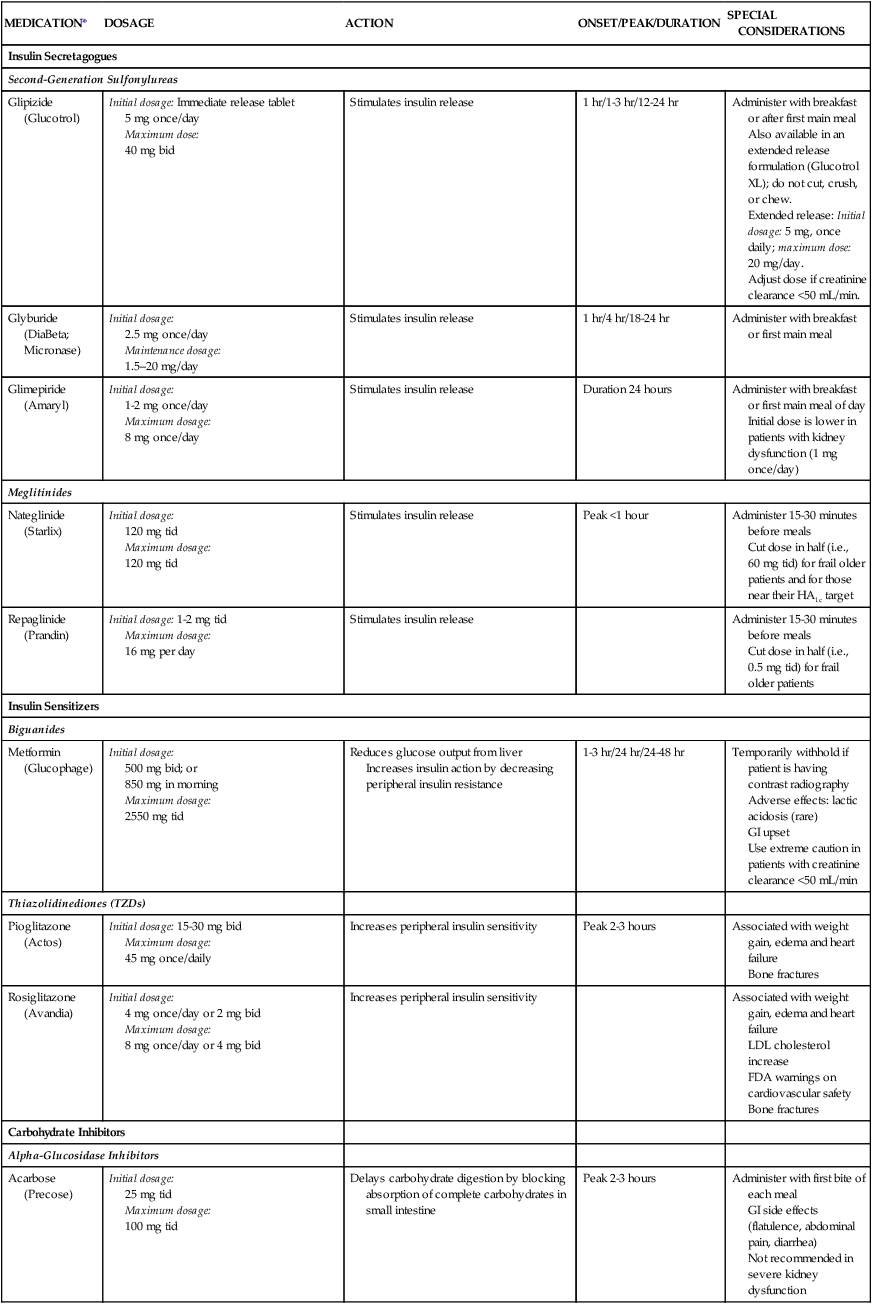
If lifestyle changes are unsuccessful in reversing the pattern of type 2 diabetes, oral noninsulin antihyperglycemic medications are prescribed (Table 33-3).16 These medications are not oral forms of insulin, because insulin would be destroyed by gastric juices. There are several types of oral agents: sulfonylureas, glinides, biguanides, thiazolidinediones, alpha-glucosidase inhibitors, incretin mimetics, and incretin enhancers. These oral antidiabetic medications work to lower plasma glucose levels by a variety of mechanisms: increasing insulin secretion, increasing sensitivity to insulin, and delaying carbohydrate absorption (Box 33-4). The pharmacologic management of type 2 diabetes is increasingly complex. Current guidelines recommend a patient-focused, individualized approach that includes pharmacologic management and risk factor modification to reduce early mortality from cardiovascular disease.19
TABLE 33-3
PHARMACOLOGIC MANAGEMENT
Medications for Type 2 Diabetes
| MEDICATION* | DOSAGE | ACTION | ONSET/PEAK/DURATION | SPECIAL CONSIDERATIONS |
| Insulin Secretagogues | ||||
| Second-Generation Sulfonylureas | ||||
| Glipizide (Glucotrol) | Initial dosage: Immediate release tablet 5 mg once/day Maximum dose: 40 mg bid |
Stimulates insulin release | 1 hr/1-3 hr/12-24 hr | Administer with breakfast or after first main meal Also available in an extended release formulation (Glucotrol XL); do not cut, crush, or chew. Extended release: Initial dosage: 5 mg, once daily; maximum dose: 20 mg/day. Adjust dose if creatinine clearance <50 mL/min. |
| Glyburide (DiaBeta; Micronase) | Initial dosage: 2.5 mg once/day Maintenance dosage: 1.5–20 mg/day |
Stimulates insulin release | 1 hr/4 hr/18-24 hr | Administer with breakfast or first main meal |
| Glimepiride (Amaryl) | Initial dosage: 1-2 mg once/day Maximum dosage: 8 mg once/day |
Stimulates insulin release | Duration 24 hours | Administer with breakfast or first main meal of day Initial dose is lower in patients with kidney dysfunction (1 mg once/day) |
| Meglitinides | ||||
| Nateglinide (Starlix) | Initial dosage: 120 mg tid Maximum dosage: 120 mg tid |
Stimulates insulin release | Peak <1 hour | Administer 15-30 minutes before meals Cut dose in half (i.e., 60 mg tid) for frail older patients and for those near their HA1c target |
| Repaglinide (Prandin) | Initial dosage: 1-2 mg tid Maximum dosage: 16 mg per day |
Stimulates insulin release | Administer 15-30 minutes before meals Cut dose in half (i.e., 0.5 mg tid) for frail older patients |
|
| Insulin Sensitizers | ||||
| Biguanides | ||||
| Metformin (Glucophage) | Initial dosage: 500 mg bid; or 850 mg in morning Maximum dosage: 2550 mg tid |
Reduces glucose output from liver Increases insulin action by decreasing peripheral insulin resistance |
