[level-membership-for-neonatal-and-perinatal-medicine-category]
13 Double-Outlet Right Ventricle
I. CASE
A. Fetal echocardiography findinges
1. The fetal echo reveals situs solitus of the atria, levocardia, left aortic arch, heart rate of 132 bpm, normal cardiac axis and position, and mild cardiomegaly (cardiothoracic ratio = 0.38).
2. The four-chamber view is abnormal, with a small posterior left ventricle.
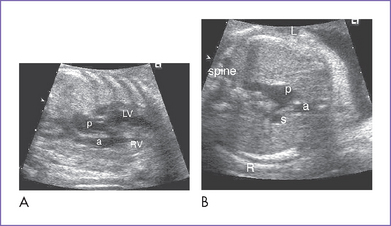
3. The outflow assessment reveals transposition of the great arteries (TGA) (aorta anterior and to the right of the pulmonary artery), mild asymmetry and the aorta slightly smaller than the pulmonary artery (Fig. 13-1).
4. The aortic valve is bicuspid, and the annulus is a normal size for gestation. The valve domes mildly in systole, with moderate flow acceleration through it by Doppler (1.5 m/s).
5. The main pulmonary artery is a good size and has confluent branch pulmonary arteries with normal Doppler velocity through them.
6. The aortic arch is leftward; however, the aortic arch is smaller than the ductal arch. Both arches have antegrade flow.
7. There is a large perimembranous ventricular septal defect (VSD) with outlet extension.
8. There is straddling of the mitral valve.
9. Flow through the foramen ovale is unrestricted, with a bidirectional shunt.
10. The pulmonary venous flow pattern and drainage are normal.
11. The Tei index (myocardial performance index) is normal in both ventricles.
D. Fetal management and counseling
1. Management: Diagnosis of any form of double-outlet right ventricle (DORV) should prompt referral for the following:
a. Thorough anatomic examination by ultrasound.
c. In this case, the abnormal maternal serum screen is an additional reason to ask for amniocentesis.
2. Follow-up includes serial antenatal studies at 4- to 6-week intervals.
a. Pulmonary and aortic outflow obstruction (bilateral outflow tract obstruction) is possible, with progressive hypoplasia of the aortic valve, ascending aorta, and arch and evolving coarctation of the aorta.
b. Check the size of the VSD (might become restrictive).
c. Monitor the direction of ductal flow.
d. Monitor ventricular size and function using the biventricular Tei index.
e. Watch for development of hydrops fetalis. It is unlikely unless there is biventricular dysfunction or significant atrioventricular (AV) valve or semilunar valve regurgitation.
F. Neonatal management
a. If there is progressive arch hypoplasia with or without aortic outflow obstruction, initiation of prostaglandin E1 (PGE1) infusion should be considered to keep the ductus open at least until a postnatal echocardiogram confirms or excludes important outflow tract and arch pathology.
b. In addition, given the potential for transposition physiology and streaming, PGE1 may be necessary until an atrial septostomy is performed to improve atrial and ventricular level mixing.
c. Administration of oxygen can increase oxygen saturation by decreasing pulmonary valve regurgitation and increasing blood flow.
d. At times, volume and inotropic support may be indicated to improve ventricular function and atrial level mixing.
2. Surgical: In this case, given the presence of a straddling mitral valve, single ventricle palliation is most likely in order.
b. Stage 2 is a bidirectional Glenn shunt at 4 to 6 months, following diagnostic cardiac catheterization.
c. Stage 3 is a modified Fontan procedure at 2 to 5 years of age.
II. YOUR HANDY REFERENCE
A. Prevalence
1. DORV accounts for less than 1% of all congenital heart disease.
2. Allan and Sharland (1992) studied a total group of 2136 fetuses with heart diseases diagnosed prenatally. These included 62 (3%) cases of DORV.
b. Survival: 17 babies survived (61% survival rate of continuing pregnancies).
3. In fetal life, DORV in the absence of ventricular hypoplasia is one of the less commonly identified lesions in utero. As in TGA, the four-chamber view may be normal, and most of these lesions go undetected at the time of obstetric ultrasound examination.
C. Associated syndromes and extracardiac anomalies
1. DORV can be associated with chromosomal abnormalities and is present in 12% of cases in one fetal series.
2. The tetralogy of Fallot (TOF) type of DORV may be associated with trisomies 13, 18, and 21 as well as with microdeletion of chromosome 22 (velocardiofacial or DiGeorge’s syndrome).
