[level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 58 Acute Pancreatitis
INCIDENCE AND BURDEN OF DISEASE
The incidence of acute pancreatitis in England, Denmark, and the United States varies from 4.8 to 38 per 100,000 patients.1–3 However, estimates of incidence are inaccurate because the diagnosis of mild disease may be missed, and death may occur before diagnosis in 10% of patients with severe disease.4
Diseases of the pancreas (acute and chronic pancreatitis) accounted for 327,000 inpatient hospital stays, 78,000 outpatient hospital visits, 195,000 emergency department visits, and 531,000 physician office visits in 1998.5 The cost of pancreatic diseases (direct and indirect costs) was estimated to be 2.5 billion dollars in the year 2000.5 In the same year, there were 2834 deaths in the United States from acute pancreatitis, making it the 14th most common cause of deaths due to gastrointestinal (GI) diseases.6 Acute pancreatitis ranks as the second most common inpatient GI diagnosis in the United States after cholelithiasis and acute cholecystitis and ahead of acute appendicitis.6
The incidence of acute pancreatitis appears to be increasing.7–9 As the population is becoming increasingly overweight, the incidence of gallstones, the most common cause of acute pancreatitis is rising. Unfortunately, during this same period, the overall mortality rate from acute pancreatitis has declined only gradually to approximately 5% to 10%.10–12
DEFINITIONS
Many pancreatologists use the 1992 Atlanta Symposium definition of acute pancreatitis,13 which is an acute inflammatory process of the pancreas with variable involvement of other regional tissues or remote organ systems. Acute pancreatitis is best defined clinically by a patient presenting with two of the following criteria12: (1) symptoms, such as epigastric pain, consistent with the disease; (2) a serum amylase or lipase greater than three times the upper limit of normal; or (3) radiologic imaging consistent with the diagnosis, usually using computed tomography (CT) or magnetic resonance imaging (MRI). Pancreatitis is classified as acute unless there are CT, MRI, or endoscopic retrograde cholangiopancreatography (ERCP) findings of chronic pancreatitis. Then pancreatitis is classified as chronic pancreatitis, and any episode of acute pancreatitis is considered an exacerbation of inflammation superimposed on chronic pancreatitis (see Chapter 59).
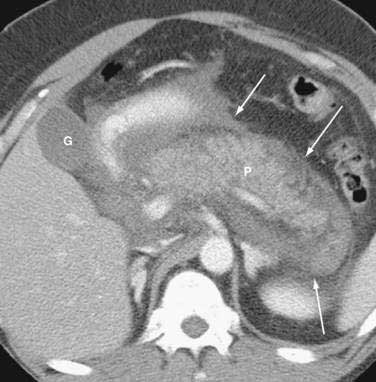
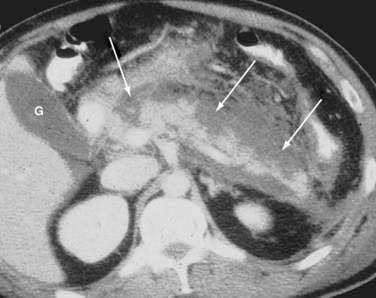
Once the diagnosis is established, patients are classified as having mild or severe pancreatitis. Mild acute pancreatitis consists of interstitial (edematous) pancreatitis on imaging, minimal or no extrapancreatic organ dysfunction, and typically an uneventful recovery. Severe pancreatitis manifests as organ failure or local complications such as necrosis, abscess, or pseudocyst. The Atlanta criteria13 (Table 58-1) defines severity by the presence of organ failure or pancreatic necrosis on dynamic contrast–enhanced CT scan (Figs. 58-1 and 58-2). Other acceptable markers of severe pancreatitis include three or more of Ranson’s 11 criteria for non-gallstone pancreatitis (Table 58-2),14 and an Acute Physiology and Chronic Health Evaluation (APACHE-II) score of greater than eight.15
Table 58-1 Atlanta Criteria for Severe Acute Pancreatitis13
| NON-GALLSTONE PANCREATITIS (1974) | GALLSTONE PANCREATITIS (1982) |
|---|---|
| At Admission | |
| Age >55 yr | Age >70 yr |
| White blood cells >16,000/mm3 | >18,000/mm3 |
| Blood glucose >200 mg/dL | >220 mg/dL |
| Serum lactate dehydrogenase >350 IU/L | >400 IU/L |
| Serum aspartate aminotransferase >250 IU/L | >250 IU/L |
| During Initial 48 hr | |
| Hematocrit decrease of >10 % | >10% |
| Blood urea nitrogen increase of >5 mg/dL | >2 mg/dL |
| Serum calcium <8 mg/dL | <8 mg/dL |
| Arterial po2 <60 mm Hg | NA |
| Serum base deficit >4 mEq/L | >5 mEq/L |
| Fluid sequestration >6 L | >4 L |
NA, not applicable.
It is important to use precise terms in describing the anatomic complications of acute pancreatitis. The ability to apply appropriate therapy depends on a clear understanding of these terms. An old term that should be used only sparingly is phlegmon. Although this term is often used by radiologists to describe an inflammatory mass, this term has carried different meaning to gastroenterologists, internists, and surgeons. Whereas patients with interstitial pancreatitis have a normally perfused gland, manifesting contrast-enhanced CT as a normal bright appearance indicating flow throughout the gland, patients with necrotizing pancreatitis have greater than 30% of the gland not perfused, with low attenuation. Pancreatic necrosis consists of focal or diffuse nonviable pancreatic parenchyma and usually peripancreatic fat necrosis. Pancreatic necrosis can be sterile or infected. Peripancreatic necrosis describes necrotic fatty and stromal tissue around the pancreas. It is more important to surgeons because this is typically not appreciated on imaging. However, the presence of peripancreatic necrosis may delineate a more complicated course for patients with acute pancreatitis. An acute fluid collection is fluid located in or near the pancreas that lacks a definite wall and typically occurs early in the course of acute pancreatitis. On CT scan these collections appear as a low attenuation mass with poor margins and no capsule. It is very difficult to distinguish acute fluid collections in the pancreatic parenchyma from pancreatic necrosis. An acute fluid collection occurs in 30% to 50% of cases of acute pancreatitis and most resolve spontaneously.16 A pseudocyst is a fluid collection that persists for 4 to 6 weeks and becomes encapsulated by a wall of fibrous or granulation tissue. Pseudocysts are located adjacent to or off the body of the pancreas. At times these enzyme-rich fluid-filled sacks can be found distantly in the pelvis and chest. When a pseudocyst is located within the body of the pancreas, the cyst may contain necrotic pancreatic debris even when the pseudocyst is fluid-appearing with low attenuation on CT. The term for a walled-off fluid-appearing pseudocyst-like structure involving the pancreas is walled off pancreatic necrosis (WOPN). A pancreatic abscess is a circumscribed intra-abdominal collection of pus occurring after an episode of acute pancreatitis or pancreatic trauma. It usually develops close to the pancreas and contains little pancreatic necrosis. Due to confusion of whether an abscess represents an infected pseudocyst or infected pancreatic necrosis, the term abscess should be used sparingly. Because of important differences in management, it is best to use the terms infected pseudocyst and infected necrosis. The term hemorrhagic pancreatitis should also be used with caution, and this term is not a synonym for necrotizing pancreatitis. Hemorrhage is more commonly associated with pseudoaneurysm, an erosion of peripancreatic blood vessels with hemoperitoneum. Unfortunately, hemorrhagic pancreatitis has more commonly been used to inappropriately describe necrotizing pancreatitis.
Of all these terms, the most important distinction is that between pancreatic necrosis and pseudocyst. WOPN is pancreatic necrosis that has liquefied after five to six weeks.17 Similar to a pseudocyst, a wall develops. However, whereas a pseudocyst always contains fluid, pancreatic necrosis, even if walled off early, contains a significant amount of debris that only becomes liquefied after five to six weeks. No attempt should be made to drain WOPN early (less than four weeks) because the debris is typically thick, often with the consistency of rubber early in the course of the disease. After five to six weeks, WOPN can be treated similar to the fluid-filled pseudocyst and drained surgically, endoscopically, or percutaneously.
NATURAL HISTORY
Acute pancreatitis appears to have two distinct stages. The first stage is related to the pathophysiology of the inflammatory cascade. This first phase usually lasts a week. During this phase, the severity of acute pancreatitis is related to extrapancreatic organ failure secondary to the patient’s systemic inflammatory response elicited by acinar cell injury. Infectious complications are uncommon at this time. Fever, tachycardia, hypotension, respiratory distress, and leukocytosis are typically related to the systemic inflammatory response syndrome (SIRS). Multiple cytokines are involved, including platelet activating factor, tumor necrosis factor-α (TNF-α) and various interleukins (ILs) (see Chapter 2).
During the first week the initial state of inflammation evolves dynamically with variable degrees of pancreatic and peripancreatic ischemia or edema to either resolution or to irreversible necrosis and liquefaction, or the development of fluid collections in and around the pancreas. The extent of the pancreatic and peripancreatic changes is usually proportional to the severity of extrapancreatic organ failure. However, organ failure may develop independent of pancreatic necrosis.17
Approximately 75% to 80%, of patients with acute pancreatitis have a resolution of the disease process (interstitial pancreatitis) and do not enter the second phase. However, in 25% of patients, a more protracted course develops, often related to the necrotizing process (necrotizing pancreatitis) lasting weeks to months. The mortality peak in the second phase is related to a combination of factors, including organ failure secondary to sterile necrosis, infected necrosis, or complications from surgical intervention.11,12,18–20 There are two peaks for mortality. Most studies in the United States and Europe reveal that about half the deaths occur within the first week or two, usually of multiorgan failure.19–21 Death can be very rapid. About one quarter of all deaths in Scotland occurred within 24 hours of admission and one third within 48 hours.21 After the second week of illness, patients succumb to pancreatic infection associated with multiorgan failure. Some studies in Europe report a very high late mortality rate from infection.22 Patients who are older and have comorbid illnesses have a substantially higher mortality rate than younger healthier patients. In those who survive their illness, severe pancreatic necrosis can scar the pancreas, resulting in a stricture of the main pancreatic duct with subsequent obstructive chronic pancreatitis and permanent diabetes and malabsorption.23
PATHOGENESIS
The initial step in the pathogenesis of acute pancreatitis is conversion of trypsinogen to trypsin within acinar cells in sufficient quantities to overwhelm normal mechanisms to remove active trypsin (see Fig. 57-3). Trypsin, in turn, catalyzes conversion of proenzymes, including trypsinogen and inactive precursors of elastase, phospholipase A2 (PLA2), and carboxypeptidase, to active enzymes. Trypsin also may activate the complement and kinin systems. Active enzymes autodigest the pancreas and initiate a cycle of releasing more active enzymes. Normally small amounts of trypsinogen are spontaneously activated within the pancreas, but intrapancreatic mechanisms quickly remove activated trypsin. Pancreatic secretory trypsin inhibitor (PSTI, now called SPINK1) binds and inactivates about 20% of the trypsin activity. Other mechanisms for removing trypsin involve mesotrypsin, enzyme Y, and trypsin itself, which splits and inactivates trypsin. The pancreas also contains nonspecific antiproteases such as α1-antitrypsin and α2-macroglobulin. Additional protective mechanisms are the sequestration of pancreatic enzymes within intracellular compartments of the acinar cell during synthesis and transport and the separation of digestive enzymes from lysosomal hydrolases as they pass through the Golgi apparatus, which is important because cathepsin B activates trypsin from trypsinogen. Low intra-acinar calcium concentrations also prevent further autoactivation of trypsin.
In experimental pancreatitis, activation of trypsin occurs within 10 minutes, and large amounts of trypsin24 and increased concentrations of trypsinogen activation peptide (TAP) accumulate within the pancreas.25,26 TAP is cleaved when trypsinogen is activated to trypsin, and concentrations of TAP in plasma, urine, and ascites correlate with the severity of the pancreatic inflammatory response, with the highest levels associated with acinar necrosis and intrapancreatic hemorrhage.27,28
Co-localization of pancreatic enzymes in lysosomes, followed by acinar cell injury, is an attractive hypothesis for the pathogenesis of acute pancreatitis, but the relevance of co-localization to the pathogenesis of acute pancreatitis is unclear. Activation of trypsinogen occurs before biochemical or morphologic injury to acinar cells, in association with co-localization of lysosomal enzymes, such as cathepsin B, and digestive enzymes, including trypsinogen within unstable vacuoles.27,28 Complete inhibition of pancreatic cathepsin B activity in vitro prevents trypsinogen activation induced by the cholecystokinin (CCK) analog cerulein,29 supporting the co-localization hypothesis. Thus, complete inhibition of cathepsin B may prevent or be a treatment for acute pancreatitis. However, enzyme co-localization may occur without inducing significant acinar cell injury.30
Two other features of experimental acute pancreatitis are early blockade of the secretion of pancreatic enzymes while enzyme synthesis continues and disruption of the paracellular barrier of acinar cells and intralobular pancreatic duct cells. The disruption facilitates the extravasation of pancreatic enzymes from acinar cells and from the duct lumen into interstitial spaces. This phenomenon may explain the rapid development of interstitial edema and the increase of pancreatic enzymes in the serum.31
As discussed in Chapter 57, the discovery of genetic mutations associated with hereditary pancreatitis also lends support to the hypothesis that intrapancreatic activation of pancreatic zymogens is central to the pathogenesis of acute pancreatitis.32–35 The mutant trypsin in hereditary pancreatitis (usually R122H or N29I mutation) causes trypsin to be resistant to lysis or causes premature trypsinogen activation (gain of function mutation) leading to autodigestion of the pancreas and episodes of acute pancreatitis.36,37
Mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene have also been implicated in pancreatitis (see Chapter 57). CFTR anion channel allows for chloride and bicarbonate secretion into the pancreatic ducts and thus allows flushing of the liberated enzymes and proenzymes into the duodenum. There are more than 1200 mutations that have been described for the CFTR gene. Some of these are considered severe and some mild. Homozygote severe mutations produce a viscid, concentrated, acidic pancreatic juice leading to ductal obstruction and pancreatic insufficiency in infanthood. Heterozygotes of minor or major mutations may lead to acute recurrent or chronic pancreatitis by altering acinar or ductal cell function (e.g., alteration of bicarbonate conductance).
A third genetic abnormality associated with pancreatitis is mutation of the SPINK1 gene.38 As noted, SPINK1 protects the pancreatic acinar cell by inhibiting prematurely activated trypsin. Mutations of this gene presumably limit the activity of this protein, but the exact mechanism is unclear.
The pathogenesis of gallstone-related pancreatitis is unknown (see Chapter 65). Factors that may initiate gallstone pancreatitis include reflux of bile into the pancreatic duct39,40 or obstruction of the pancreatic duct at the ampulla from stone(s) or edema resulting from the passage of a stone.41 Reflux of bile into the pancreatic duct could occur when the distal bile and pancreatic ducts form a common channel and a gallstone becomes impacted in the duodenal papilla. Alternatively, bile could reflux into the pancreatic duct from the duodenum through an incompetent sphincter of Oddi injured by recent passage of a gallstone.
Experimentally, reflux of bile into the pancreatic duct, particularly if infected or mixed with pancreatic enzymes, causes pancreatic injury. Mixtures of bile and pancreatic enzymes increase the permeability of the main pancreatic duct, which is associated with local parenchymal inflammation.42 The common channel theory is somewhat problematic because pancreatic duct pressure is invariably higher than bile duct pressure, making bile reflux into the pancreatic duct unlikely. Reflux of bile from the duodenum also is unlikely because pancreatitis does not occur in conditions with easily demonstrable reflux, such as after surgical sphincteroplasty or endoscopic sphincterotomy.
A popular opinion for the mechanism of gallstone pancreatitis is that an impacted gallstone in the distal bile duct obstructs the pancreatic duct, which increases pancreatic pressure, thereby damaging ductal and acinar cells. Experiments in the opossum that support this theory are the observations that ligation of the pancreatic duct causes severe necrotizing pancreatitis41 and that decompression of the ductal system within three days prevents progression to acinar cell necrosis and severe inflammation.43
PATHOPHYSIOLOGY
The release of pancreatic enzymes damages the vascular endothelium, the interstitium, and acinar cells.43–45 Acinar injury leads to expression of endothelial adhesion molecules (e.g., VCAM-1), which further propagates the inflammatory response.46 Microcirculatory changes, including vasoconstriction, capillary stasis, decreased oxygen saturation, and progressive ischemia, occur early in experimental acute pancreatitis. These abnormalities increase vascular permeability and lead to edema of the gland (edematous or interstitial pancreatitis). Vascular injury could lead to local microcirculatory failure and amplification of the pancreatic injury. It is uncertain whether ischemia-reperfusion injury occurs in the pancreas.40 Reperfusion of damaged pancreatic tissue could lead to the release of free radicals and inflammatory cytokines into the circulation, which could cause further injury. In early stages of animal and human pancreatitis, activation of complement and the subsequent release of C5a play significant roles in the recruitment of macrophages and polymorphonuclear leukocytes.47–49 Active granulocytes and macrophages release proinflammatory cytokines in response to transcription factors such as nuclear factor κB (NF-κB). Proinflammatory cytokines include TNF, IL-1, IL-6, and IL-8, and platelet-activating factor (PAF). Proinflammatory cytokines frequently are followed by anti-inflammatory cytokines (IL-2, IL-10, IL-11) that attempt to down-regulate inflammation.48 Other mediators of inflammation include arachidonic acid metabolites (prostaglandins, PAF, and leukotrienes), nitric oxide, proteolytic and lipolytic enzymes, and reactive oxygen metabolites that overwhelm scavenging by endogenous antioxidant systems. These substances also interact with the pancreatic microcirculation to increase vascular permeability, which induces thrombosis and hemorrhage and leads to pancreatic necrosis. A recent study suggests that gene polymorphisms that affect acinar cell glutathione concentrations may lead to increased oxidant stress and more severe pancreatitis.50 Meanwhile, ischemia and severe inflammation of the gland can lead to disruption of the main and secondary pancreatic ducts, leading to local fluid accumulations within and surrounding the pancreas that can eventuate into pseudocysts.51,52
Some patients with severe pancreatic damage develop systemic complications, including fever, acute respiratory distress syndrome (ARDS), pleural effusions, renal failure, shock, myocardial depression, and metabolic complications. SIRS is common in patients with acute pancreatitis and is probably mediated by activated pancreatic enzymes (phospholipase, elastase, trypsin) and cytokines (TNF, PAF) released into the portal circulation from the inflamed pancreas.53 Cytokines reaching the liver activate hepatic Kupffer cells, which, in turn, induces hepatic expression and secretion of cytokines into the systemic circulation. These cause acute phase protein synthesis (C-reactive protein [CRP], IL-6) and may cause SIRS and damage to the kidneys, lungs, and other organs leading to multiorgan dysfunction and failure.54
ARDS may be induced by active phospholipase A (lecithinase), which digests lecithin, a major component of lung surfactant. Acute renal failure has been explained on the basis of hypovolemia and hypotension. Myocardial depression and shock are likely secondary to vasoactive peptides and a myocardial depressant factor. Metabolic complications include hypocalcemia, hyperlipidemia, hyperglycemia with or without ketoacidosis, and hypoglycemia. The pathogenesis of hypocalcemia is multifactorial and includes hypoalbuminemia (the most important cause), hypomagnesemia, calcium-soap formation, hormonal imbalances (e.g., involving parathyroid hormone, calcitonin, and glucagon), binding of calcium by free fatty acid–albumin complexes, intracellular translocation of calcium, and systemic exposure to endotoxin.55
Pancreatic infection (infected necrosis and infected pseudocyst) can occur from the hematogenous route or from translocation of bacteria from the colon into the lymphatics. Under normal circumstances bacterial translocation does not occur because there are complex immunologic and morphologic barriers. However, during acute pancreatitis, these barriers break down, which can result in local and systemic infection.56 Penetration of the gut barrier by enteric bacteria is likely due to gut ischemia secondary to hypovolemia and pancreatitis-induced arteriovenous shunting in the gut.57 Indeed, in canine experimental pancreatitis, luminal Escherichia coli translocate to mesenteric lymph nodes and distant sites.58 In feline experimental pancreatitis, enclosing the colon in impermeable bags prevents translocation of bacteria from the colon to the pancreas.59
PREDISPOSING CONDITIONS
Many conditions predispose to acute pancreatitis to varying degrees (Table 58-3). This list will undoubtedly continue to grow, and the number of cases diagnosed as “idiopathic” will decrease as our understanding of the disease improves. Gallstones and chronic alcohol abuse account for 70% of acute pancreatitis in the United States.
Table 58-3 Conditions That Predispose to Acute Pancreatitis
OBSTRUCTION
Gallstones
The most common obstructive process leading to pancreatitis is gallstones, which cause approximately 40% of cases acute pancreatitis,60 although only 3% to 7% of patients with gallstones develop pancreatitis. Gallstone pancreatitis is more common in women than men because gallstones are more frequent in women.61 Acute pancreatitis occurs more frequently when stones are less than 5 mm in diameter (odds ratio, 4 to 5),62 because small stones are more likely than large stones to pass through the cystic duct and cause ampullary obstruction. Cholecystectomy and clearing the bile duct of stones prevents recurrence, confirming the cause-and-effect relationship.61
Biliary Sludge and Microlithiasis
Biliary sludge is a viscous suspension in gallbladder bile that may contain small (<3 mm) stones (i.e., microlithiasis).63 Because small stones can hide in biliary sludge, the two are commonly referred together as biliary sludge and microlithiasis. Biliary sludge is asymptomatic in most patients. It is usually composed of cholesterol monohydrate crystals or calcium bilirubinate granules.64 On ultrasonography, sludge produces a mobile, low-amplitude echo that does not produce an acoustic shadow and that layers in the most dependent part of the gallbladder.
Sludge may result from functional bile stasis, such as that associated with prolonged fasting or total parenteral nutrition, or from mechanical stasis such as occurs in distal bile duct obstruction. In addition, the cephalosporin antibiotic ceftriaxone can complex with bile to form a sludge within the biliary system when its solubility in bile is exceeded; this rarely causes stones65 and the sludge disappears after stopping the drug. Commonly, biliary sludge is associated with idiopathic acute pancreatitis. However, the association between biliary sludge and acute pancreatitis is unproved. There is no prospective, randomized study documenting that removing sludge or microcrystals by cholecystectomy prevents further attacks of pancreatitis. Nevertheless, results of two uncontrolled studies suggest that biliary sludge can lead to pancreatitis, and that cholecystectomy, papillotomy, or ursodeoxycholic acid therapy reduces recurrent attacks of acute pancreatitis.64,66 In these two studies, the incidence of biliary sludge in presumed idiopathic pancreatitis was 67% and 74%, respectively. However, other investigators have detected microlithiasis or sludge in less than 10% of patients with recurrent acute pancreatitis.67,68 Until prospective controlled studies clarify the proper treatment of sludge and microlithiasis, firm recommendations about therapy cannot be made. Choices include cholecystectomy, ursodeoxycholic acid therapy, endoscopic sphincterotomy, or watchful waiting.
Tumors
Tumors, presumably by obstructing the pancreatic duct, can cause recurrent acute pancreatitis especially in individuals older than age 40 (see Chapter 60). The most common tumor that presents in this manner is intraductal papillary mucinous neoplasm (IPMN).69 Pancreatic adenocarcinoma can also present as acute pancreatitis in a small percentage of patients.70 Metastases from other primary tumors (lung, breast) to the pancreas have also caused pancreatitis.71 Large adenomas of the major papilla can likewise occasionally be the cause of obstructive pancreatitis.
Other Obstructive Causes
Other obstructive conditions that are rarely associated with acute pancreatitis are conditions discussed elsewhere in this text and include choledochoceles,72 duodenal diverticula,73 annular pancreas,74 and parasites that obstruct the pancreatico-biliary system such as ascaris75 or clonorchis.76 Ascariasis obstructing the pancreatic duct represents the second most common cause of acute pancreatitis in Kashmir.60
ALCOHOL, OTHER TOXINS, AND DRUGS
Ethyl Alcohol
Alcohol causes at least 30% of cases of acute pancreatitis,77 and alcohol is the most common etiology of chronic pancreatitis in developed countries. Interestingly, only 10% of chronic alcoholic patients develop chronic pancreatitis. The classic teaching is that alcohol causes chronic pancreatitis, and that alcoholic patients who present with clinically acute pancreatitis have underlying chronic disease.16 However, a few patients with alcohol-induced acute pancreatitis by clinical criteria do not have or progress to chronic pancreatitis, even with continued alcohol abuse.77,78 By contrast, a small percentage of chronic alcoholic patients develop attacks of acute pancreatitis that are indistinguishable from other forms of acute pancreatitis, but eventually develop chronic pancreatitis after 10 to 20 years of alcohol abuse. Early in the course of the disease, when attacks occur, the diagnosis of underlying chronic pancreatitis is difficult without tissue specimens because the diagnosis of chronic pancreatitis is usually made after definite signs of chronic pancreatitis appear (e.g., pancreatic calcification, exocrine and endocrine insufficiency, or typical duct changes by CT or ERCP). Most of the models described suggest possible mechanisms of alcohol-related injury, including perturbations in exocrine function, changes in cellular lipid metabolism, induction of oxidative stress, and activation of stellate cells. However, the exact mechanism remains unclear and may be related to other factors.
Bordalo and colleagues79 first proposed that alcohol was directly toxic to the acinar cell through a change in cellular metabolism. Alcohol produces cytoplasmic lipid accumulation within the acinar cells, leading to fatty degeneration, cellular necrosis, and eventual widespread fibrosis. Fatty acid ethyl esters, by-products of pancreatic ethanol metabolism, may be the key factor in this “toxic metabolic” change. Bordalo and colleagues suggested that alcohol produces a stepwise progression from fatty accumulation to fibrosis (direct toxic effects on cellular metabolism). The main limitation to this toxic-metabolic theory of alcohol toxicity is the lack of proof of the steatopancreatitis precursor to fibrosis seen in liver disease.80
Sarles80 emphasized the duality of acute and chronic pancreatitis; they were separate diseases with distinct pathogenesis. Whereas acute pancreatitis can be precipitated in patients with gallstones immediately, alcoholism requires years of toxin exposure. Alcohol modulates exocrine function to increase the lithogenicity of pancreatic fluid, leading to the formation of protein plugs and stones. Chronic contact of the stones with the ductal cells produces ulceration and scarring, resulting in obstruction, stasis, and further stone formation. Eventually, atrophy and fibrosis develop as a result of this obstructive process. Several studies have provided mechanisms in which alcohol promote stone formation, including the known precipitation of GP-2 (a Tamm-Horsfall–like protein),81 increased secretion and viscosity of pancreatic juice, and hypersecretion of enzymes and lactoferrin. In addition to these ethanol-mediated perturbations in pancreatic exocrine function, specific proteins have been implicated in stone formation. Pancreatic stones consist of a calcium carbonate crystalline lattice interspersed within a gel-like matrix formed of multiple fibrillar proteins and polysaccharides.82
In contrast to the stone theory, which is based on the de novo development of fibrosis without acute pancreatitis, the necrosis-fibrosis hypothesis envisions the development of fibrosis from recurrent, perhaps subclinical, acute pancreatitis. Inflammation and necrosis from the initial episodes of acute pancreatitis produce scarring in the periductular areas and scarring leads to obstruction of the ductules leading to stasis within the duct and subsequent stone formation. Support for this theory comes from histopathologic studies that revealed mild perilobular fibrosis in resolving acute pancreatitis, with marked fibrosis with ductal distortion occurring later. It is thought that a stepwise progression occurs to fibrosis from recurrent episodes of acute pancreatitis. Support for this theory is seen in a clinical study by Ammann and colleagues.83 In this study, 254 patients were prospectively followed after the first episode of alcoholic pancreatitis. There was a direct correlation between the frequency and severity of attacks to the rate of progression to chronic pancreatitis.
