204 Emergency Management of Red Blood Cell Disorders
 Key Points
Key Points• Anemia is the absolute reduction in the amount of oxygen-carrying pigment hemoglobin (Hgb) that represents a relative decrease in the capacity of blood to carry oxygen to the tissues.
• Anemia is not a diagnosis. It is an indication of an underlying disease, disorder, or deficiency.
• Transfusion of red blood cells provides immediate correction of low Hgb levels helpful in the context of either severe anemia (in which the Hgb is <8.0 g/dL) or life-threatening anemia (in which the Hgb is <6.5 g/dL).
• Most cases of anemia (chronic) do not require acute interventions and drug therapy in the emergency department (ED). Patients can be referred for follow-up to their primary care physician or gastroenterologist.
• The cardinal features of acute chest syndrome are fever, pleuritic chest pain, referred abdominal pain, cough, lung infiltrates, and hypoxia.
• Pneumococcal sepsis is a leading cause of death among infants with sickle cell anemia because a damaged spleen cannot clear pneumococci from the blood.
• Transfusions are not needed for the usual anemia or episodes of pain associated with sickle cell disease.
• Splenic sequestration is life-threatening and requires intensive care admission with transfusion and possible splenectomy.
• ED-based pain management protocols have been shown to decrease ED visits and hospitalizations and to increase use of primary care clinics by patients with sickle cell disease.
• Patients with severe pain should be given an opiate parenterally at frequent, fixed intervals until the pain has diminished, at which time the dose of the opiate can be tapered and then stopped, and oral analgesic therapy can be instituted.
• For polycythemia vera, phlebotomy is the only therapy indicated for isolated erythrocytosis when its mechanism cannot be established.
Anemia
Epidemiology
Anemia is more common than is generally realized. The World Health Organization defines anemia as a condition characterized by hemoglobin (Hgb) levels lower than 13 g/dL in men or lower than 12 g/dL in women.1 Data from the National Center for Health Statistics that likely underestimate the frequency of anemia indicated that approximately 3.4 million U.S residents have anemia, and that the groups with the highest prevalence are women, African Americans,2 older persons, and those with the lowest incomes. Using laboratory data from the general U.S. population, the second National Health and Nutrition Survey reported anemia to be the most prevalent in infants, teenage girls, young women, and older men.3 In another survey, the prevalence of anemia declined significantly among U.S women and children from 1988 to 2002, but the cause of this decline was unknown.4 In persons 65 years and older, anemia was present in 11.0% of men and 10.2% of women, and the prevalence rose to more than 20% in people 85 years and older. One third of the cases of anemia were the result of nutritional deficiencies, and one third of cases were secondary to chronic illness, including but not limited to chronic renal disease.
Pathophysiology
Anemia is classified into three broad categories: (1) disorders of decreased RBC production, (2) disorders of increased RBC destruction, and (3) disorders resulting from RBC loss. Disorders in each of these categories may manifest differently and ultimately have their own management approaches (Table 204.1).
| CATEGORY | CLASSIFICATION | DISEASE PROCESS |
|---|---|---|
| Decreased RBC production (hypoproliferative) | Microcytic | Iron deficiency Thalassemia Sideroblastic Chronic disease (neoplasm, infection, diabetes, uremia, thyroid disease, cirrhosis) |
| Normocytic | Primary bone marrow problem (aplastic, myeloid metaplasia, myelofibrosis, myelophthisic anemia, Diamond-Blackfan anemia) | |
| Secondary bone marrow problem (uremia, liver disease, endocrinopathy, chronic inflammation) | ||
| Macrocytic | Folic acid deficiency | |
| Liver disease | ||
| Vitamin B12 deficiency | ||
| Scurvy | ||
| Hypothyroidism | ||
| Chemotherapy, immunosuppressive therapy | ||
| Increased RBC destruction (hemolytic) | Intrinsic | Membrane disorder (spherocytosis, sickle cell, stem cell disorder, elliptocytosis, spur cell) |
| Extrinsic | Hemoglobin disorder (thalassemia, autoimmune, hemoglobinopathies) | |
| Infections (hepatitis and cytomegalovirus, Epstein-Barr virus, typhoid fever, Escherichia coli) | ||
| Medications (penicillin, antimalarials, sulfa drugs, or acetaminophen) | ||
| Leukemia or lymphoma | ||
| Autoimmune disorders (systemic lupus erythematosus, rheumatoid arthritis, Wiskott-Aldrich syndrome, ulcerative colitis) | ||
| Enzyme defect (G6PD) | ||
| RBC loss (hemorrhagic) | Acute or chronic | Gastrointestinal tract |
| Traumatic | ||
| Intraperitoneal | ||
| Extraperitoneal | ||
| Gynecologic | ||
| Urinary | ||
| Pelvic | ||
| Drug related | ||
| Epistaxis, hemoptysis | ||
G6PD, Glucose-6-phosphate dehydrogenase; RBC, red blood cell.
