Dissolution testing of solid dosage forms
Ana Cristina Freire and Abdul W. Basit
Chapter contents
The relevance of drug dissolution and dissolution testing
General requirements for in-vitro dissolution testing
The design of suitable dissolution tests; QC versus predictive dissolution testing
Dissolution testing for quality control
Compendial dissolution apparatuses
Volume and composition of the dissolution medium
Predictive dissolution testing
Key points
• Dissolution testing is a key quality control test.
• Dissolution tests are normally performed under sink conditions.
The relevance of drug dissolution and dissolution testing
The biological events that occur in the body following the administration of a drug are varied and complex. Before being absorbed into the bloodstream, an orally administered drug is exposed to the dynamic conditions of the gastrointestinal lumen and much of what happens here will have a bearing on its therapeutic activity.
Only drug in its dissolved state can permeate the intestinal mucosa. If the solubility of the drug in the gastrointestinal aqueous fluids is limited, dissolution will be the rate-limiting step to absorption, and will dictate the rate and extent to which the drug becomes available in the bloodstream. Because of the link between oral bioavailability and dissolution, most oral drug products such as suspensions, granules, pellets, tablets and capsules are currently required to be tested for their dissolution characteristics. Disintegration measurements are also routinely performed on immediate-release products, although, for the most part, the results from such tests correlate poorly with bioavailability.
The concept of dissolution from a physical chemistry standpoint is well established; research into the mechanisms of dissolution of solids in liquids started over a century ago with the pioneering work of Noyes and Whitney (discussed fully in Chapter 2). However, it took several decades before the link between dissolution and oral bioavailability in the context of a pharmaceutical product was fully appreciated. This is considered to have occurred in 1951, when Edwards studied the dissolution of aspirin tablets in different media. His conclusions from that study were that “the dissolution of an aspirin tablet in the stomach and intestine is the rate process controlling the absorption of aspirin into the bloodstream”.
The clinical relevance of Edward’s observations was only realized in the mid-1960s, when several reports linking dissolution data with clinical efficacy, inferior efficacy and even toxicity of some commercial oral solid products were made available. For example, clinical inadequacies with formulations of the oral anti-diabetic and poorly water soluble drug, tolbutamide, were documented. Ineffective tablets were shown to disintegrate and dissolve at a slower rate than clinically effective ones (Fig. 35.1).
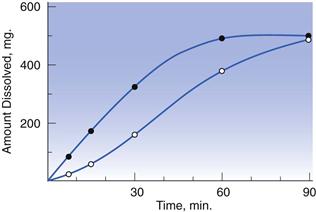
Fig. 35.1 Dissolution of tolbutamide from clinically effective (–•–) and ineffective (– –) tablets, as a function of time. (Courtesy of Levy, 1964, with permission.)
–) tablets, as a function of time. (Courtesy of Levy, 1964, with permission.)
Reports of toxicity with a formulation of phenytoin available in Australia and New Zealand followed in 1968. The substitution of the more hydrophilic excipient lactose for calcium sulfate in an immediate-release hard gelatin capsule enhanced the dissolution rate and bioavailability of phenytoin, leading to numerous cases of anticonvulsant intoxication. This is further discussed in Chapter 33 (see Fig. 33.6 and associated text).
However, undoubtedly the reference case for the impact of drug dissolution on oral bioavailability was that of digoxin tablets. In the early 1970s, several marketed formulations of digoxin were shown to yield up to seven-fold differences in blood levels of the drug. In search for an explanation, digoxin tablet formulations were studied for their dissolution properties, exposing huge differences in this parameter. Interestingly, a good correlation was noted between dissolution rate and digoxin blood levels. In other words, absorption of digoxin was higher for those formulations which dissolved quicker (Fig. 35.2). In contrast, the disintegration time of the different tablets was very similar and bore no relation to digoxin blood levels, raising questions as to the value of the disintegration test in detecting differences in the oral bioavailability of solid drug products.
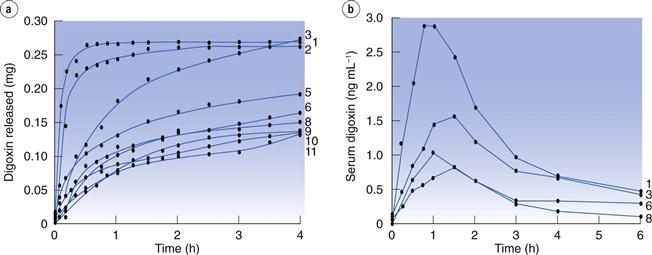
Fig. 35.2 Dissolution rates of different brands of digoxin tablets available in the UK market at the time of the study (a) and corresponding blood (serum) levels of digoxin (b). Formulation 1 (‘new Lanoxin’) and 8 (‘old Lanoxin’) are from the same brand, prepared using different manufacturing methods. (Courtesy of Fraser et al, 1973.)