Whitcomb and Schneider have proposed an interesting hypothesis for chronic pancreatitis which unifies and incorporates recent knowledge in an attempt to reconcile prior theories.84 In at-risk individuals, the pancreatic acinar cells are stimulated by alcohol. Fibrosis does not occur because a profibrotic cellular infiltrate is not yet present. A sentinel event occurs as trypsinogen is activated. This event results in a massive inflammatory response. Cytokines are then released and work with the activation of stellate cells in the late phase. The attraction and activation of stellate cells set the stage for the development of fibrosis. If the inciting factors are removed, then the pancreas returns to normal. If the inciting factor, alcohol, is not removed, the acinar cells continue to secrete cytokines in response to the oxidative stress and the stellate cells continue to be activated. This model, using a sentinel event also represents a time for disease modifying therapy, when such therapy becomes available.
Other Toxins
Methyl alcohol,85 organophosphorous insecticides,86 and the venom of the Trinidad scorpion87 have been reported to induce pancreatitis. The mechanism of the latter two is thought to be by hyperstimulation of the pancreas. Smoking increases the risk of alcoholic and idiopathic pancreatitis, but not gallstone pancreatitis.88
Drugs
Medications are an infrequent but an important cause of acute pancreatitis.89 More than 120 drugs have been implicated, mostly from anecdotal case reports. Many case reports suffer from a combination of inadequate criteria for the diagnosis of acute pancreatitis, failure to rule out more common causes, or a lack of a rechallenge with the medication. Drug-induced pancreatitis rarely is accompanied by clinical or laboratory evidence of a drug reaction, such as rash, lymphadenopathy, or eosinophilia. Although a positive rechallenge with a drug is the best evidence available for cause and effect, it is not proof. It is clear that many patients with idiopathic pancreatitis or microlithiasis have recurrent attacks of acute pancreatitis. Therefore, stopping and restarting a drug with recurrence of pancreatitis may be a coincidence and not cause and effect. Despite the lack of a rechallenge, a drug may be strongly suspected if there is a consistent latency among the case reports between initiating the drug and the onset of acute pancreatitis. Table 58-4 shows the drugs with the greatest evidence for causing acute pancreatitis, those with rechallenges or with a relatively predictable latency.89
Table 58-4 Drugs Associated with Acute Pancreatitis*
* Class 1 and class 2 drugs. For class 1 drugs: two or more case reports published, absence of other causes of acute pancreatitis, rechallenge documented in at least one report. For class 2 drugs: four or more case reports published, absence of other causes of acute pancreatitis, consistent latency in at least 75% of cases published.
From Badalov N, Baradarian R, Iswara K, et al. Drug induced acute pancreatitis: An evidence based approach. Clin Gastroenterol Hepatol 2007; 101:454-76.
METABOLIC DISORDERS
Hypertriglyceridemia
Hypertriglyceridemia is perhaps the third most common identifiable cause of pancreatitis after gallstones and alcoholism. Serum triglyceride concentrations greater than 1000 mg/dL (11 mmol/L) may precipitate attacks of acute pancreatitis. Patients may have lactescent (milky) serum owing to increased concentrations of chylomicrons.90 The pathogenesis of hypertriglyceridemic pancreatitis is unclear, but the release of free fatty acids by lipase may damage pancreatic acinar cells or capillary endothelium.91
Hypertriglyceridemia may cause up to 5% of cases of acute pancreatitis. The association between hypertriglyceridemia and acute pancreatitis is best defined in children with rare inherited disorders of lipoprotein metabolism and severe hypertriglyceridemia92,93 who develop acute pancreatitis in early childhood. These children are homozygous for lipoprotein lipase deficiency or, even less commonly, apoprotein-CII (APO-CII) deficiency. Acute pancreatitis develops in 35%, 15%, and 30% to 40% of patients with type I, IIb, and V hyperlipidemia, respectively. Lowering serum triglyceride levels to less than 200 mg/dL (2.2 mmol/L) can prevent pancreatitis.
Most adults with hyperchylomicronemia have a mild form of genetically inherited type I or type V hyperlipoproteinemia and an additional acquired condition known to raise serum lipids (e.g., alcohol abuse, obesity, diabetes mellitus, hypothyroidism, pregnancy, estrogen91 or tamoxifen therapy, glucocorticoid excess, nephrotic syndrome, thiazide or beta blocker therapy). Typically three types of patients develop hypertriglyceridemia-induced pancreatitis. The first is a poorly controlled diabetic patient with a history of hypertriglyceridemia. The second is an alcoholic patient with hypertriglyceridemia detected on hospital admission. The third (15% to 20%) is a nondiabetic, nonalcoholic, nonobese person who has drug- or diet-induced hypertriglyceridemia. Drug-induced disease is more likely to occur if there is a background of hypertriglyceridemia prior to drug exposure.
Most persons who abuse alcohol have moderate, but transient, elevations of serum triglyceride levels. This condition is likely an epiphenomenon and not the cause of their pancreatitis94 because alcohol itself not only damages the pancreas but also increases serum triglyceride concentrations in a dose-dependent manner. For example, serum triglyceride concentrations greater than 227 mg/dL (2.5 mmol/L) occurred in 10%, 14%, and 20% of people who drank three to five, six to eight, or nine or more drinks per day, respectively.95 Alcoholic patients with severe hyperlipidemia often have a coexisting primary genetic disorder of lipoprotein metabolism.
Hypercalcemia
Hypercalcemia, of any cause, is rarely associated with acute pancreatitis. Proposed mechanisms include deposition of calcium in the pancreatic duct and calcium activation of trypsinogen within the pancreatic parenchyma.96 The low incidence of pancreatitis in chronic hypercalcemia suggests that factors other than the serum calcium per se (e.g., acute elevations of serum calcium) are responsible for pancreatitis. Acute calcium infusion into rats leads to conversion of trypsinogen to trypsin, hyperamylasemia, and dose-dependent morphologic changes of acute pancreatitis such as edema and acinar cell necrosis.
Hypercalcemia due to hyperparathyroidism has been associated with pancreatitis. However, primary hyperparathyroidism causes less than 0.5% of all cases of acute pancreatitis, and the incidence of acute pancreatitis in patients with hyperparathyroidism varies from 0.4% to 1.5%.97 Rarely, pancreatitis occurs with other causes of hypercalcemia, including metastatic bone disease, total parenteral nutrition, sarcoidosis, vitamin D toxicity, and infusions of calcium in high doses perioperatively during cardiopulmonary bypass.
INFECTIONS
Many infectious agents may cause acute pancreatitis,76 but often published reports do not meet usual standards for the diagnosis of pancreatitis or the infection. Using modern criteria for diagnosis of pancreatitis, definite pancreatitis exists if there is surgical, autopsy, or radiologic evidence; probable pancreatitis exists if there is biochemical evidence (more than three times the elevation of serum lipase or amylase) plus characteristic symptoms; and possible pancreatitis exists if there is only asymptomatic biochemical evidence. The definite criterion for an infection causing pancreatitis is finding the organism in the pancreas or pancreatic duct by stain or culture. Probable criteria for infection are culture of the organism from pancreatic juice or blood or serologic evidence combined with a characteristic clinical or epidemiologic setting. The criterion for a possible infection is culture of the organism from other body sites or serologic evidence of infection.
Using these criteria, definite pancreatitis has been associated with viruses (mumps, coxsackievirus, hepatitis B, cytomegalovirus, varicella-zoster, herpes simplex, Epstein-Barr, hepatitis A, and hepatitis C); the vaccine that contains attenuated measles, mumps, and rubella (MMR); bacteria (Mycoplasma, Legionella, Leptospira, Salmonella, tuberculosis, and brucellosis); fungi (Aspergillus and Candida albicans); and parasites (Toxoplasma, Cryptosporidium, Ascaris, Clonorchis sinensis). C. sinensis and Ascaris cause pancreatitis by blocking the main pancreatic duct. In acquired immunodeficiency syndrome (AIDS), infectious agents causing acute pancreatitis include cytomegalovirus, Candida, Cryptococcus neoformans, Toxoplasma gondii, and possibly Mycobacterium avium complex.76
An infectious agent should be suspected of causing acute pancreatitis if the characteristic syndrome caused by the infectious agent is present because this occurs 70% of the time.76 Because an infectious agent may be found in the pancreas without pancreatitis, routine search for an infection in idiopathic pancreatitis is not recommended because false-positive test results may result. In addition, it is unknown whether treating an infectious agent reverses pancreatic pathology.
VASCULAR DISEASE
Rarely, pancreatic ischemia causes pancreatitis. In most cases it is mild, but fatal necrotizing pancreatitis may occur. Ischemia may result from vasculitis (systemic lupus erythematosus)98 and polyarteritis nodosa,99 atheromatous embolization of cholesterol plaques from the aorta to the pancreas after transabdominal angiography,100 intraoperative hypotension,101 hemorrhagic shock,102 ergotamine overdose, and transcatheter arterial embolization for hepatocellular carcinoma. Also, ischemia is one possible explanation for pancreatitis after cardiopulmonary bypass. In pigs, cardiogenic shock induced by pericardial tamponade causes vasospasm and selective pancreatic ischemia due to activation of the renin-angiotensin system.103 Acute pancreatitis has occurred in long-distance runners, which may be on an ischemic basis.104
TRAUMA
Either penetrating trauma (gunshot or stab wounds) or blunt trauma can damage the pancreas.105 In most cases there is also injury to adjacent viscera. Laparotomy is essential in all cases of penetrating trauma to assess and treat all intra-abdominal injuries, including those to the pancreas. Blunt trauma results from compression of the pancreas by the spine, such as in an automobile accident. In blunt trauma it is important to determine preoperatively whether there is injury to the pancreas because depending on the severity of pancreatic injury, it will be necessary to include the pancreas in the surgical plan. Secondly, even in the absence of serious injury to adjacent organs, surgery or endoscopic therapy may be necessary to treat a pancreatic ductal injury.
Diagnosis is highly dependent on CT, MRI, or magnetic resonance cholangiopancreatography (MRCP), which may show enlargement of a portion of the gland caused by a contusion or subcapsular hematoma, pancreatic inflammatory changes, or fluid within the anterior pararenal space if there is ductal disruption. The CT may be normal during the first two days despite significant pancreatic trauma. If there is a strong clinical suspicion of pancreatic injury or if the CT or MRCP scan shows an abnormality, ERCP is required to define whether there is pancreatic duct injury. If the pancreatic duct is intact and there are no other significant intra-abdominal injuries, surgery is not required. However, if ERCP reveals duct transection with extravasation of pancreatic fluid and there are no other intra-abdominal injuries, stenting of the pancreatic duct may be successful.106 Serious injuries to the pancreas can be treated with appropriate débridement. Associated injuries to the duodenum or bile duct can be treated by biliary diversion, gastrojejunostomy, and feeding jejunostomy. External pancreatic fistulas occur in approximately one third of patients after surgery for pancreatic trauma. Octreotide may be beneficial after pancreatic injury.107
POST-ERCP
Acute pancreatitis is the most common and feared complication of ERCP, associated with substantial morbidity and occasional mortality. About 500,000 ERCPs are performed annually in the United States. Asymptomatic hyperamylasemia occurs after 35% to 70% of ERCPs.108 Acute pancreatitis occurs in 5% of diagnostic ERCPs, 7% of therapeutic ERCPs, and up to 25% in those with suspected sphincter of Oddi dysfunction or in those with a history of post-ERCP pancreatitis.109 About half the cases are moderate to severe in intensity.
The mechanisms that lead to post-ERCP pancreatitis are complex and not fully understood. Rather than a single pathogenesis, post-ERCP pancreatitis is believed to be multifactorial, involving a combination of chemical, hydrostatic, enzymatic, mechanical, and thermal factors. Although there is some uncertainty in predicting which patients will develop acute pancreatitis following ERCP, a number of risk factors acting independently or in concert have been proposed as predictors of post-ERCP pancreatitis (Table 58-5).110–113 Identification of these risk factors for post-ERCP pancreatitis is essential to recognize cases in which ERCP should be avoided if possible, or in which protective endoscopic or pharmacologic interventions should be considered.
Table 58-5 Factors That Increase the Risk of Post-ERCP Pancreatitis
| Patient Related |
| Young age, female gender, suspected sphincter of Oddi dysfunction, recurrent pancreatitis, history of post-ERCP pancreatitis, normal serum bilirubin |
| Procedure Related |
| Pancreatic duct injection, difficult cannulation, pancreatic sphincterotomy, precut access, balloon dilation |
| Operator or Technical Related |
| Trainee (fellow) participation, nonuse of a guidewire for cannulation, nonuse of a pancreatic duct stent in high-risk procedures |
ERCP, endoscopic retrograde cholangiopancreatography.
In general, the more likely a patient is to have an abnormal bile duct or pancreatic duct, the less likely the patient will develop post-ERCP pancreatitis. Cheng created a 160 variable database that prospectively evaluated more than 1000 patients from 15 centers in the midwestern United States.112 Their study emphasized the role of patient factors, including age, sphincter of Oddi dysfunction (SOD), prior history of post-ERCP pancreatitis, and technical factors, including number of pancreatic duct (PD) injections, minor papilla sphincterotomy, and operator experience. The patient most at risk of developing post-ERCP pancreatitis was a woman with suspected choledocholithiasis and normal bilirubin, who underwent a sphincterotomy and no stone was found. In this patient population, more than a quarter of patients (27%) developed post-ERCP pancreatitis. MRCP and endoscopic ultrasound, which do not cause pancreatitis, can provide useful information (perhaps as accurate as ERCP) in many of these cases and are preferred modalities in the initial evaluation of such patients.
Early recognition of post-ERCP pancreatitis may be possible by evaluating serum amylase or lipase after the procedure.114,115 In a study that involved 231 patients, the two-hour serum amylase and lipase were more accurate than a clinical assessment in distinguishing nonpancreatitis abdominal pain from post-ERCP acute pancreatitis. Serum values greater than 276 IU/L for amylase and greater than 1000 IU/L for lipase obtained from serum two hours after the procedure had almost a 100% positive predictive value for post-ERCP pancreatitis.116 More recently, Ito and colleagues found that if the serum amylase was normal at three hours, only 1% of patients had post-ERCP pancreatitis compared with 39% if the amylase was greater than five times the upper limit of normal.117 A serum amylase or lipase alone should not guide a decision of whether a patient has post-ERCP pancreatitis. However, it appears that the tests may assist the clinician’s assessment of a patient with post-ERCP abdominal pain.
Although there has been an interest in developing medications that can prevent post-ERCP pancreatitis, studies have failed to identify a medication worthy of widespread use. In terms of attenuating the inflammatory response, the most promising results have been seen with nonsteroidal anti-inflammatory drugs (NSAIDs). Two clinical trials have been published evaluating the role of diclofenac in reducing the incidence of post-ERCP pancreatitis.116,118 Both trials placed patients on 100 mg of diclofenac by rectal suppository and both showed a reduction in the incidence of acute pancreatitis. However, another trial failed to show any benefit to diclofenec in preventing post-ERCP pancreatitis.119
It has been suggested that relaxation of the sphincter of Oddi following ERCP will promote pancreatic drainage and prevent acute pancreatitis. Several agents have been used in the effort to relax the sphincter of Oddi with the purpose of preventing post-ERCP pancreatitis. There have been three placebo-controlled randomized studies evaluating the use of nitroglycerin during ERCP, with negative results.120–122 Furthermore, trials of oral nifedipine,123,124 sprayed lidocaine,125 and injected botulinum toxin126 failed to demonstrated any benefit in the reduction of severity or incidence of post-ERCP pancreatitis. There have been several studies evaluating the role of glucocorticoids using a variety of agents including oral prednisolone or intravenous hydrocortisone or methylprednisolone, with essentially no benefit in reduction of severity or incidence of post-ERCP pancreatitis.
Gabexate is a protease inhibitor with anti-inflammatory properties. The in vivo effect of gabexate on inhibiting circulating trypsin is greater than most other protease inhibitors. In 1995, Messori and colleagues127 published a meta-analysis of five trials128–132 showing a statistically significant reduction in the incidence of complications in patients receiving gabexate after the development of post-ERCP pancreatitis. However, the trials were small and had a limited number of patients. Several additional trials have been published with conflicting results.133–137 Although data are conflicting, it appears that infusions of the drug would likely need to be started 1 to 2 hours pre-ERCP and continued for 12 hours following ERCP to show a beneficial effect of the drug. In patients with a low risk, the costs likely outweigh any benefit. Currently, gabexate is not available in the United States.
Theoretically, inhibition of exocrine pancreatic secretion could prevent post-ERCP pancreatitis by inducing “rest” to a damaged gland. Although an attractive concept, there is little scientific basis to support this approach. Somatostatin and its synthetic octapeptide analog, octreotide, are potent inhibitors of pancreatic secretion. Although several trials of somatostatin have demonstrated an efficacy in reducing the incidence of post-ERCP pancreatitis,138–143 the majority of the studies do not support the routine use of this medication.144–150 Octreotide, the analog of somatostatin,151 was only effective in reducing post-ERCP hyperamylasemia, and did not reduce the incidence of post-ERCP pancreatitis.
Pancreatic stent placement clearly decreases the risk of post-ERCP pancreatitis in high-risk patients.152 Placement of pancreatic duct stents has become a standard practice for patients who are thought to be at high risk for pancreatitis after the procedure (see Table 58-5).153 Pancreatic duct stent placement is effective presumably by preventing cannulation-induced edema that can cause pancreatic duct obstruction. Pancreatic sphincter hypertension is believed to be an important causative factor in post-ERCP pancreatitis and may explain the high risk of pancreatitis in patients with SOD. There is prolonged alleviation of ductal obstruction when pancreatic stents are placed. Typically, 3- to 5-French unflanged pancreatic stents are used in the following settings: SOD, difficult cannulation, biliary orifice balloon dilation, and precut sphincterotomy. In general, pancreatic duct stents are not beneficial in patients who undergo routine biliary sphincterotomy. In all reported studies, which cumulatively include 1500 high-risk patients undergoing ERCP, only 1 patient developed severe pancreatitis after a pancreatic duct stent had been placed.154 Aside from the obvious benefits in preventing post-ERCP pancreatitis regarding morbidity and mortality, prophylactic stent placement is a cost-effective strategy for the prevention of post-ERCP pancreatitis for high-risk patients.155
Guidewire cannulation, whereby the biliary or pancreatic duct is initially cannulated by a guidewire inserted through the catheter or sphincterotome, has been shown to decrease the risk of pancreatitis (see Table 58-5).156 In a study of 400 consecutive patients who underwent ERCP by a single endoscopist, randomized to initial cannulation with contrast versus initial cannulation by a guidewire under fluoroscopic control, pancreatitis rates were profoundly different. No cases of acute pancreatitis were seen in the guidewire group compared with eight cases in the standard contrast group (P < 0.001). Cannulation success rates between the standard contrast and guidewire techniques were comparable 98.5% versus 97.5%. A later study157 confirmed a decrease in post-ERCP pancreatitis in 300 patients prospectively randomized to guidewire cannulation compared with conventional radiocontrast. However, the decrease in post-ERCP pancreatitis appears to be related to a decreased need for precut sphincterotomy in patients undergoing guidewire cannulation.
POSTOPERATIVE
Postoperative pancreatitis can occur after abdominal or thoracic surgery.158 Pancreatitis occurs after 0.4% to 7.6% of cardiopulmonary bypass operations113,159 and after 6% of liver transplantations.160 Twenty-seven percent of patients undergoing cardiac surgery develop hyperamylasemia, and 1% develop necrotizing pancreatitis.113 Significant risks for pancreatitis after cardiopulmonary bypass are preoperative renal insufficiency, postoperative hypotension, and administration of calcium chloride perioperatively. Mortality from postoperative pancreatitis is said to be higher (up to 35%) than for other forms of pancreatitis. Contributors to morbidity and mortality from postoperative pancreatitis are delay in diagnosis, hypotension, medications (e.g., azathioprine/perioperative calcium chloride administration), and infections.
HEREDITARY AND GENETIC
Hereditary pancreatitis is an autosomal dominant disorder with variable penetrance.161–169 It is discussed in Chapter 57.
CONTROVERSIAL CAUSES
Pancreas Divisum
Pancreas divisum is the most common congenital malformation of the pancreas occurring in 5% to 10% of the general healthy population (see Chapter 55). Controversy continues to surround the issue as to whether pancreas divisum with otherwise normal ductular anatomy is a cause of acute recurrent pancreatitis. The presumed mechanism of action in those who develop pancreatitis is that there is relative obstruction to the flow of pancreatic juice through the minor papilla. The arguments in favor of attributing pancreatitis to pancreas divisum are the following: (1) various series from ERCP referral centers show that patients referred with recurrent acute pancreatitis have a higher frequency of pancreas divisum than would be expected from the general population170; (2) multiple observational series report that performing endoscopic sphincterotomy or placing a stent across the minor papilla reduces the rate of recurrent pancreatitis156; and (3) there is one randomized controlled study suggesting that patients with pancreas divisum who are stented for one year have a lower frequency of attacks of pancreatitis than those not stented.171 The arguments against the association are the following: (1) there are studies to show that the rate of pancreatitis in pancreas divisum patients is the same as the general population172; (2) the observational reports are flawed in that follow-up was not long enough (usually only 1 to 2 years) and that recurrent acute pancreatitis is a disease of great variability163; (3) the single randomized study163 was flawed in that it was not blinded, was small (19 patients total), and its patients probably had chronic pancreatitis in that they had multiple pain attacks in between attacks of acute pancreatitis; (4) the risk of endoscopic therapy is considerable with a high rate of post-ERCP pancreatitis in patients with pancreas divisum,111,112 therefore, making the risk-benefit ratio of treating pancreas divisum endoscopically questionable; and (5) the rate of genetic abnormalities in patients with pancreas divisum and acute recurrent pancreatitis are either the same173 or higher161 than expected in the general population or population of patients with acute pancreatitis of other etiologies, suggesting a possible genetic source. For example, there appears to be a higher incidence of CFTR mutations in patients with pancreas divisum who develop acute pancreatitis.161 Therefore, it may not be the presence of pancreas divisum alone that predisposes to acute pancreatitis but other factors may be necessary to precipitate an attack.163
Sphincter of Oddi Dysfunction (see Chapter 63)
SOD is also a controversial cause of pancreatitis. Investigators who study patients with recurrent acute pancreatitis report that SOD (usually defined as a basal pancreatic sphincter pressure > 40 mm Hg) is the most common abnormality discovered, occurring in approximately 35% to 40% of patients. The argument in favor of this entity as a cause of acute pancreatitis is the many observational series that report that endoscopic pancreatic sphincterotomy or surgical sphincteroplasty reduces or eliminates recurrent attacks of pancreatitis.174 The arguments against SOD as a cause of acute pancreatitis are (1) the lack of any prospective controlled blinded trials in the treatment of this disorder; (2) the short duration of follow-up in the observational reports; and (3) the high risk of pancreatitis (25% to 35%) associated with ERCP, sphincter of Oddi manometry, and pancreatic sphincterotomy in patients with suspected SOD. Furthermore, there is a relative dearth of data determining the normal range of pancreatic sphincter pressure.174
MISCELLANEOUS
Pancreatitis has been rarely associated with Crohn’s disease.164 A recent case control study from Denmark found a 4-fold increase in acute pancreatitis in patients with Crohn’s and a 1.5-fold increase in patients with ulcerative colitis. This has been attributed by some to the use of drugs such as aminosalicylates/sulfasalazine, azathioprine, or 6-mercaptopurine (see Table 58-4). Theories to support a relationship between idiopathic inflammatory bowel disease (IBD) and pancreatitis are that pancreatitis is an extraintestinal manifestation of IBD, that duodenal Crohn’s disease can cause obstruction to the flow of pancreatic juice, that granulomatous disease can involve the pancreas, or that there is an autoimmune process affecting the pancreas. Celiac disease165 has also been described in association with pancreatitis, but the relationship remains uncertain. It has been suggested that abnormalities in the normal barrier of the small bowel seen in patients with celiac disease may allow excessive absorption of amylase from the intestinal lumen, leading to hyperamylasemia. In the setting of abdominal pain in a patient with celiac disease, it is not uncommon to find elevations in the serum amylase in the absence of acute pancreatitis.166 Pancreatitis has been seen in patients after severe burns.167 A relationship of smoking with acute pancreatitis has been suggested. A Swedish case control study showed that there is a fourfold increased rate of acute pancreatitis in heavy smokers compared with nonsmokers.168
Autoimmune pancreatitis (discussed in the next chapter in more detail) typically presents as a mass or fullness in the pancreas. It is most commonly seen in older men, with biliary obstruction and an elevation of the serum immunoglobulin (IgG4) level. Occasionally, patients will present with signs and symptoms of chronic pancreatitis, such as stricturing of the main pancreatic duct, diabetes, exocrine pancreatic insufficiency, or as acute pancreatitis. Investigators have more recently described patients with autoimmune recurrent pancreatitis, especially in younger women often without the classic elevation of serum IgG4.169
CLINICAL FEATURES
It is difficult to diagnose acute pancreatitis by history and physical examination because clinical features are similar to those of many acute abdominal illnesses (Table 58-6).
Table 58-6 Differential Diagnosis of Acute Pancreatitis
HISTORY
Onset of pain is rapid but not as abrupt as that of a perforated viscus. Usually it is at maximal intensity in 10 to 20 minutes. Occasionally, pain gradually increases and takes several hours to reach maximum intensity. Pain is steady and moderate to very severe. There is little pain relief with changing position. Frequently, pain is unbearable, steady, and boring. Band-like radiation of the pain to the back occurs in half of patients. Pain that lasts only a few hours and then disappears suggests a disease other than pancreatitis, such as biliary colic or peptic ulcer. Pain is absent in 5% to 10% of attacks, and a painless presentation may be a feature of serious fatal disease.4
PHYSICAL EXAMINATION
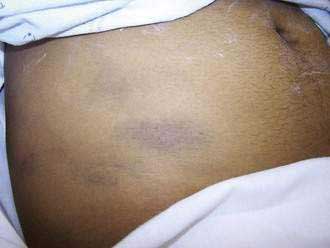
Additional abdominal findings may include ecchymosis in one or both flanks (Grey Turner’s sign; Fig. 58-3) or about the periumbilical area (Cullen’s sign), owing to extravasation of hemorrhagic pancreatic exudate to these areas. These signs occur in less than 1% of cases and are associated with a poor prognosis. Rarely there is a brawny erythema of the flanks caused by extravasation of pancreatic exudate to the abdominal wall. A palpable epigastric mass may appear during the disease from a pseudocyst or a large inflammatory mass.
