[level-membership-for-pulmolory-and-respiratory-category]18
Disorders of Ventilatory Control
The finely tuned system of ventilatory control described in Chapter 17 is altered in a variety of clinical circumstances. In some cases, a primary disorder of the nervous system affects the neurologic network involved in ventilatory control and therefore may either diminish or increase the “drive” to breathe. In other instances the controlling system undergoes a process of adaptation in response to primary lung disease, so any alteration in function is a secondary phenomenon.
Primary Neurologic Disease
Presentation with Hypoventilation
Patients with these syndromes of hypoventilation are characterized by depressed ventilatory responses to the chemical stimuli of hypercapnia and hypoxia. Measurement of arterial blood gases generally reveals an elevation in arterial PCO2 accompanied by a decrease in PO2, the latter primarily attributable to hypoventilation. As in other disorders associated with these blood gas abnormalities, cor pulmonale may result and be the presenting problem in these syndromes. The term congenital central hypoventilation syndrome or Ondine’s curse (see Chapter 17) has been applied to a rare subset of patients with congenital alveolar hypoventilation. However, an element of decreased ventilatory response to hypercapnia and hypoxia is much more commonly seen in clinical practice and probably represents a spectrum of abnormalities in ventilatory response.
In the past, treatment of alveolar hypoventilation generally centered around two modalities: drugs (most commonly the hormone progesterone) and electrical stimulation of the phrenic nerve. Progesterone is well known to be a respiratory stimulant and in some cases may improve respiratory drive and decrease CO2 retention. In the second approach, the diaphragm can be induced to contract by repetitive electrical stimulation of the phrenic nerve, which can be achieved by intermittent current applied via an implanted electrode. Although both of these modalities are still used, the most common current therapy for patients with clinically significant hypoventilation is noninvasive positive-pressure (i.e., assisted) ventilation, usually applied nocturnally. This topic is discussed in Chapter 29.
Cheyne-Stokes Breathing
Cheyne-Stokes breathing is a cyclic pattern in which periods of gradually increasing ventilation alternate with periods of gradually decreasing ventilation (even to the point of apnea). This type of ventilation is shown schematically in Figure 18-1. It has been known for many years that two main types of disorders are associated with this type of breathing: heart failure and some forms of CNS disease. Cheyne-Stokes breathing can also be seen under certain physiologic situations even in the absence of underlying disease. Examples include the onset of sleep and exposure to high altitude.
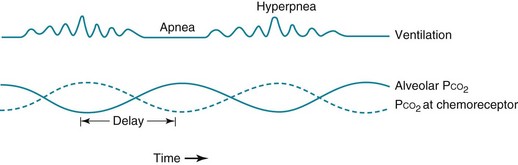
Prolongation in circulation time, which is one mechanism postulated to play a role in heart failure, results in an abnormal delay between events in the lung and sensing of PCO2 changes by the central chemoreceptors. Hence, medullary respiratory output is out of phase with gas exchange at the lungs, and oscillations in ventilation occur as the central chemoreceptor and the medullary respiratory center make belated attempts to maintain a stable PCO2 (see Fig. 18-1).
A similar type of instability of ventilatory control occurs when hypoxia is driving the feedback system, as is seen on exposure to high altitude. The ventilatory response to hypoxia is nonlinear. For the same drop in PO2, the increment in ventilation is larger at a lower absolute PO2 (see Fig. 17-3). This means that at a relatively high initial PO2, the system is less likely to respond to small changes in PO2 but then is apt to overshoot as PO2 falls further. This instability of the respiratory control system results in a widely oscillating output from the respiratory center and thus a cyclic pattern of ventilation.
