Chapter 23 Drugs to Treat Heart Failure
| Abbreviations | |
|---|---|
| ACE | Angiotensin-converting enzyme |
| ARB | Angiotensin receptor blocker |
| ATP | Adenosine triphosphate |
| AV | Atrioventricular |
| BNP | B-natriuretic peptide |
| cAMP | Cyclic adenosine monophosphate |
| CHF | Congestive heart failure |
| CNS | Central nervous system |
| Epi | Epinephrine |
| GI | Gastrointestinal |
| NE | Norepinephrine |
| NO | Nitric Oxide |
| PDE | Phosphodiesterase |
| RAAS | Renin-angiotensin-aldosterone system |
| SA | Sinoatrial |
| SNS | Sympathetic nervous system |
Therapeutic Overview
| Therapeutic Overview |
|---|
| Problem |
| Reduced force of contraction |
| Decreased cardiac output |
| Increased total peripheral resistance |
| Inadequate organ perfusion |
| Development of edema |
| Decreased exercise tolerance |
| Ischemic heart disease |
| Sudden death |
| Ventricular remodeling and decreased function |
| Goals |
| Alleviation of symptoms, improve quality of life |
| Arrest ventricular remodeling |
| Prevent sudden death |
| Nondrug Therapy |
| Reduce cardiac work; rest, weight loss, low sodium diet |
| Drug Therapy |
| Acute heart failure |
| Intravenous diuretics, inotropic agents, phosphodiesterase inhibitors, vasodilators |
| Chronic heart failure |
| ACE inhibitors, β adrenergic receptor blockers, angiotensin receptor blockers, aldosterone antagonists, digoxin, diuretics |
nearly 21%, whereas the death rate during that period declined by 2%.
Mechanisms of Action
Remodeling of the heart occurs through complex structural changes in one or more cardiac chambers, especially the ventricles. These result in an increase in end-diastolic and end-systolic volume along with changes in cardiac shape and left ventricular mass (Frank-Starling curves are shown in Figure 23-1). Impaired contractility was previously thought to be responsible for heart failure, although a specific biochemical abnormality could not be identified. This idea has given way to the concept that heart failure involves endogenous neurohormones and cytokines in response to an initial “index event,” usually an acute injury to the heart or genetic mutation. Coronary artery disease and hypertension account for most cases, with myocardial infarction being a major contributor. Any insult, whether acute myocardial infarction, essential hypertension, aortic stenosis, or volume overload caused by aortic insufficiency, idiopathic cardiomyopathy, or inflammatory disease, leads to activation of specific mediators involved in the remodeling process.
Neurohormonal systems defend against changes in intravascular volume and act to maintain regional blood flow and regulate systemic blood pressure. Initially, they compensate for the decline in ventricular function and mask the underlying deficiency. Chronic activation of multiple compensatory mechanisms perpetuates progression to irreversible myocyte injury and worsening of cardiac function. Initiating factors include: stretch of the ventricular myocardium, increased cytokine and growth factor production, nitric oxide (NO) production, tumor necrosis factor, natriuretic peptides, free radical-mediated oxidative stress, ischemia, activation of myocardial metalloproteinases, proapoptotic factors, and chronic inflammation. Heart failure is usually accompanied by an increase in sympathetic nervous system (SNS) activation along with chronic up regulation of the renin-angiotensin-aldosterone system (RAAS) and effects of aldosterone on heart, vessels, and kidneys. CHF should be viewed as a complex, interrelated sequence of events involving hemodynamic, nonhemodynamic, genetic, energetic, and neurohormonal events.
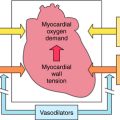
In the failing heart, the loss of contractile function leads to a decline in cardiac output and a decrease in arterial blood pressure. The baroreceptors sense the hemodynamic changes and initiate countermeasures to maintain support of the circulatory system. Activation of the SNS serves as a compensatory mechanism in response to a decline in left ventricular stroke volume. This helps maintain adequate cardiac output by increasing myocardial contractility and heart rate (β1 adrenergic receptors) and by increasing vasomotor tone (α1 adrenergic receptors) to maintain systemic blood pressure (see Chapters 11 and Chapters 19). Over the long term, this hyperadrenergic state leads to irreversible myocyte damage, cell death, and fibrosis. In addition, the augmentation in peripheral vasomotor tone increases left ventricular afterload, placing an added stress upon the left ventricle and an increase in myocardial O2 demand, factors involved in ventricular remodeling. The frequency and severity of cardiac arrhythmias are enhanced in the failing heart, in part as a result of the increased adrenergic tone.
