[level-membership-for-neurology-category]
Chapter 72 Disorders of the Cerebellum, Including the Degenerative Ataxias
Cerebellar ataxia can result from a variety of insults to the cerebellum and its connecting pathways. The cerebellum can be the seat of pathology in many well-recognized diseases of the central nervous system (CNS) and may be involved in such processes in isolation or in combination with other structures. Ataxia can either be the major feature of the disease or one of its various clinical signs. Progressive degeneration of the cerebellum and its connections resulting in ataxia can be due to a number of genetic abnormalities, classified as inherited ataxia. Lastly, the term sporadic ataxia or idiopathic ataxia is used for those diseases in which ataxia related to cerebellar degeneration occurs in the absence of a definite genetic or acquired etiology (Klockgether, 2010). In this chapter, we discuss some of the well-defined acquired causes of ataxia, summarize the current understanding of inherited ataxias, and address the issue of sporadic ataxia.
Acquired Ataxias
In many patients, progressive ataxia results from environmental insults and other well-recognized disorders involving the nervous system. In any patient presenting with cerebellar ataxia, such disorders as ischemic or hemorrhagic stroke involving the cerebellum, previous episodes of cerebral hypoxia, primary or metastatic tumors, and CNS demyelinating diseases such as multiple sclerosis can be diagnosed by appropriate history, imaging studies, and other investigations. Many of these diseases tend to have an acute or subacute evolution rather than the chronic course associated with degenerative ataxias. There are, however, diseases that can present with ataxia in which the major imaging abnormality may be an atrophic cerebellum akin to the finding in degenerative ataxias (Table 72.1). Some of these will be briefly discussed next.
| Entity | Diagnostic Process |
|---|---|
Hypothyroidism
Occasionally, patients with hypothyroidism develop a mild dysequilibrium and gait ataxia in conjunction with their systemic symptoms. This is correspondent with pathological changes in midline cerebellar structure (Barnard et al., 1971). Thyroid functions should be tested in patients with progressive ataxia, since replacement treatment may dramatically improve the symptoms (Jellinek and Kelly, 1960).
Toxic Causes
Alcohol
Alcohol remains the major exogenous agent that causes ataxia. A significant proportion of alcoholics have midline cerebellar degeneration at autopsy. Clinically this disease is characterized by a progressive trunk ataxia with gait disturbance of a cerebellar type; upper-limb ataxia, speech difficulties, or eye movement abnormalities occur only in a small proportion of patients. This may reflect the relative sparing of the cerebellar hemispheres. Imaging studies typically reveal vermian atrophy. Chronic alcoholism can also produce significant cerebellar atrophy in the absence of major clinical deficits (Hillbom et al., 1986). Gait ataxia can dramatically improve with abstinence for over a year (Diener et al., 1984).
Chemotherapeutic Agents
Some cancer chemotherapeutic agents are recognized to produce ataxia as a side effect. 5-Fluorouracil (5FU), a fluorinated pyrimidine that acts by incorporating into ribonucleic acid (RNA) and interfering with RNA function, has been used to treat breast and gastrointestinal cancer. Conventional doses of 5FU may cause cerebellar ataxia if there is an abnormality of pyrimidine metabolism in the form of dihydropyrimidine dehydrogenase deficiency. Higher doses of 5FU can also cause a pancerebebellar syndrome that has an acute to subacute evolution (Gottlieb and Luce, 1971).
When cytosine arabinoside (ara-C) is given in high doses (>3 g/m2 for 8-12 doses, as opposed to the conventional 100-200 mg/m2 for 5-7 days), a significant number of patients develop a cerebellar syndrome. Pathologically, this is characterized by loss of Purkinje cells, gliosis, loss of dentate neurons, and spongiform changes (Salinsky et al., 1983). Signs of cerebellar dysfunction mandate immediate cessation of high-dose cytosine arabinoside.
Heavy Metals
Organic mercury poisoning has occurred in epidemic form as a result of contamination from mercury-containing fungicides. Mercury appears to be particularly toxic to cerebellar granule cells and visual cortex and causes a syndrome that includes paresthesia, ataxia, and restricted visual fields. Lead toxicity causes encephalopathy that is especially common in children. It is usually associated with abdominal colic. However, cerebellar signs can be the prominent presentation. Successful chelation therapy may achieve complete neurological recovery (Mani and Chaudhary, 1998). Gait ataxia associated with other signs such as confusion and myoclonus has also been described with bismuth toxicity due to excessive intake of bismuth subsalicylate (Pepto-Bismol) (Gordon et al., 1995). This is reversible over several weeks or months when bismuth intake is stopped.
Anticonvulsants
Transient cerebellar signs associated with supratherapeutic levels of drugs have been seen with many anticonvulsants, especially phenytoin. More persistent ataxia and documented Purkinje cell loss has been primarily seen in epileptics treated with phenytoin for prolonged periods of time. Cerebellar atrophy occurs in phenytoin-treated patients but may not always be associated with overt ataxia. The pathogenesis of this syndrome remains unclear. The various hypotheses include a direct toxic effect of phenytoin, the result of repeated hypoxia related to seizures, the effect of the seizure-related electrical discharge on cerebellar Purkinje cells, and the possibility that both the seizures and the cerebellar pathology are secondary to a unifying underlying pathology such as prenatal injuries. Another intriguing possibility is that both the seizures and the progressive cerebellar syndrome could result from a single underlying gene mutation such as can be seen in SCA10 (Matsuura et al., 2000). It is probably best to avoid phenytoin in an epileptic patient if either ataxia or cerebellar atrophy is present. In a meta-analysis of second-generation anticonvulsants (gabapentin, lamotrigine, levetiracetam, oxcarbazepine, pregabalin, tiagabine, topiramate, zonisamide) used as adjunct therapy, all of them were found to increase the risk of imbalance, with the exception of gabapentin and levetiracetam. A dose-response relation was seen with almost all the drugs (Sirven et al., 2007).
Infectious and Transmissible Diseases
Ataxia can be one of the features of postinfectious encephalomyelitis but usually accompanies a more diffuse cerebral process. A more restricted cerebellar syndrome has been seen in children after a viral syndrome. Connolly et al. (1994) have proposed that this diagnosis be considered when children develop an acute ataxic disorder not associated with a more diffuse process reflected by seizures, meningismus, or obtundation. In most children, this was preceded by a nonspecific viral syndrome or varicella, with a peak incidence at the age of 5 to 6 years. A similar syndrome following Epstein-Barr virus (EBV) or vaccinations occurred in the teenage years. Cerebrospinal fluid (CSF) may show some elevation of protein and a modest mononuclear pleocytosis, and PCR may allow identification of a specific etiology such as EBV. Magnetic resonance imaging (MRI) often reveals signal density changes in the cerebellum (Fig. 72.1). Prognosis for recovery is usually excellent, with residual dysfunction in a distinct minority. However, swelling, hydrocephalus, and herniation have been described in some and may require decompressive surgery (Van Lierde et al., 2004).
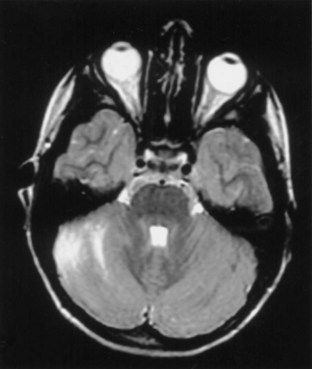
Fig. 72.1 Signal density change in the cerebellum in a child with acute cerebellar ataxia of childhood.
(Courtesy Dr. V. Vedanarayanan, Department of Pediatrics, University of Mississippi Medical Center.)
Similarly, a combination of ataxia, ophthalmoplegia, and other lower cranial nerve palsies can occur as a result of brainstem encephalitis (Bickerstaff encephalitis). There appears to be significant overlap between this disease and the Miller-Fisher variant of Guillain-Barré syndrome. In a report of 62 cases diagnosed as Bickerstaff encephalitis, most had some changes in consciousness level, 40% had Babinski signs, and 60% had tetraparesis; in addition, anti-GQ1 antibody was present in 66%. MRI brain abnormalities occurred only in 30% (Odaka et al., 2003).
Human immunodeficiency virus (HIV) infection can result in many neurological syndromes including ataxia. The majority of patients with ataxia in the presence of HIV infection have discrete, well-recognized lesions such as lymphomas, chronic meningeal infection, progressive multifocal leukoencephalopathy, or toxoplasmosis. Approximately 30% of patients with HIV dementia have an ataxic syndrome at the onset of their illness before cognitive decline begins. Other authors have described isolated progressive cerebellar ataxia that does not evolve into HIV dementia in a small number of patients (Elsheikh et al., 2010; Kwakwa and Ghobrial, 2001; Tagliati et al., 1998). These patients had a rapid evolution of their ataxia leading to chair-bound status in less than a year; MRI revealed cerebellar atrophy. Pathological examination has revealed marked granular cell loss.
Creutzfeldt-Jakob disease (CJD) should be considered in patients presenting with progressive ataxia. CJD is typically a rapidly progressive dementing illness related to the accumulation of mutant prion protein (PrP), which is a posttranslational modification of the normal prion protein. Among sporadic CJD patients, over 70% have ataxia (Parchi et al., 1999). Jellinger et al. (1974) noted that the ataxic variant of CJD began with minor behavior symptoms followed by ataxia. Upper motor neuron signs were frequent; myoclonus occurred only in 25%, and dementia evolved late. Survival among these patients was slightly longer than in typical CJD, with a mean time to death of 16 months and a range of 7 weeks to 8 years. Pathologically, the cerebellum shows striking granule cell loss. A recent review noted that both the iatrogenic and variant form of CJD (human bovine spongiform encephalopathy) usually have ataxia at the onset, as does the inherited form known as Gerstmann-Sträussler-Scheinker syndrome (GSS) (Wadsworth and Collinge, 2007). Prion protein gene mutation analysis is helpful in the familial cases. Iatrogenic cases are usually homozygous for codon 129, and the same codon is all methionine in the variant cases. Tonsillar biopsy for immunocytochemistry for PrP and Western blot studies for abnormal PrPSc (scrapie prion protein) can be useful in the variant cases as well. CSF protein biomarker studies involving 14-3-3, S100b, and tau are often useful in diagnosing CJD (Pennington et al., 2009).