3. The TGA type of DORV has a lower risk of chromosomal abnormalities.
4. DORV associated with mitral atresia and normally related great arteries can be seen in trisomies 18 and 13.
5. Other extracardiac lesions have been described including ectopia cordis, omphalocele, diaphragmatic hernia, cleft lip and palate, renal abnormalities, tracheoesophageal fistula, and central nervous system abnormalities such as Dandy–Walker malformation.
D. Clues to fetal sonographic diagnosis
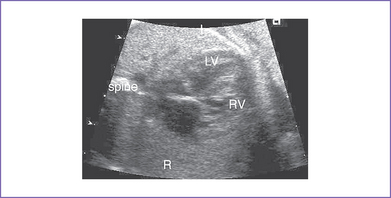
1. The four-chamber view may be normal unless there is a ventricular discrepancy, which occurs more often when there is additional intracardiac pathology (Fig. 13-2).
2. There is a subtle degree of great artery override and discontinuity between the mitral valve and either the aortic valve (in normally related great arteries) or the pulmonary valve (in TGA).
3. The great arteries may be in a transposed configuration (right or left) or normally related. The great arteries are often side by side.
4. There can be a discrepancy in the size of the aorta and the pulmonary artery in the case of either aortic or pulmonary valve stenosis. Normally, the pulmonary artery is slightly larger than the aorta.
5. In DORV in fetal life, if there is pulmonary or aortic obstruction, there is diversion of blood toward the unaffected outflow. Hence, Doppler velocities in the affected outflow tract are usually slightly elevated, with hardly any gradient.
6. Aortic arch hypoplasia and coarctation of the aorta can coexist with aortic outflow obstruction.
7. AV regurgitation may be present, particularly with an abnormality of the AV valve. Straddling of the mitral valve is usually associated with a cleft in the anterior leaflet. This may be a source of regurgitation.
8. If there is stenosis or hypoplasia of the left AV valve in situs solitus, foramen ovale and pulmonary venous flow must be assessed in predicting the potential for significant LA hypertension after birth.
9. Conversely, if the right-sided AV valve in situs solitus is smaller, assessment of the foramen ovale is important, given that foramen ovale restriction can result in hydrops fetalis.
10. Flow through the VSD in all forms of DORV is from LV to RV.
E. Progression in utero
Progression in DORV in the presence of an L-looped ventricle in situs solitus can include:
F. Immediate postnatal management
1. Medical management of patients without prenatal diagnosis of DORV and TGA.
a. Medical treatment follows the clinical picture. Often the infants have some degree of cyanosis.
b. If there is cyanosis (or if the hyperoxia test is positive), PGE1 infusion should be started to maintain pulmonary blood flow through the patent duct or, if there is arch or aortic outflow obstruction, to maintain the systemic circulation.
c. Occasionally, the systemic saturations (in the absence of a significant arterial duct) suggest that there is sufficient pulmonary blood flow. Such babies can be discharged from the hospital, can be followed closely, and can have surgical palliation or repair later in infancy.
2. Surgical management of patients without prenatal diagnosis of DORV and TGA.
a. Normally related great arteries with subaortic or doubly committed VSD and no outflow obstruction.
b. Infants with normally related great arteries with subaortic VSD are treated much like infants with TOF.
c. D-Transposed great arteries with a subpulmonary VSD.
d. If there is a remote VSD, single-ventricle palliation is the most likely course.
e. Surgical management of more complex forms of DORV is beyond the scope of this book (Box 13-1).
BOX 13-1 SURGICAL OPTIONS IN DOUBLE-OUTLET RIGHT VENTRICLE DEFECT
DORV WITH NORMALLY RELATED GREAT ARTERIES
VSD closure, baffling the LV outflow to the aorta and the RV outflow to the pulmonary artery.
DORV PLUS D-TRANSPOSED GREAT ARTERIES WITH A SUBPULMONARY VSD
Arterial switch operation with no or mild aortic or pulmonary outflow obstruction.
DORV, double-outlet right ventricle; LV, left ventricle; PS, pulmonary stenosis; RV, right ventricle; TGA, transposition of the great arteries; TOF, tetralogy of Fallot; VSD, ventricular septal defect.
G. Pathophysiology
1. Double-outlet right or left ventricle is a type of ventricular connection in which both great arteries arise from the morphological right or left ventricle. In defining the ventriculoarterial connections, a great artery is considered to be connected to a ventricle when more than half of its valve is committed to that ventricle.
