CHAPTER 71 Embryology, Anatomy, Histology, and Developmental Anomalies of the Liver
EMBRYOLOGY
The liver develops at three to four weeks’ gestation as an outgrowing bud of proliferating endodermal cells from the ventral wall of the foregut in response to signals from the adjacent developing heart (Fig. 71-1).1–3 At this stage, the liver bud is separated from the mesenchyme of the septum transversum by basement membrane.2 Shortly thereafter, the basement membrane surrounding the liver bud is lost, and cells delaminate from the bud and invade the septum transversum as cords of hepatoblasts—bipotential cells that will differentiate into hepatocytes and cholangiocytes.1,4 As they invade the septum transversum mesenchyme, the hepatoblasts intermingle with endothelial cells; this interaction is necessary to support hepatic morphogenesis.2
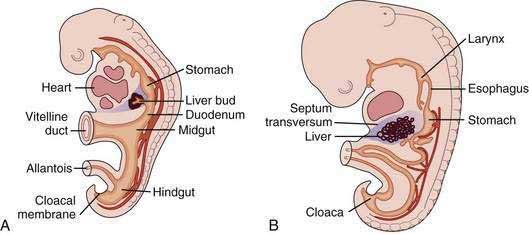
Hepatocytic differentiation involves the development of abundant rough endoplasmic reticulum and the Golgi apparatus needed for synthesis of secreted proteins. The establishment of hepatocyte cell apical-basal polarity begins as early as seven weeks.2,3 Hepatic differentiation is highly dependent on signals from the cardiogenic mesoderm and septum transversum mesenchyme, which produce fibroblast growth factor (FGF) and bone morphogenetic protein (BMP), respectively; these factors induce the expression of hepatic messenger ribonucleic acids (mRNAs).1,4 In the absence of FGF signaling from the cardiac mesoderm, the ventral endoderm develops into pancreas.2
Biliary development begins at six weeks when a subset of hepatoblasts close to the portal mesenchyme strongly express biliary-specific antigens (see Chapter 62).3,5 These biliary precursor cells form a continuous single layered ring around the portal mesenchyme, called the ductal plate. This plate becomes partly bilayered in the next step. A period of remodeling follows in which focal dilations appear between the two cell layers and eventually form lumens. The parts of the ductal plate not involved in the formation of ducts regress by apoptosis, and, around the time of birth, the remaining ducts are incorporated into the portal mesenchyme.5 This process begins in the hilum and extends along a gradient to the periphery of the liver.
The mechanism by which hepatoblasts differentiate into biliary cells is not fully understood. Hepatocyte nuclear factor (HNF)-6 is a transcription factor that regulates the number of cells that enter the biliary differentiation pathway and their localization; Hnf-6-deficient mouse embryos show numerous hepatoblasts that express biliary antigens extending into the liver parenchyma, whereas wild-type mouse embryos have far fewer cells that express biliary antigens and that are restricted to the vicinity of the portal mesenchyme.5 The Notch pathway is also involved in bile duct development; the Jagged1/Notch2 interaction may be critical for induction of biliary differentiation and repression of hepatocytic differentiation.6 Mutations in the gene that codes for the Notch ligand Jagged-1 are associated with Alagille syndrome (see Chapter 62). Longitudinal studies of patients with Alagille syndrome have shown that duct paucity develops progressively after birth, suggesting that the Notch pathway is required to maintain a differentiated phenotype rather than to initiate differentiation and morphogenesis.5 Mutations of several genes, the products of which are involved with ciliary function, lead to disrupted morphogenesis of the bile ducts and cyst formation.7 The extrahepatic bile ducts and gallbladder develop separately and become anastomosed to the intrahepatic bile ducts by an unknown mechanism.
In the initial stages of sinusoidal development, sinusoidal endothelium is continuous and non-fenestrated. This continuous fetal endothelial lining is eventually replaced by a discontinuous fenestrated endothelial lining. In the process, the sinusoidal endothelial cells lose cell markers of continuous endothelial cells, such as platelet-endothelial adhesion molecule-1 and CD34, and acquire markers of adult sinusoidal endothelial cells, such as CD4 and intracellular adhesion molecule-1.8 Thereafter, portal veins develop, followed by centrilobular veins and arteries.
