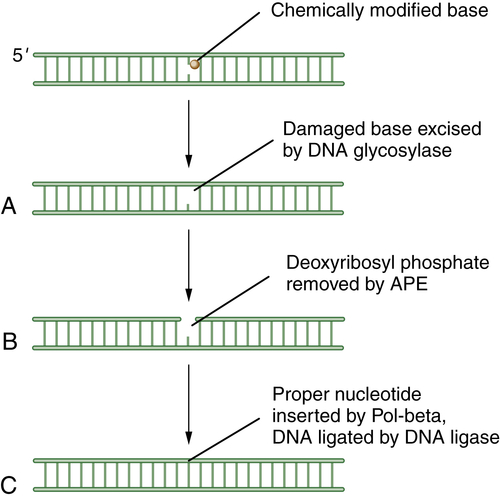
Base-Excision Repair
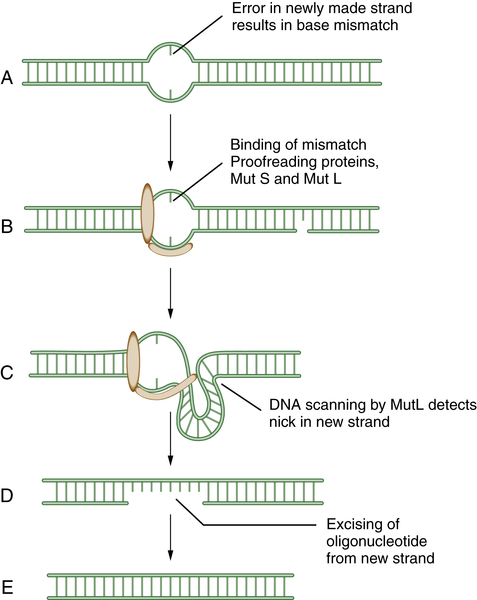
Mismatch Repair (MMR)
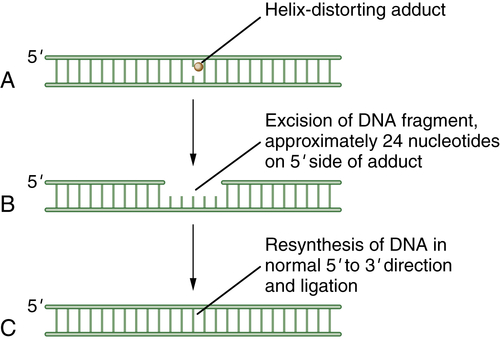
Nucleotide-Excision Repair (NER)
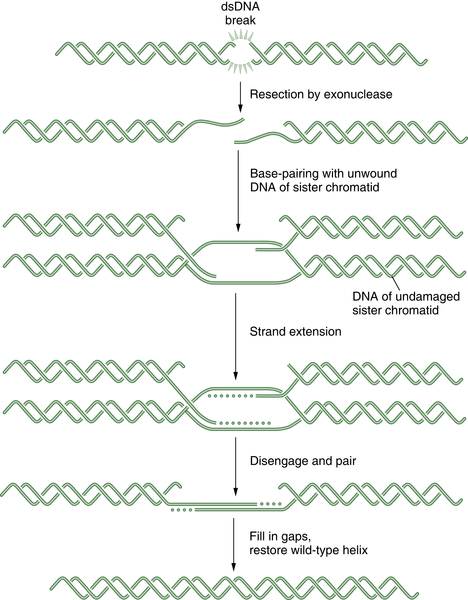
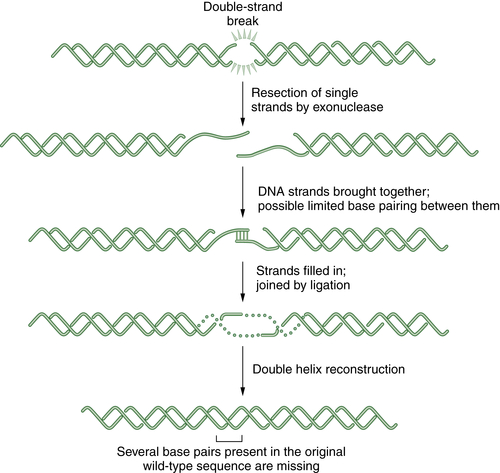
Homologous Recombination Repair
Nonhomologous End Joining (NHEJ)
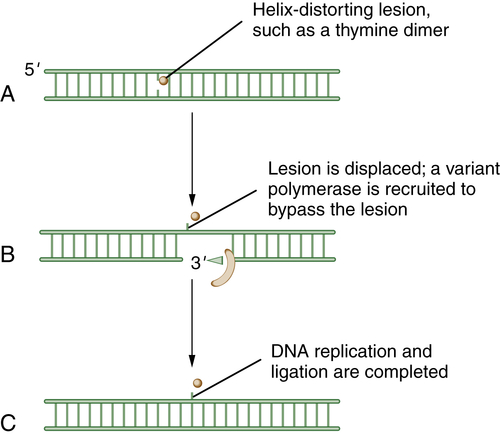
Translesion DNA Synthesis (TLS)
Examples of Redundancy among the DNA Repair Pathways
Regulation of the Six DNA Repair Pathways
Sequential Use of Three DNA Repair Pathways to Repair DNA Crosslinks
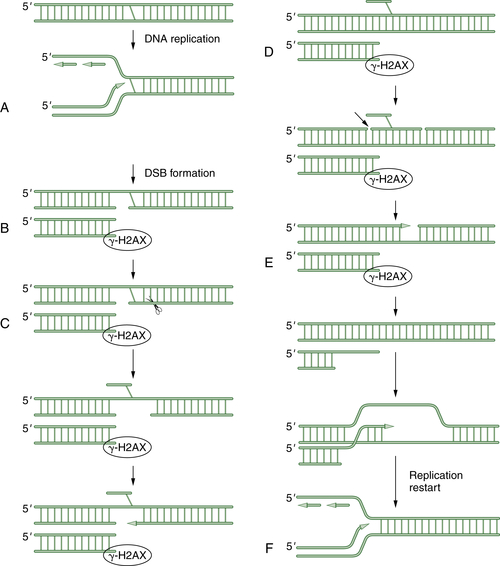
DNA Repair and the DNA Damage Response
The DNA Damage Response Is Mediated by Sensor and Effector Kinases
Phosphorylated Effector Proteins Assemble in DNA Damage Foci
Inherited Chromosome Instability Syndromes as Models for DNA Repair Defects
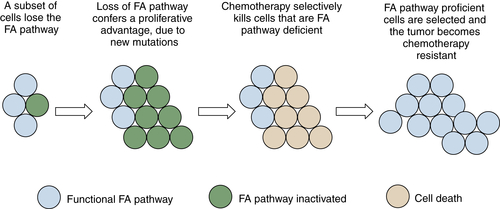
Fanconi Anemia: A Specific Inherited DNA Repair Defect
