[level-membership-for-basic-science-category]
Chapter 11 Drugs Affecting the Sympathetic Nervous System
| Therapeutic Overview |
|---|
| Mpathomimetics |
| Nasal decongestion |
| Decrease formation of aqueous humor in glaucoma |
| Neurogenic shock, cardiogenic shock |
| Paroxysmal atrial tachycardia |
| Bronchospasm, asthma |
| Decrease diffusion of local anesthetics |
| Sympatholytics |
| Essential hypertension, hypertensive emergencies |
| Benign prostatic hyperplasia |
| Cardiac dysrhythmias, angina pectoris, postmyocardial infarction |
| Essential tremor |
| Glaucoma |
Therapeutic Overview
The Therapeutic Overview Box presents a summary of the primary uses of different classes of compounds that affect the sympathetic nervous system.
Mechanisms of Action
The biochemistry and physiology of the autonomic nervous system, including a discussion of adrenergic receptors, is presented in Chapter 9. Detailed information on receptors and signaling pathways involved are in Chapter 1. This section covers topics that pertain specifically to noradrenergic neurotransmission and its modulation by drugs.
The neurochemical steps that mediate noradrenergic neurotransmission are summarized in Figure 11-1. The first step involves the transport of tyrosine into the neuron followed by its conversion to 4-dihydroxy-phenylalanine (l-DOPA) by the rate-limiting enzyme tyrosine hydroxylase. l-DOPA is rapidly decarboxylated to dopamine (DA) by aromatic l-amino acid decarboxylase (ALAAD), also known as DOPA decarboxylase. In dopaminergic neurons within the central nervous system (CNS), this is the last step in the synthetic process, and DA is released as a neurotransmitter. Noradrenergic neurons in the CNS and peripheral sympathetic nervous system contain an additional enzyme, DA β-hydroxylase, which converts DA to NE. This enzyme is located in synaptic vesicles, so DA must be actively transported into these vesicles before conversion to NE.
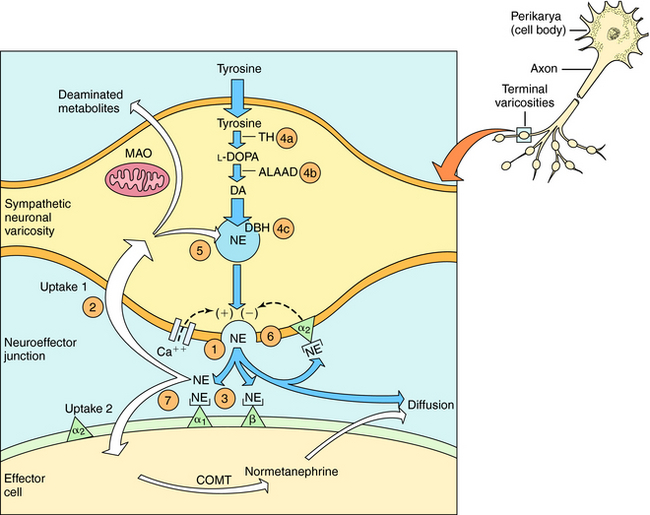
When the action potential depolarizes the nerve terminal, voltage-gated Ca++ channels open, allowing Ca++ to enter the neuron and cause the vesicles to release NE. At the neuroeffector junction, NE binds to adrenergic receptor subtypes on both the presynaptic and postsynaptic membranes, with different organs containing different receptor subtypes. Activation of α2-“autoreceptors” on the presynaptic neuronal membrane causes feedback inhibition and reduces further NE release. The signaling mechanisms occurring after adrenergic receptor activation are discussed in Chapter 9.
After NE has activated its receptors, its action is terminated primarily by a high-affinity reuptake system (termed “uptake 1” in Fig. 11-1), which transports NE back into the neuron for repackaging and eventual re-release. A smaller fraction of the NE in the synapse diffuses away from the receptors and can be taken up by a lower affinity extraneuronal process (termed “uptake 2” in Fig. 11-1). Some of the NE taken back into the sympathetic neurons by reuptake can be oxidatively deaminated by monoamine oxidase (MAO) located on the external mitochondrial membrane. The biologically inactive deaminated metabolites enter the circulation and are excreted in the urine. The NE that is transported into the postjunctional cell is O-methylated by catechol-O-methyltransferase (COMT) to normetanephrine. The high-affinity reuptake of NE into presynaptic terminals is the major mechanism that terminates its actions, although COMT and MAO also play important roles in metabolizing circulating NE and Epi and some exogenously administered sympathomimetic amines.
Drugs modify the activity of sympathetic neurons by increasing or decreasing the noradrenergic signal (see Fig. 11-1). Sympathomimetics may mimic noradrenergic transmission by acting directly on postsynaptic adrenergic receptors (e.g., phenylephrine), by facilitating NE release (e.g., amphetamine) or by blocking neuronal reuptake (e.g., cocaine). Amphetamine and cocaine have intense effects on the CNS and are drugs of abuse with severe addiction liability (Chapter 37).
Activation of Adrenergic Receptors
Over the last half century it has become clear that the actions of NE and Epi are mediated through multiple adrenergic receptor subtypes (see Chapter 9). We now know that there are nine different subtypes of adrenergic receptors, each encoded by separate genes, which are grouped into three major families (α1, α2, β), each containing three different members. Of these, only four (α1, α2, β1, and β2) are currently important in clinical pharmacology; therefore they are the major focus of this chapter. Both NE and Epi activate most adrenergic receptors with similar, but not identical, potencies, although NE is much less potent than Epi at the β2-subtype. Isoproterenol (ISO) is a synthetic analog of NE and Epi and selectively activates only β receptors. Differences in the pharmacological profiles of different adrenergic receptors are illustrated in Figure 11-2, where dose-response curves for the actions of these catecholamines on different tissues are depicted.