Mesenchymal cells from the septum transversum transform to hepatic stellate cells and fibroblasts.8 Kupffer cells presumably originate from the yolk sac because their presence in fetal liver precedes bone marrow development.1
VASCULAR DEVELOPMENT
The developing liver incorporates the omphalomesenteric (vitelline) and umbilical (placental) veins. The vitelline veins run from the yolk sac to the heart, and as the liver invades them, the midsection of the veins becomes capillarized.9 Portions of the inferior segments of the vitelline veins regress and form a single portal vein, although the right and left vitelline circulations persist as the right and left intrahepatic portal circulations in the adult liver.9 The superior segment of the left vitelline vein regresses and the superior segment of the right vitelline vein becomes the common hepatic vein, which is incorporated into the inferior vena cava.9
The umbilical veins run from the placenta to the heart. During the 6- to 7-mm stage of human development, part of the left umbilical vein becomes the ductus venosus, which shunts placenta-derived arterial blood from the umbilical vein to the inferior vena cava, bypassing the liver; the remainder of the left umbilical vein and the right umbilical vein disappear. After birth, the obliterated prehepatic segment of the left umbilical vein becomes the round ligament of the liver (ligamentum teres hepatis) in the free edge of the falciform ligament, and the ductus venosus becomes the ligamentum venosum.3
ANATOMY
Parietal peritoneum covers the liver except for the bare area, where the liver comes in direct contact with the diaphragm and is suspended by fibrous tissue and the hepatic veins.10 The peritoneal reflections that surround the bare area comprise the superior and inferior coronary ligaments and the right and left triangular ligaments, which attach the liver to the diaphragm.
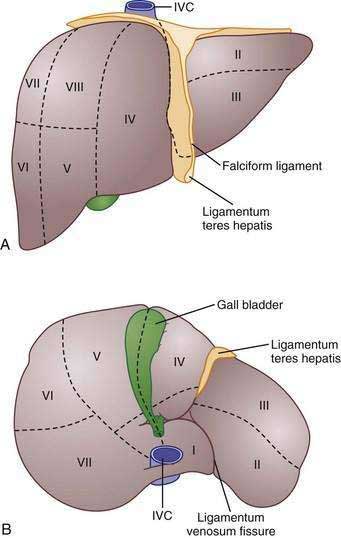
Traditionally, four lobes are distinguished in the liver based on its external appearance: right, left, caudate, and quadrate. On the anterior surface, the falciform ligament divides the liver into the right and left anatomic lobes. On the inferior surface, the quadrate lobe is defined by the gallbladder fossa, porta hepatis, and ligamentum teres hepatis. The caudate lobe is delineated by the inferior vena cava groove, porta hepatis, and ligamentum venosum fissure.11 Although these lobes are convenient and well known, these structures are not true functional lobes.
The true right and left lobes of the liver are of roughly equal size and are divided not by the falciform ligament, but by a plane passing through the bed of the gallbladder and the notch of the inferior vena cava. This plane, which has no external indications, is called the Cantlie line.10,11 Based on arterial blood supply, portal venous blood supply, biliary drainage, and hepatic venous drainage, the liver is divided into right and left functional lobes, each of which is divided into two segments, and these are further subdivided into two subsegments.10 Several systems of subdivision have been proposed, but the most widely used systems are those of Couinaud, which follows the distribution of the portal and hepatic veins,12 and Healey and Schroy, which is based on the distribution of bile ducts.13 In these systems, the subsegments are assigned numbers from 1 to 8, with the caudate lobe being subsegment 1 and the others following in a clockwise pattern (Fig. 71-2).11
The liver receives approximately 70% of its blood supply and 40% of its oxygen from the portal vein and 30% of its blood supply and 60% of its oxygen from the hepatic artery.14 The portal vein is formed from the confluence of the superior mesenteric vein and the splenic vein. At the hilum, the portal vein divides into right and left branches, upon which the right and left lobes of the liver are based.15 The hepatic artery commonly arises from the celiac trunk, although occasionally it arises from the superior mesenteric artery.15 A common variant is a left hepatic artery that branches from the left gastric artery and a right hepatic artery branch that arises from the superior mesenteric artery.15 Within the hilum, the hepatic artery lies anterior to the portal vein and to the left of the bile duct. In the liver, the arteries, portal veins, and bile ducts are surrounded by a fibrous sheath, the Glissonian sheath, whereas the hepatic veins lack this structure.10 Three major hepatic veins drain into the inferior vena cava, although in 60% to 85% of persons, the left and middle veins unite to enter the inferior vena cava as a single vein.10,15
