[level-membership-for-neurology-category]
Chapter 74 Disorders of Upper and Lower Motor Neurons
Disorders of Upper Motor Neurons
Neuroanatomy of Upper Motor Neurons
The UMN is a motor neuron, the cell body of which lies within the motor cortex of the cerebrum, and the axon of which forms the corticobulbar and corticospinal tracts. The LMNs, lying in the brainstem motor nuclei and the anterior horns of the spinal cord, directly innervate skeletal muscles. The UMNs are rostral to the LMNs and exert direct or indirect supranuclear control over the LMNs (Box 74.1).
Box 74.1 Upper Motor Neurons and Their Descending Tracts
Motor Cortex
Limbic Motor Control
The limbic system is involved in emotional experience and expression and associated with a variety of autonomic, visceral, and endocrine functions. It strongly influences the somatic motor neurons. The emotional status and experience of an individual determines overall spinal cord activity, and the limbic motor system also influences respiration, vomiting, swallowing, chewing, and licking (at least in animal studies). Furthermore, the generation of signs of pseudobulbar hyperemotionality (pseudobulbar affect, emotional incontinence) in ALS is closely related to an abnormal limbic motor control, particularly in the periaqueductal gray and nucleus retroambiguus. The latter nuclei project to the somatic motor neurons that innervate pharyngeal, soft palatal, intercostal, diaphragmatic, abdominal, and probably laryngeal muscles. Pseudobulbar hyperemotionality symptoms may appear when UMN control over these motor nuclei is impaired, and thus limbic motor control is disinhibited. There appears to be some degree of emotional regulation by the cerebellum. The “cerebellar cognitive affective syndrome” can arise when stroke, tumor, or infection interrupts connections between the cerebellum and cerebral association and paralimbic regions (Schmahmann and Sherman, 1998).
Signs and Symptoms of Upper Motor Neuron Involvement
Loss of Dexterity
Loss of dexterity is one of the most characteristic signs of UMN impairment. Voluntary skillful movements require the integrated activation of many interneuron circuits in the spinal cord; such integration is ultimately controlled by the corticospinal tract and thus by UMNs. Loss of dexterity may express itself as stiffness, slowness, and clumsiness in performing any skillful motor actions. Asking the patient to perform rapid repetitive motions such as foot or finger tapping assesses loss of dexterity at the bedside. It is useful to assess both sides of the body, as many motor neuron disorders are asymmetrical (Box 74.2).
Box 74.2 Signs and Symptoms of Upper Motor Neuron Involvement
Loss of muscle strength (mild weakness)
Pathological reflexes (Babinski, Hoffmann sign, loss of abdominal reflexes)
Increased reflexes in an atrophic limb (probable upper motor neuron sign)
Pseudobulbar (spastic bulbar) palsy (emotional lability, brisk jaw jerk, hyperactive gag, forced yawning, snout reflex, suck reflex, slow tongue movements, spastic dysarthria)
Laboratory Evidence of Upper Motor Neuron Involvement
Neuroimaging
The use of brain magnetic resonance imaging (MRI) in ALS is largely to exclude other conditions but sometimes shows abnormal signal intensity in the corticospinal and corticobulbar tracts as they descend from the motor strip via the internal capsules to the cerebral peduncles. In ALS, signal changes, best appreciated on proton density images of the internal capsules, probably represent wallerian degeneration; similar changes also appear on conventional T2-weighted and fluid-attenuated inversion recovery (FLAIR) sequences. However, these changes do not appear to be sufficiently sensitive, and efforts continue to evaluate other potential MRI techniques such as diffusion tensor and volumetric MRI, which may serve as markers of UMN disease (Agosta et al., 2010).
Magnetic Resonance Spectroscopy Imaging
Proton density magnetic resonance spectroscopy (1H-MRS) is a noninvasive nuclear magnetic resonance technique that combines the advantages of MRI with in vivo biochemical information. A significant reduction of N-acetylaspartate, a neuronal marker, relative to creatine or choline (used as internal standards) exists in the sensorimotor cortices of patients with ALS who have UMN signs. Alterations in the measured levels of these metabolites using 1H-MRS are useful in the detection of UMN dysfunction early in the evolution of ALS and are useful to monitor progression over time. MRS still requires further technological improvements before it comes into widespread use (Mitsumoto et al., 2007).
Primary Lateral Sclerosis
Primary lateral sclerosis (PLS), first described by Erb in 1875, is a rare UMN disease variant that accounts for 2% to 4% of all cases of ALS and is traditionally distinguished by a lack of LMN involvement. The Pringle criteria for PLS stipulated that disease be restricted to the UMN system for at least 3 years from the time of clinical onset (Pringle et al., 1992), but a figure of 4 years is now proposed during which there is neither clinical nor neurophysiological evidence of LMN involvement. In a recent study comparing the evolution of disease in PLS versus UMN-predominant ALS and typical ALS, the median time to development of electromyographic (EMG) LMN features after onset in those with an evolving ALS was 3.17 years; in those patients, clinical signs of LMN disease occurred on average about 6 months later. Nonetheless, later development of LMN signs may occur and require reclassification as ALS in some cases, which therefore necessitates constant longitudinal review of each case (Gordon et al., 2009).
PLS typically presents in patients in their early 50s (about a decade younger than typical MND/ALS patients) as a very slowly evolving spastic paraparesis that spreads to the upper limbs and eventually causes pseudobulbar palsy. In rare instances, onset is in the bulbar system or follows a slowly ascending or descending hemiplegic pattern (Mills hemiplegic variant), but a bulbar-onset presentation should make the clinician wary of later LMN signs elsewhere. Other features include cramps and fasciculations, but such complaints are neither prominent nor universal. Bladder dysfunction is rare and, if it occurs at all, tends to be a late feature. Although muscle weakness is present, the main deficits are due to spasticity in dexterity and gait. The rate of progression can be exceedingly slow, often progressing over many years to the point where the patient manifests a robotic gait, debilitating generalized spasticity, and prominent pseudobulbar palsy. Muscle atrophy, if it occurs at all, is a very late feature. No clinically detectable sensory changes occur. Neuropsychological test batteries may define subtle cognitive deficits due to frontal cortical involvement, but dementia is not a prominent feature. A few patients may exhibit abnormal voluntary eye movements. Breathing is usually unimpaired in PLS, and as a consequence, forced vital capacity (FVC) is not affected (Gordon et al., 2009).
The prognosis is significantly better than for MND/ALS: one series had a median disease duration of 19 years and another series exhibited a range of survival from 72 to 491 months (Murray, B., 2006). The underlying pathogenesis of PLS remains undefined. Pathological changes include a striking loss of Betz cells in layer 5 of the frontal and prefrontal motor cortex (and other smaller pyramidal cells) together with laminar gliosis of layers 3 and 5 and degeneration of the corticospinal tracts. Spinal anterior horn cells are characteristically unaffected.
Diagnosis
The diagnosis of PLS is essentially one of exclusion (Table 74.1). Rare reports exist of UMN-onset ALS exist where the interval between onset of UMN signs and subsequent LMN signs have been up to 27 years. As such, it is vital to reassess patients diagnosed with PLS, as late signs of LMN involvement may occur that would reclassify their disorder as UMN-onset ALS.
Table 74.1 Disorders of Upper Motor Neurons and Their Key Characteristics
| Disorders | Key Characteristics |
|---|---|
| Primary lateral sclerosis | A diagnosis of exclusion |
| Hereditary spastic paraplegia | Heredity, usually autosomal dominant, spastin gene mutation, other mutations (see text), “sporadic” |
| HTLV-1-associated myelopathy | Slowly progressive myelopathy, endemic, and positive HTLV-1 |
| HTLV-2-associated myelopathy | Amerindian, IV drug abuser, concomitant HIV |
| Adrenomyeloneuropathy | X-linked recessive inheritance, adrenal dysfunction, myelopathy, very long-chain fatty acid assay |
| Lathyrism | History of consumption of chickling peas |
| Konzo | Eastern African, cassava root consumption |
HIV, Human immunodeficiency virus; HTLV, human T-lymphotropic virus; IV, intravenous.
Appropriate testing must exclude all definable causes for generalized UMN involvement. These include structural abnormalities (Chiari malformation and intrinsic and extrinsic spinal cord lesions) and myelopathies such as multiple sclerosis (MS) spondylotic cervical myelopathy, human immunodeficiency virus (HIV) myelopathy, human T-lymphotropic virus type 1 (HTLV-1) myelopathy, Lyme disease, syphilis, or adrenomyeloneuropathy. Spondylotic cervical myelopathy and MS are probably the most common causes among these disorders. The family history must be negative to rule out hereditary spastic paraplegia (HSP)/familial spastic paraparesis, spinocerebellar ataxia (SCA), hexosaminidase-A (Hex-A) deficiency, familial ALS (FALS), or adrenomyeloneuropathy. It is now apparent that some spastin mutation–associated HSP may lack a family history; it is worthwhile to carry out this gene test in patients presenting with symptoms and signs that are restricted to the lower extremities (Brugman et al., 2009). Paraneoplastic syndromes (especially in association with breast cancer) and Sjögren syndrome may clinically resemble PLS. Understanding of these entities is poor.
Treatment
No specific pharmacotherapy is available, and treatment therefore focuses on symptom control and supportive care. However, antispasticity drugs such as the GABA-B agonist, baclofen, and the central α2-agonist, tizanidine, may be tried for symptomatic treatment. Severe spasticity sometimes requires the insertion of an intrathecal baclofen pump. Tricyclic antidepressants, selective serotonin reuptake inhibitors, or dextromethorphan/quinidine may control pseudobulbar affect lability (Brooks et al., 2005).
Hereditary Spastic Paraplegia
HSP (or familial spastic paraparesis) is a genetically and clinically heterogeneous group of disorders rather than a single entity. The clinical feature common to all cases is progressively worsening spasticity of the lower extremities, often with variable degrees of weakness. The characteristic pathology is retrograde degeneration of the longest nerve fibers in the corticospinal tracts and posterior columns. Its estimated prevalence is 0.5 to 11.9 in 100,000, but its worldwide prevalence may actually be underestimated because of the benign nature of the disease in many families. Although the most common mode of inheritance is autosomal dominant, it may also be inherited in a recessive or X-linked fashion, and 12% to 13% of cases with apparently sporadic spastic paraparesis have spastin mutations (Depienne et al., 2006).
Genetic linkage studies of families around the world have mapped loci to over 40 autosomes as well as the X chromosome, with 17 distinct genes identified to date (Salinas et al., 2008). Inheritance of most pure HSP is autosomal dominant, whereas complicated forms are more often autosomal recessive. Between 40% and 45% of all families link to the SPAST gene on chromosome 2p22-21, which encodes spastin, a 616-amino acid protein. Mutations of various types (missense, nonsense, frameshift, splice site) may affect this gene (McDermott, C.J., 2006). Spastin is a highly conserved member of the AAA family of proteins (adenosine triphosphatase [ATPase] associated with various cellular activities). The exact role of mutant spastin in the pathogenesis of HSP is undefined, although a disturbance in maintenance of the microtubule cytoskeleton may exists, thus disrupting axonal transport. More than half of all cases do not manifest symptoms and signs until after age 30 years. Although this is normally a pure HSP, complicated forms occur, and some cases can develop a late-onset cognitive decline. Pathologically, degeneration of the longest corticospinal tracts and, to a lesser degree, the posterior columns of the spinal cord is seen.
Mutations in the SPG3A gene on 14q11-q21 encoding the novel protein, atlastin, give rise to an autosomal dominant often early-onset (<10 years of age) pure HSP which accounts for about 10% of autosomal dominant cases. This protein shares structural homology to guanylate-binding protein 1, which is a member of the dynamin family. Dynamins are important in intracellular trafficking of various kinds of vesicles. Mutations in KIF5A (SPG10, Chr 12q), a kinesin motor domain that is critical in intracellular transport, can cause both early- and late-onset spastic paraparesis with distal amyotrophy (Blair et al., 2006). Spastic paraplegia 11 (SPG11) is an autosomal recessive complicated HSP (thin corpus callosum, neuropathy, cognitive impairment) due to mutations in the spatacsin gene on chromosome 15q. This protein is of unknown function and does not appear to interact with the Golgi apparatus or microtubules. The cause of autosomal dominant pure HSP, linked to 2q24-34, is a mutation in the SPG13 gene, which encodes a mitochondrial heat shock protein. Recessively inherited complicated HSP links to chromosome 16q and is caused by a mutation in a gene encoding a mitochondrial protein known as paraplegin; this disorder can be ether pure or complicated (cerebellar signs, pale optic discs, and peripheral neuropathy). The genes for two different X-linked complicated HSP have been identified. In the first, mutant L1 (neural) cell adhesion molecule (L1CAM) may disrupt neuronal migration or differentiation; in the second mutant proteolipid protein (PLP1) is found in association with changes in white matter (duplication mutations in this same gene can also cause Pelizaeus-Merzbacher disease). Spastic paraplegia 17 (SPG17) is caused by mutations in the seipin gene on chromosome 11q12-q14. Also known as Silver syndrome, this disorder is an autosomal dominant complicated form of HSP with distal hand and foot amyotrophy beginning in the late teens to early 30s. Mutations in this gene are also the cause of a form of distal hereditary neuropathy (Charcot-Marie-Tooth [CMT] disease type 5).
Diagnosis
The basis for diagnosis of HSP is evidence of a family history in the setting of progressive gait disturbance, evidence of lower-extremity spasticity, and sparing of craniobulbar function. However, difficulties arise when there is no clear family history in recessive or X-linked disease and in cases of sporadic spastin mutation–associated HSP. Furthermore, considerable variation in disease expression exists between and within HSP families. In the absence of a family history or a demonstration of a known mutation, it is important to consider alternative causes for the clinical presentation, including structural disease (e.g., cerebral palsy, hydrocephalus, myelopathy), degenerative/infiltrative/inflammatory disease (e.g., MS, ALS, SCA, leukodystrophy), infections (syphilis, HIV, HTLV), levodopa-responsive dystonia, metabolic/toxic damage (vitamin B12 deficiency [subacute combined degeneration of the spinal cord (SCDC)], vitamin E deficiency, copper deficiency, lathyrism), and paraneoplastic disorders. MRI may reveal that cervical and thoracic spinal cord diameters are significantly smaller in both pure and complicated HSP than in controls (Sperfeld et al., 2005). Perhaps the most important differential diagnosis is that between apparently sporadic pure HSP and PLS, especially as the later may present with a slowly evolving spastic paraparesis for many years prior to the development of upper limb or bulbar features. The only reliable way to distinguish such disorders is through genetic testing; age at onset, urgency of micturition, and signs of dorsal column involvement (clinical or abnormal somatosensory evoked potentials [SSEPs]) are not accurate indicators of HSP versus PLS (Brugman et al., 2009).
Human T-Lymphotropic Virus Type 1–Associated Myelopathy, or Tropical Spastic Paraparesis
HTLV-1 causes a chronic progressive myelopathy that is referred to as tropical spastic paraparesis (TSP) in the Caribbean or HTLV-1–associated myelopathy (HAM) in Japan. This retrovirus is endemic in the Caribbean area, southwestern Japan, equatorial Africa, South Africa, parts of Asia, Central America, and South America, where it infects between 10 and 20 million people. Transmission occurs though sexual contact, intravenous (IV) drug use and also through breastfeeding. While between 2% and 3% of those infected can develop adult-onset T-cell leukemia, an estimated 2.5% to 3.8% can develop a chronic inflammatory myelopathy, with up to 20/100,000 affected in the Caribbean population and 3/100,000 in Japan. Recent evidence implicates high levels of activated HTLV-1–specific helper T cells and cytotoxic T cells in the pathogenesis of this syndrome; these immune cells appear to activate in response to interactions with retroviral env and tax proteins with greatest activity within the thoracic cord. Increased susceptibility for neurological disease appears to depend on both viral and host factors, with differences in certain HTLV-1 subgroups, proviral load, and HLA background being important. This may also explain differences in susceptibility between ethnic populations (Saito, 2010). Mode of transmission is through contaminated blood, sexual activity, breastfeeding, and very rarely in utero.
HAM/TSP is a chronic, insidiously progressive myelopathy that typically begins after age 30 years (but can occur as early as the first decade). In addition to slowly progressive spastic paraparesis, patients complain of lower-extremity paresthesias, a painful sensory neuropathy, and bladder dysfunction, and some patients may also develop optic neuropathy. Examination reveals UMN signs in the legs (weakness, spasticity, pathological reflexes, hyperreflexia), although reflexes may also be brisk in the arms. Overall, evidence of LMN involvement may be scant, and objective sensory findings may be difficult to detect. MRI may reveal increased signal on T2-weighted sequences in periventricular white matter and atrophy of the thoracic cord, but these findings may not be specific to HTLV-1. The definitive diagnosis of HAM/TSP requires HTLV-1–positive serology in blood and cerebrospinal fluid (CSF). To be sensitive and specific, CSF should reveal a combination of a polymerase chain reaction amplification of HTLV-1 deoxyribonucleic acid (DNA), together with evidence of an increased HTLV-1–specific antibody index and oligoclonal bands (Puccioni-Sohler et al., 2001). At present, no antiviral agents effectively treat HAM/TSP, but a case report showed partial benefit of plasmapheresis (Narakawa et al., 2001). As more is learned about the molecular etiology of HAM/TSP, future therapies will likely target the pathogenic effect of HTLV-1–reactive T cells.
Human T-Lymphotropic Virus Type 2–Associated Myelopathy
Though phylogenetically similar in many respects, HTLV-1 and HTLV-2 are still antigenically distinct. Nonetheless, using enzyme-linked immunosorbent assay (ELISA) and Western blot techniques, many laboratories worldwide often report the presence of sero-indeterminate HTLV-1/2. It has long been thought that myelopathy in such sero-indeterminate cases is due to HTLV-1 rather than HTLV-2, but rare cases are now being described of a syndrome characterized by spastic paraparesis, diffuse hyperreflexia, spastic bladder, and periventricular white matter changes on MRI in patients infected with HTLV-2 but not HTLV-1. This retrovirus is endemic in some Native American tribes and now often encountered worldwide among IV drug abusers. It is worthwhile to test CSF and serum for the presence of this virus in known IV drug abusers who present with a spastic paraparesis (Silva et al., 2002). However, co-infection with HIV-1 is a confounding factor in many cases of presumed HTLV-2–associated neurological disease. It has been suggested that such co-infection, rather than infection with HTLV-2 alone, increases the likelihood of neurological manifestations (Araujo and Hall, 2004; Posada-Vergara et al., 2006).
Adrenomyeloneuropathy
Adrenomyeloneuropathy is a variant of adrenoleukodystrophy, an X-linked recessive disorder caused by mutations in the ABCD1 gene on chromosome Xq28 that encodes a ubiquitously expressed integral membrane peroxisomal ATPase-binding cassette transporter protein. Mutations in this gene lead to abnormal peroxisomal β-oxidation, which results in the harmful accumulation of very long-chain fatty acids (VLCFAs) in affected cells. Excessive levels of VLCFAs may interfere with the membrane components of both neurons and axons. The most common phenotype, adrenoleukodystrophy, is an inflammatory disorder of brain and spinal cord that affects young boys 4 to 8 years of age, who develop severe adrenal insufficiency, progressive cognitive deterioration, seizures, blindness, deafness, and spastic quadriparesis. Adrenomyeloneuropathy is a noninflammatory axonopathy of the spinal cord that involves descending corticospinal tracts in the thoracic and lumbosacral regions and the ascending posterior columns in the cervical region. The characteristic clinical picture is a slowly progressive spastic paraparesis and mild polyneuropathy in adult men (in their late 20s), with or without sensory symptoms and sphincter disturbances. Adrenal insufficiency may be present and may predate onset of neurological symptoms by several years. Adult female carriers may present with slowly progressive spastic paraparesis. Approximately 20% of men with adrenomyeloneuropathy also develop cerebral changes on MRI that may accompany cognitive/language/behavioral deterioration. Rare cases may present as a spinocerebellar degeneration. Considerable phenotypic variation exists even within individual families. Female carriers may manifest more subtle symptoms such as cramps, back pain, or arthralgias. The diagnosis should be suspected in male cases with progressive sensorimotor deficits in the legs and a family history of a myelopathy (including supposed MS). Progressive sensorimotor deficits in the lower extremities with a history of memory loss or “attention deficit disorder” should also prompt testing for adrenomyeloneuropathy, as should a history of idiopathic childhood epilepsy or primary adrenal failure (Mukherjee et al., 2006). Sural nerve biopsies show loss of both myelinated and unmyelinated axons, with some degree of onion bulb formation. Ultrastructural examination may show characteristic inclusions (empty lipid clefts) in Schwann cell cytoplasm. Nerve conduction studies and needle electrode examination may reveal a predominantly axon-loss type of sensorimotor polyneuropathy with a lesser component of demyelination, and SSEPs may show reduced or absent responses. The diagnostic test of choice is to demonstrate increased VLCFA levels in plasma, red blood cells, or cultured skin fibroblasts. No specific therapy exists for adult-onset adrenomyeloneuropathy.
