Chapter 18 Drugs Affecting the Gastrointestinal System
| Abbreviations | |
|---|---|
| 5-ASA | 5-Aminosalicylic acid |
| 5-HT | 5-Hydroxytryptamine (serotonin) |
| ACh | Acetylcholine |
| AChE | Acetylcholine Sterase |
| CB | Cannabinoid |
| CNS | Central nervous system |
| COX | Cyclooxygenase |
| CTZ | Chemoreceptor trigger zone |
| CYP | Cytochrome P450 |
| DA | Dopamine |
| GERD | Gastroesophageal reflux disease |
| GI | Gastrointestinal |
| H. pylori | Helicobacter pylori |
| IBD | Inflammatory bowel disease |
| M | Muscarinic |
| IBS | Irritable bowel syndrome |
| NK | Neurokinin (substance P) |
| NSAID | Nonsteroidal antiinflammatory drug |
| PPIs | Proton pump inhibitors |
| PUD | Peptic ulcer disease |
| TNF | Tumor necrosis factor |
Therapeutic Overview
The gastrointestinal (GI) tract stores, digests, and absorbs nutrients and eliminates wastes. Regulation of the GI organs is mediated by intrinsic nerves of the enteric nervous system, neural activity in the central nervous system (CNS), and an array of hormones. These processes are summarized in Figure 18-1.
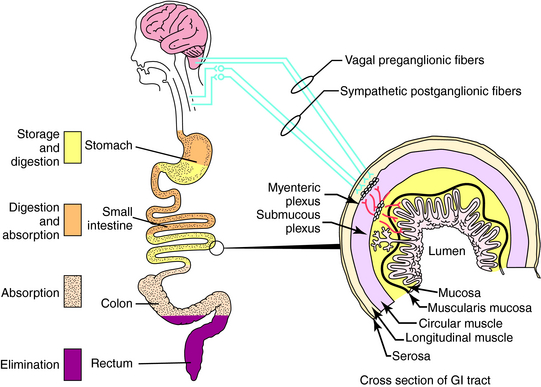
Pharmacologically treatable GI disorders include:
H. pylori infection produces inflammatory changes in the mucosa, impairs mucosal defense mechanisms (barrier function), and increases acid secretion. Although histamine H2 receptor antagonists, proton pump inhibitors (PPIs), and sucralfate heal peptic ulcers in H. pylori-positive patients, there is a high rate of ulcer recurrence upon discontinuing drug treatment. Continuous low-dose maintenance therapy reduces the risk of ulcer recurrence but does not cure the disease, because the organism has not been eliminated. Eradication of H. pylori cures the disease and in most patients eliminates the need for continuous antisecretory maintenance therapy.
Nonselective NSAIDs, including aspirin, damage the gastric mucosa by a direct topical effect or by systemic inhibition of endogenous mucosal prostaglandin synthesis. The initial topical injury is caused by the acidic property of the NSAIDs, but inhibition of protective prostaglandins is the primary cause of the ulcer. Nonselective NSAIDs inhibit cyclooxygenase (COX), which is the rate-limiting enzyme in the conversion of arachidonic acid to GI mucosal prostaglandins (see Chapter 15). Two forms of COX exist:
Nonselective NSAIDs inhibit both COX-1 and COX-2 to varying degrees. Selective COX-2 inhibitors such as celecoxib were thought to spare the protective prostaglandins and decrease the incidence of adverse GI effects, but recent evidence has questioned this idea, because COX-2 inhibitors have been observed to increase the risk of adverse cardiac events (see Chapter 36). The concomitant use of a PPI or misoprostol with a nonselective NSAID can reduce the risk of ulcers.
