Chapter 21 Diuretic Drugs
| Abbreviations | |
|---|---|
| ATP | Adenosine triphosphate |
| ECF | Extracellular fluid |
| ENaC | Epithelial Na+ channel |
| GFR | Glomerular filtration rate |
| GI | Gastrointestinal |
| IV | Intravenous |
| NCC | Na+/Cl− cotransporter protein |
| NHE3 | Na+/H+ exchange (antiport) protein |
| NKCC2 | Na+/K+/2Cl− cotransporter protein |
Therapeutic Overview
not a true tolerance but results from activation of compensatory salt-retaining mechanisms. Specifically, contraction of the ECF volume activates the sympathetic nervous system (see Chapter 19), with a resultant increase in release of angiotensin II, aldosterone, and antidiuretic hormone that may lead to a compensatory increase in Na+ reabsorption. Moreover, continued delivery of Na+ to more distal nephron segments induced by loop diuretics,
| Therapeutic Overview | |
|---|---|
| Goal: To increase excretion of salt and water | |
| Thiazide Diuretics | K+-sparing Diuretics |
| Hypertension | Chronic liver failure |
| Congestive heart failure (mild) | Congestive heart failure, when hypokalemia is a problem |
| Renal calculi | Carbonic Anhydrase Inhibitors |
| Nephrogenic diabetes insipidus | Cystinuria (to alkalinize tubular urine) |
| Chronic renal failure (as an adjunct to loop diuretic) | Glaucoma (to decrease intraocular pressure) |
| Osteoporosis | Periodic paralysis that affects muscle membrane function |
| Loop Diuretics | Acute mountain sickness (to counteract respiratory alkalosis) |
| Hypertension, in patients with impaired renal function | Metabolic alkalosis |
| Congestive heart failure (moderate to severe) | Osmotic Diuretics |
| Acute pulmonary edema | Acute or incipient renal failure |
| Chronic or acute renal failure | Reduce intraocular or intracranial pressure (preoperatively) |
| Nephrotic syndrome | |
| Hyperkalemia | |
| Chemical intoxication (to increase urine flow) | |
Mechanisms of Action
All diuretics promote natriuresis and diuresis to reduce ECF volume; however, their mechanisms and sites of action differ. The five major types of diuretics are listed in Table 21-1, and their primary sites of action on the nephron are depicted in Figure 21-1. Knowledge of the mechanisms and sites of action of these agents is important in selecting an appropriate drug and anticipating and preventing complications. In addition, because each class of drugs exerts effects at specific targets, a combination of two or more drugs will often result in additive or synergistic effects to affect the reabsorption or excretion of Na+, Cl−, 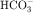 , water, and, to some extent, K+, H+, and organic ions. To understand the mechanisms and consequences of the actions of the diuretics, a basic knowledge of renal physiology is essential.
, water, and, to some extent, K+, H+, and organic ions. To understand the mechanisms and consequences of the actions of the diuretics, a basic knowledge of renal physiology is essential.
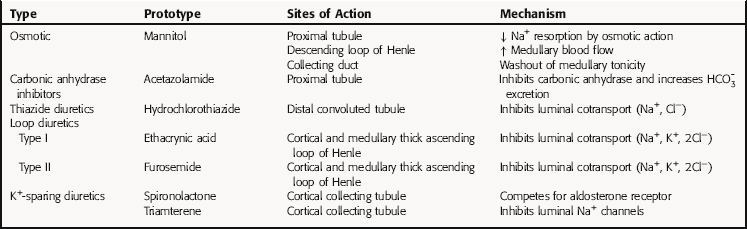
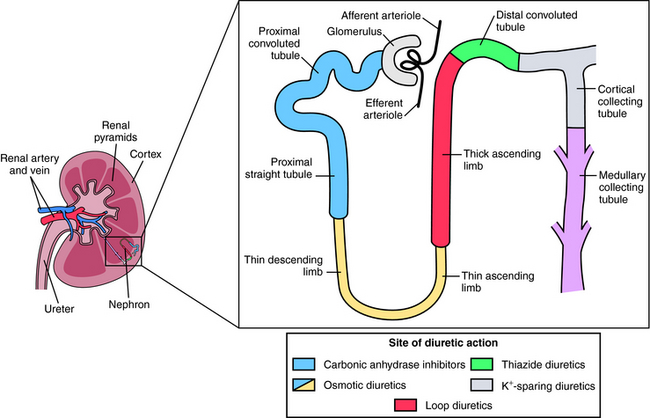
FIGURE 21–1 The nephron, depicting its location, segments, and the sites of action of different classes of diuretics.