Control Abnormalities Secondary to Lung Disease
In contrast, patients with chronic obstructive pulmonary disease (COPD) have variable levels of PCO2. Some patients with COPD do not demonstrate CO2 retention, whereas the condition of others is often characterized by hypercapnia (see Chapter 6). In the latter group, the ventilatory control mechanism appears to be recalibrated to operate at a higher setpoint for PCO2. When responsiveness to increased levels of PCO2 is measured in hypercapnic patients, it is apparent that their ventilatory response is diminished. However, these patients with chronic compensated respiratory acidosis have higher levels of plasma (as well as cerebrospinal fluid) bicarbonate because of bicarbonate retention by the kidneys. Therefore, for any increment in PCO2, the effect on pH at the medullary chemoreceptor is attenuated by the increased buffering capacity available. A “chicken and egg” question then becomes important: Is CO2 retention secondary to an underlying ventilatory control abnormality in these patients, or is the diminution in ventilatory sensitivity merely secondary to chronic CO2 retention? Although this question remains unanswered, some evidence suggests that hereditary factors may be important and that CO2 retention is more likely to develop in patients with a genetically lower respiratory sensitivity.
However, it now is known that hypoxic drive plays only a limited role in the frequently observed increase in PCO2 occurring in patients with underlying hypoventilation who are given supplemental oxygen. Three factors account for this well-recognized clinical event: changes in minute ventilation, changes in ventilation-perfusion matching, and the Haldane effect. To grasp this complicated phenomenon, each of these factors should be understood. The easiest factor to understand is the change in minute ventilation. If a patient is hypoxemic, low PaO2 is sensed by the peripheral chemoreceptors, causing stimulation of the respiratory center. When supplemental oxygen is given and the patient is no longer hypoxemic, this stimulation abates. As discussed, this was previously thought to be the sole explanation for the rise in PCO2 seen in hypoxic hypercapnic patients who were given supplemental oxygen. However, it now is known from a number of studies that the decrease in ventilation accounts for only a small proportion of the rise in PCO2. More important is a worsening of ventilation-perfusion matching. Recall that alveolar hypoxia results in decreased perfusion to the hypoxic lung segments, an effect that is mediated through hypoxic vasoconstriction of those pulmonary arterioles supplying hypoxic alveoli. However, administration of supplemental O2 may alleviate alveolar hypoxia in these poorly ventilated regions, thus aborting the compensatory localized vasoconstriction. Ventilation-perfusion mismatch becomes more marked in the absence of hypoxic vasoconstriction, leading to less efficient elimination of CO2 and increased levels of PCO2. The third factor contributing to the rise in PCO2 is the Haldane effect, in which deoxygenated hemoglobin has a higher affinity for CO2 (see Chapter 1). When supplemental oxygen is administered, the more oxygenated hemoglobin has a lower affinity for CO2, leading to enhanced release of CO2 from hemoglobin and a higher PCO2.
Sleep Apnea Syndrome
Pathophysiology
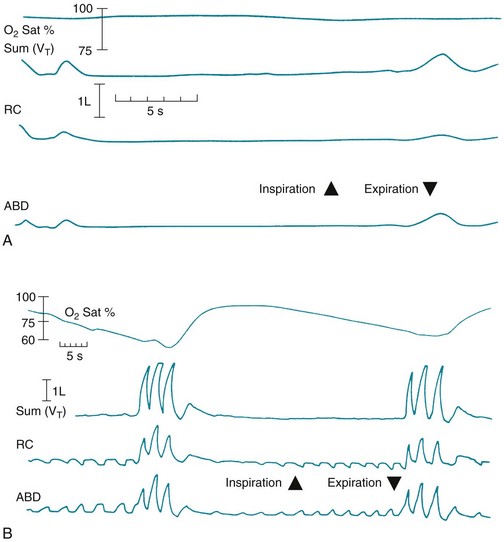
During an episode of central apnea, monitoring of chest wall motion reveals no movement, corresponding to cessation of airflow and a fall in O2 saturation (Fig. 18-2, A). With obstructive apnea, chest wall and abdominal movement can be detected during a fruitless attempt to move air through the obstructed airway. Airflow measured simultaneously is found to be absent (tidal volume = 0) and O2 saturation drops, often to profoundly low levels (Fig. 18-2, B). When O2 saturation drops significantly during sleep, disturbances in cardiac rhythm can occur, and elevation of pulmonary artery pressure may be seen as a consequence of hypoxia-induced pulmonary vasoconstriction.