In light of these cases, it became clear that the formulation and manufacturing process was linked to the therapeutic efficacy of the drug. Dissolution, and not disintegration, became an accepted indicator of oral bioavailability of solid drug products. This prompted the Food and Drug Administration (FDA) in the United States of America to make the use of dissolution testing official in 1970, and to introduce dissolution requirements in the pharmacopoeial monographs of tablets and capsules. By 1995, four different dissolution apparatuses had been included in the United States Pharmacopeia (USP).
General requirements for in-vitro dissolution testing
Today, in-vitro dissolution studies are carried out for several reasons:
• most often to ensure that preparations comply with product specifications
• to provide an indication of the performance of the preparation in vivo.
Changes to the release profile and/or dissolution rate of the drug can be brought about by the characteristics of the dosage form and by its method of manufacturing. However, dissolution becomes more complicated if the properties of the physiology of the gastrointestinal tract are taken into consideration (Chapter 19). These must be reflected in any efficient in-vitro test.
pH of the gastrointestinal luminal fluids
As it travels through the gastrointestinal tract, a drug is exposed to increasing pH conditions. Such pH conditions play an important role in the solubility of ionizable drugs, with pKa values within the pH physiological range (1–7.5), and may affect their bioavailability (discussed fully in Chapter 20). This must be simulated, particularly in predictive dissolution testing (see below).
Composition of the gastrointestinal luminal fluids
While pH may be sufficiently simulated with buffered solutions, the composition of the gastrointestinal tract fluids is not. Gastrointestinal luminal fluids are enriched with amphiphilic bile components, such as bile salts and lecithin. These substances enhance the dissolution rate of drugs either via an increase in wettability or, at higher concentrations, through the formation of micelles. Although fasted fluids already have wetting and solubilizing effects, this is further increased after a meal, due to increased secretion of bile and to the presence of degradation products of lipids contained in the meal (i.e. fatty acids and monoglycerides). For this reason, the dissolution of poorly soluble drugs is generally higher under fed than in fasted conditions. This is well-exemplified by the lipophilic drug, danazol; as seen in Figure 35.3 the bioavailability of this compound is considerably higher in the fed state.
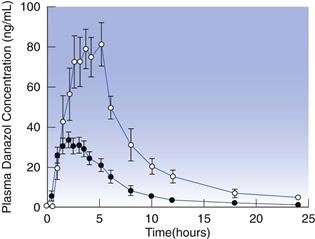
Fig. 35.3 Plasma concentration-time profiles following administration of 100 mg danazol in a hard gelatin capsule in the fed (– –) and fasted (–•–) states in humans. (Courtesy of Charman et al 1993.)
–) and fasted (–•–) states in humans. (Courtesy of Charman et al 1993.)
Two main uses of in-vitro dissolution testing are now discussed further.
As a quality control tool
Before a drug product is released onto the market place (and routinely once marketed), it must be subjected to rigorous control to ensure that the quality and performance (with regards to both safety and efficacy) of the final product are acceptable. Currently, almost all solid oral drug products require in-vitro dissolution testing as part of their quality control (QC) assessment. This involves analysing the drug release profile of different batches of the same drug product with a view to either confirm manufacturing and product consistency or to verify the stability of the product during its shelf-life.
Predictive dissolution testing
Dissolution data can also be used as a prognostic tool to predict the behaviour and performance of oral solid drug products in the gastrointestinal tract. The aim is to correlate as closely as possible measured in-vitro parameters with oral bioavailability. This type of dissolution testing is known as predictive dissolution testing. Unlike QC dissolution testing, it may require dissolution test methods which reflect more closely the physiological makeup of the gastrointestinal tract. Such test conditions are not easy to devise, but if done appropriately, they will enable the development of in vitro/in vivo predictions.
These predictions can be simple qualitative or semi-quantitative relationships (IVIVR) or quantitative correlations established using mathematical models (IVIVC). In its simplest definition, an IVIVC is a correlation (preferably linear) between an in vitro feature of the drug product (in the present context, its dissolution characteristics) and a biological parameter (e.g. the uptake of the drug in vivo and subsequent drug blood levels). The establishment of such a correlation during predictive dissolution testing is one of the most important aspects of a dissolution test for a preparation undergoing formulation development.