1-3 hr/24 hr/24-48 hr | Temporarily withhold if patient is having contrast radiography Adverse effects: lactic acidosis (rare) GI upset Use extreme caution in patients with creatinine clearance <50 mL/min |
| Thiazolidinediones (TZDs) | ||||
| Pioglitazone (Actos) | Initial dosage: 15-30 mg bid Maximum dosage: 45 mg once/daily |
Increases peripheral insulin sensitivity | Peak 2-3 hours | Associated with weight gain, edema and heart failure Bone fractures |
| Rosiglitazone (Avandia) | Initial dosage: 4 mg once/day or 2 mg bid Maximum dosage: 8 mg once/day or 4 mg bid |
Increases peripheral insulin sensitivity | Associated with weight gain, edema and heart failure LDL cholesterol increase FDA warnings on cardiovascular safety Bone fractures |
|
| Carbohydrate Inhibitors | ||||
| Alpha-Glucosidase Inhibitors | ||||
| Acarbose (Precose) | Initial dosage: 25 mg tid Maximum dosage: 100 mg tid |
Delays carbohydrate digestion by blocking absorption of complete carbohydrates in small intestine | Peak 2-3 hours | Administer with first bite of each meal GI side effects (flatulence, abdominal pain, diarrhea) Not recommended in severe kidney dysfunction |
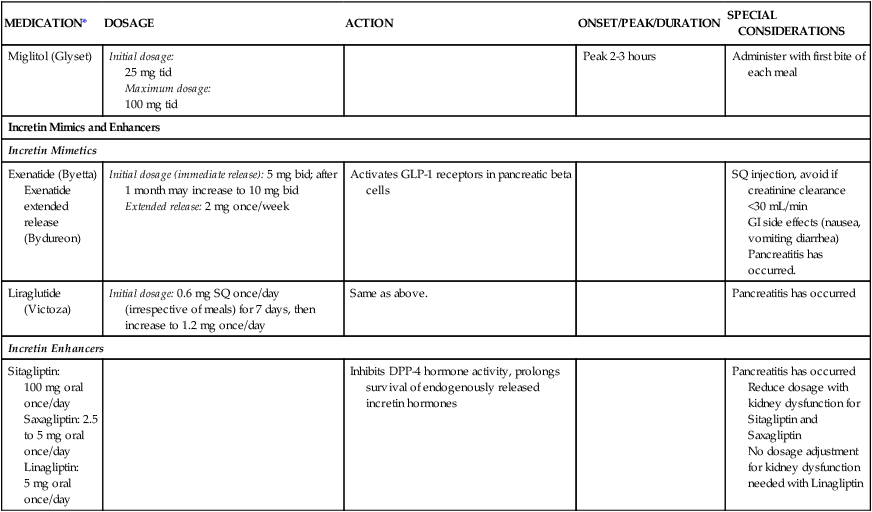
| Miglitol (Glyset) | Initial dosage: 25 mg tid Maximum dosage: 100 mg tid |
Peak 2-3 hours | Administer with first bite of each meal | |
| Incretin Mimics and Enhancers | ||||
| Incretin Mimetics | ||||
| Exenatide (Byetta) Exenatide extended release (Bydureon) |
Initial dosage (immediate release): 5 mg bid; after 1 month may increase to 10 mg bid Extended release: 2 mg once/week |
Activates GLP-1 receptors in pancreatic beta cells | SQ injection, avoid if creatinine clearance <30 mL/min GI side effects (nausea, vomiting diarrhea) Pancreatitis has occurred. |
|
| Liraglutide (Victoza) | Initial dosage: 0.6 mg SQ once/day (irrespective of meals) for 7 days, then increase to 1.2 mg once/day | Same as above. | Pancreatitis has occurred | |
| Incretin Enhancers | ||||
| Sitagliptin: 100 mg oral once/day Saxagliptin: 2.5 to 5 mg oral once/day Linagliptin: 5 mg oral once/day |
Inhibits DPP-4 hormone activity, prolongs survival of endogenously released incretin hormones | Pancreatitis has occurred Reduce dosage with kidney dysfunction for Sitagliptin and Saxagliptin No dosage adjustment for kidney dysfunction needed with Linagliptin |
||
*Combination medications are too numerous to include in this table.
Insulin secretagogues stimulate the secretion of insulin from the pancreatic beta cells. Two medication classes have this action: the second generation sulfonylureas (glyburide, glipizide, glimepiride, and gliclazide), and the meglitinides (repaglinide and nateglinide).16 The sulfonylureas reduce the A1C by 1 to 2 percentage points and have a long duration of action. Side effects include hypoglycemia and weight gain.18