Renal Epithelial Transport and Nephron Function
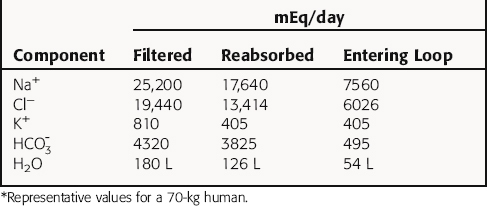
The normal human glomerular filtration rate (GFR) is approximately 180 L/day. Assuming a normal plasma Na+ concentration of 140 mmol/L, that means that 25,200 mmol of Na+ are filtered each day. To maintain Na+ balance, the kidney must reabsorb more than 99% (24,950 mmol) of the filtered load of Na+. This staggering amount of solute and water reabsorption is achieved by the actions of the million nephrons in each human kidney. Renal epithelial cells transport solute and water from the apical cell membrane to the basolateral cell membrane. The polarization of structures that differentiate the apical membrane from the basolateral membrane allows the vectorial transport of solute and water (Fig. 21-2). The basolateral cell membrane expresses the ubiquitous Na+/K+-adenosine triphosphatase (ATPase) (the Na+ pump) that exchanges 3 Na+ ions for 2 K+ ions, resulting in decreased intracellular Na+ providing a chemical gradient and an electronegative cell interior for Na+ entry from the lumen through the apical membrane (a potential difference of approximately 60 mV), which also attracts Na+. The concentration gradient then favors passive efflux of the K+ that entered the cell to the intercellular space. Because the ECF concentration of K+ is low relative to Na+, a recycling of K+ between the cell and interstitial fluid is necessary to maintain the Na+ pump.
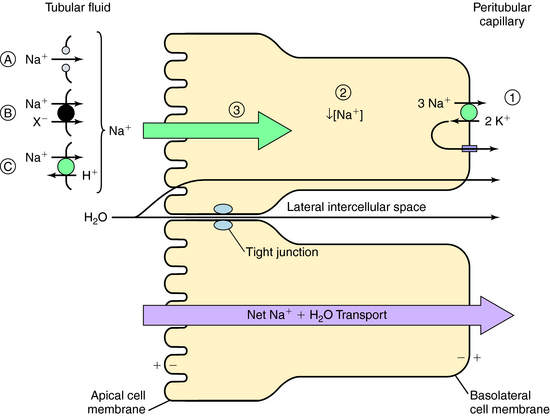
The transport of Na+ across the apical cell membrane adjacent to the tubular fluid is achieved by passive diffusion via proteins that form a pore or channel (see Fig. 21-2, A), and two types of carrier-mediated transport, a cotransport (symport) pathway that transports Na+ and another solute species (such as Cl− or amino acids) in the same direction (see Fig. 21-2, B), and a countertransport (antiport) pathway that transports Na+ and another solute species (H+) in the opposite direction (see Fig. 21-2, C). In each case the low intracellular Na+ concentration as a consequence of the action of the Na+/K+-ATPase pump provides the electrochemical gradient for Na+ entry.
This transepithelial transport causes the osmolality of the lateral intercellular spaces to increase as a result of the accumulation of solute, producing an osmotic gradient that permits water to flow by two routes (see Fig. 21-2):
Tubular Reabsorption: Transport by the Proximal Tubule
The GFR of healthy adults ranges from 1.7 to 1.8 mL/min/kg, and approximately two thirds of the water and NaCl filtered at the glomerulus is reabsorbed by the proximal tubule. 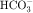 , glucose, amino acids, and other organic solutes are also reabsorbed. When GFR increases, salt and water excretion increases, but fractional reabsorption in the proximal tubule does not change. This is termed glomerulotubular balance. It moderates but does not entirely eliminate the effects of alterations in the GFR on salt and water excretion.
, glucose, amino acids, and other organic solutes are also reabsorbed. When GFR increases, salt and water excretion increases, but fractional reabsorption in the proximal tubule does not change. This is termed glomerulotubular balance. It moderates but does not entirely eliminate the effects of alterations in the GFR on salt and water excretion.
The transport of Na+, 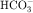 , and Cl− is important for the actions of several diuretics on the proximal tubule. Na+ is reabsorbed primarily with
, and Cl− is important for the actions of several diuretics on the proximal tubule. Na+ is reabsorbed primarily with 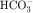 in the early proximal tubule, whereas Na+ is reabsorbed primarily with Cl− in the late proximal tubule (Fig. 21-3). At the apical cell membrane of the early proximal tubule, Na+ entry is coupled with H+ efflux via the Na+/H+ antiporter (NHE3), which is a protein containing 10 to 12 transmembrane spanning domains and a hydrophilic C-terminal domain and is subject to regulation by a variety of factors, including angiotensin II, which increases its activity. H+ extruded from the cell combines with
in the early proximal tubule, whereas Na+ is reabsorbed primarily with Cl− in the late proximal tubule (Fig. 21-3). At the apical cell membrane of the early proximal tubule, Na+ entry is coupled with H+ efflux via the Na+/H+ antiporter (NHE3), which is a protein containing 10 to 12 transmembrane spanning domains and a hydrophilic C-terminal domain and is subject to regulation by a variety of factors, including angiotensin II, which increases its activity. H+ extruded from the cell combines with 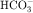 to form H2CO3, which rapidly forms CO2 and water in the presence of the enzyme carbonic anhydrase. CO2 rapidly enters the cell via simple diffusion and is rehydrated to form carbonic acid. Because the concentration of cellular H+ is low, the reaction proceeds as follows: CO2 + H2O → H2CO3 → H+ +
to form H2CO3, which rapidly forms CO2 and water in the presence of the enzyme carbonic anhydrase. CO2 rapidly enters the cell via simple diffusion and is rehydrated to form carbonic acid. Because the concentration of cellular H+ is low, the reaction proceeds as follows: CO2 + H2O → H2CO3 → H+ + 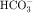 . Thus a constant supply of H+ is furnished for countertransport with Na+.
