[level-membership-for-hematology-oncology-and-palliative-medicine-category]
There is also considerable redundancy in the function of the DNA repair pathways. When one pathway is disrupted, another pathway can partially compensate, especially if the second pathway is upregulated. For instance, a cell that is deficient in HR repair may depend more on the error-prone NHEJ repair pathway for the repair of double-strand breaks. Also, thymine dimers, which are generated by UV light exposure, can be repaired by NER repair or bypassed and effectively ignored by TLS polymerases. In some cases, the absence of one DNA repair pathway results in a hyperdependence on one or more other DNA repair pathways.
11,12 This so-called synthetic lethality among DNA repair pathways has important implications for the design of new anticancer drugs (see later discussion).
The six DNA repair pathways are not constitutively activated, but instead are highly regulated. The pathways are often activated at discrete times in the cell cycle. For instance, HR repair and TLS repair are active during the S phase of the cell cycle. Also, the DNA repair pathways are differentially active in various tissues and cell types. For instance, HR and TLS are more active in rapidly growing cells, such as hematopoietic cells, whereas NHEJ is more active in postreplicative cells. Accordingly, absence of a particular DNA repair pathway may be particularly disruptive to the growth and survival of some normal tissues and some cancers. Here is a brief description of the six DNA repair pathways, with an emphasis on the enzymes in the pathways and the preference for DNA lesions repaired.
Base-Excision Repair
BER has been reviewed by Wilson.
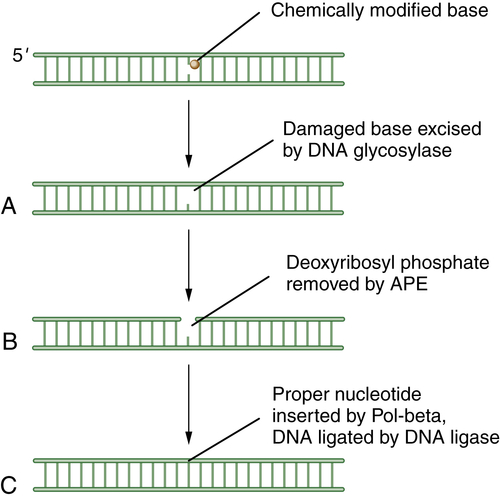
13 The cell uses BER to correct damaged DNA bases or single-strand DNA breaks. These lesions often result from spontaneous DNA damage (DNA deamination or hydroxylation of bases) or from exposure to environmental alkylating agents. In this pathway, damaged bases are removed by one of at least 10 DNA glycosylases. The resulting apurinic/apyrimidinic (AP) sites are processed first by the Ape1 AP endonuclease, leaving a 5′ deoxyribose phosphate; then by an AP lyase activity, leaving a 3′-elimination product. Single-strand breaks are then filled in by a DNA polymerase, either with a single nucleotide or with a longer repair patch, followed by ligation. The latter events in the BER pathway are regulated by the enzyme PARP1 (polyADP ribose polymerase 1). During the DNA
damage response, the PARP1 enzyme polyADP ribosylates BER enzymes and enhances BER activity. Accordingly, tumors that have high levels of BER activity may be hypersensitive to PARP inhibitors.
14 A schematic representation of BER is shown in
Figure 4-1 .
Figure 4-1 Schematic description of base-excision repair (BER) BER is focused on small DNA lesions, often from endogenous sources, resulting in minor helix distortions. Initially, the lesion is recognized by one of the cellular DNA glycosylases, which cleaves the covalent bond between the abnormal base and the deoxyribose sugar (A). This cleavage leaves a so-called apurinic (A-P) site. Next, the apurinic endonuclease (APE) is recruited to cleave the phosphodiester backbone of the DNA (B). Finally, an error-free polymerase, Pol-beta, is engaged to replace the normal nucleotide, followed by DNA ligation (C) and restoration of the normal double-stranded DNA sequence.
Mismatch Repair (MMR)
MMR has been reviewed by Modrich.
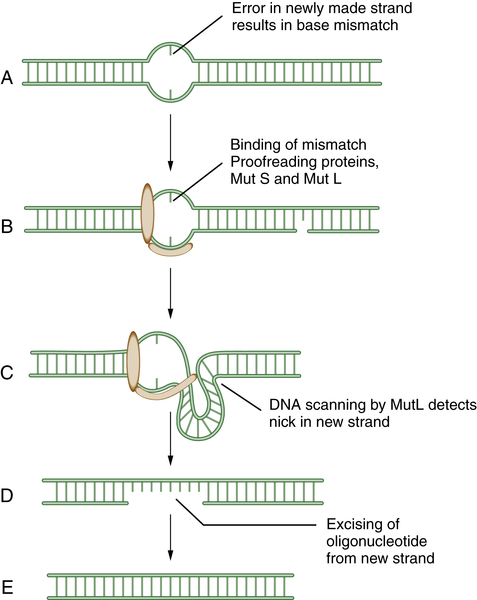
6 MMR rapidly removes mispaired nucleotides that result from replication errors and is also involved in the detection and repair of DNA adducts such as those resulting from platinum-based chemotherapeutic agents. Initially, the heterodimeric MSH complex recognizes the nucleotide mismatch, followed by its interaction with MLH1/PMS2 and MLH1/MLH3 complexes. Several proteins participate in the process of nucleotide excision and resynthesis. Tumor cells deficient in MMR have much higher mutation frequencies than normal cells and exhibit microsatellite instability, a genomic biomarker of the underlying defect. Patients with the genetic disease HNPCC (hereditary nonpolyposis colon cancer) have germline mutations of MMR genes and are predisposed to MMR-deficient colon cancers. Recent studies suggest that MMR-deficient cells may be hypersensitive to inhibitors of various DNA polymerases, such as POLB and POLG.
15 At least six genes—MSH2, MLH1, PMS2, MSH3, MSH6, and MLH3—are involved in mismatch repair. A schematic representation of MMR is shown in
Figure 4-2 .
Figure 4-2 Schematic model of mismatch repair (MMR) Mismatch repair proteins function by sensing, binding, and repairing mistakes made during DNA replication. These mistakes include misincorporated bases and errors made during replication of microsatellite sequences (A). MutS can bind to the mismatch (B) and generate a kink in the DNA (C). This allows MutL to scan the DNA for a nearby single-strand nick in the newly replicated DNA. MutL then identifies, cleaves, and removes an oligonucleotide patch from the newly replicated strand (D,E). This allows replication and the insertion of the proper DNA base at the site of the former mismatch. Mutations in the human genes encoding homologues of these bacterial proteins play a critical role in the inherited disease hereditary nonpolyposis colon cancer (HNPCC).
Nucleotide-Excision Repair (NER)
NER acts on a variety of helix-distorting DNA lesions, caused mostly by exogenous sources that interfere with normal base pairing. This pathway may be particularly important in the response to adduct-forming chemotherapeutic agents such as platinum-based chemotherapy.
16 The primary function of NER appears to be the removal of damage such as pyrimidine dimers, which are induced by UV light. Members of the NER pathway include the XPA, XPB, XPC, XPD, XPE, and XPG proteins. Two other NER proteins, XPF and ERCC1, are especially important for the
processing of DNA crosslink repair. Recent studies indicate that monitoring the levels of these proteins in tumors may provide important biomarkers for predicting crosslinker drug sensitivity. For instance, some non–small-cell lung cancers are deficient in ERCC1, and this deficiency correlates with the cisplatin sensitivity of the specific tumor.
17
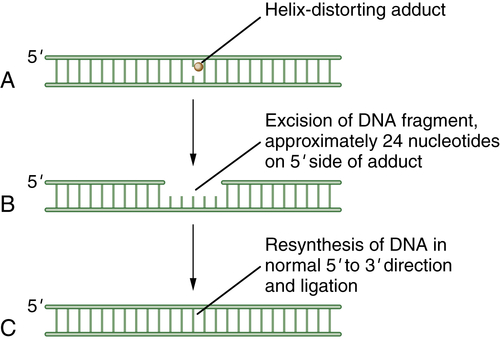
Figure 4-3 Schematic model of nucleotide excision repair (NER) NER is invoked when a base is modified by a larger helix-distorting lesion (A), such as a UV-generated thymine dimer. Initially, the bulky lesion is recognized by a sensor complex, including the XPE protein (also known as DDB2). This protein is part of a ubiquitin conjugating complex, containing Cul4A and DDB1. The complex polyubiquitinates XPC, allowing for the recruitment of the excision repair complex. Next, a patch of nucleotides is excised from the damaged DNA. In general, the excision occurs approximately 24 nucleotides 5′ to the damaged base and 3 nucleotides to the 3′ side (B). Finally, new DNA polymerization can occur, and the repaired DNA is ligated (C). The nucleotide excision repair complex is a large multisubunit complex. Mutations in genes encoding subunits of this complex underlie the human disease xeroderma pigmentosum (XP). The complex also contains proteins that can recognize and remove bases with large bulky adducts, such as those generated by polycyclic hydrocarbons and aflatoxin B1.
As for the other DNA repair pathways, these proteins cooperate to recognize and excise the damaged nucleotides and resynthesize and ligate the damaged DNA strand. In the process of NER, initially a DNA-binding component, the DDB, binds to sites of damaged DNA, such as cyclopyrimidine dimers or 6-4 photoproducts. The DDB consists of DDB1 and DDB2. Mutations in the DDB2 gene cause the genetic complementation group XPE. DDB is part of a ubiquitin E3 ligase that polyubiquitinates XPC. Polyubiquitination of XPC enhances its DNA binding. This binding sets the stage for the downstream binding of the entire excision repair complex, TFIIH, thus leading to excision of the damaged bases.
Eukaryotic NER includes two major branches, transcription-coupled repair (TCR) and global genome repair (GGR). GGR is a slow, random process of inspecting the entire genome for injuries, whereas TCR is highly specific and efficient and concentrates on damage-blocking RNA polymerase II. The two mechanisms differ in substrate specificity and recognition, and hence the enzymes involved are important nodal points for posttranslational modifications. A schematic representation of NER is shown in
Figure 4-3 .
Homologous Recombination Repair
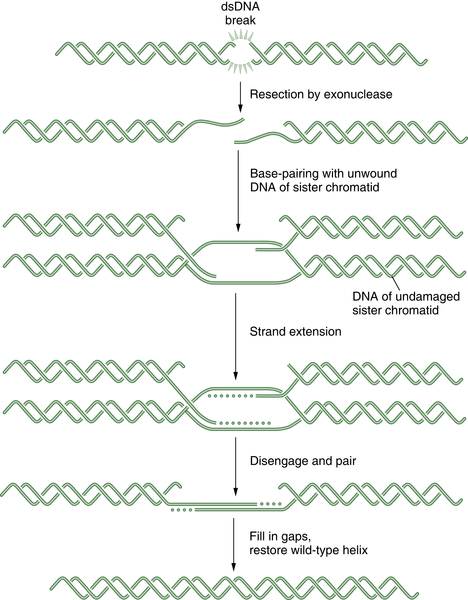
DNA double-strand breaks (DSBs) can be caused by many different environmental factors, including reactive oxygen species, IR, and certain antineoplastic drugs, such as bleomycin, anthracyclines, and topoisomerase inhibitors. Alternatively, DSBs can result from endogenous factors, especially during normal S-phase progression. Failure to repair DSBs can lead to a number of consequences, including mutations, gross chromosomal rearrangements, and other aberrations and eventually cell death. HR is a process by which DSBs are repaired through the alignment of homologous sequences of DNA and occurs primarily during the late S to M phase of the cell cycle. Initially the RAD50, MRE11, and NBS1 complex, which possesses a 3′-5′ exonuclease activity, exposes the 3′ ends on either side of the DSB, a process that may also require BRCA1. The 3′ advancing strand from the damaged chromosome then invades the complementary sequence of the homologous chromosome. The breast cancer susceptibility protein BRCA2 and the single-strand DNA binding protein RAD51 are required for the process. The 3′ end of this strand is then extended by an HR polymerase, by reading off of this complementary sequence. After replication has extended past the region of the DSB, the 3′ end of the advancing strand returns to the original chromosome, and replication continues. A schematic representation of HR is shown in
Figure 4-4 . HR repair is especially important in the repair of DSBs and DNA interstrand crosslinks. Because some tumors, particularly breast and ovarian tumors, are defective in HR repair, drugs that cause these lesions may be particularly effective in this setting.
Nonhomologous End Joining (NHEJ)
NHEJ, which has been reviewed by Lieber et al.,
18 is another major pathway for repairing DSBs. In common with HR, this pathway is important in the repair of agents that result in DSBs such as IR, bleomycin, topoisomerase II poisons, and anthracyclines. The DNA-dependent protein kinase (DNA-PK) consists of the catalytic subunit (DNA-PKcs) and the regulatory subunit (the Ku70/Ku80 heterodimer). The DNA-PKcs subunit is a serine/threonine kinase that belongs to the phosphatidylinositol-3 kinase family. The Ku80/Ku70 heterodimer (Ku) exhibits sequence-independent affinity for double-stranded termini and, on binding to DNA, recruits and activates the DNA-PKcs catalytic subunit.
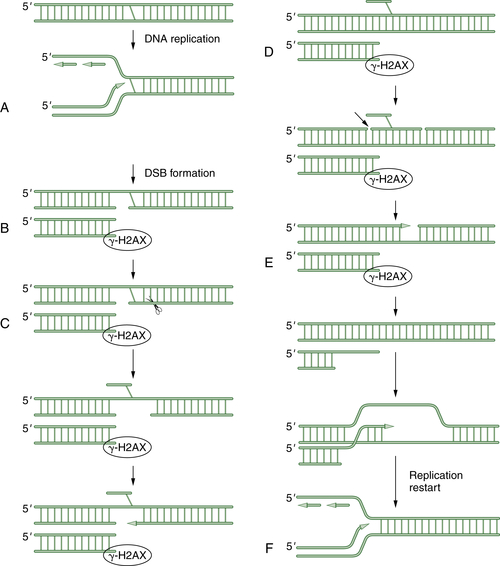
Additional proteins are required for the completion of NHEJ, including the Artemis protein and DNA ligase IV. Importantly, NHEJ is an error-prone repair pathway. Because the process does not use a complementary template, the fusion of the blunt-ended DNA duplexes may result in
deletion or insertion of base pairs. A schematic representation of NHEJ is shown in
Figure 4-5 . NHEJ has a normal function in immune cells to generate diversity at the immunoglobulin and T-cell receptor gene loci.
Figure 4-4 Schematic representation of homologous recombination (HR) HR repair is required for the normal repair of double-strand breaks (DSBs) as well as covalent interstrand DNA crosslinks. Initially, the DSB is recognized by a sensor. Some tumors have defects in HR repair, such as BRCA1 or BRCA2-deficient breast cancers. These tumor cells have prolonged time periods with unrepaired DSBs, thus leading to chromosome translocation events and a more malignant phenotype. Alternatively, the defective HR repair results in hyperdependence on the more error-prone NHEJ mechanism.