RBCs, or erythrocytes, contain fluid Hgb encased in a lipid membrane and are the predominant cellular component of blood. RBCs make up 45% of the blood volume and are responsible for carrying oxygen from the lungs to the peripheral tissues. A 70-kg person has approximately 30 trillion RBCs, resulting in approximately 300 million RBCs in each drop of blood. The normal RBC is composed of three types of Hgb: Hgb A (97%), Hgb F (1%) or fetal Hgb, and Hgb A2 (2%).5 Hgb A is composed of two β-globin chains and two α-globin chains bonded to four iron-containing heme groups. Hgb production requires iron, the synthesis of the protoporphyrin ring, and the production of the globin chains. Reductions in any of these processes result in anemia.
Presenting Signs and Symptoms
Chronic Anemia
Because anemia can be a primary disorder or can occur secondary to hypoproliferation or chronic blood loss, a careful history and physical examination provide valuable insight into the potential cause. Individuals with mild anemia are often asymptomatic and are able to sustain a relatively normal level of function at Hgb levels that are significantly lower than normal. Other patients may present with myriad nonspecific symptoms (Box 204.1). Because fatigue is nonspecific, determining the concomitant presence of a systemic inflammatory disorder, infection, or malignant disease may be critical in determining the underlying causes of anemia.
Past medical history is quite informative. For instance, a history of diabetes mellitus is associated with significantly impaired renal production of erythropoietin.6 Certain medications are associated with bone marrow depression. Therefore, all pharmacologic agents, both prescribed drugs and over-the-counter agents, including alternative medications, should be reviewed. Occupational history is relevant, as in the case of welders, who may have been exposed to lead or other agents potentially toxic to the bone marrow. Social history is important because a history of intravenous drug use may suggest the possibility of human immunodeficiency virus infection, which can be associated with anemia.7 Dietary history is relevant. For example, the finding of pica in adults (most commonly from the ingestion of nonfood items) is well known to be associated with iron deficiency anemia. A family history of anemia is important; for example, adults with congenital hereditary spherocytosis often develop symptoms later in life.
Physical findings in either acute or chronic anemia are myriad and often nonspecific, and they may relate to the underlying disease process and the duration (Table 204.2). Pathognomonic findings are not the norm. Furthermore, patients with chronic anemia usually do not have the typical physical findings associated with acute anemia.
| ORGAN | FINDING |
|---|---|
| Skin | Pallor Usefulness limited by color of skin, Hgb concentration, and fluctuation of blood flow to skin Palmar crease color a better indicator, if as pale as surrounding skin, Hgb usually <7 g/dL |
| Hematologic | Purpura, petechiae, and jaundice |
| Cardiovascular | Tachycardia Wide pulse pressure Orthostatic hypotension Hyperdynamic precordium Systolic eject murmur over pulmonic area |
| Respiratory | Tachypnea Rales |
| Gastrointestinal | Hepatomegaly and/or splenomegaly Ascites Masses Positive result on Hemoccult test |
| Ophthalmologic | Pale conjunctiva Scleral icterus Retinal hemorrhages |
| Neurologic | Peripheral neuritis or neuropathy Mental status changes |
Hgb, Hemoglobin.
Differential Diagnosis and Diagnostic Testing
The differential diagnosis of anemia is myriad, as documented in Table 204.3. Once anemia is suspected, the initial diagnosis involves the complete blood count (CBC). The variables to focus on when examining the CBC are hematocrit (as a general indicator of anemia or polycythemia), mean corpuscular volume ([MCV] a key parameter for the classification of anemias), RBC distribution width (a relatively useful parameter in the differential diagnosis of anemia), RBC count (an increased RBC count associated with anemia is characteristic in the thalassemia trait), platelet count (to detect either thrombocytopenia or thrombocytosis), and white blood cell (WBC) count with differential (usually gives important clues to the diagnosis of acute leukemia and chronic lymphoid or myeloid disorders, as well as clues to the presence of leukopenia and neutropenia).8
| CATEGORY | DIFFERENTIAL DIAGNOSIS | CBC CLUES |
|---|---|---|
| Microcytic | Iron deficiency anemia | Elevated RDW Thrombocytosis |
| Thalassemia | Normal or elevated RBC count Normal or elevated RDW |
|
| Anemia of chronic disease | Normal RDW | |
| Normocytic | Hemolysis | Normal or elevated RDW Thrombocytosis |
| Bleeding | Unchanged | |
| Nutritional anemia | Elevated RDW | |
| Anemia of chronic disease | Normal RDW | |
| Primary bone marrow disease | Elevated RDW Leukocytosis Thrombocytosis Monocytosis |
|
| Macrocytic | Alcohol use, liver disease | Normal RDW Thrombocytopenia |
| Drug induced | Elevated RDW | |
| Bone marrow disorder | Elevated RDW | |
| Hypothyroidism | Normal RDW | |
| Hemolysis | Normal or elevated RDW | |
| Nutritional | Elevated RDW |
CBC, Complete blood cell count; RBC, red blood cell; RDW, red blood cell distribution width.