Predictive dissolution data have two main applications. Firstly, they can guide the early development of a new drug product by selecting formulations that yield desired in-vivo dissolution characteristics. Secondly they serve as a surrogate for clinical studies. In order to introduce new generic drug products into the market, companies are required to demonstrate that the generic product yields statistically similar rates and extents of drug absorption to the innovator (or branded) drug product – in other words that the two products are bioequivalent. Bioequivalence has traditionally been demonstrated in appropriately designed clinical studies, which are both time-consuming and expensive. However, following the introduction of the Biopharmaceutics Classification System in 1995 (BCS, Chapter 21) it became apparent that the dissolution data of some drug products can be correlated to oral bioavailability. Since 2000, bioequivalence between immediate-release products of highly water-soluble and easily absorbed drugs can be established on the basis of dissolution data, provided that the excipients present in the formulation do not affect the absorption of the drug.
Dissolution testing
The aim of any dissolution test is to measure the rate at which the drug substance is released from the dosage form and dissolves in a particular dissolution medium. It is necessary for such tests to be performed under well-defined conditions to allow comparison of observed data. Various guidelines and protocols are available. However, in many instances it is left to the scientists to decide what are the best conditions. Choosing these conditions is never an easy task. Protocols tend to vary greatly amongst drug products and often the same product is tested under different conditions, depending on whether the test is conducted for quality control (QC) purposes or to predict the performance of the drug product in the gastrointestinal tract. Key variables to consider when designing dissolution tests are:
Type of dissolution apparatus used.
Although there are many types of dissolution apparatus available, the most commonly used are the compendial dissolution apparatus. These are standardized and robust dissolution testers described by pharmacopoeias.
Volume and composition of dissolution medium.
The selection of medium and volume is guided by the aim of the dissolution test, solubility of the drug and type of apparatus used. All tests are conducted at 37 °C to mimic body temperature.
Hydrodynamics.
This refers to the mechanical agitation provided by the dissolution apparatus which will aid in the dissolution of the drug. The various dissolution apparatuses offer different hydrodynamics and these may be varied to allow for the best results.
Number of units to be tested.
Dissolution tests designed to assess the quality of a batch of tablets are normally repeated for at least 6 units per batch or formulation, depending on test variability.
The design of suitable dissolution tests; QC versus predictive dissolution testing
QC dissolution methods are often easier to design; these make use of established compendial equipment, and the composition and volume of the dissolution medium is generally chosen according to the solubility of the drug. Devising a predictive dissolution test is more challenging as bio-relevant conditions need to be sought to mimic the physiological parameters which affect the dissolution of the drug in the GI tract. The major considerations in designing both types of dissolution test methods will be commented upon below.
Dissolution testing for quality control
QC methods are described in the monograph of the product in the various pharmacopoeias. As a general rule these methods are of simple execution, reliable, reproducible, yet sufficiently discriminatory to be able to detect small product deviations. From a QC point of view, an over-discriminatory method is sometimes preferred so as to detect any product changes before the performance in the gastrointestinal tract is affected. The other prime concern of a QC test is to use conditions under which at least 80% of the drug can be dissolved.
Compendial dissolution apparatuses
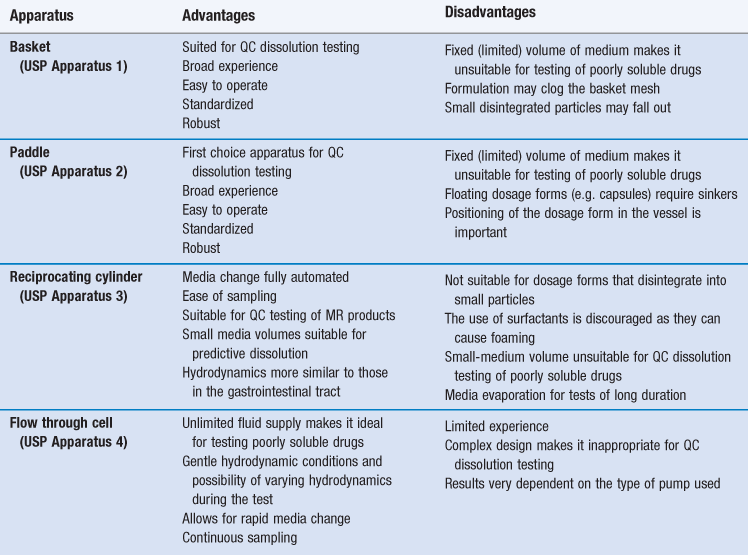
There are currently four dissolution apparatus described in the US and European Pharmacopoeias for the testing of oral solid drug products. These are the basket and paddle apparatus, the reciprocating cylinder and the flow through cell (Figures 35.4–35.7). The selection of a dissolution apparatus depends mainly on the solubility of the drug and type of dosage form. The first choice equipment for QC dissolution testing are the basket and paddle apparatuses because their simple design makes them ideal for routine use. However, due to the limited volume of medium and operational difficulties in medium change, these apparatus are often more suited to immediate-release than to modified-release products, and in particular immediate-release formulations of soluble drugs.