The extrahepatic biliary tree is composed of the common hepatic duct, cystic duct, gallbladder, and right and left hepatic ducts. The right and left hepatic ducts drain the right and left lobes of the liver, respectively. The fusion of the right and left hepatic ducts gives rise to the common hepatic duct. The caudate lobe usually drains to the origin of the left hepatic duct or to the right hepatic duct. The cystic duct usually drains into the lateral aspect of the common hepatic duct below its origin.16
NERVES
Sympathetic or adrenergic nerve fibers form a rich plexus around blood vessels. Branches from the plexus supply the lobules where terminal branches surround perisinusoidal cells and hepatocytes.17 Parasympathetic (cholinergic) nerve fibers innervate extrahepatic and intrahepatic branches of the hepatic artery, portal vein, and hepatic vein.17 Intrinsic nerves regulate hepatic blood flow, biliary physiology, and metabolism. With the advent of liver transplantation, however, the importance of the hepatic nervous system has been questioned, given the adequate functioning of the denervated allograft.17,18
LYMPHATICS
Superficial lymphatics from the convex surface of the liver run through the right or left triangular ligament and the falciform ligament. They cross the diaphragm to enter precardiac, superior phrenic, and juxtaesophageal lymph nodes or travel alongside the right or left inferior phrenic artery to the celiac nodes.19 Superficial lymphatics from the visceral surface of the liver mostly run to the hepatic lymph nodes. From the caudate lobe, lymph vessels drain into precaval nodes. Deep lymphatic vessels leave the liver at the porta hepatis to drain into the foraminal node at the epiploic foramen and the superior pancreatic nodes. Lymphatic vessels that leave the liver with the hepatic veins continue in the wall of the inferior vena cava.19
HISTOLOGY
The majority of the liver is composed of hepatocytes arranged in plates or “muralium” one or two cells thick, separated by sinusoids (see also Chapter 72). Hepatocytes appear as polygonal cells with round nuclei of varying sizes with frequent binucleate cells. Portal tracts within the parenchyma contain a branch of the hepatic arteriole, portal vein, and bile duct running together as a triad and accompanied by nerve fibers and lymphatic vessels (Fig. 71-3). Terminal hepatic arterioles and terminal portal venules originate from portal tracts and supply blood to the sinusoids. The sinusoids lead mixed portal and arterial blood from the portal tract to the terminal hepatic venules (also known as central veins). These terminal hepatic venules drain into sublobular veins, then into hepatic veins, and eventually to the vena cava.
The terminal portal venules do not possess a muscle layer and so have no inlet sphincters at their junction with sinusoids; however, large endothelial cells at that junction bulge their nuclei into the lumen and, by means of contraction, control the flow of blood into the sinusoids.18 A similar outlet sphincter-like activity occurs at the site where the sinusoid connects with the terminal hepatic vein.18 In contrast to terminal portal venules, terminal hepatic arterioles are invested by smooth muscle and are capable of forming presinusoidal sphincters.18
Sinusoidal endothelial cells are the primary barrier between blood and hepatocytes (see Fig. 72-1). They are a unique type of endothelial cell in that they have fenestrations, have a limited capacity for endocytosis, and lack a basal lamina.14 The size of the fenestrae is different in the lobular periphery and in the center of the lobule.20 Contraction of actin filaments within endothelial cells controls the pore size, which in turn is controlled by calcium and calmodulin.20 Kupffer cells are macrophages that also line sinusoids and have the capacity to phagocytose large particles.20 They are more numerous, larger, and more phagocytically active in the periportal region.14 Their major