Plant Excitotoxins
Lathyrism
Lathyrism is a chronic toxic nutritional neurological disease caused by long-term (or subacute) ingestion of flour made from the drought-resistant chickling pea (Lathyrus sativus). It is an important example of a disease in which a natural excitotoxin causes selective UMN impairment. The responsible neurotoxin is β-N-oxalylamino-l-alanine (BOAA), an α-amino-3-hydroxyl-5-methyl-4-isoxazolepropionic acid (AMPA) glutamate receptor agonist. Ingestion of this neurotoxin results in increased intracellular levels of reactive oxygen species and subsequent impairment of the mitochondrial oxidative phosphorylation chain. Degeneration is most prominent in those Betz cells of the motor cortex (and the longest corresponding pyramidal tracts) that subserve lower-extremity function. Lathyrism occurs in the indigenous populations of Bangladesh, China, Ethiopia, India, Romania, and Spain. It also occurred in regional concentration camps during World War II. The condition may occur in epidemic form when malnourished populations increase consumption of flour made from L. sativus chickling peas during times of food shortage due to droughts. An analysis of an epidemic of neurolathyrism in Ethiopia showed a higher incidence in boys aged 10 to 14 years. The increased risk was associated with cooking grass pea foods in traditional clay pots (Getahun et al., 2002). The onset of clinical toxicity is either acute or chronic, manifesting as muscle spasms, cramps, and leg weakness. In addition to spastic paraparesis, sensory (including leg formications) and bladder dysfunction may occur. Occasionally there is a coarse tremor of the upper extremities. Although irreversible, the disorder is not progressive (unless there is continuing intoxication), and lifespan is not affected.
Disorders of Lower Motor Neurons
Clinical Features of Lower Motor Neuron Involvement
Loss of Muscle Strength (Weakness)
In a disease causing chronic motor unit depletion, neighboring axons belonging to healthy motor neurons may reinnervate denervated muscle fibers belonging to a diseased motor unit by collateral sprouting. In this way, existing motor units continually modify in the face of persistent losses of motor axons to maintain muscle strength. For example, in patients who have recovered from acute poliomyelitis, depletion of more than 50% of LMNs occurs before residual muscle weakness is clinically detectable. Healthy individuals have sufficient motor units available to offset an unexpected loss of motor neurons (Box 74.3).
Fasciculations
Fasciculations are spontaneous contractions of muscle fibers belonging to a single (or part of a) motor unit. (A video of this disorder can be found at www.expertconsult.com) Clinically, fasciculations appear on the muscle surface as fine, rapid, flickering, and sometimes vermicular contractions that occur irregularly in time and location. The impulse for the fasciculation appears to arise from hyperexcitable motor axons anywhere in their course. Fasciculations can occur both in healthy individuals and in patients with LMN involvement, so fasciculations themselves do not indicate LMN disease.
 (see video). Large fasciculations may occur in muscles undergoing extensive chronic reinnervation, such as chronic spinal muscular atrophy (SMA), Kennedy disease, and the postpoliomyelitis syndrome.
(see video). Large fasciculations may occur in muscles undergoing extensive chronic reinnervation, such as chronic spinal muscular atrophy (SMA), Kennedy disease, and the postpoliomyelitis syndrome.Laboratory Evidence of Lower Motor Neuron Involvement
Electrodiagnostic Examination
The electrodiagnostic examination (EDX) consists of nerve conduction studies and needle electrode examination (see Chapter 32B). The loss of motor units reflects in the loss of the amplitude of the maximal compound muscle action potential (CMAP). In a primarily demyelinating process, conduction velocity slows, and in severe cases, block. In a primarily axon loss process, there is usually only a modest degree of conduction velocity slowing commensurate with dropout of large myelinated axons. Sensory nerve conduction studies are normal in pure LMN disorders.
Acute Poliomyelitis
Clinical Features
After a brief 3- to 6-day incubation period, a viremia occurs, during which approximately 90% of individuals remain asymptomatic. Most of the remaining individuals develop an acute flulike illness with cough, malaise, diarrhea, myalgia, headache, and fever. This self-limited “abortive” polio usually lasts 2 to 3 days, and patients do not progress to develop acute muscle weakness. Between 2% and 3% of acutely infected patients develop aseptic meningitis characterized by severe headache due to meningeal irritation. This is typically self-limited and resolves within 7 to 14 days. Less than 1% of infected patients who ingest poliovirus develop the acute paralytic syndrome, characterized by localized fasciculations, severe myalgia, hyperesthesias, usually fulminant focal and asymmetrical paralysis, and fever. Any skeletal muscle can weaken, including bulbar muscles and muscles of respiration, but the leg muscles are the most commonly affected (Howard, 2005).
Laboratory Features
Motor nerve conduction studies performed 21 or more days after the onset (see Chapter 32B) may reveal low-amplitude maximum CMAPs. No evidence of significant demyelination-related motor conduction slowing or block exists. Sensory nerve action potentials (SNAPs) are normal. EMG examination in the acute phase shows profuse axon loss in the form of positive sharp waves and fibrillation potentials. In addition, fasciculations may be prominent. As motor axon loss progresses, evidence of neurogenic motor unit potential changes may be detected. The CSF typically shows increased protein content with normal glucose and a pleocytosis, with polymorphonuclear cells predominating during the acute stages and lymphocytes predominating later in the disease. Identification of CSF poliovirus-specific immunoglobulin M (IgM) antibody allows a specific diagnosis. Stool or nasopharyngeal cultures are positive for poliovirus in nearly 90% of patients by the 10th day of illness. The diagnosis may also be established by documenting a fourfold or greater increase in serum antibody titer against poliovirus from the acute as compared to the convalescent phase. Polymerase chain reaction (PCR) is now the best technique to diagnose poliovirus subtype as well as determine if the illness is related to wild-type versus Sabin oral polio vaccine–induced disease.
Vaccination
Immunocompromised individuals and their unimmunized direct household contacts are at particular risk for this rare complication. Consequently, in Northern America, Japan, Europe, New Zealand, and Australia, where wild viral infection is almost completely eradicated, policy shifted to use of the Salk inactivated vaccine in immunization schedules (Alexander et al., 2004).
Postpolio Syndrome/Progressive Postpoliomyelitis Muscular Atrophy
In the United States alone, it is estimated that 250,000 to 640,000 people survived acute paralytic poliomyelitis; the last epidemic was in 1952. Many years after recovery from acute poliomyelitis, some patients experience progressive functional impairment, with muscle fatigue, pain, sleep disturbances, cold intolerance, depression, dysphagia, and dysarthria, called the post-polio syndrome. If progressive muscle weakness and wasting occurs in this setting, the term progressive postpoliomyelitis muscular atrophy (PPMA) is used. The reported incidence of postpolio syndrome/PPMA among polio survivors ranges from 28.5% to 64%; no accurate estimate of the incidence exists. By definition, patients with postpolio syndrome/PPMA have recovered from acute poliomyelitis, and the disease course has been stable for at least 10 years after the recovery (Box 74.4).
Box 74.4 Characteristic Features of Postpolio Syndrome/Progressive Postpoliomyelitis Muscular Atrophy
EMG, Electromyography; PPMA, postpoliomyelitis muscular atrophy.
Treatment
No specific pharmacotherapy for postpolio syndrome exists. Care focuses on symptom relief (Gonzalez et al., 2010). A randomized controlled trial of intravenous immunoglobulin (IVIG; 2 courses of IVIG at a dose of 90 g per course over 3 days, with a 3-month interval) reported a significant improvement in muscle strength but not quality of life in 135 patients (Gonzalez et al., 2006). A need for further trials in larger patient groups exists.
West Nile Virus
West Nile virus (WNV) is an arthropod-borne flavivirus that cause epidemics of meningitis, encephalitis, and in some instances an acute polio-like flaccid paralysis. Approximately 80% of those who are infected are asymptomatic, and 20% develop a flulike illness (termed West Nile fever). Less than 1% present with neuroinvasive disease. Following the 1999 outbreak in New York, an epidemic spread across the North American continent and peaked in 2002/2003. Between 1999 and 2008, almost 29,000 confirmed and probable cases of WNV infection were received in the United States, and over 40% of cases were of the neuroinvasive type. The highest incidence of neuroinvasive WNV occurred in the western north-central United States and mountain states between the months of July and September (Lindsey et al., 2010). Epidemics also occurred in Israel, Italy, Russia, Romania, Hungary, Tunisia, the Sudan, the Caribbean, and Latin America. WNV is now endemic in North America and is the leading cause of arboviral encephalitis in the United States. WNV is a zoonotic pathogen that has crossed over from birds to humans, the latter serving as incidental hosts. The prime bridge vector is the Culex mosquito, and the spread of disease appears to have followed the migration patterns of bird populations (Kilpatrick et al., 2006). Human-to-human transmission can occur via blood transfusion, organ transplant, intrauterine exposure, and breastfeeding, hence the need to screen for the virus amongst blood donors. One of the most dramatic presentations is an acute asymmetrical flaccid paralysis in a febrile patient with or without meningitis, encephalitis, and cranial neuropathies (including hearing loss). Aching pains in affected limbs often accompany the paralysis, but actual sensory loss is not a feature. Respiratory failure and death may occur, and up to one-third of cases suffer bladder and bowel dysfunction. Recovery is very slow, and from the available evidence, incomplete. Other presentations include a GBS, multifocal chorioretinitis, pancreatitis, hepatitis, myocarditis, nephritis, and splenomegaly. Risk factors for death from WNV infection include chronic renal disease, an immunosuppressed state (e.g., in organ transplant recipients), the presence of encephalitis (versus meningitis) and old age (Murray, K., et al., 2006; Tyler et al., 2006). Detection of WNV-specific IgM antibody in the serum of a patient with abnormal CSF and acute neurological illness confirms the diagnosis. The diagnosis can also be confirmed by WNV-specific IgM antibody capture ELISA in CSF. PCR has also been developed to detect the virus (Tang et al., 2006). CSF protein is typically elevated (100 mg/dL or more and often higher in encephalitis versus meningitis), glucose is typically normal, and there are increased numbers of white cells (mean of about 220/mm3 with ≈ 45% neutrophils). The number of red cells is variable and may be higher in encephalitis (Tyler et al., 2006). Neuroimaging of the brain is usually normal, but that of the spinal cord may reveal increased signal in the anterior horns. Electrodiagnostic studies reveal an acute disorder of anterior horn cells, with acute loss of motor units in affected myotomes.
Multifocal Motor Neuropathy
A complete description of multifocal motor neuropathy is in Chapter 76. The condition is believed to be autoimmune in nature, and most cases have evidence of focal demyelination in the peripheral nerves (multifocal motor neuropathy with conduction block [MMNCB]) similar to that in chronic inflammatory demyelinating peripheral neuropathy. The clinical presentation, however, is with pure LMN involvement. The condition enters into the differential diagnosis of benign focal amyotrophy and the progressive muscular atrophy variant of ALS. It is important to search for this condition, since it is treatable by high-dose immunoglobulin infusions or other immunotherapy.
Benign Focal Amyotrophy
The terms benign focal amyotrophy, brachial monomelic amyotrophy, benign calf amyotrophy, Hirayama disease, or juvenile segmental muscular atrophy are used to describe disorders characterized by LMN disease clinically restricted to one limb. The etiology is unknown. Autopsy studies have shown the affected region of spinal cord flattened, the anterior horn markedly atrophied and gliotic, and a reduction in the numbers of both large and small motor neurons. Based upon neuroradiological studies, Hirayama, who established the disease entity, has proposed a mechanically induced limited form of ischemic cervical myelopathy, being the result of local compression of the dura and spinal cord against vertebrae during repeated neck flexion/extension, in turn due to disproportionate growth between the contents of the dural sac and the vertebral column (Hirayama, 2008; Hirayama and Tokamaru, 2000). However, surgical decompression has not altered the course of the disease, and this theory is no longer widely held. Another school of thought is that this is a segmental, perhaps genetically determined, SMA, but the actual cause is still unknown.
The disease usually begins in the late teens, but many cases can present in the fourth decade. More than 60% of patients are men. Although originally described in Indian and Japanese patients, the disorder is now recognizable around the world. The most common presentation is one of an idiopathic, slowly progressive, painless weakness and atrophy in one hand or forearm. The distribution of muscle weakness varies markedly from case to case, but a characteristic feature is that the condition remains limited to only a few myotomes in the affected limb. The most common pattern is unilateral atrophy of C7-T1 innervated muscles, with sparing of the brachioradialis (the “oblique atrophy” pattern). Muscle stretch reflexes are invariably hypoactive or absent in the muscles innervated by the involved cord segment but are normal elsewhere. UMN signs are not present, and if they are, one should consider the onset of ALS instead. Approximately 20% have hyperesthesia to pinprick and touch, usually located on the dorsum of the hand. The cranial nerves, pyramidal tracts, and the autonomic nervous system are normal. Weakness and atrophy may progress steadily for the initial 2 to 3 years, but most patients have stabilized within 5 years. The arm is the affected limb in approximately 75% of the patients and the leg in the remaining 25% (benign calf amyotrophy). Spread may occur to the contralateral limb in about 20% of cases (Gourie-Devi and Nalini, 2003), and rare patients later develop an ALS-like picture.
Spinal Muscular Atrophy
The SMA are a group of disorders caused by degeneration of anterior horn cells and, in some subtypes, of bulbar motor neurons. Almost all cases are genetically determined, with most being autosomal recessive due to homozygous deletions of the survival motor neuron (SMN) gene on chromosome 5. Traditionally, SMA is classified as one of the four types based on the age at onset: SMA type 1 (infantile SMA or Werdnig-Hoffmann syndrome), SMA type 2 (intermediate SMA), SMA type 3 (juvenile SMA or Kugelberg-Welander disease), and SMA type 4 (adult-onset SMA, pseudomyopathic SMA). A very severe prenatal form of SMA (type 0 SMA) can manifest prenatally with reduced fetal movements and respiratory distress at birth. It is also important to consider the maximum function that a child achieves in terms of sitting and walking; this is of prognostic significance. In the less severe forms of the disease, there can be periods where the child will improve or plateau, but long-term studies have demonstrated a net deterioration (Russman, 2007) (Table 74.2). The estimated incidence of infantile and juvenile recessive SMA is 1 in 6000 live births, with an approximate carrier frequency of 1 in 35 of the general population, making it a leading genetic cause of infant mortality. True adult-onset disease accounts for probably less than 10% of all cases of SMA, with an estimated prevalence of 0.32 in 100,000. The mean age at onset is the mid-30s but ranges from 20 to the late 40s. Up to 95% of all childhood cases are due to deletion of the survival motor neuron (SMN1, telomeric SMN, SMNT) gene located on chromosome 5q11.2-13.3. The remaining cases are due to small SMN mutations (rather than full deletions). SMN1 is located within an inverted gene duplication, the other half of which is occupied by the almost identical SMN2 (centromeric SMN, SMNC) gene. The SMN1 protein product is functionally absent in the vast majority (95%-98%) of cases of SMN-mutated SMA, and small amounts are present in the remaining few percent. The SMN2 protein is present in all patients, but the copy number can vary considerably. Only 1% to 2% of childhood-onset SMA is unrelated to the SMN locus on chromosome 5.
SMN1 protein is a 38-kD polypeptide important in the processing of the primary transcripts of other genes. Although a ubiquitous protein, expression is great within spinal motor neurons, and this may be why the disorder manifests as a motor neuron disease. It is associated with both nuclear and cytoplasmic complexes involved in messenger RNA splicing and interacts with other proteins that are important in the regulation of ribosomal RNA processing and modification. Within the nucleus, SMN1 forms macromolecular complexes with other nuclear proteins important in the assembly of spliceosomal small nuclear ribonucleoproteins (snRNPs). It is thus possible that SMA develops because of disruption in mRNA transport and/or SMN-dependent snRNP biogenesis. SMN2 protein is almost identical to SMN1 protein but only has about 10% of the activity of SMN1 protein because of a C-to-T transition within exon 7 that alters splicing. The full-length SMN1 transcript has all 9 exons, whereas 90% of the transcripts from SMN2 lacks exon 7. Thus, only 10% of the SMN2 output is the full-length SMN transcript, the remainder being unstable and rapidly degraded. Motor neuron health requires at least 23% full-length SMN protein. The SMN genome is rather unstable and as a consequence, increased copy numbers of SMN2 are possible through a process of gene conversion from SMN1 to SMN2. This has major implications for the clinical phenotype: the infantile form is very severe because most of these children have no SMN1 and only two copies of SMN2, thus producing about 9% of full-length functional transcript, whereas multiple copies of SMN2 (3-5) are associated with mild SMA (Hirth et al., 2005; Kostova et al., 2007; Monani, U.R., 2005).
Most (about 70%) adult-onset type 4 SMA is autosomal recessive, is allelic with SMA types 1, 2, and 3, and is due to mutations or deletions in the SMN1 gene. Gene conversion events occur in some cases with SMA-4 whereby SMN1 is “converted” to SMN2. The remaining adult-onset SMA cases are autosomal dominant, autosomal recessive (but not linked to chromosome 5), or are apparently sporadic. One rare form of adult-onset SMA described in a large Brazilian family was caused by a missense mutation in the vesicle trafficking protein, VAPB. This can present with typical ALS or with a late-onset SMA (Nishimura et al., 2004).
Clinical Features
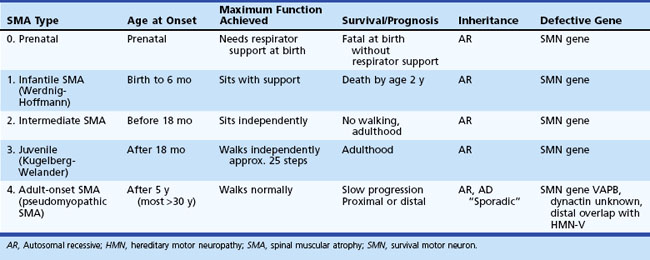
Spinal Muscular Atrophy Type 1, Infantile Form (Werdnig-Hoffmann Disease)
SMA type 1 begins within the first few months of life. By definition, children with this disease are never able to sit without support. Symptoms include severe hypotonia, a weak cry, and respiratory distress. These children are unable to lift their heads when placed prone and demonstrate severe head lag when pulled from a supine to a seated position (Fig. 74.1). The baby’s posture at rest also takes a characteristic “frog-leg” position, with the thighs externally rotated and abducted and the knees flexed (a “floppy” baby). Limb weakness is severe, generalized, and worse proximally. The infant is unable to sit and raise its arms or legs from the examining table, but there may be antigravity movements of the hands and flickering movements of the feet. Muscle stretch reflexes are usually absent, and the sensory examination is normal. Observation of the fingers may reveal fine, small-amplitude involuntary movements called minipolymyoclonus that are due to dense fasciculations. Contractures usually do not develop in the early phases but may develop after several months of immobilization. Bulbar muscle weakness makes feeding laborious, causes a continuous gurgling, and eventually leads to aspiration pneumonia. Fasciculations of the tongue occur in about 50% of affected infants. In contrast to bulbar and extremity muscles, the facial muscles are only mildly weak, giving these children an alert expression. Extraocular movements are always normal. Intercostal muscles are severely weak, but diaphragmatic strength preserves until late in the disease. This dysequilibrium of ventilatory muscle function causes outward flaring of the lower ribcage and gives rise to a bell-shaped chest deformity. Death from respiratory failure, pneumonia, and malnutrition usually occurs before age 2 years. A rare form of atypical infantile SMA, spinal muscular atrophy with respiratory distress (SMARD), is associated with respiratory distress, cardiomyopathy, and lactic acidosis. This disorder is not due to SMN1 deletion but caused by mutations in the gene for immunoglobulin mu-binding protein 2 (IGHMBP2). It is interesting that this gene has homology to SETX, the gene responsible for ALS4, which can cause a familial form of distal amyotrophy, oculomotor apraxia–cerebellar ataxia, or juvenile ALS.