The biguanides (metformin) increase insulin sensitivity in the liver and have only a minor effect on skeletal muscle. Metformin is considered first-line therapy for patients with type 2 diabetes.18,20 Metformin may be used as monotherapy or be combined with basal insulin or with other oral medications that lower blood glucose.18 Metformin is associated with a rare risk of metabolic acidosis; gastrointestinal side effects are common.
The thiazolidinediones (TZDs), also known as glitazones (pioglitazone and rosiglitazone), belong to a class of medications called peroxisome proliferator–activated receptor gamma modulators, which increase the sensitivity of muscle, fat, and liver cells to endogenous and exogenous insulin (insulin sensitizers).21 Caution is recommended when using these medications with patients who have risk factors for heart failure (pioglitazone and rosiglitazone), or risk factors for myocardial infarction (rosiglitazone).18
The alpha-glucosidase inhibitors slow digestion of ingested carbohydrates in the proximal small intestine.18 These medications delay glucose absorption and reduce postprandial hyperglycemia following meals.22 The medications in this class are acarbose and miglitol.19 A review of the physiology of carbohydrate digestion is helpful to understand how these agents work. Carbohydrates are broken down to absorbable components in the duodenum and upper jejunum. The carbohydrates are digested to oligosaccharides in the small intestine by pancreatic lipase, after which the oligosaccharides are cleaved to monosaccharides by alpha-glucosidase enzymes. The monosaccharides are then available to be absorbed from the intestine into the bloodstream. The alpha-glucosidase inhibitor medications work by decreasing the conversion of carbohydrates from oligosaccharides to monosaccharides, thus limiting the rise in blood glucose that occurs after eating. The most frequently prescribed medication in this class is acarbose, which is nonabsorbable.
A newer class of medications lowers blood glucose by acting on gastric incretin hormones. The main therapeutic target is the incretin hormone glucagon-like peptide-1 (GLP-1), which is secreted from the gastric mucosa in response to food ingestion. GLP-1 stimulates the pancreatic beta cells to produce insulin. See Table 31-3 in Chapter 31 for a description of the incretin hormones’ physiologic effects.23
Pharmacologic incretin therapies have two different mechanisms of action16:
• The incretin mimetics augment activity of GLP-1. The medications are GLP-1 receptor agonists that increase insulin secretion from the pancreatic beta-receptors. The medications in this class are administered by subcutaneous injection (exenatide and liraglutide).