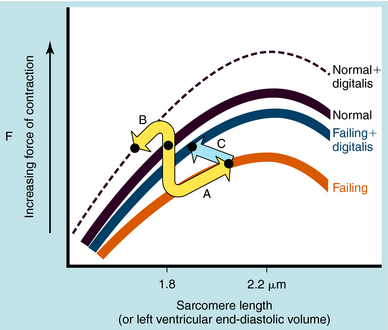
The most frequent cause for chronic systolic dysfunction is ischemic cardiomyopathy, characterized by a reduction in the ventricular ejection fraction and enlargement of the left ventricle, due to a failure of the left ventricle to empty as a result of impaired myocardial contractility or pressure overload. This results from destruction of myocytes, impaired myocyte function, or fibrosis. Chronic pressure overload, caused by untreated long-standing hypertension or aortic stenosis, decreases the left ventricular ejection fraction by increasing resistance to forward flow. Initially, the increase in the left ventricular end-diastolic pressure (volume) results in a compensatory enhancement in stroke volume due to the pressure-induced lengthening of the sarcomeres that invokes the Frank-Starling mechanism (see Fig. 23-1), thereby partially compensating for the failing ventricle. The marked diastolic derangement in filling and the decreased ventricular distensibility do not permit adequate stretch of the myocytes because it occurs under conditions that require an increase in cardiac output. Therefore, in the presence of systolic heart failure, the Frank-Starling mechanism fails to adequately increase stroke volume in response to exercise.
Angiotensin-Converting Enzyme Inhibitors
Baroreceptor-mediated activation of the SNS leads to an increase in renin release and formation of angiotensin II (see Chapter 19), which causes intense vasoconstriction and stimulates aldosterone production (Fig. 23-2). This is decreased by the angiotensin-converting enzyme (ACE) inhibitors, which inhibit formation of angiotensin II from angiotensin I, as discussed in Chapter 20.
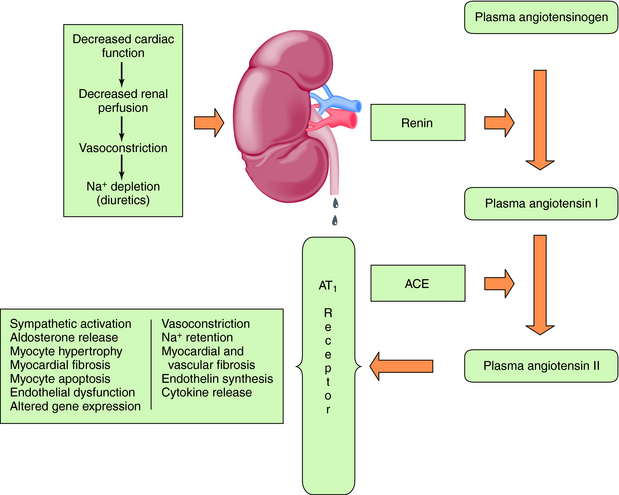
Angiotensin II acts through AT1 and AT2 receptors, although most of its actions occur through AT1 receptor activation. Although the AT2 receptor is distributed widely in fetal tissues, its distribution is limited in adults. Angiotensin II mediates cell growth, vasoconstriction, Na+ and fluid retention, and sympathetic activation (Table 23-1). The central role of the RAAS in the development and progression of cardiovascular disease and, in particular, CHF is well established. In addition to regulation of blood pressure and maintenance of fluid and electrolyte balance, short-term activation of the RAAS in heart failure improves cardiac output through fluid and Na+ retention (increased preload), whereas long-term activation results in vasoconstriction, increased afterload, and decreased cardiac output. These mechanisms, together with SNS activation, induce a vicious cycle of increased preload, afterload, and cardiac workload, leading to increased myocardial O2 consumption, loss of myocytes through apoptosis, and progressive worsening heart failure.