Diarrhea results from the presence of excessive fluid in the intestinal lumen, generating rapid, high-volume flow that overwhelms the absorptive capacity of the colon. In most diarrheas, fluid and electrolyte absorption occur at an essentially normal rate. However, diarrhea increases fluid secretion into the lumen at a rate that exceeds its absorptive capacity, thus leading to a net accumulation of luminal fluid. Diarrhea can be acute, secondary to an enteric bacterial or viral infection, or chronic, secondary to inflammatory or functional bowel disease. The most effective way to manage diarrhea is to eliminate the infection, remove the secretagogue-producing tumor, or cure the inflammation. The major hazard associated with diarrhea is loss of fluid and electrolytes. Serious sequelae of diarrhea can generally be prevented by replacement of fluid and electrolytes. However, many patients with serious acute or chronic diarrhea require antidiarrheal therapy. Diarrhea is also common with parasitic infections (see Chapter 52).
IBD describes nonspecific inflammatory disorders of the GI tract, including ulcerative colitis and Crohn’s disease, characterized by recurrent acute inflammatory episodes of diarrhea, abdominal pain, and GI bleeding. Although the exact causes of these disorders remain unknown, both appear to be immunologically mediated and influenced by genetics and environment. Treatment includes antidiarrheals, antispasmodics, and analgesics; aminosalicylates; and glucocorticoids, antibiotics, and immunomodulators (particularly azathioprine and 6-mercaptopurine as well as methotrexate, cyclosporine, and tacrolimus). The aminosalicylates have been the cornerstone of drug therapy. Infliximab, a monoclonal antibody approved for the treatment of Crohn’s disease (see Chapter 6), targets tumor necrosis factor α (TNF-α). A single infusion of infliximab induces significant improvement in patients with Crohn’s disease, and drug effects may persist for up to 12 weeks; however, long-term efficacy and safety have not been established.
| Therapeutic Overview | |
|---|---|
| Problem | Treatment |
| Peptic ulcer disease | H2 receptor antagonists, PPIs, sucralfate, misoprostol, antibiotics to eradicate Helicobacter pylori |
| Gastroesophageal reflux disease | Antacids, H2 receptor antagonists, PPIs |
| Delayed gastric emptying | Promotility agents |
| Constipation | Laxatives |
| Diarrhea | Antidiarrheals |
| Emesis | Antiemetics |
| IBS | 5-HT3 receptor antagonists |
| IBD | Aminosalicylates, immunosuppressants, TNF-α antibodies |
Mechanisms of Action
Antisecretory Drugs, Antacids, Mucosal Protectants, and Prostaglandins
The secretion of gastric acid by gastric parietal cells is regulated by histamine, acetylcholine (ACh), and gastrin (Fig. 18-2). Psychic stimuli (sight and smell of food) and the presence of food in the mouth or stomach stimulate vagally mediated acid secretion, which results from the action of ACh on parietal and paracrine cells. ACh released from secretomotor terminals of the vagus nerve acts at muscarinic M1 receptors on paracrine cells to cause the release of histamine, which acts at parietal cell H2 receptors to stimulate acid secretion. ACh also acts directly at parietal cell M3 receptors to stimulate acid production (see Chapter 10). The presence of food in the stomach, which raises the antral pH, also causes gastrin to be released from gastrin-releasing cells of the antral mucosa. Circulating gastrin stimulates gastrin receptors on paracrine cells to cause the release of histamine and on gastrin receptors on parietal cells to stimulate acid production. Thus histamine release constitutes the major event in the stimulation of acid production by ACh and gastrin, and ACh and gastrin, in turn, also act directly on parietal cells to augment the actions of histamine. Histamine, released from the paracrine cells located near parietal cells in oxyntic glands, acts at parietal cell H2 receptors to activate the H+,K+-ATPase located at the luminal membrane. Stimulation of M3 and gastrin receptors on the parietal cell also activates this H+,K+-ATPase, which serves as the so-called proton pump that secretes H+ into the gastric lumen.