• The incretin enhancers inhibit degradation of GLP-1 by dipeptidyl peptidase-4 (DPP-4). The medications are DPP-4 inhibitors and they slow the degradation of native GLP-1 hormone by the enzyme DPP-4; this extends the physiologic glucose-lowering activity of GLP-1. The DPP-4 inhibitor oral medications (sitagliptin, saxagliptin, linagliptin) are recommended in current guidelines for management of hyperglycemia in type 2 diabetes.19
Diabetic Ketoacidosis
Epidemiology and Etiology
The diagnostic criteria for DKA are as follows24:
DKA is categorized as mild, moderate, or severe depending on the severity of the metabolic acidosis (assessed by blood pH, bicarbonate, ketones) and by the presence of altered mental status (Table 33-4).24 Hospitalizations for DKA are increasing.24 DKA accounts for more than 500,000 hospital days per year, with hospital costs that exceed $2.4 billion per year.24 Infection is the most common precipitating cause of DKA.24 Symptoms of fatigue and polyuria may precede full-blown DKA, which can develop in less than 24 hours in a person with type 1 diabetes. In a patient with undiagnosed diabetes, it is unknown how long it may take for DKA to develop as the pancreatic beta cells gradually fail. Hospital admission is generally required for DKA related to new-onset type 1 diabetes. With management by experienced clinicians, the mortality rate from DKA in type 1 diabetics is less than 1%.24
TABLE 33-4
DIAGNOSTIC CRITERIA FOR DIABETIC KETOACIDOSIS (DKA) AND HYPERGLYCEMIC HYPEROSMOLAR SYNDROME (HHS)
| DKA | HHS | |||
| MILD (PLASMA GLUCOSE >250 mg/dL) | MODERATE (PLASMA GLUCOSE >250 mg/dL) | SEVERE (PLASMA GLUCOSE >250 mg/dL) | PLASMA GLUCOSE >600 mg/dL | |
| Arterial pH | 7.25-7.30 | 7.00 to <7.24 | <7.00 | >7.30 |
| Serum bicarbonate (mEq/L) | 15-18 | 10 to <15 | <10 | >18 |
| Urine ketone* | Positive | Positive | Positive | Small |
| Serum ketone* | Positive | Positive | Positive | Small |
| Effective serum osmolality† | Variable | Variable | Variable | >320 mOsm/kg |
| Anion gap‡ | >10 | >12 | >12 | Variable |
| Mental status | Alert | Alert/drowsy | Stupor/coma | Stupor/coma |
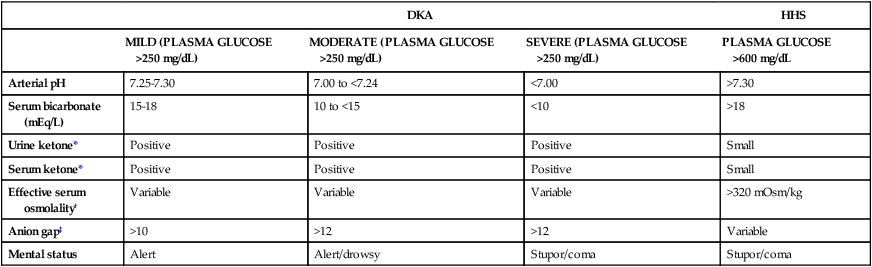
*Nitroprusside reaction method.
†Effective serum osmolality: 2[measured Na+ (mEq/L)] + glucose (mg/dL)18.
‡Anion gap: (Na+) − (Cl + HCO3−)mEq/L.
Data from Kitabchi AE, et al. Hyperglycemic crises in adult patients with diabetes: a consensus statement from the American Diabetes Association. Diabetes Care. 2009;32(7):1335.