Burton, MD, Kazemi, H. Neurotransmitters in central respiratory control. Respir Physiol. 2000;122:111–121.
Chebbo, A, Tfaili, A, Jones, SF. Hypoventilation syndromes. Med Clin North Am. 2011;95:1189–1202.
Kennedy, JD, Martin, AJ. Chronic respiratory failure and neuromuscular disease. Pediatr Clin North Am. 2009;56:261–273.
Laffey, JG, Kavanagh, BP. Hypocapnia. N Engl J Med. 2002;347:43–53.
Lustik, SJ, Chhibber, AK, Kolano, JW, et al. The hyperventilation of cirrhosis: progesterone and estradiol effects. Hepatology. 1997;25:55–58.
Mellins, RB, Balfour, HH, Turino, GM, et al. Failure of automatic control of ventilation (Ondine’s curse). Medicine. 1970;49:487–504.
Pino-Garcia, JM, García-Río, F, Díez, JJ, et al. Regulation of breathing in hyperthyroidism: relationship to hormonal and metabolic changes. Eur Respir J. 1998;12:400–407.
Piper, AJ, Grunstein, RR. Obesity hypoventilation syndrome: mechanisms and management. Am J Respir Crit Care Med. 2011;183:292–298.
Weese-Mayer, DE, Berry-Kravis, EM, Ceccherini, I, ATS Congenital Central Hypoventilation Syndrome Subcommittee. An official ATS clinical policy statement: Congenital central hypoventilation syndrome: genetic basis, diagnosis, and management. Am J Respir Crit Care Med. 2010;181:626–644.
Cherniack, NS, Longobardo, GS. Abnormalities in respiratory rhythm. In: Fishman AP, Cherniack NS, Widdicombe JG, et al, eds. Handbook of physiology. Section 3: The respiratory system, Vol. II. Control of breathing, Part 2. Bethesda, MD: American Physiological Society; 1986:729–749.
Cherniack, NS, Longobardo, GS. Cheyne-Stokes breathing: an instability in physiologic control. N Engl J Med. 1973;288:952–957.
Cherniack, NS, Longobardo, G, Evangelista, CJ. Causes of Cheyne-Stokes respiration. Neurocrit Care. 2005;3:271–279.
Naughton, MT. Pathophysiology and treatment of Cheyne-Stokes respiration. Thorax. 1998;53:514–518.
Sharma, B, Owens, R, Malhotra, A. Sleep in congestive heart failure. Med Clin North Am. 2010;94:447–464.
Control Abnormalities Secondary to Lung Disease
Aubier, M, Murciano, D, Milic-Emili, J, et al. Effects of the administration of O2 on ventilation and blood gases in patients with chronic obstructive pulmonary disease during acute respiratory failure. Am Rev Respir Dis. 1980;122:747–754.
Caruana-Montaldo, B, Gleeson, K, Zwillich, CW. The control of breathing in clinical practice. Chest. 2000;117:205–225.
Dunn, WF, Nelson, SB, Hubmayr, RD. Oxygen-induced hypercarbia in obstructive pulmonary disease. Am Rev Respir Dis. 1991;144:526–530.
Epstein, SK, Singh, N. Respiratory acidosis. Respir Care. 2001;46:366–383.
Kutty, K. Sleep and chronic obstructive pulmonary disease. Curr Opin Pulm Med. 2004;10:104–112.
Milic-Emili, J, Aubier, M. Some recent advances in the study of the control of breathing in patients with chronic obstructive lung disease. Anesth Analg. 1980;59:865–873.
Mountain, R, Zwillich, C, Weil, J. Hypoventilation in obstructive lung disease: the role of familial factors. N Engl J Med. 1978;298:521–525.