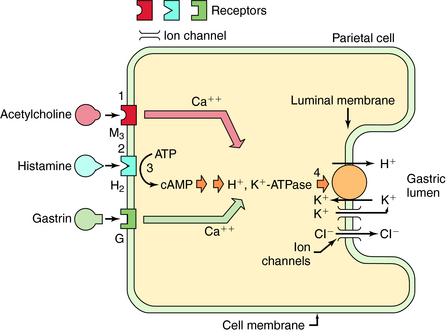
The only important role of peripheral H2 receptors in humans appears to be in the regulation of acid secretion. Drugs can decrease gastric secretion (see Fig. 18-2) by blocking H2 receptors, blocking M1 or M3 receptors, or by inhibiting the activity of the H+,K+-ATPase in the parietal cell.
The H2 receptor antagonists (cimetidine, famotidine, nizatidine, and ranitidine) block H2 receptors competitively and reversibly, diminishing basal, nocturnal, and food-stimulated gastric acid secretion (Table 18-1). Although relative antisecretory potencies vary from cimetidine, the least potent, to famotidine, the most potent, increased potency does not confer greater efficacy if the drugs are given in an equipotent antisecretory dose.
TABLE 18–1 Summary of Action of the Antisecretory Drugs, Antacids, Protectants, and Prostaglandins
| Category | Prototype | Mechanism of Action |
|---|---|---|
| Antacids | Magnesium oxide and magnesium hydroxide | Neutralize secreted acid |
| Anticholinergics | Propantheline | Block muscarinic receptors, decrease acid secretion |
| Bismuth salts | Bismuth subsalicylate | Topical antibacterial activity |
| H2 receptor antagonists | Cimetidine | Block H2 receptors, decrease acid secretion |
| Prostaglandins | Misoprostol | Inhibit mucosal prostaglandins, decrease acid secretion |
| Mucosal protectants | Sucralfate | Protect mucosal barrier |
| PPIs | Omeprazole | Inhibit H+, K+-ATPase, decrease acid secretion |
The PPIs, omeprazole, esomeprazole (the S-enantiomer of omeprazole), lansoprazole, pantoprazole, and rabeprazole, share a common mechanism of action to inhibit parietal cell H+,K+-ATPase irreversibly, decreasing basal, nocturnal, and food-stimulated gastric acid secretion. The parent drugs are inactive, but under highly acidic conditions in the parietal cell, they are protonated and converted to active compounds that react covalently with cysteine residues in the enzyme. This inactivates the pump and prevents the transport of H+ into the stomach lumen (see Fig.18-2). Because all secretory stimuli ultimately cause acid production by augmenting the activity of the H+,K+-ATPase-dependent transporter, irreversible blockade of this enzyme inhibits the final step and is the most effective way to diminish acid secretion.
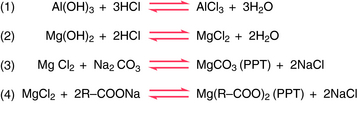
Antacids are weak bases that act primarily by neutralizing intragastric hydrochloric acid. They do not decrease acid secretion. The cations (Na+, Ca++, Mg++, and Al+++) initially form soluble chloride salts (Fig. 18-3). Although NaCl can be absorbed from the small intestine, the divalent ions form poorly soluble bicarbonates and carbonates, which precipitate and remain in the bowel to be excreted in the feces. The acid-neutralizing effects in the stomach lumen decrease total acid load to the duodenum and inhibit pepsin activity at an intragastric pH of 5 or above. Antacids also bind bile salts, and aluminum-containing antacids may enhance gastric cytoprotection.
In parietal cells, many prostaglandins (see Chapter 15) inhibit histamine-stimulated acid secretion. Misoprostol, a synthetic prostaglandin E1 analog, modestly inhibits the concentration and total amount of acid in the gastric lumen, resulting in a reduction of basal, nocturnal, and food-stimulated acid secretion. Misoprostol also increases mucus, mucosal bicarbonate secretion, and mucosal blood flow and inhibits mucosal cell turnover, all of which enhance mucosal defense.