Changes in the type of insulin, change in dosage, or increased metabolic demand can precipitate DKA in individuals with type 1 diabetes.24 Life cycle changes, such as growth spurts in the adolescent, require an increase in insulin intake, as do surgery, infection, and trauma. In young persons with diabetes, psychologic problems combined with eating disorders are a contributing factor in up to 20% of cases of recurrent ketoacidosis.24
Ketoacidosis also occurs with acute pancreatitis. In addition to elevated glucose and acidosis, the serum amylase and lipase are abnormally high, which helps to establish pancreatitis as a separate diagnosis from type 1 diabetes. Other nondiabetic causes of ketoacidosis are starvation ketosis and alcoholic ketoacidosis. These cases are distinguished from classic DKA by clinical history and usually by a plasma glucose below 200 mg/dL.24
Pathophysiology
Insulin Deficiency
Insulin is the metabolic key to the transfer of glucose from the bloodstream into the cell, where it can be used immediately for energy or stored for use at a later time. Without insulin, glucose remains in the bloodstream, and cells are deprived of their energy source. A complex pathophysiologic chain of events follows (Fig. 33-2). The release of glucagon from the liver is stimulated when insulin is ineffective in providing the cells with glucose for energy. Glucagon increases the amount of glucose in the bloodstream by breaking down stored glucose (glycogenolysis). Noncarbohydrates (fat and protein) are converted into glucose (gluconeogenesis). Plasma glucose levels for the patient in DKA typically are above 250 mg/dL. Elevated serum glucose levels alone do not define DKA; the other crucial determining factor is the presence of ketoacidosis as listed in Table 33-4.24 About 10% of patients in DKA present to the hospital with a plasma glucose below 250 mg/dL and with ketoacidosis.24
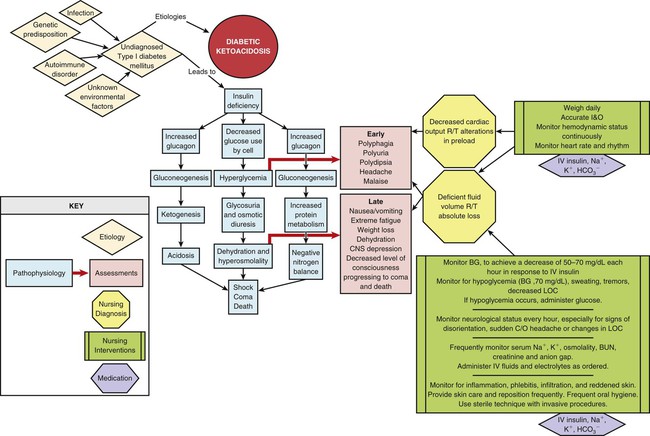
Fluid Volume Deficit
Polyuria (excessive urination) and glycosuria (sugar in the urine) occur as a result of the osmotic particle load that occurs with DKA. The excess glucose, filtered at the glomeruli, cannot be resorbed at the renal tubule and spills into the urine. The unresorbed solute exerts its own osmotic pull in the renal tubules, and less water is returned to circulation through the collecting ducts. As a result, large volumes of water, along with sodium, potassium, and phosphorus, are excreted in the urine, causing a fluid volume deficit. The serum sodium concentration may be decreased because of the movement of water from the intracellular to the extracellular (vascular) space.24
Ketoacidosis
Ketones are measurable in the bloodstream (ketonemia). Blood tests that measure the quantity of β-hydroxybutyric acid, the predominant ketone body in the blood, are the most useful.24 Because ketones are excreted by the kidney, they are also measurable in the urine (ketonuria). Ketone blood tests are preferred over urine tests for diagnosis and monitoring of DKA in critical care. When the blood and urine become clear of ketones, the DKA is resolved.
Acid–Base Balance
The acid–base balance varies depending on the severity of the DKA. The patient with mild DKA typically has a pH between 7.25 and 7.30. In severe DKA, the pH can drop below 7.00 (see Table 33-4).24 Acid ketones dissociate and yield hydrogen ions (H+), which accumulate and precipitate a fall in serum pH. The level of serum bicarbonate also decreases, consistent with a diagnosis of metabolic acidosis. Breathing becomes deep and rapid (Kussmaul respirations) to release carbonic acid in the form of carbon dioxide. Acetone is exhaled, giving the breath its characteristic fruity odor.