The first step in approaching anemia is to classify the process as microcytic (MCV < 80 fL), normocytic (MCV, 80 to 100 fL), or macrocytic (MCV > 100 fL). Clues to the diagnostic possibilities for the three major classes are listed in Table 204.3.
 Facts and Formulas
Facts and Formulas
Corrected reticulocyte count (%) = Observed count × Measured count (%) / 45%
Finch reticulation production index = Corrected reticulocyte count (%) / Expected maturation time (days)
Finch CA, Marshall PN, Brecher G, et al. Method for reticulocyte counting. NCCLS proposed standard H16-P. Villanova, Penn: National Committee for Clinical Laboratory Standards; 1985.
Treatment
 Priority Actions
Priority Actions
1. Determine the patient’s hemodynamic status. The need for transfusion is often limited to those in hypovolemic shock.
2. If the patient is unstable, initiate resuscitation with crystalloids.
3. Once the patient is stabilized, blood transfusions are widely used as a rapid and effective therapeutic intervention in the context of either severe anemia (Hgb < 8.0 g/dL) or life-threatening anemia (Hgb < 6.5 g/dL).9
Patients with long-standing or chronic anemias are able to compensate and do not require transfusion, especially if the Hgb is greater than 9.0 g/dL. Patients who are expected to respond to the administration of a specific agent such as folic acid, iron, or vitamin B12 can usually be spared transfusions. If the anemia has precipitated an episode of congestive heart failure or myocardial ischemia, prompt administration of packed RBCs is indicated. For some patients in the ED, treatment can be begun without waiting for a definitive outpatient evaluation. For example, prenatal vitamins and iron replacement can be begun in the pregnant patient with anemia. In symptomatic pregnant patients, parenteral iron is preferred.10 Megaloblastic anemia resulting from folic acid or vitamin B12 deficiency can be treated with parenteral cobalamin (1000 g/day) or oral folic acid (1 mg/day). Erythropoietin therapy remains an option for patients undergoing elective surgical procedures or receiving chemotherapy and in patients with chronic heart failure or acquired immunodeficiency syndrome. In the acute setting, however, specifically in symptomatic heart failure, the role of erythropoietin therapy remains to be defined.11,12
Sickle Cell Anemia
Epidemiology
Sickle cell disease (SCD), characterized by lifelong hemolytic anemia and many different painful and debilitating vasoocclusive events, occurs in 70,000 to 80,000 U.S. residents of African, Mediterranean, or Middle Eastern descent. In the United States, the life expectancy for patients with SCD is shortened by approximately 30 years, whereas in Africa, where comprehensive medical care is less readily available, death in early childhood is usual.13 Eight percent of African Americans are heterozygous carriers of the sickle cell trait; approximately 40% of their Hgb is Hgb S. They do not have anemia and need neither treatment nor occupational restrictions.