Patients with an Inherited Germline DNA Repair Deficiency Exhibit a Characteristic Tumor Spectrum
Somatic Disruption of DNA Repair Pathways by Methylation and Gene Silencing
Prognostic and Predictive DNA Repair Biomarkers in Cancer Treatment
Selection of Biomarkers of DNA Repair Pathways
Functional DNA Repair Biomarkers
DNA Repair Biomarkers of Gene/Protein Expression
Clinical Application of DNA Repair Biomarkers
DNA Repair Biomarkers as Predictors of Response to Conventional Therapy
DNA Repair Biomarkers to Guide Chemo- and Radiosensitization
DNA Repair Biomarkers as Predictors of Response to Targeted Monotherapy
The Development of New DNA Repair Biomarkers
DNA Repair Inhibitors as a New Area for Anticancer Drug Development
Importance of DNA Repair to Clinical Oncology: Other Specific Examples
BRCA1 and BRCA2
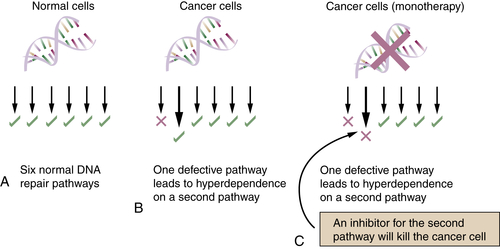
Defects in DNA Repair Pathways Can Account for the Elevated Mutation Rate of Cancer