Whipple disease (WD) is a multisystemic infectious disease caused by Tropheryma whippelii. The classic symptoms include diarrhea, weight loss, and abdominal pain. The pathognomic symptoms of CNS involvement are oculomasticatory myorhythmia (OMM) and oculo-facial-skeletal myorhythmia. Many cases of CNS WD are not diagnosed until postmortem (Gerard et al., 2002), but it is postulated that all patients have CNS involvement. Cerebellar ataxia is a common feature of CNS WD. It has been reported to be present in 45% of cases, more common than OMM (Matthews et al., 2005). Since untreated WD is fatal and therapy is available, it is paramount important to consider WD in the differential diagnosis when a patient presents with ataxia along with gastrointestinal symptoms. Those with possible CNS WD should undergo multiple duodenal biopsies if needed. Polymerase chain reaction (PCR) assay of CSF, brain MRI, and a stereotactic cerebral biopsy can also be helpful. Prolonged antibiotic therapy with drugs able to cross the blood-brain barrier is recommended. A regimen with intravenous ceftriaxone sodium, 2 g twice daily for 2 weeks, followed by oral trimethoprim-sulfamethoxazole, double strength, 1 tablet twice daily, achieved good results (Matthews et al., 2005). CSF should be analyzed repeatedly to monitor efficacy. Treatment can be discontinued only when the results of CSF PCR are negative.
Autoimmune Causes
Paraneoplastic Cerebellar Degeneration
Paraneoplastic cerebellar degeneration is a rapidly progressive pancerebellar syndrome that reaches its nadir within a few months of onset. It produces a severe ataxic disease associated with dysarthria and oscillopsia. Diplopia and vertigo may also occur. Many patients also develop other neurological signs including dementia, extrapyramidal signs, hearing loss, and dysphagia. MRI typically shows atrophy of the entire cerebellum, though some high-signal density changes may occur in the deep white matter. CSF usually shows a mononuclear pleocytosis, and oligoclonal bands may be present. It is believed that in many cases the syndrome results from an autoimmune process triggered by the cancer, but evidence for this cannot be found in every patient. Several autoantibodies have been described in these patients. The anti-Yo antibody is usually seen in ovarian cancer and primarily causes a cerebellar syndrome. The anti-Hu (type 1 antineuronal nuclear antibody [ANNA1]) antibody is seen with small-cell cancer of the lung and typically causes a multifocal disorder, the most common of which is a sensory ganglionopathy. Purkinje cell degeneration has been noted in about 25% of patients with anti-Hu antibody. The anti-Ri (ANNA2) antibody that recognizes two closely related neuronal RNA-binding proteins designated Nova-1 and Nova-2 has been seen in patients with a opsoclonus-myoclonus-ataxia syndrome in the setting of breast cancer and non–small cell lung carcinoma (Luque et al., 1991; Musunuru and Kesari, 2008). Patients who exhibit a combination of ataxia and Lambert-Eaton syndrome against a background of small-cell lung cancer may have antibodies to voltage-gated calcium channels (VGCC). Cerebellar degeneration in Hodgkin disease often occurs months to years after the lymphoma presents and is associated with the anti-Tr antibody (Bernal et al., 2003). Less common antibodies that have been related to paraneoplastic cerebellar ataxia with or without additional signs include the anti-Ta, anti-Ma, and anti-CV2 antibodies and autoantibodies against metabotropic glutamate receptor and protein kinase C gamma (Hoffmann et al., 2008; Posner and Dalmau, 2000; Sabater et al., 2006; Sillevis et al., 2000). The zic4 antibody against the neuronal zinc-finger protein is also associated with small-cell lung cancer and ataxia (Bataller et al., 2004). When routine studies are unfruitful, positron emission tomography (PET) scan may be useful to detect small cancers (Land et al., 2005). The eradication of underlying cancer usually results in improvement or resolution of ataxia (Ammar et al., 2008). Plasmapheresis has been reported effective in the reduction of autoantibodies and improvement of symptoms (Armstrong et al., 2005; Bier and Hile, 2010; Hoffmann et al., 2008; Taniguchi et al., 2006).
Ataxia with Gluten Sensitivity
Gluten sensitivity is a systemic autoimmune disease with diverse manifestations. Celiac disease is only one aspect of this entity. Celiac disease is an autoimmune disorder of the small intestine caused by an immune reaction to gliadin, a prolamin (gluten protein) found in wheat. Its prevalence is between 1 in 1750 to 1 in 105 people in the United States, varying on the definition (Rewers, 2005). Cerebellar ataxia has been reported since 1966 in this group of patients (Cooke and Smith, 1966). Vitamin deficiency from malabsorption or concomitant autoimmune diseases may account for the ataxia. A more controversial area is the association of antigliadin antibodies in the etiology of sporadic ataxia with no overt evidence for small bowel disease.
Hadjivassiliou et al. (2010) reported that 47% (101/215) of patients with idiopathic sporadic ataxia had serological evidence of gluten sensitivity. Other studies have shown a lower prevalence of gliadin antibodies in patients with ataxia or none at all (Burk et al., 2001; Combarros et al., 2000; Pellecchia et al., 1999; Wong et al., 2007). Bushara et al. (2001) and Lock et al. (2005) reported that patients with idiopathic ataxia and those with definite genetic forms of ataxia had a similar occurrence of gluten sensitivity, raising doubts about the significance of the gliadin antibodies. Contrary data have been reported by Hadjivassiliou et al. (2003), noting that while there was a 14% prevalence of gluten sensitivity among inherited cases, the figure was 41% among 132 cases of sporadic ataxia. Gluten-free diet was shown to improve measures of ataxia over a 1-year period, associated with elimination of the antigliadin antibodies, but this was not a randomized study. Patients with gluten sensitivity usually have had slowly progressive ataxia associated with brisk tendon reflexes and peripheral neuropathy. Nervous system pathology was characterized by cerebellar Purkinje cell loss, infiltration by T lymphocytes and posterior column degeneration. A variable proportion of patients had typical celiac disease on duodenal biopsy, even when there were only minor gastrointestinal symptoms; some had either normal biopsies or simply lymphocytic infiltrates, so the role of gluten sensitivity in the etiology of sporadic ataxia remains controversial. Presence of antigliadin antibodies should prompt further testing including antideaminated gliadin antibody, antitransglutaminase antibody, and small-bowel biopsy. If celiac disease is confirmed, gluten-free diet may offer a good chance for symptomatic relief (Rashtak et al., 2010).
Ataxia and Anti–Glutamic Acid Decarboxylase Antibodies
A number of reports have noted the presence of antibodies to glutamic acid decarboxylase (GAD) among a small number of patients with progressive ataxia (Bayreuther et al., 2008; Honnorat et al., 1995; Rakocevic et al., 2006; Saiz et al., 1997). The patients have usually been middle-aged women. Ataxia was often associated with peripheral neuropathy, rarely associated with neuropathy or slow saccades and in some cases with stiff person syndrome. Many patients had multiple organ specific antibodies including those directed against thyroid, parietal cells, and pancreatic islet cells, and insulin-dependent diabetes was an invariable accompaniment. The antibodies occurred in higher titers than those found in adult-onset diabetes, and there is evidence that they bind to presynaptic nerve terminals around cerebellar Purkinje cells. GAD is the enzyme that synthesizes γ-aminobutyric acid (GABA) from glutamate, and it is believed that the antibodies may be pathogenic because of their binding to GABA terminals. Intravenous immunoglobulin or immunosuppressants may cause partial remission of symptoms (Markakis et al., 2008; Nanri et al., 2009; Nociti et al., 2010; Vulliemoz et al., 2007).
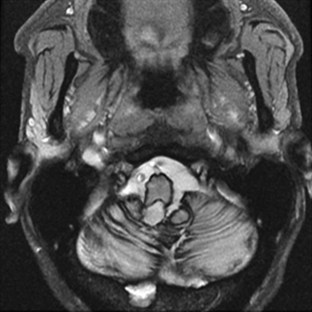
Superficial Siderosis
Superficial siderosis occurs when iron and hemosiderin are deposited in the pial and subpial regions of the brain. Clinical features include ataxia, hearing loss, and cognitive decline (Fearnley et al., 1995). Superficial siderosis results from bleeds that are often covert, such as with cryptic vascular malformations, or more obvious, such as with tumors or prior surgery. Diagnosis can be made by the characteristic hypointensity over CNS structures in T2-weighted images on MRI (Fig. 72.2). The disease can be controlled if the source of bleeding is found and obliterated.