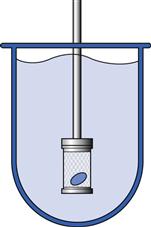
Fig. 35.4 Basket apparatus (USP Apparatus 1).
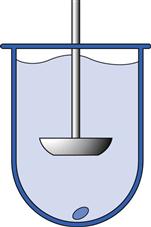
Fig. 35.5 Paddle apparatus (USP Apparatus 2).
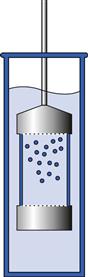
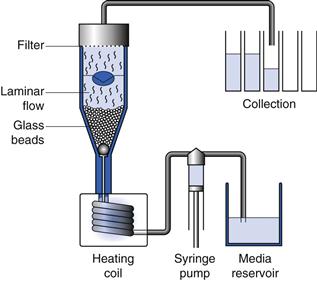
Fig. 35.7 Flow through cell (USP Apparatus 4).
The reciprocating cylinder and flow through cell system are particularly useful for testing of modified-release (MR) dosage forms and poorly soluble drugs, respectively. A brief description of each apparatus is given below. Advantages and disadvantages to their use are summarized in Table 35.1.
Note than many pharmacopoeias worldwide define detailed specifications for standard dissolution apparatus and methodologies. In the process of harmonization of these standards most pharmacopoeial specifications are extremely similar. In the interests of brevity this chapter will refer to the United State Pharmacopeia (USP) specifications. All major pharmacopoeias are very similar but in others, even if the detail varies, the principle of operation and interpretation of results is the same.
Basket apparatus (USP Apparatus 1)
The basket apparatus was the first official dissolution tester to be described in the USP in 1970 and remains one of the most commonly used methods for testing the dissolution of capsules and tablets.
In this apparatus, the dosage form is placed inside a rotating basket made of a stainless steel wire mesh and immersed in dissolution medium which has been pre-warmed at 37 °C. An outline of the apparatus is shown in Figure 35.4 with a more detailed diagram shown in Figure 30.20. During the test, the basket rotates at a constant speed, typically set between 50 and 100 rpm. The dissolution medium is contained in a glass cylindrical vessel with a spherical bottom and with a nominal capacity of no less than 1 L. The dissolution medium volume used with this method is normally 0.9 L, although lower (0.5 L) and higher (4 L) volumes may also be employed. The composition and/or pH of the medium may be changed by manually replacing it or by adding media of different composition. At pre-determined times, samples of dissolution medium are removed and analysed for drug content.
Paddle apparatus (USP Apparatus 2)
Following its introduction in the USP in 1978, the paddle apparatus became the most widely used dissolution tester. It utilizes the same dissolution vessels as the basket apparatus but here the dosage form is positioned at the centre bottom of the vessel. An outline of the apparatus is shown in Figure 35.5 with a more detailed diagram shown in Figure 30.21. Agitation is provided by a metallic paddle which rotates at speeds between 50 and 150 rpm (most often 50 to 75 rpm). To prevent dosage forms from floating (this normally occurs with capsules), the use of sinkers is recommended. Sinkers are a wire helix made of non-reactive material wherein the dosage form is placed. Changes of dissolution medium during the test are done manually as described for the basket apparatus.
Reciprocating cylinder (USP Apparatus 3)
In 1991, driven by the need to provide a controlled and automated pH and volume change of the dissolution medium during the test, the USP introduced the reciprocating cylinder apparatus. Current designs of this equipment allow for up to 6 automated medium changes per test, as well as changes to the agitation speed. This feature makes it particularly suited to estimate the drug release profile in different parts of the gastrointestinal tract as needed in the case of modified-release formulations, such as extended-release or gastro-resistant coated products. It also represents a step closer to bio-relevant conditions and to developing in vitro/in vivo correlations (IVIVCs).
The apparatus (shown in Fig. 35.6) comprises two cylinders: an inner cylinder containing the dosage form and an outer cylinder vessel, which holds around 200 to 300 mL of dissolution medium. During the test, the inner cylinder is dipped vertically into the dissolution medium several times, creating convective forces for dissolution. It is generally considered that 5 dips per minute (dpm) are equivalent to 50 rpm in the paddle apparatus. The inner cylinder is fitted with a mesh screen at the bottom and top which allows the medium to circulate freely inside it, yet prevents losses of finely disintegrated material.