The general physical examination, particularly in severe pancreatitis, may uncover markedly abnormal vital signs if there are third-space fluid losses and systemic toxicity. Commonly, the pulse is 100 to 150 beats/minute. Blood pressure can be initially higher than normal and then lower than normal with third-space losses and hypovolemia. Initially the temperature may be normal, but within one to three days it may increase to 101°F to 103°F owing to the severe retroperitoneal inflammatory process and the release of inflammatory mediators from the pancreas.175
Tachypnea and shallow respirations may be present if the subdiaphragmatic inflammatory exudate causes painful breathing. Dyspnea may accompany pleural effusions, atelectasis, ARDS, or congestive heart failure. Chest examination may reveal limited diaphragmatic excursion if abdominal pain causes splinting of the diaphragm, or dullness to percussion and decreased breath sounds at the lung bases if there is a pleural effusion. There may be disorientation, hallucinations, agitation, or coma,176 which may be due to alcohol withdrawal, hypotension, electrolyte imbalance such as hyponatremia, hypoxemia, fever, or toxic effects of pancreatic enzymes on the central nervous system. Conjunctival icterus may be present due to choledocholithiasis (gallstone pancreatitis) or bile duct obstruction from edema of the head of the pancreas, or from coexistent liver disease.
Uncommon findings include subcutaneous nodular fat necrosis,177 thrombophlebitis in the legs, and polyarthritis. Subcutaneous fat necroses are 0.5- to 2-cm tender red nodules that usually appear over the distal extremities but may occur over the scalp, trunk, or buttocks. They occasionally precede abdominal pain or occur without abdominal pain, but usually they appear during a clinical episode and disappear with clinical improvement. If they occur over a joint, they may be confused with arthritis.
DIFFERENTIAL DIAGNOSIS
The abdominal pain of biliary colic may simulate acute pancreatitis. It is frequently severe and epigastric, but it lasts for several hours rather than several days (see Chapter 65). The pain of a perforated ulcer is sudden, becomes diffuse, and precipitates a rigid abdomen; movement aggravates pain. Nausea and vomiting occur but disappear soon after onset of pain (see Chapter 52). In mesenteric ischemia or infarction, the clinical setting often is an older person with cardiac dysrhythmia or arteriosclerotic disease who develops sudden pain out of proportion to physical findings, bloody diarrhea, nausea, and vomiting. Abdominal tenderness may be mild to moderate, and muscular rigidity may not be severe despite severe pain (see Chapter 114). In intestinal obstruction, pain is cyclical, abdominal distention is prominent, vomiting persists and may become feculent, and peristalsis is hyperactive and often audible (see Chapter 119). Other conditions that enter into the differential diagnosis of acute pancreatitis are listed in Table 58-6.
LABORATORY DIAGNOSIS
PANCREATIC ENZYMES
In general, the diagnosis of acute pancreatitis relies on at least a three-fold elevation of amylase or lipase in the blood.178
Serum Amylase
A limitation of serum amylase is that it is not 100% sensitive or specific. With respect to sensitivity, which is greater than 85%, the serum amylase may be normal or minimally elevated in fatal pancreatitis,4 during a mild attack or an attack superimposed on chronic pancreatitis (because the pancreas has little acinar tissue), or during recovery from acute pancreatitis. Serum amylase also may be falsely normal in hypertriglyceridemia-associated pancreatitis179 because an amylase inhibitor may be associated with triglyceride elevations. In this case, serial dilution of serum often reveals an elevated serum amylase.
Hyperamylasemia is not specific for pancreatitis because it occurs in many conditions other than acute pancreatitis. In fact, one half of all patients with an elevated serum amylase may not have pancreatic disease.180 In acute pancreatitis, the serum amylase concentration is usually more than two to three times the upper limit of normal; it is usually less than this with other causes of hyperamylasemia.177 However, this level is not an absolute discriminator. Thus, an increased serum amylase level supports rather than confirms the diagnosis of acute pancreatitis. In addition, there are some individuals who have persistent hyperamylasemia without clinical symptoms. This has been reported due to macroamylasemia or pancreatic hyperamylasemia on a familial basis.174 Nonpancreatic diseases that cause hyperamylasemia include pathologic processes in organs (e.g., salivary glands, fallopian tubes) that normally produce amylase. Furthermore, mass lesions such as papillary cystadenocarcinoma of the ovary, benign ovarian cyst, and carcinoma of the lung, cause hyperamylasemia because they produce and secrete salivary-type isoamylase. Transmural leakage of pancreatic-type isoamylase and peritoneal absorption probably explain hyperamylasemia in intestinal infarction and in perforated viscus. Renal failure increases serum amylase up to four to five times the upper limit of normal due to decreased renal clearance of this enzyme.181 Patients on hemodialysis tend to have higher serum amylase levels than those on peritoneal dialysis. In patients with chronic kidney disease, there is no clear correlation between the creatinine clearance and serum levels of amylase, and about one third of patients with marked renal insufficiency have normal pancreatic enzyme levels.
Chronic elevations of serum amylase (without amylasuria) occur in macroamylasemia. In this condition, normal serum amylase is bound to an immunoglobulin or abnormal serum protein to form a complex that is too large to be filtered by renal glomeruli and thus has a prolonged serum half-life.182 Macroamylasemia may complicate the diagnosis of pancreatic disease, but it has no other clinical consequence. The urinary amylase-to-creatinine clearance ratio (ACCR) increases from approximately 3% to approximately 10% in acute pancreatitis.183 However, even moderate renal insufficiency interferes with the accuracy and specificity of the ACCR. Other than to diagnose macroamylasemia, which has a low ACCR, urinary amylase and the ACCR are not used clinically. Macroamylasemia can also be measured directly using serum. Deliberate contamination of urine with saliva, as in Munchausen’s syndrome, can increase the urine amylase, with the serum amylase being normal. This situation can be excluded by measuring salivary amylase in the urine.
Serum Lipase
The sensitivity of serum lipase for the diagnosis of acute pancreatitis is similar to that of serum amylase and is between 85% and 100%.174 Lipase may have greater specificity for pancreatitis than amylase as serum lipase is normal when serum amylase is elevated as in salivary gland dysfunction, tumors, gynecologic conditions, and macroamylasemia. Serum lipase always is elevated on the first day of illness and remains elevated longer than does the serum amylase.184 Consequently some suggest combining lipase with amylase as a test for acute pancreatitis. However, we and others have found that combining enzymes does not improve diagnostic accuracy.
Specificity of lipase can suffer from some of the same problems as those of amylase. In the absence of pancreatitis, serum lipase may increase less than two-fold above normal in severe renal insufficiency.185 With intra-abdominal conditions that resemble acute pancreatitis,186 lipase increases to levels less than three-fold above normal, presumably by absorption through an inflamed or perforated intestine. Rarely, a nonpancreatic abdominal condition such as small bowel obstruction can raise the amylase and lipase above three times normal. Some believe that serum lipase measurement is preferable to that of serum amylase because it is as sensitive as amylase measurement and more specific, whereas others find no clear advantage of one over the other.5
OTHER BLOOD AND URINE TESTS
Many nonenzymatic proteins are overexpressed in acute pancreatitis. Pancreatitis-associated protein (PAP), a heat shock protein, is undetectable in the normal pancreas but markedly increases in acute pancreatitis, with PAP detectable in serum. The sensitivity of serum PAP or pancreatic-specific protein (PSP) is no better than that of conventional tests,187 but PAP and PSP are as accurate as serum amylase for the detection of acute pancreatitis.
STANDARD BLOOD TESTS
The white blood cell count frequently is elevated, often markedly so in severe pancreatitis. The blood glucose also may be high and associated with high levels of serum glucagon. Serum aspartate transaminase (AST), alanine aminotransferase (ALT), alkaline phosphatase, and bilirubin also may increase, particularly in gallstone pancreatitis. Presumably, calculi in the bile duct account for these abnormalities. However, pancreatic inflammation may partially obstruct the distal bile duct in acute pancreatitis of other causes and cause abnormalities in liver biochemical tests. Nevertheless, aminotransferases may help distinguish biliary from alcoholic pancreatitis (see later).188 The erythrocyte mean corpuscular volume (MCV) has been also shown to help differentiate alcoholic from nonalcoholic acute pancreatitis.189 Alcoholic patients tend to have a higher MCV due to the toxic effects of alcohol on erythrocyte formation in the bone marrow. Serum triglyceride levels increase in acute pancreatitis, but also with alcohol use, uncontrolled diabetes mellitus, or defective triglyceride metabolism.
DIAGNOSTIC IMAGING
CHEST RADIOGRAPHY
Abnormalities visible on the chest roentgenogram occur in 30% of patients with acute pancreatitis, including elevation of a hemidiaphragm, pleural effusion(s), basal or plate-like atelectasis secondary to limited respiratory excursion, and pulmonary infiltrates. Pleural effusions may be bilateral or confined to the left side; rarely they are only on the right side.190 Patients with acute pancreatitis found to have a pleural effusion and/or infiltrate on admission are more likely to have severe disease.191 During the first 7 to 10 days, there also may be signs of congestive heart failure or acute respiratory distress syndrome. Pericardial effusion is rare.
ENDOSCOPIC ULTRASONOGRAPHY
Usually endoscopic ultrasonography (EUS) is not helpful early in acute pancreatitis. Imaging of the pancreas during an attack of acute pancreatitis and weeks following an episode reveal signals that are not normal (typically hypoechoic) and indistinguishable from chronic pancreatitis and malignancy. However, after a month, especially in patients with idiopathic interstitial pancreatitis, EUS may help determine the presence of small tumors, pancreas divisum, and bile duct stones.192 EUS is equal to MRCP and ERCP but far more sensitive than either abdominal ultrasonography or CT in detecting common duct stones.193 In a patient with biliary pancreatitis, whose serum bilirubin is rising in the setting of biliary sepsis, ERCP should not be delayed by first performing EUS (see later). Although there has been some concern that ERCP can worsen pancreatitis in such settings, ERCP appears to be safe in acute pancreatitis if needed. One caveat is that the contrast instillation into the pancreatic duct could introduce infection into necrotic areas of the pancreas. For this reason, EUS might be the best method of evaluating the bile duct in a patient with necrotizing pancreatitis.194
COMPUTED TOMOGRAPHY
CT is the most important imaging test for the diagnosis of acute pancreatitis and its intra-abdominal complications.195 The three main indications for a CT in acute pancreatitis are to exclude other serious intra-abdominal conditions, such as mesenteric infarction or a perforated ulcer; to stage the severity of acute pancreatitis; and to determine whether complications of pancreatitis are present, such as involvement of the GI tract or nearby blood vessels and organs, including liver, spleen, and kidney.196 Helical CT is the most common technique. If possible, scanning should occur after the patient receives oral contrast, followed by intravenous contrast to identify any areas of pancreatic necrosis. If there is normal perfusion of the pancreas, interstitial pancreatitis is said to be present (see Fig. 58-1). Pancreatic necrosis manifested as perfusion defects after intravenous contrast may not appear until 48 to 72 hours after onset of acute pancreatitis (see Fig. 58-2).
Contraindications to using intravenous contrast are a patient’s history of severe allergy (respiratory distress or anaphylaxis) or significant renal impairment (serum creatinine greater than 2 mg/dL). If severe renal impairment requires dialysis, intravenous contrast medium may be used.197 Hives or less severe allergic reactions with previous administration of iodinated contrast material are not absolute contraindications, but a nonionic contrast agent should be used, and 200 mg of hydrocortisone should be administered intravenously every six hours for four doses starting before the scan and 50 mg of diphenhydramine (Benadryl) should be given intramuscularly 30 minutes before the scan.198
It has been suggested that intravenous contrast media early in the course of acute pancreatitis might increase pancreatic necrosis because iodinated contrast medium given at the onset of pancreatitis increases necrosis in experimental rat acute pancreatitis.199 However, it did not do so in the opossum.200 Data in humans are conflicting. Two retrospective studies suggested that early contrast-enhanced CT worsened pancreatitis199 but this was not corroborated by a third retrospective study.198
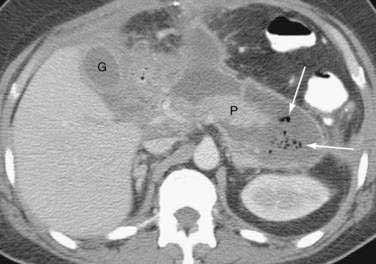
The severity of acute pancreatitis has been classified into five grades (A to E) based on findings on unenhanced CT199 (see following). Although the presence of gas in the pancreas suggests pancreatic infection with a gas-forming organism, this finding can also accompany sterile necrosis (Fig. 58-4) with microperforation of the gut or adjacent pseudocyst into the pancreas.201 Moreover, the great majority of pancreatic infections occur in the absence of gas on CT scan.
MAGNETIC RESONANCE IMAGING
MRI provides similar information regarding the severity of pancreatitis as does CT. MRI is as good as CT in detecting necrosis and fluid collections. MRI is better than CT, but equal to EUS and ERCP in detecting choledocholithiasis202 The MRCP contrast agent gadolinium, previously thought to be safe in patients with renal failure,203 can cause nephrogenic systemic fibrosis (NSF), which has raised concern.204 MRI is less accessible and more expensive than CT. MRI also requires the patient to remain still during capture of images, which typically is longer than with spiral CT. The use of intravenous secretin prior to MRCP allows a better visualization of the pancreatic ducts. This has been shown to be particularly useful in the evaluation of patients with idiopathic pancreatitis and recurrent pancreatitis.205
DISTINGUISHING ALCOHOLIC FROM GALLSTONE PANCREATITIS
Differentiation between alcoholic and gallstone pancreatitis is important because eliminating these causes may prevent further attacks of pancreatitis. Alcoholic pancreatitis occurs more frequently in men approximately 40 years old. The first clinical episode usually occurs after 5 to 10 years of heavy alcohol consumption. By contrast, biliary pancreatitis is more frequent in women, and the first clinical episode is often after the age of 40 years. Recurrent attacks of acute pancreatitis suggest an alcoholic etiology, but unrecognized gallstones may cause recurrent pancreatitis. Among patients with acute biliary pancreatitis discharged from hospital without cholecystectomy, 30% to 50% develop recurrent acute pancreatitis relatively soon after discharge (average time to recurrent pancreatitis, 108 days).206 Thus, removing the gallbladder in biliary pancreatitis is imperative.
Laboratory tests may help distinguish between these two disorders. The specificity for gallstone pancreatitis of ALT concentration greater than 150 IU/L (approximately a three-fold elevation) is 96%; the positive predictive value is 95%, but the sensitivity is only 48%.205 The AST concentration is nearly as useful as the ALT, but the total bilirubin and alkaline phosphatase concentrations are not as helpful to distinguish gallstone pancreatitis from alcoholic and other etiologies. There are differing reports as to whether a high serum lipase-to-amylase ratio can differentiate alcoholic from other causes of pancreatitis.207,208
Conventional transabdominal ultrasonography should be performed in every patient with a first attack of acute pancreatitis to search for gallstones in the gallbladder, common duct stones, or signs of extrahepatic biliary tract obstruction. However, bile duct stones are frequently missed by transabdominal ultrasonography, and most stones pass during the acute attack. ERCP is limited to patients with severe acute pancreatitis due to gallstones with persistent bile duct obstruction and to those in whom the stone could not be removed during surgery. In most patients with biliary pancreatitis the bile duct can be imaged with an operative cholangiogram at the time of laparoscopic cholecystectomy performed during the same admission. Although EUS is the most accurate method of detecting common duct stones and has been recommended for evaluating the common duct prior to cholecystectomy, it is rarely needed in this setting. MRCP is another noninvasive test that is highly accurate in determining if common duct stones are present. If a common duct stone is found at surgery, it is either removed at operation or endoscopically after surgery. Laparoscopic exploration of the bile duct is as safe and effective as postoperative ERCP in clearing stones from the common duct.209
PREDICTORS OF SEVERITY
Predicting severity of pancreatitis early in the course of disease is critical to maximize therapy and to prevent and minimize organ dysfunction and complications. Unfortunately the management of patients with acute pancreatitis is complicated by the inability to distinguish mild from severe disease during the early stages. The definition of the severity of acute pancreatitis early in the course of disease (during the first week) is typically based on clinical rather than anatomic parameters. At admission, several potential risk factors of severity and measurements that may reflect severity should be documented including age, body mass index, elevated hematocrit, elevated blood urea nitrogen (BUN), and pleural effusions or infiltrates on admission chest radiograph. The height of elevation of the serum amylase and lipase does not correlate with severity. Obese patients with pancreatitis have a higher incidence of local complications,210 respiratory failure,211 severe acute pancreatitis,212 and death from sterile necrosis213 than do nonobese patients.
Initially at presentation and over the first 48 hours, patients should be classified temporarily as having severe acute pancreatitis (and managed as such initially) based on the presence of SIRS or organ failure. SIRS is defined by two or more of the following four criteria: pulse greater than 90 beats/minute; rectal temperature less than 36°C or more than 38°C; white blood count less than 4000 or more than 12,000/mm3; and respirations greater than 20/minute or Pco2 less than 32 mm Hg. The presence of SIRS at admission and persistence of SIRS to 48 hours increases the morbidity and mortality rate. In one study, 25% of patients with persistent SIRS died from acute pancreatitis, 8% with transient SIRS, and less than 1% without SIRS.214
Although severity is now defined by the presence of organ failure or anatomic complications of acute pancreatitis, such as pancreatic necrosis, prospective systems using clinical criteria have been developed to determine severity in patients with acute pancreatitis. These systems include Ranson criteria (see Table 58-2) and APACHE score.13,14 Unfortunately these scoring systems (discussed following) are cumbersome, requiring multiple measurements. Additionally, the systems are not accurate until 48 hours after presentation.
SCORING SYSTEMS
Ranson’s Score
Ranson and colleagues identified 11 signs that had prognostic significance during the first 48 hours. The original list14 was analyzed in patients who primarily suffered from alcoholic pancreatitis and was then modified 8 years later for those with gallstone pancreatitis (see Table 58-2).215 Higher Ranson’s scores predict more severe disease. In mild pancreatitis (scores < 2), the mortality is 2.5% and in severe pancreatitis (scores > 3) the mortality is 62%.216 Also, the higher the Ranson’s score the higher the incidence of systemic complications, necrosis, and infected necrosis. These lists continue to remain in wide use in both the United States and Europe.217
The Ranson criteria have several drawbacks. First, the list is cumbersome and there are two lists to follow depending on suspected etiology (see Table 58-2). Second, an accurate Ranson’s score takes 48 hours to compute and the criteria have not been validated beyond the 48-hour time limit. Third, not all laboratories measure all the parameters in routine blood tests (e.g., serum lactate dehydrogenase [LDH]). Fourth, the overall sensitivity of the Ranson criteria (using three signs as the cutoff) for diagnosing severe disease is only 40% to 88% and the specificity is only 43% to 90%. The positive predictive value is approximately 50% and the negative predictive value around 90%.218 Therefore, the best use of Ranson’s score is to exclude severe disease. The Imrie or Glasgow score219 is a slightly simplified list (eight criteria) that is used commonly in the United Kingdom. It has similar drawbacks to the Ranson score.
APACHE-II Scores
APACHE-II is another commonly used scoring system in the United States to predict severity. It has the advantage of being able to be used on a daily basis and has similar positive and negative predictive values as the Ranson score at 48 hours after admission. The APACHE-II system assigns points for 12 physiologic variables, for age, and for chronic health status, in generating a total point score. The 12 variables are temperature; heart rate; respiratory rate; mean arterial blood pressure; oxygenation; arterial pH; serum potassium, sodium, and creatinine; hematocrit; white blood cell (WBC); and Glasgow Coma Scale. APACHE-II scores on admission and within 48 hours help distinguish mild from severe pancreatitis and to predict death.218,220 Most patients survive if APACHE-II scores are 9 or less during the first 48 hours. However, patients with APACHE-II scores of 13 or more have a high likelihood of dying. At admission, sensitivity is 34% to 70%, and specificity is 76% to 98%. At 48 hours, sensitivity remains less than 50%, but specificity is close to 90% to 100%.220 Strong drawbacks are its complexity, its low sensitivity on admission, and the fact that at 48 hours the score is no better than other scoring systems.221 Like the Ranson criteria, the APACHE-II score has its highest value in predicting mild disease.
BISAP
The problem with scoring systems is that they are cumbersome, using multiple variables. As described above, accuracy in predicting morbidity and/or mortality of the most commonly used scoring systems, Ranson and APACHE, is typically not achieved until 48 hours. By this time, it is usually apparent that the patient has developed severe disease manifested by organ failure. In order to develop a simple scoring system for patients with acute pancreatitis that would be useful within the first 12 hours from admission, the Pancreas Center at Brigham and Women’s Hospital performed a series of studies retrospectively and prospectively.221a,221b The studies were performed on a large database including almost 37,000 patients and more than 200 hospitals. After careful analysis, including a validation study, they determined that a simple system that included 5 variables could accurately determine severity early in the course of the disease. The scoring system, referred to as BISAP (Bedside Index for Severity in Acute Pancreatitis), also uses the first letter of each parameter for 1 point. The BISAP score provides a single point for 5 parameters: blood urea nitrogen (BUN) greater than 25 mg/dL, impaired mental status, systemic inflammatory response syndrome, age greater than 60, and/or the presence of a pleural effusion, for a possible total of 5 points. A BISAP score greater than 3 is associated with a seven- to twelve-fold increase in developing organ failure.221b Accurate, yet much easier to use, this new simple scoring system appears to be useful in the early identification of patients who are at risk of developing complications and mortality.221a
Blood Urea Nitrogen
Several prognostic scoring systems, including the Ranson criteria and BISAP, incorporate blood urea nitrogen (BUN) for the prediction of mortality in patients with acute pancreatitis. Hemoconcentration, as described above, has been shown to be an accurate predictor of necrosis and organ failure. Both BUN and the hematocrit or hemoglobin are routine laboratory tests that may provide information on changes in intravascular volume status. Either test may be used in monitoring the early response to initial fluid resuscitation. Wu and colleagues221c recently performed a large observational cohort study on data from 69 U.S. hospitals and found that BUN may be superior to hemoglobin (not hematocrit). For every 5 mg/dL increase in BUN during the first 24 hours, the age- and gender-adjusted odds ratio for mortality increased by 2.2. Of multiple routine laboratory tests examined, BUN yielded the highest accuracy at 24 hours and 48 hours. Although further study is needed, this paper suggests that following serial BUN measurements would be the most valuable single routine laboratory test for predicting mortality in acute pancreatitis.
ORGAN FAILURE
There is considerable interest among pancreatologists in using organ failure to predict severity. The Atlanta criteria defined which organ systems are of importance: pulmonary, renal, and cardiovascular. However, these criteria did not attempt to quantitate or prognosticate using organ failure. It has been appreciated that multiorgan failure or persistent single organ failure has a greater associated mortality than transient single organ failure. Multisystem organ failure is defined as two or more organs failing on the same day, rather than one organ failing on one day and another failing on the subsequent day. Patients with multisystem organ failure or persistent organ failure have a much higher mortality rate (approaching 50%) compared with patients with single and transient organ failure.222 Persistent organ failure is defined as lasting greater than 24 hours regardless of intervention. Survival among patients with organ failure at admission has also been shown to correlate with the duration of organ failure. When organ failure is corrected within 48 hours, mortality is close to zero. When organ failure persists for more than 48 hours, mortality is 36%.223 The Marshall Scoring System224 for organ failure is commonly used by intensivists for patients admitted to an intensive car unit. Data have not yet been generated using this system to prognosticate mortality in acute pancreatitis. Studies are needed to determine if this scoring system improves on the Ranson and APACHE scoring systems.
PERITONEAL LAVAGE
Percutaneous recovery of any volume of peritoneal fluid with a dark color or recovery of at least 20 mL of free intraperitoneal fluid of dark color portends a significant mortality.225 The sensitivity of peritoneal lavage is 36% to 72%, and the specificity is greater than 80% to 100%.226 An advantage is that it can be used any time, but it has not gained wide acceptance because it is invasive.
LABORATORY MARKERS
Because the degree of elevation of serum amylase and lipase does not distinguish mild from severe pancreatitis,219 other factors have been examined.
Hematocrit
A high hematocrit on admission, or one that fails to decrease after 24 hours of rehydration is thought to be a sign of hemoconcentration due to retroperitoneal fluid loss and thus a marker of severe disease.227 One study showed that a hematocrit greater than 44% had a sensitivity of 72% on admission and of 94% after 24 hours in detecting organ failure. The negative predictive value at 24 hours was 96%. Although one study from Germany found no correlation between admission hematocrit and organ failure, most investigators have found hematocrit to be important in the management of patients with acute pancreatitis.228 An elevated hematocrit (>44%) is a predictor for the development of necrosis. The hematocrit should be observed at admission for prognostic purposes and followed prospectively to assist in guiding the rate of intravenous hydration.12
C-Reactive Protein
CRP is an acute-phase reactant produced by the liver and is used extensively in Europe as a marker of severe pancreatitis. CRP is inexpensive to measure and readily available. The sensitivity for detecting severe disease is 60% to 100% (using cutoffs of 100 to 210 mg/L, or 10 to 21 mg/dL) and the specificity is 75% to 100%.229
Interleukin-6
IL-6 is an acute-phase-reactant cytokine that is produced by a variety of cells and induces hepatic synthesis of CRP. Several studies have shown that it is a reasonably good marker to differentiate mild from severe disease, but the test is not readily available.230
Polymorphonuclear Leukocyte Elastase
Polymorphonuclear leukocyte elastase rises very early (before CRP) in acute pancreatitis. High levels have been reported to differentiate severe from mild disease,231 but the test is not generally available.
Phospholipase A2
PLA2 is involved in the release of prostaglandins from cell membranes and degrades surfactant in the lung. It may play a role in the pulmonary dysfunction associated with acute pancreatitis. Levels of catalytic type II PLA2 have been reported to differentiate between mild and severe disease within 24 hours of admission.232
Urinary Trypsinogen Activation Peptide
Urinary TAP may serve as an early predictor of severity in patients with acute pancreatitis.233 Unlike other markers of severity, such as CRP, TAP is not a surrogate marker of inflammation. Normally trypsinogen is cleaved to trypsin in the intestinal lumen by the enzyme enterokinase. Premature intrapancreatic activation during acute pancreatitis results in the release of TAP. The degree of pancreatic necrosis and systemic inflammatory response or sepsis is directly related to TAP concentration. Elevated urinary TAP (>30 nmol/L) correlates with disease severity. The test can be applied within 12 hours of admission. The positive predictive value of an elevated TAP is 80% and the negative predictive value approaches 100%.
Procalcitonin
This propeptide is another acute-phase reactant that has been shown to differentiate mild from severe acute pancreatitis within the first 24 hours after symptom onset. A serum strip test has been developed that has a sensitivity of 86% and a specificity of 95% in detecting organ failure.234
COMPUTED TOMOGRAPHY
The finding of extensive fluid collections or extensive necrosis on CT has been correlated with severe disease. Balthazar reported that 5 of 37 (13.5%) patients who had grade D or E findings on CT died, as opposed to none of 51 who had grades B or C findings (Table 58-7).119 Using the CT severity index (CTSI score) (see Table 58-7), among those with a score of 0 to 6, 3 of 77 (3.8%) died, as compared with 2 of 11 (18%) with scores of 7 to 10. The CT grading scores correlate better with local complications (pseudocysts and abscesses) than with mortality. Among the 37 patients with a grade D or E score, 54% developed a local complication, whereas only 2 of 51 (3.9%) with grades A through C developed this problem.211 Thus, the data do not confirm that the CTSI is any more predictive than the grades A through E score.