role is to clear blood of senescent red blood cells and toxic endogenous and exogenous substances.20 Apart from phagocytosis, Kupffer cells handle low-density lipoproteins and produce lymphokine mediators that direct hepatocyte protein synthesis, inflammatory mediators, and hepatocyte-protective prostaglandins.18Hepatic stellate cells were formerly known as Ito cells, or fat-storing cells; they are perisinusoidal cells that are sites of fat metabolism and vitamin A storage.18 They encircle the sinusoidal wall and may regulate the width of the lumen. When hepatic stellate cells are activated, they transform into myofibroblasts that express desmin and smooth muscle actin.14
A perisinusoidal space, the space of Disse, remains between the sinusoidal lining and the vascular pole of hepatocytes and communicates with the sinusoidal space through multiple fenestrations.18 This space contains plasma and collagen types I, III, IV, and V, which act as the scaffolding of the organ.18 The space of Mall is a space between the periportal hepatocytes and portal connective tissue. Lymphatic fluid accumulates in the space of Disse and then passes into the space of Mall before draining into lymphatic vessels.14 Lymphatic vessels form a network in the portal spaces in association with branches of the hepatic artery.18
One aspect of the hepatocyte borders the sinusoid and another borders the bile canaliculus. The canaliculi direct bile to the terminal canals of Hering, which are lined partly by hepatocytes and partly by cholangiocytes.21 The canals of Hering do not stop at the limiting plate of the portal tract but extend into the periportal region of the lobule. The canals of Hering pass into bile ductules, which are lined entirely by cholangiocytes.21 The ductules in turn connect to the smallest interlobular bile ducts. Interlobular bile ducts connect to septal bile ducts and then into hepatic bile ducts. Histologically, the smaller ducts are lined by cuboidal cells, whereas the larger ducts are lined by columnar epithelial cells.
ORGANIZATION OF LIVER PARENCHYMA
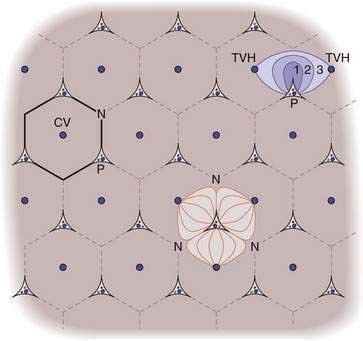
The classic lobule of the liver was described in 1833 by Kiernan as a hexagon with a central vein at its center and portal tracts at three corners. Because many glands have as the center of their functional unit a duct, Mall envisioned the basic unit of the liver to be the portal unit, defined at its center by a portal tract and at its periphery by central veins.18 The liver acinus was defined in 1954 by Rappaport as the parenchyma around terminal afferent portal and arterial vessels that supply this group of hepatocytes with blood. At the periphery of the acinus lies the terminal hepatic venule (the “central vein”) which drains several acini.18 In this model, the following three zones exist: (1) the periportal zone, or zone 1, which is supplied by blood with high oxygen content; (2) the intermediate zone (zone 2); and (3) the perivenular zone (zone 3), which receives blood that is relatively low in oxygen content.18 The acinus represents a functional and a structural unit that facilitates the description of lesions such as bridging necrosis and fibrosis (Fig. 71-4).14
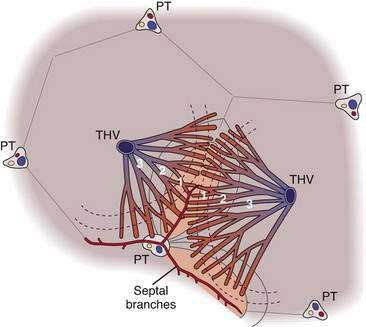
In 1982, Matsumoto and Kawakami presented a view of liver architecture based on its angioarchitecture.22 In this concept, the portal and hepatic venous systems are divided into a conducting portion, which delivers and drains blood from the parenchyma, and a parenchymal portion, which is the basis for the primary lobule. The parenchymal portion of the portal and hepatic venous systems consists of minute side branches that originate as orderly rows along the terminal branches of the conducting portion. The portal venous branches divide several times more often than the hepatic venous branches, thereby creating a larger number of portal venous channels for each hepatic venous channel. The final ramifications of the portal venous system are known as septal branches. The “central vein,” meanwhile, is actually six to eight draining venules that individually face a corresponding inflow unit. The conical cluster of hepatocytes fed by a septal branch and drained by a hepatic vein branch forms a “primary lobule.” Several primary lobules together form a classic lobule.