Assessment and Diagnosis
Clinical Manifestations
DKA has a predictable clinical presentation. It is usually preceded by patient complaints of malaise, headache, polyuria (excessive urination), polydipsia (excessive thirst), and polyphagia (excessive hunger). Nausea, vomiting, extreme fatigue, dehydration, and weight loss follow. Central nervous system depression, with changes in the level of consciousness, can lead quickly to coma.16,24
Laboratory Studies
Considering the complexity and potential seriousness of DKA, the laboratory diagnosis is straightforward. With a known type 1 diabetic patient, the presence of hyperglycemia and ketones provides rapid diagnostic confirmation of DKA. A blood gas sample can confirm the acid–base imbalance. Other clues may be gleaned from the venous blood chemistry panel. CO2, if measured, is low in the presence of uncompensated metabolic acidosis, and the anion gap is elevated. Serum sodium may be low as a result of the movement of water from the intracellular space into the extracellular (vascular) space.24 The serum potassium level is often normal; a low serum potassium level in DKA suggests a significant potassium deficiency may be present.24
Medical Management
Reversing Dehydration
The patient with DKA is dehydrated and may have lost 5% to 10% of body weight in fluids. Aggressive IV fluid replacement is provided to rehydrate the intracellular and extracellular compartments and prevent circulatory collapse (Fig. 33-3).24 Assessment of hydration is an important first step in the treatment of DKA.
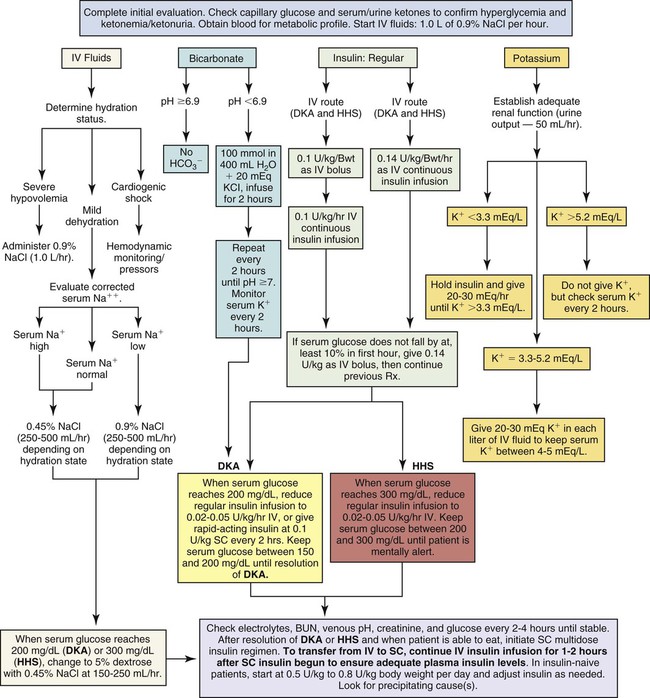
Intravenous isotonic normal saline (0.9% NaCl) is infused to replenish the vascular deficit and to reverse hypotension. For the severely dehydrated patient, 1 L of normal saline is infused immediately. Laboratory assessment of serum osmolality and the serum sodium concentration can help guide the subsequent interventions. If the serum osmolality is elevated and serum sodium is high (hypernatremia), infusions of hypotonic sodium chloride (0.45) follow the initial saline replacement. The replacement infusion typically includes 20 to 30 mEq of potassium per liter to restore the intracellular potassium debt, provided kidney function is normal (see Fig. 33-3).24 In patients without normally functioning kidneys and in those with cardiopulmonary disease, careful attention must be paid to the volume of fluid replacement to avoid fluid overload.
After the serum glucose level decreases to 200 mg/dL, the infusing solution is changed to a 50/50 mix of hypotonic saline and 5% dextrose. Dextrose is added to replenish depleted cellular glucose as the circulating serum glucose decreases to 200 mg/dL.24 Dextrose infusion also prevents unexpected hypoglycemia when the insulin infusion is continued but the patient cannot take in sufficient carbohydrate from an oral diet.
Replacing Insulin
In moderate to severe DKA, an initial IV bolus of regular insulin at 0.1 unit for each kilogram of body weight may be administered. Subsequently, a continuous infusion of regular insulin at 0.1 unit/kg/hr is infused simultaneously with IV fluid replacement (see Fig. 33-3).24 In a 70-kg adult, the infusion would be 7 units of insulin per hour. If the plasma glucose concentration does not fall by 50 to 70 mg/dL during the first hour of treatment, the glucose measurement should be rechecked. When the plasma glucose level is decreasing as expected, the insulin infusion will be increased each hour until a steady glucose decline of between 50 and 70 mg/dL per hour is achieved.24
Frequent assessment of the patient’s blood glucose concentration is mandatory in moderate to severe DKA. Initially, blood glucose tests are performed hourly. The frequency then decreases to every 2 to 4 hours as the patient’s blood glucose level stabilizes and approaches normal. After the level has decreased to 200 mg/dL, the acidosis has been corrected, and rehydration has been achieved, the insulin infusion rate may be decreased to 0.05 to 0.1 unit/kg/hr. This usually represents 3 to 6 units per hour in an adult receiving a continuous IV insulin infusion. It is important to verify that the serum potassium concentration is not lower than 3.3 mEq/L and to replace potassium if necessary, before administering the initial insulin bolus.24
Reversing Ketoacidosis
Adequate hydration and insulin replacement usually correct the acidosis, and this treatment is sufficient for many patients with DKA. As shown in Figure 33-3, replacement of bicarbonate is no longer routine except for the severely acidotic patient with a serum pH value lower than 7.0.24 An indwelling arterial line provides access for hourly sampling of arterial blood gases (ABGs) to evaluate pH, bicarbonate, and other laboratory values in the patient with severe DKA.