Administration of single antimicrobial agents is not effective in eradicating H. pylori, but a combination of antibiotics and an antisecretory drug is effective. A typical regimen for eradication of H. pylori includes two antibiotics (usually clarithromycin and amoxicillin or metronidazole) and an antisecretory drug (usually a PPI). Other regimens include bismuth subsalicylate, metronidazole, tetracycline, and either a PPI or an H2 receptor antagonist (Table 18-2). These agents can be given together or sequentially with oral probiotics to reduce adverse effects. Probiotics contain live nonpathogenic bacteria, including bifidobacteria and lactobacilli, in nonprescription products because these bacteria are found normally in the GI tract and are proposed to be beneficial in several GI disorders. The mechanism of the therapeutic effects are unknown but are proposed to improve digestive process and reduce growth of pathogenic bacteria. Probiotics induce minimal adverse effects (gas and bloating) except in immunosuppressed patients.
TABLE 18–2 Drugs Used for Eradication of H. Pylori–Associated Ulcers
| Therapeutic Category | Drug Choices |
|---|---|
| Antisecretory | PPI or H2 receptor antagonist |
| Bismuth salt | Bismuth subsalicylate |
| Nitroimidazole | Metronidazole |
| Antibiotic | Clarithromycin, amoxicillin, or tetracycline |
Promotility agents increase GI tract contractions and propulsion by increasing cholinergic stimuli at smooth muscle M3 receptors (Fig. 18-4). This can be accomplished by four different mechanisms, summarized in Table 18-3.
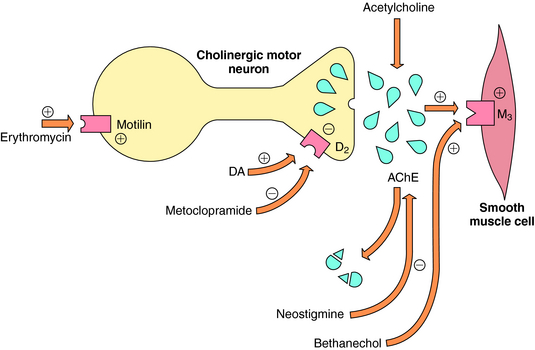

Cholinergic agonists and cholinesterase inhibitors increase ACh-mediated secretion at salivary, gastric, pancreatic, and intestinal secretory cells in addition to stimulation of smooth muscle cells (see Chapter 10). However, excessive secretory activity leads to significant side effects, and these drugs fail to produce closely coordinated contractions between the antrum of the stomach and the duodenum required for effective gastric emptying.
Motilin is a GI tract hormone that participates in the initiation of migrating motor complexes that characterize the fasting motility pattern of the stomach and small intestine. Macrolide antibiotics such as erythromycin (see Chapter 46) bind to nerve and muscle motilin receptors to enhance GI tract contractions and increase gastric emptying. This promotility effect is not related to the antimicrobial activity of these drugs.
Nausea and vomiting (emesis) result from a variety of causes, including adverse drug effects most prominently from anticancer drugs. The underlying cause of the emesis needs to be identified and treated with specific agents if they are available. The brainstem contains the CTZ located anatomically in the area postrema where the blood-brain barrier is essentially absent. Neurons in this brain area contain 5-HT3, D2, M1, and substance P or neurokinin (NK1) receptors. In addition, the vestibular nuclei of the brainstem also play a role in nausea, particularly motion sickness-induced nausea, and neurons in these sites express H1 and muscarinic and cannabinoid (CB) receptors. All of these receptors are targets for effective antiemetic compounds. Drugs useful as antiemetics that are discussed in other chapters in this text include D2 receptor antagonists such as prochlorperazine (Chapter 29), H1 receptor antagonists such as cyclizine (Chapter 14), muscarinic receptor antagonists such as scopolamine (Chapter 10), and less selective antiemetic agents including the antianxiety compounds such as diazepam (Chapter 31) and the glucocorticoids (Chapter 39). The pharmacokinetics and adverse effects of these agents are listed in the chapters covering these agents.