Inherited Ataxias
The initial recognition that progressive balance difficulties related to pathology in the cerebellum and its connecting pathways could have a genetic basis is attributed to Nicholas Friedreich, who published a series of papers describing siblings with such a disease, now known as Friedreich ataxia (FA). As early as 1893, Pierre Marie recognized the clinical and genetic heterogeneity among the inherited ataxias. During much of the 20th century, inherited ataxias that did not quite fit the Friedreich mould were often labeled as Marie ataxia. It eventually became apparent that many patients have progressive ataxia of a degenerative nature, clinically and pathologically resembling patients with inherited ataxias, and yet have no discernible genetic basis. Much of the last 100 years saw detailed clinical and pathological descriptions of patients with various forms of ataxia and attempts at understanding them on a clinicopathological basis. The classification of these diseases based on the underlying neuropathology reflects attempts at such understanding. Nevertheless, the clinical, neuropathological, and genetic heterogeneity of the disorders did not allow a universally acceptable classification. Harding (1983), based on an extensive clinical study of many families, wrote a clinical-genetic classification that served as a prelude to an increasingly gene-based classification of the inherited ataxias (Harding, 1983). This section will summarize the current information regarding inherited forms of ataxia.
Autosomal Recessive Ataxias
As the autosomal recessive ataxias have been elucidated at the molecular genetic level, a semblance of a pathogenetic classification may be emerging, albeit subject to revisions or additions. The genetic abnormalities discovered so far have either pointed to abnormalities in mitochondria, oxidative stress, abnormalities of deoxyribonucleic acid (DNA) repair mechanisms, possibly abnormal protein folding, or changes in cytoskeletal proteins. Table 72.2 lists various autosomal recessive ataxias based on specific gene loci. Most of these diseases begin in childhood or early adult life; however, onset even late in life is possible with autosomal recessive ataxias. Singleton patients may occur in many families. Typically, parents do not manifest symptoms, since they are heterozygous for the mutation. If the sibship size is large, affected siblings may be found, and the disorder will affect both males and females. Parental consanguinity may be found but is not essential.
| Entity | Gene locus/gene | Mutation |
|---|---|---|
AOA1, Ataxia with oculomotor apraxia type 1; AOA2, ataxia with oculomotor apraxia type 2; ARCA1, autosomal recessive cerebellar ataxia type 1; ARSACS, autosomal recessive ataxia of Charlevoix-Saguenay; ATLD, ataxia-telangiectasia-like disorder; IOSCA, infantile-onset spinocerebellar ataxia; MIRAS, mitochondrial recessive ataxia syndrome; MSS, Marinesco-Sjögren syndrome; SCAN1, spinocerebellar ataxia with axonal neuropathy.
Friedreich Ataxia
Clinical Features
Friedreich ataxia is the most common inherited ataxia, with a prevalence of 1 in 50,000 Caucasians. Classical descriptions of the disease include an age at onset below 25 years, typically early in adolescence (Harding, 1981). Initial symptoms include gait difficulties and clumsiness. Neurological examination reveals gait ataxia, loss of proprioceptive sense in the lower limbs, and absence of deep tendon reflexes (Pandolfo, 2003). These findings are related to early pathology in dorsal root ganglion cells, and occasional patients may be mistaken for having a hereditary sensory neuropathy. However, most will exhibit signs indicating involvement of the CNS including dysarthria, some upper motor neuron findings such as extensor plantar responses, and eye movement abnormalities such as square-wave jerks. Rarely, patients may present with cardiac disease or a spinal deformity and are later found to develop neurological disease; they tend to lose ambulation in about 9 to 15 years. At this stage, patients have increasing ataxia of both upper and lower limbs, profound proprioceptive loss, areflexia, weakness of lower-limb muscles, flexor spasms, and increasing dysarthria and dysphagia. Optic atrophy and hearing loss may occur in a minority of patients.
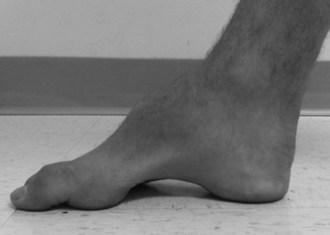
Systemic abnormalities include abnormal electrocardiograms, hypertrophic cardiomyopathy in about 50% of the patients, and diabetes in 10%. The hypertrophic cardiomyopathy may later become a dilated cardiomyopathy and rhythm problems such as atrial fibrillation may add to the cardiac morbidity. Skeletal abnormalities such as spinal deformities and foot deformities are common (Fig. 72.3). The mean age of death among patients with FA has been reported to be late in the fourth decade, but this information comes from clinical studies done prior to the availability of FA mutation analysis. Also, the availability of better diabetic and cardiac care as well as adaptive rehabilitation techniques can allow much longer survival. Cause of death is often cardiac, but respiratory compromise related to the motor difficulties and spinal deformity may also contribute.
Nerve conduction studies show early absence or reduction of sensory nerve potentials in a diffuse fashion, reflecting the loss of large sensory axons in peripheral nerves. Central motor conduction studies show abnormalities that evolve over the course of the disease and may reflect disease progression. MRI scans of the brain reveal no abnormalities in the cerebellum; rather, the upper cervical cord shows atrophy. Signal density changes may be seen in the posterior columns by MRI (Mascalchi et al., 1994). Functional imaging has been reported to show decreased cerebellar blood flow, but there is cerebral glucose hypermetabolism in ambulatory patients with FA (Gilman et al., 1990). Pathologically, there is loss of dorsal root ganglion cells, resultant degeneration of the dorsal columns, degeneration of spinocerebellar and corticospinal tracts, and loss of cells in the cerebellar dentate nucleus.
The Friedreich Ataxia Mutation
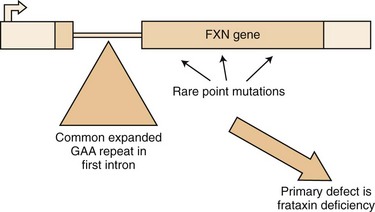
The mutation in FA is an unstable expansion of a repeated trinucleotide (GAA) sequence within the first intron of the FRDA gene on chromosome 9q13-21.1 (Campuzano et al., 1996) (Fig. 72.4). Expanded alleles have 66 to over 1000 repeats. In nearly 95% of affected persons, the GAA expansion occurs in both alleles (homozygous expansion), although the size of the expansion can be different in each.
Since the identification of the FA mutation, we have realized that many patients with a phenotype very atypical for classical FA carry the FA mutation (Durr et al., 1996; Filla et al., 1996; Montermini et al., 1997). Some 15% of patients with homozygous GAA expansion have neurological signs identical to those of classical FA but have an age at onset later than 25 years (late-onset FA [LOFA]). Other patients with the GAA expansion have the typical age at onset but retain their tendon reflexes or even have exaggerated reflexes (FA with retained reflexes [FARR]). There are patients who combine a later onset with retained reflexes as well; in addition, very late onset of mild gait ataxia, spastic paraparesis, and even chorea have been occasionally associated with the FA GAA expansion (Hou and Jankovic, 2003). The variation of expansion size may account for the clinical diversities. As with other trinucleotide repeat diseases, there is an inverse correlation between the size of the GAA repeat and the age at onset; this correlation is better with the smaller of the two expanded alleles. Cardiomyopathy and diabetes also tend to occur in patients with larger expansions (>700 GAA repeats). However, not all of the variation in age at onset and severity of disease can be correlated with GAA size alone. Other genetic and possibly environmental factors may contribute to this variation. In addition, variation in clinical picture can result from somatic mosaicism of the repeat size, with different tissues having significant differences in the size of the expansions (Bidichandani et al., 1999). Recently it has been reported that the U mitochondrial (mt)DNA haplogroup can delay the age at onset and have a lower rate of cardiomyopathy in FA (Giacchetti et al., 2004).
Point Mutations
Approximately 5% of patients with clinical findings compatible with FA have only one expanded allele; however, such patients harbor point mutations in the unexpanded allele. Sequencing of the unexpanded allele to document a point mutation will have to be done in this situation. Point mutations located in the carboxy terminal half of the mature frataxin appear to be associated with a typical FA phenotype (Cossee et al., 1999). Missense mutations in the amino terminal half of the protein, such as the G130V mutation, appear to result in a milder phenotype with less ataxia, greater spasticity, and no dysarthria. To date, all the patients with point mutations have been compound heterozygotes with one expanded allele. Homozygous point mutations will cause complete lack of frataxin, and this may be incompatible with life. This has been further supported by the fact that complete knockout of the gene in animal models is embryonically lethal.
Pathogenesis
The presence of the expanded GAA sequence in the first intron of the gene results in reduced transcriptional efficiency leading to a partial deficiency of the protein, frataxin (Lodi et al., 2001; Pandolfo, 2001; Puccio and Koenig, 2000). The reduced transcriptional efficiency of the mutated gene has been attributed to an unusual “sticky DNA” configuration of the expanded repeat (Sakamoto et al., 1999). In addition, there is evidence for chromatin condensation and remodeling, which can further suppress frataxin transcription (Pandolfo and Pastore, 2009). The exact role of frataxin in normal biology is still unclear, but many studies suggest that it is a mitochondrial protein (Fig. 72.5). Knockout of the frataxin homolog in yeast leads to accumulation of iron in the mitochondria, a finding of interest in view of the presence of iron in the cardiac muscle in human disease (Babcock et al., 1997; Pandolfo and Pastore, 2009). Activity of enzymes such as aconitase and complex I, II, and III of the respiratory chain that contain iron-sulfur clusters is reduced in cardiac muscle biopsies of patients with FA (Rotig et al., 1997). The iron-sulfur cluster synthesis defect results from the role of frataxin in the process and probably is the cause of iron accumulation. It has been hypothesized that the presence of excess iron as well as excess oxidative stress related to impaired respiratory chain function can induce oxidative stress via the Fenton reaction and cause progressive nuclear and mitochondrial damage (Karthikeyan et al., 2002, 2003; Pandolfo and Pastore, 2009).