Table 58-7 Computed Tomography (CT) Grading System of Balthazar and CT Severity Index (CTSI)
| Balthazar Grades | |
| Grade A | Normal pancreas consistent with mild pancreatitis |
| Grade B | Focal or diffuse enlargement of the gland, including contour irregularities and inhomogeneous attenuation but without peripancreatic inflammation |
| Grade C | Grade B plus peripancreatic inflammation |
| Grade D | Grade C plus associated single fluid collection |
| Grade E | Grade C plus two or more peripancreatic fluid collections or gas in the pancreas or retroperitoneum |
| CTSI = Balthazar Grade Score Plus Necrosis Score* | |
| Balthazar grade score: | |
| A | =0 |
| B | =1 |
| C | =2 |
| D | =3 |
| E | =4 |
| Necrosis score: | Absence of necrosis = 0 |
| Necrosis of up to 33% of pancreas = 2 | |
| Necrosis of 33% to 50% = 4 | |
| Necrosis of >50% = 6 | |
* Highest attainable score = 10 (Balthazar grade E + necrosis >50%).
There is controversy in the literature as to whether the extent of necrosis on CT predicts organ failure. Two studies failed to show any correlation between these two events,223,235 whereas a third study found a strong correlation.236
TREATMENT (Fig. 58-5)
GENERAL CONSIDERATIONS
Patients with acute pancreatitis require adequate intravenous hydration and adequate analgesia to eliminate or markedly reduce pain. The patient is usually on nothing per mouth until any nausea and vomiting have subsided. Abdominal pain can be treated with opiate analgesics, often by a patient-controlled anesthesia pump. Opiate dosing is monitored carefully and adjusted on a daily basis according to ongoing needs. Although morphine has been reported to increase sphincter of Oddi tone and increase serum amylase,237 its use to treat the pain of pancreatitis has not been shown to adversely effect outcome. Nasogastric intubation is not used routinely because it is not beneficial in mild pancreatitis. It is used only to treat gastric or intestinal ileus or intractable nausea and vomiting. Similarly, proton pump inhibitors or H2-receptor blocking agents69 are not beneficial and not used.
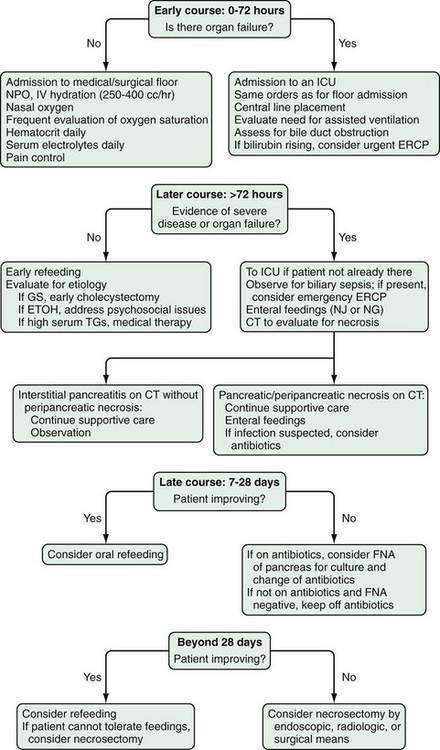
FLUID RESUSCITATION
As the inflammatory process progresses early in the course of the disease, there is an extravasation of protein-rich intravascular fluid into the peritoneal cavity resulting in hemoconcentration. The decreased perfusion pressure into the pancreas leads to microcirculatory changes that lead to pancreatic necrosis. An admission hematocrit of more than 47% and a failure of the admission hematocrit to decrease at 24 hours have been shown to be predictors of necrotizing pancreatitis.238
The relationship of hematocrit to severity of pancreatitis implies that the opposite is also true. Early vigorous intravenous hydration for the purpose of intravascular resuscitation is of foremost importance. The goal is to decrease the hematocrit. Laboratory and clinical studies with intravenous dextran to promote hemodilution have suggested efficacy in preventing severe disease.239 Too often patients with acute pancreatitis are given suboptimal intravenous hydration. One of the markers of severity of pancreatitis defined by Ranson and colleagues is intravascular losses (“fluid sequestration”). Ranson and colleagues found that a sequestration of more than 6 L of fluids during the first 48 hours was an independent predictor of disease severity in non-gallstone pancreatitis.14 If this amount of fluid (6 L) is added to the minimal intravenous fluid requirements of a 70-kg person during the first 48 hours, intravenous hydration should be at least 250 to 300 mL/hour for 48 hours. The rate of volume replacement is likely to be more important during the first 24 hours, when a rising hematocrit has been shown to correlate closely with severe disease. A study from the Mayo Clinic showed that patients with severe acute pancreatitis who do not receive at least one third of their initial 72-hour cumulative intravenous fluid volume during the first 24 hours after emergency department presentation are at risk for greater mortality than those who are initially resuscitated more aggressively.240
RESPIRATORY CARE
Hypoxemia (oxygen saturation <90%) requires supplemental oxygen, ideally by nasal prongs or by face mask if needed. If nasal oxygen fails to correct hypoxemia or if there is fatigue and borderline respiratory reserve, endotracheal intubation and assisted ventilation are required early. It is important to use a Swan-Ganz catheter to determine whether hypoxemia is due to congestive heart failure (increased pulmonary artery wedge pressure) or to primary pulmonary damage (normal or low pulmonary artery wedge pressure). Due to the common and indolent nature of hypoxemia affecting patients with acute pancreatitis, current guidelines recommend the initial routine use of nasal cannula oxygen to all patients with acute pancreatitis.12
ANTIBIOTICS
The use of prophylactic antibiotics in acute pancreatitis to prevent complications is controversial. In the 1970s, controlled studies compared intravenous antibiotics to no therapy in the treatment of mild acute alcoholic pancreatitis,241 with negative results. However, low-risk patients were studied (mild alcoholic disease with no mortality) and the wrong antibiotic (ampicillin) was used.242 In the 1980s, the bacteriology of infected pancreatic tissue was elucidated by analyzing either surgical specimens243 or fine-needle aspirations of the pancreas.242 The majority of organisms detected were gram-negative aerobic or anaerobic species (Escherichia coli, Enterobacter aerogenes, Pseudomonas aeruginosa, Proteus species, Klebsiella pneumoniae, Citrobacter freundii, and Bacteroides species), with occasional gram positives (Streptococcus faecalis, Staphylococcus aureus, Streptococcus viridans, Staphylococcus epidermidis) and rare fungi (Candida species). Studies in the early 1990s elucidated the appropriate antibiotics to use given these organisms and the level of penetration of antibiotics into necrotic pancreatic tissue.244 Imipenem, fluoroquinolones (ciprofloxacin, ofloxacin, pefloxacin), and metronidazole emerged as the drugs that achieved the highest inhibitory concentrations in pancreatic tissue, whereas aminoglycosides did not. Several randomized controlled (no placebo given), nonblinded clinical trials of prophylactic intravenous antibiotics in patients with severe pancreatitis were performed in Europe in the 1990s.245–248 One study added oral nonabsorbable antibiotics to the intravenous antibiotic regimen.249 Meta-analyses of the intravenous antibiotic trials showed that mortality of necrotizing pancreatitis was significantly reduced by antibiotics.250,251
Very little comparative information is known as to which antibiotic is the most effective. One study compared pefloxacin to imipenem in severe disease, showing imipenem to be significantly more effective in reducing pancreatic and extrapancreatic infection; however, mortality was unaffected.252 Likewise, little information is available on the length of treatment. One investigation compared one week of ciprofloxacin to three weeks and showed that longer treatment reduced the rates of pancreatic and nonpancreatic infection.253
The aforementioned studies have been criticized because they were not placebo-controlled, double-blinded studies. Furthermore, the use of prophylactic antibiotics in all patients with severe pancreatitis raises the concern that some patients will become superinfected with resistant organisms or fungi, which might lead to greater mortality in the future. Two studies have used double-blind randomized protocols to study this question. The first randomized 114 patients with severe acute pancreatitis to prophylactic ciprofloxacin plus metronidazole or to placebo. Patients suspected of having an infection were allowed open-label antibiotics. There were no differences in rates of infected necrosis or mortality. The only difference noted was that the group randomized to receive placebo eventually received more open-label antibiotics than the group randomized to receive the original antibiotics (46% vs. 28%).254 Dellinger and colleagues255 performed a multicenter, double-blind placebo-controlled randomized study in 32 centers in North America and Europe. One hundred patients were equally randomized to two groups, meropenem (1 g intravenously every eight hours) or placebo within 5 days of the onset of symptoms. Meropenem was continued for 7 to 21 days. This study demonstrated no significant difference between the treatment groups for pancreatic or peripancreatic infection, mortality, or requirement for surgical intervention. Based on these last two placebo-controlled studies, routine use of antibiotics is questionable in the absence of biliary sepsis or obvious pancreatic or peripancreatic infection. Although practice guidelines published prior to this latest paper recommended the use of prophylactic antibiotics in patients with severe necrotizing pancreatitis,256 more recent guidelines12 state that prophylactic antibiotics should not be used for the purpose of preventing infection in patients with necrotizing pancreatitis.
ENDOSCOPIC
The question of early removal of a possibly impacted gallstone in improving the outcome of gallstone pancreatitis remains a controversial issue. There have been three randomized fully published studies comparing urgent ERCP plus sphincterotomy (for any retained stones) versus conventional treatment in the management of gallstone pancreatitis.257–259 The earliest study,257 a single-center investigation from England, found that urgent ERCP within 72 hours of admission improved the outcome (complications and mortality) of patients with severe, but not mild, acute gallstone-induced pancreatitis. The second,258 another single-center study from Hong Kong, found that the urgent intervention group had a reduction in biliary sepsis and a trend toward lower mortality compared with the control group. The third and largest study from Germany259 included 22 centers and came to an opposite conclusion, namely, that there was a higher complication rate and a trend toward higher mortality in the urgently treated ERCP group compared with standard nonurgent therapy. Differences in the designs of these studies do not allow direct comparisons. However, there is consensus that severe acute gallstone pancreatitis with ascending cholangitis (jaundice and fever) is an indication for urgent ERCP (see Chapter 61).
In a prospective study of biliary pancreatitis using ERCP, Uomo and colleagues51 noted a 30.5% incidence of main pancreatic duct leakage. It has been proposed that early endoscopic stenting of the main pancreatic duct in patients with this problem may shorten hospital stay and the need for subsequent necrosectomy. Lau and colleagues260 evaluated 144 patients with severe acute pancreatitis and found that the presence of a PD leak was significantly associated with the development of necrosis. Patients with a PD leak had a longer length of stay compared with the patients with acute pancreatitis with no PD leak. In this retrospective study, patients who underwent early ERCP and had a PD stent placed were less likely to have other more invasive interventions performed, such as placement of external drains. Although PD stents have been shown to be helpful in the management of late complications related to pancreatic duct disruption, such as fistulas and pseudocysts, it remains unclear that routine use of these stents will prevent late complications. Kozarek and associates found that PD stent placement was associated with polymicrobial contamination of the pancreas.261 Although the contamination was largely asymptomatic, the benefit of placing a stent in the PD early in the course of acute pancreatitis to treat a duct disruption must be weighed against the possibility of seeding sterile necrosis with organisms that could lead to infected necrosis. Until randomized studies are performed, it is not clear whether potential advantages of early pancreatic duct stenting outweigh the risks.
NUTRITIONAL
In general, intravenous feedings are continued until patients are able to tolerate liquids or solids. The timing of early feedings is unclear. One report suggested that early refeeding improved outcome and allowed early discharge.262 However, a meta-analysis of three studies showed that early refeeding prolonged hospitalization.263 The question of whether an elevated serum amylase or lipase should influence the clinician to prolong the time to refeeding has been addressed. One hundred sixteen patients with acute pancreatitis were fed at the clinician’s discretion; 21% developed pain on refeeding 250 kcal/day.264 If the serum lipase was more than three-fold elevated, refeeding increased the clinical relapse rate compared with the group in which the lipase was less than three-fold elevated (39% vs. 16%). However, the corollary of this finding is that most patients with three-fold elevated serum lipase levels do not have a recrudescence of their pain on refeeding. Once refeeding is begun, a low-fat diet appears to be comparable to a clear liquid diet as the initial meal.265
Formerly, total parenteral nutrition (TPN) was the standard of care for refeeding patients with severe acute pancreatitis. Sax and coworkers266 randomized 54 patients with mild pancreatitis to intravenous nutrition or to TPN. The TPN group had a greater number of septic complications and longer hospitalizations. McClave267 randomized 30 patients with mild to moderate pancreatitis to receive TPN or enteral feedings administered through a nasoenteric tube, beginning 48 hours after admission. The Ranson and APACHE scores and blood glucose levels normalized more quickly in the enteral group, and the length of hospitalization showed a trend toward a shorter stay in the enteral group. Windsor268 randomized 34 patients with either mild to moderate pancreatitis to either oral feedings or TPN or with severe pancreatitis to either enteral feedings via a nasoenteric tube or TPN. The group receiving oral or enteral feedings had shorter intensive care unit (ICU) stays and improved acute phase response markers and disease severity scores compared with the TPN group. Kalfarentzos269 randomized 38 patients with severe necrotizing pancreatitis to TPN versus nasoenteric feedings. The enteral group had fewer septic complications and fewer total complications, although hospital stay, ICU stay, and days until resumption of a regular diet were similar in the two groups. Thus, these studies demonstrate that enteral nutrition is cheaper and safer and is preferable in patients with severe acute pancreatitis. It is still unclear, however, when nutrition should be initiated and for how long it needs to be continued. Furthermore it is unclear if nasoenteric feedings are needed or if nasogastric or even oral feedings are similarly effective if the patient tolerates this modality. Along those lines, a UK group randomized 50 patients with severe pancreatitis to nasogastric versus nasoenteric tube feedings. No difference was seen in the ability to tolerate feedings, in markers of inflammation, or in morbidity or mortality between the groups.270 They also noted no differences in the length of hospital stay or complications in patients with severe acute pancreatitis.
SURGICAL THERAPY
Cholecystectomy is routinely performed in patients with gallstone pancreatitis, and a consensus conference suggested that in mild or severe gallstone pancreatitis, cholecystectomy should be performed as soon as the patient has recovered and the acute inflammatory process has subsided.271 A second potential role for surgery in pancreatitis is to debride pancreatic necrosis (necrosectomy) or drain a pancreatic abscess. With regard to pancreatic necrosectomy, the data are more complicated and evolving. Studies in the 1980s suggested that early necrosectomy (within the first week of hospitalization for severe disease) reduced mortality. However, in the only randomized study comparing early (within 72 hours of admission) to late necrosectomy (12 days or more after admission), the mortality with early operation was greater than with later débridement (56% vs. 27%).272 Some investigators have reported that it is important to differentiate sterile necrosis from infected necrosis by fine-needle aspiration of the pancreas. Sterile necrosis can be managed nonoperatively because the mortality of this condition without surgery is less than 5%.273,274 Infected necrosis (as documented by fine-needle aspiration of the pancreas with Gram stain and cultures), on the other hand, has been historically regarded as an indication for surgical débridement because of the previous belief that infected necrosis treated medically has a nearly uniform fatal outcome.275,276 This has led to the recommendation that patients who are not improving on maximal medical therapy or who develop organ failure should have a fine-needle aspiration of the pancreas (Fig. 58-6). The finding of infection should then lead to a consideration of surgical intervention.
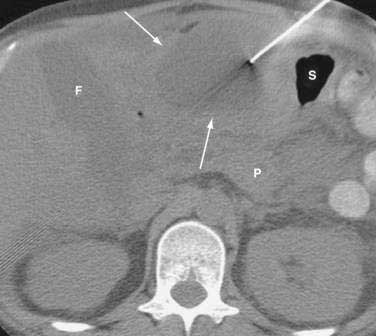
However, surgical therapy of infected pancreatic necrosis carries a substantial mortality of 15% to 73%,275–278 especially when it is carried out early within the first few weeks of the disease. Thus, it is unclear if the higher mortality rates that have been reported in patients with infected necrosis are the result of the underlying disease or the early surgical intervention. It has been shown that delaying surgical débridement beyond the fourth week in patients with pancreatic necrosis is associated with a lower mortality rate.277 The concept that infected pancreatic necrosis requires prompt surgical débridement has been challenged by several reports of patients who have been treated by antibiotic therapy alone.278–280
A series of 28 patients with infected pancreatic necrosis was treated prospectively with antibiotics rather than urgent surgical débridement.281 Among 12 patients who eventually required elective surgical intervention, there were two deaths. Among the other 16 patients who were treated with long-term antibiotic therapy, there were also two deaths. Thus, 14 of 28 patients with infected necrosis were successfully treated with no surgical, endoscopic, or radiologic drainage.
Several additional procedures have been introduced that are less invasive than standard open surgical débridement of infected necrosis. These techniques have generally been reserved for patients with infected pancreatic necrosis who are too ill to undergo prompt surgical débridement (such as those with organ failure or serious comorbid disease) and include laparoscopic necrosectomy with placement of large-caliber drains under direct surgical inspection and percutaneous catheter drainage of infected necrosis (see Chapter 26). Other minimally invasive approaches, including endoscopic (see Chapter 61) or a combined endoscopic and laparoscopic approach may become more commonly used as skill and technology advance.
OTHER THERAPEUTIC AGENTS OF POSSIBLE OR QUESTIONABLE EFFICACY
Pancreatic protease inhibitors have been used to treat established severe acute pancreatitis and to prevent post-ERCP pancreatitis (see earlier section). Gabexate mesylate is the most widely studied pancreatic protease inhibitor. A meta-analysis of five clinical trials of gabexate mesylate in acute pancreatitis found no effect on the 90-day mortality rate, but a reduced incidence of complications.282 This agent is not available in the United States. Multiple trials of the antisecretory hormone somatostatin or its synthetic analog octreotide have failed to show convincing evidence of efficacy in the treatment of acute pancreatitis.283 The use of anti-inflammatory cytokines has so far not shown efficacy. The largest experience has been with lexipafant, a PAF inhibitor. After initial promising reports,284 subsequent studies have not shown clear efficacy.285
Japanese investigators have suggested that pancreatic protease inhibitors and antibiotics can be better targeted to the affected regions in the pancreas with continuous regional arterial infusion (CRAI) into the celiac, splenic, inferior pancreaticoduodenal, and common hepatic arteries. Using CT, Anai and colleagues286 showed that with CRAI the contrast was distributed to the entire pancreas in six of nine patients with inflammation of the entire pancreas; in the remaining three patients, contrast material did not penetrate the entire area of pancreatic inflammation. Two later studies suggested that intra-arterial infusion of the protease inhibitor nafamostat mesylate plus imipenem reduces mortality when compared with intravenous infusion of the same agents.287,288 These studies warrant further investigation. Many other measures have been shown to be ineffective in randomized trials, including anticholinergics, glucagon, fresh frozen plasma, and peritoneal lavage.60,289 Infectious complications and associated mortality are major concerns in acute pancreatitis. Enteral administration of probiotics might prevent infectious complications, but convincing evidence is scarce. Although an early study suggested that probiotics might decrease infectious complications,290 other studies have suggested an increase in morbidity and mortality.291 In this more recent study, infectious complications occurred in 46 patients in the probiotics group (30%) and in 41 of those in the placebo group (28%). Twenty four patients in the probiotics group died (16%), compared with 9 (6%) in the placebo group (relative risk of death, 2.53; 95% confidence interval, 1.22 to 5.25). Nine patients in the probiotics group developed bowel ischemia (8 with fatal outcome), compared with none in the placebo group (P = 0.004). At the present time, probiotic prophylaxis should therefore not be administered in this category of patients.292
LOCAL COMPLICATIONS (Table 58-8)
PSEUDOCYST
A pseudocyst may occur secondary to acute pancreatitis, pancreatic trauma, or chronic pancreatitis. It usually contains a high concentration of pancreatic enzymes and variable amounts of tissue debris. Most are sterile. Regardless of size, an asymptomatic pseudocyst does not require treatment.293,294 It is satisfactory to monitor the pseudocyst with abdominal ultrasonography. Pseudocysts can be complicated by infection, intracystic hemorrhage (Fig. 58-7), or rupture leading to pancreatic ascites. Further, pseudocysts can migrate into the chest or other unusual locations. In patients with known pseudocysts, new symptoms, such as abdominal pain, chills, or fever, should alert the clinician to the emergence of an infected pseudocyst or abscess. Treatment choices include surgical, radiologic, and endoscopic drainage. No randomized prospective trials have compared these methods.
| Local |
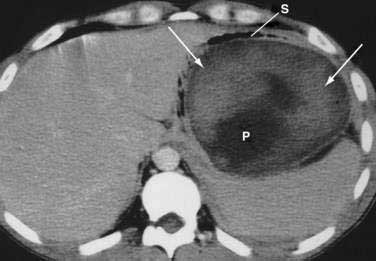
Surgical drainage of a pseudocyst is possible with a cyst-gastrostomy or cyst-duodenostomy if the pseudocyst wall is broadly adherent to the stomach or duodenum, respectively. Other procedures include a Roux-en-Y cyst-jejunostomy or pancreatic resection if the pseudocyst is in the tail. Surgical mortality is 6% or less.293–295 Pseudocysts recur after internal (surgical) drainage in 15% of cases; recurrence is more frequent if the main pancreatic duct is obstructed downstream from the surgical anastomosis. For this reason, a preoperative ERCP is usually done to determine whether there is duct obstruction. If this is the case, a resection of the pseudocyst is preferred, is possible.
Percutaneous catheter drainage is effective treatment to drain and close sterile as well as infected pseudocysts.146 As with surgical drainage, percutaneous catheter drainage may fail if there is obstruction of the main pancreatic duct downstream from the pseudocyst. Therefore, an ERCP is usually done before attempting catheter drainage (see Chapter 26).
As discussed in Chapter 61, there are two endoscopic methods to decompress a pancreatic pseudocyst: (1) insertion of a stent through the ampulla directly into the pancreatic duct and then into the pseudocyst itself,296,297 or (2) an endoscopic cyst-gastrostomy or cyst-duodenostomy. With either method, the catheter is removed after three to four weeks if closure of the pseudocyst is seen by CT scan. Failure of percutaneous or endoscopic drainage of a pancreatic pseudocyst increases morbidity and prolongs hospitalization. However, most series show long-term resolution with successful endoscopic drainage of pseudocysts.298
There are several complications of endoscopic drainage of pseudocysts (see Chapter 61). The most important is bleeding; the risk of bleeding may be reduced if endoscopic ultrasonography is used to be certain that there are no large vessels in the drainage area. Infection may occur if the double-pigtail catheter becomes occluded. A nasocystic drain to irrigate the cyst may prevent this complication. An endoscopically placed stent in the pancreatic duct may induce ductal changes identical to those of chronic pancreatitis. For this reason a stent should be removed after several weeks.
NECROTIZING PANCREATITIS AND ABSCESS
Pancreatic necrosis may either be sterile or infected. For the first 10 to 14 days, pancreatic necrosis is typically considered sterile and managed conservatively. Often patients appear ill and require ICU care. The goal during this time is to maximize supportive care and prevent infection by providing enteral feeding and minimizing potential sources of infection, such as intravenous lines. At this time, the use of antibiotics to prevent infection has been shown to be questionable and not recommended (see earlier). Surgical débridement of sterile pancreatic necrosis has also been shown not be helpful in the vast majority of patients.299 Early surgical débridement is exceedingly difficult and avoided early within the first 4 to 8 days due to the cement-like nature of the necrosis.300 However, surgical intervention is often needed in patients with infected necrosis.
When infection is suspected, the diagnosis is readily established by CT-guided fine-needle aspiration (CT-FNA). This procedure is safe and effective in establishing the diagnosis.244 The Gram stain alone has a sensitivity of almost 95% if carefully examined in a fresh specimen. The procedure is also safe, rarely introducing infection into a sterile field in the abdomen. If negative, an aspiration can be repeated every four to seven days if infection continues to be suspected.
In the past, the diagnosis of infected necrosis implied the urgent need for surgical débridement. This is no longer true. In a persistently ill patient with sepsis or organ failure found to have infected necrosis, surgical débridement should be strongly considered. However, in a stable patient with infected necrosis, maximal supportive care and the use of pancreatic-penetrating antibiotics should be provided. It is in these patients that antibiotics such as fluoroquinolones, metronidazole, and imipenem should be administered. Although the antibiotics will not likely clear the infection in most patients, the ability to allow time for the formation of a fibrous wall, creating WOPN (see Fig. 58-7), will lead to a more minimally invasive approach to draining the pancreatic necrosis. Whereas early débridement of pancreatic necrosis within the first four to five weeks of an attack will require surgery, WOPN can be treated laproscopically,301 percutaneously, or endoscopically (see Chapter 61).302 The timing and method of débridement require a clear discussion between the surgeon and gastroenterologist, but should be left at the discretion of the pancreatic surgeon.
GASTROINTESTINAL BLEEDING
GI bleeding may arise from lesions not related to the local inflammatory aspects of pancreatitis such as stress-induced mucosal gastropathy, Mallory-Weiss tear, or alcoholic gastropathy. Alternatively, bleeding can be due to the inflammatory aspects of the pancreatitis (see Table 58-8). The latter is thought to occur from the irritative effects of liberated activated enzymes on vascular structures or pressure necrosis of inflammatory debris or fluid collections on surrounding structures. Rupture of the splenic artery, splenic vein, or portal vein has been reported.303 High mortality is reported with these complications. Temporizing treatments with interventional radiologic techniques are used, followed by more definitive surgical ligation and resection. Acute and chronic inflammatory processes of the pancreas can lead to thrombosis of the adjacent splenic vein, which can lead to gastric varices, with or without esophageal varices. These varices can rupture, leading to massive bleeding. Treatment of this problem can be endoscopic with banding of varices or splenectomy, which is curative. Pseudocysts can be complicated by pseudoaneurysm formation, which can usually be seen by dynamic contrast-enhanced CT. If these bleed, arteriography with embolization is the treatment of choice. Rarely, bleeding into the pancreatic duct occurs (hemosuccus pancreaticus), but this typically occurs in chronic pancreatitis. Postnecrosectomy bleeding is common and can be caused by overly aggressive débridement or the placement or the use of noncompliant drainage tubes next to vascular structures.
SPLENIC COMPLICATIONS
Splenic complications of pancreatitis include intrasplenic pseudocysts, infarction and necrosis of the spleen, splenic rupture, and hematoma.304 Some of these complications can be life threatening and require emergency splenectomy (see Table 58-8).
BOWEL COMPRESSION OR FISTULIZATION
Pressure necrosis from inflammatory debris from the tail of the pancreas can obstruct or fistulize into the small or large bowel. The most common site is the left colon.305 Treatment is frequently surgical.
SYSTEMIC COMPLICATIONS
ORGAN DYSFUNCTION
Organ dysfunction has been discussed extensively earlier in the chapter. Respiratory insufficiency is the most common systemic complication associated with pancreatitis. The causes are multifactorial and include pleural effusions, pneumonia, atelectasis, and ARDS. Oxygen supplementation, antibiotics, thoracentesis, and assisted ventilation may be necessary. Renal complications are due to hypovolemia causing prerenal azotemia or hypotension, leading to acute tubular necrosis. These are treated by increasing intravenous fluid administration in the former case or with hemofiltration or hemodialysis in the latter situation. Shock is usually caused by hypovolemia secondary to third-space losses, vomiting, and interstitial pancreatic edema. Other uncommon causes of hypotension include myocardial infarction and pericardial effusions. Fluid replacement in severe acute pancreatitis is best accomplished using central venous– or Swan-Ganz catheter–assisted monitoring. As mentioned, mortality is substantially increased with increasing organ dysfunction especially if shock is involved.226
COAGULATION DISORDERS
Mild coagulation defects are common in acute pancreatitis as measured by elevated d-dimer levels in the blood.306 Full-blown disseminated intravascular coagulation (DIC) with a bleeding diathesis associated with a hypercoagulable state is very uncommon.