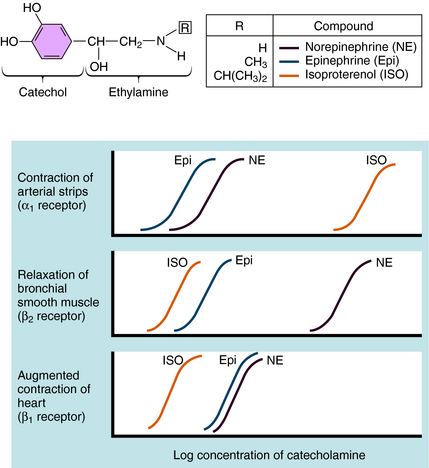
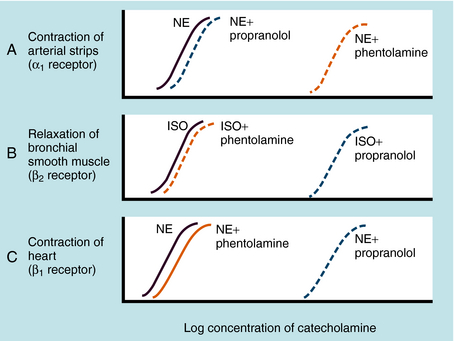
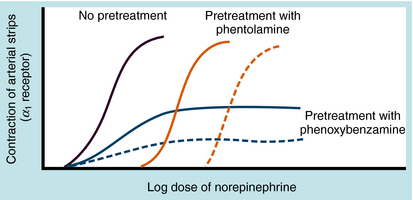
Adrenergic receptor activation increases the contraction of cardiac muscle and induces smooth muscle to either contract or relax, depending on the receptor subtype(s) present. Contraction of arterial strips is mediated by α1 receptors, where the relative potencies of the three catecholamines are Epi ≥ NE >>> ISO. Relaxation of bronchial smooth muscle is mediated by β2 receptors, with the relative potencies being ISO > Epi >>> NE. Contraction of cardiac muscle is mediated by β1 receptors, with the relative potencies being ISO > Epi = NE. The presence of different receptor subtypes in these three tissues is further demonstrated in Figure 11-3 by examining the effects of selective antagonists. Phentolamine, a competitive antagonist at α1 receptors, causes a parallel shift to the right of NE-induced contractions of arterial strips (see Fig. 11-3, A) but does not affect the other two responses. Propranolol, a competitive antagonist at both β1 and β2 receptors, causes a parallel shift to the right of responses mediated by bronchial β2 receptors (see Fig. 11-3, B) and cardiac β1 receptors (see Fig. 11-3, C), without affecting the α1 receptor response.
Adrenergic α2 receptors are found on platelets, in the CNS, and postsynaptically in several other peripheral tissues (blood vessels, pancreas, and enteric cholinergic neurons). As mentioned, activation of α2 receptors on the terminals of sympathetic neurons reduces NE release. These receptors are also found both presynaptically and postsynaptically on neurons in brain, and activation of these receptors reduces central sympathetic outflow.
Direct-Acting Sympathomimetics
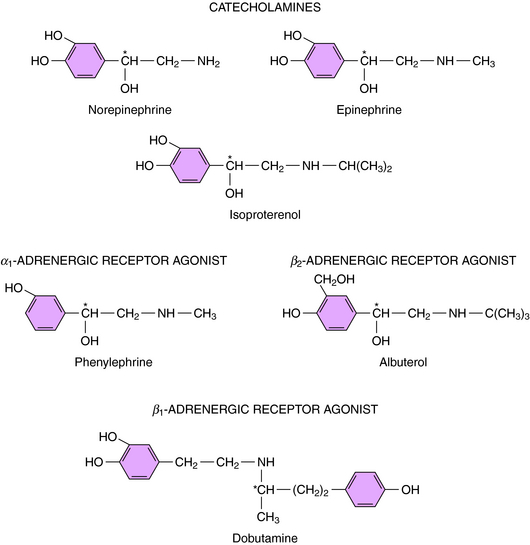
Direct-acting adrenergic receptor agonists mimic some of the effects of sympathetic nervous system activation by binding to and activating specific receptor subtypes (see Fig. 11-1, site 3). For example, as mentioned, ISO selectively activates β receptors, phenylephrine selectively activates α1 receptors, and clonidine selectively activates α2 receptors. Similarly, dobutamine and albuterol are relatively specific agonists at β1 and β2 receptors, respectively. The structures of some clinically important adrenergic receptor agonists are shown in Figure 11-4.
Indirect-Acting Sympathomimetics
Indirect-acting sympathomimetics do not activate receptors directly but facilitate the release of NE from sympathetic neurons or block high-affinity reuptake. Thus they act presynaptically to facilitate adrenergic neurotransmission. Amphetamine and related drugs produce their sympathomimetic effects by facilitating NE release (see Fig. 11-1, site 1); the effects of these drugs in the CNS are described in Chapter 37. On the other hand, cocaine and tricyclic antidepressants, such as desipramine, exert their sympathomimetic effects by blocking high-affinity reuptake (see Fig. 11-1, site 2). The structure and characteristics of cocaine are discussed in Chapter 37, with similar information for tricyclic antidepressants given in Chapter 30.
Because neuronal reuptake, and not metabolism, is the primary mechanism for terminating the actions of NE and Epi, it is not surprising that drugs that inhibit the metabolism of these amines show little or no sympathomimetic actions. On the other hand, inhibitors of MAO (e.g., pargyline) or COMT (e.g., tolcapone) can enhance the actions of other synthetic sympathomimetic amines that are substrates for these enzymes, with some important toxicological implications. For example, the actions of tyramine, a sympathomimetic amine present in a variety of foods, are greatly enhanced in patients treated with MAO inhibitors (see Chapter 30), and the pharmacokinetics of l-DOPA, used for the treatment of Parkinson’s disease, are increased when administered concurrently with a COMT inhibitor (see Chapter 28).