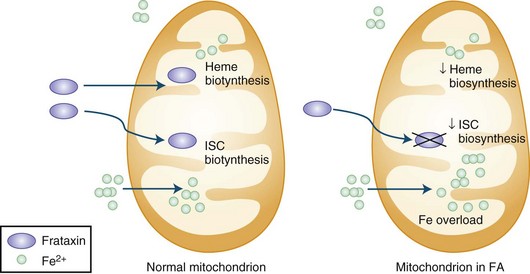
Treatment
Based on evidence for excess oxidative stress and poor defense mechanisms of FA tissue to such stress, antioxidant therapy has been tried over the last 10 years; as of 2008, at least 11 studies using idebenone in patients with FA have been reported (Meier and Buyse, 2009). This drug has been reasonably well tolerated in doses of 900 mg/day or more by FA patients, the main adverse effect being gastrointestinal. At doses of 5 mg/kg/day, it appeared to have beneficial effect on the cardiomyopathy of FA. In a 6-month study, idebenone given in high doses appeared to improve neurological function as measured by International Cooperative Ataxia Rating Scale (ICARS) scores in ambulatory FA patients (Schulz et al., 2009). Newer approaches now in the experimental phase include gene replacement (Fleming et al., 2005) and the use of molecules that may enhance frataxin production even in the presence of the GAA expansion (Hebert, 2008). In fact, recent open-label experience suggests some beneficial effect from erythropoietin (Boesch et al., 2008).
Mitochondrial Recessive Ataxia Syndrome
An autosomal recessive ataxic syndrome caused by a mutation in the polymerase γ (POLG) gene has been found to be the most common cause of recessive ataxia in Finland (Hakonen et al., 2005). Polymerase γ is encoded by the nuclear gene, POLG, in humans and is responsible for replication and repair of mtDNA. In a recent study report, 27 patients were found among 15 Finnish families with a carrier frequency of 1 : 125. In addition, patients with recessive ataxia from Norway, Belgium, and England shared the same core Finnish haplotype, suggesting a common European founder mutation and making it likely that this disease will be found in places other than Finland. The age at onset varied from 5 to 41 years. The neurological picture was heterogeneous, but ataxia, hypo- or areflexia, axonal neuropathy, and epilepsy were common. There was often mild cognitive impairment and psychiatric manifestations such as depression, anxiety, and hallucinations.
Infantile-Onset Spinocerebellar Ataxia
Infantile-onset spinocerebellar ataxia has been described among an isolated population in Finland. The disorder begins soon after children acquire the ability to walk. Neurological signs include ataxia, peripheral neuropathy with areflexia, athetosis of hands and feet, Babinski signs, and clumsiness. Speech becomes impaired, and sensorineural hearing loss occurs before the age of 5. Ophthalmoplegia and optic atrophy develop, and some patients have seizures. Patients usually have learning deficits and skeletal deformities as well. The gene responsible has recently been found to be C10ORF2, a nuclear gene encoding for Twinkle, an mtDNA-specific helicase, and a splice variant known as Twinky (Nikali et al., 2005) located on 10q24. Dominant mutations in the same gene cause a syndrome of progressive external ophthalmoplegia. In contrast to the latter, the recessive mutations in infantile-onset spinocerebellar ataxia do not appear to alter mtDNA maintenance.
Ataxia with Isolated Vitamin E Deficiency
The occurrence of a childhood-onset recessive ataxia associated with isolated vitamin E deficiency is related to mutations in the gene encoding the α-tocopherol transfer protein (α-TTP) on chromosome 8 (Ouahchi et al., 1995). A hepatic protein, α-TTP is involved in the processing of vitamin E for transport in the chylomicrons. The low vitamin E levels are related to impaired hepatic processing, not to deficient intestinal absorption. Deficiency of vitamin E, which has antioxidant properties, may cause neuronal degeneration. Purkinje cells are more vulnerable owing to their high metabolic activity and high oxygen demand. The severity of the phenotype associated with α-TTP mutations depends on residual protein activity. The childhood-onset disease has considerable resemblance to FA. There is progressive ataxia and evidence for a polyneuropathy with loss of proprioception and tendon reflexes. Retinitis pigmentosa and visual loss can accompany this syndrome. Patients often have head titubation. Cardiomyopathy can occur but is less common than in FA (Cavalier et al., 1998). Vitamin E levels should be obtained in all persons with sporadic ataxia of childhood or young-adult onset. Patients typically have less than 1.8 mg/L of vitamin E. Treatment with large doses of vitamin E stabilizes the neurological features, especially when started early in the disease, but some features may show worsening despite therapy (Mariotti et al., 2004).
Abetalipoproteinemia
Abetalipoproteinemia results from mutations in the MTTP gene, which codes for a subunit of the microsomal triglyceride transfer protein; this results in impaired lipid absorption in the intestine, including lipid-soluble vitamins like vitamin E. The ataxia resembles that in isolated vitamin E deficiency but is associated with evidence for malabsorption (Fogel and Perlman, 2007). Vitamin E and cholesterol levels are low, and apolipoprotein B is absent. Acanthocytes are found in peripheral blood smears. Transaminases may be high and INR elevated due to vitamin K deficiency. Serum lipoprotein electrophoresis can establish the diagnosis. Treatment involves dietary modification and vitamin replacement, which may prevent neurological complications.
Cayman Ataxia
This recessive ataxia of early childhood onset has been identified in the Grand Cayman island. Affected patients have been shown to carry two homozygous mutations in the gene that encodes caytaxin (ATCAY) on chromosome 19. Caytaxin contains a region with homology to a CRAL-TRIO domain, also present in α-TTP. A ligand similar to vitamin E may bind to this protein, and its deficiency may cause ataxia by a mechanism similar to that in vitamin E deficiency (Bomar et al., 2003).
Ataxia-Telangiectasia
Clinical Features
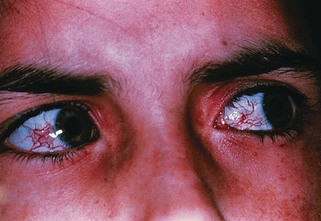
The occurrence of ataxia-telangiectasia (AT) has been estimated to be 1 in 20,000 to 100,000 live births (Swift et al., 1993). This disorder has its onset early in the first decade with truncal instability and impaired gait. This is followed by progressive ataxia associated with polyneuropathy, hypotonia, and loss of reflexes (Fogel and Perlman, 2007). Choreiform movements are frequent. Characteristic eye movement abnormalities are seen, with the need for a rapid head thrust for fixating on targets to one side, with the eyes following later (eye-head lag). This is referred to as oculomotor apraxia. Telangiectasia tends to appear at about 5 years of age and is most frequent on the conjunctivae (Fig. 72.6) but can also occur over the earlobes, popliteal fossa, and other locations. There is evidence for an immunodeficiency, with frequent bronchopulmonary infections. These children are at risk for malignancies, especially hematological tumors like lymphomas and leukemias; other types of cancers occur if the patient reaches adulthood. Infections related to immunodeficiency are also common. The thymus gland is typically small or absent in patients with AT. Many have decreased immunoglobulin (Ig)A levels; decreased IgE and IgM levels, lymphocytopenia, and skin anergy may also occur. In addition, elevation of α-fetoprotein (AFP) in the serum is a consistent finding in AT.
Mutation and Pathogenesis
AT is caused by truncating, missense, deletion, or insertion mutations of the ATM gene on chromosome 11 (Halazonetis and Shiloh, 1999; Savitsky et al., 1995). More than 300 mutations of this gene have been associated with AT. The protein product of ATM is a serine-threonine kinase and may be involved in checkpoint responses to double-stranded DNA breaks by conveying the signal of damage from sensor proteins such as MRN (MRE11-RAD50-NBS1 complex) to the effector proteins. Increased susceptibility to radiation damage has been shown to cause chromosomal translocations and has been used in a fibroblast survival assay as a diagnostic test. The degree of DNA damage determines the response: either cell-cycle arrest and repair or initiation of apoptosis (Lavin et al., 2005).
Laboratory Diagnosis
Serum AFP is usually elevated (>10 ng/mL), and immunoglobulin levels are low. Karyotyping often shows chromosomal abnormalities, especially a 7 : 14 translocation. Increased sensitivity of cultured fibroblasts to radiation can be documented by the colony survival assay. The definitive way of establishing diagnosis is immunoblotting for the ATM protein; 90% of patients have no protein, 10% have trace amounts, and rare patients have normal ATM levels but no kinase activity (so-called kinase-dead) (www.geneclinics.org). Further molecular confirmation can be achieved by detecting the mutation, but this can be difficult because of the size of the ATM gene and the lack of mutation “hot-spots.” Sequence analysis of the coding regions of the ATM gene will detect 90% of mutations but miss intronic alterations and heterozygous deletions; benign polymorphisms and pathogenic alterations may be difficult to distinguish. Other types of DNA-based tests include protein truncation tests (examining for premature stop codons), which can detect about 70% of the mutations, and mutation scanning using denaturing high-performance liquid chromatography (DHPLC), which has a sensitivity of 85%.
Ataxia–Telangiectasia-like Disorder
Ataxia–telangiectasia-like disorder (ATLD) is related to mutations in the gene coding for MRE11, another protein involved in double-stranded DNA break repair. It has a clinical picture resembling AT but has a milder course, no telangiectasia, and no elevation of serum AFP (Fogel and Perlman, 2007).