| Site of Action | Response |
|---|---|
| Vascular smooth muscle | Vasoconstriction—increased renal and peripheral resistance, increased left ventricular afterload, vessel wall hyperplasia, hypertrophy initiated by AT1 receptors |
| Heart | Positive inotropic effect by opening voltage-gated Ca++ channels, myocardial hypertrophy, activation of matrix metalloproteinases, myocardial fibrosis initiated by AT1 receptors, increase in release of norepinephrine |
| Adrenal cortex | Increased aldosterone synthesis and release, release of catecholamines from adrenal medulla |
| Kidney | Reduction in renal blood flow and excretory functions, increase in Na+ channels in the apical membrane of renal tubules, increased activity of Na+,K+-ATPase in the basal lateral membrane, increased renal tubular reabsorption of Na+, increased K+ excretion |
| Sympathetic nervous system | Increased norepinephrine release and inhibition of reuptake (increase in peripheral resistance, stimulation of renin release) |
| Central nervous system | Release of vasopressin, increased fluid retention, activation of the sympathetic nervous system |
β Adrenergic Receptor Blocking Drugs
The β receptor blockers inhibit the adverse effects of the SNS in patients with heart failure. Whereas cardiac adrenergic drive initially serves as a compensatory mechanism to support the failing heart, long-term activation leads to a down regulation of β1 receptors and an uncoupling from adenylyl cyclase (see Chapter 1), thereby reducing myocardial contractility. The β receptor blockers may be beneficial through resensitization of the down regulated receptor, improving myocardial contractility.
Another approach is to block AT1 receptors with the use of angiotensin receptor blockers (ARBs). The currently available drugs selectively block AT1 receptors and replicate many of the actions of ACE inhibitors; they do not block AT2 receptors. Activation of AT2 receptors may cause vasodilation, preventing hypertrophy of vascular smooth muscle and cardiomyocytes, production of bradykinin, and release of NO. There is also overexpression of AT2 receptors in the failing heart. There may also be non-ACE-dependent formation of angiotensin II by enzymes, such as chymase, cathepsin G, trypsin, and tissue plasminogen activator. An ARB might block the deleterious actions mediated by AT1 receptors while preserving the desirable effects of AT2 receptor activation. Although the hemodynamic and clinical effects may appear similar, ACE inhibitors and ARBs should not be regarded as being identical.
The elevated circulating angiotensin II levels in the patient with CHF lead to greatly increased production of aldosterone (see Chapter 39), an important mediator in the progressive development of CHF. Aldosterone binds to mineralocorticoid receptors in renal epithelial cells and promotes Na+ retention, Mg++ and K+ loss, sympathetic activation, parasympathetic inhibition, myocardial and vascular fibrosis, baroreceptor dysfunction, impaired arterial compliance, and vascular damage. Aldosterone antagonists include spironolactone and eplerenone, which may reduce norepinephrine (NE) release from cardiac sympathetic nerves and increase plasma K+. The elevated concentrations of aldosterone in CHF led to the concept that inhibition of aldosterone receptors could be beneficial, and competitive aldosterone antagonists are now part of the therapeutic armamentarium.
The cardiac glycosides increase the force of myocardial contraction, alter electrophysiological properties in specialized regions, and have extracardiac actions associated with toxicity. Cardiac glycosides influence the heart through a direct inhibition of membrane Na+,K+-ATPase and an indirect increase in vagal tone (Table 23-2). Their cardiotoxic effects are an overextension of the same mechanisms responsible for their positive inotropic actions.