Matsumoto and Kawakami also noted that the sinusoids that arise from the septal branches have a transverse course near the portal tract before turning radially to the central vein, and this bed of transverse sinusoids forms a sickle-shaped “inflow front” for perfusion of the lobule that differs from the linear supply proposed by the acinus model (Fig. 71-5).22 The convex aspect of the sickle abuts a portal tract, its arms extend along septal branches, and the concave aspect faces the central vein. This arrangement defines two zones: the peripheral part of the classic lobule composed of adjoining sickle-shaped areas and the centrilobular portion bound by these sickle-shaped areas. Immunohistochemical studies of hepatic enzymes and many pathologic processes, such as centrilobular necrosis in chronic passive congestion and steatosis of the liver, highlight the presence of a continuous periportal network around portal tracts and terminal afferent vessels and a distinct concentric perivenous area around the central vein, supporting the idea that the liver architecture resembles the classic lobule more than the acinus.23
DEVELOPMENTAL ANOMALIES OF THE LIVER
RIEDEL’S LOBE
Riedel’s lobe denotes a prominent right liver lobe that extends below the level of the umbilicus. Riedel’s lobe is an anatomic variation that occurs more often in women than in men. It may be mistaken for an abdominal mass. Liver biochemical test levels are normal, and the diagnosis is established by ultrasound.3
ABERNETHY MALFORMATION
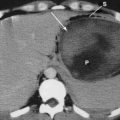
The Abernethy malformation is a congenital extrahepatic portocaval shunt. Two types of shunts are known to occur. In a type 1 shunt, portal blood is diverted completely into the inferior vena cava, with absence of the portal vein (Fig. 71-6). This type of shunt occurs more often in girls than in boys; is associated with other congenital abnormalities such as cardiac defects, biliary atresia, and polysplenia; may manifest with hypergalactosemia, hyperbilirubinemia, hyperammonemia, or variceal bleeding; and may be complicated by the formation of hepatic tumors such as focal nodular hyperplasia.24 Type 1 Abernethy malformation can be further divided into subtype 1a, in which the superior mesenteric vein and the splenic vein do not join and thus there is no anatomic portal vein, and subtype b, in which the superior mesenteric vein and splenic vein do join to form a portal vein, which then drains into a systemic vein.24,25 In type 2 Abernethy malformation, the portal vein is intact, but a side-to-side anastomosis with the inferior vena cava leads to shunting; technically, therefore, type 2 is not a true absence of the portal vein. A type 2 shunt occurs in both girls and boys and is not associated with other malformations.25,26
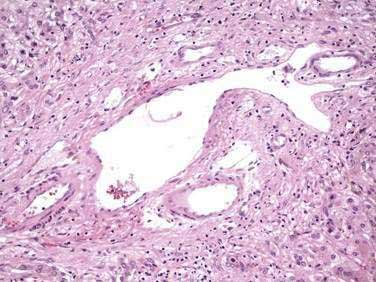
SIMPLE CYSTS
Simple cysts lined by biliary-type epithelium may arise from the biliary tree. These cysts may lose connection with the biliary tree, in which case their contents will not be bile stained (see Chapter 62).27
CHOLEDOCHAL CYSTS
Choledochal cysts are congenital dilatations of the biliary tree. They occur more often in females than in males. The location (extrahepatic, intrahepatic, or both) and character (saccular, diverticular, or fusiform) of these cysts is the basis for their classification (see Chapter 62).
DUCTAL PLATE MALFORMATION
Defects in duct morphogenesis result from a variety of mutations that affect ductal plate remodeling; they manifest as a discontinuous ring of bile duct structures encircling the portal tracts. Many of these disorders are associated with hepatic and renal cyst formation. Included in this group of disorders is congenital hepatic fibrosis, autosomal dominant and recessive polycystic kidney disease, polycystic liver disease, Caroli’s disease, and von Meyenburg complexes.3 Caroli’s disease is characterized by dilatation of the intrahepatic biliary tree. Caroli’s syndrome is defined as Caroli’s disease in conjunction with congenital hepatic fibrosis. Sporadic von Meyenburg complexes occur frequently in the periphery of the adult liver, suggesting that they represent somatic mutations that occur postnatally in a small portion of the biliary tree (see Chapter 62).27
BILIARY ATRESIA
Extrahepatic biliary atresia is obliteration of the bile duct caused by inflammation and fibrosis. Affected infants are well until several weeks of age, when they develop conjugated hyperbilirubinemia.27 Biliary atresia occurs in syndromic and nonsyndromic settings. Syndromic biliary atresia is probably of antenatal onset and is associated with cardiac and other extrahepatic malformations, often involving laterality.27 Liver biopsy specimens from syndromic cases have histologic evidence of ductal plate malformation. The predisposition to inflammation and fibrosis may be related to abnormally toxic bile or abnormal cholangiocytes that cannot withstand the detergent action of bile acids (see Chapter 62).