FAT NECROSIS
Fat necrosis occurs in subcutaneous tissue, bone, retroperitoneal tissue, peritoneum, mediastinum, pleura, and pericardium.191 Histologically fat cells are necrotic, associated with a diffuse inflammatory infiltration. The subcutaneous lesions are circumscribed tender red nodules that are adherent to the skin but are movable over deeper structures. Most commonly they are over the ankles, fingers, knees, and elbows. The lesions may drain through the skin. Rarely there is also necrosis of adjacent tendons or involvement of joints, particularly the metatarsal, interphalangeal, wrist, knee, and ankle joints. The lesions usually resolve after days to weeks.
MISCELLANEOUS
Pancreatic encephalopathy consists of a variety of central nervous system symptoms occurring in acute pancreatitis, including agitation, hallucinations, confusion, disorientation, and coma.198 A similar syndrome may be due to alcohol withdrawal, and other causes are possible, such as electrolyte disturbances (e.g., hyponatremia) or hypoxia. Purtsher’s retinopathy (discrete flame-shaped hemorrhages with cotton wool spots) can cause sudden blindness.307 It is thought to be due to microembolization in the choroidal and retinal arteries.
Badalov N, Baradarian R, Iswara K, et al. Drug induced acute pancreatitis: An evidence based approach. Clin Gastroenterol Hepatol. 2007;101:454-76. (Ref 89.)
Besselink MG, Verwer TJ, Schoenmaeckers EJ, et al. Timing of surgical intervention in necrotizing pancreatitis. Arch Surg. 2007;142:1194-201. (Ref 277.)
Freeman ML, DiSario JA, Nelson DB, et al. Risk factors for post-ERCP pancreatitis: A prospective, multicenter study. Gastrointest Endosc. 2001;54:425. (Ref 109.)
Gardner TB, Vege SS, Chari ST, et al. Fluid resuscitation in acute pancreatitis. Clin Gastroenterol Hepatol. 2008;6:1070-6. (Ref 240.)
Gerzof SG, Banks PA, Robbins AH, et al. Early diagnosis of pancreatic infection by computed tomography-guided aspiration. Gastroenterology. 1987;93:1315. (Ref 242.)
Isenmann R, Runzi M, Kron M, et al. Prophylactic antibiotic treatment in patients with predicted severe acute pancreatitis: A placebo-controlled, double blind trial. Gastroenterology. 2004;126:997. (Ref 254.)
Kalfarentzos F, Kehagias J, Mead N, et al. Enteral nutrition is superior to parenteral nutrition in severe acute pancreatitis: Results of a randomized prospective trial. Br J Surg. 1997;84:1665. (Ref 269.)
Mier J, Luque-deLeon E, Castillo A, et al. Early vs. late necrosectomy in severe necrotizing pancreatitis. Am J Surg. 1997;173:71. (Ref 272.)
Neoptolemos JP, Carr-Locke DL, London NJ, et al. Controlled trial of urgent endoscopic retrograde cholangiopancreatography and endoscopic sphincterotomy versus conservative treatment for acute pancreatitis due to gallstones. Lancet. 1988;2:979. (Ref 257.)
Opie EL. The etiology of acute hemorrhagic pancreatitis. Bull Johns Hopkins Hosp. 1901;12:182. (Ref 40.)
Ranson JHC, Rifkind RM, Roses DF. Prognostic signs and the role of operative management in acute pancreatitis. Surg Gynecol Obstet. 1975;139:69. (Ref 14.)
Runzi M, Niebel W, Goebell H, et al. Severe acute pancreatitis: Nonsurgical treatment of infected necrosis. Pancreas. 2005;30:195-9. (Ref 281.)
Singh V, Wu BU, Maurer R, et al. A prospective evaluation of the Bedside Index of Severity in Acute Pancreatitis. Am J Gastroenterol. 2009;104:966-71. (Ref 221b.)
Tenner S, Sica G, Hughes M, et al. Relationship of necrosis to organ failure in severe acute pancreatitis. Gastroenterology. 1997;113:899-903. (Ref 17.)
Wu BU, Johannes RS, Sun S, et al. Early changes in blood urea nitrogen predict mortality in acute pancreatitis. Gastroenterology. 2009;137:129-35. (Ref 221c.)
1. Go VLW, Everhart JE. Pancreatitis. In: Everhart JE, editor. Digestive diseases in the United States: Epidemiology and impact. US Department of Health and Human Services, Public Health Service, National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases. NIH Publication no. 94-1447. Washington, D.C.: Government Printing Office; 1994:693.
2. Corfield AP, Cooper MJ, Williamson RCN. Acute pancreatitis: A lethal disease of increasing significance. Gut. 1985;26:724.
3. Sinclair MT, McCarthy A, Mckay C, et al. The increasing incidence and high early mortality rate from acute pancreatitis in Scotland over the last 10 years. Gastroenterology. 1997;112:A482.
4. Lankisch PG, Schirren CA, Kunze E. Undetected fatal acute pancreatitis: Why is the disease so frequently overlooked? Am J Gastroenterol. 1991;86:322.
5. Sandler RS, Everhardt JE, Donowitz M, et al. The burden of selected digestive diseases in the United States. Gastroenterology. 2002;122:1500.
6. Russo MW, Wei JT, Thiny MT, et al. Digestive and liver diseases statistics 2004. Gastroenterology. 2004;126:1448.
7. National Center for Health Statistics. National Hospital Discharge Summary: Annual summary 1998; series report 13:203.
8. DeFrances CJ, Hall MJ, Podgornik MN. 2003 National Hospital Discharge Survey: Advance data from vital and health statistics. Hyattsville, Md: National Center for Health Statistics; 2005.
9. Frey CF, Zhou H, Harvey DJ, White RH. The incidence and case-fatality rates of acute biliary, alcoholic, and idiopathic pancreatitis in California, 1994-2001. Pancreas. 2006;33:336-44.
10. Mann DV, Hershman MJ, Hittinger R, et al. Multicentre audit of death from acute pancreatitis. Br J Surg. 1984;81:890-3.
11. Neoptolemos JP, Raraty M, Finch M, et al. Acute pancreatitis: The substantial human and financial costs. Gut. 1998;42:886-91.
12. Banks PA, Freeman ML. Practice guidelines in acute pancreatitis. Am J Gastroenterol. 2006;101:2379-2400.
13. Bradley EL3rd. A clinically based classification system for acute pancreatitis. Arch Surg. 1993;128:586.
14. Ranson JHC, Rifkind RM, Roses DF. Prognostic signs and the role of operative management in acute pancreatitis. Surg Gynecol Obstet. 1975;139:69.
15. Knaus WA, Draper EA, Wagner DP, et al. APACHE II: A severity of disease classification system. Crit Care Med. 1985;13:818.
16. Bradley EL, Gonzalez AC, Clements JLJr. Acute pancreatic pseudocysts: Incidence and implications. Ann Surg. 1976;184:734.
17. Tenner S, Sica G, Hughes M, et al. Relationship of necrosis to organ failure in severe acute pancreatitis. Gastroenterology. 1997;113:899-903.
18. Lankish PG, Blum T, Maisonneuve P, et al. Severe pancreatitis: When to be concerned? Pancreatology. 2003;3:102.
19. Mutinga M, Rosenbluth A, Tenner SM, et al. Does mortality occur early or late in acute pancreatitis? Int J Pancreatol. 2000;28:91.
20. Renner IG, Savage WT, Pantoja JL, et al. Death due to acute pancreatitis. A retrospective analysis of 405 autopsy cases. Dig Dis Sci. 1985;40:1005.
21. McKay CJ, Buter A. Natural history of organ failure in acute pancreatitis. Pancreatology. 2003;3:111.
22. Gloor B, Muller CA, Worni M, et al. Later mortality in patients with severe acute pancreatitis. Br J Surg. 2001;88:975.
23. Migliori M, Pezzilli R, Tomassetti P, et al. Exocrine pancreatic function after alcoholic or biliary acute pancreatitis. Pancreas. 2004;28:359.
24. Steer ML. Pathogenesis of acute pancreatitis. Digestion. 1997;58:46.
25. Nakae Y, Naruse S, Kitagawa M, et al. Activation of trypsinogen in experimental models of acute pancreatitis in rats. Pancreas. 1995;10:306.
26. Bettinger JR, Grendell JH. Intracellular events in the pathogenesis of acute pancreatitis. Pancreas. 1991;6:S2.
27. Fernández-del Castillo C, Schmidt J, Warshaw AL, et al. Interstitial protease activation is the central event in progression to necrotizing pancreatitis. Surgery. 1994;116:497.
28. Steer ML. How and where does acute pancreatitis begin? Arch Surg. 1992;127:1650.
29. Lerch MM, Adler G. Experimental animal models of acute pancreatitis. Int J Pancreatol. 1994;15:159.
30. Grady T, Saluja A, Kaiser A, et al. Pancreatic edema and intrapancreatic activation of trypsinogen during secretagogue-induced pancreatitis precedes glutathione depletion. Am J Physiol. 1996;271:G20.
31. Saluja AK, Donovan EA, Yamanaka K, et al. Cerulein-induced in vitro activation of trypsinogen in rat pancreatic acini is mediated by cathepsin B. Gastroenterology. 1997;113:304.
32. Lüthen R, Niederau C, Niederau M, et al. Influence of ductal pressure and infusates on activity and subcellular distribution of lysosomal enzymes in the rat pancreas. Gastroenterology. 1995;109:573.
33. Fallon MB, Gorelick FS, Anderson JM, et al. Effect of cerulein hyperstimulation on the paracellular barrier of rat exocrine pancreas. Gastroenterology. 1995;108:1863.
34. LeBodic LL, Bignon JD, Raguenes O, et al. The hereditary pancreatitis gene maps to long arm of chromosome 7. Hum Mol Genet. 1996;5:549.
35. Whitcomb C, Preston RA, Aston CE, et al. A gene for hereditary pancreatitis maps to chromosome 7q35. Gastroenterology. 1996;110:1975.
36. Whitcomb DC, Gorry MC, Preston RA, et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet. 1996;14:141.
37. Gorry MC, Gabbaizedeh D, Furey W, et al. Mutations in the cationic trypsinogen gene are associated with recurrent acute and chronic pancreatitis. Gastroenterology. 1997;113:1063.
38. Witt H, Luck W, Hennies HC, et al. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nature Genetics. 2000;25:213.
39. Lerch MM, Saluja AK, Rünzi M, et al. Pancreatic duct obstruction triggers acute necrotizing pancreatitis in the opossum. Gastroenterology. 1993;104:853.
40. Opie EL. The etiology of acute hemorrhagic pancreatitis. Bull Johns Hopkins Hosp. 1901;12:182.
41. Rünzi M, Saluja A, Lerch MM, et al. Early ductal decompression prevents the progression of biliary pancreatitis: An experimental study in the opossum. Gastroenterology. 1993;105:157.
42. Lüthen RE, Niederau C, Grendell JH. Effects of bile and pancreatic digestive enzymes on permeability of the pancreatic duct systems in rabbits. Pancreas. 1993;8:671.
43. Prinz RA. Mechanisms of acute pancreatitis: Vascular etiology. Int J Pancreatol. 1991;9:31.
44. Klar E, Messmer K, Warshaw AL, et al. Pancreatic ischemia in experimental acute pancreatitis: Mechanism, significance, and therapy. Br J Surg. 1990;77:1205.
45. Toyama MT, Lewis MP, Kusske AM, et al. Ischaemia-reperfusion mechanisms in acute pancreatitis. Scand J Gastroenterol. 1996;31:20.
46. Makhija R, Kingsnorth AN. Cytokine storm in acute pancreatitis. J Hepat Pancreatic Surg. 2002;9:401.
47. Rinderknecht H. Fatal pancreatitis, a consequence of excessive leukocyte stimulation? Int J Pancreatol. 1988;3:105.
48. Kingsnorth A. Role of cytokines and their inhibitors in acute pancreatitis. Gut. 1997;40:1.
49. Sweiry JH, Mann GE. Role of oxidative stress in the pathogenesis of acute pancreatitis. Scand J Gastroenterol. 1996;31:10.
50. Rahman S, Ibrahim K, Larvin M, et al. Association of antioxidant enzyme gene polymorphisms and glutathione status with severe acute pancreatitis. Gastroenterology. 2004;126:1312.
51. Uomo G, Molino D, Visconti M, et al. The incidence of main pancreatic duct disruption in severe biliary pancreatitis. Am J Surg. 1998;176:49.
52. Neoptolemos JP, London NJM, Carr-Locke DL. Assessment of main pancreatic duct integrity by endoscopic retrograde pancreatography in patients with acute pancreatitis. Br J Surg. 1993;80:94.
53. Agarwal N, Pitchumoni CS. Acute pancreatitis: A multisystem disease. Gastroenterologist. 1993;1:115.
54. Weber CK, Adler G. From acinar cell damage to systemic inflammatory response: Current concepts in pancreatitis. Pancreatology. 2001;1:356.
55. Ammori BJ, Barclay GR, Larvin M, et al. Hypocalcemia in patients with acute pancreatitis: A putative role for systemic endotoxin exposure. Pancreas. 2004;26:213.
56. Schmid SW, Uhl W, Friess H, et al. The role of infection in acute pancreatitis. Gut. 1999;45:311.
57. Andersson R, Wang XD. Gut barrier dysfunction in experimental acute pancreatitis. Ann Acad Med Singapore. 1999;28:141.
58. Kazantsev GB, Hecht DW, Rao R, et al. Plasmid labeling confirms bacterial translocation in pancreatitis. Am J Surg. 1994;167:201.
59. Widdison AL, Karanjia ND, Reber HA. Route of spread of pathogens into the pancreas in a feline model of acute pancreatitis. Gut. 1994;35:1306.
60. Steinberg WM, Tenner S. Acute pancreatitis. N Engl J Med. 1994;330:1198-1210.
61. Moreau JA, Zinsmeister AR, Melton LJ, et al. Gallstone pancreatitis and the effect of cholecystectomy. Mayo Clin Proc. 1988;63:466.
62. Diehl AK, Holleman DRJr, Chapman JB, et al. Gallstone size and risk of pancreatitis. Arch Intern Med. 1997;157:1674.
63. Ko CW, Sekijima JH, Lee SP. Biliary sludge. Ann Intern Med. 1999;130:301.
64. Ros E, Navarro S, Bru C, et al. Occult microlithiasis in “idiopathic” acute pancreatitis: Prevention of relapses by cholecystectomy or ursodeoxycholic acid therapy. Gastroenterology. 1991;101:1701.
65. Lopez AJ, O’Keefe P, Morrissey M, et al. Ceftriaxone-induced cholelithiasis. Ann Intern Med. 1991;115:712.
66. Lee SP, Nichols JF, Park HZ. Biliary sludge as a cause of acute pancreatitis. N Engl J Med. 1992;326:589.
67. Venu RP, Geenen JE, Hogan W, et al. Idiopathic acute pancreatitis. Dig Dis Sci. 1989;34:56.
68. Sherman S, Gottlieb K, Earle D, et al. The role of microlithiasis in idiopathic pancreatitis. Gastrointest Endosc. 1997;45:165A.
69. Choudari CP, Fogel EL, Sherman S, et al. Idiopathic pancreatitis: yield of ERCP correlated with patients age. Am J Gastroenterol. 1998;93:1654A.
70. Mujica VR, Barkin JS, Go VLW, et al. Acute pancreatitis secondary to pancreatic carcinoma. Pancreas. 2000;21:329.
71. McLatchie GR, Imrie CW. Acute pancreatitis associated with tumor metastases in the pancreas. Digestion. 1981;21:13.
72. Goldberg PB, Long WB, Oleaga JA, et al. Choledococele as a cause of recurrent pancreatitis. Gastro. 1980;78:1041.
73. Griffin M, Carey WD, Hermann R, et al. Recurrent acute pancreatitis and intussusception complicating an intraluminal duodenal diverticulum. Gastroenterology. 1981;81:345.
74. Urayama S, Kozarek R, Ball T, et al. Presentation and treatment of annular pancreas in an adult population. Am J Gastroenterol. 1995;90:995.
75. Khuroo MS, Zargar SA, Yatoo GN, et al. Ascaris-induced acute pancreatitis. Br J Surg. 1992;79:1335.
76. Parenti DM, Steinberg WM, Kang P. Infectious causes of acute pancreatitis. Pancreas. 1996;13:356.
77. Hanck C, Singer MV. Does acute alcoholic pancreatitis exist without preexisting chronic pancreatitis? Scand J Gastroenterol. 1997;32:625.
78. Ammann RW, Heitz PU, Kloeppel G. Course of alcoholic chronic pancreatitis: A prospective clinicomorphological long-term study. Gastroenterology. 1996;111:224.
79. Bordalo O, Gonclaves D, Noronha M, et al. Newer concepts for the pathogenesis of chronic alcoholic pancreatitis. Am J Gastroenterology. 1977;68:278-85.
80. Sarles H. Pathogenesis of chronic pancreatitis. Gut. 1990;31:629-32.
81. Tenner S, Freedman S. Chronic ethanol administration selectively impairs endocytosis in the rat exocrine pancreas. Pancreas. 1998;17:127-33.
82. Bockman DE, Kennedy RH, Multigner L, et al. Fine structure of the organic matrix of human pancreatic stones. Pancreas. 1986;1:204-10.
83. Ammann RW, Muelhaupt B. Progression of alcoholic acute to chronic pancreatitis. Gut. 1994;35:552-6.
84. Whitcomb DC, Schneider A. A model for inflammatory disease of the pancreas. Res Clin Gastroenterol. 2002;16:347-63.
85. Bennett ILJr, Cary FH, Mitchell GL, et al. Acute methyl alcohol poisoning: A review based on experiences in an outbreak of 323 cases. Medicine (Baltimore). 1953;32:431.
86. Lee HS. Acute pancreatitis and organophosphate poisoning—A case report and review. Singapore Med J. 1989;30:599.
87. Bartholomew C. Acute scorpion pancreatitis in Trinidad. BMJ. 1970;1:666.
88. Morton C, Klatsky AL, Udaltsova N. Smoking, coffee and pancreatitis. Am J Gastroenterol. 2004;99:731.
89. Badalov N, Baradarian R, Iswara K, et al. Drug induced acute pancreatitis: An evidence based approach. Clin Gastroenterol Hepatol. 2007;101:454-76.
90. Fortson MR, Freedman SN, Webster PD. Clinical assessment of hyperlipidemic pancreatitis. Am J Gastroenterol. 1995;90:2134.
91. Glueck CJ, Lang J, Hamer T, et al. Severe hypertriglyceridemia and pancreatitis when estrogen replacement therapy is given to hypertriglyceridemic women. J Lab Clin Med. 1994;123:18.
92. Toskes PP. Hyperlipidemic pancreatitis. Gastroenterol Clin North Am. 1990;19:783.
93. Salen S, Kessler JI, Janowitz HD. The development of pancreatic secretory insufficiency in a patient with recurrent pancreatitis and type V hyperlipoproteinemia. Mt Sinai J Med. 1970;37:103.
94. Toskes PP. Is there a relationship between hypertriglyceridemia and development of alcohol- or gallstone-induced pancreatitis? Gastroenterology. 1994;106:810.
95. Whitfield JB, Hensley WJ, Bryden D, et al. Some laboratory correlates of drinking habit. Ann Clin Biochem. 1978;15:297.
96. Mithofer K, Fernandez-del Castillo C, Frick TW. Acute hypercalcemia causes acute pancreatitis and ectopic trypsinogen activation in the rat. Gastroenterology. 1995;109:23.
97. Prinz RA, Aranha GV. The association of primary hyperparathyroidism and pancreatitis. Am Surg. 1985;51:325.
98. Takasaki M, Yorimitsu Y, Takahashi I, et al. Systemic lupus erythematosus presenting with drug-unrelated acute pancreatitis as an initial manifestation. Am J Gastroenterol. 1995;90:1172.
99. Watts RA, Isenberg DA. Pancreatic disease in the autoimmune rheumatic disorders. Semin Arthritis Rheum. 1989;19:158.
100. Orvar K, Johlin FC. Atheromatous embolization resulting in acute pancreatitis after cardiac catheterization and angiographic studies. Arch Intern Med. 1994;154:1755.
101. Fernández-del Castillo C, Harringer W, Warshaw AL, et al. Risk factors for pancreatic cellular injury after cardiopulmonary bypass. N Engl J Med. 1991;325:382.
102. Warshaw AL, O Hara PJ. Susceptibility of the pancreas to ischemic injury in shock. Ann Surg. 1978;188:197.
103. Reilly PM, Toung TJ, Miyachi M, et al. Hemodynamics of pancreatic ischemia in cardiogenic shock in pigs. Gastroenterology. 1997;113:938.
104. Ertan A, Schneider FE. Acute pancreatitis in long-distance runners. Am J Gastroenterol. 1995;90:70.
105. Wilson RH, Moorehead RJ. Current management of trauma to the pancreas. Br J Surg. 1991;78:1196.
106. Kozarek RA, Ball TJ, Patterson DJ, et al. Endoscopic transpapillary therapy for disrupted pancreatic duct and peripancreatic fluid collections. Gastroenterology. 1991;100:1362.
107. Nwariaku FE, Terracina A, Mileski WJ, et al. Is octreotide beneficial following pancreatic injury? Am J Surg. 1995;170:582.
108. Aliperti G. Complications related to diagnostic and therapeutic endoscopic retrograde cholangiopancreatography. Gastrointest Endosc Clin North Am. 1996;6:379.
109. Freeman ML, DiSario JA, Nelson DB, et al. Risk factors for post-ERCP pancreatitis: A prospective, multicenter study. Gastrointest Endosc. 2001;54:425.
110. Freeman ML, Guda NM. Prevention of post-ERCP pancreatitis: A comprehensive review. Gastrointest Endosc. 2004;59:845-64.
111. Freeman ML, DiSario JA, Nelson DB, et al. Risk factors for post-ERCP pancreatitis: A prospective, multicenter study. Gastrointest Endosc. 2001;54:425-34.
112. Cheng CL. Risk factors for post ERCP pancreatitis: A prospective multicenter study. Am J Gastroenterol. 2006;101:139-47.
113. Mehta SN, Pavone E, Barkun JS, et al. Predictors of post-ERCP complications in patients with suspected choledocholithiasis. Endoscopy. 1998;30:457-63.
114. Testoni PA, Bagnolo F, Caporuscio S, Lella F. Serum amylase measured four hours after endoscopic sphincterotomy is a reliable predictor of post-procedure pancreatitis. Am J Gastroenterology. 1999;94:1235-52.
115. Gottlieb K, Sherman S, Pezzi J, et al. Early recognition of post-ERCP pancreatitis by clinical assessment and serum pancreatic enzymes. Am J Gastroenterol. 1996;91:1553-61.
116. Murray B, Carter R, Imrie C, et al. Diclofenac reduces the incidence of acute pancreatitis after endoscopic retrograde cholangiopancreatography. Gastroenterology. 2003;124:1786-91.
117. Ito K, Fujita J, Noda Y, et al. Relationship between post-ERCP pancreatitis and the change of serum amylase level after the procedure. World J Gastroenterol. 2007;13:3855-60.
118. Khoshbaten M, Khorram H, Madad L, et al. Role of diclofenac in reducing post-endoscopic retrograde chalangiopancreatography pancreatitis. J Gastroenterol Hepatol. 2007.
119. Cheon YK, Cho KB, Watkins JL. Efficacy of diclofenac in the prevention of post-ERCP pancreatitis in predominately high risk patients: A randomized, double blind, prospective trial. Gastrointest Endosc. 2007;66:1126-32.
120. Sudhindran S, Bromwich E, Edwards PR. Prospective randomized double-blind placebo-controlled trial of glyceryl trinitrate in endoscopic retrograde cholangiopancreatography-induced pancreatitis. Br J Surg. 2001;88:1178-82.
121. Moreto M, Zaballa M, Casado I, et al. Transdermal glyceryl trinitrate for prevention of post-ERCP pancreatitis: A randomized double-blind trial. Gastrointest Endosc. 2003;57:1-7.
122. Kaffles AJ, Bourke MJ, Ding S, et al. A prospective, randomized, placebo-controlled trial of glyceryl trinitrate in ERCP: Effects on technical success and post ERCP pancreatitis. Gastrointest Endosc. 2006;64:351-7.
123. Prat F, Amaris J, Ducot B, et al. Nifedipine for prevention of post-ERCP pancreatitis: A prospective, double-blind randomized study. Gastrointest Endosc. 2002;56:202-8.
124. Sand J, Nordback I. Prospective randomized trial of the effect of nifedipine on pancreatic irritation after endoscopic retrograde cholangiopancreatography. Digestion. 1993;54:179-84.
125. Schwartz JJ, Lew RJ, Ahmad NA, et al. The effect of lidocaine sprayed on the major duodenal papilla on the frequency of post-ERCP pancreatitis. Gastrointest Endosc. 2004;59:179-84.
126. Gorelick A, Barnett J, Chey W, et al. Botulinum toxin injection after biliary sphincterotomy. Endoscopy. 2004;36:170-3.
127. Messori A, Rampazzo R, Scroccaro G, et al. Effectiveness of gabexate mesilate in acute pancreatitis. A metaanalysis. Dig Dis Sci. 1995;40:734-8.
128. Pederzoli P, Cavallini G, Falconi M, Bassi C. Gabexate mesilate vs aprotinin in human acute pancreatitis (GA.ME.P.A.). A prospective, randomized, double-blind multicenter study. Int J Pancreatol. 1993;14:117-24.
129. Buchler M, Malfertheiner P, Uhl W, et al. Gabexate mesilate in human acute pancreatitis. German Pancreatitis Study Group. Gastroenterology. 1993;104:1165-70.
130. Goebell H, German Pancreatitis Study Group. Multicenter double-blind study of gabexate-mesilate (FOY), given intravenously in low dose in acute pancreatitis and the Digestion, 40, 1998:83
131. Valderrama R, Pérez-Mateo M, Navarro S, et al. Multicenter double-blind trial of gabexate mesylate (FOY) in unselected patients with acute pancreatitis. Digestion. 1992;51:65-70.
132. Young CY, Chang-Chien CS, Liaw YF. Controlled trial of protease inhibitor gabexate mesilate (FOY) in the treatment of acute pancreatitis. Pancreas. 1987;2:698-700.
133. Manes G, Ardizzone S, Lombardi G, et al. Efficacy of postprocedure administration of gabexate mesylate in the prevention of post-ERCP pancreatitis: A randomized, controlled, multicenter study. Gastrointest Endosc. 2007;65:982-7.
134. Fujishiro H, Adachi K, Imaoka T, et al. Ulinastatin shows preventive effect on post-endoscopic retrograde cholangiopancreatography pancreatitis in a multicenter prospective randomized trial. J Gastroenterol Hepatol. 2006;21:1065-9.
135. Lee JK, Park JK, Yoon WJ, et al. Risk for post-ERCP pancreatitis after needle knife precut sphincterotomy following repeated cannulation attempts. J Clin Gastroenterol. 2007;41:427-31.