2. Pathophysiology of DORV is determined primarily by the position of the VSD and the great artery relationship and the presence of outflow tract obstruction (Figs. 13-3 to 13-5).
a. With normally related great arteries without outflow obstruction and a subaortic VSD, oxygenated blood from the LV is directed to the aorta, and desaturated systemic venous blood is directed to the pulmonary artery, producing mild or no cyanosis. The pulmonary blood flow is increased in the absence of pulmonary stenosis, resulting in CHF.
b. With normally related great arteries and pulmonary outflow obstruction, the presentation is much like that of TOF, with varying degrees of desaturation depending upon the severity of pulmonary outflow obstruction.
c. When the great arteries are D-transposed, with a subpulmonary VSD, there is usually transposition physiology, with the highly saturated blood being directed mostly toward the pulmonary artery and the desaturated blood being directed toward the aorta. A balloon atrial septostomy might be necessary in this condition to resuscitate the infant and improve atrial-level mixing.
d. With the VSD close to both semilunar valves (doubly committed VSD) or remotely located from these valves (remote VSD), cyanosis of a mild degree is present and pulmonary blood flow is increased. Cyanosis is related to either transposition mixing or severe pulmonary stenosis.
e. When VSD is remote, the degree of desaturation is largely determined by the degree of pulmonary outflow obstruction. If there is restriction of the atrial septum, however, left atrial hypertension can result in cyanosis and pulmonary edema.
f. Rarely, the ventricular septum is intact.
g. In situs solitus, the aorta is usually located to the right of the pulmonary artery. The great arteries tend to lie side by side (Box 13-2). In the past, bilaterally present muscular infundibulum or conus (subpulmonary and subaortic) was thought to be the hallmark of DORV. This results in absence of the continuity of either arterial valve with either of the AV valves. This is not consistently present in DORV, and conversely, DORV is not necessarily present with bilateral conus.
h. There has been some debate about the relationship between TOF and DORV. The term tetralogy describes the infundibular morphology, and the term double-outlet right ventricle describes the type of ventriculoarterial connection. Thus, a case of tetralogy with more than 50% aortic override (>50% of the aortic orifice in the right ventricle) is unequivocally a DORV.
i. In DORV, the AV valves can be of normal and equal in size or can be discrepant, with one valve hypoplastic and stenotic or even atretic. In this scenario, the ventricle corresponding to the hypoplastic or atretic valve is hypoplastic: The right ventricle with the right (tricuspid) AV valve and the left ventricle with the left (mitral) AV valve. These additional lesions have a major impact on the surgical options.
j. DORV can also be associated with either left or right isomerism (heterotaxy syndromes) (see Section VI: Heterotaxy Syndromes).
k. Double-outlet left ventricle is extremely rare. The VSD is most commonly subaortic and less commonly subpulmonary. (Box 13-3).
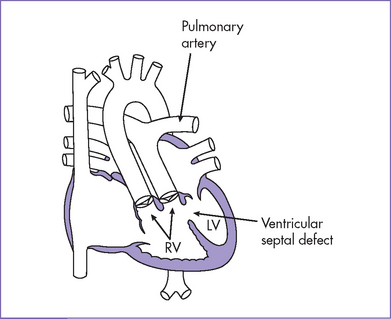
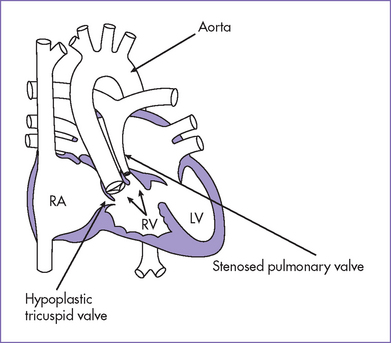
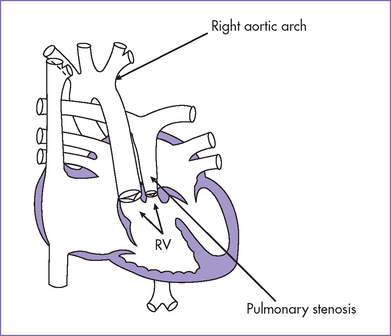
BOX 13-2 POSSIBLE ANATOMICAL ARRANGEMENTS OF GREAT ARTERIES IN DOUBLE-OUTLET RIGHT VENTRICLE (SITUS SOLITUS)
DORV, double-outlet right ventricle.
Subaortic VSD (usually perimembranous type)
Subpulmonary VSD (perimembranous or anterior malalignment type)
Doubly committed VSD (sitting under both semilunar valves)
Noncommitted VSD (remote in the muscular septum or in the inlet septum)
DORV, double-outlet right ventricle; PS, pulmonary stenosis; TGA, transposition of the great arteries; VSD, ventricular septal defect.