Pathophysiology
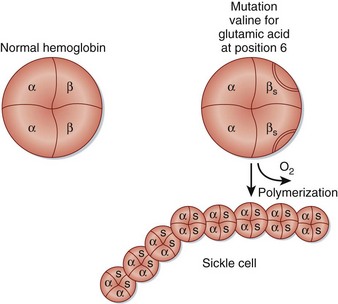
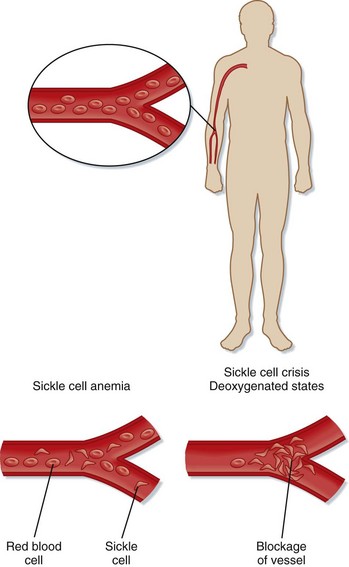
SCD is an inherited condition caused by a point mutation in the β-globin gene (Hgb B) that causes the substitution of valine for glutamic acid at position 6 of the β-globin chain (Glu6Val). This mutation results in the abnormal Hgb S14 (Fig. 204.1). When deoxygenated, Hgb S polymerizes, thus damaging the sickle RBC. These sickle cells are short-lived and interact with endothelial cells, WBCs, platelets, and other plasma components to initiate the vasoocclusive manifestations associated with SCD.15 Among hemolytic anemias, the vasoocclusive features of SCD are unique. By occluding small blood vessels and sometimes large vessels, sickle cells cause vascular injury (Fig. 204.2).
Presenting Signs and Symptoms
Vasoocclusion, which is responsible for most of the severe complications of SCD, can occur wherever blood flows. The clinical features of SCD are outlined in Table 204.4.
| TYPE | CLINICAL FEATURES |
|---|---|
| Vasoocclusive complications | Pain crises Acute chest syndrome |
| Splenic sequestration | |
| Cerebrovascular crisis | |
| Priapism | |
| Liver disease | |
| Leg ulcers | |
| Spontaneous abortion | |
| Osteonecrosis | |
| Renal crisis | |
| Retinopathy | |
| Infectious complications | Osteomyelitis |
| Escherichia coli sepsis | |
| Streptococcus pneumoniae sepsis | |
| Hemolytic complications | Cholelithiasis |
| Anemia | |
| Aplastic anemia |
Acute Chest Syndrome
The acute chest syndrome is the leading cause of death and hospitalization among patients with SCD. Affecting approximately 40% of all patients with sickle cell anemia, it is the second most common reason for hospitalization and the leading cause of intensive care unit admission and premature death in patients with SCD.16 Its cardinal features are fever, pleuritic chest pain, referred abdominal pain, cough, lung infiltrates, and hypoxia.17 It is most common but least severe in children, can occur postoperatively, and when recurrent, can lead to pulmonary hypertension, restrictive lung disease, and eventually right-sided heart failure and death.16
Asthma has been associated with multiple complications of SCD, including acute chest syndrome. Asthma is an independent risk factor for mortality in children with SCD, and it confers a twofold higher risk of death.18
Right Upper Quadrant Syndrome
The three acute hepatic syndromes seen in SCD are acute hepatic cell crisis, acute hepatic sequestration crisis, and sickle cell intrahepatic cholestasis. Intrahepatic cholestasis is benign and may be associated with severe hyperbilirubinemia that resolves in 7 to 10 days, especially in children.19 A syndrome more common in adults is associated with fever, leukocytosis, abdominal pain, and deteriorating liver function, as indicated by measurement of liver enzymes. This hepatic crisis usually progresses to hepatic failure, coagulopathy, encephalopathy, and death.
Priapism
Priapism is a sustained, painful, and unwanted erection of the penis that pathophysiologically is the result of an accumulation of sickled RBCs in the corpora cavernosa that cause ischemia or low flow. Approximately 30% of male patients with SCD who are less than 20 years old report at least one episode of priapism, whereas frequencies of 30% to 45% are estimated for adult men. This condition is most common in patients with the greatest amount of hemolysis. Postpubertal male patients tend to have more prolonged episodes of priapism and have a less favorable prognosis for future potency.20 One sequela is impotence; therefore, for the EP, the utmost priority in these patients is detumescence, especially within the first 12 hours.
Differential Diagnosis and Diagnostic Testing
 Priority Actions
Priority Actions
Diffuse or Isolated Pain?
Does the patient have any identifiable triggers (i.e., infection, dehydration, hypoxia, pregnancy, cold exposure, acidosis, recent flight in unpressurized aircraft)?
If yes, assess and treat underlying cause. If not, consider whether this is a usual vasoocclusive crisis; inquire about onset, duration, location, and a history of previous episodes. Treat with analgesics, preferably opiates.
Abdominal Pain, Nausea, and Vomiting?
Does the patient have pancreatitis, appendicitis, peptic ulcer disease, diverticulitis, colitis, or renal colic?
If not, consider symptomatic cholelithiasis, acute hepatic crisis, or splenic sequestration. Obtain right upper quadrant ultrasound imaging, check transaminases, and consider surgical consultation. Consider transfusion for severe anemia.
Productive Cough with Fever?