. Thus a constant supply of H+ is furnished for countertransport with Na+. 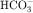 that accumulates is cotransported with Na+ across the basolateral cell membrane into the interstitial fluid and, subsequently, into the blood. Although the cytoplasmic hydration reaction occurs spontaneously, the rate is inadequate to allow reabsorption of the
that accumulates is cotransported with Na+ across the basolateral cell membrane into the interstitial fluid and, subsequently, into the blood. Although the cytoplasmic hydration reaction occurs spontaneously, the rate is inadequate to allow reabsorption of the 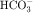 load filtered (approximately 4000 mEq/day). However, little or none of the filtered
load filtered (approximately 4000 mEq/day). However, little or none of the filtered 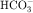 is excreted because of the presence of carbonic anhydrase. The net effect of coupling of the Na+/H+ antiporter to the carbonic anhydrase-mediated hydration and rehydration of CO2 is preservation of
is excreted because of the presence of carbonic anhydrase. The net effect of coupling of the Na+/H+ antiporter to the carbonic anhydrase-mediated hydration and rehydration of CO2 is preservation of 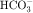 . The final step is its transfer from the interstitial fluid into peritubular capillaries.
. The final step is its transfer from the interstitial fluid into peritubular capillaries.
In the late proximal tubule (see Fig. 21-3), Na+ is reabsorbed primarily with Cl−. Reabsorption is secondary to activation of both the NHE3, which couples inward Na+ transport with outward H+ transport, and a Cl− base (formate) exchanger that transports Cl− from lumen to cell in exchange for a base. The parallel operation of both exchangers results in net Na+ and Cl− absorption by the late proximal tubule. Passive transport of Na+ and Cl− also occurs between cells through the paracellular pathway.
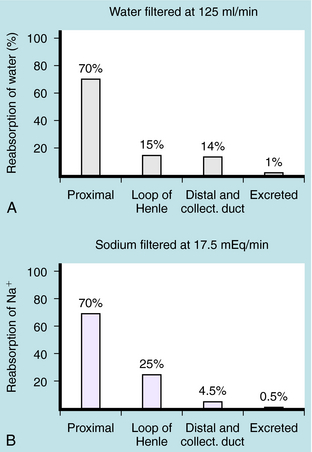
In summary, the proximal tubule reabsorbs approximately 70% of filtered water, Na+, and Cl−, 85% of filtered 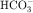 , and 50% of filtered K+ (Table 21-2). These percentages are relatively constant, even when filtered quantities increase or decrease. As a result, minor fluctuations in GFR do not influence fluid and electrolyte excretion very much. The driving force for reabsorption of water and electrolytes is the Na+/K+-ATPase. Passive movements of other ions and water are initiated and sustained by active transport of Na+ across basolateral cell membranes. Osmotic equilibrium with plasma is maintained to the end of the proximal tubule. Most of the filtered
, and 50% of filtered K+ (Table 21-2). These percentages are relatively constant, even when filtered quantities increase or decrease. As a result, minor fluctuations in GFR do not influence fluid and electrolyte excretion very much. The driving force for reabsorption of water and electrolytes is the Na+/K+-ATPase. Passive movements of other ions and water are initiated and sustained by active transport of Na+ across basolateral cell membranes. Osmotic equilibrium with plasma is maintained to the end of the proximal tubule. Most of the filtered  is not actually reabsorbed directly from the lumen; rather, it is converted to CO2 and water in the vicinity of the brush border membranes, within which large concentrations of carbonic anhydrase are located. The direction of this reaction is H2CO3 → CO2 + H2O (established by the high concentration of carbonic acid in luminal fluid resulting from the secretion of H+). Carbonic anhydrase in the cytoplasm catalyzes formation of carbonic acid. Cellular H+ is then exchanged for luminal Na+, and
is not actually reabsorbed directly from the lumen; rather, it is converted to CO2 and water in the vicinity of the brush border membranes, within which large concentrations of carbonic anhydrase are located. The direction of this reaction is H2CO3 → CO2 + H2O (established by the high concentration of carbonic acid in luminal fluid resulting from the secretion of H+). Carbonic anhydrase in the cytoplasm catalyzes formation of carbonic acid. Cellular H+ is then exchanged for luminal Na+, and 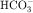 is reabsorbed across the basolateral cell membranes. In this indirect way, filtered
is reabsorbed across the basolateral cell membranes. In this indirect way, filtered 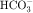 is reabsorbed.
is reabsorbed.