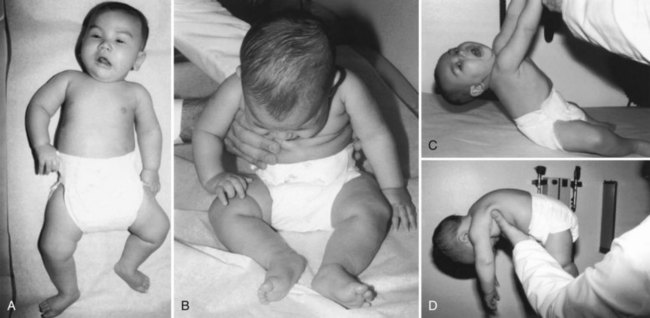
Spinal Muscular Atrophy Type 4, Adult-Onset
Most cases of autosomal recessive adult-onset SMA appear to affect proximal muscles. The characteristic clinical presentation is that of a slowly progressive limb-girdle weakness leading to difficulty in walking, climbing stairs, and rising from a chair or the floor. Fasciculations are an important finding and occur in 75% of patients. Quadriceps muscle weakness is often a prominent feature. Muscle cramps occur but are not prominent. Bulbar signs, bony deformities such as scoliosis, and respiratory weakness are rare. Many cases have a distribution of weakness reminiscent of the limb-girdle muscular dystrophies, leading to the older term, pseudomyopathic SMA (Fig. 74.2). Many cases of autosomal dominant adult-onset SMA (also known as Finkel-type SMA) are clinically similar to the recessive form described earlier. Finkel-type SMA usually begins in the third decade of life, is proximal in distribution, is very slowly progressive, and involves the legs before the arms. Most patients remain ambulatory for decades after clinical onset. One of the autosomal dominant missense mutations causing adult-onset Finkel-type SMA affects a vesicle trafficking protein called VAPB. It is interesting that some patients with this mutation develop the clinical features of ALS rather than SMA (Nishimura et al., 2004).
A new class of adult-onset SMA has recently emerged and is sometimes referred to as SMA-5 to help distinguish a distal rather than proximal pattern of slowly progressive muscular atrophy. The classification of these rare disorders is rather vague, and considerable overlap with distal CMT (see later discussion) exists. Several patterns of inheritance occur, including autosomal dominant, autosomal recessive, and X-linked recessive. Some lack any apparent pattern of inheritance. Distal-predominant adult-onset SMA and some of the neuronal forms of CMT disease appear to overlap both clinically and genetically: indeed, the difference may be purely semantic. Motor-predominant CMT variants such as hereditary motor neuronopathy type 5 (HMN-5), itself a heterogeneous group of conditions, present with a slowly progressive LMN-predominant disorder affecting distal limb muscles. Mutations in the glycyl-tRNA synthetase (GARS) gene, for example, were identifiable in multiple families around the world. Patients usually present with very indolent symmetrical or asymmetrical weakness, clumsiness, and wasting of intrinsic hand muscles (with a particular predilection for thenar muscles) in the absence of any proximal weakness or sensory findings. There is little functional disability (Del Bo et al., 2006; Dubourg et al., 2006). Mutations in the p150Glued subunit of the dynactin gene, a microtubule protein important in axonal transport, cause another distal-predominant atrophic disorder that also has a predilection for thenar muscles. Unlike the GARS-associated disorder, involvement of the face and vocal cords may occur (Puls et al., 2005).
Treatment
No disease-specific pharmacotherapy is yet available for any form of SMA. However, small molecules that have the capacity to increase SMN expression are undergoing investigation, as is therapeutic RNA-based modulation of SMN2 and stem cell therapy (Lorson et al., 2010). The mainstay of current treatment focuses on supportive care including physiotherapy, respiratory care, nutritional support, orthotics, and orthopedic interventions. Typical Werdnig-Hoffmann disease is almost uniformly fatal by age 2 years. However, because some affected infants survive beyond infancy and live into childhood, aggressive management including physiotherapy and respiratory therapy is essential in all cases.
Kennedy Disease (X-Linked Recessive Bulbospinal Neuronopathy)
In 1968, Kennedy and colleagues reported a new X-linked recessive SMA with bulbar involvement and gynecomastia. The primary pathology was thought to be in the LMNs, but sensory system involvement was later recognized, which led to the term bulbospinal neuronopathy. Molecular genetics research has shown Kennedy disease to be a trinucleotide repeat expansion disease. Though rare, it is more common than adult-onset SMA (Box 74.5).
Clinical Features
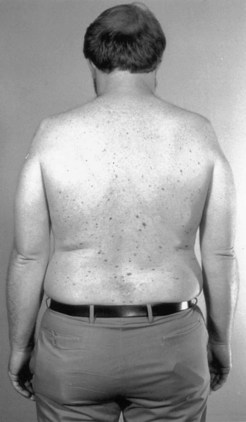
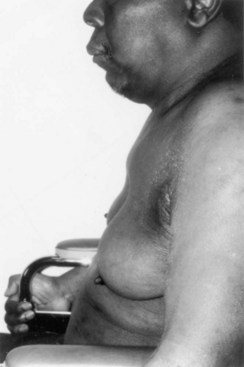
As is often seen in an X-linked syndrome, this is a disorder of men, who remain largely asymptomatic until after age 30 years. Hand tremor and subtle speech disturbance are early features that are followed by LMN muscle weakness, initially involving either the proximal hip extensor or shoulder girdle muscles, and associated with decreased or absent reflexes, muscle atrophy, and occasionally calf pseudohypertrophy. Kennedy disease usually causes no respiratory muscle weakness. Coarse muscle fasciculations, often with cramps, can be prominent in the extremities and trunk. Facial and perioral fasciculations are present in more than 90% of patients. The tongue shows chronic atrophy, often as a longitudinal midline furrow. However, despite weakness of facial and tongue muscles, significant bulbar symptoms are usually a relatively late feature. Neurological examination of the sensory system may reveal only modest impairment. Progression is slow, with most cases remaining independent of assist devices until late into the fifth decade of life (Atsuna et al., 2006). If bulbar dysfunction is severe, the prognosis becomes less favorable. Partial androgen insensitivity is an important element of this condition, and gynecomastia is one of the unique features of Kennedy disease that can be found in 60% to 90% of patients (Fig. 74.3). Other endocrine abnormalities include testicular atrophy, infertility (40%), and diabetes mellitus (10%-20%). It is now recognized that female carriers may manifest subtle neurological deficits such as late-onset bulbar dysfunction.
Laboratory Studies
Motor nerve conduction study results are generally normal, although one-third of the patients have reduced amplitudes of CMAPs. EMG examination of these patients is always abnormal and shows modest acute but prominent chronic denervation changes in motor units. EDX reveals a sensory neuronopathy in 95% of patients (Ferrante and Wilbourn, 1997). Another unique change is the presence of prominent fasciculation potentials in the face (especially in the perioral region) and limbs.
Progressive Muscular Atrophy
PMA, first described by Aran in 1850, is a clinical LMN disorder during its entire clinical course and comprises approximately 5% to 8% of all adult-onset motor neuron diseases. It is an overwhelmingly sporadic disease, but rare genetic diseases, such as those due to mutations in dynactin, VAPB, and A4V SOD1, may present with a pure LMN disorder, so a careful family history is important. Although PMA occurs in both sexes, men are more often affected than women. In a recent study, the average age of onset of PMA was about 3 years older than that of ALS, but other studies report a younger age at onset (Kim et al., 2009; Murray, B., 2006). Several studies have demonstrated that PMA progresses more slowly than ALS, so the average survival is significantly longer. The mean duration of disease was 159 months in one series of cases, and in another study, the 5-year survival was 63.7% in PMA versus 36.8% in ALS. Most of the very longest-duration cases of “ALS” have the PMA variant. Interestingly, the recent paper by Kim et al. showed that the development of UMN signs was unrelated to survival time after diagnosis (Kim et al., 2009; Murray, B., 2006).
It has been questioned whether PMA is an independent disease or represents one end of the spectrum of ALS. However, if followed over time, many patients with PMA go on to develop clinical features of upper motor neuron disease, which allows reclassification to ALS (and thus eligibility for entry into clinical trials). In a recent retrospective study of 916 cases diagnosed with ALS at a major neurological center, in 91, the original diagnosis was PMA; 20 of these developed UMN signs within 61 months of the original diagnosis. Autopsy studies have also demonstrated UMN involvement in some cases classified as PMA in life. Studies using magnetic resonance spectroscopy and/or transcranial magnetic stimulation reveal evidence for upper motor neuron involvement in PMA patients (Kim et al., 2009; Maragakis et al., 2010; Rowland, 2006).
Etiology
All hypotheses about the cause of ALS are also applicable to PMA (see Etiology, under Amyotrophic Lateral Sclerosis, later in this chapter).
Differential Diagnosis
PMA is usually a fatal disease and has no cure. Therefore, the diagnosis of PMA requires the exclusion of all other potentially treatable or definable diseases. In a previous review, 17 of 89 patients originally diagnosed with PMA were later diagnosed with MMNCB, CIDP, inflammatory myopathy, and myasthenia gravis (Visser et al., 2002). MMNCB is the most important of the alternative conditions that may present with focal and asymmetrical weakness in the absence of UMN signs (for more detailed description, see Chapter 76). The classic form is associated with EDX evidence of multifocal demyelination conduction blocks and elevated titers of antibodies against GM1 gangliosides. Clinically, patients develop slowly progressive multifocal muscle weakness but less prominent muscle atrophy. The treatment of choice is human IVIG (Van den Berg-Vos et al., 2000). The clinical and electrodiagnostic findings of sensory involvement, high CSF protein levels, and response to immunotherapy readily separate CIDP and PMA. Important clues that should lead one to suspect inclusion body myositis (IBM) are elevated serum CK to levels more than expected in typical PMA and a selective weakness in wrist flexors, finger flexors, and quadriceps muscles, without fasciculations. EMG in IBM should show evidence of a primary myopathy with increased insertional activity but without fasciculations. In IBM, additional neurogenic changes are common, and quantitative EMG may be required to clearly identify the myopathic nature of this disorder. Muscle biopsy characteristically reveals rimmed vacuoles and nuclear inclusions. Adult-onset SMA is a far more indolent disorder than PMA, and the very chronic process of denervation and reinnervation in SMA leads to fiber-type grouping on muscle biopsy, which is not a prominent feature of the less-protracted PMA. It is important to carry out regular follow-up examinations on patients with PMA to search for signs of UMN involvement that indicate the diagnosis of ALS. The pure motor neuropathy forms of CMT (especially hereditary motor neuropathy type V), present with a slowly progressive distal pattern of weakness and wasting, with no sensory changes. A familial pattern is usual, and genetic testing may reveal mutations in different genes such as GARS or seipin. A paraneoplastic motor neuronopathy has been described with clinical features that are similar to PMA, albeit with more rapid progression and with later development of nonmotor features. Many such cases have anti-Hu antineuronal antibodies in the setting of solid cancers (especially small-cell lung cancer). A similar subacute presentation may also occur in patients with lymphoma or other lymphoproliferative disorders, although signs of corticospinal tract dysfunction may become apparent in over 50% of cases. The onset of lymphoma may or may not coincide with onset of motor features.
Subacute Motor Neuronopathy in Lymphoproliferative Disorders
A subacute, progressive, and painless motor neuron syndrome may rarely develop in patients who have Hodgkin and non-Hodgkin lymphoma with or without a paraproteinemia (Rowland, 2006; Rudnicki and Dalmau, 2000). The lymphoma may or may not temporally coincide with the motor neuron disorder, and one or other disorder may present first. Although UMN signs may develop later in more than half of all cases, a LMN-onset syndrome is typical, with patchy, asymmetrical, lower extremity–predominant muscle weakness and wasting. Neuropathology shows a loss of anterior horn cells and ventral root nerve fibers; some have evidence of inflammation in the anterior horns of the spinal cord, and half have corticospinal tract degeneration. In some patients, the disease may be relatively benign. The rate of progression of muscle weakness and atrophy tends to slow down with time, and in rare instances, the motor syndrome may respond to treatment of the underlying lymphoproliferative disorder. However, the prognosis appears to be less favorable in those who develop a combined UMN and LMN disorder. Twenty percent of all cases so far reported with motor neuron presentations in the setting of lymphoproliferative disease had myeloma or macroglobulinemia. The pathogenesis of this ALS-like disorder is undetermined, but an immune mechanism may be at play; small patient series and case reports reveal that some patients who develop this LMN syndrome may have various autoantibodies (such as antisulfatide antibody), paraproteinemia, increased CSF protein, and/or oligoclonal bands.
Disorders of Both Upper and Lower Motor Neurons
Amyotrophic Lateral Sclerosis
ALS is a neurodegenerative disorder of undetermined etiology that primarily affects the motor neuron cell populations in the motor cortex, brainstem, and spinal cord. It is progressive, and most patients eventually succumb to respiratory failure. The first detailed description was by Jean Martin Charcot in 1869, in which he discussed the clinical and pathological characteristics of “la sclérose latérale amyotrophique,” a disorder of muscle wasting (amyotrophy) and gliotic hardening (sclerosis) of the anterior and lateral corticospinal tracts (Gordon, 2006) involving both upper and lower motor neurons. ALS is known by several other names including Charcot disease, motor neuron disease, and in the United States, “Lou Gehrig disease” in remembrance of the famous “Iron Horse” of baseball who was diagnosed with ALS in 1939.
The World Federation of Neurology Research Group on Neuromuscular Disorders has classified ALS as a disorder of motor neurons of undetermined cause, and several variants are recognized. Included in this group are PLS and PBP. As previously mentioned, PMA is also thought to be a variant of ALS, despite its exclusion from current clinical research trial criteria. It is important to recognize that ALS is a progressive dynamic disorder. Some cases present with the classic combination of UMN and LMN signs, but others may have UMN onset, LMN onset, bulbar onset, or dyspnea at onset and only later develop signs of involvement of the other parts of the motor system (Box 74.6).
Box 74.6 Practical Classification of Amyotrophic Lateral Sclerosis
The incidence and prevalence rates for sporadic ALS are surprisingly uniform throughout the world. The estimated incidence in North America and Europe is about 2 per 100,000, and the prevalence is about 6 per 100,000. In sporadic spinal ALS, the male-to-female ratio is 1.2 to 1.4 : 1, but a slight female predominance exists in the bulbar-onset variety. ALS may occur as early as in the second decade of life, but the peak incidence is in the 65- to 74-year-old age bracket (McGuire and Nelson, 2006). The mean disease duration from symptom onset to death is approximately 3 years, but roughly 1 in 5 patients survive to 5 years, and 1 in 10 patients survive to 10 years (Murray B., 2006). No specific environmental, occupational, or physical factors link with absolute certainty to an increased risk of ALS. Areas of interest include chronic exposure to electromagnetic fields, high levels of physical activity, high dietary intake of glutamate, environmental toxins, and a history of military service in the Persian Gulf War. Smoking appears to be an independent risk factor for sporadic ALS, with a higher risk for those who have smoked for many years (Armon, 2009; Gallo et al., 2009). Several environmental trace elements have been evaluated as potential causative agents for ALS, including selenium, aluminum, iron, manganese, copper, zinc, cadmium, and lead, but there is no convincing evidence that any one of these plays a major part in ALS pathogenesis.
Etiology
The cause for sporadic ALS is unknown. A significant body of basic and clinical research lends strong support to a theory of ALS pathogenesis which proposes selective motor neuron damage from a complex chain of injurious events involving excitotoxins, oxidative stress, neurofilament dysfunction, altered calcium homeostasis, mitochondrial dysfunction, enhanced motor neuron apoptosis, and proinflammatory cytokines (Cleveland and Rothstein, 2001). Genetic factors may play a role in “sporadic” disease: several proposed ALS susceptibility genes include APOE, SMN, peripherin, apex nuclease gene, and vascular endothelial growth factor (VEGF) gene. A polymorphism in the kinesin-associated protein-3 gene (KIFAP) appears to increase survival (Landers et al., 2009).
Glutamate Excitotoxicity and Free Radical Injury
Glutamate released from presynaptic axon terminals into the synaptic cleft binds to its receptors, causing signal transduction to occur. After signal transduction, interstitial glutamate reabsorbs into its main reservoir, the surrounding astrocytic glial cell processes. This absorption process involves specific transporter proteins known as GLT (glutamate transporter) or EAAT (excitatory amino acid transporter) proteins. Among these, the astrocytic glutamate transporter, termed GLT1 or EAAT2, is markedly reduced in the motor cortex and anterior horn cells of patients with ALS, a fact that has been supported by the finding of significantly raised levels of glutamate in CSF in ALS (Spreux-Varoquaux et al., 2002). Rothstein et al. found intriguing abnormalities in the DNA encoding GLT1 in more than 60% of patients with ALS (predominantly the sporadic form) (Rothstein et al., 1995). However, subsequent research suggests that GLT1 does not appear to be a candidate gene for FALS or SALS.
Impaired glutamate transport reduces clearance of glutamate from the synaptic cleft, which may leave excessive amounts of free excitatory neurotransmitter to repeatedly stimulate the glutamate receptor and thus allow calcium ions to enter the neuron. Excess calcium ions are usually buffered by intracellular calcium-buffering proteins, such as parvalbumin or calbindin, and by mitochondria that may also function as an extra calcium reservoir. Low levels of parvalbumin, calbindin, and altered mitochondrial function are detectable in ALS models. When calcium ion levels exceed this reduced buffering capacity, they may catalyze activity in specific destructive enzymes that do not activate under normal conditions including xanthine oxidase, phospholipase, and nitric oxide synthase. These enzymes produce free radicals, including reactive oxygen and nitrogen species, which cause harmful nitration of tyrosine residues on key neuronal proteins and ultimately may cause apoptosis. Regional differences in the levels of activity of buffering systems and in glutamate receptor subtype expression may explain the selective vulnerability of certain motor neuron pools within the CNS. Excitotoxic NMDA receptor co-agonists such as d-serine may play a significant role in motor neuron death in ALS; glutamate toxicity appears enhanced by high levels of d-serine produced from activated glial cells (Sasabe et al., 2007).
Immunological and Inflammatory Abnormalities
Several pieces of evidence implicate an immune process in the pathogenesis, if not the initiation, of ALS. Immune complexes have been identified in gut and renal tissue from patients with ALS. A small number of patients with ALS may have a monoclonal gammopathy (usually IgM), and some may have low-level titers of anti-GM1 antibody (but notably rare in patients with UMN features). However, IgM gammopathy and anti-GM1 have similar frequency in non-ALS/MND populations, and GM1 antibody may not be relevant to classic ALS but rather to the pure LMN disorder, MMNCB. Activated spinal cord microglial cells, reactive astrocytes, elevated inflammatory cytokine levels, and increased expression of cyclooxygenase (COX)-2 have also been found in ALS tissue samples (Boillée et al., 2006; Yiangou et al., 2006). It is well established that viral infection on the CNS, such as with poliovirus, WNV, HTLV 1/2, and HIV can cause motor neuron injury through a process of secondary immune activation. However, all prior immunotherapies and antiinflammatory therapies, including cyclophosphamide, IVIG, plasmapheresis, corticosteroids, COX-2 inhibitors, phosphodiesterase/TNF-α inhibitors, and total lymphoid irradiation, have failed to alter the course of ALS. Although this might indicate that immune mechanisms are not of primary importance in the pathogenesis of ALS, evidence exists from animal studies of ALS to suggest that cell-targeted immune therapy and antiinflammatory therapy might be useful.
Mitochondrial Dysfunction
Disturbances in mitochondrial function and structure occur in both human ALS and in transgenic animal models of the SOD1-associated disease, which suggests a role for aberrant redox chemistry in the earlier stages of disease. In effect, mitochondrial damage may impair the cellular energy production system. Mutant SOD1 aggregates appear to clump together on mitochondrial membranes and may also interfere with chaperone-assisted mitochondrial protein folding. Furthermore, axonal transport of mitochondria along axons may be disrupted (Shi et al., 2010).
Neurofilament and Microtubule Dysfunction
Mutations in the genes for neurofilament subunits appear to confer increased risk for the later development of SALS (Cleveland and Rothstein, 2001). The neurofilament heavy chain is thought to be important in the correct spacing of neurofilaments from each other and thus in the regulation of axonal diameter. In rare cases of SALS (and very rarely FALS), mutations have been found in the heavy-chain gene segment that encodes an amino acid repeat motif. Overexpression of another intermediate motor neuron-specific protein called peripherin may lead to accumulation of toxic intraneuronal aggregates, as has been demonstrated in patients with SALS and in mice with SOD1 mutations. In the setting of peripherin overexpression, selective motor neuron toxicity appears to occur in mice that lack light subunits, which implies that the light subunit may somehow prevent a harmful interaction between peripherin and other neurofilament subunits (Beaulieu et al., 2002; Leung et al., 2004).
Aberrant Rna Processing
RNA metabolism refers to the various steps in the life of RNA molecules from pre–messenger RNA splicing through RNA editing and processing, and then on to transport, reassembly into polyribosomes, and degradation. An evolving theory in ALS pathogenesis is that mutations in such proteins as TDP43, FUS/TLS, senataxin, peripherin, SMN1, SOD1, and angiogenin may result in aberrant interactions in RNA metabolism. SMN1 protein, for example, participates in the assembly of small nuclear ribonuclear proteins that are important in the manufacture of the spliceosome. Angiogenin promotes ribosomal RNA transcription and can also act as a ribonuclease to inhibit translation when the cell is under stress. TDP43 has RNA recognition motifs as well as nuclear export and localization sequences and appears to play a part in RNA trafficking and translation. TDP43 modulates neurofilament light-chain messenger RNA stability and may play a role in the formation of neurofilament light-chain aggregates in ALS (Strong, 2010).