Translesion DNA Synthesis (TLS)
The process of TLS is another mechanism for dealing with thymine dimers and bases with bulky chemical adducts. At a DNA replication fork, DNA adducts may cause a replicative polymerase such as DNA polymerase delta to stall. Cells have, therefore, developed sophisticated mechanisms for switching off the replicative polymerase and switching on alternative polymerases (i.e., a polymerase such as pol eta, which will replicate past certain DNA lesions with high fidelity).
19 Interestingly, human cells have at least 15 DNA polymerases, although the situations and mechanisms of their deployment are largely unknown.
20 Cancer may have a heightened dependence on one of the error-prone TLS polymerases, such as polymerases β or kappa, accounting for high rates of mutagenesis. A schematic representation of TLS is shown in
Figure 4-6 .
Examples of Redundancy among the DNA Repair Pathways
Specific DNA repair pathways can antagonize the activity of anticancer agents. The status of a particular DNA repair pathway in a tumor may therefore predict the best antitumor therapy. As described earlier, at least two DNA repair pathways, BER and NER, are dedicated to the removal of DNA bases modified by monofunctional alkylating agents. BER can cleave the bond linking the modified base to the deoxyribose. NER, in contrast, will remove the entire modified nucleotide, along with a small stretch of surrounding nucleotides. In either case, the undamaged DNA can be used to synthesize the normal DNA sequence, followed by ligation of the segments.
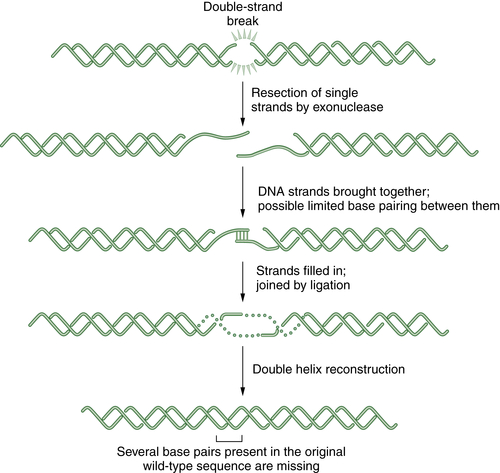
Figure 4-5 Schematic representation of nonhomologous end joining (NHEJ) NHEJ is an error-prone alternative to HR repair that can also be employed to repair double strand breaks. Since NHEJ does not use a homologous DNA template, such as a sister chromatid or a homologous chromosome, it often results in the insertion or deletion of new nucleotides at the fused DSB junction. In NHEJ, the DSBs are coated by the blunt end binding protein Ku. In some cases, the blunt ends may be brought together by limited microsequence homology. The enzymes DNA-PK, XRCC4, Artemis, and DNA ligase IV are required for the successful religation of the free ends. Interestingly, NHEJ appears to be the repair mechanism used for the cleavage and religation of immunoglobulin gene variable regions; thus the error-prone religation adds to the diversity of the somatically generated Ig gene repertoire. Germline mutations in some NHEJ genes, such as DNA-PK and Artemis, result in an inherited defect in NHEJ and a severe combined immunodeficiency syndrome.
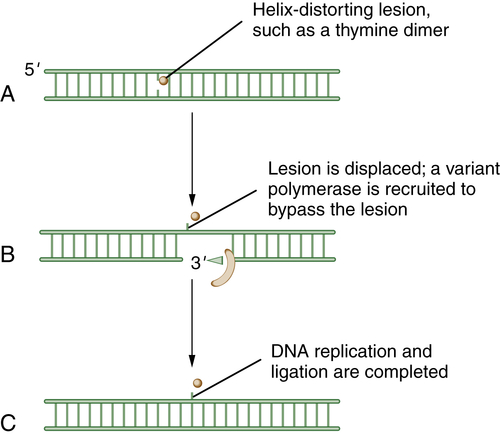
Figure 4-6 Schematic model of translesion DNA synthesis (TLS) repair TLS is not a DNA repair pathway per se; it is instead a mechanism of DNA damage bypass. In this process, an advancing replication fork encounters a damaged DNA base (A). Although the replicative polymerase (the Pol delta complex) cannot read through the damaged base, a variant polymerase such as Pol eta can bypass the lesion. Cells have developed sophisticated mechanisms for switching polymerases (B). For instance, in response to UV damage and the generation of a CPD (cyclopyrimidine dimer), the processivity factor, PCNA, becomes monoubiquitinated by RAD18. Modified PCNA now excludes Pol delta binding and has preferred binding for Pol eta. Pol eta is recruited, and it has the ability to “read through” the damaged base and insert the proper nucleotide (i.e., AA residues are replaced opposite the TT residues of a thymine dimer). Less is known about the regulation of TLS than about other DNA repair pathways. Depending on the kind of DNA damage, it is becoming increasingly clear that there are biochemical “switching” mechanisms for recruiting one of the other 12 TLS polymerases, as needed.
Cancer cells have other mechanisms for coping with modified bases. One enzyme, MGMT (0-6 methylguanine DNA methyltransferase), is capable of catalyzing the reversal of the chemical modification. Interestingly, this enzyme is switched off by MGMT gene promoter methylation in some solid tumors (gliomas and colorectal tumors), accounting, at least in part, for the hypersensitivity of these tumors to some monofunctional alkylating agents such as temozolamide.
21 In addition, damaged bases in the DNA can be bypassed through the use of TLS. Through this mechanism, the modified lesions are sensed, the normal replicative polymerase is removed from the replication fork, and a new polymerase is invoked to bypass the lesions. Rapid TLS (damage avoidance) is essential in order for a cell to transverse S phase rapidly, without succumbing to replication arrest and apoptosis. Interestingly, however, TLS is an error-prone process, and the promiscuous use of TLS by cancer cells may result in their increased mutation frequency (see later discussion).
Some of the 15 variant polymerases can extend a nascent DNA strand past a thymine dimer or past a bulky DNA lesion. Other variant polymerases can replace a single nucleotide at the site of an unpaired base. One of these variant polymerases, referred to as POLeta, is mutated in the autosomal recessive human disease XP-variant (xeroderma pigmentosum variant). Absence of the POLeta enzyme results in UV light hypersensitivity, an inability to replicate
past thymine dimers, and a predisposition to squamous cell cancers.
The variant polymerases exhibit a variable level of fidelity. Important unanswered questions in the TLS research field include the following: (1) What are the circumstances and mechanisms for recruiting the variant polymerase to a specific damaged DNA site? and (2) Are any of these variant polymerases overexpressed or dysregulated in cancer, accounting for the elevated mutation frequency of solid tumors?
As described previously, IR causes double-strand breaks as well as a wide range of oxidative DNA damage. Two redundant DNA repair pathways, HR repair and NHEJ repair, are particularly adept at dealing with double-strand break damage in a cancer cell. In clinical oncology, some tumors that have defects in these DNA repair processes are particularly sensitive to the cytolytic activity of IR. Also, radiation resistance can emerge through the induction of these DNA repair activities in treated tumor cells. Tumor cells that grow in a more hypoxic environment may also be more resistant to the killing effect of IR, perhaps because of the decrease in oxidative damage generated in these cells.
Regulation of the Six DNA Repair Pathways
As described earlier, the major proteins involved with DNA repair include sensory (DNA binding) proteins, enzymes that remove damaged bases, and enzymes that restore the normal DNA sequence. A large number of regulatory enzymes also control each DNA repair pathway. These enzymes are required for switching DNA repair on and off, as needed by the cells. Regulatory enzymes, such as helicases, serve to load DNA repair complexes at the sites of DNA damage. Other regulatory enzymes, such as topoisomerases, serve to unwind damaged DNA, in order to facilitate DNA repair complex assembly, loading into chromatin, and disassembly.
A major subclass of regulatory enzymes add critical posttranslational modifications to DNA repair enzymes. For instance, in BER, a sumoylating enzyme modifies one of the glycosylases, TDG, thereby enhancing the activity of the glycosylase in removing damages bases.
22,23 In NER, an E3 ligase complex (Cul4A, DDB1, DDB2) activates the polyubiquitination of the XPC protein. This XPC modification is a necessary event for the downstream activity of the nucleotide excision repair complex.
24 In TLS, an E3 ligase, RAD18, monoubiquitinates the DNA processivity factor, PCNA, and allows this clamp to interact with the downstream DNA polymerase Pol eta. Many of these regulatory processes have recently been reviewed.
25 Regulatory enzymes are also required to disassemble DNA repair enzymes after a repair pathway has been completed. For instance, the negative regulatory phosphatase, PP2A, removes phosphate from ATM substrates and thereby switches off the DNA damage response.
26 The deubiquitinating enzyme, USP1, can remove ubiquitin from activated FANCD2 and thereby switch off HR repair.
27 USP1 can also deubiquitinate PCNA and switch off TLS repair.
28,29 These negative regulatory events have also recently been reviewed.
25 The function of these regulatory enzymes underscores the dynamic nature of DNA repair. Loss of these regulatory mechanisms may result in the failure to (1) activate an error-free DNA repair pathway or (2) inactivate an error-prone DNA repair pathway. In either case, the consequence may be a heightened mutation frequency of the dysregulated cell and a predisposition to cancer.
Recent studies, generated through human cancer genome sequencing projects,
30 have demonstrated that some tumors, primarily melanomas and squamous cell lung cancers, have elevated levels of random point mutagenesis. This elevated point mutation signature indicates that these tumors (1) may have an underlying dysregulation of DNA repair and (2) may be more sensitive to specific therapies (see later discussion of synthetic lethality relationships). Finally, the regulation of DNA repair is a major focus of the DNA repair research field. For instance, it is unknown how DNA repair pathways are activated in specific cell types or at specific stages of the cell cycle. Because some DNA repair processes, such as HR repair, are specifically activated in S phase, it is likely that these pathways are activated by the cdk family of cyclin-dependent kinases. Several recent studies have performed shRNA screens to identify other regulatory genes that control DNA repair.
31–33 Many of these genes encode proteins that control the posttranslational modification of known DNA repair proteins or that regulate chromatin disassembly. Disruption of these gene products with shRNAs or small molecules can block DNA repair and further sensitize tumors to conventional cytotoxic cancer therapies.
Sequential Use of Three DNA Repair Pathways to Repair DNA Crosslinks
Interstrand DNA crosslinks (ICLs) make up a particular subtype of DNA lesions, and these lesions have an especially potent biological effect. Because ICLs involve the covalent modification of both strands of DNA, the lesions can prevent DNA strand separation during DNA replication. The lesions can also prevent the access of some DNA repair enzymes and transcription factors that normally require DNA strand separation for DNA binding to occur. DNA crosslinking agents, such as cisplatin derivatives (carboplatin
and oxaliplatin) and mitomycin C, are especially cytotoxic to tumor cells, and their therapeutic index derives, at least in part, from the high proliferative rate of tumor cells versus normal cells.
Figure 4-7 A schematic model of DNA crosslink repair DNA crosslink repair is believed to occur primarily during the S phase of the cell cycle. When a replication fork encounters an interstrand crosslink (A), DNA replication arrests. Initially, a double-strand break is generated by an unknown endonuclease (B). This DSB is next surrounded by phosphorylated histone 2AX. Next, another endonuclease (perhaps ERCC1/XPF) cleaves on the opposite side of the crosslink, allowing extrusion of the crosslinked bases from the double helix (C). Next, a series of three DNA repair pathways act sequentially. Translesion synthesis (TLS) repair allows bypass of the damaged bases (D). Then nucleotide excision repair (NER) excises the damaged oligonucleotide and allows gap filling (E). Finally, end resection occurs, and the double-strand break can be repaired by homologous recombination (HR). The replication fork is regenerated in an error-free mechanism (F).
The mechanism of DNA crosslink repair in human cells is poorly understood, and our understanding to date is derived more from the study of crosslink repair in prokaryotes and in the yeast
Saccharomyces cerevisiae. As shown in
Figure 4-7 , crosslink repair in human cells probably requires multiple DNA repair pathways. According to this model, the ICL is only repaired during S phase progression. Initially, an advancing replication fork encounters an ICL. An unknown endonuclease cleaves the DNA, thus generating a double-strand break (DSB). Next, a second endonuclease is invoked to cleave the DNA after the DNA crosslink. Recent data suggest that this endonuclease may be made up of the ERCC1/XPF proteins.
Now that an endonucleolytic event has occurred on each side of the crosslink, the crosslinked single-strand fragment can be flipped out of the helix. This allows three of the normal DNA repair pathways to work sequentially. First, TLS allows bypass of the crosslink and replication and ligation of the upper double helix. Recent studies indicate that some variant polymerases, such as POL eta, are particularly important to the translesion synthesis across MMC adducts. Next, the NER pathway can excise a stretch of damaged DNA and allow gap filling of the excised oligonucleotide. Finally, HR repair can be used for the error-free, template-driven repair of the damage. The end result of this sequential use of three independent DNA repair pathways is to resume DNA replication and restart the replication fork.
Consistent with this model of crosslink repair, some repair-deficient cells are especially prone to the cytotoxic effects of DNA crosslinking drugs. For instance, cells that are
deficient in ERCC1/XPF generate the first double-strand break upstream of the DNA crosslink. However, these double-strand breaks, as measured indirectly by the presence of histone 2AX foci, persist in the repair-deficient cells, suggesting that ERCC1/XPF may work farther downstream in the pathway. Similarly, cells deficient in the FA pathway have persistent double-strand breaks after MMC exposure.
34 Thus, the presence or absence of the double-strand break intermediates is helpful in determining the level at which a repair process is disrupted and the sequence of repair events in the pathway.
DNA Repair and the DNA Damage Response
DNA repair is, in fact, only one class of a broader set of cellular responses referred to as the DNA damage response. DNA damage responses include the activation of cell cycle checkpoints, the activation of apoptosis, and the activation of DNA damage tolerance. This last mechanism allows a cell to “accept” DNA damage and continue DNA replication even in the setting of a heightened mutation frequency. The DNA damage response is therefore a highly coordinated set of signaling events. These responses require a DNA damage sensor (such as a sensor kinase, ATM, or ATR) and an effector kinase, as well as downstream protein machines dedicated to DNA repair, apoptosis, or checkpoint activities.
35
The DNA Damage Response Is Mediated by Sensor and Effector Kinases
The DNA damage response can be activated by a wide range of environmental exposures or drug interactions. An important early player in the damage response is the molecular “sensor” of DNA damage. A local distortion in the DNA double helix, perhaps resulting from a DNA adduct or a thymine dimer, can activate a sensor kinase, such as ATM, ATR, or DNA-PK. These kinases are believed to autophosphorylate
36,37 and go on to phosphorylate a large number of substrates thereafter. The ATM kinase is the product of the ATM gene, the gene mutated in the cancer susceptibility disorder ataxia-telangiectasia.
Activated ATM and ATR proteins phosphorylate additional downstream “effector” kinases, such as the checkpoint kinases, Chk1 and Chk2. Activated Chk1 and Chk2 then go on to phosphorylate a wide array of protein targets involved in the machinery of DNA repair or DNA damage checkpoints.