Tubular Reabsorption: Transport by the Loop of Henle
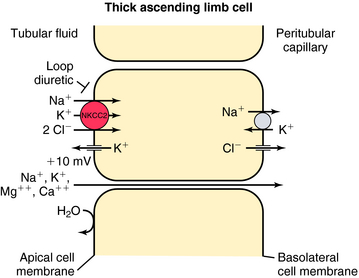
In contrast, the thick ascending limb and its transport functions are an important site of action of the loop (also called high-ceiling) diuretics (Fig. 21-4). Approximately 25% to 35% of filtered Na+ and Cl− is reabsorbed by the loop of Henle. The Na+/K+-ATPase in the basolateral membrane provides the gradient for Na+ and Cl− absorption. Na+ entry across the apical membrane is mediated by an electroneutral transport protein that binds one Na+, one K+, and two Cl− ions and is referred to as the Na+/K+/2Cl− (NKCC2) cotransporter. Although the ascending limb is highly permeable to Na+, K+, and Cl−, it is impermeable to water. Thus the continuous reabsorption of these ions without reabsorption of water dilutes the luminal fluid, thus the name diluting segment. Na+ entry down an electrochemical gradient drives the uphill transport of K+ and Cl−. This system depends on the simultaneous presence of these three ions in the luminal fluid. Once inside the cell, K+ passively reenters the lumen (K+ recycling) via conductive K+ channels in the apical membrane. Cl−, on the other hand, exits the cell via conductive Cl− channels in the basolateral membrane. Depolarization of the basolateral membrane occurs as a consequence of Cl− efflux, creating a lumen-positive (relative to the interstitial fluid) transcellular potential difference of approximately 10 mV. This drives paracellular cation transport, including Na+, Ca++, and Mg++. Inhibition of the NKCC2 cotransporter not only results in excretion of Na+ and Cl− but also in excretion of divalent cations, such as Ca++ and Mg++.
Tubular Reabsorption: Transport by the Distal Convoluted Tubule
In contrast to the proximal tubule and loop of Henle, there is less reabsorption of water and electrolytes in the distal convoluted tubule. It reabsorbs approximately 10% of the filtered load of NaCl. Similar to the thick ascending limb, this segment is impermeable to water, and the continuous reabsorption of NaCl further dilutes tubular fluid. NaCl entry across the apical membrane is mediated by the electroneutral NCC cotransporter sensitive to thiazide diuretics (Fig. 21-5). Unlike the NKCC2 cotransporter of the thick ascending limb, this cotransporter does not require participation of K+. As in other segments, the basolateral Na+/K+-ATPase provides the low intracellular Na+ concentration that facilitates downhill transport of Na+. The distal tubule does not have a pathway for K+ recycling, and therefore the transepithelial voltage is near zero. Therefore the reabsorption of Ca++ and Mg++ is not driven by electrochemical forces. Instead, Ca++ crosses the apical membrane via a Ca++ channel and exits the basolateral membrane via the NCC exchanger. Thus, by inhibiting the NCC cotransporter, thiazide diuretics indirectly affect Ca++ transport through changes in intracellular Na+. Another mechanism of increased Ca++ reabsorption with thiazide diuretics is an increase in the intracellular concentrations of Ca++-binding proteins.
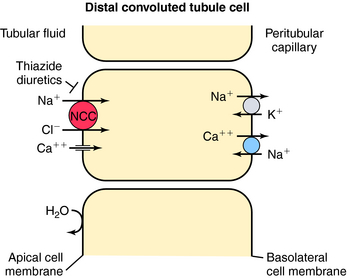
Recently, inactivating mutations have been found in the human gene encoding the NCC transporter in patients with Gitelman’s syndrome, characterized by hypotension, hypokalemia, hypomagnesemia, and hypocalciuria, similar to the effects of thiazides. Pseudohypoaldosteronism type II is an autosomal dominant disease characterized by hypertension, hyperkalemia, and sensitivity to thiazide diuretics. It has been suggested that an activating mutation of the NCC cotransporter is responsible. Recently, two protein kinases have also been linked to the pathogenesis of this syndrome. They are found in the distal nephron and are thought to control the activity of the cotransporters.
Tubular Reabsorption: Transport by the Collecting Tubule
The collecting tubule is the final site of Na+ reabsorption, and approximately 3% of filtered Na+ is reabsorbed by this segment. Although the collecting tubule reabsorbs only a small percentage of the filtered load, two characteristics are important for diuretic action. First, this segment is the site of action of aldosterone, a hormone controlling Na+ reabsorption and K+ secretion (see Chapter 39). Second, virtually all K+ excreted results from its secretion by the collecting tubule. Thus the collecting tubule contributes to the hypokalemia induced by diuretics.
The collecting tubule is composed of two cell types with separate functions. Principal cells are responsible for the transport of Na+, K+, and water, whereas intercalated cells are primarily responsible for the secretion of H+ or 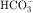 . Intercalated cells are of two types, A and B, the former responsible for secretion of H+ via an H+-ATPase (primary active ion pump) in the apical cell membrane, and the latter responsible for secretion of
. Intercalated cells are of two types, A and B, the former responsible for secretion of H+ via an H+-ATPase (primary active ion pump) in the apical cell membrane, and the latter responsible for secretion of 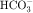 via a Cl−/
via a Cl−/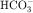 exchanger in the apical membrane. In contrast to more proximal cells, the apical membrane of principal cells does not express cotransport or countertransport systems; rather it expresses separate channels that permit selective conductive transport of Na+ and K+ (Fig. 21-6). Na+ is reabsorbed through a conductive Na+ channel. The low intracellular Na+ as a result of the basolateral Na+/K+-ATPase generates a favorable electrochemical gradient for Na+ entry through epithelial Na+ channels (ENaCs). Because Na+ channels are present only in the apical cell membrane of principal cells, Na+ conductance causes depolarization, resulting in an asymmetrical voltage across the cell and a lumen-negative transepithelial potential difference. This, together with a high intracellular-to-lumen K+ gradient, provides the driving force for K+ secretion.