136. Ueki T, Otani K, Kawamoto K, et al. Comparison between ulinastatin and gabexate mesylate for the prevention of post-endoscopic retrograde cholangiopancreatography pancreatitis: A prospective, randomized trial. J Gastroenterol. 2007;42:161-7.
137. Testoni PA, Mariani A, Masci E, Curioni S. Frequency of post-ERCP pancreatitis in a single tertiary referral centre without and with routine prophylaxis with gabexate: A 6-year survey and cost-effectiveness analysis. Dig Liver Dis. 2006;38:588-95.
138. Guelrud M, Mendoza S, Viera L, Gelrud D. Somatostatin prevents acute pancreatitis after pancreatic duct sphincter hydrostatic balloon dilation in patients with idiopathic recurrent pancreatitis. Gastrointest Endosc. 1990;36:44-7.
139. Persson B, Slezak P, Efendic S, Haggmark A. Can somatostatin prevent injection pancreatitis after ERCP? Hepatogastroenterology. 1992;39:259-61.
140. Bordas JM, Toledo-Pimentel V, Llach J, et al. Effects of bolus somatostatin in preventing pancreatitis after endoscopic pancreatography: Results of a randomized study. Gastrointest Endosc. 1998;47:230-4.
141. Poon RTP, Yeung C, Lo CM, et al. Prophylactic effect of somatostatin on post-ERCP pancreatitis: A randomized controlled trial. Gastrointest Endosc. 1999;49:593-8.
142. Poon RT, Yeung C, Liu CL, et al. Intravenous bolus somatostatin after diagnostic cholangio-pancreatography reduces the incidence of pancreatitis associated with therapeutic endoscopic retrograde cholangiopancreatography procedures: A randomized controlled trial. Gut. 2003;52:1768-73.
143. Arvanitidis D, Anagnostopoulos GK, Giannopoulos D, et al. Can somatostatin prevent post-ERCP pancreatitis? Results of a randomized controlled trial. J Gastroenterol Hepatol. 2004;19:278-82.
144. Borsch G, Bergbauer M, Nebel W, Sabin G. Der einfluß von somatostatin auf die amylasespiegel und pankreatitistrate nach ERCP. Med Welt. 1984;35:109-12.
145. Bordas M, Toledo V, Mondelo F, Rodès J. Prevention of pancreatic reactions by bolus somatostatin administration in patients undergoing endoscopic retrograde cholangio-pancreatography and endoscopic sphincterotomy. Hormone Res. 1988;29:106-8.
146. Testoni PA, Masci E, Bagnolo F, Tittobello A. Endoscopic papillo-sphincterotomy: prevention of pancreatic reaction by somatostatin. Ital J Gastroenterol. 1988;20:70-3.
147. Flati G, Porowska B, Negro P, et al. Efecto de la somatostatina sobre las secuelas de la colangiopancreatografia retrograda endoscopica. Rev Esp Enf Ap Digest. 1988;74:17-19.
148. Deschner K, Kalloo A, Collen M, et al. Somatostatin: an evaluation of its utility as adjunctive medication for ERCP [abstract]. Gastrointest Endosc. 1989;35:A149.
149. Vila JJ, Jimenez FJ, Prieto C, et al. Utility of bolus somatostatin administration in preventing pancreatitis after endoscopic retrograde cholangiopancreatography: A controlled, non-randomized study. Gastroenterol Hepatol. 2006;29:231-6.
150. Manolakopoulos S, Avgerinos A, Vlachogiannakos J, et al. Octreotide vs hydrocortisone versus placebo in the prevention of post-ERCP pancreatitis: A multicenter randomized controlled trial. Gastrointest Endosc. 2002;62:245-50.
151. Andriulli A, Leandro G, Niro G, et al. Pharmacologic treatment can prevent pancreatic injury after ERCP: A meta-analysis. Gastrointest Endosc. 2000;51:1-7.
152. Lella F, Bagnolo F, Colombo E, Bonassi U. A simple way of avoiding post-ERCP pancreatitis. Gastrointest Endosc. 2004;59:830-4.
153. Freeman ML, Overby C, Qi D. Pancreatic stent insertion: Consequence of failure and results of a modified technique to maximize success. Gastrointest Endosc. 2004;5:8-14.
154. Singh P, Sivak MV, Agarwal D, et al. Prophylactic pancreatic stenting for prevention of post- ERCP acute pancreatitis: A meta-analysis of controlled trials [abstract]. Gastrointest Endosc. 2003;57:AB89.
155. Das A, Singh P, Sivak MVJr, Chak A. Pancreatic-stent placement for prevention of post-ERCP pancreatitis: A cost-effectiveness analysis. Gastrointest Endosc. 2007;65:960-8.
156. Steinberg WM, Chari ST, Forsmark CE, et al. Controversies in clinical pancreatology: Management of acute idiopathic recurrent pancreatitis. Pancreas. 2003;27:103.
157. Artifon ELA, Sakai P, Cunha JEM, et al. Guidewire cannulation reduces risk of post-ERCP pancreatitis and facilitates bile duct cannulation. Am J Gastroenterol. 2007;102:1-7.
158. Bragg LE, Thompson JS, Burnett DA, et al. Increased incidence of pancreas-related complications in patients with postoperative pancreatitis. Am J Surg. 1985;150:694.
159. Lefor AT, Vuocolo P, Parker FB, et al. Pancreatic complications following cardiopulmonary bypass: Factors influencing mortality. Arch Surg. 1992;127:1225.
160. Camargo CA, Greig PD, Levy GA, et al. Acute pancreatitis following liver transplantation. J Am Coll Surg. 1995;181:249.
161. Gelrud A, Sheth S, Banerjee S, et al. Analysis of cystic fibrosis gene product (CFTR) function in patients with pancreas divisum and recurrent acute pancreatitis. Am J Gastroenterol. 2004;99:1557-62.
162. Fogel EL, Toth TG, Lehman GA, et al. Does endoscopic therapy favorably affect the outcome of patients who have recurrent acute pancreatitis and pancreas divisum? Pancreas. 2007;34:21-45.
163. Steinberg WM. Controversies in pancreatology. Should the sphincter of Oddi pressure be measured in patients with idiopathic recurrent acute pancreatitis and should sphincterotomy be performed if the pressure is high? Pancreas. 2003;27:118.
164. Munk EM, Pedersen L, Floyd A, et al. Inflammatory bowel diseases, 5-aminosalicylic acid and sulfasalazine treatment and risk of acute pancreatitis: A population based case control study. Am J Gastroenterol. 2004;99:884.
165. Patel RS, Johlin FC, Murray JA. Celiac disease and recurrent pancreatitis. Gastrointest Endosc. 1999;50:823.
166. Carroccio A, Di Prima L, Scalici C, et al. Unexplained elevated serum pancreatic enzymes: A reason to suspect celiac disease. Clin Gastroenterol Hepatol. 2006;4:455-9.
167. Ryan CM, Sheridan RL, Schoenfeld DA, et al. Postburn pancreatitis. Ann Surg. 1995;222:163.
168. Blomgren KB, Sundstrom A, Steinexk G, et al. A Swedish case control network for studies of drug-induced morbidity-acute pancreatitis. Eur J Clin Pharmacol. 2002;58:275.
169. Baggenstoss BR, Freeman ML. Autoimmune pancreatitis presenting as acute recurrent pancreatitis. Pancreas. 2008;37:461A.
170. Bernard JP, Sahel J, Giovanni M, et al. Pancreas divisum is a probable cause of acute pancreatitis: A report of 137 cases. Pancreas. 1990;5:248.
171. Lans JL, Geenen JE, Johanson JF, et al. Endoscopic therapy in patients with pancreas divisum and acute pancreatitis. A prospective, randomized, controlled clinical trial. Gastrointest Endosc. 1992;38:430.
172. Delhaye M, Engelholm L, Cremer M. Pancreas divisum: Congenital anatomic variant or anomaly? Contribution of endoscopic retrograde dorsal pancreatography. Gastroenterology. 1985;89:951.
173. Choudari CP, Fogel EL, Sherman S, et al. Pancreas divisum, pancreatitis and CFTR mutations. Gastrointest Endosc. 1999;49:AB187.
174. Gullo L. Familial pancreatic hyperenzymemia. Pancreas. 2000;20:158.
175. Bohidar NP, Garg PK, Khanna S, et al. Incidence, etiology and impact of fever in patients with acute pancreatitis. Pancreatology. 2003;3:9.
176. Boon P, de Rueck J, Achten E, et al. Pancreatic encephalopathy: A case report and review of the literature. Clin Neuro Neurosurg. 1991;93:137.
177. Potts DE, Mass MF, Iseman MD. Syndrome of pancreatic disease, subcutaneous fat necrosis and polyserositis. Am J Med. 1975;58:417.
178. Steinberg WM, Goldstein SS, Davis N, et al. Diagnostic assays in acute pancreatitis. Ann Intern Med. 1985;102:576.
179. Agarwal N, Pitchumoni CS, Sivaprasad AV. Evaluating tests for acute pancreatitis. Am J Gastroenterol. 1990;85:356.
180. Sternby B, O’Brien JF, Zinsmeister AR, et al. What is the best biochemical test to diagnose acute pancreatitis? A prospective clinical study. Mayo Clin Proc. 1996;71:1138.
181. Kimmel P, Tenner S, Habwe VQ, et al. Trypsinogen and other pancreatic enzymes in patients with renal disease: A comparison of high efficiency hemodialysis and continuous ambulatory peritoneal dialysis. Pancreas. 1995;10:325.
182. Sachdeva CK, Bank S, Greenberg R, et al. Fluctuations in serum amylase in patients with macroamylasemia. Am J Gastroenterol. 1995;90:800.
183. Schmidt J, Hotz HG, Foitzik T, et al. Intravenous contrast medium aggravates the impairment of pancreatic microcirculation in necrotizing pancreatitis in the rat. Ann Surg. 1995;221:257.
184. Gwodz GP, Steinberg WM, Werner M, et al. Comparative evaluation of the diagnosis of acute pancreatitis based on serum and urine enzyme assays. Clin Chim Acta. 1990;187:243.
185. Seno T, Harada H, Ochi K, et al. Serum levels of six pancreatic enzymes as related to the degree of renal dysfunction. Am J Gastroenterol. 1995;90:2002.
186. Gumaste VV, Roditis N, Mehta D, et al. Serum lipase levels in nonpancreatic abdominal pain versus acute pancreatitis. Am J Gastroenterol. 1993;88:2051.
187. Pezzilli R, Billi P, Migliori M, et al. Clinical value of pancreatitis-associated protein in acute pancreatitis. Am J Gastroenterol. 1997;92:1887.
188. Tenner S, Dubner H, Steinberg W. Predicting gallstone pancreatitis with laboratory parameters: A meta analysis. Am J Gastroenterol. 1994;89:1863.
189. Stimac D, Lenac T, Marusic Z. A scoring system for early differentiation of the etiology of acute pancreatitis. Scand J Gastroenterology. 1998;33:209-11.
190. Lankisch PG, Dröge M, Becher R. Pleural effusions: A new negative prognostic parameter for acute pancreatitis. Am J Gastroenterol. 1994;89:1849.
191. Heller S, Tenner SM, Hughes M, Banks PA. Pleural effusion as a predictor of severity in patients with acute pancreatitis. Pancreas. 1997;15:222-5.
192. Lee JK, Enns R. Review of idiopathic pancreatitis. World J Gastroenterol. 2007;13:6296-313.
193. Amouyal P, Amouyal G, Levy P, et al. Diagnosis of choledocholithiasis by endoscopic ultrasonography. Gastroenterology. 1994;106:1062.
194. Arguedas M, Dupont AW, Wilcox CM. Where do ERCP, endoscopic ultrasound, magnetic resonance cholangiopancreatography, and intraoperative cholangiography fit in the management of acute biliary pancreatitis? A decision analysis model. Am J Gastroenterol. 2001;96:2892-9.
195. Balthazar EJ, Freeny PC, van Sonnenberg E. Imaging and intervention in acute pancreatitis. Radiology. 1994;193:297.
196. Freeny PC. Incremental dynamic bolus computed tomography of acute pancreatitis. Int J Pancreatol. 1993;13:147.
197. McMenamin DA, Gates LK. A retrospective analysis of the effect of contrast-enhanced CT on the outcome of acute pancreatitis. Am J Gastroenterol. 1996;91:861-8.
198. Uhl W, Roggo A, Kirschstein T, et al. Influence of contrast-enhanced computed tomography on course and outcome in patients with acute pancreatitis. Pancreas. 2002;24:191.
199. Balthazar EJ, Ranson JH, Naidich DP, et al. Acute pancreatitis: Prognostic value of CT. Radiology. 1985;156:767.
200. Carmen-Sanchez R, Uscanga L, Bezaury-Rivas P, et al. Potential harmful effect of iodinated intravenous contrast medium on the clinical course of mild acute pancreatitis. Arch Surg. 2000;135:1280.
201. White M, Simeone JF, Wittenberg J. Air within a pancreatic inflammatory mass: Not necessarily a sign of abscess. J Clin Gastroenterol. 1983;5:173.
202. Arvanitakis M, Delhaye M, De Maertelaere V. Computed tomography and magnetic resonance imaging in the assessment of acute pancreatitis. Gastroenterology. 2004;126:715.
203. Haustein J, Niendorf HP, Krestin G, et al. Renal clearance of gadolinium-DTPA/dimeglumine in patients with chronic renal failure. Invest Radiol. 1992;27:153.
204. Cavanese C, Mereu MC, Aime S, et al. Gadolinium associated nephrogenic systemic fibrosis: The need for nephrologists’ awareness. J Nephrology. 2008;21:324-36.
205. Testoni PA, Mariani A, Curioni S, et al. MRCP-secretin test-guided management of idiopathic recurrent pancreatitis: Long-term outcomes. Gastrointest Endosc. 2007;143:165-9.
206. Ranson JH. The timing of biliary surgery in acute pancreatitis. Ann Surg. 1979;189:654.
207. Tenner SM, Steinberg W. The admission serum lipase:amylase ratio differentiates alcoholic from nonalcoholic acute pancreatitis. Am J Gastroenterol. 1992;87:1755.
208. King LG, Seelig CB, Ranney JE. The lipase to amylase ratio in acute pancreatitis. Am J Gastroenterol. 1995;90:67.
209. Rhodes M, Sussman L, Cohen L, et al. Randomised trial of laparoscopic exploration of common bile duct versus postoperative endoscopic retrograde cholangiography for common bile duct stones. Lancet. 1998;351:159.
210. Tsai C-J. Is obesity a significant prognostic factor in acute pancreatitis? Dig Dis Sci. 1998;43:2251.
211. Porter KA, Banks PA. Obesity as a predictor of severity in acute pancreatitis. Int J Pancreatol. 1991;10:247.
212. Funnell IC, Bornman PC, Weakley SP, et al. Obesity: An important prognostic factor in acute pancreatitis. Br J Surg. 1993;80:484.
213. Karimgani I, Porter KA, Langevin RE, et al. Prognostic factors in sterile pancreatic necrosis. Gastroenterology. 1992;103:1636.
214. Mofidi R, Duff MD, Wigmore SJ, et al. Association between early systemic inflammatory response, severity of multiorgan dysfunction and death in acute pancreatitis. Br J Surg. 2006;93:738-44.
215. Ranson JHC. Etiological and prognostic factors in human acute pancreatitis: A review. Am J Gastroenterol. 1982;77:633-8.
216. Leese T, Shaw D, Holliday M. Prognostic markers in acute pancreatitis: Can pancreatic necrosis be predicted? Ann R Coll Surg Engl. 1988;70:227.
217. King NKK, Siriwardena AK. European survey of surgical strategies for the management of severe acute pancreatitis. Am J Gastroenterol. 2004;99:719.
218. Dominguez-Munoz JE, Carballo F, García MJ, et al. Evaluation of the clinical usefulness of APACHE-II and SAPS systems in the initial prognostic classification of acute pancreatitis: A multicenter study. Pancreas. 1993;8:682.
219. Blamey SL, Imrie CW, O’Neill J, et al. Prognostic factors in acute pancreatitis. Gut. 1984;25:1340-6.
220. Wilson C, Health DI, Imrie CW. Prediction of outcome in acute pancreatitis: A comparative study of APACHE-II, clinical assessment and multiple factor scoring system. Br J Surg. 1990;77:1260.
221. Malfertheiner P, Dominguez-Munoz JE. Prognostic factors in acute pancreatitis. Int J Pancreatol. 1993;14:1.
221a. Wu BU, Johannes RS, Sun X, et al. The early prediction of mortality in acute pancreatitis: A large population based study. Gut. 2008;57:1698-703.
221b. Singh V, Wu BU, Maurer R, et al. A prospective evaluation of the Bedside Index of Severity in Acute Pancreatitis. Am J Gastroenterol. 2009;104:966-71.
221c. Wu BU, Johannes RS, Sun X, et al. Early changes in blood urea nitrogen predict mortality in acute pancreatitis. Gastroenterology. 2009;137:129-35.
222. Johnson CD, Abu-Hillal A. Persistent organ failure during the first week as a marker of fatal outcome in acute pancreatitis. Gut. 2004;53:1340-4.
223. Tenner S, Sica G, Hughes M, et al. Relationship of necrosis to organ failure in severe acute pancreatitis. Gastroenterology. 1997;113:899.
224. Marshall JC, Cook DJ, Christou NV, et al. Multiple organ dysfunction score: A reliable descriptor of a complex clinical outcome. Crit Care Med. 1995;23:1638-52.
225. McMahon MJ, Playworth MJ, Pickford IR. A comparative study of methods for the prediction of severity of attacks of acute pancreatitis. Br J Surg. 1980;67:22-5.
226. Baillargeon JD, Orav J, Ramagopal V, et al. Hemoconcentration as an early risk factor for necrotizing pancreatitis. Am J Gastroenterol. 1998;93:2130.
227. Brown A, Orav J, Banks PA. Hemoconcentration is an early marker for organ failure and necrotizing pancreatitis. Pancreas. 2000;20:367.
228. Lankisch PG, Mahlke R, Blum T, et al. Hemoconcentration: An early marker of severe and/or necrotizing pancreatitis. Am J Gastrenterol. 2001;96:2081.
229. Mayer AD, McMahon MJ, Bowen M, et al. C reactive protein: An aid to assessment and monitoring of acute pancreatitis. J Clin Pathol. 1984;37:207.
230. Heath DI, Cruickshank A, Gudgeon S. Role of interleukin 6 in mediating the acute phase protein response and potential as an early means of severity assessment in acute pancreatitis. Gut. 1993;34:41.
231. Uhl W, Buchler MW, Malfertheiner P, et al. PMN elastase in comparison with CRP, antiproteases and LDH as indicators of necrosis in human acute pancreatitis. Pancreas. 1991;6:253.
232. Werner J, Hartwig W, Uhl W, et al. Useful markers for predicting severity and monitoring progression of acute pancreatitis. Pancreatology. 2003;3:115-27.
233. Khan Z, Vlodov J, Horovitz J, et al. Urinary trypsinogen activation peptide is more accurate than HCT in determining severity in patients with acute pancreatitis. A prospective study. Am J Gastroenterol. 2002;97:1973-7.
234. Kylanpaa-Back ML, Takala A, Kemppainen EA, et al. Procalcitonin strip test in the early detection of severe acute pancreatitis. Br J Surg. 2001;88:222.
235. Lankisch PG, Pflichthofer D, Lehnick D. No strict correlation between necrosis and organ failure in acute pancreatitis. Pancreas. 2000;3:319-22.
236. Isenmann R, Rau B, Beger HG. Bacterial infection and extent of necrosis are determinants of organ failure in patients with acute necrotizing pancreatitis. Br J Surg. 1999;86:1020.
237. Mathieson DR, Gross JB, Power MH. Elevated values for serum amylase and lipase following the administration of opiates, a preliminary report. Mayo Clin Proc. 1951;26:81.
238. Foitzik T, Eibl G, Holz H, et al. Persistent multiple organ microcirculatory disorders in severe acute pancreatitis: experimental findings and clinical implications. Dig Dis Sci. 2002;47:130-8.
239. Klar E, Herfarth C, Messmer K. Therapeutic effect of isovolemic hemodilution with dextran 60 on the impairment of pancreatic microcirculation in acute biliary pancreatitis. Ann Surg. 1990;211:346-53.
240. Gardner TB, Vege SS, Chari ST, et al. Fluid resuscitation in acute pancreatitis. Clin Gastroenterol Hepatol. 2008;6:1070-6.
241. Finch WT, Sawyers JL, Shenker S. A prospective study to determine the efficacy of antibiotics in acute pancreatitis. Ann Surg. 1976;183:667.
242. Gerzof SG, Banks PA, Robbins AH, et al. Early diagnosis of pancreatic infection by computed tomography-guided aspiration. Gastroenterology. 1987;93:1315.
243. Beger HG, Bittner R, Block S, et al. Bacterial contamination of pancreatic necrosis—a prospective clinical study. Gastroenterology. 1986;91:433.
244. Buchler M, Malfertheiner P, Fries H, et al. Human pancreatic tissue concentration of bactericidal antibiotics. Gastroenterology. 1992;103:1902.
245. Pederzoli P, Bassi C, Vesentini RM, et al. A randomized multicenter clinical trial of antibiotic prophylaxis of septic complications in acute necrotizing pancreatitis with imipenem. Surg Gynecol Obstet. 1993;176:480.
246. Sainio V, Kemppainen E, Puolakkainen P, et al. Early antibiotic treatment in acute necrotizing pancreatitis. Lancet. 1995;346:663.
247. Delcenserie R, Yzet T, Ducroix JP. Prophylactic antibiotics in the treatment of severe alcoholic pancreatitis. Pancreas. 1996;13:198.
248. Schwarz M, Isenmann R, Meyer H, et al. Antibiotika bei nekrotisierender pancreatitis. Ergebnisse einer kontrollierten studie. Dtsch Med Wochenschr. 1997;122:356.
249. Luiten EJT, Hop WCJ, Lange JF, et al. Controlled clinical trial of selective decontamination for the treatment of severe acute pancreatitis. Ann Surg. 1995;222:57.
250. Golub R, Siddiqi F, Pohl D. Role of antibiotics in acute pancreatitis: A meta analysis. J Gastrointest Surg. 1998;2:496.
251. Sharma VK, Howden C. Prophylactic antibiotic administration reduces sepsis and mortality in acute necrotizing pancreatitis: A meta analysis. Pancreas. 2001;22:28.
252. Bassi C, Falconi M, Talamini G, et al. Controlled clinical trial of pefloxacin versus imipenem in severe acute pancreatitis. Gastroenterology. 1998;115:1513.
253. Delcenserie C, Dellion-Lozinguez M-P, Pagenault M, et al. Prophylactic ciprofloxacin treatment in acute necrotizing pancreatitis: A prospective, randomized multicenter clinical trial. Gastroenterology. 2001;120:A25.
254. Isenmann R, Runzi M, Kron M, et al. Prophylactic antibiotic treatment in patients with predicted severe acute pancreatitis: A placebo-controlled, double blind trial. Gastroenterology. 2004;126:997.
255. Dellinger EP, Tellado JM, Soto NE, et al. Early antibiotic treatment for severe acute necrotizing pancreatitis: Randomized, double blind, placebo controlled study. Ann Surg. 2007;245:673-83.
256. Bradley EL. Guiding the reluctant: A primer on guidelines in general and pancreatitis in particular. Pancreatology. 2003;3:139.
257. Neoptolemos JP, Carr-Locke DL, London NJ, et al. Controlled trial of urgent endoscopic retrograde cholangiopancreatography and endoscopic sphincterotomy versus conservative treatment for acute pancreatitis due to gallstones. Lancet. 1988;2:979.
258. Fan ST, Lai ECS, Mok FPT, et al. Early treatment of acute biliary pancreatitis by endoscopic papillotomy. N Engl J Med. 1993;328:228.
259. Folsch UR, Nitsche R, Ludtke R, et al. Early ERCP and papillotomy compared with conservative treatment for acute biliary pancreatitis. N Engl J Med. 1997;336:237.
260. Lau ST, Simchuk EJ, Kozarek RA, et al. A pancreatic ductal leak should be sought to direct treatment in patients with acute pancreatitis. Am J Surg. 2001;181:411-15.
261. Kozarek R, Hovde O, Attia F, et al. Do pancreatic stents cause or prevent pancreatic sepsis. Gastrointest Endosc. 2003;58:505-9.
262. Jacobson B, Vander V, Liet MA, et al. A prospective randomized trial of clear liquids vs low fat solid diet as the initial meal in mild acute pancreatitis. J Clin Gastroenterol Hepatol. 2007;5:946-51.
263. Petrov MS, Santvoort HC, Besselink MGH, et al. Early oral feeding in mild acute pancreatitis: A meta-analysis. Am J Gastroenterol. 2007;102:2079-84.
264. Levy P, Heresbach D, Pariente EA, et al. Frequency and risk factors of recurrent pain during refeeding in patients with acute pancreatitis: A multivariate multicentre prospective study of 116 patients. Gut. 1997;40:262.
265. Eckerwall GE, Tingstedt BBA, Bergenzaun PE, Andersson RG. Immediate oral feeding in patients with mild acute pancreatitis is safe and may accelerate recovery—A randomized clinical study. Clin Nutr. 2007;18:139-47.
266. Sax HC, Warner BW, Talamini MA, et al. Early total parenteral nutrition in acute pancreatitis: Lack of beneficial effect. Am J Surg. 1987;153:117.
267. McClave SA, Greene LM, Snider HL, et al. Comparison of the safety of early vs. parenteral nutrition in mild acute pancreatitis. JPEN. 1997;21:14.
268. Windsor ACJ, Kanwar S, Li AGK, et al. Compared with parenteral nutrition, enteral feeding attenuates the acute phase response and improves the disease severity in acute pancreatitis. Gut. 1998;42:431.
269. Kalfarentzos F, Kehagias J, Mead N, et al. Enteral nutrition is superior to parenteral nutrition in severe acute pancreatitis: Results of a randomized prospective trial. Br J Surg. 1997;84:1665.
270. Eatock FC, Chong PS, Menezes N, et al. Nasogastric feeding in severe acute pancreatitis is safe and avoids the risks associated with the nasojejunal route: A randomized controlled trial. Am J Gastroenterology. 2005;100:432-9.
271. Uhl W, Warshaw A, Imrie C, et al. IAP guidelines for the surgical management of acute pancreatitis. Pancreatology. 2002;2:565.
272. Mier J, Luque-deLeon E, Castillo A, et al. Early vs. late necrosectomy in severe necrotizing pancreatitis. Am J Surg. 1997;173:71.
273. Bradley EL, Allen K. A prospective longitudinal study of observation versus surgical intervention in the management of necrotizing pancreatitis. Am J Surg. 1991;161:19.
274. Buchler MW, Gloor B, Muller CA, et al. Acute necrotizing pancreatitis: Treatment strategy according to the status of infection. Ann Surg. 2000;232:619.
275. Widdison AL, Karanjia ND. Pancreatic infection complicating acute pancreatitis. Br J Surg. 1993;80:148.
276. Beger HG, Krautzberger W, Bittner R, et al. Results of surgical treatment of necrotizing pancreatitis. World J Surg. 1985;9:972.
277. Besselink MG, Verwer TJ, Schoenmaeckers EJ, et al. Timing of surgical intervention in necrotizing pancreatitis. Arch Surg. 2007;142:1194-201.