Clinical Features
The typical clinical picture in ALS is that of a patient with a progressive motor deterioration manifesting with both UMN and LMN symptoms and signs. Thus, one should consider this diagnosis when a patient presents with a combination of marked weakness and wasting but with brisk reflexes, spasticity, and pathological reflexes. Of course, not all patients present with this classic pattern: muscle weakness in ALS usually begins in a focal area, first spreading to contiguous muscles in the same region before involvement of another region. The first presentation may appear very similar to a focal mononeuropathy, sometimes called the pseudoneuritic or flail leg presentation (Wijesekera et al., 2009). More commonly, however, single-limb weakness appears to occur in muscles derived from more than one peripheral nerve and/or nerve root distribution; this is a monomelic presentation. Onset of muscle weakness is more common in the upper than the lower extremities (classic, spinal ALS), but in approximately 25% of patients, weakness begins in bulbar-innervated muscles (bulbar-onset ALS). On rare occasions (1% or 2% of patients, more often male), the weakness starts in the respiratory muscles (dyspnea or respiratory-onset). Some patients present with weakness that is restricted to one side of the body (Mills hemiplegic variant), and up to 10% of patients appear with bilateral upper-extremity wasting, which is known as the flail arm or flail person in the barrel variant. The latter is more commonly seen in males and typically presents in proximal muscles of the upper limb before spreading distally into the hands, and then much later (one study used a 12-month interval) into other regions. Reflexes may be retained or even brisk in the markedly atrophic limbs.
Symptoms of muscle weakness vary, depending on which motor function is impaired. For example, when weakness begins in the hand and fingers, patients report difficulty in turning a key, buttoning, opening a bottle cap, or turning a doorknob (Fig. 74.4). When weakness begins in the lower leg, foot drop may be the first symptom, or the patient may complain of instability of gait, falling, or fatigue when walking (Fig. 74.5). When bulbar muscles are affected, the first symptoms may be slurred speech, hoarseness, or an inability to sing or shout, soon followed by progressive dysphagia (Fig. 74.6). Patients with bulbar-onset ALS often initially consult ENT specialists and not only experience progressive impairment in bulbar function but also excessive drooling (sialorrhea) and weight loss. Pseudobulbar palsy may present with inappropriate or forced crying or laughter (see Signs and Symptoms of Upper Motor Neuron Involvement, earlier in this chapter), which is often a source of great emotional distress for patients. Excessive forced yawning may also be a manifestation of pseudobulbar palsy. In the rare patient who presents with progressive respiratory muscle weakness, the first consultation may be with a pulmonologist or even admission to the intensive care unit; the diagnosis of ALS may be established when the patient fails weaning from the ventilator. Head drop (or droop) may be a feature in ALS, caused by weakness of cervical and thoracic paraspinal muscles (Fig. 74.7). Fasciculations are not commonly the presenting feature of ALS, but they develop in almost all patients soon after onset, and their absence should prompt one to reconsider the diagnosis. In some patients, waves of fasciculations spread across the chest or back. Muscle cramps are one of most common symptoms in patients with ALS and often precede other symptoms by many months. In ALS they can occur in unusual muscles such as in the thigh, abdomen, back, or tongue. Spasticity develops in wasted muscles, and patients may suffer painful flexor spasms in limbs.
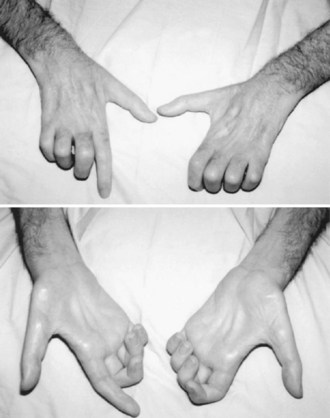
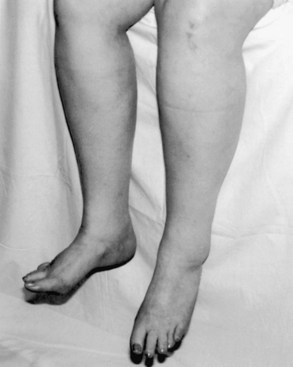
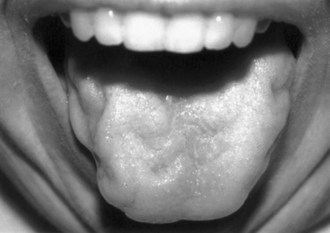

As dysphagia worsens, reduced caloric intake worsens fatigue and accelerates muscle weakness. Aspiration of liquids, secretions, and food becomes a risk. Patients may complain that they produce copious amounts of abnormally thick oral secretions, which may drool excessively from the mouth. This sialorrhea is made worse as perioral muscles weaken and/or head drop develops. Weight loss is often rapidly progressive; this does not simply reflect poor caloric intake but represents a form of ALS cachexia. Marked loss of muscle bulk exposes joints and associated connective tissues to abnormal mechanical stresses that can lead to joint contractures, joint deformities, painful shoulder pericapsulitis, and bursitis. Sleep disturbances in the form of increased awakenings from hypopnea and hypoxia are common in ALS and contribute to daytime sleepiness, morning headaches, and fatigue. As respiratory difficulty worsens, patients may be unable to lie supine because of worsening diaphragmatic weakness and thus compensate by using multiple pillows. In more advanced stages, patients are unable to lie in bed at all. Other manifestations of ventilatory failure include dyspnea on exertion and eventually dyspnea at rest. As the disease advances, motor function is progressively impaired, and activities of daily living (e.g., self-hygiene, bathing, dressing, toileting, walking, feeding, and verbal communication) become difficult. Accordingly, a patient’s quality of life progressively deteriorates. It may be difficult to distinguish daytime fatigue, broken sleep, affect lability, and sighing from depression, but it is vitally important to be aware of the latter, as both fatigue and depression may occur in ALS (McElhiney et al., 2009).
Frontotemporal-type dementia (FTD) and/or cognitive impairment is present in many patients with ALS, albeit on a spectrum from apparently normal to a florid FTD. These observations lend support to the notion that ALS is not a pure disorder of motor neurons, but rather a disorder that primarily affects motor neurons, with the potential to involve nonmotor systems. The presence of ubiquitinated TDP43 inclusions in ALS patients with and without FTD supports the idea that ALS and ALS-FTD may be a continuum. One needs to be cautious when assessing apparently cognitively normal patients with ALS because the deficits may be so subtle as to require specific assessments of personality, behavior, praxis, verbal fluency, visual attention, and verbal reasoning. Dysarthria may mask language disturbances (especially anomia). With appropriate testing, cognitive deficits may be found in about 50% of patients with ALS, but the full clinical (Neary) criteria for a diagnosis of FTD are met in only about 20% of cases (Lomen-Hoerth and Strong, 2006; Murphy, J., 2007). Conversely, subclinical motor neuron degeneration may be found in up to 50% of patients who have FTD (Lipton et al., 2004; MacKenzie and Feldman, 2005).
Natural History of the Disease
Evidence exists for a preclinical phase in ALS. Patients lose motor neurons before they became aware of weakness. Wohlfart (1958) estimated that collateral reinnervation could offset the development of clinical weakness until at least 30% of anterior horn cell motor neurons had been lost. Swash and Ingram described a case of sporadic ALS who complained of muscle fatigue for 6 years before onset of weakness, wasting, and fasciculations. However, once the clinical phase is evident, a generally linear decline in motor function occurs over time. The pattern of disease spread is predictable. When onset is in one arm, spread is often first to the contralateral side, then the ipsilateral leg, the contralateral leg, and finally the bulbar region. Onset in the leg often follows a similar pattern, yet again with final involvement of the bulbar region (Brooks et al., 1994). Bulbar-onset ALS tends to spread to the hands first, with spread to thoracic myotomes, and then the legs. Overall, the pattern suggests that rostral-caudal involvement is faster than caudal-rostral spread. During the course of the disease, transitory improvement, plateaus, or sudden worsening can occur, but spontaneous improvement, although reported, is exceedingly rare.
Prognosis
The median duration of ALS from clinical onset ranges from 22 to 52 months and the mean duration from 23 to 43 months, with an average 5-year survival rate of 22% (roughly 1 in 5) and a 10-year survival rate of 9.4% (roughly 1 in 10) (Murray B., 2006). The most robust poor prognostic factors in ALS are older age at onset and bulbar-onset pattern (Chio et al., 2009). Other important poor prognostic factors include short interval between onset and clinical diagnosis (correlating with a more aggressive presentation), rapid progression rate as assessed on return visits, low body mass index, FTD-ALS presentation, dyspnea at onset, and rapid rate of decline in pulmonary function. PLS and PMA (clinically UMN or LMN only presentations) usually portend a better prognosis, whereas several other clinical subtypes including Mills hemiplegic variant, the pseudoneuritic presentation (flail leg), and the flail-arm variant harbor a better prognosis. Those who have younger age at onset and those who are psychologically well adjusted have a better prognosis. Those who have low-amplitude CMAPs in the setting of normal sensory potentials (the generalized low motor-normal sensory pattern) as revealed by nerve conduction studies appear to have a poor prognosis. Low serum chloride levels are associated with a short-term survival without ventilatory support because they reflect accumulation of bicarbonate due to respiratory failure.
Laboratory Studies
The electrodiagnostic examination is an invaluable tool in the investigation of ALS and its variants (see Chapter 32B). It serves as an adjunct to the clinical examination and is particularly useful in determining the presence or extent of LMN disease. Again, none of the EDX findings are ALS specific, but they can strongly support the diagnosis. Furthermore, repeated investigations at intervals monitor disease progression. Sensory nerve conduction studies are characteristically normal unless the patient happens to have a coincidental mononeuropathy or polyneuropathy. Motor nerve conduction study results may be normal, although the conduction velocity and CMAP amplitude may diminish in keeping with the extent of motor axon loss. There should be no evidence of conduction slowing or block, which would suggest a primarily demyelinating disorder. Severe motor axon loss may give rise to the “generalized low motor-normal sensory” EDX pattern, which may portend a poorer prognosis.
The most important role for neuroimaging studies in ALS is to exclude structural, inflammatory, or infiltrative disorders that may mimic this disease, and therefore all patients should undergo appropriate imaging of brain and spinal cord. On occasion, one may discern abnormal signal in the motor tracts when viewed with proton density–weighted MRI scans of brain; this signal change is due to wallerian degeneration and if seen occurs in patients with more severe disease. FLAIR and T2-weighted fast-spin echo sequences are less specific in their ability to detect such corticospinal tract signal changes. Nonspecific atrophy of the frontal and parietal cortex may also occur. The search for ALS biomarkers has led to the investigation of other imaging techniques such as magnetization transfer ratio (MTR) imaging, magnetic resonance voxel-based morphometry, magnetic resonance spectroscopy, and diffusion tensor MRI (DTI). Functional imaging studies with blood oxygenation level–dependent (BOLD) functional MRI and magnetoencephalography may reveal abnormal activity in motor and non-motor areas in ALS, but further studies are needed to determine their role in UMN assessment (Agosta et al., 2010; Turner et al., 2009). Similarly, additional research is necessary to clarify the role of transcranial magnetic stimulation (TMS), whether used alone or in combination with DTI in the evaluation of the UMN system (Mitsumoto et al., 2007).
While erect forced vital capacity is the most commonly measured index of pulmonary function in ALS, supine FVC provides a more accurate assessment of diaphragmatic weakness. The maximal inspiratory pressure (MIP) and nocturnal oximetry are possibly more effective for the detection of nocturnal hypoventilation. Trans-diaphragmatic sniff pressure (sniff Pdi) and the sniff nasal pressure (SNP) are also useful indicators of hypercapnia and nocturnal hypoxemia (Miller et al., 2009).
Diagnosis
In May 1990, at El Escorial, Spain, the World Federation of Neurology established diagnostic criteria for ALS, which were later modified at Airlie House, Virginia (1998) (www.wfnals.org). These criteria (Table 74.3) include clinical, electrodiagnostic, and pathological components. The clinical criteria divide candidates into those with definite, probable, lab-supported probable, possible, and FALS based on a careful history and examination of four regions of the neuraxis: bulbar, cervical, thoracic, and lumbosacral. The purpose of establishing these criteria was to facilitate entry of appropriate candidates into clinical research trials, but they prove invaluable in the assessment of all patients with ALS.
Table 74.3 Diagnostic Criteria for the Clinical Diagnosis of Amyotrophic Lateral Sclerosis
| Definite ALS | UMN and LMN signs in at least 3 regions (bulbar and 2 spinal regions or 3 spinal regions without bulbar) |
| Probable ALS | UMN and LMN signs in 2 regions, with some UMN signs rostral to LMN signs |
| Probable ALS, lab-supported | UMN and LMN signs in 1 region, with UMN signs alone in another region and EMG evidence of LMN involvement in at least 2 limbs |
| Possible ALS | UMN and LMN signs in 1 region; or UMN signs alone in 2 or more regions; or LMN signs are rostral to UMN signs |
| Familial ALS, lab-supported | Otherwise unexplained UMN or LMN signs in at least 1 region, with SOD1 gene mutation in the proband or a positive family history of family member with a disease-causing SOD1 gene mutation |
ALS, Amyotrophic lateral sclerosis; LMN, lower motor neuron; UMN, upper motor neuron.
Adapted from revised World Federation of Neurology Criteria for the Diagnosis of ALS. Available at www.wfnals.org.
A patient is referred to as having “definite ALS” if there is clinical evidence of both UMN and LMN signs in three or more regions. “Probable ALS” is UMN and LMN signs in two regions. “Possible ALS” implies that a patient either has UMN and LMN signs in one region only or has UMN signs alone in two regions. In addition, “possible ALS” may be applied to those with LMN signs in two regions as long as these are detected rostrally to the UMN signs. “Probable ALS-laboratory supported” refers to those patients who have clinical evidence of possible ALS but also have EDX evidence of more widespread LMN involvement. The proposal is that one should apply the Awaji neurophysiological algorithm to the revised El Escorial criteria to clarify the El Escorial electrodiagnostic criteria and improve diagnostic sensitivity. The Awaji algorithm has increased the importance of fasciculation potentials as being representative of acute denervation as long as there is evidence of chronic denervation in the same muscles. Using both sets of criteria together, Carvalho and Swash recently demonstrated an increased sensitivity in the diagnosis of bulbar-onset ALS from 38% with revised El Escorial alone to 87% when both sets of criteria were used. Another group achieved a specificity of over 95% when using both sets of criteria together (Carvalho and Swash, 2009; Douglass et al., 2010). Follow-up examinations may be helpful in assessing patients with ALS, as disease progression may move a patient up a category, which not only may clarify the diagnosis but also may allow entry of that patient into research trials.
Treatment
Treatment of ALS is outlined in Box 74.7 and Table 74.4.
Box 74.7 Comprehensive Care and Management for Patients with Amyotrophic Lateral Sclerosis
Table 74.4 Symptomatic Treatment in Amyotrophic Lateral Sclerosis
| Symptoms | Pharmacotherapy | Other Therapy |
|---|---|---|
| Fatigue | Pyridostigmine bromideAntidepressants MethylphenidateAmantadine Modafinil |
Energy conservationWork modification Sleep study: BiPAP if abnormal |
| Spasticity | BaclofenTizanidineDantrolene sodiumDiazepam | Physical therapyRange-of-motion exercisesBotulinum toxin injections |
| Jaw clenching | Benzodiazepines | Botulinum toxin injections into masseters |
| Cramps | Quinine sulfateBaclofenVitamin E Clonazepam |
MassagePhysical therapy |
| Fasciculations | Carbamazepine | Assurance |
| Sialorrhea | Hyoscyamine sulphate Diphenhydramine Scopolamine patch Glycopyrrolate Atropine TCA |
Suction machine Botulinum toxin injection into salivary glands Parotid gland radiation therapy Steam inhalation Nebulization Dark grape juice |
| Pseudobulbar laughing or crying | TCAsSSRIsl-Dopa/carbidopaLithium Mirtazapine Venlafaxine Quinidine/dextromethorphan |
|
| Thick phlegm | Guaifenesin Nebulized N-acetylcysteine Nebulized saline Propranolol |
Insufflation-exsufflation High-flow chest wall oscillation therapy Cool mist humidifier Rehydration Pineapple or papaya juice Reduced intake of dairy products, caffeine, alcohol |
| Aspiration | Cisapride | Modified food consistencyTracheostomyModified laryngectomy and tracheal diversion |
| Joint pains | Antiinflammatory drugsAnalgesics | Range-of-motion exercisesHeat |
| Depression | TCAsSSRIs, venlafaxine, mirtazapine, bupropion | CounselingSupport group meetings, psychiatry |
| Insomnia | Zolpidem tartrateLorazepamOpioids TCAs |
Pressure air pad/gel mattress Noninvasive positive pressure ventilation where appropriate |
| Laryngospasm | Sublingual lorazepam | |
| Respiratory failure | Bronchodilators Morphine sulfate |
Hospital bed Nocturnal noninvasive ventilator IPPB |
| Constipation | Increase oral liquidMetamucilDulcolax suppositoriesLactulose and other laxative | Exercise“Power pudding”: prune juice, prunes, applesauce, bran |
IPPB, Intermittent positive-pressure breathing; SSRI, selective serotonin reuptake inhibitor; TCA, tricyclic antidepressant.
Presentation of the Diagnosis of Amyotrophic Lateral Sclerosis
The first step in the management of ALS is to present the diagnosis in a compassionate yet informative manner. Allow adequate time to present the diagnosis. Whenever possible, the patient should not be alone. A second appointment a short time later is often required because many patients and their families find it difficult to absorb the information at first. At the appropriate time, it is important to bring up issues such as advance directives and issues regarding terminal care (Mitsumoto and Rabkin, 2007). Providing information on progress in research, available pharmacotherapies, and the possibility of active participation in clinical trials may increase hope for patients. It is also important to convey the concept of the multidisciplinary care team. Owing to the serious nature of the diagnosis, it is important to facilitate a second opinion if so requested. ALS is almost invariably a relentlessly progressive and terminal disorder, and physicians must raise the issues of the living will and durable power of attorney for health care relatively early after diagnosis to allow the patient and family to prepare ahead. Such decisions are not final and are reversible at any time. Furthermore, some patients either do not wish to or cannot make such decisions.
Specific Pharmacotherapy
In 1996, the U.S. Food and Drug Administration (FDA) approved riluzole (Rilutek) as the first specific drug for the treatment of ALS. It principally functions as an antiglutamate agent, but its mechanism of action is uncertain. The two studies that led to riluzole approval showed that survival was significantly longer in patients with ALS who took 50 mg of riluzole twice a day compared with those who took placebo, although this survival benefit was only modest and was disproportionately beneficial in bulbar-onset disease. A Cochrane meta-analysis of the controlled riluzole trials have shown that 100 mg daily results in a 9% increase in the probability of survival for 1 year and prolongs median survival by 2 to 3 months when taken for 18 months (Miller et al., 2007). Side effects are relatively uncommon and include fatigue, gastrointestinal upset, dizziness, and an increase in liver function tests. To minimize side effects, we recommend 50 mg per day in the evening, and after a week or two, the patient can increase to the regular dose of 50 mg twice a day. Not all patients with ALS receive riluzole therapy; the cost of the drug is one of the main factors in this regard.
Assessment of several agents with antiglutamate activity (talampanel, lamotrigine, branched-chain amino acids, topiramate, dextromethorphan, and gabapentin) revealed no clinical benefit, although dextromethorphan-quinidine has benefit in the treatment of pseudobulbar emotional lability. Small studies of agents with antioxidant and/or neuroprotectant properties (vitamin E, l-deprenyl, N-acetylcysteine, and calcium-channel blockers) show no definite benefit. Neurotrophic factors are a heterogeneous group of basic peptides belonging to the cytokine family produced in regulated amounts by various tissues and are important in cellular proliferation, differentiation, maintenance, maturation, and repair. Several recombinant neurotrophic factors were studied in well-designed trials including ciliary neurotrophic factor, brain-derived neurotrophic factor, insulin-like growth factor (IGF-1, myotropin), and glial cell–derived neurotrophic factor. Results from these trials were generally disappointing and did not appear to achieve adequate tissue levels, while causing unpleasant side effects. IGF-1 polypeptide is currently being studied in a phase III trial. Xaliproden (Sanofi SR57746A), a novel small nonpeptide molecule with both neurotrophic and neuroprotectant properties, held promise because of its good CNS penetration after oral administration, but two large phase III trials did not report sufficient effect on survival to warrant clinical use. Other agents that have been studied and found ineffective in ALS include creatine, coenzyme Q10, and minocycline. Ceftriaxone is currently in a clinical trial. In summary, over the last 15 years, more than 30 different pharmacotherapeutic agents have been rigorously assessed in clinical trials, but apart from riluzole, none to date have shown definite clinical efficacy. A recent randomized double-blind placebo-controlled trial comparing riluzole therapy alone to riluzole in combination with lithium carbonate showed no additional benefit over riluzole alone (Aggarwal et al., 2010). Perhaps one of the reasons for so many disappointing results is that the promising preclinical work was based upon the mutant SOD1 mouse model, which many authorities believe is not representative of the pathogenesis of most ALS. It remains to be seen whether novel agents with relevance to TDP43 or FUS mislocalization might show more promise, and the search continues for new agents and techniques for the specific treatment of ALS.