A summary of the action of laxatives is presented in Table 18-4.
| Category | Prototype | Mechanism of Action |
|---|---|---|
| Fiber supplements | Psyllium | Increases colonic residue, stimulating peristalsis |
| Emollients | Docusate Na+ | Lowers surface tension, allowing H2O to interact with stool |
| Lubricants | Mineral oil | Lubricates the stool |
| Hyperosmolar agents | Lactulose | Increases stool osmolarity |
| Saline laxatives | Magnesium hydroxide | Draws H2O into the intestine along osmotic gradient |
| Stimulant laxatives | Senna | Stimulates intestinal secretion and motility |
Stimulant laxatives include anthraquinones such as senna, diphenylmethanes such as bisacodyl, and castor oil. The anthraquinones are converted by colonic bacteria to their pharmacologically active form, which increases fluid accumulation in the distal ileum and colon. Bisacodyl has a similar action. Castor oil is hydrolyzed by lipase in the small intestine to ricinoleic acid, which increases intestinal secretion, decreases glucose absorption, and stimulates colonic motor function through the release of neurotransmitters from mucosal enterochromaffin cells.
Transport of fluid and electrolytes by the intestinal mucosa is regulated by neurons of the enteric nervous system and by the composition of the luminal contents. It is believed that neurons of the submucosal plexus of the intestine terminate near mucosal epithelial cells and act to increase or decrease absorption by villus cells and secretion by crypt cells. A summary of the action of antidiarrheal drugs is presented in Table 18-5.
| Category | Prototypes | Mechanism of Action |
|---|---|---|
| Opioids | Loperamide, diphenoxylate | Increase resistance to flow, decrease propulsion, decrease net fluid secretion |
| Antisecretory agents | Bismuth subsalicylate* | Decrease net fluid secretion |
| Gel-forming adsorbents | Hydrated aluminum silicate, pectin, kaolin | Increase resistance to flow, increase formed stools |
| Ion-exchange resins | Cholestyramine | Bind H2O and bile salts |
* This agent has multiple actions, including antibacterial effects.
Opioids act on enteric neurons to decrease secretion and promote mucosal transport from the lumen. In addition, opioids act in the CNS to alter extrinsic neural influences on the intestine and promote a net absorption of fluid and electrolytes in addition to their analgesic effects on the CNS (see Chapter 36). Opioids also convert propulsive patterns of motility to segmenting patterns, thereby increasing resistance to flow. These actions result in slowed transit through the GI tract, allowing time for more-complete fluid absorption and leading to an increased viscosity of the luminal content. Morphine and codeine cross the blood-brain barrier and act in the brain and spinal cord to decrease transit and fluid accumulation in the intestinal lumen. Loperamide and diphenoxylate do not cross the blood-brain barrier and act locally at neural and smooth muscle sites, primarily in the submucosal plexus, to increase segmenting contractions. The increased segmenting contractions in the proximal duodenum decrease the gastroduodenal pressure gradient and delay gastric emptying.
Pharmacokinetics
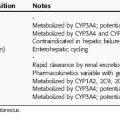
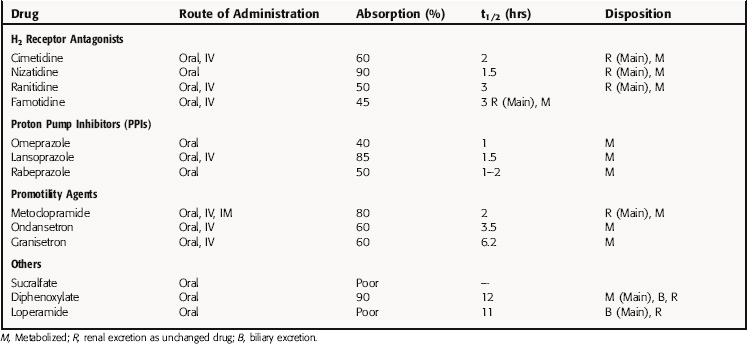
Pharmacokinetic parameters for selected drugs are given in Table 18-6.