One of the best-characterized DNA damage checkpoints is regulated by the ATM/Chk2/Cdc25A axis.
38,39 In response to IR, a double-strand break is generated, and this break activates ATM. ATM subsequently phosphorylates Chk2, which in turn phosphorylates the cell cycle activator cdc25A. Cdc25A phosphorylation leads to its rapid degradation and to cell-cycle arrest. This appears to be an important mechanism by which a cell can respond to DNA damage by arresting its cell cycle progression in S phase. By stopping S-phase entry, a cell allows itself the opportunity to slow down and to repair its DNA or, in the setting of severe damage, to undergo apoptosis. Importantly, a failure to activate this checkpoint response, as in ATM-deficient cells, results in S-phase progression even in the setting of DNA damage. Continuing to replicate DNA in the setting of DNA damage has dire consequences for the cells. The cell may have an elevated mutation rate or may complete DNA replication, only to experience a mitotic catastrophe at the end of the cell cycle.
Failure of ATM to activate the intra–S-phase checkpoint results in a characteristic cellular phenotype. When ATM-deficient cells are exposed to IR, they fail to arrest in S phase but instead continue to replicate their DNA and to incorporate tritiated thymidine in the postradiation period. This phenotype is known as radioresistant DNA synthesis (RDS), and it is the hallmark of a cell with a defect in the ATM/Chk2/cdc25A axis. An active area of DNA repair research is the identification of other CHK1 and CHK2 phosphorylated substrates.
Phosphorylated Effector Proteins Assemble in DNA Damage Foci
An important downstream event in the DNA damage response is the assembly of proteins in subnuclear foci.
40,41 These foci are often referred to as IRIFs (ionizing radiation inducible foci). Multiple ATM and ATR phosphorylated substrates, such as Chk1, BRCA1, and BARD1, assemble in foci following DNA damage. The assembly of these large protein complexes is mediated, at least in part, by the phosphorylated SQ or TQ sequences of the ATM/ATR substrates. Recent studies have indicated that these phosphorylated amino acid residues bind directly to phosphoamino acid receptors found on other adaptor proteins. For instance, phosphorylated BACH1 can bind directly to the BRCT domain (a phosphoserine receptor) of the BRCA1 protein.
42,43 The precise structure and function of these protein foci in eukaryote nuclei is not known. Clearly, the number of foci correlates with the number of unprocessed double-strand DNA breaks, and the foci are widely believed to be sites of double-strand break repair.
From immunofluorescence analysis, it is clear that multiple phosphorylated DNA-damage activated proteins colocalize in these foci. The foci have been helpful to researchers in the establishment of signaling pathways. For instance, pATM, pBRCA1, and pFANCD2 colocalize in IRIFs. Disruption of one upstream protein, say, by a germline or acquired mutation in the upstream signaling protein, ATM, results in loss of downstream proteins in the foci. The assembly of the foci therefore has become a useful tool in understanding the interrelationships of DNA response proteins.
A few DNA damage-response proteins deserve special attention here. Bonner and colleagues
40 have identified a variant histone protein, histone 2AX, that is rapidly phosphorylated by ATM after radiation damage. H2AX is an important early signaling protein in the DNA damage response. The phospho H2AX protein is incorporated in chromatin in vast stretches emanating from the site of the DNA DSB. An absence of histone 2AX, as in an H2AX knockout mouse model, results in chromosome instability and cancer predisposition,
44,45 apparently due to failure to mount the proper DNA damage response.
Another important DNA damage response protein is RAD51, which is phosphorylated by the Chk1 kinase during normal S-phase progression.
46 RAD51 is a single-strand DNA binding protein that plays a critical role in DNA repair by HR. Phosphorylated RAD51 also assembles in foci during normal S-phase progression. These “replication foci” are believed to be sites of DNA repair by HR between sister chromatids, which occurs during normal DNA replication.
A comprehensive analysis of proteins that are rapidly phosphorylated after DNA damage (and that form nuclear foci) has provided an important database for laboratories studying the DNA damage response. These phosphorylated proteins, and foci, provide a useful set of biomarkers for DNA repair activities. For instance, cells that are defective in the formation of DNA repair foci are themselves defective in DNA repair. The specific kind of focus that is absent correlates with the particular kind of DNA repair deficiency. For instance, cells deficient in RAD51 foci are defective in HR repair and are hypersensitive to IR. Cells defective in the assembly of polyADP ribose (PAR) foci are defective in the repair of single-strand breaks and may therefore have an underlying defect in BER. As such, tumor cells missing particular types of DNA repair foci may be more sensitive to certain kinds of chemotherapy or radiation.
Importantly, human cancers are often deficient in the DNA damage response. Germline mutations in DNA damage response genes, such as ATM, NBS1, FANCD2, BRCA1, and BRCA2, can result in an increased susceptibility to cancer. Individuals who inherit a single mutant allele of, say, BRCA1, have a high risk of developing a breast, ovarian, or prostate cancer during their lifetime. The tumor results from the inactivation of the second BRCA1 allele, through deletion and loss of heterozygosity, thus resulting in a tumor with a specific DNA repair defect. BRCA(−/−) tumors therefore have genomic instability, but also have increased sensitivity to some DNA-damaging agents such as IR and DNA crosslinkers.
Study of the DNA damage response reveals that cells have highly regulated responses to different levels and types of DNA damage. Whereas some DNA repair pathways may be viewed as constitutive, housekeeping pathways, other pathways are highly controlled. Some DNA repair pathways, such as ATR and CHK1, are activated primarily at the site of the advancing replication fork, leading to the activation of HR repair.
47 Other DNA repair processes are activated in nondividing cells, such as in postmitotic neurons. For instance, NHEJ is hyperactive in nondividing cells and functions as the major mechanism of double-strand break repair in these cells.
The cellular context of the DNA repair pathway is also important. Germline or somatic disruption of a pathway may result in a strikingly different phenotype, depending on the cell and tissue of origin. For instance, gene line disruption of a DNA damage response, as in the inherited disease ataxia-telangiectasia, may lead to a characteristic constellation of clinical findings, including cerebellar degeneration and lymphoma predisposition. A somatic disruption of the same pathway (say, ATM CHk2-p53) may lead to a very different set of cancers, such as solid tumors of the bladder and ovary.
48,49 Recent studies indicate that the DNA damage response provides an important “barrier” to the transformation of a normal cell to a malignant cell.
48,50 Specifically, early premalignant cells have heightened constitutive activation of the DNA damage response pathways, as exemplified by increased immunohistochemical staining with antibodies to activated ATM and to the activated checkpoint kinase CHK2. Interestingly, as cells progress from the premalignant state to the malignant state, they lose these DNA damage responses, perhaps through acquired disruptions of ATM or CHK2 activity. Because individuals with genetic diseases such as ataxia-telangiectasia already have a defect in the checkpoint response, they may be prone to earlier onset of cancers for this reason.
Inherited Chromosome Instability Syndromes as Models for DNA Repair Defects
Rare pediatric chromosome instability disorders, such as FA and XP, provide important insights into the function
of DNA repair pathways and their role in cancers in the general population. Children born with these syndromes generally have congenital abnormalities, cellular hypersensitivity to DNA-damaging agents, genomic instability, and an increased risk of specific cancers. Although these syndromes are rare, the DNA repair pathways disrupted by germline mutations in these individuals are often the same pathways disrupted by somatic mutation or epigenetic inactivation in cancers from the general population. For these sporadic cancers, a knowledge of which DNA repair mechanism is disrupted provides important clues to the behavior of the cancer or its drug sensitivity spectrum.
At least five of the major DNA repair pathways have corresponding inherited human diseases (
Table 4-1). HR and TLS repair is defective in FA cells.
51 NER repair is defective in XP cells, Cockayne syndrome (CS) cells, and trichothiodystrophy cells.
52 MMR repair is defective in children with Turcot syndrome and in tumor cells derived from adult patients with HNPCC. TLS repair is defective in patients with the XP-V (xeroderma pigmentosum variant) disease. Most of these pediatric diseases exhibit autosomal recessive inheritance, such as XP, FA, and CS. Turcot syndrome has been reported to exhibit autosomal dominant or autosomal recessive inheritance depending on the particular mutation affecting mismatch repair. Inherited mutations in BER genes have not been observed in humans, suggesting that this pathway is essential for human development.
It is interesting that patients with inherited DNA repair syndromes, such as CS and FA, have congenital abnormalities. For instance, CS patients have developmental abnormalities of the skin and skeletal system. FA patients have skeletal, kidney, cardiac, and bone marrow defects. Consistent with these findings, the NER and FA pathways appear to play dual roles. For instance, the NER excision repair complex, TFIIH, plays an important transcriptional role during embryonic development. Germline dysfunction therefore leads to defects during embryonic organogenesis. The NER complex also plays a critical role in DNA repair in somatic cells after organism development. Similarly, the FA pathway appears to have a dual role in both development and DNA repair in somatic cells.
The systematic study of these rare diseases has led to (1) a better understanding of the genes and proteins involved in the six major DNA repair pathways, (2) how an inherited (or germline) defect in a DNA repair pathway can lead to genomic instability, cancer progression, and drug hypersensitivity, and (3) how an acquired (or somatic) defect in a DNA repair pathway can influence tumor progression and drug sensitivity of tumors in the general population. Although the specific details of these individual inherited diseases is beyond the scope of this review, an example of how a study of these rare diseases can lead to general insights to tumor biology can be appreciated from recent insights into the FA pathway.
Fanconi Anemia: A Specific Inherited DNA Repair Defect
FA is an autosomal recessive or X-linked recessive cancer susceptibility syndrome characterized by multiple congenital abnormalities, progressive bone marrow failure, and cellular hypersensitivity to DNA crosslinking agents, such as cisplatin and mitomycin C (MMC). FA patients are prone to developing acute myeloid leukemia as well as squamous cell carcinomas of the head and neck or gynecologic system.
53 The study of FA cells has led to the elucidation of a DNA repair pathway for interstrand crosslinks. Clinically, this pathway is particularly important because many DNA crosslinking agents such as cisplatin or mitomycin C are used for cancer treatment. The FA defect results from biallelic mutation of any one of 15 known FA genes (A, B, C, D1, D2, E, F, G, I, J, L, M, N, O, P). The proteins encoded by these FA genes cooperate in a common DNA repair pathway, referred to as the FA/BRCA pathway.
53,54 A central event in this pathway is the monoubiquitination of the FANCD2 protein, and this event is a useful biomarker for DNA repair activity (see later discussion). Disruption of this pathway results in the characteristic clinical and cellular phenotype of FA patients.
Patients with an Inherited Germline DNA Repair Deficiency Exhibit a Characteristic Tumor Spectrum
Patients with inherited DNA repair deficiency syndromes are prone to the development of specific tumors. FA patients, for example, are predisposed to acute myeloid leukemia and squamous cell carcinomas, primarily of the head and neck or gynecologic system. Patients with XP are prone to skin squamous cell carcinomas, primarily on body surfaces with more sunlight exposure. Patients with HNPCC and an inherited MMR deficiency are prone to colon cancer and ovarian cancer.
Tumors arising from somatic disruptions of DNA repair pathways may arise in other organ systems. A specific oncogenic lesion, such as the activation of an oncogene or the disruption of a tumor suppressor gene, may have a vastly different effect depending on the cellular context of the lesion. For instance, a germline mutation in the Rb gene may result in an embryonal tumor, such as a retinoblastoma or a pineoblastoma, but a somatic disruption of the Rb gene may lead to the development of a sarcoma.
Similarly, disruptions of a DNA repair pathway by a germline mechanism, rather than a somatic mechanism, may yield a very different spectrum and behavior. Examples are shown in
Table 4-1. Somatic disruption of the FA pathway results in a wide range of tumor types, including tumors of the ovary, lung, and cervix.
51,55 Moreover, somatic disruptions result from methylation and silencing of an upstream FA gene (FANCF). Germline disruption of the same genes results from inherited mutations, such as missense mutations or nonsense mutations. Somatic disruption of the NER pathway plays a role in the development of testicular cancer and appears to account for the hypersensitivity of this tumor to the drug cisplatin. Paradoxically, somatic disruption of a DNA repair pathway can also result in chemotherapy resistance. Studies indicate that methylation and silencing of the MLH1 gene may account, at least in part, for the cisplatin resistance of some ovarian tumors.
Disruption of the other DNA repair pathways has been observed in sporadic human tumors, accounting, at least in part, for the specific drug and radiation sensitivity spectrum of these tumors and their clinical outcome. HR is disrupted in breast and ovarian cancer, NER is disrupted in testicular cancer, and MMR is disrupted in sporadic colon cancer. A few studies suggest that TLS may be disrupted in human cancers. Human cancer cells exhibit an elevation in spontaneous and damage-inducible point mutagenesis, compared to nonmalignant cells, suggesting an underlying TLS defect. Recent studies indicate that an elevation in the expression and activity of the error-prone polymerase Pol beta accounts for the increase in cisplatin resistance and mutagenesis of these cancers. Consistent with this hypothesis, inhibition of Pol beta in these cells results in resensitization to cisplatin.
56
Somatic Disruption of DNA Repair Pathways by Methylation and Gene Silencing
One of the most common mechanisms of inactivation of DNA repair pathways in sporadic cancer is the epigenetic silencing of a critical gene through methylation of the promoter region. Increasing evidence shows that the FA/BRCA pathway is one of the DNA repair mechanisms that is targeted in sporadic cancers.
FANCF methylation occurs in 24% of ovarian granulosa cell tumors, 30% of cervical cancer, 14% of squamous cell head and neck cancers, 6.7% of germ cell tumors of the testis, and 15% of non–small-cell lung cancers, where it correlates with a worse prognosis. An example of how methylation of a DNA repair gene can promote tumor progression is shown in
Figure 4-8 .
By regulating the activity of DNA repair pathways, cancer cells have a propensity to progress to a more malignant state. According to this model, early in the course of tumorigenesis, a premalignant cell may undergo a methylation and silencing of a DNA repair gene. In the case of the FA/BRCA pathway, the gene most commonly silenced by methylation is the
FANCF gene on chromosome 11p15. Inactivation of
FANCF results in a disruption of DNA repair and in genomic instability. The premalignant cell is therefore prone to multiple oncogenic events, such as the upregulation of a tyrosine kinase oncogene or the disruption of p53. A tumor with multiple somatic mutations eventually develops (see
Figure 4-8), but this tumor still has a defective DNA repair pathway and is hypersensitive to genotoxic chemotherapy. After antitumor therapy, however, there is a selective pressure for tumor cells with an intact FA/BRCA
pathway. Tumor cells with a demethylated
FANCF gene are selected, and a drug-resistant tumor emerges. By following this pattern, tumors can silence and reactivate DNA repair pathways, leading to drug resistance and tumor progression.