Multiple Mechanisms of Cisplatin Resistance
DNA Repair Gene Polymorphisms as Predictors of Chemotherapy Responsiveness
Conclusion
1. Genetic instabilities in human cancers . Nature . 1998 ; 396 ( 6712 ) : 643 – 649 .
2. The hallmarks of cancer . Cell . 2000 ; 100 ( 1 ) : 57 – 70 .
3. Defective repair replication of DNA in xeroderma pigmentosum . Nature . 1968 ; 218 ( 5142 ) : 652 – 656 .
4. Xeroderma pigmentosum: a human disease in which an initial stage of DNA repair is defective . Proc Natl Acad Sci U S A . 1969 ; 63 ( 2 ) : 428 – 435 .
5. Molecular cross-talk among chromosome fragility syndromes . Genes Dev . 2004 ; 18 ( 12 ) : 1359 – 1370 .
6. Mechanisms in eukaryotic mismatch repair . J Biol Chem . 2006 ; 281 ( 41 ) : 30305 – 30309 .
7. Trading places: how do DNA polymerases switch during translesion DNA synthesis? Mol Cell . 2005 ; 18 ( 5 ) : 499 – 505 .
8. Cellular functions of the BRCA tumour-suppressor proteins . Biochem Soc Trans . 2006 ; 34 ( Pt 5 ) : 633 – 645 .
9. Multiple repair pathways mediate tolerance to chemotherapeutic cross-linking agents in vertebrate cells . Cancer Res . 2005 ; 65 ( 24 ) : 11704 – 11711 .
10. DNA damage and repair . Nature . 2003 ; 421 ( 6921 ) : 436 – 440 .
11. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase . Nature . 2005 ; 434 ( 7035 ) : 913 – 917 .
12. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy . Nature . 2005 ; 434 ( 7035 ) : 917 – 921 .
13. DNA polymerase beta and mammalian base excision repair . Cold Spring Harb Symp Quant Biol . 2000 ; 65 : 143 – 155 .
14. Targeting EGFR induced oxidative stress by PARP1 inhibition in glioblastoma therapy . PLoS One . 2010 ; 5 ( 5 ) : e10767 .
15. DNA polymerases as potential therapeutic targets for cancers deficient in the DNA mismatch repair proteins MSH2 or MLH1 . Cancer Cell . 2010 ; 17 ( 3 ) : 235 – 248 .
16. How nucleotide excision repair protects against cancer . Nat Rev Cancer . 2001 ; 1 ( 1 ) : 22 – 33 .
17. DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy . N Engl J Med . 2006 ; 355 ( 10 ) : 983 – 991 .
18. Mechanism and regulation of human non-homologous DNA end-joining . Nat Rev Mol Cell Biol . 2003 ; 4 ( 9 ) : 712 – 720 .
19. How are specialized (low-fidelity) eukaryotic polymerases selected and switched with high-fidelity polymerases during translesion DNA synthesis? DNA Repair (Amst) . 2005 ; 4 ( 2 ) : 279 – 283 .
20. Measuring the fidelity of translesion DNA synthesis . Methods Enzymol . 2006 ; 408 : 341 – 355 .
21. MGMT gene silencing and benefit from temozolomide in glioblastoma . N Engl J Med . 2005 ; 352 ( 10 ) : 997 – 1003 .
22. Functionality of human thymine DNA glycosylase requires SUMO-regulated changes in protein conformation . Curr Biol . 2005 ; 15 ( 7 ) : 616 – 623 .