Aprepitant is well absorbed orally, reaches maximum plasma concentrations within 4 hours, and is highly (95%) bound to plasma proteins. It undergoes extensive metabolism primarily by CYP3A4 and is also a weak-to moderate inducer of both CYP3A4 and CYP2C9. Aprepitant is eliminated by metabolism; it is not excreted via the kidneys.
Relationship of Mechanisms of Action to Clinical Response
Antisecretory Drugs, Antacids, Mucosal Protectants, and Prostaglandins
Treatment of H. pylori is susceptible to many antimicrobial agents in vitro, but it has proved difficult to eradicate the infection with single agents in humans. The PPI-based three-and four-drug regimens (see Table 18-2) are successful in approximately 80% to 90% of patients. H. pylori organisms have been shown to develop resistance to nitroimidazoles (e.g., metronidazole) and macrolides (e.g., clarithromycin), but resistance to tetracycline and amoxicillin is uncommon. Therefore eradication regimens should contain at least two antimicrobial agents.
Antacid neutralization provides almost immediate relief of symptoms, but large volumes and frequent dosing are necessary for mucosal healing. The neutralizing effects occur for as long as the antacids are present in the stomach. Because of their side effects, disagreeable taste, and poor compliance, antacids are not used as single agents to heal peptic ulcers or esophagitis. They are used primarily for the occasional relief of acid indigestion, epigastric pain, and heartburn.
The opioid antidiarrheal drugs are remarkably effective in the management of acute diarrhea. Those with CNS activity should be used cautiously for acute diarrhea and should not be used for the management of chronic diarrhea. The most effective antidiarrheal drugs are morphine, codeine, loperamide, and diphenoxylate. Morphine and codeine are highly addictive controlled substances (Chapter 37), whereas the synthetic agent loperamide, which is not a controlled substance and is available by prescription, is widely used and is effective for the control of diarrhea caused by IBS or IBD. Opioid antidiarrheal agents should not be used in the symptomatic treatment of diarrhea caused by enteric infections, especially those caused by Shigella or Salmonella.
Sulfasalazine and the newer aminosalicylate forms are effective in treating mild to moderate ulcerative colitis and Crohn’s disease. The forms that release 5-ASA in the small intestine are more likely to be effective in patients with ileal involvement. Symptomatic improvement in abdominal pain and diarrhea is seen in approximately 3 weeks. Lower daily dosages are effective in maintaining remission. Topical 5-ASA rectal enemas are effective in treating ulcerative proctitis and proctosigmoiditis.
Pharmacovigilance: Side Effects, Clinical Problems, and Toxicity
Cholinergic agonists produce a variety of side effects typically associated with cholinergic stimulation (see Chapter 10).
Metoclopramide can induce dystonia or parkinsonian side effects because of its activity as a DA receptor antagonist (see Chapter 28). DA receptor antagonists also can induce symptoms of hyperprolactinemia, consisting of gynecomastia, galactorrhea, and breast tenderness. Metoclopramide also often induces sedation.
As mentioned, cisapride and tegaserod produce severe cardiovascular adverse effects.
The cannabinoid dronabinol has effects on the CNS to increase sympathetic activity and may lead to tachycardia; orthostatic hypotension is not uncommon. Dose-related effects on appetite, mood, cognition, and memory have also been reported and appear to be highly individual-dependent. Dronabinol is a schedule III drug and has been shown to produce psychological and minor degree of physiological dependence (see Chapter 37).
The adverse effects of the natural (morphine and codeine) and synthetic (loperamide and diphenoxylate) opioids are discussed in Chapter 36. Diphenoxylate crosses the blood-brain barrier poorly under normal conditions and in usual therapeutic doses does not produce CNS side effects. However, in an overdose it can cause respiratory depression, which can be reversed by naloxone. Diphenoxylate is available in combination with atropine, the latter added to deter abuse. Loperamide traverses the blood-brain barrier poorly and therefore has virtually no CNS effects and a low abuse potential.