An indirect-acting sympathomimetic amine that releases NE must penetrate the noradrenergic neuron before it can act. Nonpolar, lipid-soluble drugs such as amphetamine can diffuse across neuronal membranes, whereas polar, water-soluble compounds such as tyramine must rely on high-affinity uptake by the NE transporter. Thus drugs that inhibit the NE transporter will reduce or block the effects of tyramine but will enhance the effects of Epi, which acts directly on the adrenergic receptors. Cocaine also causes a prompt increase in the response to Epi by blocking the reuptake system, whereas reserpine disrupts amine transport from the cytoplasm into synaptic vesicles (see Fig. 11-1, site 5). Therefore reserpine has only small effects on responses to direct-acting sympathomimetics.
Inhibition of Synthesis, Storage, or Release of NE
Catecholamine synthesis can be disrupted at several steps, but effective in vivo blockade is obtained only when tyrosine hydroxylase, the enzyme catalyzing the first and rate-limiting step, is inhibited (see Fig. 11-1, site 4a). This can be produced clinically with α-methyltyrosine (metyrosine). Several compounds can inhibit other biosynthetic enzymes (e.g., DOPA decarboxylase is inhibited by carbidopa and DA β-hydroxylase is inhibited by disulfiram (see Fig. 11-1, sites 4b and 4c, respectively). Although these drugs do not effectively block endogenous catecholamine synthesis when administered, they do have clinical utility by affecting other systems. By virtue of its ability to inhibit peripheral DOPA decarboxylase, carbidopa is used in combination with l-DOPA for patients with Parkinson’s disease, to increase the amount of l-DOPA available to the CNS to enhance DA synthesis (see Chapter 28). Disulfiram is used in treating chronic alcoholism (see Chapter 32) because it blocks aldehyde dehydrogenase; however, its ability to inhibit catecholamine synthesis leads to significant adverse effects such as hypotension.
Disruption of vesicular storage also modifies noradrenergic transmission (see Fig. 11-1, site 5). As discussed, reserpine disrupts the ability of the synaptic vesicles to transport and store DA and NE. Adrenergic responses can also be impeded by inhibiting NE release (see Fig. 11-1, site 6). Drugs such as bretylium and guanethidine, which accumulate in noradrenergic nerve terminals, prevent NE release.
Adrenergic Receptor Antagonists
Many adrenergic receptor antagonists exert different subtype-selective effects (see Fig. 11-1, site 7). The early antagonists such as phenoxybenzamine and propranolol blocked α or β receptors, respectively, whereas drugs are now available that selectively block α1 (prazosin), α2 (yohimbine), or β1 (metoprolol) receptors. These selective antagonists have important therapeutic advantages over the original broad-spectrum adrenergic receptor blockers and are used for various indications, such as the control of blood pressure (see Chapter 20).
Pharmacokinetics
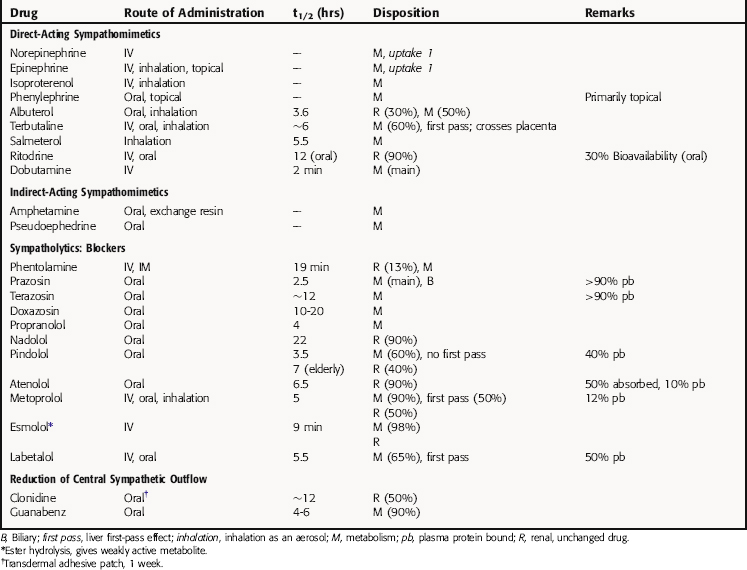
Detailed pharmacokinetics are not known for many of these drugs in humans. Some known pharmacokinetic parameters for major drugs are summarized in Table 11-1.
Relationship of Mechanisms of Action to Clinical Response
Direct and Reflex Cardiovascular Actions of Adrenergic Agents
The sympathetic nervous system plays an important role in regulating the cardiovascular system; thus adrenergic drugs have pronounced effects on this system. These drugs alter the rate and force of contraction of the heart and the tone of blood vessels (and, consequently, blood pressure) through activation of adrenergic receptors on cardiac and vascular smooth muscle cells. Compensatory reflex adjustments occur as a result of these responses, and these reflexes must be considered to understand the overall actions of adrenergic drugs on the heart and blood vessels.