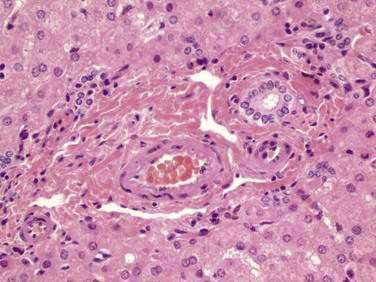
ALAGILLE SYNDROME
Alagille syndrome is characterized by bile duct paucity and chronic cholestasis (Fig. 71-7; see also Fig. 62-11), cardiac disease (most often peripheral pulmonary stenosis), characteristic facies (broad forehead, deep set eyes, straight nose, pointed chin) (see Figure 62-10), ocular abnormalities (typically posterior embryotoxon), and skeletal abnormalities (commonly butterfly vertebrae).28 The syndrome is caused by mutations or deletions of the jagged1 (JAG1) gene, which encodes the ligand for the Notch transmembrane receptor. The Notch signaling pathway functions in many cell types to regulate cell fate decisions during development (see Chapter 62).29
Bezerra JA. Liver development: A paradigm for hepatobiliary disease in later life. Semin Liver Dis. 1998;18:203-16. (Ref 3.)
Couinaud C. Le Foie. Etudes Anatomiques et Chirurgicales. Paris: Masson & Cie; 1957. (Ref 12.)
David H, Reinke P. The concept of the “perisinusoidal functional unit” of the liver—importance to pathological processes. Exp Pathol. 1987;32:193-224. (Ref 20.)
Deshpande RR, Heaton ND, Rela M. Surgical anatomy of segmental liver transplantation. Br J Surg. 2002;89:1078-88. (Ref 15.)
Duncan SA. Mechanisms controlling early development of the liver. Mech Dev. 2003;120:19-33. (Ref 2.)
Enzan H, Himeno H, Hiroi M, et al. Development of hepatic sinusoidal structure with special reference to the Ito cells. Microsc Res Tech. 1997;39:336-49. (Ref 8.)
Healey JJ, Schroy P. Anatomy of the biliary ducts within the human liver: Analysis of the prevailing pattern of branching and the major variations of the biliary ducts. Arch Surg. 1953;66:599-616. (Ref 13.)
Howard ER, Davenport M. Congenital extrahepatic portocaval shunts—the Abernethy malformation. J Pediatr Surg. 1997;32:494-7. (Ref 25.)
Knisely AS. Biliary tract malformations. Am J Med Genet. 2003;122A:343-50. (Ref 27.)
Malarkey DE, Johnson K, Ryan L, et al. New insights into functional aspects of liver morphology. Toxicol Pathol. 2005;33:27-34. (Ref 14.)
Matsumoto T, Kawakami M. The unit-concept of hepatic parenchyma—a re-examination based on angioarchitectural studies. Acta Pathol Jpn. 1982;32(Suppl 2):285-314. (Ref 22.)
Rutkauskas S, Gedrimas V, Pundzius J, et al. Clinical and anatomical basis for the classification of the structural parts of liver. Medicina (Kaunas, Lithuania). 2006;42:98-106. (Ref 11.)
Sasse D, Spornitz UM, Maly IP. Liver architecture. Enzyme. 1992;46:8-32. (Ref 18.)
Saxena R, Theise ND, Crawford JM. Microanatomy of the human liver-exploring the hidden interfaces. Hepatology. 1999;30:1339-46. (Ref 17.)
Skandalakis JE, Skandalakis LJ, Skandalakis PN, et al. Hepatic surgical anatomy. Surg Clin North Am. 2004;84:413-35. (Ref 10.)