exchanger in the apical membrane. In contrast to more proximal cells, the apical membrane of principal cells does not express cotransport or countertransport systems; rather it expresses separate channels that permit selective conductive transport of Na+ and K+ (Fig. 21-6). Na+ is reabsorbed through a conductive Na+ channel. The low intracellular Na+ as a result of the basolateral Na+/K+-ATPase generates a favorable electrochemical gradient for Na+ entry through epithelial Na+ channels (ENaCs). Because Na+ channels are present only in the apical cell membrane of principal cells, Na+ conductance causes depolarization, resulting in an asymmetrical voltage across the cell and a lumen-negative transepithelial potential difference. This, together with a high intracellular-to-lumen K+ gradient, provides the driving force for K+ secretion.
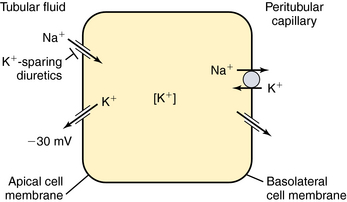
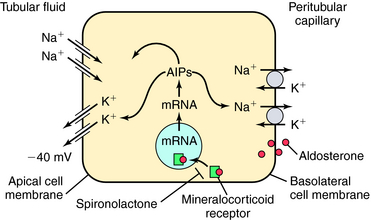
Mutations of ENaC could result in either gain of function, as in Liddle’s syndrome, associated with hypertension and hypokalemia, or loss of function, as in pseudohypoaldosteronism, associated with hypotension and hyperkalemia. The amount of Na+ and K+ in the urine is tightly controlled by aldosterone, which acts on principal cells after release from the adrenal cortex. Aldosterone penetrates the basolateral membrane of principal cells and binds to a cytosolic mineralocorticoid receptor (Fig. 21-7), where its activation causes the receptor-aldosterone complex to translocate to the nucleus to induce formation of specific messenger ribonucleic acids (RNAs) encoding proteins that enhance Na+ conductance in apical cell membranes and Na+/K+-ATPase activity in basolateral cell membranes. As a result, transepithelial Na+ transport is increased, further depolarizing the apical membrane. An increase in the lumen-negative potential, in turn, enhances K+ secretion through K+ channels in the apical membrane. The final equilibratory steps take place in medullary collecting tubules, where small amounts of NaCl and K+ are reabsorbed. In the presence of antidiuretic hormone, water is transported out of the lumen into the interstitium. The direction of water movement is determined by the medullary tonicity established by the countercurrent mechanism. A quantitative summary of the fractional reabsorption of water and Na+ of each tubule segment is shown in Figure 21-8. The proximal tubule reabsorbs more Na+ than water; the entire distal tubule and medullary collecting system reabsorbs less than 5% of filtered Na+.
Tubular Secretion and Bidirectional Transport of Organic Acids and Bases
The proximal tubular secretion of diuretic anions and cations into tubular fluid illustrates the influence of electrical charge on drug delivery to their sites of action and on their rapid decline in plasma. Two generic systems that transport organic ions from blood to urine reside in the proximal tubule. One transports organic acids (anions as the A− form of acid HA), and the other transports organic bases (cations as BH+ form) of base B (see Chapter 2). Their chief characteristics are as follows:
The tubular transport of organic acids and bases is illustrated in Figure 21-9. Organic anions (OA−) are taken up by the basolateral cell membrane through indirect coupling to Na+ (see Fig. 21-9, A). The Na+/K+-ATPase maintains a steep inward Na+ gradient, which provides energy for entry of Na+-coupled dicarboxylate (α-ketoglutarate). The operation of a parallel dicarboxylate/OA− exchange drives the uphill movement of OA− into the cell. The cell is now loaded with OA−, which enters the lumen by facilitated diffusion. The mechanism may involve anion exchange or conductive transport.
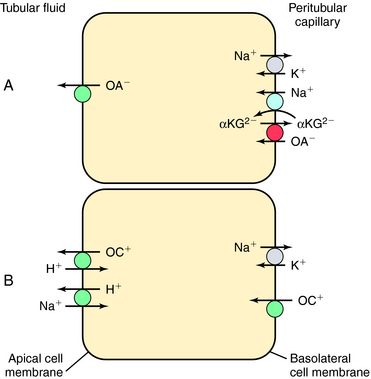
Organic cations (OC+) gain entry from the interstitial fluid across basolateral cell membranes (Fig. 21-9, B). Their entry is aided by carrier-facilitated diffusion. Once inside the cell, OC+ enter the lumen through countertransport with H+. The operation of this cation exchange is dependent on the parallel operation of the Na+/H+ antiporter, NHE3.