Flow through cell (USP Apparatus 4)
The flow through cell was adopted by the USP in 1995, primarily for the testing of modified-release products. In this apparatus, the dosage form is positioned in a small-volume cell, on a glass bead bed or on a clip holder. The sample under test is subjected to a continuous flow of media in an upward direction. The medium is pumped from a reservoir at a flow rate which may vary from 5 to 20 mL/minute. The pulsating movement of the pump creates gentler hydrodynamics compared to other compendial apparatus (arguably more similar to the movement that would be experienced by a dosage form in the gut). The dissolution medium can be changed during the test by exchanging the media reservoirs.
This apparatus (shown in Fig. 35.7) can be configured to use a fixed volume (closed system) or unlimited volumes of dissolution medium (open system). In the latter set-up, fresh dissolution medium is delivered continuously by the pump and collected for analysis after passing though the sample cell; this system is particularly suitable for testing the dissolution of poorly soluble drugs.
Volume and composition of the dissolution medium
The choice of volume and composition of the dissolution medium is very much dependent on the solubility of the drug. As previously mentioned, a QC dissolution method must allow for at least 80% of the drug to dissolve during the duration of the test. To achieve this, QC methods are normally operated under ‘sink conditions’. A dissolution test is said to be performed under sink conditions if the concentration of the drug in the bulk of the dissolution medium does not exceed 10% of the solubility of the drug. Under sink conditions, the concentration gradient between the diffusion layer surrounding the solid drug particles and the concentration in the dissolution medium is assumed to be constant (Chapter 2). It must be noted, that sink conditions may represent an important deviation from what happens in the gastrointestinal tract, where such conditions are not always present and dissolution of poorly soluble drugs is often incomplete.
Sink conditions are easy to achieve with highly water-soluble drugs but pose a considerable problem for those drugs which have limited aqueous solubility. The basket and paddle apparatus are normally operated with a volume of 0.9 L and although this may be increased to 4 L, it may still not permit sink conditions to be reached for some drugs. Increasing the volume beyond 4 L is possible only with the flow through cell system. However, this apparatus is unsuitable for routine use; therefore choosing a medium, wherein the drug is soluble, is of paramount importance. Other considerations in choosing a QC dissolution medium are:
• must not affect the stability of the drug
• simple composition required to permit automation of the method
Not surprisingly, very few media meet such requirements to be regarded as suitable for QC dissolution testing. Although, pure water would appear to be an obvious choice, its use is discouraged because of the inability of this medium to withstand pH changes. The most commonly used dissolution media are either dilute acid solutions or aqueous buffers of a higher pH. Weakly basic drugs, by virtue of their higher solubility under acid conditions, are most often tested in diluted acid solutions (e.g. dilute 0.1 M hydrochloric acid or simulated gastric fluids). These are also the media of choice for immediate-release dosage forms of highly soluble drugs. Phosphate buffer solutions (which can be manipulated to have a pH between 5.0 and 8.0) are suited for the testing of weakly acidic drugs. If the solubility of the drug in these media is low, surfactants can be added to the media to enhance drug solubility and meet sink conditions.
Dissolution limits
Performing the dissolution test is only one part of QC testing. The data obtained must be checked against set dissolution limits to ensure that the drug product meets the required specifications. Dissolution limits for immediate-release and gastro-resistant coated products are defined in the pharmacopoeias, with slight variations between the various pharmacopoeias.
For immediate-release products, a single-point specification is used to ensure prompt dissolution; normally no less than 75% of the drug must be dissolved within 45 minutes.
Dissolution limits for gastro-resistant products are based on a two-point specification to ensure limited drug dissolution in the acidic conditions of the stomach and fast dissolution under the conditions of the small intestine. The specifications are typically no more than 10% drug dissolved in 0.1M hydrochloric acid within 2 hours (the typical maximum residence time of non-disintegrating tablets in the fasted stomach), followed by no less than 75% dissolution within 45 minutes in phosphate buffer (pH 6.8).
As for extended-release products, the specifications may be set by the product’s manufacturer. A minimum of two specification points are normally required; the earlier specification point provides assurance against premature drug dissolution, while the second should ensure complete drug dissolution.
Predictive dissolution testing
Predictive dissolution tests are designed to give a close account of the product’s performance in the gastrointestinal tract (rather than a simple yes/no result as in the case of QC dissolution tests). The best way to achieve this is by using dissolution conditions which mirror, as closely as possible, the physiology of the gastrointestinal tract. However, recreating such conditions is a great challenge. This is because the gastrointestinal tract is a very complex and dynamic environment; its physiology changes every day and throughout our lives, is affected by a number of diseases, by the food we consume, and above all, it is still poorly understood.