Aggressive Symptomatic Treatment
Although specific pharmacotherapy is still markedly limited for the treatment of ALS, symptomatic treatment can substantially improve a patient’s symptoms and discomfort. Table 74.4 summarizes specific pharmacological and nonpharmacological symptomatic treatments (Miller et al., 2009).
Multidisciplinary Team Approach At Amyotrophic Lateral Sclerosis Clinic
The care of patients with ALS has become increasingly complex. As a consequence, many patients receive care by a multidisciplinary team in a specialized ALS center rather than by a single treating physician (Mitsumoto et al., 2006). The team often consists of neurologists, a nurse coordinator, physical therapists, occupational therapists, dietitians, speech pathologists, and social workers. Pulmonary specialists and other health professionals should also be available. Using this holistic approach, the aim is to maintain physical independence for as long as possible and to provide psychosocial support to patients and families. As such, specialized multidisciplinary clinic referrals should be offered to patients to optimize and improve quality of life and possibly prolong survival (Miller et al., 2009).
Nutritional Care
Percutaneous endoscopic gastroscopy (PEG) is a standard minor surgical procedure that may not only improve quality of life but also prolong survival by several months. PEG is also probably effective in helping patients maintain weight/body mass index (Miller et al., 2009). Although it is a relatively simple surgery for otherwise healthy patients who have dysphagia, patients with ALS pose particular difficulties and often have impending respiratory failure that may complicate the procedure. Guidelines advocate placement of a PEG tube in consenting patients with dysphagia whose seated predicted forced vital capacity is more than 50%, but PEG may be performed for patients with a forced vital capacity of less than 50% predicted if NIPPV is also used during the procedure (Gregory et al., 2002).
Respiratory Care
Respiratory failure is the most common cause of death in ALS. Indeed, dyspnea-onset ALS presents with obvious ventilatory difficulties, and it is for this reason that it harbors a particularly poor prognosis. It is important to make patients and family members aware that almost all forms of ALS will eventually end by ventilatory failure, although symptoms may go largely unnoticed until relatively late in the disease course. The patient must be made aware that although ventilation via a tracheostomy may indefinitely prolong life, there is no effect on the disease itself. In fact, by prolonging the natural history of the disorder, there is a strong possibility that atypical symptoms may arise, such as visual changes or even sensory loss. Nonetheless, some patients choose to have a tracheostomy and invasive ventilation; this option should be a consideration to improve the quality of life of such individuals (Albert et al., 2009). Most patients and their physicians opt for the noninvasive ventilation (NIV) approach. Evidence exists that patients should be offered NIV at the onset of dyspnea when the forced vital capacity falls to less than 50% predicted or a when a rapid, progressive weakness and wasting of perioral muscles may prevent adequate use of the NIV mask. Nasal pillows can be helpful in this circumstance and are often better tolerated than the mask at all stages. Several factors should be borne in mind when offering this form of treatment. Evidence exists that NIV improves quality of life and may prolong survival in ALS (Bourke et al., 2006; Miller et al., 2009), but NIV does not prolong life indefinitely, and these patients still face the difficult decision of whether to use an invasive ventilator. When making the decision to withdraw ventilatory support or when noninvasive means of ventilatory assistance are insufficient, it is imperative that all attempts focus on effective and compassionate palliative end-of-life care. Hospice care and judicious amounts of opioids, oxygen, and anxiolytics should be prescribed to allow patients to live their final days with dignity and in as much comfort as possible.
Home Care and Hospice Care
When the patient’s condition deteriorates, home hospice care or admission to a residential hospice care facility is required (Mitsumoto et al., 2005). Close collaboration between patients, their caregivers, home-care nurses, and ideally the ALS clinic team will ensure effective and satisfying home care. When a patient has no caregiver, choose a site other than the home for extended care. Hospice care provides highly effective palliative services to patients and their families. Just as important, hospice philosophy strongly affirms life, so that patients who are in the terminal stages of their disease can maintain their independence and dignity to the greatest degree possible (Albert et al., 2005; Mitsumoto, H., 2009).
Familial Amyotrophic Lateral Sclerosis
Between 5% and 10% of all ALS is an inherited trait, in which case it is termed familial ALS (FALS). It is quite possible that the true frequency of FALS is higher, because anything less than a detailed family history may fail to identify an affected family member, and reduced penetrance may account for some apparently sporadic disease. There are autosomal dominant, autosomal recessive, and X-linked dominant forms, some being juvenile onset and others being adult onset (see Box 74.6). The clinical presentation varies considerably, not just in terms of age and site of onset but also in terms of disease duration. FALS is currently classified from ALS1 to ALS11. X-linked dominant ALS, featuring a combination of both UMN and LMN signs, has been reported, the gene locus being on chromosome Xp11-Xq12 (Siddique and Dellefave, 2006).
ALS1 is a form of late-onset (usually >age 30) motor neuron disorder that accounts for 15% to 20% of all cases of FALS (and thus 1%-2% of all ALS). It is associated with mutations in the gene that encodes superoxide dismutase 1 (SOD1) located on chromosome 21q21 (Rosen et al., 1993). Inheritance in most is in an autosomal dominant pattern, but a recessive variant occurs (Andersen et al., 1996).
ALS2 is a rare recessively inherited disorder mapped to a gene on chromosome 2q33 that encodes a novel protein called alsin. Analysis of the original families revealed that all were due to truncated protein product from frameshift or nonsense mutations, but missense mutations occur. This juvenile ALS, originally described in consanguineous families from Tunisia, was also discovered in families from Saudi Arabia and Kuwait. The phenotype of this disorder varies according to the family of origin; in the Tunisian family, it presents as a slowly progressive ALS-like disorder with mean age of onset at age 12 years; in the Kuwaiti family, the phenotype is similar to early-onset PLS. Sequence homology analysis of this protein suggests that it is a guanine-nucleotide exchange factor for Rab 5 and is thus important in intracellular cell signaling, endosomal dynamics, mitochondrial trafficking, and cytoskeleton organization. In contrast to the toxic gain-of-function theory of pathogenesis in ALS1, it appears that loss of function of the gene product is responsible for the selective injury to, and dysfunction of, the corticospinal tract in ALS2. Alternate splicing of the alsin gene results in both long and short transcripts. Frameshift and nonsense mutations cause an ALS phenotype when there is homozygous loss of both the short and long forms, whereas the PLS presentation occurs with homozygous loss of the long form alone. Missense mutations may cause an unstable protein product or lead to a protein product that is directly toxic to the cell via aberrant regulation of the apoptotic pathway. Certain mutations in the alsin gene also give rise to an infantile ascending hereditary spastic paraparesis, which demonstrates the link between ALS and related motor neuron diseases (Eymard-Pierre et al., 2006; Panzeri et al., 2006).
ALS3 describes a large European family with adult-onset autosomal dominant ALS linked to chromosome 18q21 (Hand et al., 2002). The gene for this disorder has not yet been identified. ALS4 is a juvenile-onset, slowly progressive, dominantly inherited distal amyotrophy with UMN signs but sparing bulbar features. It is caused by mutations in the senataxin gene (SETX) on chromosome 9q34, which has a DNA/RNA helicase domain suggesting a role in DNA repair and RNA processing. SETX has homology to IGHMBP2, the gene responsible for SMARD1, a rare form of SMA. Mutations in SETX also cause a recessively inherited disorder called oculomotor apraxia and cerebellar ataxia. A recent paper expanded the phenotype by describing a novel senataxin mutation presenting as a classical case of sporadic ALS (Zhao et al., 2009). ALS5 is a recessive juvenile ALS described in North African and European families. The clinical syndrome is similar to SALS except for a younger age at onset. It links to chromosome 15q15-q22, but the pathogenic gene is unidentified. ALS6 is an autosomal dominant ALS that can also be associated with frontotemporal dementia and hallucinations. Mutations on the FUS/TLS (fused in sarcoma/translocated in liposarcoma) gene on chromosome 16q12 have the pathological characteristic of FUS-immunoreactive skein-like cytoplasmic inclusion bodies. FUS is normally a nuclear protein, so this is yet another example of protein mislocalization in neurodegenerative disease. FUS mutations have a worldwide distribution and account for about 5% of FALS; they are also detectable in about 1% of SALS (Lai et al., 2010). Furthermore, it has recently been shown that FUS-immunoreactive cytoplasmic inclusions are common in both SALS and non-SOD1 FALS and are also immunoreactive to TDP 43 and ubiquitin (Deng et al., 2010). ALS7 is a rare, late-onset, autosomal dominant disorder linked to chromosome 20ptel. The cause for ALS8 is a mutation in a vesicle trafficking protein gene called VAPB (vesicle-associated membrane protein/synaptobrevin-associated membrane protein B) on chromosome 20q13.3. The clinical presentation displays quite marked heterogeneity: some patients develop a slowly progressive ALS-like picture with prominent tremor and onset between the ages of 31 and 45 years, whereas others present with a late-onset SMA or a severe, rapidly progressive ALS (Nishimura et al., 2004). ALS9 results from mutations on the angiogenin gene on chromosome 14q. This is an autosomal dominant disorder of adults (from the fourth to the eighth decade). Angiogenin may help protect motor neurons from excitotoxic- and hypoxia-induced injury, but it may also play an important role in RNA transcription as well as interact with cellular cytoskeletal proteins. ALS10, which accounts for 2% to 5% of FALS, is caused by mutations in the TDP43 gene on chromosome 1 and presents clinically as ALS with either limb or bulbar onset. Despite the association of TDP inclusions with some forms of FTD, cognitive deficits do not occur in TDP43 mutation–associated ALS. TDP43 immunoreactive ubiquitinated inclusions are present in degenerating neurons and glial cells just as they in sporadic ALS (Kuhnlein et al., 2008; Van Deerlin et al., 2008). The TDP43 gene product is a dual DNA/RNA binding protein mainly expressed in the nucleus and may play an important part in the regulation of RNA trafficking and translation. TDP43 can modulate human low-molecular-weight neurofilament messenger ribonucleic acid (mRNA) stability, which in turn underlies neurofilament aggregates, sometimes seen in ALS (Strong et al., 2006). TDP43 inclusion bodies are in fact seen in several disorders apart from ALS, including FTD, frontotemporal lobar degeneration with motor neuron degeneration, corticobasal degeneration, Guamanian ALS-PD complex, and hippocampal sclerosis. While this might suggest that TDP43 inclusion bodies are a nonspecific marker of neuronal injury or indeed representative of a physiological cell response to injury, the frequency of TDP43 mutations (30 to date) in both SALS and FALS suggests a pathogenic role.
ALS11 is due to mutations of the FIG4 gene on chromosome 6q21, which encodes a phosphoinositide 5-phosphatase, a regulator of a signaling lipid on the surface of endosomes. Mutations in this gene are known to cause a recessively inherited severe axonal sensorimotor form of Charcot-Marie-Tooth known as CMT4J, which is usually early onset (although one family presented with an adult disorder resembling ALS). FIG4 mutations can cause an ALS or PLS presentation, and additional personality changes were noted in two cases. Of the nine cases with FIG4 mutations, only three were FALS, the rest apparently being sporadic (Chow et al., 2009). Although not formally classified as a familial ALS (it has been described as a spinal and bulbar muscular atrophy), it is apparent that mutations in the p150Glued subunit of the dynactin gene may cause ALS-like presentations. Furthermore, a family history is not always evident (Munch et al., 2004). Dynactin 1 is a vital component of the dynein-dynactin motor complex and is important in retrograde axonal transport. Puls et al. have described a particular LMN disorder caused by a mutation in the p150Glued subunit of dynactin 1. The clinical phenotype is distinctive, with early bilateral vocal cord paralysis followed by prominent involvement of intrinsic hand muscles (especially those of the thenar eminence), the legs, and the face (Puls et al., 2005). The recent paper by Maruyama and colleagues described ALS in unrelated Japanese families caused by mutations in the OPTN gene for optineurin, a negative regulator of tumor necrosis factor alpha (TNF-α)–induced activation of NF-κB. OPTN may also play a role membrane trafficking, exocytosis, and maintenance of the Golgi apparatus. Mutations in this gene are known to cause primary open angle glaucoma. For patients presenting with ALS, the inheritance patterns were both autosomal recessive and autosomal dominant, with onset between the ages of 30 and 60 years and long disease duration. One case which came to autopsy showed optineurin-positive cytoplasmic inclusion bodies in anterior horn cells; the investigators also showed that TDP43- and SOD1-positive inclusions in SALS and SOD1 FALS were co-labeled with optineurin, suggesting a broader role for this protein in the pathogenesis of ALS (Maruyama et al., 2010).
Amyotrophic Lateral Sclerosis–Parkinsonism-Dementia Complex (Western Pacific Amyotrophic Lateral Sclerosis)
In 1954, Mulder and colleagues described an unusually high incidence of ALS in the adult native Chamorro population on the Western Pacific island of Guam. Soon afterwards, a related disorder of high incidence characterized by dementia and parkinsonism was also found in this population, with some patients displaying overlap features between ALS, parkinsonism, and dementia. A similar disorder was subsequently described in western New Guinea and the Kii peninsula of Japan, with an ALS incidence between 50 and 150 times higher than elsewhere. Clinically, about 5% of patients develop a predominantly ALS type of disorder, whereas 38% manifest principally with a combination of parkinsonism and dementia. The pathology of this unusual disorder bears similarities to that of Alzheimer disease, with prominent loss of CNS neurons and the presence of abundant tau-immunoreactive neurofibrillary tangles. However, the characteristic pathology of Guamanian ALS and PDC is by TDP43-positive inclusions in neurons and glial cells. α-Synuclein pathology also is detectable in the amygdala of affected brain tissue (Forman et al., 2002). Multiple members of a single family may be affected, and it has recently been shown that first-degree relatives of patients with ALS-PDC have a significantly higher risk of developing the disease than controls. Despite these observations and a genetic association study implicating the tau gene as a susceptibility gene for ALS-PDC, accumulated epidemiological evidence strongly suggests that an environmental factor rather than a genetic factor is more important in disease pathogenesis. Various environmental toxins have been implicated in the pathogenesis of ALS-PDC, chief among them being neurotoxins derived from the native cycad seed. This seed contains β-methylamino-l-alanine (BMAA), an amino acid that is toxic to cortical and spinal motor neurons and thought to be the product of cyanobacterial activity in the roots of the cycad palm. Cycad seed also contains a carcinogenic substance called cycasin that may act either alone or in concert with BMAA to damage motor neurons. Toxic sterol glucosides have also been isolated from washed cycad flour, and they can cause the release of glutamate (Khabazian et al., 2002). However, the role of cycad seeds in neurotoxicity is still subject to debate (Snyder et al., 2011). The cyanobacteria/BMAA hypothesis has wider implications for research in SALS worldwide. It has been recently shown that protein-bound BMAA is present in the brains of North American patients dying with ALS and Alzheimer disease and it has been hypothesized that such patients may be genetically susceptible to BMAA-induced neurodegeneration (Bradley and Mash, 2009). The cycad seed has many uses: in West Papua and Guam as a topical medicine for skin lesions and in Japan as an oral medicine (Spencer et al., 2005). Cox and Sacks (2002) proposed a process of biomagnification of cycad toxins in Guam through the Chamorro practice of eating flying foxes, which themselves feed on cycad seeds. The incidence of the Guamanian ALS variant has rapidly declined over the past several decades, a process thought to reflect the Westernization of the region. The decline in the incidence of ALS-PDC in Guam may reflect the dwindling flying fox population on Guam through a massive increase in commercial hunting using firearms introduced to the island in the decades following World War II. However, other social and dietary shifts have occurred in Guam that might be responsible for the decrease.
Spinocerebellar Ataxia Type 3 (Machado-Joseph disease)
Machado-Joseph disease is an autosomal dominant syndrome with onset varying from the third to seventh decade of life. Although cerebellar ataxia is the predominant clinical feature, patients often present with slowly progressive generalized spasticity, cramps, muscle wasting, and fasciculations of the face and tongue. Other characteristic findings include extrapyramidal signs such as dystonia and rigidity, protuberant eyes, and progressive external ophthalmoparesis. Affected patients have a twofold to threefold expansion of a CAG trinucleotide repeat on the ataxin-3 gene on chromosome 14q32.1. The expanded triplet repeat results in a mutant gene product containing an expanded polyglutamine tract. This appears to aggregate into intranuclear neuronal inclusion bodies and may interfere with the function of the cellular proteosome in degradation of proteins (Schmidt et al., 2002). The Machado-Joseph disease phenotype may also occur in SCA-2, with slowly progressive ataxia, eyelid retraction, and facial fasciculations. Patients often have slow saccades or ophthalmoparesis and may have reduced or absent deep tendon reflexes. The cause of SCA-2 is an expanded polyglutamine-encoding CAG triple repeat sequence on chromosome 12q.
Adult Hexosaminidase-A Deficiency
The adult form has a mean of onset of about 18 years and usually presents as slowly progressive weakness of predominantly proximal muscles of the upper and lower extremities (Neudorfer et al., 2005). In some patients, severe cramps may present in association with muscle weakness, mimicking SMA. In others, however, a combination of dysarthria, spasticity, and LMN signs may resemble ALS. Additional sensory, cerebellar, cognitive, psychiatric, and extrapyramidal features may later develop. The EDX may reveal prominent complex repetitive discharges and abnormal SNAPs. Generally, this constellation of symptoms and signs is not easily mistaken for ALS, but in the relatively early stages, patients with Hex-A deficiency may not manifest many features other than motor system dysfunction. Genetic counseling is important before assaying a patient’s serum or leukocytes for deficiency of Hex-A activity.
Allgrove Syndrome (Four-A Syndrome)
Four-A syndrome (Allgrove syndrome) is a very rare autosomal recessive disorder that derives its name from the combination of achalasia, alacrima, adrenocorticotrophic insufficiency, and amyotrophy. The AAAS gene is located on chromosome 12q13 and encodes a ubiquitous protein called aladin which heavily expresses in the neuroendocrine system and may be important in regulation of the cell cycle, cell signaling, intracellular transport, and the cell cytoskeleton. The syndrome can manifest from the first decade of life with dysphagia and adrenocortical insufficiency, but a wide a range of neurological problems can arise later in life, including cognitive deterioration, optic atrophy, seizures, autonomic disturbance (dry mouth, postural hypotension, and syncope), and bulbospinal amyotrophy (amyotrophy of limbs and tongue, with tongue fasciculations and pyramidal signs) (Kimber et al., 2003).
Autosomal Dominant Frontotemporal Dementia with Motor Neuron Disease
For more than half a century, various observers have pointed out that symptoms and signs of ALS and FTD appear to occur together in some families with increased frequency. One such disorder, disinhibition-dementia-parkinsonism-amyotrophy complex (DDPAC), is an autosomal dominant FTD due to a mutation on the microtubule-associated protein tau gene on chromosome 17. This disorder, first described in a large Irish-American family (family Mo) in the United States, is characterized clinically by disinhibited behavior (including excessive eating and inappropriate sexual behavior), personality changes, dementia, parkinsonian manifestations, and in two cases, amyotrophy with fasciculations. Autopsy studies showed widespread neuronal loss in the substantia nigra, cerebral cortex, and anterior horn of the spinal cord, together with extensive spongy degeneration in the temporal and frontal lobes and tau-immunoreactive inclusion bodies in affected regions of the CNS (Lynch et al., 1994). In 2006, it was discovered that mutations in the progranulin gene, also on chromosome 17, can cause frontotemporal degeneration. Progranulin is a growth factor with important functions in inflammation, wound repair, tumorigenesis, and cell development. Most cases with this mutation develop a late-onset progressive disorder of personality, behavior, and especially language, without clinical features of motor neuron disease. However, TDP43-positive, tau-negative ubiquitinated inclusions are present in neurons and glial cells of affected individuals, thus linking this form of frontotemporal dementia to the pathology of ALS (Baker et al., 2006; Cruts et al., 2006; Gass et al., 2006; van Swieten and Heutink, 2008). Mutations in the TDP43 gene itself have been shown to be a rare cause of ALS, but cognitive impairment is not seen (Kuhnlein and Sperfeld, 2008; Van Deerlin et al., 2008).