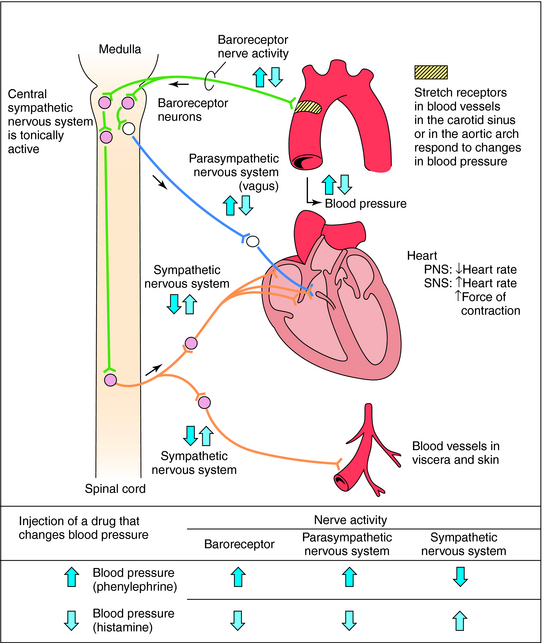
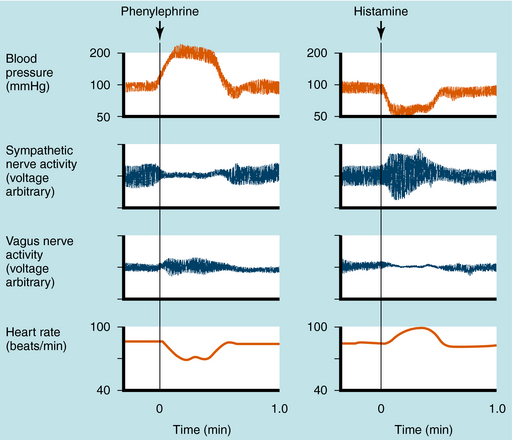
Mean arterial blood pressure does not fluctuate widely because of feedback mechanisms that evoke compensatory responses to maintain homeostasis (Fig. 11-5). Baroreceptors are stretch receptors located in the walls of the heart and blood vessels that are activated by distention of the blood vessels. Increased blood pressure increases impulse traffic in afferent baroreceptor neurons that project to vasomotor centers in the medulla. Impulses generated in the baroreceptors inhibit the tonic discharge of sympathetic neurons projecting to the heart and blood vessels and activate vagal fibers projecting to the heart. When phenylephrine, which contracts vascular smooth muscle, is administered, peripheral resistance and blood pressure increase (Fig. 11-6). In turn, this increase in pressure increases afferent baroreceptor neuronal activity, thereby reducing sympathetic nerve activity and increasing vagal nerve activity. Consequently, heart rate decreases (bradycardia). Drugs such as histamine, which relax vascular smooth muscle, decrease blood pressure, reducing impulse traffic in afferent buffer neurons. Consequently, sympathetic nerve activity increases and vagal nerve activity decreases, resulting in an increased heart rate (tachycardia).
Direct-Acting Sympathomimetics
Epi is the prototype of direct-acting sympathomimetic drugs because it activates all known adrenergic receptor subtypes. The effects of direct-acting sympathomimetic drugs on selected tissues and organ systems are discussed in this section, with the effects of the prototype Epi first, followed by those of more selective sympathomimetics as compared with the prototype.
Epi also activates conducting tissues, thereby increasing conduction velocity and reducing the refractory period in the atrioventricular node, the bundle of His, Purkinje fibers, and ventricular muscle. These changes, and the activation of latent pacemaker cells, may lead to alterations in the rhythm of the heart. Large doses of Epi may cause tachycardia, premature ventricular systole, and possibly fibrillation; these effects are more likely to occur in hearts that are diseased or have been sensitized by halogenated hydrocarbon anesthetics (see Chapter 35).
Epi is a potent bronchodilator, relaxing bronchial smooth muscle by activating β2 receptors. It dramatically reduces responses to endogenous bronchoconstrictors and can be lifesaving in acute asthmatic attacks (see Chapter 16). Epi also relaxes smooth muscle in other organs through β2 receptors. It reduces the frequency and amplitude of gastrointestinal (GI) contractions, decreases the tone and contractions of the pregnant uterus, and relaxes the detrusor muscle of the urinary bladder. However, it causes the smooth muscle of the prostate and splenic capsule and of GI and urinary sphincters to contract by activating α1 receptors. Epi can foster urinary retention by relaxing the detrusor muscle and contracting the trigone and sphincter of the urinary bladder.
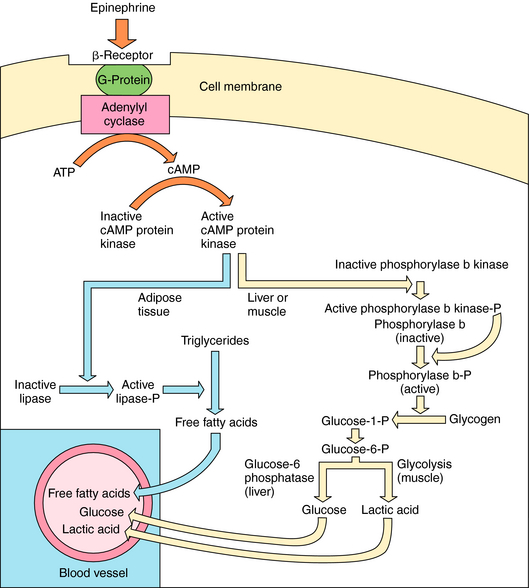
The major metabolic effects of Epi are increased circulating concentrations of glucose, lactic acid, and free fatty acids. In humans these effects are attributable to the activation of β receptors at liver, skeletal muscle, heart, and adipose cells (Fig. 11-7). Adrenergic β receptor activation results in Gs protein-mediated activation of adenylyl cyclase. The resulting increase in cyclic adenosine monophosphate (cAMP) leads to a series of phosphorylation events, culminating in activation of phosphorylase and lipase. Lipase catalyzes breakdown of triglycerides in fat to free fatty acids. The characteristic “calorigenic action” of Epi, which is reflected in a 20% to 30% increase in O2 consumption, is caused partly by the breakdown of triglycerides in brown adipose tissue and subsequent oxidation of the resulting fatty acids. In liver, phosphorylase catalyzes the breakdown of glycogen to glucose. In muscle, glycogenolysis and glycolysis produce lactic acid, which is released into the blood. Release of glucose from the liver is accompanied by the efflux of K+, so that Epi induces hyperglycemia and a brief period of hyperkalemia due to activation of hepatic α adrenergic receptors. The hyperkalemia is followed by a more pronounced hypokalemia, as the K+ released from the liver is taken up by skeletal muscle as a result of activation of muscle β2 adrenergic receptors.