Does the patient have high fever, leukocytosis, positive Gram stain, or evidence of pneumonia on radiographs?
If not, consider acute chest syndrome, especially with low-grade temperature and vasoocclusive symptoms elsewhere. Obtain a chest radiograph and blood cultures. Admit for observation and possible exchange transfusion.
Child with Fever Greater Than 101.3° F?
Does the patient have pneumonia, pyelonephritis, cholangitis, cholecystitis, osteomyelitis, or meningitis?
If not, consider sepsis. Obtain a complete blood count, urinalysis, urine and blood cultures, chest radiograph, and lumbar puncture. Pneumococcal sepsis is a leading cause of childhood mortality, with a 14% mortality rate.
Painful, Erect Penis?
Does the patient have paraphimosis, phimosis, or recent trauma?
If not, consider priapism. Obtain urology consultation, and initiate oral terbutaline or pseudoephedrine. Intracavernosal injection with α-adrenergic agonist is warranted. Also, treat with hydration, analgesia, ice packs, and exchange transfusions.
Treatment
Treatment of SCD is evolving. The description of barriers to effective pain management is interesting and has been well documented.21 EPs tend to undertreat their patients because they fear patients’ dependence on pain medication, which in reality is present in only 1% to 3% of patients.22 Patients with SCD who are in pain are also misunderstood because they display a different attitude to their severe pain than do trauma or oncology patients. Although patients with SCD complain of severe pain, they may engage in activities that are inconsistent with the traditional image of the patient in severe pain, such as watching television or talking on the telephone. These patients are therefore often perceived as exaggerating their pain to receive additional narcotics, whereas these activities may actually be learned distractions or coping mechanisms. Another example is the sleeping patient who, when awakened, reports unrelenting pain. This situation may stem from an imbalance between the sedative and analgesic effects of opiates or a need for sleep despite the pain. The result is a lack of trust between patients and health care providers. In centers specializing in sickle cell crises, the attitudes toward pain tend to be better understood, and treatment outcomes are superior, compared with EDs.
Oral analgesics suffice for treating mild to moderate pain. Patients with mild to moderate pain seem to find no difference between intravenous and oral morphine. Most opiates have comparable efficacy and safety profiles, but morphine (0.1 mg/kg) is considered the drug of choice for treatment of acute sickle cell pain. Hydromorphone (1.5 mg) may be used if morphine is unable to achieve effective analgesia. In children, studies emphasize oral dosing of potent opioids (weight-based dosing) and nonsteroidal antiinflammatory drugs, home treatment, and reduced reliance on EDs departments or inpatient admission.23 A pain protocol, if available, should be used (Box 204.2). ED-based pain management protocols have been shown to decrease ED visits and hospitalizations and to increase use of primary care clinics by patients with SCD.24
Box 204.2 Emergency Department–Based Pain Protocol
• Determine patients’ previous known requirement for analgesia.
• Determine drug allergies and document the type of reaction.
• All patients should receive ibuprofen, 600 mg PO × one dose, unless a contraindication exists.
• Morphine is the drug of choice; hydromorphone is preferred in morphine-allergic patients.
• Evaluate pain: patients with severe pain requiring IV opioids or moderate pain able to take PO opioids.
Severe Pain
• Administer morphine, 5 to 10 mg IV initial dose, then 4 to 6 mg every 5 to 10 minutes, or hydromorphone, 1.5 mg IV initial dose, then 0.5 to 1 mg IV every 5 to 10 minutes.
• If PCA demands fewer than 3, wean off IV PCA, and discharge with a standing dose of ibuprofen, Percocet (acetaminophen and oxycodone), morphine SR, or hydromorphone.
Moderate Pain
• Administer morphine liquid, 10 mg PO × one dose or hydromorphone tablet, 2 mg PO × one dose.
• If no relief occurs, treat with IV opioids.
• If relief occurs, discharge the patient with a standing dose of ibuprofen, Percocet (acetaminophen and oxycodone), morphine SR, or hydromorphone.
IV, Intravenous(ly); PCA, patient-controlled analgesia; PO, oral(ly); SR, sustained release.
Patients in severe pain should be given an opiate parenterally and preferably initiated within 15 to 20 minutes. The opiate should be dosed at frequent (15 to 30 minutes), fixed intervals, not as needed, until the pain has diminished, at which time the dose of the opiate can be tapered and then stopped, and oral analgesic therapy can be instituted.25
When available and appropriate for the treatment of acute pain, patients prefer patient-controlled analgesia (PCA) to scheduled dose or continuous infusion of morphine.26 When used to treat acute pain episodes, PCA results in similar pain relief with lower morphine consumption when compared with continuous infusion of morphine. Furthermore, when introduced in the ED for the treatment of acute pain, PCA use was associated with a shorter elapsed time between onset of pain and treatment.27 These data, along with clinical experience, suggest that PCA has emerged as a standard for the treatment of acute pain, and, if possible, it should be started in the ED.