Park, SS. Respiratory control in chronic obstructive pulmonary diseases. Clin Chest Med. 1980;1:73–84.
Weinberger, SE, Schwartzstein, RM, Weiss, JW. Hypercapnia. N Engl J Med. 1989;321:1223–1231.
American Academy of Sleep Medicine Task Force. Sleep-related breathing disorders in adults: recommendations for syndrome definition and measurement techniques in clinical research. Sleep. 1999;22:667–689.
Basner, RC. Continuous positive airway pressure for obstructive sleep apnea. N Engl J Med. 2007;356:1751–1758.
Bradley, TD, Floras, JS. Obstructive sleep apnoea and its cardiovascular consequences. Lancet. 2009;373:82–93.
Caples, SM, Garcia-Touchard, A, Somers, VK. Sleep-disordered breathing and cardiovascular risk. Sleep. 2007;30:291–303.
Casey, KR, Cantillo, KO, Brown, LK. Sleep-related hypoventilation/hypoxemic syndromes. Chest. 2007;131:1936–1948.
Chan, AS, Lee, RW, Cistulli, PA. Dental appliance treatment for sleep apnea. Chest. 2007;132:693–699.
Drager, LF, Polotsky, VY, Lorenzi-Filho, G. Obstructive sleep apnea: an emerging risk factor for atherosclerosis. Chest. 2010;140:534–542.
Eckert, DJ, Jordan, AS, Merchia, P, et al. Central sleep apnea pathophysiology and treatment. Chest. 2007;131:595–607.
Flemons, WW. Obstructive sleep apnea. N Engl J Med. 2002;347:498–504.
George, CF. Sleep apnea, alertness, and motor vehicle crashes. Am J Respir Crit Care Med. 2007;176:954–956.
Gifford, AH, Leiter, JC, Manning, HL. Respiratory function in an obese patient with sleep-disordered breathing. Chest. 2010;138:704–715.
Gozal, D, Kheirandish-Gozal, L. Cardiovascular morbidity in obstructive sleep apnea: oxidative stress, inflammation and much more. Am J Respir Crit Care Med. 2008;177:369–375.
Holty, JE, Guilleminault, C. Surgical options for the treatment of obstructive sleep apnea. Med Clin North Am. 2010;94:479–515.
Kakkar, RK, Berry, RB. Positive airway pressure treatment for obstructive sleep apnea. Chest. 2007;132:1057–1072.
Kaw, R, Hernandez, AV, Walker, E, et al. Determinants of hypercapnia in obese patients with obstructive sleep apnea: a systematic review and metaanalysis of cohort studies. Chest. 2009;136:787–796.
Park, JG, Ramar, K, Olson, EJ. Updates on definition, consequences, and management of obstructive sleep apnea. Mayo Clin Proc. 2011;86:549–554.
Patil, SP, Schneider, H, Schwartz, AR, et al. Adult obstructive sleep apnea: pathophysiology and diagnosis. Chest. 2007;132:325–337.
Primhak, R, Kingshott, R. Sleep physiology and sleep-disordered breathing: the essentials. Arch Dis Child. 2012;97:54–58.
Randerath, WJ, Verbraecken, J, Andreas, S, et al. Non-CPAP therapies in obstructive sleep apnoea. European Respiratory Society task force on non-CPAP therapies in sleep apnoea. Eur Respir J. 2011;37:1000–1028.
Roldan, G, Ang, RC. Overview of sleep disorders. Respir Care Clin North Am. 2006;12:31–54.
Teran-Santos, J, Jimenez-Gomez, A, Cordero-Guevera, J. The association between sleep apnea and the risk of traffic accidents. N Engl J Med. 1999;340:847–851.
Ulualp, SO. Snoring and obstructive sleep apnea. Med Clin North Am. 2010;94:1047–1055.