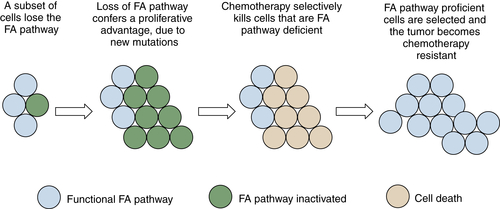
Figure 4-8 Tumor progression by serial inactivation and reactivation of DNA repair pathways According to this model, early in the course of carcinogenesis, a DNA repair pathway becomes inactivated. For instance, the FANCF gene may undergo biallelic methylation and silencing. This loss of the FA/BRCA pathway results in a state of chromosome instability, leading to secondary mutations (activation of K-Ras, inactivation of p53, for example). A tumor evolves, and the tumor is initially hypersensitive to cisplatin, as is often the case for ovarian epithelial cancer. Cisplatin causes rapid cytolysis of the tumor; however, rare tumor cells undergo a restoration of FANCF expression. Restoration may result either from an active demethylation of the FANCF gene or from positive selection of rare cells that experienced a stochastic demethylation event. Tumor cells regrow, and these cells are now cisplatin-resistant. In principle, an inhibitor of the FA/BRCA pathway can resensitize the tumor cells to cisplatin, as described in the text.
The converse scenario may occur for the MMR pathway. MMR-proficient cells are hypersensitive to the DNA crosslinking drug cisplatin. In this case, it is believed that the active MMR pathway generates a cisplatin-inducible lesion that is tumoricidal. Inactivation of MMR by methylation of the MSH2 gene therefore provides the tumor with a mechanism for achieving cisplatin resistance. Based on these examples, it appears that understanding the methylation state of various DNA repair genes may allow the prediction of drug responsiveness of some tumors.
Epigenetic silencing of BRCA1 through methylation occurs in 13% of breast cancers, 23% of advanced ovarian cancers, 6% of cervical cancers, and 4% of non–small-cell lung cancers. Epigenetic disruption of the FA pathway may also be important in the development of sporadic acute myeloid leukemia (AML), where absent or reduced expression of the FA proteins FANCA, FANCC, FANCF, and FANCG has been reported. Loss of BRCA2 mRNA and protein expression has been reported in 13% of ovarian adenocarcinomas; in contrast to the other FA genes described earlier, this loss does not result from promoter methylation.
Prognostic and Predictive DNA Repair Biomarkers in Cancer Treatment
Both hereditary cancer syndromes and sporadic cancers can arise from abnormalities in DNA repair pathways. This may be clinically important because these tumors are expected to be hypersensitive to DNA-damaging therapeutic agents or strategies that inhibit alternative DNA repair pathways. In the case of sporadic cancers, the patient’s normal cells, such as those in the bone marrow, possess a functional DNA repair pathway and are predicted to be resistant to these targeted treatments. Assessment of the status of the FA pathway or other DNA repair pathways requires the use of diagnostic biomarkers.
Selection of Biomarkers of DNA Repair Pathways
Biomarkers of DNA repair pathways can be divided into two major groups: functional biomarkers that characterize the activity of a pathway following damage and expression biomarkers that measure the availability of pathway components before damage.
Functional DNA Repair Biomarkers
These are biomarkers that indicate an intact DNA repair pathway. These biomarkers have the advantage of giving a functional measure of a particular pathway and will detect repair defects due to epigenetic events or gene mutations. Moreover, they give a global measurement of a particular pathway’s function without the need to know the identities of all the components. They could also be used to differentiate between insignificant single-nucleotide polymorphisms (SNPs) and functionally important point mutations in DNA repair pathway genes. Functional biomarkers can be applied to serial tumor samples from the same patient, at diagnosis and at the time of relapse. In this way, one can determine whether the tumor remains drug sensitive or has restored its DNA repair mechanisms. However, these markers rely on tumor tissue having been exposed to some form of DNA damage in vivo or in vitro before the assay. Functional biomarkers of DNA repair pathways include the monoubiquitination of the FANCD2 protein (a biomarker for HR repair) and the phosphorylation of DNA-PK (a biomarker of a functional NHEJ pathway). Abnormal DNA-damage–induced nuclear foci may identify disruption of the downstream events in the pathway, such as that observed in BRCA1- or BRCA2-deficient cells.
DNA Repair Biomarkers of Gene/Protein Expression
These biomarkers indicate the preexisting function of a DNA damage pathway before damage. Examples are real-time polymerase chain reaction (rt-PCR) or immunohistochemistry to test for epigenetic silencing of critical DNA repair genes. Some studies have used a microarray approach to look for genetic expression profiles indicative of abnormal DNA repair gene function. Because some DNA repair genes, such as MLH1 and MSH2, are inactivated by methylation, measurement of gene methylation via the methylation-PCR assay can also be applied as a biomarker assay. These approaches have the advantage of not requiring prior DNA damage and can be performed on fixed specimens. However, they provide only an indirect measurement of the functional capabilities of a DNA repair pathway. In addition, mutant genes can express normal levels of mRNA, and mutant protein and would not be detected by this method.
Clinical Application of DNA Repair Biomarkers
DNA Repair Biomarkers as Predictors of Response to Conventional Therapy
Loss or increased activity of particular DNA repair pathways may influence the response to DNA-damaging therapeutic strategies. For instance, a failure of a pathway
involved in the repair of DNA crosslinks such as HR would be predicted to sensitize a tumor to DNA crosslinking agents such as alkylating chemotherapeutic drugs. Indeed, BRCA1 expression levels as measured by rt-PCR have been used as a biomarker of survival following cisplatin-based chemotherapy for non–small-cell lung cancer. Methylation-specific PCR, which indicates loss of gene expression through promoter methylation, has been used to correlate loss of BRCA1 function with cisplatin sensitivity in ovarian cancer. Loss of
BRCA2/FANCD1 function through mutation in breast or ovarian cancer has also been reported to correlate with a high response to DNA-damaging chemotherapeutic agents. Absence of FANCD2 monoubiquitination may be a biomarker for loss of function of upstream FA pathway components and could be expected to predict sensitivity to DNA crosslinkers such as cisplatin or cyclophosphamide.
A recent example of the use of DNA repair biomarker in clinical medicine is the evaluation of ERCC1 protein expression levels in lung cancer. The NER pathway is important for the correction of UV-light–induced thymine dimers and for the excision of small single-base adducts. In addition, two of the proteins involved in NER, ERCC1 and XP-F, appear to have special relevance to the repair of DNA interstrand crosslinks. Primary cells derived from XP patients with germline mutations in ERCC1 or XPF are hypersensitive to UV and to DNA crosslinking agents.
57 Recent studies also suggest that the protein level of ERCC1 in cell lines correlates with the level of functional DNA crosslink repair in the cell.
Several groups have performed retrospective analyses of non–small-cell lung cancer patients who had been treated with adjuvant chemotherapy, including cisplatin. The banked primary tumor samples, which were stored in paraffin blocks, were evaluated for the level of ERCC1 protein by immunohistochemistry (IHC). Interestingly, the patients with tumors exhibiting low ERCC1 levels were more sensitive to cisplatin, based on their longer average time to relapse after cisplatin, compared to patients whose tumors had high levels of ERCC1. Together these studies indicate that ERCC1 protein expression may be a useful predictive biomarker for assessing tumor response to cisplatin.
DNA Repair Biomarkers to Guide Chemo- and Radiosensitization
Resistance to DNA-damaging chemotherapy or radiotherapy may be due to enhanced repair of DNA lesions. Therefore, a possible therapeutic strategy is to use drugs that specifically inhibit DNA repair pathways. Theoretically, this strategy may be limited because the drug may also increase the toxicity of therapeutic DNA damage in normal tissue. A therapeutic index can be achieved, in principle, by (1) selective uptake of the DNA damage sensitizers by the tumor cell, versus the normal cell, or (2) delivering one of the modalities (such as the radiation) directly to the tumor.
An understanding of the precise molecular mechanisms of new classes of sensitizing agents has important implications. First, if an agent functions by inhibiting a specific DNA repair pathway, then active derivatives of this agent should function similarly. DNA repair pathway inhibition provides an important biomarker for determining the proper dosing of the drug. Second, the chemosensitizer would be predicted to be more efficacious when used in combination with specific classes of DNA damage drugs.
DNA Repair Biomarkers as Predictors of Response to Targeted Monotherapy
Another important application for biomarkers of DNA repair pathway integrity is the potential to develop nontoxic monotherapy for tumors with specific DNA repair defects. The upregulated DNA repair pathway is the “Achilles heel” of the cancer. In principle, a nontoxic inhibitor of this second pathway, delivered as a monotherapy, may selectively kill the cancer cell. A normal cell, in comparison, may be able to tolerate the loss of this second pathway because other pathways are functioning and there is more redundancy in its DNA repair capacity.
This principle has recently been demonstrated by the use of PARP inhibitors in BRCA1- and BRCA2-deficient cells.
11,12 As discussed earlier, under normal physiological conditions DNA is being damaged continuously. The result of these stresses is the development of damaged bases or regions of single-strand DNA breaks that are repaired through the BER pathway. Part of the BER pathway requires PARP, a DNA-binding zinc finger protein that catalyzes the transfer of ADP-ribose residues from NAD
+ to itself and different chromatin constituents, forming branched ADP-ribose polymers. Initially it was observed that PARP-deficient (and therefore BER-deficient) mice develop normally but have high levels of sister chromatid exchange, a feature of HR. This observation suggested that HR could compensate for a loss of PARP-dependent BER. Consequently it was demonstrated in preclinical models that BRCA1- and BRCA2-deficient human and murine cells were sensitive to PARP-inhibiting drugs, whereas cells expressing normal levels of BRCA1 or BRCA2 were unaffected. PARP1 inhibitors are well tolerated in preclinical murine models and, in addition to being a potential treatment for BRCA1 and BRCA2 mutant tumors, may also represent an attractive strategy for chemoprevention of malignancies in mutation carriers. Clearly a biomarker that indicates a failure of BRCA1 or BRCA2 function in tumor cells may allow the application of PARP inhibitors to a wider spectrum of sporadic human malignancies.
The Development of New DNA Repair Biomarkers
Few biomarkers exist at present for evaluating the integrity of the other DNA repair pathways. Several studies have attempted to assay these pathways, using expression biomarkers (i.e., testing the expression levels of known DNA repair proteins in the pathways.) Better functional biomarkers are needed. Recent studies have indicated that posttranslational modifications of DNA repair proteins in these pathways are also required for pathway activity. For instance, polyubiquitination of XP-C is required for functional NER,
24 and sumoylation of thymine-DNA glycosylase
22 is required for function of BER. The development of antibodies specific for these activated states and the testing of these biomarkers may allow the rapid assessment of drug sensitivity and acquired resistance in clinical samples.
DNA Repair Inhibitors as a New Area for Anticancer Drug Development
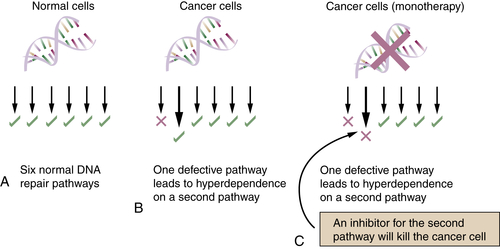
As shown in
Figure 4-9 , normal human cells may have six functional DNA repair pathways, whereas a tumor cell may have disruptions of one pathway. In the tumor, disruption of one pathway, such as HR repair, results in genomic instability and hyperdependence on a second pathway, such as BER. Because of this hyperdependent state, the tumor cell may be hypersensitive to an inhibitor of BER, such as a PARP1 inhibitor.
In this case, the tumor may respond to the PARP1 inhibitor as a single agent (monotherapy).
58–60 DNA damage, activated by the hyperproliferative state of the tumor cell, may be sufficient to kill the tumor cell. Alternatively, the PARP1 inhibitor may have a greater tumoricidal effect when it is used in combination with another cytotoxic agent, such as IR or an alkylating agent (TMZ).
Based on the early success with PARP1 inhibitor therapy, there is increasing interest in the identification of inhibitors of other DNA repair pathways.
61 For instance, an inhibitor of the sensor kinase ATM has been shown to have potent tumoricidal effects.
62 Also, inhibitors of the Chk1 kinase UCNO1 have been used in clinical trials.
35,63,64 Investigators have also begun to screen for inhibitors of HR repair that may potentially sensitize tumor cells to IR or to crosslinker damage.
65,66 Although DNA repair inhibitors may sensitize a tumor to the cytotoxic activity of conventional IR or chemotherapy, they may also enhance the toxicity of these therapies to normal human cells.
Importance of DNA Repair to Clinical Oncology: Other Specific Examples
BRCA1 and BRCA2
The importance of DNA repair to the pathogenesis and treatment of cancer is exemplified by studies of the breast cancer susceptibility gene BRCA1. Approximately 10% of women who develop breast cancer in their lifetime have a strong family history of (inherited) breast cancer. Of these women, approximately half are heterozygous carriers for mutations in either the BRCA1 or BRCA2 gene.
The
BRCA1 gene was originally mapped to human chromosome 17,
67 and it was subsequently cloned by position.
68 Strong evidence emerged that
BRCA1 is a tumor suppressor gene, since breast carriers have loss of heterozygosity at the
BRCA1 locus.
69 BRCA1-deficient breast tumor cells are hypersensitive to IR and to DNA crosslinking agents,
suggesting that
BRCA1 may function in the regulation of HR repair.
Figure 4-9 Principle of DNA inhibitor monotherapy (A). Normal human cells have six DNA repair pathways. (B) Tumor cells, in contrast, have disrupted one DNA repair pathway through somatic mutation, loss of heterozygosity (LOH), or epigenetic silencing of a DNA repair gene in that pathway. The tumor cell has genomic instability and has partially compensated for its DNA repair defect by upregulating a second pathway. (C) The tumor is hyperdependent on this second pathway, and a specific inhibitor kills the tumor cells but has little effect on the normal cells. An example of this monotherapy approach has been described for PARP inhibitors. 11,12
Studies with the BRCA1 protein indicate that, during the DNA damage response following cellular exposure to a genotoxic stress, BRCA1 is phosphorylated and accumulates in subnuclear foci that colocalize with BRCA2 and RAD51 proteins.
70,71 These foci are required for competent DNA repair. The precise role of BRCA1 in DNA repair is unknown. Because BRCA1 is itself an E3 ubiquitin ligase,
72 it may function by ubiquitinating other DNA repair proteins and regulating DNA repair indirectly. Recent studies have identified key ubiquitinated substrates of BRCA1, including the protein CTIP.
73 Because BRCA1 (and BRCA2) tumors are deficient in HR repair, this genotype may be useful in the selection of chemotherapy, as described earlier for these tumors. Recent studies indicate that BRCA1-deficient tumors are hyperdependent on BER and have elevated PARP1 activity.
11,12 Accordingly, BRCA1- and BRCA2-deficient tumors appear to be hypersensitive to PARP1 inhibitors.
Defects in DNA Repair Pathways Can Account for the Elevated Mutation Rate of Cancer
Cancer cells have an increased mutation rate compared to normal cells, and this phenotype has important clinical consequences. The increased mutation frequency can lead to point mutation and inactivation of tumor suppressor genes or to increased tumor cell resistance to chemotherapy. The increased mutation rate may also account for the increased spontaneous cell death observed in solid tumor samples (i.e., some of the mutations may be lethal to individual tumor cells), but may also enhance the outgrowth of a more malignant clone.