The use of meperidine is discouraged because of the risk of seizures. Many opioid side effects can be ameliorated by drug therapy directed at the side effect (e.g., antiemetics to treat nausea and vomiting, antihistamines to treat itching, laxatives to treat constipation).28 Antiinflammatory drugs and intravenous methylprednisolone may provide an opiate-sparing effect, but concern exists about their negative effects on bone healing. In addition, painful crises seem to recur frequently after treatment with methylprednisolone.
Urgent replacement of blood is often required for sudden severe anemia occurring in children when blood is sequestered in an enlarged spleen or when parvovirus B19 infection causes transient aplastic crisis. For aplastic crisis, clinical management is supportive and depends on the degree of anemia and cardiovascular compromise. Simple transfusions are administered to raise the Hgb to approximately 10 g/dL and the hematocrit to approximately 30% if the reticulocyte count is less than 1% to 2% with no signs of spontaneous recovery. Increasing the Hgb level to more than 11 g/dL is not recommended because of increased viscosity and risk of vasoocclusion. For shock caused by splenic sequestration, emergency management is aimed at restoring circulating blood volume and hemodynamic stability through the infusion of crystalloids and volume expanders and by repeated simple or exchange blood transfusions. Ultimately, splenectomy may be performed because sequestration has been shown to recur in 50% of patients and represents a life-threatening event. Admission is required for patients with aplastic crisis and splenic sequestration. Transfusions are not needed for the usual anemia or episodes of pain associated with SCD.28
Treatment of acute chest syndrome is supportive and may include supplemental oxygen to maintain arterial oxygen saturation at more than 92%. Analgesia and incentive spirometry can minimize chest wall splinting and thus prevent atelectasis and hypoxemia.28 Pulse oximetry in patients with SCD has been shown to correlate with arterial oxygen content. Antibiotics should be given to treat infections with S. pneumoniae, Haemophilus influenzae, and atypical organisms such as Mycoplasma, Legionella, and Chlamydia. Frequently, a macrolide with a third-generation cephalosporin is chosen. In acute chest syndrome, simple transfusion has been demonstrated to be more effective than exchange transfusion.29 Given the increased proclivity of patients with SCD to develop alloantibodies, the potential negative effects of a higher Hgb level after exchange, and the time and the expense of both the pheresis procedure and the vascular access insertion, EPs should initiate simple transfusions first in the event that the Hgb is less than 30%.
Novel therapies may include dipyridamole (Persantine), which has been shown to be a powerful inhibitor of the deoxygenation-induced fluxes of sickled cell polymerization, especially in dehydrated cells in vitro. However, more clinical trials are needed to demonstrate this benefit in vivo. Low-dose, longer-acting glucocorticoids, especially dexamethasone, have shown a benefit in the management of acute chest syndrome. However, more research is also needed. More recent studies and ongoing clinical trials have hypothesized that inhaled nitric oxide may be beneficial in managing various clinical conditions, including sickle cell anemia.30 However, because the delivery of inhaled nitric oxide may have more limited applicability in the clinical setting as a result of inherent administration problems, the oral administration of L-arginine (precursor of nitric oxide) shows promise as a potential treatment for vasoocclusive crises and acute chest syndrome.31
Disposition
 Red Flags
Red Flags
Indications for Hospital Admission
Polycythemia Vera
Epidemiology
PV is traditionally classified as a myeloproliferative disorder, which is a broad category of clonal stem cell diseases that include myelofibrosis with myeloid metaplasia and chronic myeloid leukemia.30 The true incidence and prevalence of PV are unknown. PV is relatively rare, occurring in 0.6 to 1.6 persons per million population. The disease has been recognized since the early twentieth century, and the initial description as presented by Osler has not changed. Fortunately, PV has the survival characteristics of a benign disease, and much still needs to be learned. For the EP, understanding the complications of the disease ultimately aids in its management.