Ataxia with Oculomotor Apraxia Type 1
Ataxia with oculomotor apraxia type 1 (AOA1) is an AR ataxia of childhood onset (usually <10 years), and many patients have oculomotor apraxia akin to that in AT (Le Ber et al., 2003). It is the commonest recessive ataxia in Japan and second only to FA in Portugal. In the report from Le Ber et al., AOA1 made up 5% of non-Friedreich recessive ataxias. It has other similarities to AT including peripheral neuropathy (which may predominate the clinical picture in later stages), choreoathetosis, and dystonia. Cognitive decline is frequent. However, radiosensitivity and malignancy risk are not elevated. Serum creatine kinase (CK) and cholesterol are often high and serum albumin decreased. Mutations in the aprataxin (APTX) gene have been described in AOA1 (Date et al., 2001; Moreira et al., 2001). APTX is a nuclear protein that has an N-terminal forkhead-associated (FHA) domain resembling PNKP, a histidine triad (HIT) domain, and a C-terminal zinc finger domain. Most of the mutations affect the HIT domain. The protein may function in base excision repair, a form of single-strand DNA repair mechanism. The C terminal is also known to interact with XRCC1, which plays a role in single-strand break repair. It is unclear how this functional role is related to neurodegeneration in the cerebellum.
Ataxia with Oculomotor Apraxia Type 2
Ataxia with oculomotor apraxia type 2 (AOA2) is a progressive AR ataxia associated with cerebellar atrophy and an axonal polyneuropathy and elevation of serum AFP (Anheim et al., 2009). AOA2 is the most frequent AR ataxia next to FA in European series (Anheim et al., 2010; Le Ber et al., 2004). The age at onset is around 15 years, and the disease progresses to a stage requiring significant ambulation devices in about 15 years. Oculomotor apraxia is not universal, being seen in about 50% cases (Anheim et al., 2009; Le Ber et al., 2004). Other clinical signs noted are upper motor neuron signs in about 20%, dystonia, chorea, head tremor, and strabismus. Elevation of serum AFP to over 7 µg/L may be predictive of AOA2. Other features that have been noted infrequently include elevation of serum CK and hypogonadotropic hypogonadism. No other systemic features such as high risk for malignancy, telangiectasia, or radiation sensitivity are seen.
AOA2 has been linked to mutations in the senataxin gene (SETX) that codes for senataxin (Moreira et al., 2004). The normal function of senataxin is unclear; its C-terminal region has homology to the superfamily of helicases. DNA/RNA helicase function may contribute to the pathogenesis of neurological dysfunction (Chen et al., 2004).
Other DNA Repair Defects Causing Ataxia
Mutations in tyrosyl-DNA phosphodiesterase 1 (TDP1), a DNA repair enzyme, causes spinocerebellar ataxia with axonal neuropathy. These patients have none of the systemic features that characterize AT or ATLD (Takashima et al., 2002). Cockayne syndrome and xeroderma pigmentosum are rare diseases caused by DNA repair defects in which systemic disease predominates the phenotype, but CNS features including ataxia are often present. Mutations in genes encoding nucleotide excision repair pathways occur in these diseases (Chu and Mayne, 1996).
Autosomal Recessive Ataxia of Charlevoix-Saguenay
Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) was initially identified in children with spastic ataxia among French Canadians in the Charlevoix-Saguenay province of Quebec, Canada, an inbred population with a high carrier frequency for the gene. ARSACS has since then been documented in other geographic areas such as Turkey, Japan, Spain, and Italy. Clinically it is characterized by very early onset, prominent spasticity followed by ataxia, and amyotrophy of distal muscles (Takiyama, 2007). Progression is slow, with a mean age at death in the late 50s. The mutation causing this disease has been shown to involve a gene on chromosome 13. The protein product of this gene has similarities to chaperone proteins and has been named sacsin (Engert et al., 2000). The C-terminal region of this protein has a DnaJ motif that can interact with Hsp 70, and the N terminus has homology to HSp90. Thus SACSIN may have a chaperone function involved in protein folding.
Marinesco-Sjögren Syndrome
This rare disease characterized by ataxia, early cataracts, mental subnormality, and myopathy has been linked to mutations in the SIL1 gene. SIL1 encodes a nucleotide exchange factor for the chaperone protein, Hsp70 (Anttonen et al., 2005; Senderek et al., 2005).
Autosomal Recessive Cerebellar Ataxia Type 1
The term autosomal recessive cerebellar ataxia type 1 (ARCA1) has been used to describe an autosomal recessive ataxia in a population isolate of French Canadians from Quebec. The disorder begins in young adult life (17-46 years) and causes progressive ataxia, dysarthria, and mild eye movement problems. Reflexes were normal or slightly brisk and there was cerebellar atrophy on imaging studies. Molecular studies have revealed that this is related to mutations in the SYNE1 gene. It is thought that SYNE1 is a member of the spectrin family of cytoskeletal proteins that may be involved in anchoring actin to the plasma membrane and appears enriched in Purkinje cells (Dupre et al., 2007).
Autosomal Recessive Cerebellar Ataxia Type 2
The term autosomal recessive cerebellar ataxia type 2 (ARCA2) has been used to denote cerebellar ataxia associated with coenzyme Q10 (CoQ10) deficiency (Anheim et al., 2010). CoQ10 is a lipid-soluble component of cell membranes and is involved in transport of electrons between complex I and II of the electron transport chain to complex III. Ataxia associated with deficiency of muscle CoQ10 is heterogeneous. This combination can be seen in some patients with AOA1 as well as in some with defined abnormalities in CoQ10 metabolism. Ataxia is associated with pyramidal signs and mild cognitive decline. Some patients may have hypogonadism. CoQ10 deficiency can be associated with other phenotypes such as an infantile multisystem disease, encephalomyopathy with ragged red fibers, and Leigh syndrome. Some gene mutations have been identified in the infantile cases. The syndrome may respond to CoQ10 replacement therapy.
Childhood and Young Adult–Onset Metabolic Disorders in Which Ataxia Can Be Prominent
Many childhood-onset and young adult–onset ataxias are related to metabolic errors that can be diagnosed by specific laboratory tests rather than by gene-based tests. In many of these diseases, ataxia forms only a part of the phenotype. Table 72.3 lists some of these diseases. Some of them are amenable to various forms of therapy including dietary manipulations.
Table 72.3 Ataxias in Which Specific Abnormalities May Confirm or Point to the Diagnosis*
| Disorder | Diagnosis |
|---|---|
AOA, Ataxia with oculomotor apraxia; AT, ataxia-telangiectasia; CSF, cerebrospinal fluid; MRI, magnetic resonance imaging; MRS, magnetic resonance spectroscopy.
* Some also appear in Table 72.2, and many have additional CNS features.
Clinically Defined but (Likely) Genetically Heterogeneous Recessive Syndromes
Many children and young adults with progressive ataxia still have undefined genotypes (Table 72.4). The term early-onset ataxia with retained reflexes was originally used to describe a syndrome of ataxia with onset in childhood and some resemblance to FA but with preservation of deep tendon reflexes. We now know that many of these patients carry the FA mutation; in others, however, the genotype remains unknown. Similarly, Gordon Holmes originally described the syndrome of recessive ataxia associated with hypogonadism in 1907. This may also be genetically heterogeneous, though in some patients with such a disorder, mitochondrial mutations have been described. The term Ramsay Hunt syndrome is used for the association of ataxia and myoclonus or myoclonic epilepsy. One form of this, Unverricht-Lundborg syndrome, is related to mutations in the cystatin B gene. Others have mtDNA mutations or other disorders of intermediary metabolism such as ceroid lipofuscinosis or sialidosis.
Table 72.4 Childhood or Young Adult–Onset Ataxias with Distinctive Clinical Features*
| Entity | Clinical/Laboratory Clues |
|---|---|
ARSACS, autosomal recessive ataxia of Charlevoix-Saguenay; FA, Friedrich ataxia; mtDNA, mitochondrial deoxyribonucleic acid.
Mitochondrial Diseases and Ataxia
Nuclear gene defects that secondarily cause qualitative or quantitative defects of mtDNA or code for protein subunits of the respiratory chain or are involved in the assembly of respiratory complexes are increasingly being recognized. In many of these, ataxia can be part of usually a complex phenotypical picture (Zeviani and Di Donato, 2004).
Autosomal Dominant Ataxias
Autosomal dominant (AD) ataxias are broadly grouped into those with progressive and those with episodic features. Those with a progressive course are labeled spinocerebellar ataxia (SCA) followed by a number to indicate the gene locus. The episodic ataxias (EAs) are also numbered (EA1, EA2, etc.) to indicate distinct gene loci. The genetic heterogeneity among the dominant ataxias has been more than amply documented and continues to be a fertile source for new gene discoveries. Currently, SCA1 through SCA31 are recognized as well as EA1 through EA6, with certain caveats: there is no SCA9 (initially reserved but never substantiated); SCA15 and 16 are now known to be related to mutations in the same gene; SCA19 and 22 may be allelic diseases, but this is not yet settled; SCA24 was assigned to a family which likely has a recessive mode of inheritance, and this is now called spinocerebellar ataxia with saccadic intrusions (SCASI); Machado-Joseph disease (MJD) is the preferred name for SCA3; and dentatorubral-pallidoluysian atrophy (DRPLA) is usually classified as a dominant ataxia but has no SCA designation. Jackson et al. (1977) and Harding (1982) can be credited with the initiation of modern studies of the SCAs, the former by being the first to localize one form to chromosome 6 (SCA1) and the latter by setting up clear clinical criteria as a prelude to genetic studies. Earlier terminology for these ataxias included olivopontocerebellar atrophies and Marie ataxia. The disease occurs in each generation of the pedigree, with the offspring of affected parents having a 50% risk of inheriting the disease in general. Male-to-male transmission of the disease is definite evidence for autosomal dominant inheritance. The absence of symptomatic disease in either of the affected parents with autosomal dominant ataxia is rare but can occur for a number of reasons. Examples include reduced penetrance of the disease, onset of disease in a child before onset of symptoms in the affected parent because of anticipation, death of the involved parent before onset of symptoms, and wrong paternity. De novo mutations and expansion from an intermediate size allele to a pathogenic allele in disorders related to repetitive nucleotide sequences may be other explanations.