TABLE 23–2 Effects of Cardiac Glycosides on Electrophysiological Properties of the Heart
| Direct | Indirect (increased vagal tone) | |
|---|---|---|
| SA node | No effect at therapeutic dose | No effect at therapeutic dose |
| Atrial muscle | High dose increases rate of spontaneous depolarization | High dose decreases rate of spontaneous depolarization |
| AV node | Increased refractory period | Decreased conduction velocity |
| Decreased conduction velocity | Increased refractory period | |
| His-Purkinje system | Increased refractory period | Increased refractory period |
| Decreased conduction velocity | Decreased conduction velocity | |
| High dose increases triggered activity | None | |
| Toxic doses enhance pacemaker |
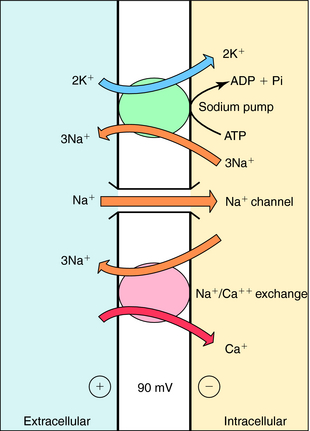
Cardiac glycosides increase contractile force; however, unlike catecholamines, they do not increase the rate of relaxation. By decreasing the activity of the Na+,K+-ATPase, they cause a progressive gain in intracellular Na+ with each cardiac cycle. This increase promotes Ca++ influx by Na+/Ca++ exchange (Fig. 23-3). The net result is an increased intracellular Ca++, enhancing the Ca++ transient resulting from an augmented Ca++ loading of the sarcoplasmic reticulum. In the presence of a digitalis glycoside, a new steady state is achieved where an increased amount of Ca++ is released after depolarization, increasing force development (stroke volume; see Fig. 23-1).
Electrophysiological effects of cardiac glycosides vary among different regions of the heart. They decrease automaticity within the sinoatrial (SA) and atrioventricular (AV) nodes as a result of an increase in parasympathetic tone along with a concomitant decrease in sympathetic tone. The increase in parasympathetic tone on the AV node leads to a decrease in conduction velocity and an increase in effective refractory period. Thus digitalis glycosides indirectly decrease heart rate and impair impulse transmission across the AV node.
The major direct effects are in the atrial muscle, AV node, and ventricles (see Table 23-2). In atria, they prolong the effective refractory period and decrease conduction velocity, effects opposite to those elicited by their indirect actions. However, the direct effects in the AV node summate with the indirect actions to further impair conduction velocity and increase the refractory period.
Diuretics are widely used in treatment of congestive heart failure to reduce extracellular fluid volume (see Chapter 21). Their primary parenteral use is in patients with acute heart failure for correction of volume overload. In the most severe cases, intravenous infusions of a loop diuretic (e.g., furosemide) can initiate a rapid, predictable, and sustained diuresis. The diuretic is titrated according to an estimated “dry” weight, based on optimal filling pressures and symptoms, without exacerbating symptomatic hypotension.
Epinephrine (Epi) and NE are β receptor agonists that produce a marked positive inotropic response and have α1 receptor agonist activity that elicits peripheral vasoconstriction (see Chapter 11). Both agents have limited utility in patients with severe heart failure but can offer significant inotropic support for short-term intervention (minutes to hours) in life-threatening situations while more definitive measures are initiated.
Dopamine acts on prejunctional D2 receptors to inhibit release of NE (see Chapter 19), resulting in vasodilation. Dopamine also acts on cardiac β1 receptors to elicit a positive inotropic action and on vascular smooth muscle to cause vasodilation, improving blood flow to renal, mesenteric, coronary, and cerebral vascular beds.
Inamrinone and milrinone exert their positive inotropic actions by inhibiting cyclic adenosine monophosphate (cAMP) phosphodiesterase (PDE), the enzyme that hydrolyzes and inactivates cAMP. The effects of these drugs differ from those of other PDE inhibitors, such as caffeine or theophylline, in that they are selective for a particular isozyme, PDE type 3. They increase cardiac cAMP concentrations but not cyclic guanosine monophosphate concentrations. This promotes cAMP-dependent protein kinase A-mediated phosphorylation of the Ca++ channel in the heart, enhancing Ca++ influx and resulting in a positive inotropic action similar to that caused by catecholamines.
Nitroprusside was among the earliest vasodilators to show improvement in cardiac output in patients with decompensated heart failure. Nitroprusside and other nitrovasodilators are discussed in Chapter 24. Nitroprusside reduces ventricular filling pressures by directly increasing venous compliance, resulting in a redistribution of blood from central to peripheral veins. In addition, the action of nitroprusside on the arterial side of the circulation makes it one of the most effective agents for reducing left ventricular afterload. Nitroprusside dilates the pulmonary arterioles, thereby decreasing right ventricular afterload. Reducing both preload and afterload improves myocardial energetics because of a reduction in wall stress. In contrast, nitroglycerin shows specificity for venodilation, thereby increasing venous capacitance and pooling of blood in the more dependent regions of the body.