Pathophysiology
The Greek term polycythemia is synonymous with the word erythrocytosis, and it literally translates as “many cells in the blood.” Absolute polycythemia is a condition with increased RBC mass. Numerous primary and secondary polycythemic disorders lead to absolute polycythemia.32
Primary and secondary polycythemias can be either acquired or congenital. Congenital polycythemias may result from inherited appropriate responses to tissue hypoxia, acquired conditions characterized by autonomous erythropoietin production (secondary polycythemias), defects in hypoxia sensing (either primary or secondary polycythemia), or inherited intrinsic defects in RBC precursors that render erythroid progenitors hypersensitive to erythropoietin (primary familial and congenital polycythemia).32
Presenting Signs and Symptoms
Bleeding manifestations in PV involve primarily the skin and mucous membranes, findings suggesting defective primary hemostasis, and include ecchymosis, epistaxis, menorrhagia, and gingival hemorrhage. Gastrointestinal hemorrhage occurs less frequently but can be severe, necessitating hospitalization and blood transfusion, and it is often associated with the use of aspirin.33 This type of bleeding pattern is consistent with platelet defects (quantitative or qualitative) or von Willebrand disease.
Differential diagnosis and Diagnostic Testing
PV is a clinical diagnosis. Diagnostic tests are nonspecific, sometimes uninformative, and none of them establish clonality. The diagnosis is currently facilitated through the laboratory measurement of RBC mass, plasma volume, and arterial oxygen saturation and determination of oxygen pressure at 50% Hgb saturation. In the ED, elevated RBC counts and hematocrit values (including Hgb levels) are used to make this diagnosis. Generally, Hgb concentrations of at least 20 g/dL or hematocrit values of at least 60% in male patients and 56% in female patients can be presumed to indicate a myeloproliferative disorder. Direct measurement of the RBC mass should show an increase, with a normal or slightly decreased plasma volume. However, this nuclear medicine test uses radiochromium-labeled RBCs to measure actual RBC and plasma volume and is not readily available. If RBC mass results are available, the Polycythemia Vera Study Group diagnostic criteria can be used (Table 204.5).
Table 204.5 Polycythemia Vera Study Group Criteria for the Diagnosis of Polycythemia Vera
| DIAGNOSTIC GROUP | CRITERIA |
|---|---|
| Category A | Total red blood cell mass In male patients, ≥36 mL/kg; in female patients, ≥32 mL/kg |
| Arterial oxygen saturation ≥ 92% | |
| Splenomegaly | |
| Category B | Thrombocytosis with a platelet count >400,000/mL |
| Leukocytosis with a white blood cell count >12,000/mL | |
| Leukocyte alkaline phosphatase > 100 units/L | |
| Serum vitamin B12 concentration > 900 pg/mL or binding capacity > 2200 pg/mL | |
| Diagnosis | A1 plus A2 plus A3 |
| A1 plus A2 plus any two criteria from category B |
Field JJ, Knight-Perry JE, Debaun MR. Acute pain in children and adults with sickle cell disease: management in the absence of evidence-based guidelines. Curr Opin Hematol. 2009;16:173–178.
Givens M, Rutherford C, Joshi G, Delaney K. Impact of an emergency department pain management protocol on the pattern of visits by patients with sickle cell disease. J Emerg Med. 2007;32:239–243.
Goodnough LT, Bach RG. Anemia, transfusion, and mortality. N Engl J Med. 2001;345:1272–1274.
Prchal JT. Polycythemia vera and other primary polycythemias. Curr Opin Hematol. 2005;12:112–116.
Tefferi A, Hanson CA, Inwards DJ. How to interpret and pursue an abnormal complete blood cell count in adults. Mayo Clin Proc. 2005;80:923–936.
1 DeMaeyer E, Adiels-Tegman M. The prevalence of anaemia in the world. World Health Stat Q. 1985;38:302–316.
2 Zakai NA, McClure LA, Prineas R, et al. Correlates of anemia in American blacks and whites: the REGARDS Renal Ancillary Study. Am J Epidemiol. 2009;169:355–364.
3 Goodnough LT, Nissenson AR. Anemia and its clinical consequences in patients with chronic diseases. Am J Med. 2004;116(suppl 7A):1S–2S.
4 Cusick SE, Mei Z, Freedman DS, et al. Unexplained decline in the prevalence of anemia among US children and women between 1988-1994 and 1999-2002. Am J Clin Nutr. 2008;88:1611–1617.
5 Weiss G, Goodnough LT. Anemia of chronic disease. N Engl J Med. 2005;352:1011–1023.
6 Thomas MC, Cooper ME, Tsalamandris C, et al. Anemia with impaired erythropoietin response in diabetic patients. Arch Intern Med. 2005;165:466–469.
7 Saif MW, Bona R, Greenberg B. AIDS and thrombosis: retrospective study of 131 HIV-infected patients. AIDS Patient Care STDS. 2001;15:311–320.