H. Risk of recurrence
1. Conotruncal heart malformations may be components of certain syndromes, such as DiGeorge’s syndrome, the velocardiofacial syndrome, and the genitopalatocardiac syndrome. Using DNA markers, Emanuel and colleagues (1992) found loss of heterozygosity indicating microdeletions of 22q11.2 in 30% of isolated conotruncal anomalies. These results were confirmed by FISH.
2. If the patient is positive for 22 q11.2 deletion, the parents should be tested to be certain neither has the deletion. If one parent has the deletion, the recurrence risk is 50% for each offspring, given the autosomal dominant inheritance. If neither parent has the deletion, the recurrence risk is fairly low at 2% to 3%.
3. This deletion can manifest with a variety of phenotypes: DiGeorge’s syndrome, Shprintzen’s syndrome, or velocardiofacial syndrome; conotruncal anomaly face syndrome (Takao syndrome); and isolated outflow tract defects of the heart including TOF, DORV, common arterial trunk, and interrupted aortic arch and other heart defects.
III. TAKE-HOME MESSAGE
A. Diagnosis
1. DORV can occur with any atrial situs or AV connection. It is often associated with heterotaxy syndrome, yet it occurs commonly with normal situs and concordant AV connection.
2. An abnormal heart can be identified in most fetuses with DORV. However, it may be difficult to distinguish this particular defect from other conotruncal abnormalities such as TOF or TGA, depending upon the great artery relationship.
3. Prenatal counseling for DORV in the fetus will be influenced by the complexity of cardiac lesions, the postnatal surgical options, and the presence of extracardiac lesions, including aneuploidy.
4. When one great artery is significantly smaller than the other in the three-vessel view, subaortic or subpulmonary obstruction can be suspected.
5. More subtle pathology that can importantly affect postnatal outcome can be found in this disease, which should also be considered in counseling of affected pregnancies.
B. Treatment
1. In the presence of significant chromosomal abnormalities—trisomy 18 and 13—or severe extracardiac lesions that would significantly influence the long-term outcome, aggressive perinatal management might not be appropriate or desired by the family, and the option of termination should be discussed.
2. Detailed evaluation of the right and left ventricular size, AV valve morphology, VSD size and location, great artery relationship and evidence of outflow obstruction, and evaluation of both arches is critical for providing accurate assessment regarding the most likely postnatal course.
Allan LD. Sonographic detection of parallel great arteries in the fetus. AJR Am J Roentgenol. 1997;168(5):1283-1286.
Allan LD, Sharland GK. Prognosis in fetal tetralogy of Fallot. Pediatr Cardiol. 1992;13(1):1-4.
Copel JA, Pilu G, Kleinman CS. Congenital heart disease and extracardiac anomalies: Associations and indications for fetal echocardiography. Am J Obstet Gynecol. 1986;154(5):1121-1132.
Emanuel BS, Budarf ML, Sellinger B, et al. Detection of microdeletions of 22q11.2 with fluorescence in situ hybridization (FISH): Diagnosis of DiGeorge syndrome (DGS), velo-cardio-facial (VCF) syndrome, CHARGE association and conotruncal cardiac malformations. Am J Hum Genet. 1992;51(suppl):A3.
Hornberger LK, Sahn DJ, Kleinman CS, et al. Antenatal diagnosis of coarctation of the aorta: A multicenter experience. J Am Coll Cardiol. 1994;23(2):417-423.
Huhta JC, Hagler DJ, Seward JB, et al. Two-dimensional echocardiographic assessment of dextrocardia: A segmental approach. Am J Cardiol. 1982;50(6):1351-1360.
Ozkutlu S, Elshershari H, Akcoren Z, et al. Visceroatrial situs solitus with atrioventricular alignment discordance double outlet right ventricle and superoinferior ventricles: Fetal and neonatal echocardiographic findings. J Am Soc Echocardiogr. 2002;15(7):749-752.
Patel CR, Muise KL, Redline RW. Double-outlet right ventricle with intact ventricular septum in a fetus with trisomy-18. Cardiol Young. 1999;9(4):419-422.
Smith RS, Comstock CH, Kirk JS, et al. Double-outlet right ventricle: An antenatal diagnostic dilemma. Ultrasound Obstet Gynecol. 1999;14(5):315-319.
Tennstedt C, Chaoui R, Korner H, Dietel M. Spectrum of congenital heart defects and extracardiac malformations associated with chromosomal abnormalities: Results of a seven year necropsy study. Heart. 1999;82(1):34-39.