Analysis of multiple pedigrees identified a locus for autosomal dominant ALS and FTD on chromosome 9q21-22. The mean age at disease onset in these families was about 54 years (range 40-62 years). Some patients developed symptoms and signs of ALS alone, but others also developed inappropriate behavior, impulsiveness, and eventually impaired memory. Both neuroimaging and pathological studies revealed frontotemporal atrophy, and histology identified gliosis, vacuoles, rare Pick bodies, and some neurofibrillary tangles and senile plaques (Hosler et al., 2000). Another familial ALS and FTD locus has been described on chromosome 9p13.2-21.3, but the cause of this disorder is unknown (Vance et al., 2006). Mutations in the p150 subunit of dynactin gene cause a LMN spinal and bulbar syndrome, but one of the mutations can cause either an autosomal dominant ALS or an FTD (Munch et al., 2005).
Adult Polyglucosan Body Disease
Polyglucosan body disease is a very rare, late-onset, slowly progressive disorder characterized by a combination of UMN and LMN signs, cognitive decline, distal sensory loss, and disturbances of bladder and bowel function. MRI of the brain may reveal diffuse white-matter signal increase on T2-weighted images. The diagnosis is clinched by the finding of characteristic pathological changes in tissue from peripheral nerve, cerebral cortex, spinal cord, or skin. Axons and neural sheath cells contain non-membrane-bound cytoplasmic periodic acid–Schiff-positive polyglucosan bodies. Ultrastructural examination shows that the inclusions consist of 6- to 8-nm branched filaments and are most abundant in myelinated nerve fibers. In Ashkenazi Jewish patients (and one reported non-Ashkenazi Jewish patient), the disorder was caused by mutations of the glycogen-branching enzyme (GBE) gene, with subsequent deficiency of the protein product. However, adult polyglucosan body disease (APBD) occurs in many different populations, and considerable molecular heterogeneity has been noted, with otherwise typical cases lacking GBE mutations despite deficiency of enzyme activity (Klein et al., 2004). The recent (albeit inadvertent) generation of muscle polyglucosan bodies in a transgenic mouse engineered to overexpress glycogen synthase in the presence of normal levels of glycogen-branching enzyme suggests that an imbalance in the activities of these two enzymes is the possible molecular mechanism underlying this unusual disorder (Raben et al., 2001). It is interesting to note that two types of polyglucosan body may be seen in ALS—Lafora bodies and corpora amylacea—although neither is considered a characteristic pathological feature.
Human Immunodeficiency Virus Type 1–Associated Motor Neuron Disorder
A retrospective review of 1700 cases of HIV-1-infected patients with neurological symptoms identified 6 cases presenting as a reversible ALS-like syndrome (Moulignier et al., 2001), representing a 27-fold increased risk of developing an ALS-like disorder in that particular HIV-1 patient population. Overall, patients were somewhat younger than the normal ALS population, all but one being younger than 40 years at the time of diagnosis. Onset was characteristically in a monomelic pattern followed by a very rapid spread to other regions over a period of weeks. There were clinical features of both UMN and LMN involvement, with fasciculations, cramps, and bulbar symptoms. Two patients also had rapidly progressive dementia, with other features suggesting an additional diagnosis of AIDS-dementia complex. Sensory and sphincter disturbances were not apparent. CSF protein levels were sometimes mildly increased, and a lymphocytic pleocytosis was evident in three patients, but all remaining laboratory results (HIV-1 seropositivity apart) were negative. EDX revealed a widespread disorder of anterior horn cells in the absence of demyelinating conduction block, and MRI in one patient showed diffuse white-matter signal increase suggestive of AIDS-dementia complex. In each case, antiretroviral therapy was beneficial either in stabilizing or (in two instances) curing the disease. No similar cases have been identified in this particular study population since the introduction of highly active antiretroviral combination chemotherapy in the management of HIV infection. Another case report found similar clinical features in a 32-year-old HIV-positive patient who also enjoyed a complete response to antiretroviral therapy. MRI of brain showed increased T2-weighted signal in the brachium pontis with some minimal contrast enhancement. The resolution of motor symptoms coincided with a lack of detectable HIV in plasma and CSF. In addition, the abnormal MRI signal almost completely resolved (MacGowan et al., 2001). Flail-arm ALS-like variants have also been described, with MRI signal changes in the anterior cervical spinal cord (Henning and Hewlett, 2008; Nalini et al., 2009). Other forms of HIV may also relate to the pathogenesis of motor neuron disease; a pure LMN syndrome occurred in a woman who was seropositive for HIV-2. Overall, there seems to be sufficient evidence to implicate HIV as a potential cause of an ALS-like disorder, but one must also consider the possibility of coincidental HIV infection in patients who have true sporadic ALS.
Aggarwal S.P., Zinman L., Simpson E., et al. Safety and efficacy of lithium carbonate in combination with riluzole for treatment of amyotrophic lateral sclerosis; a randomized, double-blind, placebo-controlled trial. Lancet Neurol. 2010;9:481-488.
Agosta F., Chiò A., Cosottini M., et al. The present and the future of neuroimaging in amyotrophic lateral sclerosis. Am J Neuroradiol. 2010;31:1769-1777.
Albert S.M., Whitaker A., Rabkin J.G., et al. Medical and supportive care among people with ALS in the months before death or tracheostomy. J Pain Symptom Manage. 2009;38(4):546-553.
Alexander L.N., Seward J.F., Santibanez T.A., et al. Vaccine policy changes and epidemiology of poliomyelitis in the United States. JAMA. 2004;2004(292):1696-1701.
Andersen P.M., Forsgren L., Binger M., et al. Autosomal recessive adult-onset amyotrophic lateral sclerosis associated with homozygosity of Asp90Ala Cu,Zn-superoxide dismutase mutation. A clinical and genealogical study of 36 patients. Brain. 1996;119:1153-1172.
Araujo A., Hall W.W. Human T-lymphotrophic virus type II and neurological disease. Ann Neurol. 2004;56(1):10-19.
Armon C. Smoking may be considered an established risk factor for sporadic ALS. Neurology. 2009;73(20):1693-1698.
Atsuna N., Watanabe H., Ito M., et al. Natural history of spinal and bulbar muscular atrophy (SBMA): a study of 223 Japanese patients. Brain. 2006;129:1446-1455.
Baker M., Mackenzie I.R., Pickering-Brown S.M., et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442(7105):916-919.
Beaulieu J.M., Kriz J., Julien J.P. Induction of peripherin expression in subsets of brain neurons after lesion injury or cerebral injury. Brain Res. 2002;946(2):153-161.
Blair M.A., Ma S., Hedera P. Mutation in KIF5A can also cause adult-onset hereditary spastic paraplegia. Neurogenetics. 2006;7(1):47-50.
Boillée S., Yamanaka K., Lobsiger C.S., et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389-1392.
Bourke S.C., Tomlinson M., Williams T.L., et al. Effects of noninvasive ventilation on survival and quality of life in patients with amyotrophic lateral sclerosis: a randomized controlled trial. Lancet Neurol. 2006;5:140-147.
Bradley W.G., Mash D.C. Beyond Guam: the cyanobacteria/BMAA hypothesis of the cause of ALS and other neurodegenerative diseases. Amyotroph Lateral Scler. 2009;10:7-20.
Brooks B.R., Lewis D., Rawling J., et al. The natural history of amyotrophic lateral sclerosis. In: Williams A.C., editor. Motor Neuron Disease. London: Chapman & Hall Medical, 1994.
Brooks B.R., Thisted R.A., Appel S.H., et al. Treatment of pseudobulbar affect in ALS with dextromethorphan/quinidine: a randomized trial. Neurology. 2005;63(8):1364-1370.
Brugman F., Veldink J.H., Franssen H., et al. Differentiation of hereditary spastic paraparesis from primary lateral sclerosis in sporadic adult-onset upper motor neuron syndromes. Arch Neurol. 2009;66(4):509-514.
Carvalho M.D., Swash M. Awaji diagnostic algorithm increases sensitivity of El Escorial criteria fro ALS diagnosis. Amyotroph Lateral Scler. 2009;10:53-57.
Chio A., Logroscino G., Hardiman O., et al. Prognostic factors in ALS: a critical review. Amyotrophic Lateral Scler. 2009;10(5-6):310-323.
Chow C.Y., Landers J.E., Bergren S.K., et al. Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am J Hum Genet. 2009;84:85-88.
Cleveland D.W., Rothstein J.D. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2(11):806-819.
Cox P.A., Sacks O.W. Cycad neurotoxins, consumption of flying foxes, and ALS-PDC disease in Guam. Neurology. 2002;58:956-959.
Cruts M., Gijselinck I., van der Zee J., et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920-924.
Del Bo R., Locatelli F., Corti S., et al. Coexistence of CMT-2D and distal SMA-V phenotypes in an Italian family with a GARS gene mutation. Neurology. 2006;66:752-754.
Deng H.X., Zhai H., Bigio E.H., et al. FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann Neurol. 2010;67:739-748.
Depienne C., Tallaksen C., Lephay J.Y., et al. Spastin mutations are frequent in sporadic spastic paraparesis and their spectrum is different from that observed in familial cases. J Med Genet. 2006;43:259-265.
Douglass C.P., Kandler R.H., Shaw P.J., et al. An evaluation of neurophysiological crieteria used in the diagnosis of motor neuron disease. J Neurol, Neurosurg Psychiatry. 2010;81(6):646-649.
Dubourg O., Azzedine H., Ben Yaou R., et al. The G526R glycyltRNA synthetase gene mutation in distal hereditary motor neuropathy type V. Neurology. 2006;66:1721-1726.
Eymard-Pierre E., Yamanaka K., Haeussler M., et al. Novel missense mutation in ALS2 gene results in infantile hereditary spastic paraparesis. Ann Neurol. 2006;59(6):976-980.
Ferrante M.A., Wilbourn A.J. The characteristic electrodiagnostic features of Kennedy’s disease. Muscle Nerve. 1997;20(3):323-329.
Forman M.S., Schmidt M.L., Kasturi S., et al. Tau and alpha-synuclein pathology in amygdala of parkinsonism-dementia complex patients of Guam. Am J Pathol. 2002;160(5):1725-1731.
Gallo V., Bueno-De-Mesquita H.B., Vermeulen R., et al. Smoking and risk for amyotrophic lateral sclerosis: analysis of the EPIC cohort. Ann Neurol. 2009;65:378-385.
Gass J., Cannon A., Mackenzie I.R., et al. Mutations in the progranulin gene are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Med Genet. 2006;15:2988-3001.
Getahun H., Lambein F., Vanhoorne M., et al. Pattern and associated factors of the neurolathyrism epidemic in Ethiopia. Trop Med Int Health. 2002;7(2):118-124.
Gonzalez H., Olsson T., Borg K. Management of postpolio syndrome. Lancet Neurol. 2010;9:634-642.
Gonzalez H., Stibrant-Sunnerhagen H., Sjoberg I., et al. Intravenous immunoglobulin for post-polio syndrome: a randomized controlled trial. Lancet Neurol. 2006;5(6):493-500.
Gordon P. History of ALS. In: Mitsumoto H., Przedborski S., Gordon P.H. Amyotrophic Lateral Sclerosis. New York: Taylor & Francis Group; 2006:1-16.
Gordon P.H., Cheng B., Katz I.B., et al. Clinical features that distinguish PLS, upper motor neuron dominant ALS, and typical ALS. Neurology. 2009;72(5):1948-1952.
Gourie-Devi M., Nalini A. Long-term follow up of 44 patients with brachial monomelic amyotrophy. Acta Neurol Scand. 2003;107(3):215-220.
Gregory S., Siderowf A., Golaszewski A.L., et al. Gastrostomy insertion in ALS patients with low vital capacity: Respiratory support and survival. Neurology. 2002;58(3):485-487.
Hand C.K., Khoris J., Salachas F., et al. A novel locus for familial amyotrophic lateral sclerosis on chromosome 18q. Am J Hum Genet. 2002;70:251-256.
Henning F., Hewlett R.H. Brachial amyotrophic diplegia (segmental proximal spinal muscular atrophy) associated with HIV infection. J Neurol Neurosurg Psychiatry. 2008;79:1392-1394.
Hirayama K. [Juvenile muscular atrophy of unilateral upper extremity (Hirayama disease)–half century progress and establishment since its discovery]. [Article in Japanese] Brain Nerve. 2008;60(1):17-29.
Hirayama K., Tokamaru Y. Cervical dural sac and spinal cord in juvenile muscular atrophy of distal upper extremity. Neurology. 2000;54(10):1922-1926.
Hirth D., Iannaccone S., Heeskerk J., et al. Challenges and opportunities in clinical trials for spinal muscular atrophy. Neurology. 2005;65(9):1352-1357.
Hosler B.A., Siddique T., Sapp P.C., et al. Linkage of familial amyotrophic lateral sclerosis with frontotemporal dementia to chromosome 9q21-22. JAMA. 2000;284:1664-1669.
Howard R.S. Poliomyelitis and the postpolio syndrome. BMJ. 2005;330:1314-1319.
Khabazian I., Bains J.S., Williams D.E., et al. Isolation of various forms of sterol beta-d-glucoside from the seed of Cycas circinalis: Neurotoxicity and implications for ALS-parkinsonism dementia complex. J Neurochem. 2002;82(3):516-528.
Kilpatrick A.M., Kramer L.D., Jones M.J., et al. West Nile Virus epidemics in North America are driven by shifts in mosquito feeding behavior. PLoS Biol. 2006;4(4):e82.
Kim W.K., Liu X., Sandner J., et al. Study of 962 patients indicates progressive muscular atrophy is a form of ALS. Neurology. 2009;73(20):1686-1692.
Kimber J., McLean B.N., Prevett M., et al. Allgrove or 4 A syndrome: an autosomal recessive syndrome causing multisystem neurological disease. J Neurol Neurosurg Psychiatry. 2003;74(5):654-657.
Klein C.J., Boes C.J., Chapin J.E., et al. Adult polyglucosan body disease: case description of an expanding genetic and clinical syndrome. Muscle Nerve. 2004;29(2):323-328.
Kostova F.V., Williams V.C., Heemskerk J., et al. Spinal muscular atrophy: classification, diagnosis, management, pathogenesis, and future research directions. J Child Neurol. 2007;22:926-945.
Kuhnlein P., Sperfeld A-D., Vanmassenove B., et al. Two German kindreds with familial amyotrophic lateral sclerosis due to TARDBP mutations. Arch Neurol. 2008;65:1185-1189.
Lai S.L., Abramzon Y., Schymick J.C., et al. FUS mutations in sporadic amyotrophic lateral sclerosis. Neurobiol Aging. 2010;32:550.
Landers J.E., Melki J., Meininger V., et al. Reduced expression of the kinesin-associated protein3 (KIFAP3) gene increases survival in sporadic amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2009;106:9004-9009.
Leung C.L., He C.Z., Kaufmann P., et al. A pathogenic peripherin gene mutation in a patient with amyotrophic lateral sclerosis. Brain Pathol. 2004;14:290-296.
Lindsey N.P., Staples J.E., Lehman J.A., et al. Surveillance for human West Nile virus disease-United States, 1999-2008, Centers for Disease Control and Prevention (CDC). MMWR Surveill Sum, 59. 2010;2:1-17.
Lipton A.M., White C.J.3rd, Bigio E.H. Frontotemporal lobar degeneration with motor neuron disease-type inclusions predominates in 76 cases of frontotemporal degeneration. Acta Neuropathol. 2004;108:379-385.
Lomen-Hoerth C., Strong M.J. Frontotemporal dysfunction in amyotrophic lateral sclerosis. In: : Mitsumoto H., Przedborski S., Gordon P.H. Amyotrophic Lateral Sclerosis. New York: Taylor & Francis Group; 2006:117-140.
Lorson C.L., Rindt R., Shababi M. Spinal muscular atrophy: mechanisms and therapeutic strategies. Hum Mol Genet. 2010;19:R111-R118.
Lynch T., Sano M., Marder K.S., et al. Clinical characteristics of a family with chromosome 17-linked disinhibition-dementia-parkinsonism-amyotrophy complex. Neurology. 1994;44:1878-1884.
MacGowan D.J.L., Scelsa S.N., Waldron M. An ALS-like syndrome with new HIV infection and complete response to antiretroviral therapy. Neurology. 2001;57:1094-1097.
MacKenzie I.R., Feldman H.H. Ubiquitin immunohistochemistry suggests classic motor neuron disease, motor neuron disease with dementia, and frontotemporal dementia of the motor neuron disease type represent a clinicopathologic spectrum. J Neuropathol Exp Neurol. 2005;64:730-739.
Maruyama H., Morino H., Ito H., et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465:223-227.
McDermott C.J. Clinical features of hereditary spastic paraplegia due to spastin mutation. Neurology. 2006;67:45-51.
McElhiney M.C., Rabkin J.G., Gordon P., et al. Prevalence of fatigue and depression in ALS and change over time. J Neurol Neurosurg Psychiatry. 2009;80(10):1146-1149.
McGuire V., Nelson L.M. Epidemiology of ALS. In: Mitsumoto H., Przedborski S., Gordon P. Amyotrophic Lateral Sclerosis. New York: Taylor & Francis; 2006:17-21.
Miller R.G., Jackson C.E., Kasarskis E.J. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: multidisciplinary care, symptom, and cognitive/behavioral impairment (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009;73(15):1227-1233.
Miller, R.G., Mitchell, J.D., Lyon, M., et al., 2007. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev; CD001447.
Mitsumoto H., editor. Amyotrophic Lateral Sclerosis. Patient and Family Guide to Management and Care, third ed, New York: Demos, 2009.
Mitsumoto H., Borasio G.D., Genge A.L., et al. The multidisciplinary care clinic: the principles and an international perspective. In: Mitsumoto H., Przedborski S., Gordon P.H. Amyotrophic Lateral Sclerosis. New York: Taylor & Francis Group; 2006:605-631.
Mitsumoto H., Bromberg M., Johnston W., et al. Promoting excellence in end-of-life care in ALS. Amyotroph Lateral Scler. 2005;6:145-154.
Mitsumoto H., Rabkin J.G. Palliative care for patients with ALS: prepare for the worst and hope for the best. JAMA. 2007;298:207-216.
Mitsumoto H., Ulug A.M., Pullman S.L. Quantitative objective markers for upper and lower motor neuron dysfunction in ALS. Neurology. 2007;68(17):1402-1410.
Monani U.R. Spinal muscular atrophy: a deficiency in a ubiquitous protein; a motor neuron-specific disease. Neuron. 2005;48:885-896.
Moulignier A., Moulonguet A., Pialoux G., et al. Reversible ALS-like disorder in HIV infection. Neurology. 2001;57(6):995-1001.
Mukherjee S., Newby E., Harvey J.N. Adrenomyelopathy in patients with “Addison’s disease”: genetic case analysis. J R Soc Med. 2006;99:245-249.
Munch C., Rosenbohm A., Sperfeld A.D., et al. Heterozygous R1101K mutation of the DCTN1 gene in a family with ALS and FTD. Ann Neurol. 2005;58(5):777-780.
Munch C., Sedlmeier R., Meyer T., et al. Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology. 2004;63(4):724-726.
Murphy J.M., Henry R.G., Langmore S., et al. Continuum of frontal lobe impairment in amyotrophic lateral sclerosis. Arch Neurol. 2007;64:530-534.
Murray B. Natural history and prognosis in amyotrophic lateral sclerosis. In: Mitsumoto H., Przedborski S., Gordon P.H. Amyotrophic Lateral Sclerosis. New York: Taylor & Francis Group; 2006:227-255.
Murray K., Baraniuck S., Resnick M., et al. Risk factors for encephalitis and death from West Nile virus infection. Epidemiol Infect. 2006;134:1325-1332.
Nalini A., Desai A., Mahato S.K. Flail arm-like syndrome associated with HIV-1 infection. Ann Indian Acad Neurol. 2009;12:127-130.
Narakawa N., Shiizaki K., Kitabata Y., et al. Plasma exchange for the treatment of human T-cell lymphotropic virus type 1 associated myelopathy. Ther Apher. 2001;5(6):491-493.
Neudorfer O., Pastores G.M., Zeng B.J., et al. Late-onset Tay-Sachs disease: phenotype characterization and genotype correlations in 21 affected patients. Genet Med. 2005;7(2):119-123.
Nishimura A.L., Mitne-Neto M., Silva H.C.A., et al. A mutation in the vesicle- trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet. 2004;75:822-831.
Panzeri C., DePalma C., Martinuzzi A., et al. The first ALS2 missense mutation in JPLS reveals new aspects of alsin biological function. Brain. 2006;129(7):1710-1719.