8 Tefferi A, Hanson CA, Inwards DJ. How to interpret and pursue an abnormal complete blood cell count in adults. Mayo Clin Proc. 2005;80:923–936.
9 Al RA, Unlubilgin E, Kandemir O, et al. Intravenous versus oral iron for treatment of anemia in pregnancy: a randomized trial. Obstet Gynecol. 2005;106:1335–1340.
10 Ghali JK, Anand IS, Abraham WT, et al. Randomized double-blind trial of darbepoetin alfa in patients with symptomatic heart failure and anemia. Circulation. 2008;117:526–535.
11 Palazzuoli A, Silverberg D, Iovine F, et al. Erythropoietin improves anemia exercise tolerance and renal function and reduces B-type natriuretic peptide and hospitalization in patients with heart failure and anemia. Am Heart J. 2006;152:1096.e9–1096.e15.
12 Scott RB. Sickle-cell anemia: high prevalence and low priority. N Engl J Med. 1970;282:164–165.
13 Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med. 1997;337:762–769.
14 Ataga KI, Orringer EP. Hypercoagulability in sickle cell disease: a curious paradox. Am J Med. 2003;115:721–728.
15 Vij R, Machado RF. Pulmonary complications of hemoglobinopathies. Chest. 2010;138:973–983.
16 Vichinsky EP, Neumayr LD, Earles AN, et al. Causes and outcomes of the acute chest syndrome in sickle cell disease: National Acute Chest Syndrome Study Group. N Engl J Med. 2000;342:1855–1865.
17 Boyd JH, Macklin EA, Strunk RC, DeBaun MR. Asthma is associated with increased mortality in individuals with sickle cell anemia. Haematologica. 2007;92:1115–1118.
18 Edwards CQ. Anemia and the liver: hepatobiliary manifestations of anemia. Clin Liver Dis. 2002;6:891–907.
19 Molitierno JA, Jr., Carson CC, 3rd. Urologic manifestations of hematologic disease sickle cell, leukemia, and thromboembolic disease. Urol Clin North Am. 2003;30:49–61.
20 Silbergleit R, Jancis MO, McNamara RM. Management of sickle cell pain crisis in the emergency department at teaching hospitals. J Emerg Med. 1999;17:625–630.
21 Solomon LR. Treatment and prevention of pain due to vaso-occlusive crises in adults with sickle cell disease: an educational void. Blood. 2008;111:997–1003.
22 Field JJ, Knight-Perry JE, Debaun MR. Acute pain in children and adults with sickle cell disease: management in the absence of evidence-based guidelines. Curr Opin Hematol. 2009;16:173–178.
23 Givens M, Rutherford C, Joshi G, Delaney K. Impact of an emergency department pain management protocol on the pattern of visits by patients with sickle cell disease. J Emerg Med. 2007;32:239–243.
24 Benjamin LJ, Dampier CD, Jacox AK. Guideline for the management of acute and chronic pain in sickle-cell disease. APS clinical practice guideline series. Glenville, Ill: American Pain Society; 1999.
25 Gonzalez ER, Bahal N, Hansen LA, et al. Intermittent injection vs patient-controlled analgesia for sickle cell crisis pain: comparison in patients in the emergency department. Arch Intern Med. 1991;151:1373–1378.
26 Melzer-Lange MD, Walsh-Kelly CM, Lea G, et al. Patient-controlled analgesia for sickle cell pain crisis in a pediatric emergency department. Pediatr Emerg Care. 2004;20:2–4.
27 Steinberg MH. Management of sickle cell disease. N Engl J Med. 1999;340:1021–1030.
28 Griffiths MJ, Evans TW. Inhaled nitric oxide therapy in adults. N Engl J Med. 2005;353:2683–2695.
29 Morris CR, Vichinsky EP, van Warmerdam J, et al. Hydroxyurea and arginine therapy: impact on nitric oxide production in sickle cell disease. J Pediatr Hematol Oncol. 2003;25:629–634.
30 Prchal JT. Polycythemia vera and other primary polycythemias. Curr Opin Hematol. 2005;12:112–116.
31 Landolfi R, Marchioli R, Kutti J, et al. Efficacy and safety of low-dose aspirin in polycythemia vera. N Engl J Med. 2004;350:114–124.
32 Goodnough LT, Bach RG. Anemia, transfusion, and mortality. N Engl J Med. 2001;345:1272–1274.
33 Turner JM, Kaplan JB. Exchange versus simple transfusion for acute chest syndrome in sickle cell anemia adults. Transfusion. 2009;49:863–868.