Dissolution data indicative of performance in the gastrointestinal tract can be obtained by adding surfactants into the dissolution medium to better simulate the solubility of the drug in the gastrointestinal luminal fluids. Although this is a very common strategy, it must be tested carefully, as many artificial surfactants, when used in high concentrations, may solubilize the drug too well and over-estimate the solubility of the drug in the gastrointestinal luminal fluids. For extended-release drug products and whenever the solubility of the drug is pH-dependent, using different buffered solutions with varying pH values in order to simulate transit through different segments of the gastrointestinal tract may be useful. These changing pH conditions are easy to recreate with the USP Apparatus 3 and 4 but not with the USP Apparatus 1 and 2. When these simple test methods fail, more advanced ones are required. Next we discuss the bio-relevant media and non-compendial dissolution apparatuses that are being used increasingly to develop predictive dissolution tests.
Bio-relevant dissolution media
The composition of the gastrointestinal tract luminal fluids could not be any more different from the simple buffered aqueous solutions that are relied on for the QC dissolution testing of drugs. Ingested food and endogenous fluids are mixed together in the lumen of the gastrointestinal tract, yielding a compositionally complex medium; the pH, composition and volume thereof are constantly changing in response to external and internal stimuli. Understandably its complexity and dynamic nature cannot be completely simulated; rather only a few parameters which are more likely to affect the solubility of drugs are often incorporated into the design of a bio-relevant dissolution medium.
Milk and nutritional liquid products
Dissolution testing may be carried out in media simulating the contents of a meal with a view to predicting food effects. For example, full fat milk (with 3.5 % fat) and the product Ensure® Plus have been successfully used to simulate the initial stages of the fed stomach. Both media contain protein, fat and carbohydrates in ratios which are comparable to those found in the typical Western diet. Fat and proteins aid the dissolution of poorly soluble drugs, whereas the higher pH of these media favours the dissolution of weak acidic drugs. Apart from the effect on solubility, milk may also delay the disintegration of slow-disintegrating tablets.
Simulated gastric and intestinal fluids
Simulated gastric or intestinal fluids are aqueous based solutions containing one or more biological components, which are known to influence the dissolution of drugs in vivo.
Media containing enzymes (pepsin or pancreatin) and artificial surfactants, in concentrations greater than those found in the gastrointestinal tract, were amongst the first to be developed to simulate the fasted gastric and small intestinal fluids. These media were soon found to over-estimate the solubility of many drugs in the gastrointestinal fluids.
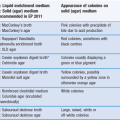
In a later development, fasted gastric and intestinal simulated media containing physiological concentrations of natural bile salts (sodium taurocholate) and lecithin were introduced. The fed versions of these media are more complex; they contain milk or lipolysis products to adequately simulate the influence of meal digestion on the solubility of drugs. The full compositions of these media are given in Tables 35.2 and 35.3.
Table 35.2
Composition of fasted (FaSSGF) and fed (FeSSGF) state simulated gastric fluids
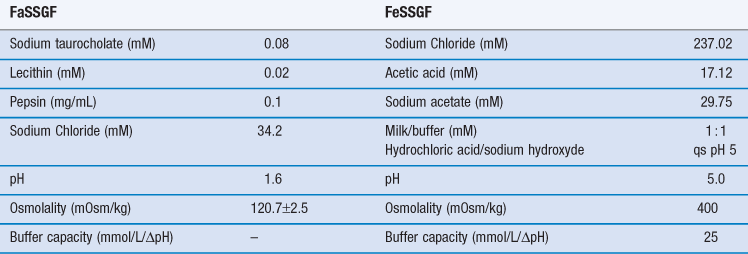
(Courtesy of Jantratid et al 2008, with permission.)
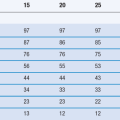
Table 35.3
Composition of fasted (FaSSIF) and fed (FeSSIF) state simulated intestinal fluids
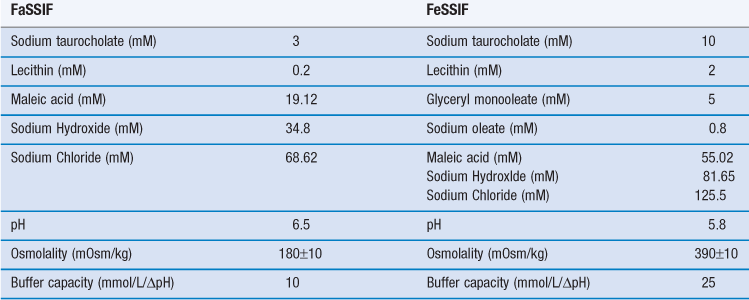
(Courtesy of Jantratid et al 2008, with permission.)