Veasey, SC, Guilleminault, C, Strohl, KP, et al. Medical therapy for obstructive sleep apnea: a review by the Medical Therapy for Obstructive Sleep Apnea Task Force of the Standards of Practice Committee of the American Academy of Sleep Medicine. Sleep. 2006;29:1036–1044.
Walker, RP. Surgical treatment for snoring and obstructive sleep apnea. Dis Mon. 2011;57:403–413.
White, DP. Pathogenesis of obstructive and central sleep apnea. Am J Respir Crit Care Med. 2005;172:1363–1370.
Witmans, M, Young, R. Update on pediatric sleep-disordered breathing. Pediatr Clin North Am. 2011;58:571–589.
[/level-membership-for-pulmolory-and-respiratory-category][not-level-membership-for-pulmolory-and-respiratory-category]18
Disorders of Ventilatory Control
The finely tuned system of ventilatory control described in Chapter 17 is altered in a variety of clinical circumstances. In some cases, a primary disorder of the nervous system affects the neurologic network involved in ventilatory control and therefore may either diminish or increase the “drive” to breathe. In other instances the controlling system undergoes a process of adaptation in response to primary lung disease, so any alteration in function is a secondary phenomenon.
Primary Neurologic Disease
Presentation with Hypoventilation
Patients with these syndromes of hypoventilation are characterized by depressed ventilatory responses to the chemical stimuli of hypercapnia and hypoxia. Measurement of arterial blood gases generally reveals an elevation in arterial PCO2 accompanied by a decrease in PO2, the latter primarily attributable to hypoventilation. As in other disorders associated with these blood gas abnormalities, cor pulmonale may result and be the presenting problem in these syndromes. The term congenital central hypoventilation syndrome or Ondine’s curse (see Chapter 17) has been applied to a rare subset of patients with congenital alveolar hypoventilation. However, an element of decreased ventilatory response to hypercapnia and hypoxia is much more commonly seen in clinical practice and probably represents a spectrum of abnormalities in ventilatory response.
In the past, treatment of alveolar hypoventilation generally centered around two modalities: drugs (most commonly the hormone progesterone) and electrical stimulation of the phrenic nerve. Progesterone is well known to be a respiratory stimulant and in some cases may improve respiratory drive and decrease CO2 retention. In the second approach, the diaphragm can be induced to contract by repetitive electrical stimulation of the phrenic nerve, which can be achieved by intermittent current applied via an implanted electrode. Although both of these modalities are still used, the most common current therapy for patients with clinically significant hypoventilation is noninvasive positive-pressure (i.e., assisted) ventilation, usually applied nocturnally. This topic is discussed in Chapter 29.
Cheyne-Stokes Breathing
Cheyne-Stokes breathing is a cyclic pattern in which periods of gradually increasing ventilation alternate with periods of gradually decreasing ventilation (even to the point of apnea). This type of ventilation is shown schematically in Figure 18-1. It has been known for many years that two main types of disorders are associated with this type of breathing: heart failure and some forms of CNS disease. Cheyne-Stokes breathing can also be seen under certain physiologic situations even in the absence of underlying disease. Examples include the onset of sleep and exposure to high altitude.
Prolongation in circulation time, which is one mechanism postulated to play a role in heart failure, results in an abnormal delay between events in the lung and sensing of PCO2 changes by the central chemoreceptors. Hence, medullary respiratory output is out of phase with gas exchange at the lungs, and oscillations in ventilation occur as the central chemoreceptor and the medullary respiratory center make belated attempts to maintain a stable PCO2 (see Fig. 18-1).
A similar type of instability of ventilatory control occurs when hypoxia is driving the feedback system, as is seen on exposure to high altitude. The ventilatory response to hypoxia is nonlinear. For the same drop in PO2, the increment in ventilation is larger at a lower absolute PO2 (see Fig. 17-3). This means that at a relatively high initial PO2, the system is less likely to respond to small changes in PO2 but then is apt to overshoot as PO2 falls further. This instability of the respiratory control system results in a widely oscillating output from the respiratory center and thus a cyclic pattern of ventilation.