278. Baril NB, Ralls PW, Wren SM, et al. Does an infected peripancreatic fluid collection or abscess mandate operation? Ann Surg. 2000;231:361-7.
279. Dubner H, Steinberg WM, Hill M, et al. Infected pancreatic necrosis and peripancreatic fluid collections: Serendipitous response to antibiotics and medical therapy in three patients. Pancreas. 1996;12:298-302.
280. Vege SS, Baron TH. Management of pancreatic necrosis in severe acute pancreatitis. Clin Gastroenterol Hepatol. 2005;3:192-6.
281. Runzi M, Niebel W, Goebell H, et al. Severe acute pancreatitis: Nonsurgical treatment of infected necrosis. Pancreas. 2005;30:195-9.
282. Messori A, Rampazzo R, Scroccaro G, et al. Effectiveness of gabexate mesilate in acute pancreatitis. A meta-analysis. Dig Dis Sci. 1995;40:734.
283. Cavallini G, Frulloni L. Somatostatin and octreotide in acute pancreatitis: The never ending story. Dig Liver Dis. 2001;33:192.
284. McKay C, Curran FJM, Sharples CE, et al. Prospective placebo-controlled randomized trial of lexipafant in predicted severe acute pancreatitis. Br J Surg. 1997;84:1239.
285. Johnson CD, Kingsnorth AN, Imrie CW, et al. Double-blind, randomized, placebo controlled study of a platelet activating factor antagonist, lexipafant, in the treatment and prevention of organ failure in predicted severe acute pancreatitis. Gut. 2001;48:62.
286. Anai H, Sakaguchi H, Uchida H, et al. Continuous arterial infusion therapy for severe acute pancreatitis: Correlation between CT arteriography and therapeutic effect. J Vasc Interv Radiol. 1999;10:1335.
287. Takeda K, Matsung S, Ogawa M, et al. Continuous regional arterial infusion (CRAI) therapy reduces the mortality rate of acute necrotizing pancreatitis: Results of a cooperative survey in Japan. J Hepat Pancreat Surg. 2001;8:216.
288. Imaizumi H, Kida M, Nishimaki H, et al. Efficacy of continuous regional arterial infusion of a protease inhibitor and antibiotic for severe acute pancreatitis in patients admitted to an intensive care unit. Pancreas. 2004;28:369.
289. Steinberg WA, Schlesselman SE. Treatment of pancreatitis: Comparison of animal and human studies. Gastroenterology. 1987;93:1420.
290. Olah A, Belagyi T, Issekutz A, et al. Randomized clinical trial of specific lactobacillus and fibre supplement to early enteral nutrition in patients with acute pancreatitis. Br J Surg. 2002;89:1103-7.
291. Besselink MG, van Santvoort HC, Buskens E, et al. Probiotic prophylaxis in predicted severe acute pancreatitis: A randomised, double-blind, placebo-controlled trial. Lancet. 2008;371:651-9.
292. Bjarnason A, Adler SN, Bjarnason I. Probiotic prophylaxis in predicted severe acute pancreatitis. Lancet. 2008;372:114-15.
293. Vitas GJ, Sarr MG. Selected management of pancreatic pseudocysts: Operative versus expectant management. Surgery. 1992;111:123.
294. Yeo CJ, Bastidas JA, Lynch-Nyhan A, et al. The natural history of pancreatic pseudocysts documented by computed tomography. Surg Gynecol Obstet. 1990;170:411.
295. Weltz C, Pappas TN. Pancreatography and the surgical management of pseudocysts. Gastrointest Endosc Clin North Am. 1995;5:269.
296. Catalano MF, Geenen JE, Schmalz MJ, et al. Treatment of pancreatic pseudocysts with ductal communication by transpapillary pancreatic duct endoprosthesis. Gastrointest Endosc. 1995;42:214.
297. Lawson JM, Baillie J. Endoscopic therapy for pancreatic pseudocysts. Gastrointest Endosc Clin North Am. 1995;5:181.
298. Sharma SS, Bhargawa N, Govil A. Endoscopic management of pancreatic pseudocyst: A long-term follow-up. Endoscopy. 2002;34:203-7.
299. Hartwig W, Maksan SM, Foitzik T, et al. Reduction in mortality with delayed surgical therapy of severe pancreatitis. J Gastrointest Surg. 2002;6:481-7.
300. Beger HG, Rau B, Isenmann R. Natural history of necrotizing pancreatitis. Pancreatology. 2003;3:93-101.
301. Zhou ZG, Zheng YC, Shu Y, et al. Laparoscopic management of severe acute pancreatitis. Pancreas. 2003;27:46-50.
302. Baron TH, Harewood GC, Morgan DE, et al. Outcome differences after endoscopic drainage of pancreatic necrosis, acute pancreatic pseudocysts, and chronic pancreatic pseudocysts. Gastrointest Endosc. 2002;56:7-17.
303. Flati G, Andren-Sandberg A, La Pinta M, et al. Potentially fatal bleeding in acute pancreatitis: Pathophysiology, prevention and treatment. Pancreas. 2003;26:8-14.
304. Lankisch PG. The spleen in inflammatory pancreatic disease. Gastroenterology. 1990;98:509-18.
305. Alridge MC, Francis ND, Glazer G, et al. Colonic complications of severe acute pancreatitis. Br J Surg. 1989;76:362-7.
306. Salamone T, Tosi P, Palateri G, et al. Coagulative disorders in human acute pancreatitis: Role for the D-dimer. Pancreas. 2003;26:111.
307. Campo SM, Gasparri V, Catarinelli G, Sepe M. Acute pancreatitis with Purtscher’s retinopathy: Case report and review of the literature. Digest Liver Dis. 2000;32:729-32.
[/level-membership-for-gastroenterology-and-hepatology-category][not-level-membership-for-gastroenterology-and-hepatology-category]
CHAPTER 58 Acute Pancreatitis
INCIDENCE AND BURDEN OF DISEASE
The incidence of acute pancreatitis in England, Denmark, and the United States varies from 4.8 to 38 per 100,000 patients.1–3 However, estimates of incidence are inaccurate because the diagnosis of mild disease may be missed, and death may occur before diagnosis in 10% of patients with severe disease.4
Diseases of the pancreas (acute and chronic pancreatitis) accounted for 327,000 inpatient hospital stays, 78,000 outpatient hospital visits, 195,000 emergency department visits, and 531,000 physician office visits in 1998.5 The cost of pancreatic diseases (direct and indirect costs) was estimated to be 2.5 billion dollars in the year 2000.5 In the same year, there were 2834 deaths in the United States from acute pancreatitis, making it the 14th most common cause of deaths due to gastrointestinal (GI) diseases.6 Acute pancreatitis ranks as the second most common inpatient GI diagnosis in the United States after cholelithiasis and acute cholecystitis and ahead of acute appendicitis.6
The incidence of acute pancreatitis appears to be increasing.7–9 As the population is becoming increasingly overweight, the incidence of gallstones, the most common cause of acute pancreatitis is rising. Unfortunately, during this same period, the overall mortality rate from acute pancreatitis has declined only gradually to approximately 5% to 10%.10–12
DEFINITIONS
Many pancreatologists use the 1992 Atlanta Symposium definition of acute pancreatitis,13 which is an acute inflammatory process of the pancreas with variable involvement of other regional tissues or remote organ systems. Acute pancreatitis is best defined clinically by a patient presenting with two of the following criteria12: (1) symptoms, such as epigastric pain, consistent with the disease; (2) a serum amylase or lipase greater than three times the upper limit of normal; or (3) radiologic imaging consistent with the diagnosis, usually using computed tomography (CT) or magnetic resonance imaging (MRI). Pancreatitis is classified as acute unless there are CT, MRI, or endoscopic retrograde cholangiopancreatography (ERCP) findings of chronic pancreatitis. Then pancreatitis is classified as chronic pancreatitis, and any episode of acute pancreatitis is considered an exacerbation of inflammation superimposed on chronic pancreatitis (see Chapter 59).
Once the diagnosis is established, patients are classified as having mild or severe pancreatitis. Mild acute pancreatitis consists of interstitial (edematous) pancreatitis on imaging, minimal or no extrapancreatic organ dysfunction, and typically an uneventful recovery. Severe pancreatitis manifests as organ failure or local complications such as necrosis, abscess, or pseudocyst. The Atlanta criteria13 (Table 58-1) defines severity by the presence of organ failure or pancreatic necrosis on dynamic contrast–enhanced CT scan (Figs. 58-1 and 58-2). Other acceptable markers of severe pancreatitis include three or more of Ranson’s 11 criteria for non-gallstone pancreatitis (Table 58-2),14 and an Acute Physiology and Chronic Health Evaluation (APACHE-II) score of greater than eight.15
Table 58-1 Atlanta Criteria for Severe Acute Pancreatitis13
| NON-GALLSTONE PANCREATITIS (1974) | GALLSTONE PANCREATITIS (1982) |
|---|---|
| At Admission | |
| Age >55 yr | Age >70 yr |
| White blood cells >16,000/mm3 | >18,000/mm3 |
| Blood glucose >200 mg/dL | >220 mg/dL |
| Serum lactate dehydrogenase >350 IU/L | >400 IU/L |
| Serum aspartate aminotransferase >250 IU/L | >250 IU/L |
| During Initial 48 hr | |
| Hematocrit decrease of >10 % | >10% |
| Blood urea nitrogen increase of >5 mg/dL | >2 mg/dL |
| Serum calcium <8 mg/dL | <8 mg/dL |
| Arterial po2 <60 mm Hg | NA |
| Serum base deficit >4 mEq/L | >5 mEq/L |
| Fluid sequestration >6 L | >4 L |
NA, not applicable.
It is important to use precise terms in describing the anatomic complications of acute pancreatitis. The ability to apply appropriate therapy depends on a clear understanding of these terms. An old term that should be used only sparingly is phlegmon. Although this term is often used by radiologists to describe an inflammatory mass, this term has carried different meaning to gastroenterologists, internists, and surgeons. Whereas patients with interstitial pancreatitis have a normally perfused gland, manifesting contrast-enhanced CT as a normal bright appearance indicating flow throughout the gland, patients with necrotizing pancreatitis have greater than 30% of the gland not perfused, with low attenuation. Pancreatic necrosis consists of focal or diffuse nonviable pancreatic parenchyma and usually peripancreatic fat necrosis. Pancreatic necrosis can be sterile or infected. Peripancreatic necrosis describes necrotic fatty and stromal tissue around the pancreas. It is more important to surgeons because this is typically not appreciated on imaging. However, the presence of peripancreatic necrosis may delineate a more complicated course for patients with acute pancreatitis. An acute fluid collection is fluid located in or near the pancreas that lacks a definite wall and typically occurs early in the course of acute pancreatitis. On CT scan these collections appear as a low attenuation mass with poor margins and no capsule. It is very difficult to distinguish acute fluid collections in the pancreatic parenchyma from pancreatic necrosis. An acute fluid collection occurs in 30% to 50% of cases of acute pancreatitis and most resolve spontaneously.16 A pseudocyst is a fluid collection that persists for 4 to 6 weeks and becomes encapsulated by a wall of fibrous or granulation tissue. Pseudocysts are located adjacent to or off the body of the pancreas. At times these enzyme-rich fluid-filled sacks can be found distantly in the pelvis and chest. When a pseudocyst is located within the body of the pancreas, the cyst may contain necrotic pancreatic debris even when the pseudocyst is fluid-appearing with low attenuation on CT. The term for a walled-off fluid-appearing pseudocyst-like structure involving the pancreas is walled off pancreatic necrosis (WOPN). A pancreatic abscess is a circumscribed intra-abdominal collection of pus occurring after an episode of acute pancreatitis or pancreatic trauma. It usually develops close to the pancreas and contains little pancreatic necrosis. Due to confusion of whether an abscess represents an infected pseudocyst or infected pancreatic necrosis, the term abscess should be used sparingly. Because of important differences in management, it is best to use the terms infected pseudocyst and infected necrosis. The term hemorrhagic pancreatitis should also be used with caution, and this term is not a synonym for necrotizing pancreatitis. Hemorrhage is more commonly associated with pseudoaneurysm, an erosion of peripancreatic blood vessels with hemoperitoneum. Unfortunately, hemorrhagic pancreatitis has more commonly been used to inappropriately describe necrotizing pancreatitis.
Of all these terms, the most important distinction is that between pancreatic necrosis and pseudocyst. WOPN is pancreatic necrosis that has liquefied after five to six weeks.17 Similar to a pseudocyst, a wall develops. However, whereas a pseudocyst always contains fluid, pancreatic necrosis, even if walled off early, contains a significant amount of debris that only becomes liquefied after five to six weeks. No attempt should be made to drain WOPN early (less than four weeks) because the debris is typically thick, often with the consistency of rubber early in the course of the disease. After five to six weeks, WOPN can be treated similar to the fluid-filled pseudocyst and drained surgically, endoscopically, or percutaneously.
NATURAL HISTORY
Acute pancreatitis appears to have two distinct stages. The first stage is related to the pathophysiology of the inflammatory cascade. This first phase usually lasts a week. During this phase, the severity of acute pancreatitis is related to extrapancreatic organ failure secondary to the patient’s systemic inflammatory response elicited by acinar cell injury. Infectious complications are uncommon at this time. Fever, tachycardia, hypotension, respiratory distress, and leukocytosis are typically related to the systemic inflammatory response syndrome (SIRS). Multiple cytokines are involved, including platelet activating factor, tumor necrosis factor-α (TNF-α) and various interleukins (ILs) (see Chapter 2).
During the first week the initial state of inflammation evolves dynamically with variable degrees of pancreatic and peripancreatic ischemia or edema to either resolution or to irreversible necrosis and liquefaction, or the development of fluid collections in and around the pancreas. The extent of the pancreatic and peripancreatic changes is usually proportional to the severity of extrapancreatic organ failure. However, organ failure may develop independent of pancreatic necrosis.17
Approximately 75% to 80%, of patients with acute pancreatitis have a resolution of the disease process (interstitial pancreatitis) and do not enter the second phase. However, in 25% of patients, a more protracted course develops, often related to the necrotizing process (necrotizing pancreatitis) lasting weeks to months. The mortality peak in the second phase is related to a combination of factors, including organ failure secondary to sterile necrosis, infected necrosis, or complications from surgical intervention.11,12,18–20 There are two peaks for mortality. Most studies in the United States and Europe reveal that about half the deaths occur within the first week or two, usually of multiorgan failure.19–21 Death can be very rapid. About one quarter of all deaths in Scotland occurred within 24 hours of admission and one third within 48 hours.21 After the second week of illness, patients succumb to pancreatic infection associated with multiorgan failure. Some studies in Europe report a very high late mortality rate from infection.22 Patients who are older and have comorbid illnesses have a substantially higher mortality rate than younger healthier patients. In those who survive their illness, severe pancreatic necrosis can scar the pancreas, resulting in a stricture of the main pancreatic duct with subsequent obstructive chronic pancreatitis and permanent diabetes and malabsorption.23
PATHOGENESIS
The initial step in the pathogenesis of acute pancreatitis is conversion of trypsinogen to trypsin within acinar cells in sufficient quantities to overwhelm normal mechanisms to remove active trypsin (see Fig. 57-3). Trypsin, in turn, catalyzes conversion of proenzymes, including trypsinogen and inactive precursors of elastase, phospholipase A2 (PLA2), and carboxypeptidase, to active enzymes. Trypsin also may activate the complement and kinin systems. Active enzymes autodigest the pancreas and initiate a cycle of releasing more active enzymes. Normally small amounts of trypsinogen are spontaneously activated within the pancreas, but intrapancreatic mechanisms quickly remove activated trypsin. Pancreatic secretory trypsin inhibitor (PSTI, now called SPINK1) binds and inactivates about 20% of the trypsin activity. Other mechanisms for removing trypsin involve mesotrypsin, enzyme Y, and trypsin itself, which splits and inactivates trypsin. The pancreas also contains nonspecific antiproteases such as α1-antitrypsin and α2-macroglobulin. Additional protective mechanisms are the sequestration of pancreatic enzymes within intracellular compartments of the acinar cell during synthesis and transport and the separation of digestive enzymes from lysosomal hydrolases as they pass through the Golgi apparatus, which is important because cathepsin B activates trypsin from trypsinogen. Low intra-acinar calcium concentrations also prevent further autoactivation of trypsin.
In experimental pancreatitis, activation of trypsin occurs within 10 minutes, and large amounts of trypsin24 and increased concentrations of trypsinogen activation peptide (TAP) accumulate within the pancreas.25,26 TAP is cleaved when trypsinogen is activated to trypsin, and concentrations of TAP in plasma, urine, and ascites correlate with the severity of the pancreatic inflammatory response, with the highest levels associated with acinar necrosis and intrapancreatic hemorrhage.27,28
Co-localization of pancreatic enzymes in lysosomes, followed by acinar cell injury, is an attractive hypothesis for the pathogenesis of acute pancreatitis, but the relevance of co-localization to the pathogenesis of acute pancreatitis is unclear. Activation of trypsinogen occurs before biochemical or morphologic injury to acinar cells, in association with co-localization of lysosomal enzymes, such as cathepsin B, and digestive enzymes, including trypsinogen within unstable vacuoles.27,28 Complete inhibition of pancreatic cathepsin B activity in vitro prevents trypsinogen activation induced by the cholecystokinin (CCK) analog cerulein,29 supporting the co-localization hypothesis. Thus, complete inhibition of cathepsin B may prevent or be a treatment for acute pancreatitis. However, enzyme co-localization may occur without inducing significant acinar cell injury.30
Two other features of experimental acute pancreatitis are early blockade of the secretion of pancreatic enzymes while enzyme synthesis continues and disruption of the paracellular barrier of acinar cells and intralobular pancreatic duct cells. The disruption facilitates the extravasation of pancreatic enzymes from acinar cells and from the duct lumen into interstitial spaces. This phenomenon may explain the rapid development of interstitial edema and the increase of pancreatic enzymes in the serum.31
As discussed in Chapter 57, the discovery of genetic mutations associated with hereditary pancreatitis also lends support to the hypothesis that intrapancreatic activation of pancreatic zymogens is central to the pathogenesis of acute pancreatitis.32–35 The mutant trypsin in hereditary pancreatitis (usually R122H or N29I mutation) causes trypsin to be resistant to lysis or causes premature trypsinogen activation (gain of function mutation) leading to autodigestion of the pancreas and episodes of acute pancreatitis.36,37
Mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene have also been implicated in pancreatitis (see Chapter 57). CFTR anion channel allows for chloride and bicarbonate secretion into the pancreatic ducts and thus allows flushing of the liberated enzymes and proenzymes into the duodenum. There are more than 1200 mutations that have been described for the CFTR gene. Some of these are considered severe and some mild. Homozygote severe mutations produce a viscid, concentrated, acidic pancreatic juice leading to ductal obstruction and pancreatic insufficiency in infanthood. Heterozygotes of minor or major mutations may lead to acute recurrent or chronic pancreatitis by altering acinar or ductal cell function (e.g., alteration of bicarbonate conductance).
A third genetic abnormality associated with pancreatitis is mutation of the SPINK1 gene.38 As noted, SPINK1 protects the pancreatic acinar cell by inhibiting prematurely activated trypsin. Mutations of this gene presumably limit the activity of this protein, but the exact mechanism is unclear.
The pathogenesis of gallstone-related pancreatitis is unknown (see Chapter 65). Factors that may initiate gallstone pancreatitis include reflux of bile into the pancreatic duct39,40 or obstruction of the pancreatic duct at the ampulla from stone(s) or edema resulting from the passage of a stone.41 Reflux of bile into the pancreatic duct could occur when the distal bile and pancreatic ducts form a common channel and a gallstone becomes impacted in the duodenal papilla. Alternatively, bile could reflux into the pancreatic duct from the duodenum through an incompetent sphincter of Oddi injured by recent passage of a gallstone.
Experimentally, reflux of bile into the pancreatic duct, particularly if infected or mixed with pancreatic enzymes, causes pancreatic injury. Mixtures of bile and pancreatic enzymes increase the permeability of the main pancreatic duct, which is associated with local parenchymal inflammation.42 The common channel theory is somewhat problematic because pancreatic duct pressure is invariably higher than bile duct pressure, making bile reflux into the pancreatic duct unlikely. Reflux of bile from the duodenum also is unlikely because pancreatitis does not occur in conditions with easily demonstrable reflux, such as after surgical sphincteroplasty or endoscopic sphincterotomy.
A popular opinion for the mechanism of gallstone pancreatitis is that an impacted gallstone in the distal bile duct obstructs the pancreatic duct, which increases pancreatic pressure, thereby damaging ductal and acinar cells. Experiments in the opossum that support this theory are the observations that ligation of the pancreatic duct causes severe necrotizing pancreatitis41 and that decompression of the ductal system within three days prevents progression to acinar cell necrosis and severe inflammation.43
PATHOPHYSIOLOGY
The release of pancreatic enzymes damages the vascular endothelium, the interstitium, and acinar cells.43–45 Acinar injury leads to expression of endothelial adhesion molecules (e.g., VCAM-1), which further propagates the inflammatory response.46 Microcirculatory changes, including vasoconstriction, capillary stasis, decreased oxygen saturation, and progressive ischemia, occur early in experimental acute pancreatitis. These abnormalities increase vascular permeability and lead to edema of the gland (edematous or interstitial pancreatitis). Vascular injury could lead to local microcirculatory failure and amplification of the pancreatic injury. It is uncertain whether ischemia-reperfusion injury occurs in the pancreas.40 Reperfusion of damaged pancreatic tissue could lead to the release of free radicals and inflammatory cytokines into the circulation, which could cause further injury. In early stages of animal and human pancreatitis, activation of complement and the subsequent release of C5a play significant roles in the recruitment of macrophages and polymorphonuclear leukocytes.47–49 Active granulocytes and macrophages release proinflammatory cytokines in response to transcription factors such as nuclear factor κB (NF-κB). Proinflammatory cytokines include TNF, IL-1, IL-6, and IL-8, and platelet-activating factor (PAF). Proinflammatory cytokines frequently are followed by anti-inflammatory cytokines (IL-2, IL-10, IL-11) that attempt to down-regulate inflammation.48 Other mediators of inflammation include arachidonic acid metabolites (prostaglandins, PAF, and leukotrienes), nitric oxide, proteolytic and lipolytic enzymes, and reactive oxygen metabolites that overwhelm scavenging by endogenous antioxidant systems. These substances also interact with the pancreatic microcirculation to increase vascular permeability, which induces thrombosis and hemorrhage and leads to pancreatic necrosis. A recent study suggests that gene polymorphisms that affect acinar cell glutathione concentrations may lead to increased oxidant stress and more severe pancreatitis.50 Meanwhile, ischemia and severe inflammation of the gland can lead to disruption of the main and secondary pancreatic ducts, leading to local fluid accumulations within and surrounding the pancreas that can eventuate into pseudocysts.51,52
Some patients with severe pancreatic damage develop systemic complications, including fever, acute respiratory distress syndrome (ARDS), pleural effusions, renal failure, shock, myocardial depression, and metabolic complications. SIRS is common in patients with acute pancreatitis and is probably mediated by activated pancreatic enzymes (phospholipase, elastase, trypsin) and cytokines (TNF, PAF) released into the portal circulation from the inflamed pancreas.53 Cytokines reaching the liver activate hepatic Kupffer cells, which, in turn, induces hepatic expression and secretion of cytokines into the systemic circulation. These cause acute phase protein synthesis (C-reactive protein [CRP], IL-6) and may cause SIRS and damage to the kidneys, lungs, and other organs leading to multiorgan dysfunction and failure.54
ARDS may be induced by active phospholipase A (lecithinase), which digests lecithin, a major component of lung surfactant. Acute renal failure has been explained on the basis of hypovolemia and hypotension. Myocardial depression and shock are likely secondary to vasoactive peptides and a myocardial depressant factor. Metabolic complications include hypocalcemia, hyperlipidemia, hyperglycemia with or without ketoacidosis, and hypoglycemia. The pathogenesis of hypocalcemia is multifactorial and includes hypoalbuminemia (the most important cause), hypomagnesemia, calcium-soap formation, hormonal imbalances (e.g., involving parathyroid hormone, calcitonin, and glucagon), binding of calcium by free fatty acid–albumin complexes, intracellular translocation of calcium, and systemic exposure to endotoxin.55
Pancreatic infection (infected necrosis and infected pseudocyst) can occur from the hematogenous route or from translocation of bacteria from the colon into the lymphatics. Under normal circumstances bacterial translocation does not occur because there are complex immunologic and morphologic barriers. However, during acute pancreatitis, these barriers break down, which can result in local and systemic infection.56 Penetration of the gut barrier by enteric bacteria is likely due to gut ischemia secondary to hypovolemia and pancreatitis-induced arteriovenous shunting in the gut.57 Indeed, in canine experimental pancreatitis, luminal Escherichia coli translocate to mesenteric lymph nodes and distant sites.58 In feline experimental pancreatitis, enclosing the colon in impermeable bags prevents translocation of bacteria from the colon to the pancreas.59
PREDISPOSING CONDITIONS
Many conditions predispose to acute pancreatitis to varying degrees (Table 58-3). This list will undoubtedly continue to grow, and the number of cases diagnosed as “idiopathic” will decrease as our understanding of the disease improves. Gallstones and chronic alcohol abuse account for 70% of acute pancreatitis in the United States.
Table 58-3 Conditions That Predispose to Acute Pancreatitis
OBSTRUCTION
Gallstones
The most common obstructive process leading to pancreatitis is gallstones, which cause approximately 40% of cases acute pancreatitis,60 although only 3% to 7% of patients with gallstones develop pancreatitis. Gallstone pancreatitis is more common in women than men because gallstones are more frequent in women.61 Acute pancreatitis occurs more frequently when stones are less than 5 mm in diameter (odds ratio, 4 to 5),62 because small stones are more likely than large stones to pass through the cystic duct and cause ampullary obstruction. Cholecystectomy and clearing the bile duct of stones prevents recurrence, confirming the cause-and-effect relationship.61
Biliary Sludge and Microlithiasis
Biliary sludge is a viscous suspension in gallbladder bile that may contain small (<3 mm) stones (i.e., microlithiasis).63 Because small stones can hide in biliary sludge, the two are commonly referred together as biliary sludge and microlithiasis. Biliary sludge is asymptomatic in most patients. It is usually composed of cholesterol monohydrate crystals or calcium bilirubinate granules.64 On ultrasonography, sludge produces a mobile, low-amplitude echo that does not produce an acoustic shadow and that layers in the most dependent part of the gallbladder.
Sludge may result from functional bile stasis, such as that associated with prolonged fasting or total parenteral nutrition, or from mechanical stasis such as occurs in distal bile duct obstruction. In addition, the cephalosporin antibiotic ceftriaxone can complex with bile to form a sludge within the biliary system when its solubility in bile is exceeded; this rarely causes stones65 and the sludge disappears after stopping the drug. Commonly, biliary sludge is associated with idiopathic acute pancreatitis. However, the association between biliary sludge and acute pancreatitis is unproved. There is no prospective, randomized study documenting that removing sludge or microcrystals by cholecystectomy prevents further attacks of pancreatitis. Nevertheless, results of two uncontrolled studies suggest that biliary sludge can lead to pancreatitis, and that cholecystectomy, papillotomy, or ursodeoxycholic acid therapy reduces recurrent attacks of acute pancreatitis.64,66 In these two studies, the incidence of biliary sludge in presumed idiopathic pancreatitis was 67% and 74%, respectively. However, other investigators have detected microlithiasis or sludge in less than 10% of patients with recurrent acute pancreatitis.67,68 Until prospective controlled studies clarify the proper treatment of sludge and microlithiasis, firm recommendations about therapy cannot be made. Choices include cholecystectomy, ursodeoxycholic acid therapy, endoscopic sphincterotomy, or watchful waiting.