This increased mutation rate results in large part through the disruption of DNA repair pathways. The MMR pathway normally functions to improve the fidelity of DNA replication by quickly identifying and excising mismatched bases generated by faulty DNA replication. Loss of the MMR pathway by germline or somatic mutation can lead to a “mutator” phenotype. This phenotype can be readily detected by microsatellite instability in the genome of the cancer cell.
This increase in mutation rate can also be accounted for by an increase in error-prone DNA repair mechanisms.
74 In the setting of elevated translesion synthesis, some error-prone polymerases, such as Rev3, may increase the frequency of point mutations in the genome of the human cancer. Also, an elevation in the error-prone NHEJ pathway may account for the elevated complex mutations (insertions and deletions) observed in some cancers. Many human tumors have recently been found to express abnormal levels of polymerase β,
75,76 which may also contribute to their increased mutation frequency.
Multiple Mechanisms of Cisplatin Resistance
Recent studies indicate that the status of DNA repair pathways in human tumors may be highly predictive of cisplatin sensitivity. As mentioned previously, defects in the NER pathway may account for cisplatin sensitivity of some testicular and non–small-cell lung cancers.
Defects in HR repair may account for the cisplatin sensitivity of ovarian and head and neck carcinomas. Other cellular mechanisms may also account for the intrinsic cisplatin resistance of many human tumors. Cisplatin-mediated tumoricidal activity can be affected by (1) the expression of cell surface P-glycoprotein, an efflux mechanism for removing cisplatin, and (2) the relative antiapoptotic state of the tumor cell, based at least in part of the level of BCL-2 and BCL-X expression. Primary cisplatin resistance may therefore rely on the systematic assay of many of these mechanisms in a given tumor cell.
77
DNA Repair Gene Polymorphisms as Predictors of Chemotherapy Responsiveness
As described earlier, disruption of the NER pathway appears to account, at least in part, for the cisplatin hypersensitivity of testicular cancers and of some ERCC1-deficient non–small-cell lung cancers.
17 The disruption of the NER pathway may result from definitive mutations (i.e., frame-shift or nonsense mutations) in NER genes or from epigenetic changes, such as methylation and silencing of NER genes. In some cases, the disruption of the NER pathway may be partial, and it may result from DNA repair gene polymorphisms carried in the germline of the cancer patient.
In principle, DNA repair polymorphisms may result in a subtler DNA repair defect. Such a defect may increase the risk that an individual develops a cancer or may increase the likelihood that the resulting tumor is sensitive to a specific genotoxic agent. Based on this idea, investigators have screened large tumor sets for the enrichment of particular SNPs in DNA repair genes. Common SNPs are known for the NER genes XPD, ERCC1, and XRCC1. SNPs in multiple NER genes appear to account, at least in part, for the cisplatin hypersensitivity of some squamous cell carcinomas and lung cancers. Whether these SNPs will serve as
predictive biomarkers for chemotherapy or radiation sensitivity remains unproven.
Conclusion
Genomic instability is characteristic of most human malignancies, and this phenotype can arise from acquired defects in any one of six DNA repair pathways. These pathways are MMR, BER, NHEJ, NER, HR, and TLS. The germline disruption of these pathways accounts for the pathogenesis of several inherited DNA repair disorders including FA, XP, and HNPCC. The somatic disruption of these pathways can account for the genomic instability and drug sensitivity of many tumor types. The six pathways differ significantly in their ability to repair modified DNA bases and DNA crosslinks. Different cell types and tumor cell types have differential dependence on these pathways for growth and survival.
In the future, the development of biomarkers for the function of other DNA repair pathways may allow the better targeting of conventional agents or the use of monotherapies designed to inhibit specific repair pathways. The biomarkers can also be used as screening tools to find inhibitors of DNA repair that function as chemosensitizers. We predict that these approaches should reduce the toxicity of existing cancer treatments by eliminating the use of noneffective agents and by directing the development of novel treatment strategies.
Understanding the status of DNA repair pathways in tumor cells will have considerable use in clinical oncology. If a tumor is defective in one pathway (say, NHEJ), it may be directly sensitive to IR, a modality that generates double-strand breaks in DNA. If a tumor is defective in another pathway (say, HR) it may be hyperdependent on a second pathway (say, BER) for its survival. Accordingly, a drug, such as a PARP1 inhibitor, that targets the BER pathway may be selectively toxic to these tumors. Recent studies indicate new synthetic lethal relationships among DNA repair pathways. MMR-deficient tumors are hypersensitive to ATM inhibition.
78 A complete understanding of the synthetic lethal relationships of all six of the major DNA repair pathways is an important future goal of DNA repair research in cancer.
66
References
1. Lengauer C. , Kinzler K.W. , Vogelstein B. Genetic instabilities in human cancers . Nature . 1998 ; 396 ( 6712 ) : 643 – 649 .
2. Hanahan D. , Weinberg R.A. The hallmarks of cancer . Cell . 2000 ; 100 ( 1 ) : 57 – 70 .
3. Cleaver J.E. Defective repair replication of DNA in xeroderma pigmentosum . Nature . 1968 ; 218 ( 5142 ) : 652 – 656 .
4. Cleaver J.E. Xeroderma pigmentosum: a human disease in which an initial stage of DNA repair is defective . Proc Natl Acad Sci U S A . 1969 ; 63 ( 2 ) : 428 – 435 .
5. Surralles J. , Jackson S.P. , Jasin M. , Kastan M.B. , West S.C. , Joenje H. Molecular cross-talk among chromosome fragility syndromes . Genes Dev . 2004 ; 18 ( 12 ) : 1359 – 1370 .
6. Modrich P. Mechanisms in eukaryotic mismatch repair . J Biol Chem . 2006 ; 281 ( 41 ) : 30305 – 30309 .
7. Friedberg E.C. , Lehmann A.R. , Fuchs R.P. Trading places: how do DNA polymerases switch during translesion DNA synthesis? Mol Cell . 2005 ; 18 ( 5 ) : 499 – 505 .
8. Boulton S.J. Cellular functions of the BRCA tumour-suppressor proteins . Biochem Soc Trans . 2006 ; 34 ( Pt 5 ) : 633 – 645 .
9. Nojima K. , Hochegger H. , Saberi A. et al. Multiple repair pathways mediate tolerance to chemotherapeutic cross-linking agents in vertebrate cells . Cancer Res . 2005 ; 65 ( 24 ) : 11704 – 11711 .
10. Friedberg E.C. DNA damage and repair . Nature . 2003 ; 421 ( 6921 ) : 436 – 440 .
11. Bryant H.E. , Schultz N. , Thomas H.D. et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase . Nature . 2005 ; 434 ( 7035 ) : 913 – 917 .
12. Farmer H. , McCabe N. , Lord C.J. et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy . Nature . 2005 ; 434 ( 7035 ) : 917 – 921 .
13. Wilson S.H. , Sobol R.W. , Beard W.A. , Horton J.K. , Prasad R. , Vande Berg B.J. DNA polymerase beta and mammalian base excision repair . Cold Spring Harb Symp Quant Biol . 2000 ; 65 : 143 – 155 .
14. Nitta M. , Kozono D. , Kennedy R. et al. Targeting EGFR induced oxidative stress by PARP1 inhibition in glioblastoma therapy . PLoS One . 2010 ; 5 ( 5 ) : e10767 .
15. Martin S.A. , McCabe N. , Mullarkey M. et al. DNA polymerases as potential therapeutic targets for cancers deficient in the DNA mismatch repair proteins MSH2 or MLH1 . Cancer Cell . 2010 ; 17 ( 3 ) : 235 – 248 .
16. Friedberg E.C. How nucleotide excision repair protects against cancer . Nat Rev Cancer . 2001 ; 1 ( 1 ) : 22 – 33 .
17. Olaussen K.A. , Dunant A. , Fouret P. et al. DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy . N Engl J Med . 2006 ; 355 ( 10 ) : 983 – 991 .
18. Lieber M.R. , Ma Y. , Pannicke U. , Schwarz K. Mechanism and regulation of human non-homologous DNA end-joining . Nat Rev Mol Cell Biol . 2003 ; 4 ( 9 ) : 712 – 720 .
19. Fischhaber P.L. , Friedberg E.C. How are specialized (low-fidelity) eukaryotic polymerases selected and switched with high-fidelity polymerases during translesion DNA synthesis? DNA Repair (Amst) . 2005 ; 4 ( 2 ) : 279 – 283 .
20. McCulloch S.D. , Kunkel T.A. Measuring the fidelity of translesion DNA synthesis . Methods Enzymol . 2006 ; 408 : 341 – 355 .
21. Hegi M.E. , Diserens A.C. , Gorlia T. et al. MGMT gene silencing and benefit from temozolomide in glioblastoma . N Engl J Med . 2005 ; 352 ( 10 ) : 997 – 1003 .
22. Steinacher R. , Schar P. Functionality of human thymine DNA glycosylase requires SUMO-regulated changes in protein conformation . Curr Biol . 2005 ; 15 ( 7 ) : 616 – 623 .
23. Ulrich H.D. SUMO modification: wrestling with protein conformation . Curr Biol . 2005 ; 15 ( 7 ) : R257 – R259 .
24. Sugasawa K. UV-induced ubiquitylation of XPC complex, the UV-DDB-ubiquitin ligase complex, and DNA repair . J Mol Histol . 2006 ; 37 ( 5-7 ) : 189 – 202 .
25. Huang T.T. , D’Andrea A.D. Regulation of DNA repair by ubiquitylation . Nat Rev Mol Cell Biol . 2006 ; 7 ( 5 ) : 323 – 334 .
26. Chowdhury D. , Keogh M.C. , Ishii H. , Peterson C.L. , Buratowski S. , Lieberman J. Gamma-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair . Mol Cell . 2005 ; 20 ( 5 ) : 801 – 809 .
27. Nijman S.M. , Huang T.T. , Dirac A.M. et al. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway . Mol Cell . 2005 ; 17 ( 3 ) : 331 – 339 .
28. Huang T.T. , Nijman S.M. , Mirchandani K.D. et al. Regulation of monoubiquitinated PCNA by DUB autocleavage . Nat Cell Biol . 2006 ; 8 ( 4 ) : 339 – 347 .
29. Yang K. , Moldovan G.L. , Vinciguerra P. , Murai J. , Takeda S. , D’Andrea A.D. Regulation of the Fanconi anemia pathway by a SUMO-like delivery network . Genes Dev . 2011 ; 25 ( 17 ) : 1847 – 1858 .
30. Stratton M.R. Exploring the genomes of cancer cells: progress and promise . Science . 2011 ; 331 ( 6024 ) : 1553 – 1558 .
31. Smogorzewska A. , Matsuoka S. , Vinciguerra P. et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair . Cell . 2007 ; 129 ( 2 ) : 289 – 301 .
32. Paulsen R.D. , Soni D.V. , Wollman R. et al. A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability . Mol Cell . 2009 ; 35 ( 2 ) : 228 – 239 .
33. O’Connell B.C. , Adamson B. , Lydeard J.R. et al. A genome-wide camptothecin sensitivity screen identifies a mammalian MMS22L-NFKBIL2 complex required for genomic stability . Mol Cell . 2010 ; 40 ( 4 ) : 645 – 657 .
34. Rothfuss A. , Grompe M. Repair kinetics of genomic interstrand DNA cross-links: evidence for DNA double-strand break-dependent activation of the Fanconi anemia/BRCA pathway . Mol Cell Biol . 2004 ; 24 ( 1 ) : 123 – 134 .
35. Kastan M.B. , Bartek J. Cell-cycle checkpoints and cancer . Nature . 2004 ; 432 ( 7015 ) : 316 – 323 .
36. Bakkenist C.J. , Kastan M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation . Nature . 2003 ; 421 ( 6922 ) : 499 – 506 .
37. Bakkenist C.J. , Kastan M.B. Initiating cellular stress responses . Cell . 2004 ; 118 ( 1 ) : 9 – 17 .
38. Falck J. , Mailand N. , Syljuasen R.G. , Bartek J. , Lukas J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis . Nature . 2001 ; 410 ( 6830 ) : 842 – 847 .
39. Sorensen C.S. , Syljuasen R.G. , Falck J. et al. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A . Cancer Cell . 2003 ; 3 ( 3 ) : 247 – 258 .
40. Nakamura A. , Sedelnikova O.A. , Redon C. et al. Techniques for gamma-H2AX detection . Methods Enzymol . 2006 ; 409 : 236 – 250 .
41. Sedelnikova O.A. , Horikawa I. , Zimonjic D.B. , Popescu N.C. , Bonner W.M. , Barrett J.C. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks . Nat Cell Biol . 2004 ; 6 ( 2 ) : 168 – 170 .
42. Manke I.A. , Lowery D.M. , Nguyen A. , Yaffe M.B. BRCT repeats as phosphopeptide-binding modules involved in protein targeting . Science . 2003 ; 302 ( 5645 ) : 636 – 639 .
43. Yu X. , Chini C.C. , He M. , Mer G. , Chen J. The BRCT domain is a phospho-protein binding domain . Science . 2003 ; 302 ( 5645 ) : 639 – 642 .
44. Celeste A. , Petersen S. , Romanienko P.J. et al. Genomic instability in mice lacking histone H2AX . Science . 2002 ; 296 ( 5569 ) : 922 – 927 .
45. Franco S. , Gostissa M. , Zha S. et al. H2AX prevents DNA breaks from progressing to chromosome breaks and translocations . Mol Cell . 2006 ; 21 ( 2 ) : 201 – 214 .
46. Sorensen C.S. , Hansen L.T. , Dziegielewski J. et al. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair . Nat Cell Biol . 2005 ; 7 ( 2 ) : 195 – 201 .
47. Andreassen P.R. , D’Andrea A.D. , Taniguchi T. ATR couples FANCD2 monoubiquitination to the DNA-damage response . Genes Dev . 2004 ; 18 ( 16 ) : 1958 – 1963 .
48. Bartkova J. , Horejsi Z. , Koed K. et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis . Nature . 2005 ; 434 ( 7035 ) : 864 – 870 .
49. Bartkova J. , Rezaei N. , Liontos M. et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints . Nature . 2006 ; 444 ( 7119 ) : 633 – 637 .
50. Gorgoulis V.G. , Vassiliou L.V. , Karakaidos P. et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions . Nature . 2005 ; 434 ( 7035 ) : 907 – 913 .
51. Kennedy R.D. , D’Andrea A.D. DNA repair pathways in clinical practice: lessons from pediatric cancer susceptibility syndromes . J Clin Oncol . 2006 ; 24 ( 23 ) : 3799 – 3808 .
52. Giglia-Mari G. , Coin F. , Ranish J.A. et al. A new, tenth subunit of TFIIH is responsible for the DNA repair syndrome trichothiodystrophy group A . Nat Genet . 2004 ; 36 ( 7 ) : 714 – 719 .