23. SUMO modification: wrestling with protein conformation . Curr Biol . 2005 ; 15 ( 7 ) : R257 – R259 .
24. UV-induced ubiquitylation of XPC complex, the UV-DDB-ubiquitin ligase complex, and DNA repair . J Mol Histol . 2006 ; 37 ( 5-7 ) : 189 – 202 .
25. Regulation of DNA repair by ubiquitylation . Nat Rev Mol Cell Biol . 2006 ; 7 ( 5 ) : 323 – 334 .
26. Gamma-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair . Mol Cell . 2005 ; 20 ( 5 ) : 801 – 809 .
27. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway . Mol Cell . 2005 ; 17 ( 3 ) : 331 – 339 .
28. Regulation of monoubiquitinated PCNA by DUB autocleavage . Nat Cell Biol . 2006 ; 8 ( 4 ) : 339 – 347 .
29. Regulation of the Fanconi anemia pathway by a SUMO-like delivery network . Genes Dev . 2011 ; 25 ( 17 ) : 1847 – 1858 .
30. Exploring the genomes of cancer cells: progress and promise . Science . 2011 ; 331 ( 6024 ) : 1553 – 1558 .
31. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair . Cell . 2007 ; 129 ( 2 ) : 289 – 301 .
32. A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability . Mol Cell . 2009 ; 35 ( 2 ) : 228 – 239 .
33. A genome-wide camptothecin sensitivity screen identifies a mammalian MMS22L-NFKBIL2 complex required for genomic stability . Mol Cell . 2010 ; 40 ( 4 ) : 645 – 657 .
34. Repair kinetics of genomic interstrand DNA cross-links: evidence for DNA double-strand break-dependent activation of the Fanconi anemia/BRCA pathway . Mol Cell Biol . 2004 ; 24 ( 1 ) : 123 – 134 .
35. Cell-cycle checkpoints and cancer . Nature . 2004 ; 432 ( 7015 ) : 316 – 323 .
36. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation . Nature . 2003 ; 421 ( 6922 ) : 499 – 506 .
37. Initiating cellular stress responses . Cell . 2004 ; 118 ( 1 ) : 9 – 17 .
38. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis . Nature . 2001 ; 410 ( 6830 ) : 842 – 847 .
39. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A . Cancer Cell . 2003 ; 3 ( 3 ) : 247 – 258 .
40. Techniques for gamma-H2AX detection . Methods Enzymol . 2006 ; 409 : 236 – 250 .
41. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks . Nat Cell Biol . 2004 ; 6 ( 2 ) : 168 – 170 .
42. BRCT repeats as phosphopeptide-binding modules involved in protein targeting . Science . 2003 ; 302 ( 5645 ) : 636 – 639 .
43. The BRCT domain is a phospho-protein binding domain . Science . 2003 ; 302 ( 5645 ) : 639 – 642 .
44. Genomic instability in mice lacking histone H2AX . Science . 2002 ; 296 ( 5569 ) : 922 – 927 .
45. H2AX prevents DNA breaks from progressing to chromosome breaks and translocations . Mol Cell . 2006 ; 21 ( 2 ) : 201 – 214 .
46. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair . Nat Cell Biol . 2005 ; 7 ( 2 ) : 195 – 201 .
47. ATR couples FANCD2 monoubiquitination to the DNA-damage response . Genes Dev . 2004 ; 18 ( 16 ) : 1958 – 1963 .
48. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis . Nature . 2005 ; 434 ( 7035 ) : 864 – 870 .
49. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints . Nature . 2006 ; 444 ( 7119 ) : 633 – 637 .
50. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions . Nature . 2005 ; 434 ( 7035 ) : 907 – 913 .
51. DNA repair pathways in clinical practice: lessons from pediatric cancer susceptibility syndromes . J Clin Oncol . 2006 ; 24 ( 23 ) : 3799 – 3808 .
52. A new, tenth subunit of TFIIH is responsible for the DNA repair syndrome trichothiodystrophy group A . Nat Genet . 2004 ; 36 ( 7 ) : 714 – 719 .