Posada-Vergara M.P., Monatheiro P., Fukumori L.M., et al. Clinical and epidemiological aspects of HTLV-II infection in Sao Paulo, Brazil: presence of tropical spastic paraparesis/HTLV-associated myelopathy (TSP/HAM) simile diagnosis in HIV-1 co-infected subjects. Rev Inst Med Trop Sao Paulo. 2006;48(4):207-210.
Pringle C.E., Hudson A.J., Munoz D.G., et al. Primary lateral sclerosis. Clinical features, neuropathology and diagnostic criteria. Brain. 1992;115:495-520.
Puccioni-Sohler M., Rios M., Carvalho S.M.F., et al. Diagnosis of HAM/TSP based on CSF proviral HTLV-I DNA and HTLV-I antibody index. Neurology. 2001;57:725-727.
Puls I., Oh S.J., Sumner C.J., et al. Distal spinal and bulbar muscular atrophy caused by dynactin mutation. Ann Neurol. 2005;57(5):687-694.
Raben N., Danon M., Lu N., et al. Surprises of genetic engineering. A possible model of polyglucosan body disease. Neurology. 2001;56:1739-1745.
Rosen D.R., Siddique T., Patterson D., et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59-62.
Rothstein J.D., Van Kammen M., Levey A.I., et al. Selective loss of glial glutamate transporter GLT1 in amyotrophic lateral sclerosis. Ann Neurol. 1995;38:73-84.
Rowland L.P. The causes of sporadic amyotrophic lateral sclerosis. In: Mitsumoto H., Przedborski S., Gordon P.H. Amyotrophic Lateral Sclerosis. New York: Taylor & Francis Group; 2006:81-98.
Rudnicki S.A., Dalmau J. Paraneoplastic syndromes of the spinal cord, nerve, and muscle. Muscle Nerve. 2000;23:1800-1818.
Russman B.S. Spinal muscular atrophy: clinical classification and disease heterogeneity. J Child Neurol. 2007;22:946-951.
Saito M. Immunogenetics and the pathological mechanisms of human T-cell leukemia type 1 (HTLV-1-) associated myelopathy/tropical spastic paraparesis (HAM/TSP). Interdiscip Perspect Infect Dis. 2010;10:478461.
Salinas S., Proukakis C., Crosby, et al. Hereditary spastic paraplegia: clinical features and pathogenic mechanisms. Lancet Neurol. 2008;7:1127-1138.
Sasabe J., Chiba T., Yamada M., et al. d-Serine is a key determinant of glutamate toxicity in amyotrophic lateral sclerosis. EMBO J. 2007;26:4149-4159.
Schmahmann J.D., Sherman J.C. The cerebellar cognitive affective syndrome. Brain. 1998;121:561-579.
Schmidt T., Lindenberg K.S., Krebs A., et al. Protein surveillance machinery in brains with spinocerebellar ataxia type 3: redistribution and differential recruitment of 26S proteasome subunits and chaperones to neuronal intranuclear inclusions. Ann Neurol. 2002;51:302-310.
Shi P., Wei Y., Zhang J., et al. Mitochondrial dysfunction is a converging point of multiple pathological pathways in amyotrophic lateral sclerosis. J Alzheimers Dis. 2010;20:S311-S324.
Siddique T., Dellefave L. Familial ALS and genetic approaches to ALS. In: Mitsumoto H., Przeborski S., Gordon P.H. Amyotrophic Lateral Sclerosis. New York: Taylor & Francis Group; 2006:141-166.
Silva E.A., Otsuki K., Leite A.C., et al. HTLV-II infection associated with a chronic neurodegenerative disease: clinical and molecular analysis. J Med Virol. 2002;66(2):253-257.
Snyder L.R., Marler T.E. Rethinking cycad metabolism research. Comm Integr Biol. 2011;4:86-88.
Spencer P.S., Palmer V.S., Ludolph A.C. On the decline and etiology of high incidence motor system disorder in West Papua (southwest New Guinea). Mov Disord. 2005;20:S119-S126.
Sperfeld A-D., Baumgartner A., Kassubek J. Magnetic resonance investigation of the upper spinal cord in pure and complicated hereditary spastic paraparesis. Eur Neurol. 2005;54:181-185.
Spreux-Varoquaux O., Bensimon G., Lacomblez L., et al. Glutamate levels in cerebrospinal fluid in amyotrophic lateral sclerosis; a reappraisal using a new HPLC method with coulometric detection in a large cohort of patients. J Neurol Sci. 2002;193:73-78.
Strong M.J. The evidence for altered RNA metabolism in amyotrophic lateral sclerosis (ALS). J Neurol Sci. 2010;288:1-12.
Strong M.J., McClean J., Julien J-P. Cytoskeletal proteins in the pathogenesis of ALS. In: Mitsumoto H., Przedborski S., Gordon P.H. Amyotrophic Lateral Sclerosis. New York: Taylor & Francis Group; 2006:395-416.
Tang Y., Anne Hapip C., Liu B., et al. Highly sensitive TaqMan RT-PCR assay for detection and quantification of both lineages of West Nile virus RNA. J Clin Virol. 2006;36(3):177-182.
Turner R.T., Kiernan M.C., Leigh P.N., et al. Biomarkers in amyotrophic lateral sclerosis. Lancet Neurol. 2009;8:94-109.
Tyler K.L., Pape J., Goody R.J., et al. CSF findings in 250 patients with serologically confirmed West Nile virus meningitis and encephalitis. Neurology. 2006;66:361-365.
Van Deerlin V.M., Leverenz J.B., Bekris L.M. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology; a genetic and histopathological analysis. Lancet Neurol. 2008;7:409-416.
Van den Berg-Vos R.M., Franssen H., Wokke J.H., et al. Multifocal motor neuropathy: diagnostic criteria that predict the response to immunoglobulin treatment. Ann Neurol. 2000;48:919-926.
van Swieten J.C., Heurtink P. Mutations in progranulin (GRN) within the spectrum of clinical and pathological phenotypes of frontotemporal dementia. Lancet Neurol. 2008;10:965-974.
Vance C., Al-Chalabi A., Ruddy D., et al. Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2-21.3. Brain. 2006;129(4):868-876.
Visser J., van den Berg-Vos R.M., Franssen H., et al. Mimic syndromes in sporadic cases of progressive spinal muscular atrophy. Neurology. 2002;58:1593-1596.
Wijesekera L.C., Mathers S., Talman P. Natural history and the clinical features of the flail arm and flail leg ALS variants. Neurology. 2009;72(12):1087-1094.
Wohlfart G. Collateral regeneration in partially denervated muscles. Neurology. 1958;8:175-180.
Yiangou Y., Facer P., Durrenberger P., et al. COX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol. 2006;6:12.
Zhao Z-H., Chen W-Z., Wu Z-Y., et al. A novel mutation in senataxin gene identified in a Chinese patient with sporadic amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2009;10:118-122.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 74 Disorders of Upper and Lower Motor Neurons
Disorders of Upper Motor Neurons
Neuroanatomy of Upper Motor Neurons
The UMN is a motor neuron, the cell body of which lies within the motor cortex of the cerebrum, and the axon of which forms the corticobulbar and corticospinal tracts. The LMNs, lying in the brainstem motor nuclei and the anterior horns of the spinal cord, directly innervate skeletal muscles. The UMNs are rostral to the LMNs and exert direct or indirect supranuclear control over the LMNs (Box 74.1).
Box 74.1 Upper Motor Neurons and Their Descending Tracts
Motor Cortex
Limbic Motor Control
The limbic system is involved in emotional experience and expression and associated with a variety of autonomic, visceral, and endocrine functions. It strongly influences the somatic motor neurons. The emotional status and experience of an individual determines overall spinal cord activity, and the limbic motor system also influences respiration, vomiting, swallowing, chewing, and licking (at least in animal studies). Furthermore, the generation of signs of pseudobulbar hyperemotionality (pseudobulbar affect, emotional incontinence) in ALS is closely related to an abnormal limbic motor control, particularly in the periaqueductal gray and nucleus retroambiguus. The latter nuclei project to the somatic motor neurons that innervate pharyngeal, soft palatal, intercostal, diaphragmatic, abdominal, and probably laryngeal muscles. Pseudobulbar hyperemotionality symptoms may appear when UMN control over these motor nuclei is impaired, and thus limbic motor control is disinhibited. There appears to be some degree of emotional regulation by the cerebellum. The “cerebellar cognitive affective syndrome” can arise when stroke, tumor, or infection interrupts connections between the cerebellum and cerebral association and paralimbic regions (Schmahmann and Sherman, 1998).
Signs and Symptoms of Upper Motor Neuron Involvement
Loss of Dexterity
Loss of dexterity is one of the most characteristic signs of UMN impairment. Voluntary skillful movements require the integrated activation of many interneuron circuits in the spinal cord; such integration is ultimately controlled by the corticospinal tract and thus by UMNs. Loss of dexterity may express itself as stiffness, slowness, and clumsiness in performing any skillful motor actions. Asking the patient to perform rapid repetitive motions such as foot or finger tapping assesses loss of dexterity at the bedside. It is useful to assess both sides of the body, as many motor neuron disorders are asymmetrical (Box 74.2).
Box 74.2 Signs and Symptoms of Upper Motor Neuron Involvement
Loss of muscle strength (mild weakness)
Pathological reflexes (Babinski, Hoffmann sign, loss of abdominal reflexes)
Increased reflexes in an atrophic limb (probable upper motor neuron sign)
Pseudobulbar (spastic bulbar) palsy (emotional lability, brisk jaw jerk, hyperactive gag, forced yawning, snout reflex, suck reflex, slow tongue movements, spastic dysarthria)
Laboratory Evidence of Upper Motor Neuron Involvement
Neuroimaging
The use of brain magnetic resonance imaging (MRI) in ALS is largely to exclude other conditions but sometimes shows abnormal signal intensity in the corticospinal and corticobulbar tracts as they descend from the motor strip via the internal capsules to the cerebral peduncles. In ALS, signal changes, best appreciated on proton density images of the internal capsules, probably represent wallerian degeneration; similar changes also appear on conventional T2-weighted and fluid-attenuated inversion recovery (FLAIR) sequences. However, these changes do not appear to be sufficiently sensitive, and efforts continue to evaluate other potential MRI techniques such as diffusion tensor and volumetric MRI, which may serve as markers of UMN disease (Agosta et al., 2010).
Magnetic Resonance Spectroscopy Imaging
Proton density magnetic resonance spectroscopy (1H-MRS) is a noninvasive nuclear magnetic resonance technique that combines the advantages of MRI with in vivo biochemical information. A significant reduction of N-acetylaspartate, a neuronal marker, relative to creatine or choline (used as internal standards) exists in the sensorimotor cortices of patients with ALS who have UMN signs. Alterations in the measured levels of these metabolites using 1H-MRS are useful in the detection of UMN dysfunction early in the evolution of ALS and are useful to monitor progression over time. MRS still requires further technological improvements before it comes into widespread use (Mitsumoto et al., 2007).
Primary Lateral Sclerosis
Primary lateral sclerosis (PLS), first described by Erb in 1875, is a rare UMN disease variant that accounts for 2% to 4% of all cases of ALS and is traditionally distinguished by a lack of LMN involvement. The Pringle criteria for PLS stipulated that disease be restricted to the UMN system for at least 3 years from the time of clinical onset (Pringle et al., 1992), but a figure of 4 years is now proposed during which there is neither clinical nor neurophysiological evidence of LMN involvement. In a recent study comparing the evolution of disease in PLS versus UMN-predominant ALS and typical ALS, the median time to development of electromyographic (EMG) LMN features after onset in those with an evolving ALS was 3.17 years; in those patients, clinical signs of LMN disease occurred on average about 6 months later. Nonetheless, later development of LMN signs may occur and require reclassification as ALS in some cases, which therefore necessitates constant longitudinal review of each case (Gordon et al., 2009).
PLS typically presents in patients in their early 50s (about a decade younger than typical MND/ALS patients) as a very slowly evolving spastic paraparesis that spreads to the upper limbs and eventually causes pseudobulbar palsy. In rare instances, onset is in the bulbar system or follows a slowly ascending or descending hemiplegic pattern (Mills hemiplegic variant), but a bulbar-onset presentation should make the clinician wary of later LMN signs elsewhere. Other features include cramps and fasciculations, but such complaints are neither prominent nor universal. Bladder dysfunction is rare and, if it occurs at all, tends to be a late feature. Although muscle weakness is present, the main deficits are due to spasticity in dexterity and gait. The rate of progression can be exceedingly slow, often progressing over many years to the point where the patient manifests a robotic gait, debilitating generalized spasticity, and prominent pseudobulbar palsy. Muscle atrophy, if it occurs at all, is a very late feature. No clinically detectable sensory changes occur. Neuropsychological test batteries may define subtle cognitive deficits due to frontal cortical involvement, but dementia is not a prominent feature. A few patients may exhibit abnormal voluntary eye movements. Breathing is usually unimpaired in PLS, and as a consequence, forced vital capacity (FVC) is not affected (Gordon et al., 2009).
The prognosis is significantly better than for MND/ALS: one series had a median disease duration of 19 years and another series exhibited a range of survival from 72 to 491 months (Murray, B., 2006). The underlying pathogenesis of PLS remains undefined. Pathological changes include a striking loss of Betz cells in layer 5 of the frontal and prefrontal motor cortex (and other smaller pyramidal cells) together with laminar gliosis of layers 3 and 5 and degeneration of the corticospinal tracts. Spinal anterior horn cells are characteristically unaffected.
Diagnosis
The diagnosis of PLS is essentially one of exclusion (Table 74.1). Rare reports exist of UMN-onset ALS exist where the interval between onset of UMN signs and subsequent LMN signs have been up to 27 years. As such, it is vital to reassess patients diagnosed with PLS, as late signs of LMN involvement may occur that would reclassify their disorder as UMN-onset ALS.
Table 74.1 Disorders of Upper Motor Neurons and Their Key Characteristics
| Disorders | Key Characteristics |
|---|---|
| Primary lateral sclerosis | A diagnosis of exclusion |
| Hereditary spastic paraplegia | Heredity, usually autosomal dominant, spastin gene mutation, other mutations (see text), “sporadic” |
| HTLV-1-associated myelopathy | Slowly progressive myelopathy, endemic, and positive HTLV-1 |
| HTLV-2-associated myelopathy | Amerindian, IV drug abuser, concomitant HIV |
| Adrenomyeloneuropathy | X-linked recessive inheritance, adrenal dysfunction, myelopathy, very long-chain fatty acid assay |
| Lathyrism | History of consumption of chickling peas |
| Konzo | Eastern African, cassava root consumption |
HIV, Human immunodeficiency virus; HTLV, human T-lymphotropic virus; IV, intravenous.
Appropriate testing must exclude all definable causes for generalized UMN involvement. These include structural abnormalities (Chiari malformation and intrinsic and extrinsic spinal cord lesions) and myelopathies such as multiple sclerosis (MS) spondylotic cervical myelopathy, human immunodeficiency virus (HIV) myelopathy, human T-lymphotropic virus type 1 (HTLV-1) myelopathy, Lyme disease, syphilis, or adrenomyeloneuropathy. Spondylotic cervical myelopathy and MS are probably the most common causes among these disorders. The family history must be negative to rule out hereditary spastic paraplegia (HSP)/familial spastic paraparesis, spinocerebellar ataxia (SCA), hexosaminidase-A (Hex-A) deficiency, familial ALS (FALS), or adrenomyeloneuropathy. It is now apparent that some spastin mutation–associated HSP may lack a family history; it is worthwhile to carry out this gene test in patients presenting with symptoms and signs that are restricted to the lower extremities (Brugman et al., 2009). Paraneoplastic syndromes (especially in association with breast cancer) and Sjögren syndrome may clinically resemble PLS. Understanding of these entities is poor.
Treatment
No specific pharmacotherapy is available, and treatment therefore focuses on symptom control and supportive care. However, antispasticity drugs such as the GABA-B agonist, baclofen, and the central α2-agonist, tizanidine, may be tried for symptomatic treatment. Severe spasticity sometimes requires the insertion of an intrathecal baclofen pump. Tricyclic antidepressants, selective serotonin reuptake inhibitors, or dextromethorphan/quinidine may control pseudobulbar affect lability (Brooks et al., 2005).
Hereditary Spastic Paraplegia
HSP (or familial spastic paraparesis) is a genetically and clinically heterogeneous group of disorders rather than a single entity. The clinical feature common to all cases is progressively worsening spasticity of the lower extremities, often with variable degrees of weakness. The characteristic pathology is retrograde degeneration of the longest nerve fibers in the corticospinal tracts and posterior columns. Its estimated prevalence is 0.5 to 11.9 in 100,000, but its worldwide prevalence may actually be underestimated because of the benign nature of the disease in many families. Although the most common mode of inheritance is autosomal dominant, it may also be inherited in a recessive or X-linked fashion, and 12% to 13% of cases with apparently sporadic spastic paraparesis have spastin mutations (Depienne et al., 2006).
Genetic linkage studies of families around the world have mapped loci to over 40 autosomes as well as the X chromosome, with 17 distinct genes identified to date (Salinas et al., 2008). Inheritance of most pure HSP is autosomal dominant, whereas complicated forms are more often autosomal recessive. Between 40% and 45% of all families link to the SPAST gene on chromosome 2p22-21, which encodes spastin, a 616-amino acid protein. Mutations of various types (missense, nonsense, frameshift, splice site) may affect this gene (McDermott, C.J., 2006). Spastin is a highly conserved member of the AAA family of proteins (adenosine triphosphatase [ATPase] associated with various cellular activities). The exact role of mutant spastin in the pathogenesis of HSP is undefined, although a disturbance in maintenance of the microtubule cytoskeleton may exists, thus disrupting axonal transport. More than half of all cases do not manifest symptoms and signs until after age 30 years. Although this is normally a pure HSP, complicated forms occur, and some cases can develop a late-onset cognitive decline. Pathologically, degeneration of the longest corticospinal tracts and, to a lesser degree, the posterior columns of the spinal cord is seen.
Mutations in the SPG3A gene on 14q11-q21 encoding the novel protein, atlastin, give rise to an autosomal dominant often early-onset (<10 years of age) pure HSP which accounts for about 10% of autosomal dominant cases. This protein shares structural homology to guanylate-binding protein 1, which is a member of the dynamin family. Dynamins are important in intracellular trafficking of various kinds of vesicles. Mutations in KIF5A (SPG10, Chr 12q), a kinesin motor domain that is critical in intracellular transport, can cause both early- and late-onset spastic paraparesis with distal amyotrophy (Blair et al., 2006). Spastic paraplegia 11 (SPG11) is an autosomal recessive complicated HSP (thin corpus callosum, neuropathy, cognitive impairment) due to mutations in the spatacsin gene on chromosome 15q. This protein is of unknown function and does not appear to interact with the Golgi apparatus or microtubules. The cause of autosomal dominant pure HSP, linked to 2q24-34, is a mutation in the SPG13 gene, which encodes a mitochondrial heat shock protein. Recessively inherited complicated HSP links to chromosome 16q and is caused by a mutation in a gene encoding a mitochondrial protein known as paraplegin; this disorder can be ether pure or complicated (cerebellar signs, pale optic discs, and peripheral neuropathy). The genes for two different X-linked complicated HSP have been identified. In the first, mutant L1 (neural) cell adhesion molecule (L1CAM) may disrupt neuronal migration or differentiation; in the second mutant proteolipid protein (PLP1) is found in association with changes in white matter (duplication mutations in this same gene can also cause Pelizaeus-Merzbacher disease). Spastic paraplegia 17 (SPG17) is caused by mutations in the seipin gene on chromosome 11q12-q14. Also known as Silver syndrome, this disorder is an autosomal dominant complicated form of HSP with distal hand and foot amyotrophy beginning in the late teens to early 30s. Mutations in this gene are also the cause of a form of distal hereditary neuropathy (Charcot-Marie-Tooth [CMT] disease type 5).
Diagnosis
The basis for diagnosis of HSP is evidence of a family history in the setting of progressive gait disturbance, evidence of lower-extremity spasticity, and sparing of craniobulbar function. However, difficulties arise when there is no clear family history in recessive or X-linked disease and in cases of sporadic spastin mutation–associated HSP. Furthermore, considerable variation in disease expression exists between and within HSP families. In the absence of a family history or a demonstration of a known mutation, it is important to consider alternative causes for the clinical presentation, including structural disease (e.g., cerebral palsy, hydrocephalus, myelopathy), degenerative/infiltrative/inflammatory disease (e.g., MS, ALS, SCA, leukodystrophy), infections (syphilis, HIV, HTLV), levodopa-responsive dystonia, metabolic/toxic damage (vitamin B12 deficiency [subacute combined degeneration of the spinal cord (SCDC)], vitamin E deficiency, copper deficiency, lathyrism), and paraneoplastic disorders. MRI may reveal that cervical and thoracic spinal cord diameters are significantly smaller in both pure and complicated HSP than in controls (Sperfeld et al., 2005). Perhaps the most important differential diagnosis is that between apparently sporadic pure HSP and PLS, especially as the later may present with a slowly evolving spastic paraparesis for many years prior to the development of upper limb or bulbar features. The only reliable way to distinguish such disorders is through genetic testing; age at onset, urgency of micturition, and signs of dorsal column involvement (clinical or abnormal somatosensory evoked potentials [SSEPs]) are not accurate indicators of HSP versus PLS (Brugman et al., 2009).