1. Zhao R, Duncan SA. Embryonic development of the liver. Hepatology. 2005;41:956-67.
2. Duncan SA. Mechanisms controlling early development of the liver. Mech Dev. 2003;120:19-33.
3. Bezerra JA. Liver development: A paradigm for hepatobiliary disease in later life. Semin Liver Dis. 1998;18:203-16.
4. Lemaigre F, Zaret KS. Liver development update: New embryo models, cell lineage control, and morphogenesis. Curr Opin Genet Dev. 2004;14:582-90.
5. Lemaigre FP. Development of the biliary tract. Mech Dev. 2003;120:81-7.
6. Tanimizu N, Miyajima A. Notch signaling controls hepatoblast differentiation by altering the expression of liver-enriched transcription factors. J Cell Sci. 2004;117:3165-74.
7. Everson GT, Taylor MR, Doctor RB. Polycystic disease of the liver. Hepatology. 2004;40:774-82.
8. Enzan H, Himeno H, Hiroi M, et al. Development of hepatic sinusoidal structure with special reference to the Ito cells. Microsc Res Tech. 1997;39:336-49.
9. Strasberg SM. Terminology of liver anatomy and liver resections: Coming to grips with hepatic Babel. J Am Coll Surg. 1997;184:413-34.
10. Skandalakis JE, Skandalakis LJ, Skandalakis PN, et al. Hepatic surgical anatomy. Surg Clin North Am. 2004;84:413-35.
11. Rutkauskas S, Gedrimas V, Pundzius J, et al. Clinical and anatomical basis for the classification of the structural parts of liver. Medicina (Kaunas, Lithuania). 2006;42:98-106.
12. Couinaud C. Le Foie. Etudes Anatomiques et Chirurgicales. Paris: Masson & Cie; 1957.
13. Healey JJ, Schroy P. Anatomy of the biliary ducts within the human liver: Analysis of the prevailing pattern of branching and the major variations of the biliary ducts. Arch Surg. 1953;66:599-616.
14. Malarkey DE, Johnson K, Ryan L, et al. New insights into functional aspects of liver morphology. Toxicol Pathol. 2005;33:27-34.
15. Deshpande RR, Heaton ND, Rela M. Surgical anatomy of segmental liver transplantation. Br J Surg. 2002;89:1078-88.
16. Catalano OA, Singh AH, Uppot RN, et al. Vascular and biliary variants in the liver: Implications for liver surgery. Radiographics. 2008;28:359-78.
17. Saxena R, Theise ND, Crawford JM. Microanatomy of the human liver—exploring the hidden interfaces. Hepatology. 1999;30:1339-46.
18. Sasse D, Spornitz UM, Maly IP. Liver architecture. Enzyme. 1992;46:8-32.
19. Trutmann M, Sasse D. The lymphatics of the liver. Anat Embryol (Berl). 1994;190:201-9.
20. David H, Reinke P. The concept of the “perisinusoidal functional unit” of the liver—importance to pathological processes. Exp Pathol. 1987;32:193-224.
21. Roskams TA, Theise ND, Balabaud C, et al. Nomenclature of the finer branches of the biliary tree: Canals, ductules, and ductular reactions in human livers. Hepatology. 2004;39:1739-45.
22. Matsumoto T, Kawakami M. The unit-concept of hepatic parenchyma—a re-examination based on angioarchitectural studies. Acta Pathol Jpn. 1982;32(Suppl 2):285-314.
23. Gebhardt R. Metabolic zonation of the liver: Regulation and implications for liver function. Pharmacol Ther. 1992;53:275-354.
24. Niwa T, Aida N, Tachibana K, et al. Congenital absence of the portal vein: Clinical and radiologic findings. J Comput Assist Tomogr. 2002;26:681-6.
25. Howard ER, Davenport M. Congenital extrahepatic portocaval shunts—the Abernethy malformation. J Pediatr Surg. 1997;32:494-7.
26. Alvarez AE, Ribeiro AF, Hessel G, et al. Abernethy malformation: One of the etiologies of hepatopulmonary syndrome. Pediatr Pulmonol. 2002;34:391-4.
27. Knisely AS. Biliary tract malformations. Am J Med Genet. 2003;122A:343-50.
28. Kamath BM, Bason L, Piccoli DA, et al. Consequences of JAG1 mutations. J Med Genet. 2003;40:891-5.
29. Harper JA, Yuan JS, Tan JB, et al. Notch signaling in development and disease. Clin Genet. 2003;64:461-72.