53. The Fanconi Anemia/BRCA pathway: new faces in the crowd . Genes Dev . 2005 ; 19 ( 24 ) : 2925 – 2940 .
54. Expanded roles of the Fanconi anemia pathway in preserving genomic stability . Genes Dev . 2010 ; 24 ( 16 ) : 1680 – 1694 .
55. Molecular pathogenesis of Fanconi anemia: recent progress . Blood . 2006 ; 107 ( 11 ) : 4223 – 4233 .
56. Modulation of cellular response to cisplatin by a novel inhibitor of DNA polymerase beta . Mol Pharmacol . 2005 ; 67 ( 5 ) : 1485 – 1492 .
57. Fanconi anemia (cross)linked to DNA repair . Cell . 2005 ; 123 ( 7 ) : 1191 – 1198 .
58. Targeting the double-strand DNA break repair pathway as a therapeutic strategy . Clin Cancer Res . 2006 ; 12 ( 15 ) : 4463 – 4468 .
59. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition . Cancer Res . 2006 ; 66 ( 16 ) : 8109 – 8115 .
60. Exploiting the DNA repair defect in BRCA mutant cells in the design of new therapeutic strategies for cancer . Cold Spring Harb Symp Quant Biol . 2005 ; 70 : 139 – 148 .
61. DNA repair inhibitors in cancer treatment . Clin Transl Oncol . 2006 ; 8 ( 9 ) : 642 – 646 .
62. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM . Cancer Res . 2004 ; 64 ( 24 ) : 9152 – 9159 .
63. Inhibition of Chk1 by CEP-3891 accelerates mitotic nuclear fragmentation in response to ionizing radiation . Cancer Res . 2004 ; 64 ( 24 ) : 9035 – 9040 .
64. Targeting Chk1 in p53-deficient triple-negative breast cancer is therapeutically beneficial in human-in-mouse tumor models . J Clin Invest . 2012 ; 122 ( 4 ) : 1541 – 1552 .
65. Chemosensitization to cisplatin by inhibitors of the Fanconi anemia/BRCA pathway . Mol Cancer Ther . 2006 ; 5 ( 4 ) : 952 – 961 .
66. Targeting the Fanconi anemia pathway to identify tailored anticancer therapeutics . Anemia . 2012 ; 2012 : 481 – 483 .
67. Population genetics of BRCA1 and BRCA2 . Am J Hum Genet . 1997 ; 60 ( 5 ) : 1013 – 1020 .
68. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1 . Science . 1994 ; 266 ( 5182 ) : 66 – 71 .
69. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability . Oncogene . 2006 ; 25 ( 43 ) : 5864 – 5874 .
70. Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage . Cell . 1997 ; 90 ( 3 ) : 425 – 435 .
71. Association of BRCA1 with Rad51 in mitotic and meiotic cells . Cell . 1997 ; 88 ( 2 ) : 265 – 275 .
72. RING fingers mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination . Proc Natl Acad Sci U S A . 1999 ; 96 ( 20 ) : 11364 – 11369 .
73. BRCA1 ubiquitinates its phosphorylation-dependent binding partner CtIP . Genes Dev . 2006 ; 20 ( 13 ) : 1721 – 1726 .
74. Elevated mutation rates in the germline of Polkappa mutant male mice . DNA Repair (Amst) . 2006 ; 5 ( 7 ) : 860 – 862 .
75. Is base excision repair a tumor suppressor mechanism? Cell Cycle . 2006 ; 5 ( 3 ) : 250 – 259 .
76. DNA polymerases and human diseases . Radiat Res . 2006 ; 166 ( 5 ) : 693 – 714 .
77. Cisplatin and the sensitive cell . Nat Med . 2003 ; 9 ( 5 ) : 513 – 514 .
78. Fanconi anemia pathway-deficient tumor cells are hypersensitive to inhibition of ataxia telangiectasia mutated . J Clin Invest . 2007 ; 117 ( 5 ) : 1440 – 1449 .