Clinical Features of Spinocerebellar Ataxias
Overall, SCAs related to different gene mutations have overlapping clinical features (Subramony, 2005). There is more experience with some of the SCAs and EAs (e.g., SCA1, 2, 3, 6, 7) than others, and the knowledge about phenotypical features in some of the SCAs is based on a limited number of families. In general, onset is in early or middle adult life, but the range of age at onset is vast, and onset in early childhood or at older ages is possible. In the better-known SCAs which are related to unstable nucleotide repeat expansions, anticipation in age at onset is seen and is usually more prominent with paternal transmission of disease; in these, age at onset is inversely correlated with the expansion size. Some of the more recently described SCAs related to more conventional mutations have childhood onset and very slow progression. Patients with SCA exhibit progressive ataxia of gait associated with clinical signs of limb ataxia such as dysmetria and dysdiadochokinesia. Speech is dysarthric. Eye movement abnormalities related to cerebellar dysfunction are frequent and include abnormal pursuit and inaccurate saccades in addition to nystagmus.
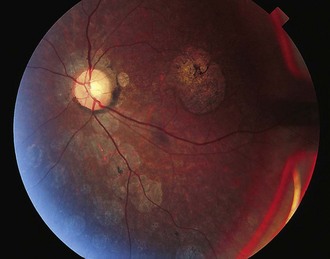
In many SCAs, ataxia is the only clinical feature. In others, clinical signs referable to pathology in structures other than cerebellum can be seen. Table 72.5 summarizes some clinical features that may help in the clinical differentiation of various SCAs. However, even in those SCAs in which non-cerebellar signs have been described, patients with a later onset may exhibit a pure cerebellar phenotype. Oculomotor abnormalities unrelated to cerebellar dysfunction include nuclear and supranuclear gaze palsy, slow saccades, ptosis, blepharospasm, and “ocular stare.” Patients may exhibit bulbar deficits such as facial atrophy, facial fasciculations, tongue atrophy and fasciculations, and poor ability to cough. Upper motor neuron signs such as brisk reflexes, with or without spasticity, and Babinski signs occur early in many SCAs. Extrapyramidal signs including akinetic-rigid syndromes, hypomimetic facies, chorea, and dystonia tend to occur variably in SCAs in different stages of the illness (Fig. 72.7). Evidence of peripheral nerve disease occurs frequently and includes distal sensory loss and loss of deep tendon reflexes as well as amyotrophy, cramps, and fasciculations. In selected SCAs, there can be associated cerebral signs such as cognitive decline or seizures; in others, there is evidence of retinal disease with visual loss (Fig. 72.8). Restless legs and sleep disturbances can occur in many. Other clinical signs that have been noted in certain genotypes include focal dystonia, postural and rest tremor, head tremor, palatal tremor, and psychiatric features such as aggressive outbursts, schizophrenic symptoms, severe depression, and apathy.
Table 72.5 Phenotypic Clues to Specific Gene Mutations in the Dominant Ataxias
| Clue | Related Dominant Ataxia |
|---|---|
DRPLA, Dentatorubral-pallidoluysian ataxia; EA, episodic ataxia; MJD, Machado-Joseph disease; SCA, spinocerebellar ataxia.
Imaging and Other Laboratory Studies in Dominant Ataxias
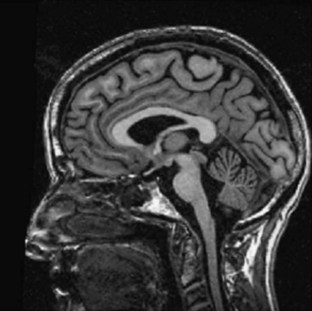
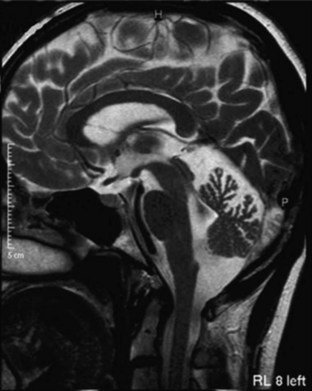
MRI and computed tomography (CT) scans of the brain are useful to exclude many disorders causing ataxia other than the inherited causes, such as strokes, tumors, and multiple sclerosis. The SCAs are associated with progressive atrophy of the cerebellum; in addition, there may also be atrophy of the pons, medulla, middle cerebellar peduncles, and upper cervical cord. In general, those SCAs that have noncerebellar features have both cerebellar and brainstem atrophy, and those that have isolated cerebellar features exhibit cerebellar atrophy only (Figs. 72.9 and 72.10). T2 hyperintensity in the pontine base, leading to a “hot cross bun” sign, can be seen in many SCAs with brainstem involvement, though it was originally thought to be seen only in sporadic ataxia associated with multiple system atrophy. There are recent reports of more quantitative imaging using such techniques as voxel-based volumetry, tractography, and diffusivity mapping (Dohlinger et al., 2008). These indicate some degree of genotype-based differences in the regions affected, but the sample sizes are small, and questions remain about the image manipulations needed to achieve these results. Such studies suggest that SCA6 has relatively isolated cerebellar atrophy; SCA2 tends to exhibit the greatest degree of brainstem atrophy, and SCA1 and 3 reveal pontocerebellar atrophy. Supratentorial atrophy tends to be milder and variable. Evidence of an axonal polyneuropathy on nerve conduction studies occurs in SCA1, 2, and 3 and less commonly in other SCAs. Systemic organ involvement and abnormal lab studies are lacking in the SCAs (with rare exceptions).
Neuropathology
In many SCAs, the cerebellum may bear the brunt of the pathology, a situation correlated with relatively pure cerebellar ataxia as the clinical feature. For example, SCA6 is characterized by such relatively isolated cerebellar pathology. In many SCAs, additional structures are involved, including a variety of brainstem and spinal cord structures. Most of the published reports have dealt with the common SCAs such as SCA1, 2, 3, and 6 and even in these the sample sizes have been small and the rigor of examination variable. In the other genetically defined SCAs, such reports are scanty. Recently, Rub and his colleagues (2003) have contributed rigorous reports on the neuropathology of many SCAs using a standard approach to the examination, and these have added significantly to knowledge regarding the extent of lesions in many diseases. However, the degree of neuropathological variation within the same families is still poorly understood; it undoubtedly exists.
Gene Mutations and Phenotype-Genotype Correlations in Dominant Ataxias
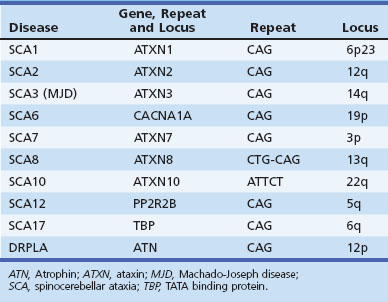
Both expansions of repeated nucleotide sequences and conventional mutations have been related to dominant ataxias (Durr, 2010) (Fig. 72-11 and Tables 72.6 and 72.7). At least seven dominant ataxias are related to unstable expansions of CAG repeats within coding sequences of specific genes inherited in a heterozygous fashion. Since CAG codes for glutamine, and the protein product of the mutated gene contains excess numbers of glutamines, these diseases are examples of polyglutamine disorders. In the case of SCA12, the CAG expansion occurs in a non-coding promoter region of the gene, PPP2R2B. The CAG expansion diseases are characterized by an inverse correlation between the number of repeats in the expanded allele and the age at onset. Larger repeat sizes may be associated with more rapid progression of the disease and with more rapid atrophy of posterior fossa structures. Thus the phenotypical heterogeneity within single genotypes is related to a complex interaction between the size of the repeats in a particular individual, the duration of the disease, and additional environmental and genetic factors of unknown nature. The unstable nature of these expansions causes a change in the size of the repeats from one generation to the next, accounting for the variability in age at onset as well as some of the phenotypical heterogeneity. The observed anticipation in age at onset of many dominant ataxias can be at least partly explained by the expansion of these unstable repeats during transmission to the next generation. Typically, paternal transmission of the disease is associated with a greater tendency for expansion. The SCA6 CAG expansion, which involves the α1 subunit of the neuronal calcium channel gene, is exceptional in that it is transmitted in a stable fashion for the most part. This appears to be true for the SCA17 CAG expansion in TATA-binding protein as well, though there are exceptions to the rule. The following section briefly describes the SCAs in which the gene has been identified and is based both on personal observations and many recent reviews that summarize the clinical features (Durr, 2010; Manto and Marmolino, 2009; Paulson, 2009).
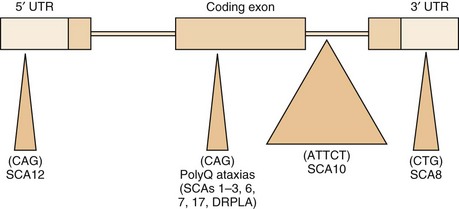
Fig. 72.11 Schematic representation of various repetitive nucleotide expansions in dominant ataxias.
(Courtesy Dr. H. Paulson, University of Iwo.)
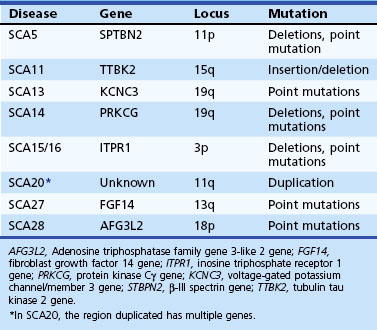
Spinocerebellar Ataxia Type 1
Age at onset of SCA1 is variable from childhood to over 70 years of age, but the mean is in the 30s (Sasaki et al., 1996). The early clinical picture is one of a cerebellar syndrome associated with upper motor neuron signs. Later, ophthalmoparesis, slow saccades, a sensory predominant polyneuropathy, amyotrophy, and some extrapyramidal features such as chorea and dystonia can develop. Dysarthria is early and significant dysphagia a later symptom. There can be ineffective cough and facial twitches. Cognitive changes such as executive dysfunction occur, and the usual course before death is about 15 to 20 years. The gene mutation is an unstable CAG expansion in the ataxin-1 gene on chromosome 6, and there is an overlap between the upper end of the normal and the lower end of the pathogenic range. However, the large normal alleles can be differentiated from the small pathogenic alleles by the presence of CAT interruptions in the former, detected by the presence of a restriction site for the enzyme, SfaNI (Chung et al., 1993).