Pharmacokinetics
The pharmacokinetics of the ACE inhibitors and ARBs are discussed in Chapter 20, of the β receptor blockers and sympathomimetics in Chapter 11, of the diuretics in Chapter 21, and of the nitrovasodilators in Chapter 24.
Pharmacokinetic parameters of cardiac glycosides are presented in Table 23-3. Digitoxin is rarely used in the United States, although it continues to be used in Europe. Absorption of digoxin given orally varies from 45% to 85%, and because of such wide variations, patients should be maintained on a specific brand. However, bioavailability of digitoxin is consistently high.
TABLE 23–3 Pharmacokinetic Parameters for Cardiac Glycosides, Phosphodiesterase Inhibitors, and Nesiritide
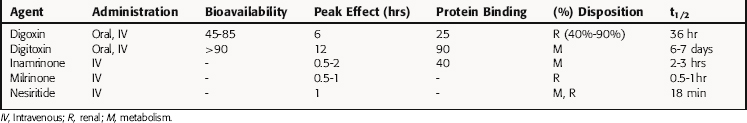
Variations may be minimized by maintenance of predetermined concentrations in plasma. However, a given plasma concentration may be therapeutic in some patients and toxic in others because of differences in sensitivity caused by various factors (Table 23-4). Because monitoring the positive inotropic effect is impractical, clinical evaluation often involves the electrocardiogram. A slight (approximately 10%) increase in PR interval is not alarming; however, a greater delay in AV nodal conduction time and conduction block, or development of ventricular bigeminy or trigeminy, is a harbinger of serious toxicity.
TABLE 23–4 Factors Leading to Altered Sensitivity to Digoxin
| Influence | Effects |
|---|---|
| Physiological influences | Increased vagal and sympathetic tone, age |
| Pathophysiological influences | Chronic pulmonary disease, renal dysfunction, myocardial ischemia or infarction, rheumatic or viral myocarditis, hyperthyroidism, or hypothyroidism |
| Abnormal plasma electrolytes | Hypokalemia or hyperkalemia, hypomagnesemia, hypercalcemia or hypocalcemia |
| Drug-drug interactions | Increased or decreased therapeutic effects or toxicity |
Because digitoxin has a long t1/2, steady-state is not achieved until 20 days after starting therapy without a loading dose; thus a loading dose is usually given. Digoxin, with its shorter t1/2, may be administered without a loading dose; steady-state concentration is reached in 5 to 7 days when administered orally daily. Switching from maintenance doses of digoxin to digitoxin results in a temporary loss of effect, because digoxin is excreted from the body rapidly, whereas digitoxin accumulates slowly. Conversely, switching from maintenance doses of digitoxin to digoxin causes a transient overdose. Recent studies suggest that “ideal” or therapeutic digoxin serum concentrations are 1.1 to 1.2 ng/mL, whereas others favor a range of 0.5 to 1.5 ng/mL.
Relationship of Mechanisms of Action to Clinical Response
β Adrenergic Receptor Blocking Drugs
The β receptor blockers that have been shown to be effective in treatment of heart failure include those that selectively block β1 receptors, such as metoprolol, and those that block both α1, β1, and β2 receptors, such as carvedilol. Their properties are discussed in Chapter 11.
In practice, β receptor blockers are almost always used together with ACE inhibitors (and usually with digoxin). Patients need not be taking high doses of ACE inhibitors before being considered for treatment with β receptor blockers. In patients taking low doses of an ACE inhibitor, addition of a β receptor antagonist produces a greater improvement in symptoms and reduces the risk of death. The β receptor blockers should not be prescribed without diuretics in patients with a history of fluid retention, because diuretics are needed to maintain Na+ balance and prevent development of fluid retention that can accompany β receptor blocker therapy. Doses should be increased gradually, until side effects associated with lower doses have disappeared. Clinical trials show that 85% of patients could tolerate short-and long-term treatment with β receptor blockers.
Cardiac glycosides increase the force of cardiac contraction in either normal or failing hearts. Although originally thought to be effective only in patients with heart failure, they also increase force of contraction and reduce end-diastolic volume in normal hearts, which in turn decreases the force of contraction, canceling their positive inotropic effects (see Fig. 23-1, B, arrow). In the failing dilated heart, the increased force of contraction and decrease in end-diastolic volume make the heart’s operation more nearly normal (see Fig. 23-1, C, arrow). Therefore, despite direct positive inotropic effects on both failing and nonfailing hearts, hemodynamic improvements are obtained only in the failing heart.