Mannitol, urea, glycerol, and isosorbide are the primary osmotic diuretics, with mannitol most widely used (Fig. 21-10). Mannitol produces a diuresis secondary to (1) an increase in osmotic pressure in the proximal tubule fluid and loop of Henle, which retards passive reabsorption of water, and (2) an increase in renal blood flow and washout of medullary tonicity. Glomerular filtration of mannitol into the tubular fluid retards passive water reabsorption primarily by the proximal tubule and thin limbs of the loop of Henle. In effect, the osmotic force of nonreabsorbable solute in the lumen opposes the osmotic force of reabsorbable Na+. The isosmolality of urine is preserved because mannitol molecules replace reabsorbed Na+. The reabsorbed fraction of water is reduced, increasing the amount of water entering the loop of Henle. The luminal concentration of Na+ decreases when Na+ is transported and water fails to follow it, resulting in a change in Na+ concentration gradient and a backward flux of Na+ into the lumen, with ultimately a small increase in excretion. Over zealous administration of mannitol may result in hypernatremia, hyperkalemia, and volume depletion.
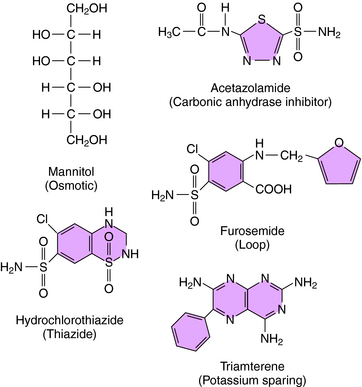
Acetazolamide, the prototypical carbonic anhydrase inhibitor (see Fig. 21-10), has limited use as a diuretic. It is used primarily to reduce intraocular pressure in glaucoma and to treat metabolic alkalosis because of its ability to enhance 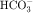 excretion.
excretion.
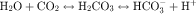
Acetazolamide inhibition of carbonic anhydrase reduces H+ concentration in the tubule lumen and decreases availability of H+ for Na+/H+ exchange. There is a resulting increase in 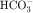 and Na+ in the proximal portion of the lumen (see Figs. 21-1 and 21-3). Although some
and Na+ in the proximal portion of the lumen (see Figs. 21-1 and 21-3). Although some 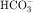 is reabsorbed at other tubular sites, approximately 30% of the filtered load appears in the urine after carbonic anhydrase inhibition. A hyperchloremic metabolic acidosis results from
is reabsorbed at other tubular sites, approximately 30% of the filtered load appears in the urine after carbonic anhydrase inhibition. A hyperchloremic metabolic acidosis results from 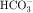 depletion, which renders subsequent doses of acetazolamide ineffective.
depletion, which renders subsequent doses of acetazolamide ineffective.
Thiazide diuretics, such as hydrochlorothiazide (see Fig. 21-10), were developed to identify compounds that increase excretion of Na+ and Cl− rather than Na+ and 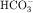 , as occurs with carbonic anhydrase inhibitors. The major site of action of the thiazides is the distal convoluted tubule (see Figs. 21-1 and 21-5), where they inhibit electroneutral NaCl absorption. The distal convoluted tubules also express high-affinity receptors for thiazides.
, as occurs with carbonic anhydrase inhibitors. The major site of action of the thiazides is the distal convoluted tubule (see Figs. 21-1 and 21-5), where they inhibit electroneutral NaCl absorption. The distal convoluted tubules also express high-affinity receptors for thiazides.
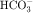 transport by the proximal tubule as a consequence of its ability to inhibit carbonic anhydrase; most other thiazides are only weak carbonic anhydrase inhibitors.
transport by the proximal tubule as a consequence of its ability to inhibit carbonic anhydrase; most other thiazides are only weak carbonic anhydrase inhibitors.Loop diuretics generate larger responses than those produced by thiazides. Acting on the thick ascending limb, loop diuretics can inhibit the reabsorption of as much as 25% of the glomerular filtrate (Fig. 21-11) and are often effective when thiazides do not suffice. Despite their efficacy, loop diuretics are remarkably safe when used properly.
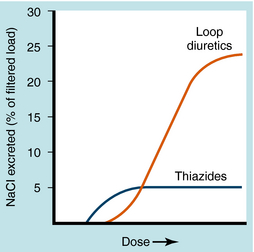
FIGURE 21–11 Dose-response curves comparing Na+ excretion after administration of thiazides or loop diuretics.
Four loop diuretics are available in the United States: ethacrynic acid, furosemide, torsemide, and bumetanide. Ethacrynic acid and furosemide are prototypes of loop I and II drugs, respectively. Bumetanide is considerably more potent and differs pharmacokinetically but is otherwise similar to the older drugs. Furosemide (see Fig. 21-10) inhibits reabsorption of Na+ and Cl− by the thick ascending limb by competing with Cl− for a binding site on the NKCC2 cotransporter. Ethacrynic acid reacts with sulfhydryl groups, a reaction formerly considered to precede diuresis. However, this is no longer thought to be the case, because several natriuretic compounds with similar structures do not react with sulfhydryl groups. Ethacrynic acid also inhibits Na+/K+-ATPase but only in excessive concentrations. Because the loop of Henle is responsible for accomplishing the countercurrent multiplication that generates a concentrated medullary interstitium, loop diuretics prevent formation of a concentrated urine.