Spinocerebellar Ataxia Type 2
SCA2 resembles SCA1 but was originally distinguished based on genetic studies of a large inbred population in Eastern Cuba (Orozco Diaz et al., 1990; Sasaki et al., 1996; Wadia et al., 1998). The presence of slow saccades early in the disease as well as peripheral neuropathy with generalized areflexia may point to SCA2. Other features that have been described in SCA2 include dystonia, l-dopa-responsive parkinsonism, and cognitive decline (Burk et al., 1999; Cancel et al., 1997). An infantile form resulting from a hyperexpanded allele has been described. The mutation is a CAG expansion in the ataxin-2 gene on chromosome 12, with normal alleles varying from 15 to 31 repeats. While normal alleles usually have CAA interruptions, pathogenic alleles are usually contiguous, but exceptions have been reported (Costanzi-Porrini et al. 2000).
Machado-Joseph Disease
MJD, or SCA3, is probably the most prevalent of the SCAs worldwide (Riess et al., 2008). Once again, the disorder has many similarities to SCA1 and 2, with an age at onset and course not too different from that of SCA1. Like SCA2, it appears to have a wider phenotypical spectrum. Apart from cerebellar signs, patients develop many brainstem signs such as facial and tongue fasciculations or myokymia, poor cough, facial atrophy, and dysphagia. Non-cerebellar eye signs such as slow saccades, disconjugate movements, ophthalmoparesis, ptosis, eyelid retraction, and blepharospasm have all been seen in MJD. In many patients, extrapyramidal features such as parkinsonism responsive to l-dopa or dystonia can dominate the clinical picture (Subramony, 1993; Subramony et al., 2002). Later-onset cases may be characterized by ataxia, amyotrophy, and peripheral neuropathy and areflexia. Other features include a cerebellar cognitive decline, sleep disturbances including restless legs, and autonomic deficits (Friedman et al., 2003; Kawai et al., 2004; Yeh et al., 2005). Impaired temperature discrimination in a generalized fashion can be a sign of MJD (Schols et al., 1996). The mutation is an unstable CAG expansion in the ataxin-3 gene on chromosome 14, and the pathogenic range from 53 to 86 is usually easily distinguished from the upper normal limit of 47 repeats.
Spinocerebellar Ataxia Type 4
SCA4 has been localized to chromosome 16q and causes a combination of sensory motor neuropathy and ataxia (Flanigan et al., 1996).
Spinocerebellar Ataxia Type 5
This dominant ataxia described in the descendents of the paternal grandparents of American President Abraham Lincoln (so-called Lincoln ataxia) has also been reported in French and German families. Age at onset is wide (10-68 years) with apparent anticipation. The disease is slowly progressive with mostly cerebellar deficits with added minor pyramidal signs. Mutations in the β-III spectrin gene (SPTBN2) have been recently described (Ikeda et al., 2006). In two of the families, small exonic deletions were found; the third family had a missense mutation (758 T to C). It is believed that β-III spectrin may have a role in stabilizing the glutamate transporter, EAAT4, at the plasma membrane.
Spinocerebellar Ataxia Type 7
This is the only SCA in which prominent visual loss related to a maculopathy occurs. Otherwise the clinical picture resembles that of SCA1 and 2 and MJD, with a combination of cerebellar ataxia and spasticity, often of an extreme nature. Slow saccades are frequent; extrapyramidal signs and peripheral neuropathy are uncommon. In different family members, visual loss and CNS features can occur in a variable combination. Subclinical visual impairment can be detected by a tritan axis defect found on the Farnsworth 15 color vision test or by electroretinogram. SCA7 is related to a CAG expansion in the ataxin-7 gene on chromosome 3. This expansion tends to be more unstable than other CAG expansions, and childhood-onset cases, usually inherited by paternal transmission, have been documented. Onset younger than age 12 was seen with repeat expansion sizes of over 67 (Johansson et al., 1998), and early-onset cases often have visual loss preceding the ataxia, a situation different from adult-onset cases. Such cases may present before the affected parent becomes symptomatic and have a more florid clinical picture including cognitive changes and seizures with a more rapid course. Similarly, a devastating infantile form of the disease due to a very large expansion can occur, and the expansion may escape detection by routine PCR testing.
Spinocerebellar Ataxia Type 8
SCA8 has been related to an unstable expansion of a CTG tract (110-250; normal, <73) in the 3′ untranslated region of the gene on chromosome 13 (Koob et al., 1999). Patients have a slowly progressive ataxia and variable spasticity and brisk tendon reflexes. Tremor and fluctuation of symptoms were noted in Finnish patients with the SCA8 expansion (Juvonen et al., 2000). The primary role of this expansion in causing ataxia has been controversial because the same CTG expansion has also been found in patients with other disorders such as stroke and Parkinson disease and even in general population samples (Stevanin et al., 2000; Worth et al., 2000). The vertical transmission of the expansion has been extensively studied by Koob et al. (1999) in only one family in which a significant logarithm of odds (LOD) score was calculated. Even in this family, there were several nonpenetrant individuals who had expansions of the repeat into larger than normal but presumably nonpathogenic range (250-800 repeats). Thus the interpretation of an expanded CTG repeat in an individual with ataxia remains controversial, and this test is probably not useful for predictive testing of at-risk individuals. However, the SCA8 expansion may be one of the more common mutations identified among “sporadic” ataxia patients.
Spinocerebellar Ataxia Type 10
Still another form of repeat expansion occurs in SCA10, a disorder typified by progressive ataxia and epilepsy. The mutation is a large unstable expansion (800-4500 repeats; normal <29) of a pentanucleotide repeat (ATTCT) on chromosome 22 (Matsuura et al., 2000). Age at onset is 14 to 45 years. Ataxia is a uniform feature and is slowly progressive. Seizures, both generalized and complex partial, occur in 25% to 85% of affected families. Variable pyramidal, peripheral nerve, and neuropsychiatric features occur (Lin and Ashizawa, 2005).
Spinocerebellar Ataxia Type 11
SCA11 has been described in a limited number of families and causes ataxia, upper motor neuron signs, and abnormal eye movements. Mutations have been described in the tubulin tau kinase gene (TTBK2) (Houlden et al., 2007).
Spinocerebellar Ataxia Type 12
SCA12 was originally described in an American family of German descent, but most of the other families have been reported from the Indian subcontinent (Holmes et al., 2003). The usual features include an action tremor of hands and head, bradykinesia, mild ataxia, and psychiatric features, with onset usually in the 40s. MRI of the brain shows more generalized atrophy. The CAG expansion that is related to SCA12 occurs in the promoter region of the PPP2R2B gene on chromosome 5 and is not apparently coded into a polyglutamine tract.
Spinocerebellar Ataxia Type 13
Two families, one of French and another of Filipino descent, have been found to have this mutation so far. The French family had childhood onset of very slowly progressive ataxia and mild developmental delay (Herman-Bert et al., 2000). The Filipino family had a later onset age and no mental retardation (Waters, Fee et al. 2005). Both had missense mutations in exon 2 of the voltage-gated potassium channel (KCNC3), 1154G→A and 1639C→A (Waters et al., 2006).
Spinocerebellar Ataxia Type 14
SCA14 is characterized by a complex and variable clinical picture including ataxia, axial myoclonus, chorea, head tremor, and rippling muscles in some (Klebe et al., 2005; Yabe et al., 2003). Facial myokymia, diplopia, dystonia rigidity, and proprioceptive loss have also been noted. SCA14 has been linked to several missense mutations in both the regulatory and catalytic domains of the protein kinase (PK)Cγ gene on chromosome 19.
Spinocerebellar Ataxia Types 15 and 16
SCA15 and 16 were originally reported from Australia and Japan, respectively, but more recent findings suggest that the same gene mutation underlies these entities (Iwaki et al., 2008; van de Leemput et al., 2007). The disease causes pure cerebellar ataxia, sometimes with associated postural tremor. Deletions involving two contiguous genes, SUMF1 and ITPR1, were found in the Australian kindred, but in the Japanese patients, only the ITPR1 gene was deleted, suggesting that this is the critical gene.
Spinocerebellar Ataxia Type 17
SCA17 has an extremely variable phenotype that includes ataxia, upper motor neuron signs, early and severe cognitive decline with significant behavioral changes, dyskinesias, dystonia, and parkinsonian signs. This is considered to be a polyglutamine disease and is linked to an expanded CAG repeat in the TATA-binding protein (TBP) gene (pathogenic range 45-66 repeats; normal, <42). Inheritance is autosomal dominant, but occasional sporadic cases have been reported (Hagenah et al., 2004; Rolfs et al., 2003).
Spinocerebellar Ataxia Type 18
Brkanac et al. (2002) described an autosomal dominant condition characterized by both ataxia and a sensory motor neuropathy in a five-generation American family of Irish ancestry and linked this to chromosome 7q22-q32.
Spinocerebellar Ataxia Type 19
SCA19 has been reported in a family of Dutch origin with a mild ataxia, myoclonus, irregular postural tremor, and cognitive impairment. Among 11 family members with clinical data, many also had decreased proprioception and tendon reflexes. The disorder in this family was mapped to chromosome 1p21-q21 by Verbeek et al. (2002).