Digoxin is especially useful in CHF patients with atrial fibrillation, because it slows ventricular rate, allowing for improved filling and increasing ejection fraction or stroke volume. The net result is a reduced need for heightened sympathetic tone. This unique property makes digoxin useful in CHF patients in sinus rhythm. An additional benefit is a reduction in ventricular size in the failing heart, reducing ventricular wall tension, an important determinant of O2 consumption. This is beneficial in patients with CHF caused by ischemic heart disease. In patients with chronic CHF and abnormal systolic function, digoxin in combination with diuretics and ACE inhibitors reduces the frequency of hospitalizations and overall mortality.
The diuretics are discussed in Chapter 21. Prospective clinical trial data do not exist for evaluating their overall efficacy on mortality in patients with heart failure. However, there is little doubt that they are useful and necessary adjuncts for relief of CHF symptoms resulting from Na+ and H2O retention in patients with acute or chronic cardiac decompensation.
Unfortunately, in more advanced CHF, the use of a single diuretic may have limited efficacy, and combination therapy may be required. With “diuretic resistance,” it is common to use a combination of a loop and a distal tubular diuretic (see Chapter 21).
Heart rate may increase during dobutamine administration. This is of particular concern in patients with atrial fibrillation, where β receptor activation at the AV node will increase atrial impulses to the ventricular conducting system. The short t1/2 of dobutamine is advantageous when unexpected hypotension, tachycardia, or tachyarrhythmia results. As with all β receptor agonists, dobutamine will be ineffective in patients being treated with β receptor blockers.
Vasodilators used for acute or chronic treatment of heart failure should be given in doses that reduce peripheral resistance but do not cause a sharp decrease in blood pressure, that is, in doses at which most blood pressure effects are compensated for by homeostatic mechanisms. These drugs relax venous and arterial smooth muscle, thereby reducing resistance to ventricular ejection and increasing the capacity of the venous reservoir. This causes relief of symptoms and an increase in exercise tolerance in patients with a dilated ventricle. These changes can be achieved acutely by intravenous nitroprusside, nitroglycerin, and nesiritide, or by chronic administration of hydralazine together with isosorbide dinitrate or an ACE inhibitor (see Chapter 24).
Pharmacovigilance: Side-Effects, Clinical Problems, and Toxicity
Digoxin toxicity remains an important clinical problem that demands vigilance for the early recognition of disturbances of cardiac impulse formation and conduction abnormalities, along with more subtle signs related to the central nervous and gastrointestinal systems (see Clinical Problems Box). Toxic effects of digoxin may occur at any serum concentration due to factors that affect sensitivity (see Table 23-4) because of its low therapeutic index.
When cardiac muscle is exposed to toxic concentrations of a glycoside, Na+ pump inhibition and cellular Ca++ loading become excessive. The cytoplasmic membrane becomes unstable for a short time immediately after each membrane repolarization. In normal ventricular muscle cells, membrane depolarization is followed by repolarization, so that the membrane potential reaches approximately -90 mV and remains there until the next wave of depolarization (Fig. 23-4, A). In digoxin toxicity, however, the membrane becomes more permeable to Na+, Ca++, and K+ immediately after repolarization. Movements of Na+ and Ca++ are particularly prominent, because these ions are driven by both chemical and electrical gradients.
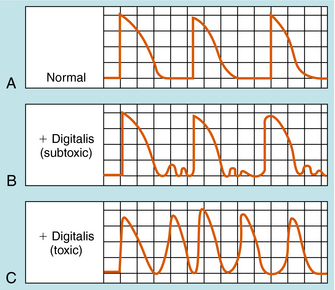
In “mild” toxicity the transient inward current subsides, causing the transmembrane potential to return to its resting level. This may be repeated several times, causing oscillatory delayed afterpotentials (Fig. 23-4, B). These small oscillatory delayed afterpotentials are most readily observed in cardiac Purkinje fibers and do not propagate beyond the individual cell. When their magnitude increases in advanced digoxin toxicity, the threshold potential is reached, causing the cell to be depolarized (i.e., to trigger action potentials; see Fig. 23-4, C). Such triggered action potentials propagate from Purkinje fiber cells to ventricular muscle, causing the muscle to contract repetitively and no longer be under the control of the SA node. This may lead to life-threatening ventricular tachycardia, fibrillation, or both.