Other Direct-Acting Sympathomimetics
Metaproterenol, terbutaline, albuterol, bitolterol, salmeterol, and ritodrine are relatively specific agonists at β2 receptors. Because these drugs are less potent at β1 receptors, they have less tendency to stimulate the heart. Nevertheless, their selectivity for β2 receptors is not absolute, and at higher doses these drugs stimulate the heart directly. These drugs also differ from ISO because they are effective orally and have a longer duration of action. Selective β2 receptor agonists relax vascular smooth muscle in skeletal muscle and smooth muscle in bronchi and uterus. Although the pharmacological properties of all β2 receptor agonists are similar, ritodrine is marketed as a tocolytic agent; that is, it relaxes uterine smooth muscle and thereby arrests premature labor. All other drugs are marketed for the treatment of bronchospasm and bronchial asthma (see Chapter 16). By activating β2 receptors, these drugs cause bronchodilation and may inhibit the release of inflammatory and bronchoconstrictor mediators (histamine, leukotrienes, prostaglandins) from mast cells in the lungs. The compounds are most effective when delivered by inhalation, which results in the least systemic adverse effects (tachycardia, skeletal muscle tremor). When used orally, selective β2 receptor agonists have an advantage over ephedrine (see later discussion) because they lack CNS stimulant properties.
DA and dobutamine are relatively specific for β1 receptors and are used to stimulate the heart. DA is an endogenous catecholamine with important actions as a neurotransmitter in the brain (see Chapter 27). When administered by intravenous infusion, DA produces a positive inotropic action on the heart by directly stimulating β1 receptors, and indirectly, by releasing NE. DA relaxes smooth muscle in some vascular beds, specifically in the kidney, increasing glomerular filtration rate, Na+ excretion, and urinary output. The latter effect is mediated by DA D1 receptors in the renal vasculature that can be blocked by many antipsychotic drugs (see Chapter 29). DA is also administered by intravenous infusion for treatment of shock, resulting from myocardial infarction, or trauma.
Indirect-Acting Sympathomimetics
The sympathomimetic actions of some drugs stem from their ability to cause the release of NE from sympathetic neurons or block the neuronal reuptake of NE. Some of these drugs (e.g., amphetamine, cocaine) have noticeable CNS stimulant actions (see Chapter 37).
Tyramine is present in a variety of foods (e.g., ripened cheese, fermented sausage, wines) and is also formed in the liver and GI tract by the decarboxylation of tyrosine. Tyramine is an indirect-acting agent that is taken up by sympathetic nerve terminals and displaces NE from vesicles into the synaptic cleft via reverse transport of the NE uptake 1 transporter (not by exocytosis). Normally, significant quantities of tyramine are not found in blood or tissues, because tyramine is rapidly metabolized by MAO in the GI tract, liver and other tissues including sympathetic neurons. However, in patients treated with MAO inhibitors for depression, increased circulating concentrations of tyramine may be achieved, particularly after the ingestion foods containing large concentrations of tyramine, leading to the massive release of NE and a severe hypertensive response. Thus patients treated with MAO inhibitors should avoid eating foods containing tyramine (see Chapter 30).
Ephedrine and amphetamine are related chemically to Epi (Fig. 11-8) but exert their sympathomimetic effects mainly by facilitating the release of NE. Ephedrine is a mixture of four isomers: D- and l-ephedrine and d– and l-pseudoephedrine. l-Ephedrine is the most potent, but the racemic mixture is often used, as is d-pseudoephedrine. Ephedrine is not metabolized by COMT or MAO and thus has a prolonged duration of action. Ephedrine exerts a direct action on β2 receptors and has some limited usefulness as a bronchodilator. It also readily crosses the blood-brain barrier. Ephedrine was widely used as a dietary supplement for its stimulant and appetite-suppressant properties (see Chapter 7) but has now been removed from the market in the United States because of increasing reports of adverse effects.
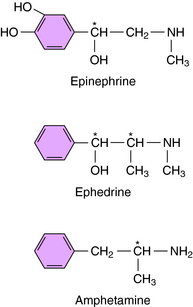
Methamphetamine and amphetamine exist as d– and l-optical isomers. On peripheral sympathetic neurons, d– and l-amphetamine are equipotent, but in the CNS, the d-isomer is three to four times more potent than the l-isomer. Amphetamine is used therapeutically only for its central stimulant action (see Chapter 37), and only the d-isomer is used to minimize peripheral sympathomimetic actions. Amphetamine facilitates the release and blocks the reuptake of NE. However, the central stimulant actions of amphetamine appear to result from its ability to cause DA release (see Chapter 37). Cocaine is structurally distinct but has similar central stimulant and sympathomimetic actions. It also blocks the neuronal reuptake of NE and DA; however, unlike amphetamine, it does not facilitate neurotransmitter release.
Phentolamine is a prototypical competitive antagonist at α receptors, meaning that blockade can be surmounted by increasing agonist concentration (Fig. 11-9). Phenoxybenzamine, on the other hand, binds covalently to α receptors and produces an irreversible blockade that cannot be overcome by addition of more agonist (see Fig. 11-9).
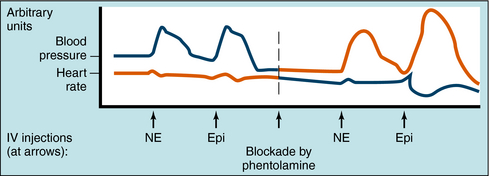
The effects of α receptor blockade on mean blood pressure and heart rate in response to NE and Epi are illustrated in Figure 11-10. The intravenous injection of NE and Epi produces a brief increase in blood pressure by activating α1 receptors in blood vessels. A reflex decrease in sympathetic and increase in vagal tone to the heart occur (see Fig. 11-5), but reflex bradycardia may be masked by direct activation of cardiac β1 receptors. In the case of NE, bradycardia is often seen because systolic and diastolic pressure both increase. Because of such complex and opposing actions, the effects of these compounds on heart rate can vary. Blockade of α receptors with phentolamine lowers blood pressure but is accompanied by a reflex increase in heart rate. NE now has little effect on blood pressure and does not cause reflex bradycardia, whereas heart rate is increased by stimulation of cardiac β1 receptors. When Epi is administered, the former pressor response is converted to a strong depressor response because vasodilatation resulting from β2 receptor activation is unmasked. Epi now causes pronounced tachycardia by direct activation of cardiac β1 receptors as well as a reflex tachycardia after the blood pressure decrease. Thus, when α receptors are blocked, the actions of Epi resemble those of the pure β receptor agonist ISO.