53. Kennedy R.D. , D’Andrea A.D. The Fanconi Anemia/BRCA pathway: new faces in the crowd . Genes Dev . 2005 ; 19 ( 24 ) : 2925 – 2940 .
54. Kee Y. , D’Andrea A.D. Expanded roles of the Fanconi anemia pathway in preserving genomic stability . Genes Dev . 2010 ; 24 ( 16 ) : 1680 – 1694 .
55. Taniguchi T. , D’Andrea A.D. Molecular pathogenesis of Fanconi anemia: recent progress . Blood . 2006 ; 107 ( 11 ) : 4223 – 4233 .
56. Boudsocq F. , Benaim P. , Canitrot Y. et al. Modulation of cellular response to cisplatin by a novel inhibitor of DNA polymerase beta . Mol Pharmacol . 2005 ; 67 ( 5 ) : 1485 – 1492 .
57. Niedernhofer L.J. , Lalai A.S. , Hoeijmakers J.H. Fanconi anemia (cross)linked to DNA repair . Cell . 2005 ; 123 ( 7 ) : 1191 – 1198 .
58. Lord C.J. , Garrett M.D. , Ashworth A. Targeting the double-strand DNA break repair pathway as a therapeutic strategy . Clin Cancer Res . 2006 ; 12 ( 15 ) : 4463 – 4468 .
59. McCabe N. , Turner N.C. , Lord C.J. et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition . Cancer Res . 2006 ; 66 ( 16 ) : 8109 – 8115 .
60. Tutt A.N. , Lord C.J. , McCabe N. et al. Exploiting the DNA repair defect in BRCA mutant cells in the design of new therapeutic strategies for cancer . Cold Spring Harb Symp Quant Biol . 2005 ; 70 : 139 – 148 .
61. Sanchez-Perez I. DNA repair inhibitors in cancer treatment . Clin Transl Oncol . 2006 ; 8 ( 9 ) : 642 – 646 .
62. Hickson I. , Zhao Y. , Richardson C.J. et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM . Cancer Res . 2004 ; 64 ( 24 ) : 9152 – 9159 .
63. Syljuasen R.G. , Sorensen C.S. , Nylandsted J. , Lukas C. , Lukas J. , Bartek J. Inhibition of Chk1 by CEP-3891 accelerates mitotic nuclear fragmentation in response to ionizing radiation . Cancer Res . 2004 ; 64 ( 24 ) : 9035 – 9040 .
64. Ma C.X. , Cai S. , Li S. et al. Targeting Chk1 in p53-deficient triple-negative breast cancer is therapeutically beneficial in human-in-mouse tumor models . J Clin Invest . 2012 ; 122 ( 4 ) : 1541 – 1552 .
65. Chirnomas D. , Taniguchi T. , de la Vega M. et al. Chemosensitization to cisplatin by inhibitors of the Fanconi anemia/BRCA pathway . Mol Cancer Ther . 2006 ; 5 ( 4 ) : 952 – 961 .
66. Jenkins C. , Kan J. , Hoatlin M.E. Targeting the Fanconi anemia pathway to identify tailored anticancer therapeutics . Anemia . 2012 ; 2012 : 481 – 483 .
67. Szabo C.I. , King M.C. Population genetics of BRCA1 and BRCA2 . Am J Hum Genet . 1997 ; 60 ( 5 ) : 1013 – 1020 .
68. Miki Y. , Swensen J. , Shattuck-Eidens D. et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1 . Science . 1994 ; 266 ( 5182 ) : 66 – 71 .
69. Gudmundsdottir K. , Ashworth A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability . Oncogene . 2006 ; 25 ( 43 ) : 5864 – 5874 .
70. Scully R. , Chen J. , Ochs R.L. et al. Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage . Cell . 1997 ; 90 ( 3 ) : 425 – 435 .
71. Scully R. , Chen J. , Plug A. et al. Association of BRCA1 with Rad51 in mitotic and meiotic cells . Cell . 1997 ; 88 ( 2 ) : 265 – 275 .
72. Lorick K.L. , Jensen J.P. , Fang S. , Ong A.M. , Hatakeyama S. , Weissman A.M. RING fingers mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination . Proc Natl Acad Sci U S A . 1999 ; 96 ( 20 ) : 11364 – 11369 .
73. Yu X. , Fu S. , Lai M. , Baer R. , Chen J. BRCA1 ubiquitinates its phosphorylation-dependent binding partner CtIP . Genes Dev . 2006 ; 20 ( 13 ) : 1721 – 1726 .
74. Burr K.L. , Velasco-Miguel S. , Duvvuri V.S. , McDaniel L.D. , Friedberg E.C. , Dubrova Y.E. Elevated mutation rates in the germline of Polkappa mutant male mice . DNA Repair (Amst) . 2006 ; 5 ( 7 ) : 860 – 862 .
75. Sweasy J.B. , Lang T. , DiMaio D. Is base excision repair a tumor suppressor mechanism? Cell Cycle . 2006 ; 5 ( 3 ) : 250 – 259 .
76. Sweasy J.B. , Lauper J.M. , Eckert K.A. DNA polymerases and human diseases . Radiat Res . 2006 ; 166 ( 5 ) : 693 – 714 .
77. Bagby G.C. , Olson S.B. Cisplatin and the sensitive cell . Nat Med . 2003 ; 9 ( 5 ) : 513 – 514 .
78. Kennedy R.D. , Chen C.C. , Stuckert P. et al. Fanconi anemia pathway-deficient tumor cells are hypersensitive to inhibition of ataxia telangiectasia mutated . J Clin Invest . 2007 ; 117 ( 5 ) : 1440 – 1449 .
[/level-membership-for-hematology-oncology-and-palliative-medicine-category][not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
There is also considerable redundancy in the function of the DNA repair pathways. When one pathway is disrupted, another pathway can partially compensate, especially if the second pathway is upregulated. For instance, a cell that is deficient in HR repair may depend more on the error-prone NHEJ repair pathway for the repair of double-strand breaks. Also, thymine dimers, which are generated by UV light exposure, can be repaired by NER repair or bypassed and effectively ignored by TLS polymerases. In some cases, the absence of one DNA repair pathway results in a hyperdependence on one or more other DNA repair pathways.
11,12 This so-called synthetic lethality among DNA repair pathways has important implications for the design of new anticancer drugs (see later discussion).
The six DNA repair pathways are not constitutively activated, but instead are highly regulated. The pathways are often activated at discrete times in the cell cycle. For instance, HR repair and TLS repair are active during the S phase of the cell cycle. Also, the DNA repair pathways are differentially active in various tissues and cell types. For instance, HR and TLS are more active in rapidly growing cells, such as hematopoietic cells, whereas NHEJ is more active in postreplicative cells. Accordingly, absence of a particular DNA repair pathway may be particularly disruptive to the growth and survival of some normal tissues and some cancers. Here is a brief description of the six DNA repair pathways, with an emphasis on the enzymes in the pathways and the preference for DNA lesions repaired.
Base-Excision Repair
BER has been reviewed by Wilson.
13 The cell uses BER to correct damaged DNA bases or single-strand DNA breaks. These lesions often result from spontaneous DNA damage (DNA deamination or hydroxylation of bases) or from exposure to environmental alkylating agents. In this pathway, damaged bases are removed by one of at least 10 DNA glycosylases. The resulting apurinic/apyrimidinic (AP) sites are processed first by the Ape1 AP endonuclease, leaving a 5′ deoxyribose phosphate; then by an AP lyase activity, leaving a 3′-elimination product. Single-strand breaks are then filled in by a DNA polymerase, either with a single nucleotide or with a longer repair patch, followed by ligation. The latter events in the BER pathway are regulated by the enzyme PARP1 (polyADP ribose polymerase 1). During the DNA
damage response, the PARP1 enzyme polyADP ribosylates BER enzymes and enhances BER activity. Accordingly, tumors that have high levels of BER activity may be hypersensitive to PARP inhibitors.
14 A schematic representation of BER is shown in
Figure 4-1 .
Figure 4-1 Schematic description of base-excision repair (BER) BER is focused on small DNA lesions, often from endogenous sources, resulting in minor helix distortions. Initially, the lesion is recognized by one of the cellular DNA glycosylases, which cleaves the covalent bond between the abnormal base and the deoxyribose sugar (A). This cleavage leaves a so-called apurinic (A-P) site. Next, the apurinic endonuclease (APE) is recruited to cleave the phosphodiester backbone of the DNA (B). Finally, an error-free polymerase, Pol-beta, is engaged to replace the normal nucleotide, followed by DNA ligation (C) and restoration of the normal double-stranded DNA sequence.
Mismatch Repair (MMR)
MMR has been reviewed by Modrich.
6 MMR rapidly removes mispaired nucleotides that result from replication errors and is also involved in the detection and repair of DNA adducts such as those resulting from platinum-based chemotherapeutic agents. Initially, the heterodimeric MSH complex recognizes the nucleotide mismatch, followed by its interaction with MLH1/PMS2 and MLH1/MLH3 complexes. Several proteins participate in the process of nucleotide excision and resynthesis. Tumor cells deficient in MMR have much higher mutation frequencies than normal cells and exhibit microsatellite instability, a genomic biomarker of the underlying defect. Patients with the genetic disease HNPCC (hereditary nonpolyposis colon cancer) have germline mutations of MMR genes and are predisposed to MMR-deficient colon cancers. Recent studies suggest that MMR-deficient cells may be hypersensitive to inhibitors of various DNA polymerases, such as POLB and POLG.
15 At least six genes—MSH2, MLH1, PMS2, MSH3, MSH6, and MLH3—are involved in mismatch repair. A schematic representation of MMR is shown in
Figure 4-2 .
Figure 4-2 Schematic model of mismatch repair (MMR) Mismatch repair proteins function by sensing, binding, and repairing mistakes made during DNA replication. These mistakes include misincorporated bases and errors made during replication of microsatellite sequences (A). MutS can bind to the mismatch (B) and generate a kink in the DNA (C). This allows MutL to scan the DNA for a nearby single-strand nick in the newly replicated DNA. MutL then identifies, cleaves, and removes an oligonucleotide patch from the newly replicated strand (D,E). This allows replication and the insertion of the proper DNA base at the site of the former mismatch. Mutations in the human genes encoding homologues of these bacterial proteins play a critical role in the inherited disease hereditary nonpolyposis colon cancer (HNPCC).
Nucleotide-Excision Repair (NER)
NER acts on a variety of helix-distorting DNA lesions, caused mostly by exogenous sources that interfere with normal base pairing. This pathway may be particularly important in the response to adduct-forming chemotherapeutic agents such as platinum-based chemotherapy.
16 The primary function of NER appears to be the removal of damage such as pyrimidine dimers, which are induced by UV light. Members of the NER pathway include the XPA, XPB, XPC, XPD, XPE, and XPG proteins. Two other NER proteins, XPF and ERCC1, are especially important for the
processing of DNA crosslink repair. Recent studies indicate that monitoring the levels of these proteins in tumors may provide important biomarkers for predicting crosslinker drug sensitivity. For instance, some non–small-cell lung cancers are deficient in ERCC1, and this deficiency correlates with the cisplatin sensitivity of the specific tumor.
17
Figure 4-3 Schematic model of nucleotide excision repair (NER) NER is invoked when a base is modified by a larger helix-distorting lesion (A), such as a UV-generated thymine dimer. Initially, the bulky lesion is recognized by a sensor complex, including the XPE protein (also known as DDB2). This protein is part of a ubiquitin conjugating complex, containing Cul4A and DDB1. The complex polyubiquitinates XPC, allowing for the recruitment of the excision repair complex. Next, a patch of nucleotides is excised from the damaged DNA. In general, the excision occurs approximately 24 nucleotides 5′ to the damaged base and 3 nucleotides to the 3′ side (B). Finally, new DNA polymerization can occur, and the repaired DNA is ligated (C). The nucleotide excision repair complex is a large multisubunit complex. Mutations in genes encoding subunits of this complex underlie the human disease xeroderma pigmentosum (XP). The complex also contains proteins that can recognize and remove bases with large bulky adducts, such as those generated by polycyclic hydrocarbons and aflatoxin B1.
As for the other DNA repair pathways, these proteins cooperate to recognize and excise the damaged nucleotides and resynthesize and ligate the damaged DNA strand. In the process of NER, initially a DNA-binding component, the DDB, binds to sites of damaged DNA, such as cyclopyrimidine dimers or 6-4 photoproducts. The DDB consists of DDB1 and DDB2. Mutations in the DDB2 gene cause the genetic complementation group XPE. DDB is part of a ubiquitin E3 ligase that polyubiquitinates XPC. Polyubiquitination of XPC enhances its DNA binding. This binding sets the stage for the downstream binding of the entire excision repair complex, TFIIH, thus leading to excision of the damaged bases.
Eukaryotic NER includes two major branches, transcription-coupled repair (TCR) and global genome repair (GGR). GGR is a slow, random process of inspecting the entire genome for injuries, whereas TCR is highly specific and efficient and concentrates on damage-blocking RNA polymerase II. The two mechanisms differ in substrate specificity and recognition, and hence the enzymes involved are important nodal points for posttranslational modifications. A schematic representation of NER is shown in
Figure 4-3 .
Homologous Recombination Repair
DNA double-strand breaks (DSBs) can be caused by many different environmental factors, including reactive oxygen species, IR, and certain antineoplastic drugs, such as bleomycin, anthracyclines, and topoisomerase inhibitors. Alternatively, DSBs can result from endogenous factors, especially during normal S-phase progression. Failure to repair DSBs can lead to a number of consequences, including mutations, gross chromosomal rearrangements, and other aberrations and eventually cell death. HR is a process by which DSBs are repaired through the alignment of homologous sequences of DNA and occurs primarily during the late S to M phase of the cell cycle. Initially the RAD50, MRE11, and NBS1 complex, which possesses a 3′-5′ exonuclease activity, exposes the 3′ ends on either side of the DSB, a process that may also require BRCA1. The 3′ advancing strand from the damaged chromosome then invades the complementary sequence of the homologous chromosome. The breast cancer susceptibility protein BRCA2 and the single-strand DNA binding protein RAD51 are required for the process. The 3′ end of this strand is then extended by an HR polymerase, by reading off of this complementary sequence. After replication has extended past the region of the DSB, the 3′ end of the advancing strand returns to the original chromosome, and replication continues. A schematic representation of HR is shown in
Figure 4-4 . HR repair is especially important in the repair of DSBs and DNA interstrand crosslinks. Because some tumors, particularly breast and ovarian tumors, are defective in HR repair, drugs that cause these lesions may be particularly effective in this setting.