The K+-sparing diuretics comprise three pharmacologically distinct groups: steroid aldosterone antagonists, pteridines, and pyrazinoylguanidines. Their site of action is the collecting tubule, where they interfere with Na+ reabsorption and indirectly with K+ secretion (see Figs. 21-1 and 21-6). Their diuretic activity is weak because fractional Na+ reabsorption in the collecting tubule usually does not exceed 3% of the filtered load. For this reason K+-sparing drugs are ordinarily used in combination with thiazides or loop diuretics to restrict K+ loss and sometimes augment diuretic action.
Spironolactone and eplerenone, analogs of aldosterone and its major metabolite, canrenone, bind to mineralocorticoid receptors in the kidney and elsewhere, acting as competitive inhibitors of aldosterone (see Chapter 39). Aldosterone antagonists decrease Na+ conductance at the apical membrane of principal cells, thereby reducing the lumen-negative potential. This results in a decrease in the electrical gradient for K+ secretion.
Triamterene (see Fig. 21-10) and amiloride are structurally different from spironolactone but have the same functional effects. Both drugs are organic bases secreted into the lumen by proximal tubular cells, and both block the apical membrane Na+ channel of principal cells and reduce Na+ conductance (see Fig. 21-7). Similar to spironolactone, they cause the lumen-negative potential and the electrical gradient for K+ secretion to be abolished. Although they are weak diuretics and natriuretics, K+ is conserved. Amiloride also blocks Na+/H+ exchange, Na+/Ca++ exchange, and Na+/K+-ATPase, but it blocks the Na+ channel at therapeutic doses only. Amiloride also decreases Ca++ and H+ excretion, also as a consequence of a decrease in the lumen-negative potential.
Pharmacokinetics
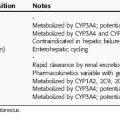
The pharmacokinetic parameters of the diuretic agents are summarized in Table 21-3.
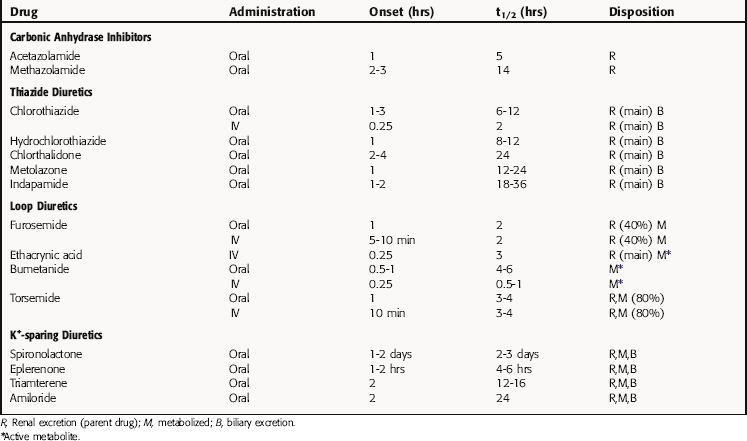
The highly lipid-soluble members of the thiazide family possess larger apparent volumes of distribution and lower renal clearances. Indapamide, bendroflumethiazide, and polythiazide are primarily metabolized in the liver, and the major route of elimination is by glomerular filtration and proximal tubular secretion of unchanged drug.
Spironolactone is discussed in Chapter 39. Triamterene has good bioavailability and is metabolized in the liver to an active metabolite, which is excreted in the urine. The half-life of this active metabolite increases in renal insufficiency but is unchanged in liver disease.
Relationship of Mechanisms of Action to Clinical Response
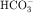 , PO4−3, Ca++, and Mg++.
, PO4−3, Ca++, and Mg++.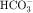 concentration, it is used to reduce aqueous humor formation. Acetazolamide is also used to prevent or treat acute mountain sickness, to alkalinize the urine, and to treat metabolic alkalosis. Carbonic anhydrase inhibitors are occasionally used to treat epilepsy. During the past several years the popularity of carbonic anhydrase inhibitors as diuretics has waned because tolerance develops rapidly, increased urinary excretion of
concentration, it is used to reduce aqueous humor formation. Acetazolamide is also used to prevent or treat acute mountain sickness, to alkalinize the urine, and to treat metabolic alkalosis. Carbonic anhydrase inhibitors are occasionally used to treat epilepsy. During the past several years the popularity of carbonic anhydrase inhibitors as diuretics has waned because tolerance develops rapidly, increased urinary excretion of 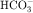 results in development of systemic acidemia, and more effective and less toxic agents have been developed. Nevertheless, acetazolamide is administered for short-term therapy, especially in combination with other diuretics, to patients who are resistant to other agents.
results in development of systemic acidemia, and more effective and less toxic agents have been developed. Nevertheless, acetazolamide is administered for short-term therapy, especially in combination with other diuretics, to patients who are resistant to other agents.In general, thiazide diuretics are used in treatment of hypertension, congestive heart failure, and other conditions in which a reduction in ECF volume is beneficial. Many large clinical studies have proved the efficacy and tolerability of these agents in treatment of hypertension. As a result, this class of diuretics is recommended as monotherapy or in combination with other agents in treatment of hypertension. The blood pressure reduction in patients with hypertension results in part from contraction of the ECF volume (see Chapter 20). This occurs acutely, leading to a decrease in cardiac output with a compensatory elevation in peripheral resistance. Vasoconstriction then subsides, enabling cardiac output to return to normal. Augmented synthesis of vasodilator prostaglandins has been reported and may be a crucial factor for long-term maintenance of a lower pressure, even though ECF volume tends to return toward normal.