Human T-Lymphotropic Virus Type 1–Associated Myelopathy, or Tropical Spastic Paraparesis
HTLV-1 causes a chronic progressive myelopathy that is referred to as tropical spastic paraparesis (TSP) in the Caribbean or HTLV-1–associated myelopathy (HAM) in Japan. This retrovirus is endemic in the Caribbean area, southwestern Japan, equatorial Africa, South Africa, parts of Asia, Central America, and South America, where it infects between 10 and 20 million people. Transmission occurs though sexual contact, intravenous (IV) drug use and also through breastfeeding. While between 2% and 3% of those infected can develop adult-onset T-cell leukemia, an estimated 2.5% to 3.8% can develop a chronic inflammatory myelopathy, with up to 20/100,000 affected in the Caribbean population and 3/100,000 in Japan. Recent evidence implicates high levels of activated HTLV-1–specific helper T cells and cytotoxic T cells in the pathogenesis of this syndrome; these immune cells appear to activate in response to interactions with retroviral env and tax proteins with greatest activity within the thoracic cord. Increased susceptibility for neurological disease appears to depend on both viral and host factors, with differences in certain HTLV-1 subgroups, proviral load, and HLA background being important. This may also explain differences in susceptibility between ethnic populations (Saito, 2010). Mode of transmission is through contaminated blood, sexual activity, breastfeeding, and very rarely in utero.
HAM/TSP is a chronic, insidiously progressive myelopathy that typically begins after age 30 years (but can occur as early as the first decade). In addition to slowly progressive spastic paraparesis, patients complain of lower-extremity paresthesias, a painful sensory neuropathy, and bladder dysfunction, and some patients may also develop optic neuropathy. Examination reveals UMN signs in the legs (weakness, spasticity, pathological reflexes, hyperreflexia), although reflexes may also be brisk in the arms. Overall, evidence of LMN involvement may be scant, and objective sensory findings may be difficult to detect. MRI may reveal increased signal on T2-weighted sequences in periventricular white matter and atrophy of the thoracic cord, but these findings may not be specific to HTLV-1. The definitive diagnosis of HAM/TSP requires HTLV-1–positive serology in blood and cerebrospinal fluid (CSF). To be sensitive and specific, CSF should reveal a combination of a polymerase chain reaction amplification of HTLV-1 deoxyribonucleic acid (DNA), together with evidence of an increased HTLV-1–specific antibody index and oligoclonal bands (Puccioni-Sohler et al., 2001). At present, no antiviral agents effectively treat HAM/TSP, but a case report showed partial benefit of plasmapheresis (Narakawa et al., 2001). As more is learned about the molecular etiology of HAM/TSP, future therapies will likely target the pathogenic effect of HTLV-1–reactive T cells.
Human T-Lymphotropic Virus Type 2–Associated Myelopathy
Though phylogenetically similar in many respects, HTLV-1 and HTLV-2 are still antigenically distinct. Nonetheless, using enzyme-linked immunosorbent assay (ELISA) and Western blot techniques, many laboratories worldwide often report the presence of sero-indeterminate HTLV-1/2. It has long been thought that myelopathy in such sero-indeterminate cases is due to HTLV-1 rather than HTLV-2, but rare cases are now being described of a syndrome characterized by spastic paraparesis, diffuse hyperreflexia, spastic bladder, and periventricular white matter changes on MRI in patients infected with HTLV-2 but not HTLV-1. This retrovirus is endemic in some Native American tribes and now often encountered worldwide among IV drug abusers. It is worthwhile to test CSF and serum for the presence of this virus in known IV drug abusers who present with a spastic paraparesis (Silva et al., 2002). However, co-infection with HIV-1 is a confounding factor in many cases of presumed HTLV-2–associated neurological disease. It has been suggested that such co-infection, rather than infection with HTLV-2 alone, increases the likelihood of neurological manifestations (Araujo and Hall, 2004; Posada-Vergara et al., 2006).
Adrenomyeloneuropathy
Adrenomyeloneuropathy is a variant of adrenoleukodystrophy, an X-linked recessive disorder caused by mutations in the ABCD1 gene on chromosome Xq28 that encodes a ubiquitously expressed integral membrane peroxisomal ATPase-binding cassette transporter protein. Mutations in this gene lead to abnormal peroxisomal β-oxidation, which results in the harmful accumulation of very long-chain fatty acids (VLCFAs) in affected cells. Excessive levels of VLCFAs may interfere with the membrane components of both neurons and axons. The most common phenotype, adrenoleukodystrophy, is an inflammatory disorder of brain and spinal cord that affects young boys 4 to 8 years of age, who develop severe adrenal insufficiency, progressive cognitive deterioration, seizures, blindness, deafness, and spastic quadriparesis. Adrenomyeloneuropathy is a noninflammatory axonopathy of the spinal cord that involves descending corticospinal tracts in the thoracic and lumbosacral regions and the ascending posterior columns in the cervical region. The characteristic clinical picture is a slowly progressive spastic paraparesis and mild polyneuropathy in adult men (in their late 20s), with or without sensory symptoms and sphincter disturbances. Adrenal insufficiency may be present and may predate onset of neurological symptoms by several years. Adult female carriers may present with slowly progressive spastic paraparesis. Approximately 20% of men with adrenomyeloneuropathy also develop cerebral changes on MRI that may accompany cognitive/language/behavioral deterioration. Rare cases may present as a spinocerebellar degeneration. Considerable phenotypic variation exists even within individual families. Female carriers may manifest more subtle symptoms such as cramps, back pain, or arthralgias. The diagnosis should be suspected in male cases with progressive sensorimotor deficits in the legs and a family history of a myelopathy (including supposed MS). Progressive sensorimotor deficits in the lower extremities with a history of memory loss or “attention deficit disorder” should also prompt testing for adrenomyeloneuropathy, as should a history of idiopathic childhood epilepsy or primary adrenal failure (Mukherjee et al., 2006). Sural nerve biopsies show loss of both myelinated and unmyelinated axons, with some degree of onion bulb formation. Ultrastructural examination may show characteristic inclusions (empty lipid clefts) in Schwann cell cytoplasm. Nerve conduction studies and needle electrode examination may reveal a predominantly axon-loss type of sensorimotor polyneuropathy with a lesser component of demyelination, and SSEPs may show reduced or absent responses. The diagnostic test of choice is to demonstrate increased VLCFA levels in plasma, red blood cells, or cultured skin fibroblasts. No specific therapy exists for adult-onset adrenomyeloneuropathy.
Plant Excitotoxins
Lathyrism
Lathyrism is a chronic toxic nutritional neurological disease caused by long-term (or subacute) ingestion of flour made from the drought-resistant chickling pea (Lathyrus sativus). It is an important example of a disease in which a natural excitotoxin causes selective UMN impairment. The responsible neurotoxin is β-N-oxalylamino-l-alanine (BOAA), an α-amino-3-hydroxyl-5-methyl-4-isoxazolepropionic acid (AMPA) glutamate receptor agonist. Ingestion of this neurotoxin results in increased intracellular levels of reactive oxygen species and subsequent impairment of the mitochondrial oxidative phosphorylation chain. Degeneration is most prominent in those Betz cells of the motor cortex (and the longest corresponding pyramidal tracts) that subserve lower-extremity function. Lathyrism occurs in the indigenous populations of Bangladesh, China, Ethiopia, India, Romania, and Spain. It also occurred in regional concentration camps during World War II. The condition may occur in epidemic form when malnourished populations increase consumption of flour made from L. sativus chickling peas during times of food shortage due to droughts. An analysis of an epidemic of neurolathyrism in Ethiopia showed a higher incidence in boys aged 10 to 14 years. The increased risk was associated with cooking grass pea foods in traditional clay pots (Getahun et al., 2002). The onset of clinical toxicity is either acute or chronic, manifesting as muscle spasms, cramps, and leg weakness. In addition to spastic paraparesis, sensory (including leg formications) and bladder dysfunction may occur. Occasionally there is a coarse tremor of the upper extremities. Although irreversible, the disorder is not progressive (unless there is continuing intoxication), and lifespan is not affected.
Disorders of Lower Motor Neurons
Clinical Features of Lower Motor Neuron Involvement
Loss of Muscle Strength (Weakness)
In a disease causing chronic motor unit depletion, neighboring axons belonging to healthy motor neurons may reinnervate denervated muscle fibers belonging to a diseased motor unit by collateral sprouting. In this way, existing motor units continually modify in the face of persistent losses of motor axons to maintain muscle strength. For example, in patients who have recovered from acute poliomyelitis, depletion of more than 50% of LMNs occurs before residual muscle weakness is clinically detectable. Healthy individuals have sufficient motor units available to offset an unexpected loss of motor neurons (Box 74.3).
Fasciculations
Fasciculations are spontaneous contractions of muscle fibers belonging to a single (or part of a) motor unit. (A video of this disorder can be found at www.expertconsult.com) Clinically, fasciculations appear on the muscle surface as fine, rapid, flickering, and sometimes vermicular contractions that occur irregularly in time and location. The impulse for the fasciculation appears to arise from hyperexcitable motor axons anywhere in their course. Fasciculations can occur both in healthy individuals and in patients with LMN involvement, so fasciculations themselves do not indicate LMN disease.
Laboratory Evidence of Lower Motor Neuron Involvement
Electrodiagnostic Examination
The electrodiagnostic examination (EDX) consists of nerve conduction studies and needle electrode examination (see Chapter 32B). The loss of motor units reflects in the loss of the amplitude of the maximal compound muscle action potential (CMAP). In a primarily demyelinating process, conduction velocity slows, and in severe cases, block. In a primarily axon loss process, there is usually only a modest degree of conduction velocity slowing commensurate with dropout of large myelinated axons. Sensory nerve conduction studies are normal in pure LMN disorders.
Acute Poliomyelitis
Clinical Features
After a brief 3- to 6-day incubation period, a viremia occurs, during which approximately 90% of individuals remain asymptomatic. Most of the remaining individuals develop an acute flulike illness with cough, malaise, diarrhea, myalgia, headache, and fever. This self-limited “abortive” polio usually lasts 2 to 3 days, and patients do not progress to develop acute muscle weakness. Between 2% and 3% of acutely infected patients develop aseptic meningitis characterized by severe headache due to meningeal irritation. This is typically self-limited and resolves within 7 to 14 days. Less than 1% of infected patients who ingest poliovirus develop the acute paralytic syndrome, characterized by localized fasciculations, severe myalgia, hyperesthesias, usually fulminant focal and asymmetrical paralysis, and fever. Any skeletal muscle can weaken, including bulbar muscles and muscles of respiration, but the leg muscles are the most commonly affected (Howard, 2005).
Laboratory Features
Motor nerve conduction studies performed 21 or more days after the onset (see Chapter 32B) may reveal low-amplitude maximum CMAPs. No evidence of significant demyelination-related motor conduction slowing or block exists. Sensory nerve action potentials (SNAPs) are normal. EMG examination in the acute phase shows profuse axon loss in the form of positive sharp waves and fibrillation potentials. In addition, fasciculations may be prominent. As motor axon loss progresses, evidence of neurogenic motor unit potential changes may be detected. The CSF typically shows increased protein content with normal glucose and a pleocytosis, with polymorphonuclear cells predominating during the acute stages and lymphocytes predominating later in the disease. Identification of CSF poliovirus-specific immunoglobulin M (IgM) antibody allows a specific diagnosis. Stool or nasopharyngeal cultures are positive for poliovirus in nearly 90% of patients by the 10th day of illness. The diagnosis may also be established by documenting a fourfold or greater increase in serum antibody titer against poliovirus from the acute as compared to the convalescent phase. Polymerase chain reaction (PCR) is now the best technique to diagnose poliovirus subtype as well as determine if the illness is related to wild-type versus Sabin oral polio vaccine–induced disease.
Vaccination
Immunocompromised individuals and their unimmunized direct household contacts are at particular risk for this rare complication. Consequently, in Northern America, Japan, Europe, New Zealand, and Australia, where wild viral infection is almost completely eradicated, policy shifted to use of the Salk inactivated vaccine in immunization schedules (Alexander et al., 2004).
Postpolio Syndrome/Progressive Postpoliomyelitis Muscular Atrophy
In the United States alone, it is estimated that 250,000 to 640,000 people survived acute paralytic poliomyelitis; the last epidemic was in 1952. Many years after recovery from acute poliomyelitis, some patients experience progressive functional impairment, with muscle fatigue, pain, sleep disturbances, cold intolerance, depression, dysphagia, and dysarthria, called the post-polio syndrome. If progressive muscle weakness and wasting occurs in this setting, the term progressive postpoliomyelitis muscular atrophy (PPMA) is used. The reported incidence of postpolio syndrome/PPMA among polio survivors ranges from 28.5% to 64%; no accurate estimate of the incidence exists. By definition, patients with postpolio syndrome/PPMA have recovered from acute poliomyelitis, and the disease course has been stable for at least 10 years after the recovery (Box 74.4).
Box 74.4 Characteristic Features of Postpolio Syndrome/Progressive Postpoliomyelitis Muscular Atrophy
EMG, Electromyography; PPMA, postpoliomyelitis muscular atrophy.
Treatment
No specific pharmacotherapy for postpolio syndrome exists. Care focuses on symptom relief (Gonzalez et al., 2010). A randomized controlled trial of intravenous immunoglobulin (IVIG; 2 courses of IVIG at a dose of 90 g per course over 3 days, with a 3-month interval) reported a significant improvement in muscle strength but not quality of life in 135 patients (Gonzalez et al., 2006). A need for further trials in larger patient groups exists.
West Nile Virus
West Nile virus (WNV) is an arthropod-borne flavivirus that cause epidemics of meningitis, encephalitis, and in some instances an acute polio-like flaccid paralysis. Approximately 80% of those who are infected are asymptomatic, and 20% develop a flulike illness (termed West Nile fever). Less than 1% present with neuroinvasive disease. Following the 1999 outbreak in New York, an epidemic spread across the North American continent and peaked in 2002/2003. Between 1999 and 2008, almost 29,000 confirmed and probable cases of WNV infection were received in the United States, and over 40% of cases were of the neuroinvasive type. The highest incidence of neuroinvasive WNV occurred in the western north-central United States and mountain states between the months of July and September (Lindsey et al., 2010). Epidemics also occurred in Israel, Italy, Russia, Romania, Hungary, Tunisia, the Sudan, the Caribbean, and Latin America. WNV is now endemic in North America and is the leading cause of arboviral encephalitis in the United States. WNV is a zoonotic pathogen that has crossed over from birds to humans, the latter serving as incidental hosts. The prime bridge vector is the Culex mosquito, and the spread of disease appears to have followed the migration patterns of bird populations (Kilpatrick et al., 2006). Human-to-human transmission can occur via blood transfusion, organ transplant, intrauterine exposure, and breastfeeding, hence the need to screen for the virus amongst blood donors. One of the most dramatic presentations is an acute asymmetrical flaccid paralysis in a febrile patient with or without meningitis, encephalitis, and cranial neuropathies (including hearing loss). Aching pains in affected limbs often accompany the paralysis, but actual sensory loss is not a feature. Respiratory failure and death may occur, and up to one-third of cases suffer bladder and bowel dysfunction. Recovery is very slow, and from the available evidence, incomplete. Other presentations include a GBS, multifocal chorioretinitis, pancreatitis, hepatitis, myocarditis, nephritis, and splenomegaly. Risk factors for death from WNV infection include chronic renal disease, an immunosuppressed state (e.g., in organ transplant recipients), the presence of encephalitis (versus meningitis) and old age (Murray, K., et al., 2006; Tyler et al., 2006). Detection of WNV-specific IgM antibody in the serum of a patient with abnormal CSF and acute neurological illness confirms the diagnosis. The diagnosis can also be confirmed by WNV-specific IgM antibody capture ELISA in CSF. PCR has also been developed to detect the virus (Tang et al., 2006). CSF protein is typically elevated (100 mg/dL or more and often higher in encephalitis versus meningitis), glucose is typically normal, and there are increased numbers of white cells (mean of about 220/mm3 with ≈ 45% neutrophils). The number of red cells is variable and may be higher in encephalitis (Tyler et al., 2006). Neuroimaging of the brain is usually normal, but that of the spinal cord may reveal increased signal in the anterior horns. Electrodiagnostic studies reveal an acute disorder of anterior horn cells, with acute loss of motor units in affected myotomes.
Multifocal Motor Neuropathy
A complete description of multifocal motor neuropathy is in Chapter 76. The condition is believed to be autoimmune in nature, and most cases have evidence of focal demyelination in the peripheral nerves (multifocal motor neuropathy with conduction block [MMNCB]) similar to that in chronic inflammatory demyelinating peripheral neuropathy. The clinical presentation, however, is with pure LMN involvement. The condition enters into the differential diagnosis of benign focal amyotrophy and the progressive muscular atrophy variant of ALS. It is important to search for this condition, since it is treatable by high-dose immunoglobulin infusions or other immunotherapy.
Benign Focal Amyotrophy
The terms benign focal amyotrophy, brachial monomelic amyotrophy, benign calf amyotrophy, Hirayama disease, or juvenile segmental muscular atrophy are used to describe disorders characterized by LMN disease clinically restricted to one limb. The etiology is unknown. Autopsy studies have shown the affected region of spinal cord flattened, the anterior horn markedly atrophied and gliotic, and a reduction in the numbers of both large and small motor neurons. Based upon neuroradiological studies, Hirayama, who established the disease entity, has proposed a mechanically induced limited form of ischemic cervical myelopathy, being the result of local compression of the dura and spinal cord against vertebrae during repeated neck flexion/extension, in turn due to disproportionate growth between the contents of the dural sac and the vertebral column (Hirayama, 2008; Hirayama and Tokamaru, 2000). However, surgical decompression has not altered the course of the disease, and this theory is no longer widely held. Another school of thought is that this is a segmental, perhaps genetically determined, SMA, but the actual cause is still unknown.
The disease usually begins in the late teens, but many cases can present in the fourth decade. More than 60% of patients are men. Although originally described in Indian and Japanese patients, the disorder is now recognizable around the world. The most common presentation is one of an idiopathic, slowly progressive, painless weakness and atrophy in one hand or forearm. The distribution of muscle weakness varies markedly from case to case, but a characteristic feature is that the condition remains limited to only a few myotomes in the affected limb. The most common pattern is unilateral atrophy of C7-T1 innervated muscles, with sparing of the brachioradialis (the “oblique atrophy” pattern). Muscle stretch reflexes are invariably hypoactive or absent in the muscles innervated by the involved cord segment but are normal elsewhere. UMN signs are not present, and if they are, one should consider the onset of ALS instead. Approximately 20% have hyperesthesia to pinprick and touch, usually located on the dorsum of the hand. The cranial nerves, pyramidal tracts, and the autonomic nervous system are normal. Weakness and atrophy may progress steadily for the initial 2 to 3 years, but most patients have stabilized within 5 years. The arm is the affected limb in approximately 75% of the patients and the leg in the remaining 25% (benign calf amyotrophy). Spread may occur to the contralateral limb in about 20% of cases (Gourie-Devi and Nalini, 2003), and rare patients later develop an ALS-like picture.
Spinal Muscular Atrophy
The SMA are a group of disorders caused by degeneration of anterior horn cells and, in some subtypes, of bulbar motor neurons. Almost all cases are genetically determined, with most being autosomal recessive due to homozygous deletions of the survival motor neuron (SMN) gene on chromosome 5. Traditionally, SMA is classified as one of the four types based on the age at onset: SMA type 1 (infantile SMA or Werdnig-Hoffmann syndrome), SMA type 2 (intermediate SMA), SMA type 3 (juvenile SMA or Kugelberg-Welander disease), and SMA type 4 (adult-onset SMA, pseudomyopathic SMA). A very severe prenatal form of SMA (type 0 SMA) can manifest prenatally with reduced fetal movements and respiratory distress at birth. It is also important to consider the maximum function that a child achieves in terms of sitting and walking; this is of prognostic significance. In the less severe forms of the disease, there can be periods where the child will improve or plateau, but long-term studies have demonstrated a net deterioration (Russman, 2007) (Table 74.2
[/not-level-membership-for-neurology-category]