The tachycardia that occurs after administration of α receptor antagonists is attributable in part to blockade of presynaptic α2 receptors on sympathetic neurons (Fig. 11-11). Activation of these receptors inhibits NE release, and this feedback inhibition is disrupted when α2 receptors are blocked. This has little consequence when the postsynaptic receptors are α1, because the α receptor antagonists mentioned also block these receptors. In the heart, however, the postsynaptic receptors are β1. Thus effects of sympathetic activation are enhanced when α2 receptors are blocked, increasing NE release and enhancing reflex tachycardia (see Fig. 11-11, B).
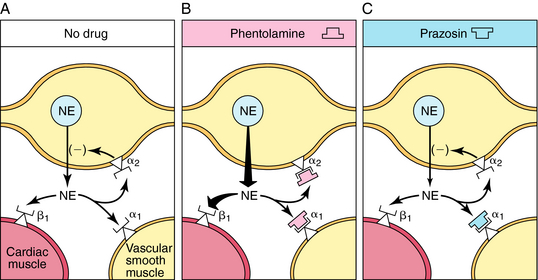
Prazosin, terazosin, doxazosin, tamsulosin, and alfuzosin are selective α1 receptor antagonists with similar pharmacological profiles but some differences in pharmacokinetics. By blocking α1 receptors in arterioles and veins, these drugs reduce peripheral vascular resistance and lower blood pressure; accordingly, they are used in treating hypertension. Selective α1 receptor antagonists cause less tachycardia than nonselective α receptor antagonists, because the α2 receptors, which reduce NE release, are not blocked by these drugs (see Fig. 11-11, C). Adrenergic α1 receptor antagonists also relax smooth muscle in the prostate and bladder neck and thereby relieve urinary retention in benign prostatic hyperplasia. Tamsulosin, terazosin, and alfuzosin are commonly prescribed for this condition.
Propranolol is the prototypical nonselective β receptor antagonist. Newer compounds differ primarily in their duration of action and subtype selectivity. Propranolol is a competitive antagonist at both β1 and β2 receptors. Like all adrenergic receptor blocking drugs, its pharmacological effects depend on the activity of the sympatho-adrenal system. When impulse traffic in sympathetic neurons and circulating concentrations of NE and Epi are high (e.g., during exercise), the effects of the drugs are more pronounced. The most profound effects of propranolol are on the cardiovascular system. Propranolol blocks the positive chronotropic and inotropic responses to β receptor agonists and to sympathetic activation. It reduces the rate and contractility of the heart at rest, but these effects are more dramatic during exercise. The drug may precipitate acute failure in an uncompensated heart. Propranolol, but not some other β receptor antagonists, also has a direct membrane-stabilizing action (local anesthetic action), which may contribute to its cardiac antiarrhythmic effect.
Drugs that Reduce Central Sympathetic Outflow
The activity of peripheral sympathetic neurons is regulated in a complex manner by neuronal systems located in the lower brainstem. These central neurons, in turn, are regulated in part by α2 receptors. Clonidine is the prototype α2 receptor agonist. It is lipid soluble and penetrates the blood-brain barrier to activate α2 receptors in the medulla, resulting in diminished sympathetic outflow. Clonidine lowers blood pressure by reducing total peripheral resistance, heart rate, and cardiac output. It does not interfere with baroreceptor reflexes and does not produce noticeable orthostatic hypotension. Accordingly, clonidine lowers blood pressure in patients with moderate to severe hypertension and produces less orthostatic hypotension than drugs acting in the periphery. However, side effects of clonidine may include dry mouth, sedation, dizziness, nightmares, anxiety, and mental depression. Various signs and symptoms related to sympathetic nervous system overactivity (hypertension, tachycardia, sweating) may occur after withdrawal of long-term clonidine therapy; thus the dose of clonidine should be reduced gradually. Clonidine is also used to ameliorate signs and symptoms associated with increased activity of the sympathetic nervous system that accompany withdrawal from long-term opioid use.
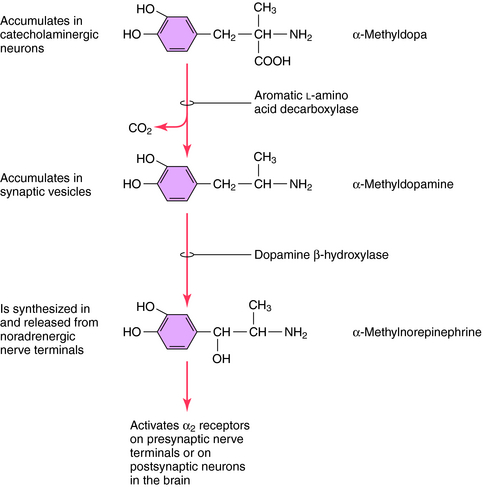
Methyldopa is an analog of the catecholamine precursor l-DOPA that is transported into noradrenergic neurons, where it is converted to α-methyl-NE (Fig. 11-12). α-Methyl-NE partially displaces NE in synaptic vesicles, where it is released in place of NE. This compound selectively activates α2 receptors to cause hypotensive actions, which appear to be attributable to α-methyl-NE in the brain not in the periphery.