In addition to their use in the treatment of edematous disorders and hypertension, thiazide diuretics are also used in other disorders. Because they decrease renal Ca++ excretion, they are used in the treatment of Ca++ nephrolithiasis and osteoporosis. Thiazide diuretics are also used in the treatment of nephrogenic diabetes insipidus, where tubules are unresponsive to vasopressin and patients undergo a water diuresis. Often the volume of dilute urine excreted is large enough to lead to intravascular volume depletion, if it is not offset by an adequate intake of fluid. Chronic administration of thiazides increases urine osmolality and reduces flow. The mechanism hinges on excretion of Na+ and its removal from the ECF, which contracts ECF volume. The proximal tubule then avidly reabsorbs Na+. Urine flow rate diminishes and urine osmolality increases when Na+ transport in the distal convoluted tubule is inhibited. Drug therapy in this instance is most effective when used in combination with dietary salt restriction.
The value of loop diuretics in pulmonary edema may be attributed in part to their stimulation of prostaglandin synthesis in kidney and lung. Furosemide and ethacrynic acid increase renal blood flow for brief intervals, promoting the urinary excretion of prostaglandin E. IV injection of furosemide also reduces pulmonary arterial pressure and peripheral venous compliance. Indomethacin, an inhibitor of prostaglandin synthesis (see Chapter 15), interferes with these actions.
Pharmacovigilance: Side Effects, Clinical Problems, and Toxicity
Repeated use of diuretics is frequently associated with shifts in acid-base balance and changes in serum electrolytes. Shifts frequently encountered in patients on continuous diuretic therapy include K+ depletion and hyperuricemia. Patients at risk include the elderly, those with severe disease, those taking cardiac glycosides, and the malnourished. Such changes are difficult to avoid in most patients unless counteractive measures are taken. Supplemental intake of K+ (dietary or oral KCl) or concomitant use of K+-sparing with thiazide or loop diuretics is often used to circumvent this problem.
Vertigo and deafness sometimes develop in patients receiving large IV doses of loop diuretics; the coadministration of an aminoglycoside antibiotic produces additive effects. Higher rates of ototoxicity are associated with ethancrynic acid, limiting its use. Additional drug interactions occur with indomethacin (decreased activity), warfarin (displacement from plasma protein), and lithium (decreased clearance and increased risk of toxicity). All diuretics are contraindicated in anuric patients.
is highest in patients with limited renal function. Additional complications include elevated serum blood urea nitrogen and uric acid, glucose intolerance, and GI tract disturbances. Triamterene may contribute to, or initiate, formation of renal stones, and hypersensitivity reactions may occur in patients receiving it. Some drug-drug interactions involving diuretics are presented in Table 21-4.
| Diuretic | Drug Class or Agent | Problem |
|---|---|---|
| Thiazide diuretics | β Adrenergic blockers | Increase in blood glucose, urates, and lipids |
| Chlorpropamide | Hyponatremia | |
| Thiazides and loop diuretics | Digitalis glycosides | Hypokalemia resulting in increased digitalis binding and toxicity |
| Adrenal steroids | Enhanced hypokalemia | |
| Loop diuretics | Aminoglycosides | Ototoxicity, nephrotoxicity |
| K+-sparing diuretics | Angiotensin-converting enzyme inhibitors | Hyperkalemia, cardiac effects |
New Horizons
Recent clinical studies have targeted the natriuretic peptide family in mediating natriuresis in disorders of heart failure (see Chapter 23). The renal hemodynamic effects of A and B natriuretic peptides include increased GFR, afferent arteriolar dilation, and efferent arteriolar constriction. In addition, they have direct effects to block Na+ transport in the inner medullary collecting duct and block aldosterone release. Nesiritide, a recombinant human B natriuretic peptide, is currently used for the treatment of fluid retention in congestive heart failure.
Nielsen et al. 2002 Nielsen S, Frøkiær J, Marples D, et al. Aquaporins in the kidney: From molecules to medicine. Physiol Rev. 2002;82:205-244.
Okusa MD, Ellison DH. Diuretics: Physiology and pathophysiology. In: Seldin DW, Giebisch G, editors. The kidney. 3. Philadelphia: Lippincott Williams & Wilkins; 2000:2877-2922.
Supuran CT. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov. 2008;7:168-181.
Wang DJ, Gottlieb SS. Diuretics: Still the mainstay of treatment. Crit Care Med. 2008;36(1 Suppl):S89-S94.