Hyperglycemia usually resolves before the ketoacidemia does.24 Type 1 diabetes patients with DKA can require 6 L of IV fluid replacement even for mild DKA.25 In one clinical report, patients with previously diagnosed type 1 diabetes in DKA took an average of 21 hours after being started on an IV insulin protocol to clear ketones from the urine. The insulin infusion was continued for 36 hours until the patients could tolerate an oral diet; during this time, the patients received a total of 9.5 L of normal saline for rehydration.26 Patients who are newly diagnosed with type 1 diabetes take longer to clear urine ketones and require more insulin to achieve normal glycemic control, compared with long-term diabetics.26
Nursing Management
Nursing management of the patient with DKA incorporates a variety of nursing diagnoses (Box 33-5). The goals of nursing management are to administer prescribed fluids, insulin, and electrolytes; monitor response to therapy; maintain surveillance for complications; and provide patient education.
 Box 33-5 Nursing Diagnoses
Box 33-5 Nursing Diagnoses
Diabetic Ketoacidosis
• Decreased Cardiac Output related to alterations in preload, p. 1128
• Deficient Fluid Volume related to absolute loss, p. 1132
• Anxiety related to threat to biologic, psychologic, and social integrity, p. 1125
• Disturbed Body Image related to functional dependence on life-sustaining technology, p. 1136
• Ineffective Coping related to situational crisis and personal vulnerability, p. 1150
• Powerlessness related to lack of control over current situation or disease progression, p. 1152
• Deficient Knowledge related to lack of previous exposure to information, p. 1134 (see Box 33-9, Patient Education for Diabetic Ketoacidosis)
Administering Fluids, Insulin, and Electrolytes
Rapid IV fluid replacement requires the use of a volumetric pump. Insulin is administered intravenously to patients who are severely dehydrated or have poor peripheral perfusion, to ensure effective absorption. Patients with DKA are kept on NPO status (nothing by mouth) until the hyperglycemia is under control. The critical care nurse is responsible for monitoring the rate of plasma glucose decline in response to insulin. The goal is to achieve a fall in glucose levels of approximately 50 to 70 mg/dL each hour.24 The coordination involved in monitoring blood glucose, potassium, and often blood gases on an hourly basis is considerable.
When the blood glucose level falls to 200 mg/dL, a 5% dextrose solution (D5W) with 0.45% NaCl solution is infused to prevent hypoglycemia.24 At this time, it is likely that the insulin dose per hour will also be decreased. The regular insulin drip is not discontinued until the ketoacidosis subsides, as identified by absence of ketones and a normal pH by arterial blood gas analysis.24
Monitoring Response to Therapy
Accurate intake and output (I&O) measurements must be maintained to monitor reversal of dehydration. Hourly urine output is an indicator of kidney function and provides information to prevent overhydration or insufficient hydration. Vital signs, especially heart rate (HR), hemodynamic values, and blood pressure (BP), are continuously monitored to assess response to the fluid replacement. Evidence that fluid replacement is effective includes normal central venous pressure (CVP), decreased HR, and normal BP. Box 33-6 lists the standard features to be included in an assessment of hydration status. More invasive hemodynamic monitoring, such as a pulmonary artery catheter, is rarely needed. Further evidence of hydration improvement includes a change from a previously weak, thready pulse to a pulse that is strong and full, and a change from hypotension to a gradual elevation of systolic BP. Respirations are assessed frequently for changes in rate, depth, and presence of the fruity acetone odor.
Surveillance for Complications
Hypoglycemia.
Hypoglycemia is defined as a serum glucose level lower than 70 mg/dL.24 Most acute care hospitals have specific procedures for management of the hypoglycemic patient (Box 33-7). For example, if hypoglycemia is detected by finger-stick point-of-care testing at the bedside, a blood sample is sent to the laboratory for verification, the physician is notified immediately, and replacement glucose is given intravenously or orally, depending on the patient’s clinical condition, diagnosis, and level of consciousness.