Bicarbonate buffers
Bicarbonate is the main buffer species in the luminal fluids of the small intestine. However, thus far its use in dissolution media has been very limited. Dissolution media are typically composed of phosphate ions, whose presence in the gastrointestinal luminal fluids is insignificant. A change from phosphate- to bicarbonate-based buffers should therefore better resemble the ionic composition and buffer capacity of the fasted jejunal and ileal fluids. The very few dissolution studies that have made use of bicarbonate buffers found these to be superior to phosphate buffers in predicting the dissolution behaviour of gastro-resistant polymers and the solubility of ionizable drugs in the gastrointestinal tract (Liu et al, 2011).
Unlike the fasted and fed simulated fluids, these buffers are easy to prepare and their composition is very simple. However, due to the difficulty in stabilizing the pH, bicarbonate buffers must be continuously purged with carbon dioxide, making the experimental set up more difficult.
Non-compendial apparatuses
The various compendial dissolution apparatus bear little resemblance to the gastrointestinal tract physiology and recreating such complexity in vitro may be too great of a task. The gastrointestinal tract simply does not ‘paddle’ in the same way or at the same speed as the compendial dissolution apparatus. Attempts to render flow rates more bio-relevant led to the development of the USP Apparatus 4. However, its unidirectional flow pattern does not account for retropulsion and the flow patterns employed may still be too high compared to the gastrointestinal tract. Apart from poor hydrodynamics, the closed and static environment of compendial dissolution apparatus does not account for drug transport across the intestinal mucosa. Combining drug dissolution with absorption may improve the predictive power of dissolution testing. Some examples of non-compendial apparatus, which are increasingly being used to establish IVIVCs, are described below.
Stress test apparatus
This novel apparatus simulates the irregular motility patterns of the gastrointestinal tract and the physical stress experienced by the dosage form during gastric emptying and transit through the gut. Its design allows the dosage form to be either submerged in liquid or exposed to air in an attempt to recreate the discontinuous distribution of fluid in the intestines.
This apparatus has been shown to successfully reproduce the dissolution profile of an extended release formulation of diclofenac in the gastrointestinal tract, which was not possible using the USP Apparatus 2 (Garbacz et al, 2008).
Dynamic gastric model (DGM)
The DGM is a computer-controlled simulator of the digestion patterns of the stomach. It consists of three mains parts: the main body (or fundus), the valve assembly and the distal region of the stomach or antrum. The simulator models the mechanical and enzymatic events occurring in these regions and which lead to the digestion of meals. Heterogeneous gastric mixing in the main body of the stomach is provided by gentle contractions. Gastric secretions (acid and enzymes) are delivered via a dispenser, from the sides of the simulator to recreate the secretion of these compounds by the gastrointestinal mucosa. The rate at which secretions are released into the main body is controlled by the volume and pH of the contents in a similar manner to what occurs in the gastrointestinal tract (Vardakou et al, 2011).
The contents are moved from the main body into the antrum and vice-versa through the valve assembly. The forces which coexist in the antrum are considerably stronger than those in the main body; these represent the physiological grinding forces responsible for the breakdown of food particles. Once ready, the processed bolus is ejected through a valve and collected for further analysis.
As a stand-alone equipment, the DGM is unlikely to give a useful estimate of the performance of dosage forms in the gastrointestinal tract. However, if combined to an equally bio-relevant simulator of the small intestine, the DGM may provide good correlations to oral bioavailability.
Simulator of the gastrointestinal tract (TIM-1)
The dynamic system, known as TIM-1, is undoubtedly the most complete simulator of the gastrointestinal tract. The TIM-1 models the upper gastrointestinal tract and is composed of interconnected segments representing the stomach, duodenum, jejunum and ileum. Most physiological parameters such as body temperature, pH, peristaltic mixing, gastrointestinal transit and main secretions (saliva, lipase, pepsin, HCl, pancreatic juice, bile and sodium bicarbonate) are simulated using this system. Transit is regulated by peristaltic valves which exist at the end of each segment and mixing is generated by consecutive cycles of compression and relaxation (Blanquet et al, 2004).
Its unique feature is the ability to simulate passive absorption of water and small molecules, such as dissolved drug, via dialysis membranes. Although, this represents a vast improvement compared to most simulators which do not account for absorption, it is still far from the conditions present in the gastrointestinal tract, where active transport, efflux and intestinal wall metabolism are also present.