Nonhomologous End Joining (NHEJ)
NHEJ, which has been reviewed by Lieber et al.,
18 is another major pathway for repairing DSBs. In common with HR, this pathway is important in the repair of agents that result in DSBs such as IR, bleomycin, topoisomerase II poisons, and anthracyclines. The DNA-dependent protein kinase (DNA-PK) consists of the catalytic subunit (DNA-PKcs) and the regulatory subunit (the Ku70/Ku80 heterodimer). The DNA-PKcs subunit is a serine/threonine kinase that belongs to the phosphatidylinositol-3 kinase family. The Ku80/Ku70 heterodimer (Ku) exhibits sequence-independent affinity for double-stranded termini and, on binding to DNA, recruits and activates the DNA-PKcs catalytic subunit.
Additional proteins are required for the completion of NHEJ, including the Artemis protein and DNA ligase IV. Importantly, NHEJ is an error-prone repair pathway. Because the process does not use a complementary template, the fusion of the blunt-ended DNA duplexes may result in
deletion or insertion of base pairs. A schematic representation of NHEJ is shown in
Figure 4-5 . NHEJ has a normal function in immune cells to generate diversity at the immunoglobulin and T-cell receptor gene loci.
Figure 4-4 Schematic representation of homologous recombination (HR) HR repair is required for the normal repair of double-strand breaks (DSBs) as well as covalent interstrand DNA crosslinks. Initially, the DSB is recognized by a sensor. Some tumors have defects in HR repair, such as BRCA1 or BRCA2-deficient breast cancers. These tumor cells have prolonged time periods with unrepaired DSBs, thus leading to chromosome translocation events and a more malignant phenotype. Alternatively, the defective HR repair results in hyperdependence on the more error-prone NHEJ mechanism.
Translesion DNA Synthesis (TLS)
The process of TLS is another mechanism for dealing with thymine dimers and bases with bulky chemical adducts. At a DNA replication fork, DNA adducts may cause a replicative polymerase such as DNA polymerase delta to stall. Cells have, therefore, developed sophisticated mechanisms for switching off the replicative polymerase and switching on alternative polymerases (i.e., a polymerase such as pol eta, which will replicate past certain DNA lesions with high fidelity).
19 Interestingly, human cells have at least 15 DNA polymerases, although the situations and mechanisms of their deployment are largely unknown.
20 Cancer may have a heightened dependence on one of the error-prone TLS polymerases, such as polymerases β or kappa, accounting for high rates of mutagenesis. A schematic representation of TLS is shown in
Figure 4-6 .
Examples of Redundancy among the DNA Repair Pathways
Specific DNA repair pathways can antagonize the activity of anticancer agents. The status of a particular DNA repair pathway in a tumor may therefore predict the best antitumor therapy. As described earlier, at least two DNA repair pathways, BER and NER, are dedicated to the removal of DNA bases modified by monofunctional alkylating agents. BER can cleave the bond linking the modified base to the deoxyribose. NER, in contrast, will remove the entire modified nucleotide, along with a small stretch of surrounding nucleotides. In either case, the undamaged DNA can be used to synthesize the normal DNA sequence, followed by ligation of the segments.
Figure 4-5 Schematic representation of nonhomologous end joining (NHEJ) NHEJ is an error-prone alternative to HR repair that can also be employed to repair double strand breaks. Since NHEJ does not use a homologous DNA template, such as a sister chromatid or a homologous chromosome, it often results in the insertion or deletion of new nucleotides at the fused DSB junction. In NHEJ, the DSBs are coated by the blunt end binding protein Ku. In some cases, the blunt ends may be brought together by limited microsequence homology. The enzymes DNA-PK, XRCC4, Artemis, and DNA ligase IV are required for the successful religation of the free ends. Interestingly, NHEJ appears to be the repair mechanism used for the cleavage and religation of immunoglobulin gene variable regions; thus the error-prone religation adds to the diversity of the somatically generated Ig gene repertoire. Germline mutations in some NHEJ genes, such as DNA-PK and Artemis, result in an inherited defect in NHEJ and a severe combined immunodeficiency syndrome.
Figure 4-6 Schematic model of translesion DNA synthesis (TLS) repair TLS is not a DNA repair pathway per se; it is instead a mechanism of DNA damage bypass. In this process, an advancing replication fork encounters a damaged DNA base (A). Although the replicative polymerase (the Pol delta complex) cannot read through the damaged base, a variant polymerase such as Pol eta can bypass the lesion. Cells have developed sophisticated mechanisms for switching polymerases (B). For instance, in response to UV damage and the generation of a CPD (cyclopyrimidine dimer), the processivity factor, PCNA, becomes monoubiquitinated by RAD18. Modified PCNA now excludes Pol delta binding and has preferred binding for Pol eta. Pol eta is recruited, and it has the ability to “read through” the damaged base and insert the proper nucleotide (i.e., AA residues are replaced opposite the TT residues of a thymine dimer). Less is known about the regulation of TLS than about other DNA repair pathways. Depending on the kind of DNA damage, it is becoming increasingly clear that there are biochemical “switching” mechanisms for recruiting one of the other 12 TLS polymerases, as needed.
Cancer cells have other mechanisms for coping with modified bases. One enzyme, MGMT (0-6 methylguanine DNA methyltransferase), is capable of catalyzing the reversal of the chemical modification. Interestingly, this enzyme is switched off by MGMT gene promoter methylation in some solid tumors (gliomas and colorectal tumors), accounting, at least in part, for the hypersensitivity of these tumors to some monofunctional alkylating agents such as temozolamide.
21 In addition, damaged bases in the DNA can be bypassed through the use of TLS. Through this mechanism, the modified lesions are sensed, the normal replicative polymerase is removed from the replication fork, and a new polymerase is invoked to bypass the lesions. Rapid TLS (damage avoidance) is essential in order for a cell to transverse S phase rapidly, without succumbing to replication arrest and apoptosis. Interestingly, however, TLS is an error-prone process, and the promiscuous use of TLS by cancer cells may result in their increased mutation frequency (see later discussion).
Some of the 15 variant polymerases can extend a nascent DNA strand past a thymine dimer or past a bulky DNA lesion. Other variant polymerases can replace a single nucleotide at the site of an unpaired base. One of these variant polymerases, referred to as POLeta, is mutated in the autosomal recessive human disease XP-variant (xeroderma pigmentosum variant). Absence of the POLeta enzyme results in UV light hypersensitivity, an inability to replicate
past thymine dimers, and a predisposition to squamous cell cancers.
The variant polymerases exhibit a variable level of fidelity. Important unanswered questions in the TLS research field include the following: (1) What are the circumstances and mechanisms for recruiting the variant polymerase to a specific damaged DNA site? and (2) Are any of these variant polymerases overexpressed or dysregulated in cancer, accounting for the elevated mutation frequency of solid tumors?
As described previously, IR causes double-strand breaks as well as a wide range of oxidative DNA damage. Two redundant DNA repair pathways, HR repair and NHEJ repair, are particularly adept at dealing with double-strand break damage in a cancer cell. In clinical oncology, some tumors that have defects in these DNA repair processes are particularly sensitive to the cytolytic activity of IR. Also, radiation resistance can emerge through the induction of these DNA repair activities in treated tumor cells. Tumor cells that grow in a more hypoxic environment may also be more resistant to the killing effect of IR, perhaps because of the decrease in oxidative damage generated in these cells.
Regulation of the Six DNA Repair Pathways
As described earlier, the major proteins involved with DNA repair include sensory (DNA binding) proteins, enzymes that remove damaged bases, and enzymes that restore the normal DNA sequence. A large number of regulatory enzymes also control each DNA repair pathway. These enzymes are required for switching DNA repair on and off, as needed by the cells. Regulatory enzymes, such as helicases, serve to load DNA repair complexes at the sites of DNA damage. Other regulatory enzymes, such as topoisomerases, serve to unwind damaged DNA, in order to facilitate DNA repair complex assembly, loading into chromatin, and disassembly.
A major subclass of regulatory enzymes add critical posttranslational modifications to DNA repair enzymes. For instance, in BER, a sumoylating enzyme modifies one of the glycosylases, TDG, thereby enhancing the activity of the glycosylase in removing damages bases.
22,23 In NER, an E3 ligase complex (Cul4A, DDB1, DDB2) activates the polyubiquitination of the XPC protein. This XPC modification is a necessary event for the downstream activity of the nucleotide excision repair complex.
24 In TLS, an E3 ligase, RAD18, monoubiquitinates the DNA processivity factor, PCNA, and allows this clamp to interact with the downstream DNA polymerase Pol eta. Many of these regulatory processes have recently been reviewed.
25 Regulatory enzymes are also required to disassemble DNA repair enzymes after a repair pathway has been completed. For instance, the negative regulatory phosphatase, PP2A, removes phosphate from ATM substrates and thereby switches off the DNA damage response.
26 The deubiquitinating enzyme, USP1, can remove ubiquitin from activated FANCD2 and thereby switch off HR repair.
27 USP1 can also deubiquitinate PCNA and switch off TLS repair.
28,29 These negative regulatory events have also recently been reviewed.
25 The function of these regulatory enzymes underscores the dynamic nature of DNA repair. Loss of these regulatory mechanisms may result in the failure to (1) activate an error-free DNA repair pathway or (2) inactivate an error-prone DNA repair pathway. In either case, the consequence may be a heightened mutation frequency of the dysregulated cell and a predisposition to cancer.
Recent studies, generated through human cancer genome sequencing projects,
30 have demonstrated that some tumors, primarily melanomas and squamous cell lung cancers, have elevated levels of random point mutagenesis. This elevated point mutation signature indicates that these tumors (1) may have an underlying dysregulation of DNA repair and (2) may be more sensitive to specific therapies (see later discussion of synthetic lethality relationships). Finally, the regulation of DNA repair is a major focus of the DNA repair research field. For instance, it is unknown how DNA repair pathways are activated in specific cell types or at specific stages of the cell cycle. Because some DNA repair processes, such as HR repair, are specifically activated in S phase, it is likely that these pathways are activated by the cdk family of cyclin-dependent kinases. Several recent studies have performed shRNA screens to identify other regulatory genes that control DNA repair.
31–33 Many of these genes encode proteins that control the posttranslational modification of known DNA repair proteins or that regulate chromatin disassembly. Disruption of these gene products with shRNAs or small molecules can block DNA repair and further sensitize tumors to conventional cytotoxic cancer therapies.
Sequential Use of Three DNA Repair Pathways to Repair DNA Crosslinks
Interstrand DNA crosslinks (ICLs) make up a particular subtype of DNA lesions, and these lesions have an especially potent biological effect. Because ICLs involve the covalent modification of both strands of DNA, the lesions can prevent DNA strand separation during DNA replication. The lesions can also prevent the access of some DNA repair enzymes and transcription factors that normally require DNA strand separation for DNA binding to occur. DNA crosslinking agents, such as cisplatin derivatives (carboplatin
and oxaliplatin) and mitomycin C, are especially cytotoxic to tumor cells, and their therapeutic index derives, at least in part, from the high proliferative rate of tumor cells versus normal cells.
Figure 4-7 A schematic model of DNA crosslink repair DNA crosslink repair is believed to occur primarily during the S phase of the cell cycle. When a replication fork encounters an interstrand crosslink (A), DNA replication arrests. Initially, a double-strand break is generated by an unknown endonuclease (B). This DSB is next surrounded by phosphorylated histone 2AX. Next, another endonuclease (perhaps ERCC1/XPF) cleaves on the opposite side of the crosslink, allowing extrusion of the crosslinked bases from the double helix (C). Next, a series of three DNA repair pathways act sequentially. Translesion synthesis (TLS) repair allows bypass of the damaged bases (D). Then nucleotide excision repair (NER) excises the damaged oligonucleotide and allows gap filling (E). Finally, end resection occurs, and the double-strand break can be repaired by homologous recombination (HR). The replication fork is regenerated in an error-free mechanism (F).
The mechanism of DNA crosslink repair in human cells is poorly understood, and our understanding to date is derived more from the study of crosslink repair in prokaryotes and in the yeast
Saccharomyces cerevisiae. As shown in
Figure 4-7 , crosslink repair in human cells probably requires multiple DNA repair pathways. According to this model, the ICL is only repaired during S phase progression. Initially, an advancing replication fork encounters an ICL. An unknown endonuclease cleaves the DNA, thus generating a double-strand break (DSB). Next, a second endonuclease is invoked to cleave the DNA after the DNA crosslink. Recent data suggest that this endonuclease may be made up of the ERCC1/XPF proteins.
Now that an endonucleolytic event has occurred on each side of the crosslink, the crosslinked single-strand fragment can be flipped out of the helix. This allows three of the normal DNA repair pathways to work sequentially. First, TLS allows bypass of the crosslink and replication and ligation of the upper double helix. Recent studies indicate that some variant polymerases, such as POL eta, are particularly important to the translesion synthesis across MMC adducts. Next, the NER pathway can excise a stretch of damaged DNA and allow gap filling of the excised oligonucleotide. Finally, HR repair can be used for the error-free, template-driven repair of the damage. The end result of this sequential use of three independent DNA repair pathways is to resume DNA replication and restart the replication fork.
Consistent with this model of crosslink repair, some repair-deficient cells are especially prone to the cytotoxic effects of DNA crosslinking drugs. For instance, cells that are
deficient in ERCC1/XPF generate the first double-strand break upstream of the DNA crosslink. However, these double-strand breaks, as measured indirectly by the presence of histone 2AX foci, persist in the repair-deficient cells, suggesting that ERCC1/XPF may work farther downstream in the pathway. Similarly, cells deficient in the FA pathway have persistent double-strand breaks after MMC exposure.
34 Thus, the presence or absence of the double-strand break intermediates is helpful in determining the level at which a repair process is disrupted and the sequence of repair events in the pathway.
DNA Repair and the DNA Damage Response
DNA repair is, in fact, only one class of a broader set of cellular responses referred to as the DNA damage response. DNA damage responses include the activation of cell cycle checkpoints, the activation of apoptosis, and the activation of DNA damage tolerance. This last mechanism allows a cell to “accept” DNA damage and continue DNA replication even in the setting of a heightened mutation frequency. The DNA damage response is therefore a highly coordinated set of signaling events. These responses require a DNA damage sensor (such as a sensor kinase, ATM, or ATR) and an effector kinase, as well as downstream protein machines dedicated to DNA repair, apoptosis, or checkpoint activities.
35
The DNA Damage Response Is Mediated by Sensor and Effector Kinases
The DNA damage response can be activated by a wide range of environmental exposures or drug interactions. An important early player in the damage response is the molecular “sensor” of DNA damage. A local distortion in the DNA double helix, perhaps resulting from a DNA adduct or a thymine dimer, can activate a sensor kinase, such as ATM, ATR, or DNA-PK. These kinases are believed to autophosphorylate
36,37 and go on to phosphorylate a large number of substrates thereafter. The ATM kinase is the product of the ATM gene, the gene mutated in the cancer susceptibility disorder ataxia-telangiectasia.
Activated ATM and ATR proteins phosphorylate additional downstream “effector” kinases, such as the checkpoint kinases, Chk1 and Chk2. Activated Chk1 and Chk2 then go on to phosphorylate a wide array of protein targets involved in the machinery of DNA repair or DNA damage checkpoints.
One of the best-characterized DNA damage checkpoints is regulated by the ATM/Chk2/Cdc25A axis.