 Box 33-7
Box 33-7
NIC
Hypoglycemia Management
Preventing and treating low blood glucose levels
Identify patient at risk for hypoglycemia
Determine recognition of hypoglycemia signs and symptoms
Monitor blood glucose levels, as indicated
Monitor for signs and symptoms of hypoglycemia (e.g., shakiness, tremor, sweating, nervousness, anxiety, irritability, impatience, tachycardia, palpitation, chills, clamminess, lightheadedness, pallor, hunger, nausea, headache, tiredness, drowsiness, weakness, warmth, dizziness, faintness, blurred vision, nightmares, crying out in sleep, paresthesia, difficulty concentrating, difficulty speaking, incoordination, behavior change, confusion, coma, seizure)
Provide simple carbohydrate, as indicated
Provide complex carbohydrate and protein, as indicated
Administer glucagon, as indicated
Contact emergency medical services, as necessary
Administer intravenous glucose, as indicated
Maintain IV access, as appropriate
Maintain patent airway, as necessary
Protect from injury, as necessary
Review events prior to hypoglycemia to determine probable cause
Provide feedback regarding appropriateness of self-management of hypoglycemia
Instruct patient and significant others on signs and symptoms, risk factors, and treatment of hypoglycemia
Instruct patient to have simple carbohydrates available at all times
Instruct patient to obtain and carry/wear appropriate emergency identification
Instruct significant others on the use and administration of glucagon, as appropriate
Instruct on interaction of diet, insulin or oral agents, and exercise
Provide assistance in making self-care decisions to prevent hypoglycemia (e.g., reducing insulin/oral agents and/or increasing food intake for exercise)
Encourage self-monitoring of blood glucose levels
Encourage ongoing telephone contact with diabetes care team for consultation regarding adjustments in treatment regimen
Collaborate with patient and diabetes care team to make changes in insulin regimen (e.g., multiple daily injections), as indicated
Modify blood glucose goals to prevent hypoglycemia in the absence of hypoglycemia symptoms
Inform patient of increased risk of hypoglycemia with intensive therapy and normalization of blood glucose levels
Instruct patient regarding probable changes in hypoglycemia symptoms with intensive therapy and normalization of blood glucose levels
From Bulechek GM, et al. Nursing Interventions Classification (NIC). 6th ed. St. Louis: Mosby; 2013.
Unexpected behavior change or decreased level of consciousness, diaphoresis, and tremors are physical warning signs that the patient has become hypoglycemic. These symptoms are especially important to recognize if the frequency of glucose testing has lengthened to 2- to 4-hour intervals. A comparison between the physical symptoms expected with hypoglycemia and those of hyperglycemia is provided in Box 33-8.

Hypokalemia and Hyperkalemia.
Hyperkalemia occurs with acidosis or with overaggressive administration of potassium replacement in patients with renal insufficiency. Severe hyperkalemia is demonstrated on the cardiac monitor by a large, peaked T wave; flattened P wave; and widened QRS complex. See Figure 14-75 in Chapter 14. Ventricular fibrillation can follow.
Patient Education
It is important to be aware of the knowledge level and adherence history of patients with previously diagnosed diabetes to formulate an appropriate teaching plan. Learning objectives include a discussion of target glucose levels, definition of hyperglycemia and its causes, harmful effects, symptoms, and how to manage insulin and diet when one is unwell and unable to eat. Additional objectives include a definition of DKA and its causes, symptoms, and harmful consequences. The patient and family are also expected to learn the principles of diabetes management. Universal precautions must be emphasized for all family caregivers. The patient and family must also learn the warning signs of DKA to report to the attention of a health care practitioner. Education of the patient, family, or other support persons to achieve knowledge-based, independent self-management of blood glucose level and avoidance of diabetes-related complications are the ultimate goals of the teaching process (Box 33-9).
Collaborative Management
In all aspects of patient care management, health care professionals work as a team with the major collaborative goal of providing the best possible outcome for each patient. Current guidelines related to Collaborative Management of patients with hyperglycemia crisis are listed Box 33-10.
 Summary of evidence and evidence-based recommendations for controlling symptoms related to diabetic ketoacidosis (DKA)
Summary of evidence and evidence-based recommendations for controlling symptoms related to diabetic ketoacidosis (DKA)[/level-membership-for-critical-care-medicine-category][not-level-membership-for-critical-care-medicine-category]<
[/not-level-membership-for-critical-care-medicine-category]