Drugs that Inhibit Catecholamine Synthesis
Carbidopa, a hydrazine derivative of methyldopa, inhibits ALAAD. Unlike methyldopa, carbidopa does not penetrate the blood-brain barrier and therefore has no effect in the CNS. Because ALAAD is ubiquitous, is present in excess, and does not control the rate-limiting step in catecholamine synthesis, clinical doses of carbidopa have no appreciable effect on the endogenous synthesis of NE in sympathetic neurons. They do, however, reduce the conversion of exogenously administered l-DOPA to DA outside the brain, rendering carbidopa useful for the adjunctive treatment of Parkinson’s disease (see Chapter 28).
Schaak S, Mialet-Perez J, Flordellis C, Paris H. Genetic variation of human adrenergic receptors: From molecular and functional properties to clinical and pharmacogenetic implications. Curr Top Med Chem. 2007;7:217-231.
Shin J, Johnson JA. Pharmacogenetics of beta-blockers. Pharmacotherapy. 2007;27:874-887.
[/level-membership-for-basic-science-category][not-level-membership-for-basic-science-category]
Chapter 11 Drugs Affecting the Sympathetic Nervous System
| Therapeutic Overview |
|---|
| Mpathomimetics |
| Nasal decongestion |
| Decrease formation of aqueous humor in glaucoma |
| Neurogenic shock, cardiogenic shock |
| Paroxysmal atrial tachycardia |
| Bronchospasm, asthma |
| Decrease diffusion of local anesthetics |
| Sympatholytics |
| Essential hypertension, hypertensive emergencies |
| Benign prostatic hyperplasia |
| Cardiac dysrhythmias, angina pectoris, postmyocardial infarction |
| Essential tremor |
| Glaucoma |
Therapeutic Overview
The Therapeutic Overview Box presents a summary of the primary uses of different classes of compounds that affect the sympathetic nervous system.
Mechanisms of Action
The biochemistry and physiology of the autonomic nervous system, including a discussion of adrenergic receptors, is presented in Chapter 9. Detailed information on receptors and signaling pathways involved are in Chapter 1. This section covers topics that pertain specifically to noradrenergic neurotransmission and its modulation by drugs.
The neurochemical steps that mediate noradrenergic neurotransmission are summarized in Figure 11-1. The first step involves the transport of tyrosine into the neuron followed by its conversion to 4-dihydroxy-phenylalanine (l-DOPA) by the rate-limiting enzyme tyrosine hydroxylase. l-DOPA is rapidly decarboxylated to dopamine (DA) by aromatic l-amino acid decarboxylase (ALAAD), also known as DOPA decarboxylase. In dopaminergic neurons within the central nervous system (CNS), this is the last step in the synthetic process, and DA is released as a neurotransmitter. Noradrenergic neurons in the CNS and peripheral sympathetic nervous system contain an additional enzyme, DA β-hydroxylase, which converts DA to NE. This enzyme is located in synaptic vesicles, so DA must be actively transported into these vesicles before conversion to NE.
When the action potential depolarizes the nerve terminal, voltage-gated Ca++ channels open, allowing Ca++ to enter the neuron and cause the vesicles to release NE. At the neuroeffector junction, NE binds to adrenergic receptor subtypes on both the presynaptic and postsynaptic membranes, with different organs containing different receptor subtypes. Activation of α2-“autoreceptors” on the presynaptic neuronal membrane causes feedback inhibition and reduces further NE release. The signaling mechanisms occurring after adrenergic receptor activation are discussed in Chapter 9.
After NE has activated its receptors, its action is terminated primarily by a high-affinity reuptake system (termed “uptake 1” in Fig. 11-1), which transports NE back into the neuron for repackaging and eventual re-release. A smaller fraction of the NE in the synapse diffuses away from the receptors and can be taken up by a lower affinity extraneuronal process (termed “uptake 2” in Fig. 11-1). Some of the NE taken back into the sympathetic neurons by reuptake can be oxidatively deaminated by monoamine oxidase (MAO) located on the external mitochondrial membrane. The biologically inactive deaminated metabolites enter the circulation and are excreted in the urine. The NE that is transported into the postjunctional cell is O-methylated by catechol-O-methyltransferase (COMT) to normetanephrine. The high-affinity reuptake of NE into presynaptic terminals is the major mechanism that terminates its actions, although COMT and MAO also play important roles in metabolizing circulating NE and Epi and some exogenously administered sympathomimetic amines.
Drugs modify the activity of sympathetic neurons by increasing or decreasing the noradrenergic signal (see Fig. 11-1). Sympathomimetics may mimic noradrenergic transmission by acting directly on postsynaptic adrenergic receptors (e.g., phenylephrine), by facilitating NE release (e.g., amphetamine) or by blocking neuronal reuptake (e.g., cocaine). Amphetamine and cocaine have intense effects on the CNS and are drugs of abuse with severe addiction liability (Chapter 37).
Activation of Adrenergic Receptors
Over the last half century it has become clear that the actions of NE and Epi are mediated through multiple adrenergic receptor subtypes (see Chapter 9). We now know that there are nine different subtypes of adrenergic receptors, each encoded by separate genes, which are grouped into three major families (α1, α2, β), each containing three different members. Of these, only four (α1, α2, β1, and β2) are currently important in clinical pharmacology; therefore they are the major focus of this chapter. Both NE and Epi activate most adrenergic receptors with similar, but not identical, potencies, although NE is much less potent than Epi at the β2-subtype. Isoproterenol (ISO) is a synthetic analog of NE and Epi and selectively activates only β receptors. Differences in the pharmacological profiles of different adrenergic receptors are illustrated in Figure 11-2, where dose-response curves for the actions of these catecholamines on different tissues are depicted.