38,39 In response to IR, a double-strand break is generated, and this break activates ATM. ATM subsequently phosphorylates Chk2, which in turn phosphorylates the cell cycle activator cdc25A. Cdc25A phosphorylation leads to its rapid degradation and to cell-cycle arrest. This appears to be an important mechanism by which a cell can respond to DNA damage by arresting its cell cycle progression in S phase. By stopping S-phase entry, a cell allows itself the opportunity to slow down and to repair its DNA or, in the setting of severe damage, to undergo apoptosis. Importantly, a failure to activate this checkpoint response, as in ATM-deficient cells, results in S-phase progression even in the setting of DNA damage. Continuing to replicate DNA in the setting of DNA damage has dire consequences for the cells. The cell may have an elevated mutation rate or may complete DNA replication, only to experience a mitotic catastrophe at the end of the cell cycle.
Failure of ATM to activate the intra–S-phase checkpoint results in a characteristic cellular phenotype. When ATM-deficient cells are exposed to IR, they fail to arrest in S phase but instead continue to replicate their DNA and to incorporate tritiated thymidine in the postradiation period. This phenotype is known as radioresistant DNA synthesis (RDS), and it is the hallmark of a cell with a defect in the ATM/Chk2/cdc25A axis. An active area of DNA repair research is the identification of other CHK1 and CHK2 phosphorylated substrates.
Phosphorylated Effector Proteins Assemble in DNA Damage Foci
An important downstream event in the DNA damage response is the assembly of proteins in subnuclear foci.
40,41 These foci are often referred to as IRIFs (ionizing radiation inducible foci). Multiple ATM and ATR phosphorylated substrates, such as Chk1, BRCA1, and BARD1, assemble in foci following DNA damage. The assembly of these large protein complexes is mediated, at least in part, by the phosphorylated SQ or TQ sequences of the ATM/ATR substrates. Recent studies have indicated that these phosphorylated amino acid residues bind directly to phosphoamino acid receptors found on other adaptor proteins. For instance, phosphorylated BACH1 can bind directly to the BRCT domain (a phosphoserine receptor) of the BRCA1 protein.
42,43 The precise structure and function of these protein foci in eukaryote nuclei is not known. Clearly, the number of foci correlates with the number of unprocessed double-strand DNA breaks, and the foci are widely believed to be sites of double-strand break repair.
From immunofluorescence analysis, it is clear that multiple phosphorylated DNA-damage activated proteins colocalize in these foci. The foci have been helpful to researchers in the establishment of signaling pathways. For instance, pATM, pBRCA1, and pFANCD2 colocalize in IRIFs. Disruption of one upstream protein, say, by a germline or acquired mutation in the upstream signaling protein, ATM, results in loss of downstream proteins in the foci. The assembly of the foci therefore has become a useful tool in understanding the interrelationships of DNA response proteins.
A few DNA damage-response proteins deserve special attention here. Bonner and colleagues
40 have identified a variant histone protein, histone 2AX, that is rapidly phosphorylated by ATM after radiation damage. H2AX is an important early signaling protein in the DNA damage response. The phospho H2AX protein is incorporated in chromatin in vast stretches emanating from the site of the DNA DSB. An absence of histone 2AX, as in an H2AX knockout mouse model, results in chromosome instability and cancer predisposition,
44,45 apparently due to failure to mount the proper DNA damage response.
Another important DNA damage response protein is RAD51, which is phosphorylated by the Chk1 kinase during normal S-phase progression.
46 RAD51 is a single-strand DNA binding protein that plays a critical role in DNA repair by HR. Phosphorylated RAD51 also assembles in foci during normal S-phase progression. These “replication foci” are believed to be sites of DNA repair by HR between sister chromatids, which occurs during normal DNA replication.
A comprehensive analysis of proteins that are rapidly phosphorylated after DNA damage (and that form nuclear foci) has provided an important database for laboratories studying the DNA damage response. These phosphorylated proteins, and foci, provide a useful set of biomarkers for DNA repair activities. For instance, cells that are defective in the formation of DNA repair foci are themselves defective in DNA repair. The specific kind of focus that is absent correlates with the particular kind of DNA repair deficiency. For instance, cells deficient in RAD51 foci are defective in HR repair and are hypersensitive to IR. Cells defective in the assembly of polyADP ribose (PAR) foci are defective in the repair of single-strand breaks and may therefore have an underlying defect in BER. As such, tumor cells missing particular types of DNA repair foci may be more sensitive to certain kinds of chemotherapy or radiation.
Importantly, human cancers are often deficient in the DNA damage response. Germline mutations in DNA damage response genes, such as ATM, NBS1, FANCD2, BRCA1, and BRCA2, can result in an increased susceptibility to cancer. Individuals who inherit a single mutant allele of, say, BRCA1, have a high risk of developing a breast, ovarian, or prostate cancer during their lifetime. The tumor results from the inactivation of the second BRCA1 allele, through deletion and loss of heterozygosity, thus resulting in a tumor with a specific DNA repair defect. BRCA(−/−) tumors therefore have genomic instability, but also have increased sensitivity to some DNA-damaging agents such as IR and DNA crosslinkers.
Study of the DNA damage response reveals that cells have highly regulated responses to different levels and types of DNA damage. Whereas some DNA repair pathways may be viewed as constitutive, housekeeping pathways, other pathways are highly controlled. Some DNA repair pathways, such as ATR and CHK1, are activated primarily at the site of the advancing replication fork, leading to the activation of HR repair.
47 Other DNA repair processes are activated in nondividing cells, such as in postmitotic neurons. For instance, NHEJ is hyperactive in nondividing cells and functions as the major mechanism of double-strand break repair in these cells.
The cellular context of the DNA repair pathway is also important. Germline or somatic disruption of a pathway may result in a strikingly different phenotype, depending on the cell and tissue of origin. For instance, gene line disruption of a DNA damage response, as in the inherited disease ataxia-telangiectasia, may lead to a characteristic constellation of clinical findings, including cerebellar degeneration and lymphoma predisposition. A somatic disruption of the same pathway (say, ATM CHk2-p53) may lead to a very different set of cancers, such as solid tumors of the bladder and ovary.
48,49 Recent studies indicate that the DNA damage response provides an important “barrier” to the transformation of a normal cell to a malignant cell.
48,50 Specifically, early premalignant cells have heightened constitutive activation of the DNA damage response pathways, as exemplified by increased immunohistochemical staining with antibodies to activated ATM and to the activated checkpoint kinase CHK2. Interestingly, as cells progress from the premalignant state to the malignant state, they lose these DNA damage responses, perhaps through acquired disruptions of ATM or CHK2 activity. Because individuals with genetic diseases such as ataxia-telangiectasia already have a defect in the checkpoint response, they may be prone to earlier onset of cancers for this reason.
Inherited Chromosome Instability Syndromes as Models for DNA Repair Defects
Rare pediatric chromosome instability disorders, such as FA and XP, provide important insights into the function
of DNA repair pathways and their role in cancers in the general population. Children born with these syndromes generally have congenital abnormalities, cellular hypersensitivity to DNA-damaging agents, genomic instability, and an increased risk of specific cancers. Although these syndromes are rare, the DNA repair pathways disrupted by germline mutations in these individuals are often the same pathways disrupted by somatic mutation or epigenetic inactivation in cancers from the general population. For these sporadic cancers, a knowledge of which DNA repair mechanism is disrupted provides important clues to the behavior of the cancer or its drug sensitivity spectrum.
At least five of the major DNA repair pathways have corresponding inherited human diseases (
Table 4-1). HR and TLS repair is defective in FA cells.
51 NER repair is defective in XP cells, Cockayne syndrome (CS) cells, and trichothiodystrophy cells.
52 MMR repair is defective in children with Turcot syndrome and in tumor cells derived from adult patients with HNPCC. TLS repair is defective in patients with the XP-V (xeroderma pigmentosum variant) disease. Most of these pediatric diseases exhibit autosomal recessive inheritance, such as XP, FA, and CS. Turcot syndrome has been reported to exhibit autosomal dominant or autosomal recessive inheritance depending on the particular mutation affecting mismatch repair. Inherited mutations in BER genes have not been observed in humans, suggesting that this pathway is essential for human development.
It is interesting that patients with inherited DNA repair syndromes, such as CS and FA, have congenital abnormalities. For instance, CS patients have developmental abnormalities of the skin and skeletal system. FA patients have skeletal, kidney, cardiac, and bone marrow defects. Consistent with these findings, the NER and FA pathways appear to play dual roles. For instance, the NER excision repair complex, TFIIH, plays an important transcriptional role during embryonic development. Germline dysfunction therefore leads to defects during embryonic organogenesis. The NER complex also plays a critical role in DNA repair in somatic cells after organism development. Similarly, the FA pathway appears to have a dual role in both development and DNA repair in somatic cells.
The systematic study of these rare diseases has led to (1) a better understanding of the genes and proteins involved in the six major DNA repair pathways, (2) how an inherited (or germline) defect in a DNA repair pathway can lead to genomic instability, cancer progression, and drug hypersensitivity, and (3) how an acquired (or somatic) defect in a DNA repair pathway can influence tumor progression and drug sensitivity of tumors in the general population. Although the specific details of these individual inherited diseases is beyond the scope of this review, an example of how a study of these rare diseases can lead to general insights to tumor biology can be appreciated from recent insights into the FA pathway.
Fanconi Anemia: A Specific Inherited DNA Repair Defect
FA is an autosomal recessive or X-linked recessive cancer susceptibility syndrome characterized by multiple congenital abnormalities, progressive bone marrow failure, and cellular hypersensitivity to DNA crosslinking agents, such as cisplatin and mitomycin C (MMC). FA patients are prone to developing acute myeloid leukemia as well as squamous cell carcinomas of the head and neck or gynecologic system.
53 The study of FA cells has led to the elucidation of a DNA repair pathway for interstrand crosslinks. Clinically, this pathway is particularly important because many DNA crosslinking agents such as cisplatin or mitomycin C are used for cancer treatment. The FA defect results from biallelic mutation of any one of 15 known FA genes (A, B, C, D1, D2, E, F, G, I, J, L, M, N, O, P). The proteins encoded by these FA genes cooperate in a common DNA repair pathway, referred to as the FA/BRCA pathway.
53,54 A central event in this pathway is the monoubiquitination of the FANCD2 protein, and this event is a useful biomarker for DNA repair activity (see later discussion). Disruption of this pathway results in the characteristic clinical and cellular phenotype of FA patients.
Patients with an Inherited Germline DNA Repair Deficiency Exhibit a Characteristic Tumor Spectrum
Patients with inherited DNA repair deficiency syndromes are prone to the development of specific tumors. FA patients, for example, are predisposed to acute myeloid leukemia and squamous cell carcinomas, primarily of the head and neck or gynecologic system. Patients with XP are prone to skin squamous cell carcinomas, primarily on body surfaces with more sunlight exposure. Patients with HNPCC and an inherited MMR deficiency are prone to colon cancer and ovarian cancer.
Tumors arising from somatic disruptions of DNA repair pathways may arise in other organ systems. A specific oncogenic lesion, such as the activation of an oncogene or the disruption of a tumor suppressor gene, may have a vastly different effect depending on the cellular context of the lesion. For instance, a germline mutation in the Rb gene may result in an embryonal tumor, such as a retinoblastoma or a pineoblastoma, but a somatic disruption of the Rb gene may lead to the development of a sarcoma.
Similarly, disruptions of a DNA repair pathway by a germline mechanism, rather than a somatic mechanism, may yield a very different spectrum and behavior. Examples are shown in
Table 4-1. Somatic disruption of the FA pathway results in a wide range of tumor types, including tumors of the ovary, lung, and cervix.
51,55 Moreover, somatic disruptions result from methylation and silencing of an upstream FA gene (FANCF). Germline disruption of the same genes results from inherited mutations, such as missense mutations or nonsense mutations. Somatic disruption of the NER pathway plays a role in the development of testicular cancer and appears to account for the hypersensitivity of this tumor to the drug cisplatin. Paradoxically, somatic disruption of a DNA repair pathway can also result in chemotherapy resistance. Studies indicate that methylation and silencing of the MLH1 gene may account, at least in part, for the cisplatin resistance of some ovarian tumors.
Disruption of the other DNA repair pathways has been observed in sporadic human tumors, accounting, at least in part, for the specific drug and radiation sensitivity spectrum of these tumors and their clinical outcome. HR is disrupted in breast and ovarian cancer, NER is disrupted in testicular cancer, and MMR is disrupted in sporadic colon cancer. A few studies suggest that TLS may be disrupted in human cancers. Human cancer cells exhibit an elevation in spontaneous and damage-inducible point mutagenesis, compared to nonmalignant cells, suggesting an underlying TLS defect. Recent studies indicate that an elevation in the expression and activity of the error-prone polymerase Pol beta accounts for the increase in cisplatin resistance and mutagenesis of these cancers. Consistent with this hypothesis, inhibition of Pol beta in these cells results in resensitization to cisplatin.
56
Somatic Disruption of DNA Repair Pathways by Methylation and Gene Silencing
One of the most common mechanisms of inactivation of DNA repair pathways in sporadic cancer is the epigenetic silencing of a critical gene through methylation of the promoter region. Increasing evidence shows that the FA/BRCA pathway is one of the DNA repair mechanisms that is targeted in sporadic cancers.
FANCF methylation occurs in 24% of ovarian granulosa cell tumors, 30% of cervical cancer, 14% of squamous cell head and neck cancers, 6.7% of germ cell tumors of the testis, and 15% of non–small-cell lung cancers, where it correlates with a worse prognosis. An example of how methylation of a DNA repair gene can promote tumor progression is shown in
Figure 4-8 .
By regulating the activity of DNA repair pathways, cancer cells have a propensity to progress to a more malignant state. According to this model, early in the course of tumorigenesis, a premalignant cell may undergo a methylation and silencing of a DNA repair gene. In the case of the FA/BRCA pathway, the gene most commonly silenced by methylation is the
FANCF gene on chromosome 11p15. Inactivation of
FANCF results in a disruption of DNA repair and in genomic instability. The premalignant cell is therefore prone to multiple oncogenic events, such as the upregulation of a tyrosine kinase oncogene or the disruption of p53. A tumor with multiple somatic mutations eventually develops (see
Figure 4-8), but this tumor still has a defective DNA repair pathway and is hypersensitive to genotoxic chemotherapy. After antitumor therapy, however, there is a selective pressure for tumor cells with an intact FA/BRCA
pathway. Tumor cells with a demethylated
FANCF gene are selected, and a drug-resistant tumor emerges. By following this pattern, tumors can silence and reactivate DNA repair pathways, leading to drug resistance and tumor progression.
Figure 4-8 Tumor progression by serial inactivation and reactivation of DNA repair pathways
Buy Membership for Hematology, Oncology and Palliative Medicine Category to continue reading.
Learn more here
[/not-level-membership-for-hematology-oncology-and-palliative-medicine-category]
The Molecular Basis of Cancer Expert Consult - Online