The TIM-1 can be used to predict the performance of dosage forms in the gastrointestinal tract, yet this is not without some technical problems. For instance, each experiment takes considerably longer than a standard dissolution test and the conditions in which the test is conducted are often drug- and formulation-specific. Problems aside, this simulator has successfully established IVIVC where compendial dissolution apparatuses have failed.
Conclusions
Testing of the dissolution properties of oral solid drug products has been practiced in the pharmaceutical industry for many decades, yet the role of this test is still very much evolving. What was once a simple test to differentiate a ‘good’ batch of a drug product from a ‘bad’ one is now developing into a key tool for predicting bioavailability. In early stage research and development, dissolution data aid in the selection of candidate formulations for further development, a role of increasing relevance due to the growing number of poorly water-soluble investigational drugs. From a regulatory point of view, dissolution testing is used to establish bioequivalence between drug products, obviating the need for expensive and lengthy clinical studies.
Given the many uses and benefits of dissolution testing in various stages of the drug product’s lifecycle, it is not surprising that considerable efforts have been made into developing appropriate dissolution methods. As there is no absolute method for dissolution testing, pharmaceutical scientists are constantly being challenged to design new methods based on the goal of the test (i.e., QC, establishing IVIVC or demonstrating bioequivalence) and on the characteristics of the drug product. The key challenge remains that of designing predictive dissolution tests because of the difficulties in simulating the interactions between the drug and the complex environment of the gastrointestinal tract. However, as understanding of these interactions improves, so does the predictive power of dissolution tests. The recent development of bio-relevant dissolution media and the design of sophisticated simulators of the gastrointestinal tract are key examples of this.
References
1. Blanquet S, Zeijdner E, Beyssac E, et al. A dynamic artificial gastrointestinal system for studying the behavior of orally administered drug dosage forms under various physiological conditions. Pharmaceutical Research. 2004;21:585–591.
2. Charman WN, Rogge MC, Boddy AH, Berger BM. Effect of food and a monoglyceride emulsion formulation on danazol bioavailability. Journal of Clinical Pharmacolology. 1993;33:381–386.
3. Fraser EJ, Leach R,H, Poston JW, Bold AM, Culank LS, Lipede AB. Dissolution and bioavailability of digoxin tablets. Journal of Pharmacy and Pharmacology. 1973;25:968–973.
4. Garbacz G, Wedemeyer R-S, Nagel S, et al. Irregular absorption profiles observed from diclofenac extended release tablets can be predicted using a dissolution test apparatus that mimics in vivo physical stresses. European Journal of Pharmaceutics and Biopharmaceutics. 2008;70:421–428.
5. Jantratid E, Janssen N, Reppas C, Dressman JB. Dissolution media simulating conditions in the proximal human gastrointestinal tract: an update. Pharmaceutical Research. 2008;25:1663–1676.
6. Levy G. Effect of dosage form properties on therapeuctic efficacy of tolbutamide tablets. Canadian Medical Association Journal. 1964;90:978–979.
7. Liu F, Merchant HA, Kulkarni RP, Alkademi M, Basit AW. Evolution of a physiological pH 6.8 bicarbonate buffer system: Application to the dissolution testing of enteric coated products. European Journal of Pharmaceutics and Biopharmaceutics. 2011;78:151–157.
8. Vardakou M, Mercuri A, Naylor TA, et al. Predicting the human in vivo performance of different oral capsule shell types using a novel in vitro dynamic gastric model. International Journal of Pharmaceutics. 2011;419:192–199.
Bibliography
1. Dokoumetzidis A, Macheras P. A century of dissolution research: From Noyes and Whitney to the Biopharmaceutics Classification System. International Journal of Pharmaceutics. 2006;321:1–11.
2. Dressman JB, Vertzoni M, Goumas K, Reppas C. Estimating drug solubility in the gastrointestinal tract. Advanced Drug Delivery Reviews. 2007;59:591–602.
3. Freire AC, Basit AW, Choudhary R, Piong CW, Merchant HA. Does sex matter? The influence of gender on gastrointestinal physiology and drug delivery. International Journal of Pharmaceutics. 2011;415:15–28.
4. McAllister M. Dynamic dissolution: a step closer to predictive dissolution testing? Molecular Pharmaceutics. 2010;7:1374–1387.
5. McConnell EL, Fadda HM, Basit AW. Gut instincts: explorations in intestinal physiology and drug delivery. International Journal of Pharmaceutics. 2008;364:213–226.
6. Mudie DM, Amidon GL, Amidon GE. Physiological parameters for oral delivery and in vitro testing. Molecular Pharmaceutics. 2010;7:1388–1405.