Tumors
Tumors, presumably by obstructing the pancreatic duct, can cause recurrent acute pancreatitis especially in individuals older than age 40 (see Chapter 60). The most common tumor that presents in this manner is intraductal papillary mucinous neoplasm (IPMN).69 Pancreatic adenocarcinoma can also present as acute pancreatitis in a small percentage of patients.70 Metastases from other primary tumors (lung, breast) to the pancreas have also caused pancreatitis.71 Large adenomas of the major papilla can likewise occasionally be the cause of obstructive pancreatitis.
Other Obstructive Causes
Other obstructive conditions that are rarely associated with acute pancreatitis are conditions discussed elsewhere in this text and include choledochoceles,72 duodenal diverticula,73 annular pancreas,74 and parasites that obstruct the pancreatico-biliary system such as ascaris75 or clonorchis.76 Ascariasis obstructing the pancreatic duct represents the second most common cause of acute pancreatitis in Kashmir.60
ALCOHOL, OTHER TOXINS, AND DRUGS
Ethyl Alcohol
Alcohol causes at least 30% of cases of acute pancreatitis,77 and alcohol is the most common etiology of chronic pancreatitis in developed countries. Interestingly, only 10% of chronic alcoholic patients develop chronic pancreatitis. The classic teaching is that alcohol causes chronic pancreatitis, and that alcoholic patients who present with clinically acute pancreatitis have underlying chronic disease.16 However, a few patients with alcohol-induced acute pancreatitis by clinical criteria do not have or progress to chronic pancreatitis, even with continued alcohol abuse.77,78 By contrast, a small percentage of chronic alcoholic patients develop attacks of acute pancreatitis that are indistinguishable from other forms of acute pancreatitis, but eventually develop chronic pancreatitis after 10 to 20 years of alcohol abuse. Early in the course of the disease, when attacks occur, the diagnosis of underlying chronic pancreatitis is difficult without tissue specimens because the diagnosis of chronic pancreatitis is usually made after definite signs of chronic pancreatitis appear (e.g., pancreatic calcification, exocrine and endocrine insufficiency, or typical duct changes by CT or ERCP). Most of the models described suggest possible mechanisms of alcohol-related injury, including perturbations in exocrine function, changes in cellular lipid metabolism, induction of oxidative stress, and activation of stellate cells. However, the exact mechanism remains unclear and may be related to other factors.
Bordalo and colleagues79 first proposed that alcohol was directly toxic to the acinar cell through a change in cellular metabolism. Alcohol produces cytoplasmic lipid accumulation within the acinar cells, leading to fatty degeneration, cellular necrosis, and eventual widespread fibrosis. Fatty acid ethyl esters, by-products of pancreatic ethanol metabolism, may be the key factor in this “toxic metabolic” change. Bordalo and colleagues suggested that alcohol produces a stepwise progression from fatty accumulation to fibrosis (direct toxic effects on cellular metabolism). The main limitation to this toxic-metabolic theory of alcohol toxicity is the lack of proof of the steatopancreatitis precursor to fibrosis seen in liver disease.80
Sarles80 emphasized the duality of acute and chronic pancreatitis; they were separate diseases with distinct pathogenesis. Whereas acute pancreatitis can be precipitated in patients with gallstones immediately, alcoholism requires years of toxin exposure. Alcohol modulates exocrine function to increase the lithogenicity of pancreatic fluid, leading to the formation of protein plugs and stones. Chronic contact of the stones with the ductal cells produces ulceration and scarring, resulting in obstruction, stasis, and further stone formation. Eventually, atrophy and fibrosis develop as a result of this obstructive process. Several studies have provided mechanisms in which alcohol promote stone formation, including the known precipitation of GP-2 (a Tamm-Horsfall–like protein),81 increased secretion and viscosity of pancreatic juice, and hypersecretion of enzymes and lactoferrin. In addition to these ethanol-mediated perturbations in pancreatic exocrine function, specific proteins have been implicated in stone formation. Pancreatic stones consist of a calcium carbonate crystalline lattice interspersed within a gel-like matrix formed of multiple fibrillar proteins and polysaccharides.82
In contrast to the stone theory, which is based on the de novo development of fibrosis without acute pancreatitis, the necrosis-fibrosis hypothesis envisions the development of fibrosis from recurrent, perhaps subclinical, acute pancreatitis. Inflammation and necrosis from the initial episodes of acute pancreatitis produce scarring in the periductular areas and scarring leads to obstruction of the ductules leading to stasis within the duct and subsequent stone formation. Support for this theory comes from histopathologic studies that revealed mild perilobular fibrosis in resolving acute pancreatitis, with marked fibrosis with ductal distortion occurring later. It is thought that a stepwise progression occurs to fibrosis from recurrent episodes of acute pancreatitis. Support for this theory is seen in a clinical study by Ammann and colleagues.83 In this study, 254 patients were prospectively followed after the first episode of alcoholic pancreatitis. There was a direct correlation between the frequency and severity of attacks to the rate of progression to chronic pancreatitis.
Whitcomb and Schneider have proposed an interesting hypothesis for chronic pancreatitis which unifies and incorporates recent knowledge in an attempt to reconcile prior theories.84 In at-risk individuals, the pancreatic acinar cells are stimulated by alcohol. Fibrosis does not occur because a profibrotic cellular infiltrate is not yet present. A sentinel event occurs as trypsinogen is activated. This event results in a massive inflammatory response. Cytokines are then released and work with the activation of stellate cells in the late phase. The attraction and activation of stellate cells set the stage for the development of fibrosis. If the inciting factors are removed, then the pancreas returns to normal. If the inciting factor, alcohol, is not removed, the acinar cells continue to secrete cytokines in response to the oxidative stress and the stellate cells continue to be activated. This model, using a sentinel event also represents a time for disease modifying therapy, when such therapy becomes available.
Other Toxins
Methyl alcohol,85 organophosphorous insecticides,86 and the venom of the Trinidad scorpion87 have been reported to induce pancreatitis. The mechanism of the latter two is thought to be by hyperstimulation of the pancreas. Smoking increases the risk of alcoholic and idiopathic pancreatitis, but not gallstone pancreatitis.88
Drugs
Medications are an infrequent but an important cause of acute pancreatitis.89 More than 120 drugs have been implicated, mostly from anecdotal case reports. Many case reports suffer from a combination of inadequate criteria for the diagnosis of acute pancreatitis, failure to rule out more common causes, or a lack of a rechallenge with the medication. Drug-induced pancreatitis rarely is accompanied by clinical or laboratory evidence of a drug reaction, such as rash, lymphadenopathy, or eosinophilia. Although a positive rechallenge with a drug is the best evidence available for cause and effect, it is not proof. It is clear that many patients with idiopathic pancreatitis or microlithiasis have recurrent attacks of acute pancreatitis. Therefore, stopping and restarting a drug with recurrence of pancreatitis may be a coincidence and not cause and effect. Despite the lack of a rechallenge, a drug may be strongly suspected if there is a consistent latency among the case reports between initiating the drug and the onset of acute pancreatitis. Table 58-4 shows the drugs with the greatest evidence for causing acute pancreatitis, those with rechallenges or with a relatively predictable latency.89
Table 58-4 Drugs Associated with Acute Pancreatitis*
* Class 1 and class 2 drugs. For class 1 drugs: two or more case reports published, absence of other causes of acute pancreatitis, rechallenge documented in at least one report. For class 2 drugs: four or more case reports published, absence of other causes of acute pancreatitis, consistent latency in at least 75% of cases published.
From Badalov N, Baradarian R, Iswara K, et al. Drug induced acute pancreatitis: An evidence based approach. Clin Gastroenterol Hepatol 2007; 101:454-76.
METABOLIC DISORDERS
Hypertriglyceridemia
Hypertriglyceridemia is perhaps the third most common identifiable cause of pancreatitis after gallstones and alcoholism. Serum triglyceride concentrations greater than 1000 mg/dL (11 mmol/L) may precipitate attacks of acute pancreatitis. Patients may have lactescent (milky) serum owing to increased concentrations of chylomicrons.90 The pathogenesis of hypertriglyceridemic pancreatitis is unclear, but the release of free fatty acids by lipase may damage pancreatic acinar cells or capillary endothelium.91
Hypertriglyceridemia may cause up to 5% of cases of acute pancreatitis. The association between hypertriglyceridemia and acute pancreatitis is best defined in children with rare inherited disorders of lipoprotein metabolism and severe hypertriglyceridemia92,93 who develop acute pancreatitis in early childhood. These children are homozygous for lipoprotein lipase deficiency or, even less commonly, apoprotein-CII (APO-CII) deficiency. Acute pancreatitis develops in 35%, 15%, and 30% to 40% of patients with type I, IIb, and V hyperlipidemia, respectively. Lowering serum triglyceride levels to less than 200 mg/dL (2.2 mmol/L) can prevent pancreatitis.
Most adults with hyperchylomicronemia have a mild form of genetically inherited type I or type V hyperlipoproteinemia and an additional acquired condition known to raise serum lipids (e.g., alcohol abuse, obesity, diabetes mellitus, hypothyroidism, pregnancy, estrogen91 or tamoxifen therapy, glucocorticoid excess, nephrotic syndrome, thiazide or beta blocker therapy). Typically three types of patients develop hypertriglyceridemia-induced pancreatitis. The first is a poorly controlled diabetic patient with a history of hypertriglyceridemia. The second is an alcoholic patient with hypertriglyceridemia detected on hospital admission. The third (15% to 20%) is a nondiabetic, nonalcoholic, nonobese person who has drug- or diet-induced hypertriglyceridemia. Drug-induced disease is more likely to occur if there is a background of hypertriglyceridemia prior to drug exposure.
Most persons who abuse alcohol have moderate, but transient, elevations of serum triglyceride levels. This condition is likely an epiphenomenon and not the cause of their pancreatitis94 because alcohol itself not only damages the pancreas but also increases serum triglyceride concentrations in a dose-dependent manner. For example, serum triglyceride concentrations greater than 227 mg/dL (2.5 mmol/L) occurred in 10%, 14%, and 20% of people who drank three to five, six to eight, or nine or more drinks per day, respectively.95 Alcoholic patients with severe hyperlipidemia often have a coexisting primary genetic disorder of lipoprotein metabolism.
Hypercalcemia
Hypercalcemia, of any cause, is rarely associated with acute pancreatitis. Proposed mechanisms include deposition of calcium in the pancreatic duct and calcium activation of trypsinogen within the pancreatic parenchyma.96 The low incidence of pancreatitis in chronic hypercalcemia suggests that factors other than the serum calcium per se (e.g., acute elevations of serum calcium) are responsible for pancreatitis. Acute calcium infusion into rats leads to conversion of trypsinogen to trypsin, hyperamylasemia, and dose-dependent morphologic changes of acute pancreatitis such as edema and acinar cell necrosis.
Hypercalcemia due to hyperparathyroidism has been associated with pancreatitis. However, primary hyperparathyroidism causes less than 0.5% of all cases of acute pancreatitis, and the incidence of acute pancreatitis in patients with hyperparathyroidism varies from 0.4% to 1.5%.97 Rarely, pancreatitis occurs with other causes of hypercalcemia, including metastatic bone disease, total parenteral nutrition, sarcoidosis, vitamin D toxicity, and infusions of calcium in high doses perioperatively during cardiopulmonary bypass.
INFECTIONS
Many infectious agents may cause acute pancreatitis,76 but often published reports do not meet usual standards for the diagnosis of pancreatitis or the infection. Using modern criteria for diagnosis of pancreatitis, definite pancreatitis exists if there is surgical, autopsy, or radiologic evidence; probable pancreatitis exists if there is biochemical evidence (more than three times the elevation of serum lipase or amylase) plus characteristic symptoms; and possible pancreatitis exists if there is only asymptomatic biochemical evidence. The definite criterion for an infection causing pancreatitis is finding the organism in the pancreas or pancreatic duct by stain or culture. Probable criteria for infection are culture of the organism from pancreatic juice or blood or serologic evidence combined with a characteristic clinical or epidemiologic setting. The criterion for a possible infection is culture of the organism from other body sites or serologic evidence of infection.
Using these criteria, definite pancreatitis has been associated with viruses (mumps, coxsackievirus, hepatitis B, cytomegalovirus, varicella-zoster, herpes simplex, Epstein-Barr, hepatitis A, and hepatitis C); the vaccine that contains attenuated measles, mumps, and rubella (MMR); bacteria (Mycoplasma, Legionella, Leptospira, Salmonella, tuberculosis, and brucellosis); fungi (Aspergillus and Candida albicans); and parasites (Toxoplasma, Cryptosporidium, Ascaris, Clonorchis sinensis). C. sinensis and Ascaris cause pancreatitis by blocking the main pancreatic duct. In acquired immunodeficiency syndrome (AIDS), infectious agents causing acute pancreatitis include cytomegalovirus, Candida, Cryptococcus neoformans, Toxoplasma gondii, and possibly Mycobacterium avium complex.76
An infectious agent should be suspected of causing acute pancreatitis if the characteristic syndrome caused by the infectious agent is present because this occurs 70% of the time.76 Because an infectious agent may be found in the pancreas without pancreatitis, routine search for an infection in idiopathic pancreatitis is not recommended because false-positive test results may result. In addition, it is unknown whether treating an infectious agent reverses pancreatic pathology.
VASCULAR DISEASE
Rarely, pancreatic ischemia causes pancreatitis. In most cases it is mild, but fatal necrotizing pancreatitis may occur. Ischemia may result from vasculitis (systemic lupus erythematosus)98 and polyarteritis nodosa,99 atheromatous embolization of cholesterol plaques from the aorta to the pancreas after transabdominal angiography,100 intraoperative hypotension,101 hemorrhagic shock,102 ergotamine overdose, and transcatheter arterial embolization for hepatocellular carcinoma. Also, ischemia is one possible explanation for pancreatitis after cardiopulmonary bypass. In pigs, cardiogenic shock induced by pericardial tamponade causes vasospasm and selective pancreatic ischemia due to activation of the renin-angiotensin system.103 Acute pancreatitis has occurred in long-distance runners, which may be on an ischemic basis.104
TRAUMA
Either penetrating trauma (gunshot or stab wounds) or blunt trauma can damage the pancreas.105 In most cases there is also injury to adjacent viscera. Laparotomy is essential in all cases of penetrating trauma to assess and treat all intra-abdominal injuries, including those to the pancreas. Blunt trauma results from compression of the pancreas by the spine, such as in an automobile accident. In blunt trauma it is important to determine preoperatively whether there is injury to the pancreas because depending on the severity of pancreatic injury, it will be necessary to include the pancreas in the surgical plan. Secondly, even in the absence of serious injury to adjacent organs, surgery or endoscopic therapy may be necessary to treat a pancreatic ductal injury.
Diagnosis is highly dependent on CT, MRI, or magnetic resonance cholangiopancreatography (MRCP), which may show enlargement of a portion of the gland caused by a contusion or subcapsular hematoma, pancreatic inflammatory changes, or fluid within the anterior pararenal space if there is ductal disruption. The CT may be normal during the first two days despite significant pancreatic trauma. If there is a strong clinical suspicion of pancreatic injury or if the CT or MRCP scan shows an abnormality, ERCP is required to define whether there is pancreatic duct injury. If the pancreatic duct is intact and there are no other significant intra-abdominal injuries, surgery is not required. However, if ERCP reveals duct transection with extravasation of pancreatic fluid and there are no other intra-abdominal injuries, stenting of the pancreatic duct may be successful.106 Serious injuries to the pancreas can be treated with appropriate débridement. Associated injuries to the duodenum or bile duct can be treated by biliary diversion, gastrojejunostomy, and feeding jejunostomy. External pancreatic fistulas occur in approximately one third of patients after surgery for pancreatic trauma. Octreotide may be beneficial after pancreatic injury.107
POST-ERCP
Acute pancreatitis is the most common and feared complication of ERCP, associated with substantial morbidity and occasional mortality. About 500,000 ERCPs are performed annually in the United States. Asymptomatic hyperamylasemia occurs after 35% to 70% of ERCPs.108 Acute pancreatitis occurs in 5% of diagnostic ERCPs, 7% of therapeutic ERCPs, and up to 25% in those with suspected sphincter of Oddi dysfunction or in those with a history of post-ERCP pancreatitis.109 About half the cases are moderate to severe in intensity.
The mechanisms that lead to post-ERCP pancreatitis are complex and not fully understood. Rather than a single pathogenesis, post-ERCP pancreatitis is believed to be multifactorial, involving a combination of chemical, hydrostatic, enzymatic, mechanical, and thermal factors. Although there is some uncertainty in predicting which patients will develop acute pancreatitis following ERCP, a number of risk factors acting independently or in concert have been proposed as predictors of post-ERCP pancreatitis (Table 58-5).110–113 Identification of these risk factors for post-ERCP pancreatitis is essential to recognize cases in which ERCP should be avoided if possible, or in which protective endoscopic or pharmacologic interventions should be considered.
Table 58-5 Factors That Increase the Risk of Post-ERCP Pancreatitis
| Patient Related |
| Young age, female gender, suspected sphincter of Oddi dysfunction, recurrent pancreatitis, history of post-ERCP pancreatitis, normal serum bilirubin |
| Procedure Related |
| Pancreatic duct injection, difficult cannulation, pancreatic sphincterotomy, precut access, balloon dilation |
| Operator or Technical Related |
| Trainee (fellow) participation, nonuse of a guidewire for cannulation, nonuse of a pancreatic duct stent in high-risk procedures |
ERCP, endoscopic retrograde cholangiopancreatography.
In general, the more likely a patient is to have an abnormal bile duct or pancreatic duct, the less likely the patient will develop post-ERCP pancreatitis. Cheng created a 160 variable database that prospectively evaluated more than 1000 patients from 15 centers in the midwestern United States.112 Their study emphasized the role of patient factors, including age, sphincter of Oddi dysfunction (SOD), prior history of post-ERCP pancreatitis, and technical factors, including number of pancreatic duct (PD) injections, minor papilla sphincterotomy, and operator experience. The patient most at risk of developing post-ERCP pancreatitis was a woman with suspected choledocholithiasis and normal bilirubin, who underwent a sphincterotomy and no stone was found. In this patient population, more than a quarter of patients (27%) developed post-ERCP pancreatitis. MRCP and endoscopic ultrasound, which do not cause pancreatitis, can provide useful information (perhaps as accurate as ERCP) in many of these cases and are preferred modalities in the initial evaluation of such patients.
Early recognition of post-ERCP pancreatitis may be possible by evaluating serum amylase or lipase after the procedure.114,115 In a study that involved 231 patients, the two-hour serum amylase and lipase were more accurate than a clinical assessment in distinguishing nonpancreatitis abdominal pain from post-ERCP acute pancreatitis. Serum values greater than 276 IU/L for amylase and greater than 1000 IU/L for lipase obtained from serum two hours after the procedure had almost a 100% positive predictive value for post-ERCP pancreatitis.116 More recently, Ito and colleagues found that if the serum amylase was normal at three hours, only 1% of patients had post-ERCP pancreatitis compared with 39% if the amylase was greater than five times the upper limit of normal.117 A serum amylase or lipase alone should not guide a decision of whether a patient has post-ERCP pancreatitis. However, it appears that the tests may assist the clinician’s assessment of a patient with post-ERCP abdominal pain.
Although there has been an interest in developing medications that can prevent post-ERCP pancreatitis, studies have failed to identify a medication worthy of widespread use. In terms of attenuating the inflammatory response, the most promising results have been seen with nonsteroidal anti-inflammatory drugs (NSAIDs). Two clinical trials have been published evaluating the role of diclofenac in reducing the incidence of post-ERCP pancreatitis.116,118 Both trials placed patients on 100 mg of diclofenac by rectal suppository and both showed a reduction in the incidence of acute pancreatitis. However, another trial failed to show any benefit to diclofenec in preventing post-ERCP pancreatitis.119
It has been suggested that relaxation of the sphincter of Oddi following ERCP will promote pancreatic drainage and prevent acute pancreatitis. Several agents have been used in the effort to relax the sphincter of Oddi with the purpose of preventing post-ERCP pancreatitis. There have been three placebo-controlled randomized studies evaluating the use of nitroglycerin during ERCP, with negative results.120–122 Furthermore, trials of oral nifedipine,123,124 sprayed lidocaine,125 and injected botulinum toxin126 failed to demonstrated any benefit in the reduction of severity or incidence of post-ERCP pancreatitis. There have been several studies evaluating the role of glucocorticoids using a variety of agents including oral prednisolone or intravenous hydrocortisone or methylprednisolone, with essentially no benefit in reduction of severity or incidence of post-ERCP pancreatitis.
Gabexate is a protease inhibitor with anti-inflammatory properties. The in vivo effect of gabexate on inhibiting circulating trypsin is greater than most other protease inhibitors. In 1995, Messori and colleagues127 published a meta-analysis of five trials128–132 showing a statistically significant reduction in the incidence of complications in patients receiving gabexate after the development of post-ERCP pancreatitis. However, the trials were small and had a limited number of patients. Several additional trials have been published with conflicting results.133–137 Although data are conflicting, it appears that infusions of the drug would likely need to be started 1 to 2 hours pre-ERCP and continued for 12 hours following ERCP to show a beneficial effect of the drug. In patients with a low risk, the costs likely outweigh any benefit. Currently, gabexate is not available in the United States.
Theoretically, inhibition of exocrine pancreatic secretion could prevent post-ERCP pancreatitis by inducing “rest” to a damaged gland. Although an attractive concept, there is little scientific basis to support this approach. Somatostatin and its synthetic octapeptide analog, octreotide, are potent inhibitors of pancreatic secretion. Although several trials of somatostatin have demonstrated an efficacy in reducing the incidence of post-ERCP pancreatitis,138–143 the majority of the studies do not support the routine use of this medication.144–150 Octreotide, the analog of somatostatin,151 was only effective in reducing post-ERCP hyperamylasemia, and did not reduce the incidence of post-ERCP pancreatitis.
Pancreatic stent placement clearly decreases the risk of post-ERCP pancreatitis in high-risk patients.152 Placement of pancreatic duct stents has become a standard practice for patients who are thought to be at high risk for pancreatitis after the procedure (see Table 58-5).153 Pancreatic duct stent placement is effective presumably by preventing cannulation-induced edema that can cause pancreatic duct obstruction. Pancreatic sphincter hypertension is believed to be an important causative factor in post-ERCP pancreatitis and may explain the high risk of pancreatitis in patients with SOD. There is prolonged alleviation of ductal obstruction when pancreatic stents are placed. Typically, 3- to 5-French unflanged pancreatic stents are used in the following settings: SOD, difficult cannulation, biliary orifice balloon dilation, and precut sphincterotomy. In general, pancreatic duct stents are not beneficial in patients who undergo routine biliary sphincterotomy. In all reported studies, which cumulatively include 1500 high-risk patients undergoing ERCP, only 1 patient developed severe pancreatitis after a pancreatic duct stent had been placed.154 Aside from the obvious benefits in preventing post-ERCP pancreatitis regarding morbidity and mortality, prophylactic stent placement is a cost-effective strategy for the prevention of post-ERCP pancreatitis for high-risk patients.155
Guidewire cannulation, whereby the biliary or pancreatic duct is initially cannulated by a guidewire inserted through the catheter or sphincterotome, has been shown to decrease the risk of pancreatitis (see Table 58-5).156 In a study of 400 consecutive patients who underwent ERCP by a single endoscopist, randomized to initial cannulation with contrast versus initial cannulation by a guidewire under fluoroscopic control, pancreatitis rates were profoundly different. No cases of acute pancreatitis were seen in the guidewire group compared with eight cases in the standard contrast group (P < 0.001). Cannulation success rates between the standard contrast and guidewire techniques were comparable 98.5% versus 97.5%. A later study157 confirmed a decrease in post-ERCP pancreatitis in 300 patients prospectively randomized to guidewire cannulation compared with conventional radiocontrast. However, the decrease in post-ERCP pancreatitis appears to be related to a decreased need for precut sphincterotomy in patients undergoing guidewire cannulation.
POSTOPERATIVE
Postoperative pancreatitis can occur after abdominal or thoracic surgery.158 Pancreatitis occurs after 0.4% to 7.6% of cardiopulmonary bypass operations113,159 and after 6% of liver transplantations.160 Twenty-seven percent of patients undergoing cardiac surgery develop hyperamylasemia, and 1% develop necrotizing pancreatitis.113 Significant risks for pancreatitis after cardiopulmonary bypass are preoperative renal insufficiency, postoperative hypotension, and administration of calcium chloride perioperatively. Mortality from postoperative pancreatitis is said to be higher (up to 35%) than for other forms of pancreatitis. Contributors to morbidity and mortality from postoperative pancreatitis are delay in diagnosis, hypotension, medications (e.g., azathioprine/perioperative calcium chloride administration), and infections.
HEREDITARY AND GENETIC
Hereditary pancreatitis is an autosomal dominant disorder with variable penetrance.161–169 It is discussed in Chapter 57.
CONTROVERSIAL CAUSES
Pancreas Divisum
Pancreas divisum is the most common congenital malformation of the pancreas occurring in 5% to 10% of the general healthy population (see Chapter 55). Controversy continues to surround the issue as to whether pancreas divisum with otherwise normal ductular anatomy is a cause of acute recurrent pancreatitis. The presumed mechanism of action in those who develop pancreatitis is that there is relative obstruction to the flow of pancreatic juice through the minor papilla. The arguments in favor of attributing pancreatitis to pancreas divisum are the following: (1) various series from ERCP referral centers show that patients referred with recurrent acute pancreatitis have a higher frequency of pancreas divisum than would be expected from the general population170; (2) multiple observational series report that performing endoscopic sphincterotomy or placing a stent across the minor papilla reduces the rate of recurrent pancreatitis156; and (3) there is one randomized controlled study suggesting that patients with pancreas divisum who are stented for one year have a lower frequency of attacks of pancreatitis than those not stented.171 The arguments against the association are the following: (1) there are studies to show that the rate of pancreatitis in pancreas divisum patients is the same as the general population172; (2) the observational reports are flawed in that follow-up was not long enough (usually only 1 to 2 years) and that recurrent acute pancreatitis is a disease of great variability163; (3) the single randomized study163 was flawed in that it was not blinded, was small (19 patients total), and its patients probably had chronic pancreatitis in that they had multiple pain attacks in between attacks of acute pancreatitis; (4) the risk of endoscopic therapy is considerable with a high rate of post-ERCP pancreatitis in patients with pancreas divisum,111,112 therefore, making the risk-benefit ratio of treating pancreas divisum endoscopically questionable; and (5) the rate of genetic abnormalities in patients with pancreas divisum and acute recurrent pancreatitis are either the same173 or higher161 than expected in the general population or population of patients with acute pancreatitis of other etiologies, suggesting a possible genetic source. For example, there appears to be a higher incidence of CFTR mutations in patients with pancreas divisum who develop acute pancreatitis.161 Therefore, it may not be the presence of pancreas divisum alone that predisposes to acute pancreatitis but other factors may be necessary to precipitate an attack.163