Adrenergic receptor activation increases the contraction of cardiac muscle and induces smooth muscle to either contract or relax, depending on the receptor subtype(s) present. Contraction of arterial strips is mediated by α1 receptors, where the relative potencies of the three catecholamines are Epi ≥ NE >>> ISO. Relaxation of bronchial smooth muscle is mediated by β2 receptors, with the relative potencies being ISO > Epi >>> NE. Contraction of cardiac muscle is mediated by β1 receptors, with the relative potencies being ISO > Epi = NE. The presence of different receptor subtypes in these three tissues is further demonstrated in Figure 11-3 by examining the effects of selective antagonists. Phentolamine, a competitive antagonist at α1 receptors, causes a parallel shift to the right of NE-induced contractions of arterial strips (see Fig. 11-3, A) but does not affect the other two responses. Propranolol, a competitive antagonist at both β1 and β2 receptors, causes a parallel shift to the right of responses mediated by bronchial β2 receptors (see Fig. 11-3, B) and cardiac β1 receptors (see Fig. 11-3, C), without affecting the α1 receptor response.
Adrenergic α2 receptors are found on platelets, in the CNS, and postsynaptically in several other peripheral tissues (blood vessels, pancreas, and enteric cholinergic neurons). As mentioned, activation of α2 receptors on the terminals of sympathetic neurons reduces NE release. These receptors are also found both presynaptically and postsynaptically on neurons in brain, and activation of these receptors reduces central sympathetic outflow.
Direct-Acting Sympathomimetics
Direct-acting adrenergic receptor agonists mimic some of the effects of sympathetic nervous system activation by binding to and activating specific receptor subtypes (see Fig. 11-1, site 3). For example, as mentioned, ISO selectively activates β receptors, phenylephrine selectively activates α1 receptors, and clonidine selectively activates α2 receptors. Similarly, dobutamine and albuterol are relatively specific agonists at β1 and β2 receptors, respectively. The structures of some clinically important adrenergic receptor agonists are shown in Figure 11-4.
Indirect-Acting Sympathomimetics
Indirect-acting sympathomimetics do not activate receptors directly but facilitate the release of NE from sympathetic neurons or block high-affinity reuptake. Thus they act presynaptically to facilitate adrenergic neurotransmission. Amphetamine and related drugs produce their sympathomimetic effects by facilitating NE release (see Fig. 11-1, site 1); the effects of these drugs in the CNS are described in Chapter 37. On the other hand, cocaine and tricyclic antidepressants, such as desipramine, exert their sympathomimetic effects by blocking high-affinity reuptake (see Fig. 11-1, site 2). The structure and characteristics of cocaine are discussed in Chapter 37, with similar information for tricyclic antidepressants given in Chapter 30.
Because neuronal reuptake, and not metabolism, is the primary mechanism for terminating the actions of NE and Epi, it is not surprising that drugs that inhibit the metabolism of these amines show little or no sympathomimetic actions. On the other hand, inhibitors of MAO (e.g., pargyline) or COMT (e.g., tolcapone) can enhance the actions of other synthetic sympathomimetic amines that are substrates for these enzymes, with some important toxicological implications. For example, the actions of tyramine, a sympathomimetic amine present in a variety of foods, are greatly enhanced in patients treated with MAO inhibitors (see Chapter 30), and the pharmacokinetics of l-DOPA, used for the treatment of Parkinson’s disease, are increased when administered concurrently with a COMT inhibitor (see Chapter 28).
An indirect-acting sympathomimetic amine that releases NE must penetrate the noradrenergic neuron before it can act. Nonpolar, lipid-soluble drugs such as amphetamine can diffuse across neuronal membranes, whereas polar, water-soluble compounds such as tyramine must rely on high-affinity uptake by the NE transporter. Thus drugs that inhibit the NE transporter will reduce or block the effects of tyramine but will enhance the effects of Epi, which acts directly on the adrenergic receptors. Cocaine also causes a prompt increase in the response to Epi by blocking the reuptake system, whereas reserpine disrupts amine transport from the cytoplasm into synaptic vesicles (see Fig. 11-1, site 5). Therefore reserpine has only small effects on responses to direct-acting sympathomimetics.
Inhibition of Synthesis, Storage, or Release of NE
Catecholamine synthesis can be disrupted at several steps, but effective in vivo blockade is obtained only when tyrosine hydroxylase, the enzyme catalyzing the first and rate-limiting step, is inhibited (see Fig. 11-1, site 4a). This can be produced clinically with α-methyltyrosine (metyrosine). Several compounds can inhibit other biosynthetic enzymes (e.g., DOPA decarboxylase is inhibited by carbidopa and DA β-hydroxylase is inhibited by disulfiram (see Fig. 11-1, sites 4b and 4c, respectively). Although these drugs do not effectively block endogenous catecholamine synthesis when administered, they do have clinical utility by affecting other systems. By virtue of its ability to inhibit peripheral DOPA decarboxylase, carbidopa is used in combination with l-DOPA for patients with Parkinson’s disease, to increase the amount of l-DOPA available to the CNS to enhance DA synthesis (see Chapter 28). Disulfiram is used in treating chronic alcoholism (see Chapter 32) because it blocks aldehyde dehydrogenase; however, its ability to inhibit catecholamine synthesis leads to significant adverse effects such as hypotension.
Disruption of vesicular storage also modifies noradrenergic transmission (see Fig. 11-1, site 5). As discussed, reserpine disrupts the ability of the synaptic vesicles to transport and store DA and NE. Adrenergic responses can also be impeded by inhibiting NE release (see Fig. 11-1, site 6). Drugs such as bretylium and guanethidine, which accumulate in noradrenergic nerve terminals, prevent NE release.
Adrenergic Receptor Antagonists
Many adrenergic receptor antagonists exert different subtype-selective effects (see Fig. 11-1, site 7). The early antagonists such as phenoxybenzamine and propranolol blocked α or β receptors, respectively, whereas drugs are now available that selectively block α1 (prazosin), α2 (yohimbine), or β1 (metoprolol) receptors. These selective antagonists have important therapeutic advantages over the original broad-spectrum adrenergic receptor blockers and are used for various indications, such as the control of blood pressure (see Chapter 20).