Other Drugs for Congestive Heart Failure
The side effects of β receptor blockers are discussed in detail in Chapter 11. Adverse effects are generally an extension of their therapeutic actions and include cardiac decompensation, bradycardia, hypoglycemia, and cold extremities. Initiation of treatment with a β receptor blocker has produced four types of adverse reactions that require careful attention and management:
Contraindications for use of ACE inhibitors include bilateral renal artery stenosis and known allergies. High serum creatinine is a contraindication for use of ACE inhibitors or ARBs, as is the presence of hyperkalemia that will worsen on drug therapy. Side effects of ACE inhibitors and ARBs are discussed in more detail in Chapter 20.
Adverse effects associated with aldosterone antagonists include hyperkalemia, agranulocytosis, anaphylaxis, hepatotoxicity, and renal failure (see Chapter 39). Spironolactone has the added features of inducing gynecomastia, sexual dysfunction, and menstrual irregularities. Severe adverse effects associated with eplerenone include severe arrhythmias, life-threatening myocardial infarction, and myocardial ischemia (angina).
The sympathomimetics Epi and NE may cause restlessness, headache, tremor, and cardiac palpitations, as well as cerebral hemorrhage and cardiac arrhythmias (see Chapter 11). They should be used with caution in patients receiving nonselective β receptor blockers, because their unopposed actions on vascular α1 receptors can cause an acute hypotensive crisis and possible cerebral hemorrhage. The adverse effects of dopamine and dobutamine are similar and attributable to excessive sympathomimetic activity related to overdose (see Chapter 11).
Adverse effects of nitrovasodilators, most commonly hypotension, are discussed in Chapter 24. Aggressive treatment with nitroprusside may result in a precipitous fall in left ventricular end-diastolic pressure, marked hypotension, and myocardial ischemia. The accompanying
pulmonary vasodilation may lead to an increased ventilation-perfusion mismatch and hypoxia. Nitroprusside is contraindicated in patients with severe obstructive valvular heart disease (aortic, mitral, or pulmonic stenosis or obstructive cardiomyopathy).
New Horizons
The recognition that the activation of the SNS has an important role in the pathophysiology of heart failure encouraged a reexamination of the potential usefulness of β receptor blockers in treatment. For many years these drugs were contraindicated in treatment of severe left ventricular dysfunction. However, several clinical trials in patients after myocardial infarction showed that β receptor blockers led to significant reductions in mortality in patients with mild to moderate heart failure. Clinical experience with β receptor blockers in treatment of ventricular arrhythmias has also strengthened the case for their use in heart failure. Clinical trials with carvedilol, a nonselective β receptor blocker with α1 receptor-blocking activity and antioxidant properties, showed a reduction in both disease progression and mortality.
Despite recent approaches to delay the progression of heart failure and prolong life, the important issue of prevention remains. Ischemic heart disease is an important contributor to development of chronic heart failure. Lifestyle changes discussed in Chapter 25 and the introduction of new antiatherogenic interventions will reduce the number of patients who develop chronic heart failure. Because the mode of death in patients with CHF is sudden, there is the need for novel antiarrhythmic agents (see Chapter 22) that function during the normal cardiac cycle. Pharmacological inhibition of membrane currents activated during an ischemic event that heralds the onset of the lethal arrhythmic episode could provide a major benefit.
Drugs for treatment of heart failure. Treat Guidel Med Lett. 2006;4:1-4.
Braunwald E, Bristow MR. Congestive heart failure: Fifty years of progress. Circulation. 2000;102(suppl IV):IV-14-IV23.
Gheorghiade et al. 2005 Gheorghiade M, Teerlink JR, Mebazaa A. Pharmacology of new agents for acute heart failure syndromes. Am J Cardiol. 2005;96:68G-73G.
Tavares et al. 2008 Tavares M, Rezlan E, Vostroknoutova I, et al. New pharmacologic therapies for acute heart failure. Crit Care Med. 2008;36(1 Suppl):S112-120.
Teerlink JR. Overview of randomized clinical trials in acute heart failure syndromes. Am J Cardiol. 2005;96:59G-67G.