[/level-membership-for-neonatal-and-perinatal-medicine-category][not-level-membership-for-neonatal-and-perinatal-medicine-category]
13 Double-Outlet Right Ventricle
I. CASE
A. Fetal echocardiography findinges
1. The fetal echo reveals situs solitus of the atria, levocardia, left aortic arch, heart rate of 132 bpm, normal cardiac axis and position, and mild cardiomegaly (cardiothoracic ratio = 0.38).
2. The four-chamber view is abnormal, with a small posterior left ventricle.
3. The outflow assessment reveals transposition of the great arteries (TGA) (aorta anterior and to the right of the pulmonary artery), mild asymmetry and the aorta slightly smaller than the pulmonary artery (Fig. 13-1).
4. The aortic valve is bicuspid, and the annulus is a normal size for gestation. The valve domes mildly in systole, with moderate flow acceleration through it by Doppler (1.5 m/s).
5. The main pulmonary artery is a good size and has confluent branch pulmonary arteries with normal Doppler velocity through them.
6. The aortic arch is leftward; however, the aortic arch is smaller than the ductal arch. Both arches have antegrade flow.
7. There is a large perimembranous ventricular septal defect (VSD) with outlet extension.
8. There is straddling of the mitral valve.
9. Flow through the foramen ovale is unrestricted, with a bidirectional shunt.
10. The pulmonary venous flow pattern and drainage are normal.
11. The Tei index (myocardial performance index) is normal in both ventricles.
D. Fetal management and counseling
1. Management: Diagnosis of any form of double-outlet right ventricle (DORV) should prompt referral for the following:
a. Thorough anatomic examination by ultrasound.
c. In this case, the abnormal maternal serum screen is an additional reason to ask for amniocentesis.
2. Follow-up includes serial antenatal studies at 4- to 6-week intervals.
a. Pulmonary and aortic outflow obstruction (bilateral outflow tract obstruction) is possible, with progressive hypoplasia of the aortic valve, ascending aorta, and arch and evolving coarctation of the aorta.
b. Check the size of the VSD (might become restrictive).
c. Monitor the direction of ductal flow.
d. Monitor ventricular size and function using the biventricular Tei index.
e. Watch for development of hydrops fetalis. It is unlikely unless there is biventricular dysfunction or significant atrioventricular (AV) valve or semilunar valve regurgitation.
F. Neonatal management
a. If there is progressive arch hypoplasia with or without aortic outflow obstruction, initiation of prostaglandin E1 (PGE1) infusion should be considered to keep the ductus open at least until a postnatal echocardiogram confirms or excludes important outflow tract and arch pathology.
b. In addition, given the potential for transposition physiology and streaming, PGE1 may be necessary until an atrial septostomy is performed to improve atrial and ventricular level mixing.
c. Administration of oxygen can increase oxygen saturation by decreasing pulmonary valve regurgitation and increasing blood flow.
d. At times, volume and inotropic support may be indicated to improve ventricular function and atrial level mixing.
2. Surgical: In this case, given the presence of a straddling mitral valve, single ventricle palliation is most likely in order.
b. Stage 2 is a bidirectional Glenn shunt at 4 to 6 months, following diagnostic cardiac catheterization.
c. Stage 3 is a modified Fontan procedure at 2 to 5 years of age.
II. YOUR HANDY REFERENCE
A. Prevalence
1. DORV accounts for less than 1% of all congenital heart disease.
2. Allan and Sharland (1992) studied a total group of 2136 fetuses with heart diseases diagnosed prenatally. These included 62 (3%) cases of DORV.
b. Survival: 17 babies survived (61% survival rate of continuing pregnancies).
3. In fetal life, DORV in the absence of ventricular hypoplasia is one of the less commonly identified lesions in utero. As in TGA, the four-chamber view may be normal, and most of these lesions go undetected at the time of obstetric ultrasound examination.
C. Associated syndromes and extracardiac anomalies
1. DORV can be associated with chromosomal abnormalities and is present in 12% of cases in one fetal series.
2. The tetralogy of Fallot (TOF) type of DORV may be associated with trisomies 13, 18, and 21 as well as with microdeletion of chromosome 22 (velocardiofacial or DiGeorge’s syndrome).
3. The TGA type of DORV has a lower risk of chromosomal abnormalities.
4. DORV associated with mitral atresia and normally related great arteries can be seen in trisomies 18 and 13.
[/not-level-membership-for-neonatal-and-perinatal-medicine-category]