Spinocerebellar Ataxia Type 20
SCA20 was described in an Australian family with a wide range of age at onset, prominent dysarthria and dysphonia, and the presence of calcification in the cerebellar dentate (Knight et al., 2004). The disease is localized to chromosome 11 and appears related to the duplication of a 260-kb segment on chromosome 11q. Its pathogenesis remains unclear.
Spinocerebellar Ataxia Type 21
In a family with autosomal dominant ataxia with onset in childhood and young-adult life, a new gene locus was established at chromosome 7p (Vuillaume et al., 2002). In addition to ataxia, the patients had tremor, rigidity, and cognitive impairment.
Spinocerebellar Ataxia Type 22
Chung et al. (2003) reported a Chinese family with dominant ataxia, hyporeflexia, onset between 10 and 46 years of age, and MRI showing isolated cerebellar atrophy; locus was established at chromosome 1p, overlapping that reported in SCA19.
Spinocerebellar Ataxia Type 23
In a single family with adult-onset dominant ataxia, the locus was mapped to chromosome 20p (Verbeek 2009).
Spinocerebellar Ataxia Type 25
In a single family from France, Stevanin et al. (2004) noted childhood onset of a dominant ataxia with variable progression and an associated peripheral neuropathy. Linkage studies localized the disorder to chromosome 2p.
Spinocerebellar Ataxia Type 26
Yu et al. (2005) have described a single family of Norwegian descent with relatively pure cerebellar ataxia with little in the way of extracerebellar signs. Age at onset was from 33 to 60 years. MRI showed cerebellar atrophy, but brainstem was spared. Genetic mapping has localized this disease to chromosome 19p13.3 adjacent to the SCA6 locus.
Spinocerebellar Ataxia Type 27
In a Dutch family with ataxia, early hand tremor, and psychiatric symptoms such as aggressive outbursts, a missense mutation in the fibroblast growth factor 14 gene has been identified (van Swieten et al., 2003).
Dentatorubral Pallidoluysian Atrophy
DRPLA is most prevalent in Japan, but molecularly proven DRPLA has been reported from Europe and the United States. The original African American family in which the typical neuropathology was described many years ago is now known to carry the same mutation as the Japanese cases (Burke et al., 1994). DRPLA has a very variable age at onset from infancy to old age, and anticipation in age at onset is common. With onset younger than 20 years of age, the clinical picture is one of seizures, myoclonus, ataxia, and intellectual decline. With older onset, the disease is characterized by ataxia, chorea, dementia, and psychiatric features. Thus the differential diagnosis varies with age and includes myoclonic epilepsy syndromes in children and ataxia and Huntington disease in older persons. Diagnosis can be established by documenting an expanded CAG repeat in the atrophin-1 gene.
Episodic Ataxias
The EAs (Table 72.8) are characterized by brief reversible attacks of ataxia of variable duration, often associated with other neurological features. However, in some of them, a more persistent neurological deficit may develop or may be present from the very onset, blurring the distinction from the SCAs. On the flip side, some of the SCAs (such as SCA6) may have features reminiscent of episodic attacks. Many of the EA syndromes have pediatric onset.
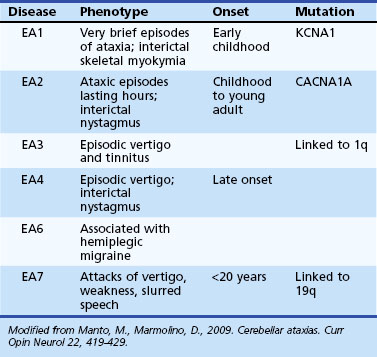
EA1 is characterized by brief episodes of ataxia lasting seconds to minutes, associated with a coarse tremor and dysarthria beginning in childhood. Interictal skeletal muscle myokymia may be detected clinically or only by electrophysiological studies. Partial epilepsy, transient postural abnormalities, and tight heel cords have been seen in some children (Jen et al., 2007). Other variant phenotypes have included myokymia with no ataxia and also persistent ataxia (Demos et al., 2009). Mutations in the potassium channel gene, KCNA1, have been identified in EA1.
Episodes of ataxia in EA2 last many hours and are associated with other symptoms such as headache, nausea, vomiting, diplopia, and dysarthria (Jen et al., 2007). Interictally, many EA2 patients have mild cerebellar deficits such as nystagmus (often downbeating) and mild gait problems. EA2 patients may have a progressive ataxia later in life. Unusual features have been described, including children with features of benign paroxysmal vertigo and cognitive decline associated with attention-deficit disorder (Bertholon et al., 2009). EA2 mutations may be associated with seizures, coma, and cerebral edema after mild head trauma. EA2 is allelic to two other diseases, familial hemiplegic migraine and SCA6.
Pathogenesis of Dominant Ataxias
The pathogenesis of the dominant ataxias has been recently reviewed (Carlson et al., 2009; Matilla-Duenas et al., 2010). The CAG expansion disorders have been considered to be examples of “toxic gain-of-function” mutations related to the misfolding and aggregation of the protein product of the expanded allele. The presence of nuclear (Fig. 72.12) or cytoplasmic aggregates containing the protein in question has been identified in all these diseases. Such misfolded protein can affect numerous other nuclear and cytoplasmic processes. Important pathways that have been noted to be affected include the ubiquitin proteasome pathway and molecular chaperones, components of both of which can be seen to co-localize with the aggregated proteins. Also, manipulations of both systems have been shown to alter the toxicity of the mutant protein. Transcriptional dysregulation due to misfolded protein within the nucleus may be a major factor. In addition, many ataxins may participate in transcriptional regulation in a more direct fashion (Carlson et al., 2009). Abnormalities of calcium signaling may play a role in some SCAs. Many genes involved in calcium handling may be down regulated and calcium homeostasis disturbed. The SCA6 mutation involves the calcium channel and can affect channel function. Secondary changes in mitochondrial function leading to oxidative stress and activation of apoptotic pathways involving caspase 3 and 9 and mitochondrial apoptotic mechanisms have been noted. The gain-of-function hypothesis has led to the possibility of therapy by switching off the mutated allele using RNA interference (Xia et al., 2004). Other potential treatment approaches include targeting many of the downhill events including protein aggregation, abnormal mitochondrial function, activation of caspases, and altered calcium homeostasis.
Conventional mutations leading to SCAs have further shed light on possible pathogenic mechanisms of these diseases. Many of these diseases cause prominent cerebellar dysfunction. The cerebellum has a major role in coordination of motor activities and may have a similar role in cognitive processing as well. The Purkinje cells function as intrinsic parts of neural circuits such as the cerebro-cerebellar loop (cerebral cortex-pons-cerebellum-thalamus-cerebral cortex) and the cerebello-olivary loop (cerebellum-red nucleus-inferior olive-cerebellum) in addition to receiving peripheral input via the spinocerebellar pathways. They have a spontaneous firing rate often modified by input from the olivary neurons via the climbing fiber input and in turn inhibit the deep cerebellar nuclei which provide the output of the cerebellum. The various inputs to the Purkinje cells allow modulation of motor activities based on motor programs from the cerebrum as well as peripheral input by appropriate modulation of deep cerebellar nuclei by Purkinje cells. In addition, these inputs allow synaptic plasticity changes that may underlie motor learning. Purkinje cells use the inositol triphosphate receptor pathway to link synaptic input to long-term depression and long-term potentiation (Schorge et al., 2010). Parallel fibers that excite Purkinje cells activate the GluR1 receptor, which in turn releases inositol triphosphate, leading to activation of the ITPR1 receptor (a calcium channel in the endoplasmic reticulum) and calcium release. The climbing fiber input acts via the GluR2 receptor, and the PQ-type calcium channels coordinate modulation of ITPR1 by climbing fibers. Thus, the ITPR1 pathway is critical for adequate Purkinje cell activity. Many of the mutations in SCAs disrupt this pathway. The β-spectrin mutations in SCA5 alter the stability of glutamate transporters such as EAAT4, possibly causing alterations in glutamate signaling. In SCA13, changes in presynaptic potassium channel function may lead to slower inactivation of parallel fiber input to Purkinje cells. Mutations in the ITPR1 itself underlie SCA15/16. Activation of ITPR1 normally activates PKCγ, mutations of which lead to SCA14. The PQ channels that modulate climbing fiber input to the ITPR1 system are mutated in EA2 and SCA6. It is possible that some of the many secondary cellular alterations seen in polyglutamine ataxias also involve this critical pathway. Alterations in synaptic, ion channel, and signaling pathways may potentially lead to significant neurological deficits in the absence of total neuronal loss, allowing for potential pharmacotherapy (Shakkottai and Paulson, 2009).
X-Linked Ataxias
Ataxia inherited in an X-linked fashion has been described in the literature. Of more interest is the fragile X tremor-ataxia syndrome (FXTAS) that has been reported among grandfathers of patients with fragile X mental retardation with an expansion in the FMR1 gene to a permutation range (Leehey, 2009). The full mutation is an expansion of a CGG repeat in the gene to over 200; normal chromosomes have fewer than 54 repeats. The maternal grandfathers of patients may carry a permutation in the 55 to 200 range. It has been estimated that about a third of these men can develop neurological disease in later life. The phenotype of this syndrome has included ataxia, tremor, frontal executive dysfunction, and global brain atrophy. Dysautonomia, mild parkinsonian features, and psychiatric disturbances may occur as well. It may be worthwhile to look for the permutation in patients with idiopathic ataxia and tremor or multiple system atrophy phenotype, especially if there is a family history of mental retardation. Many of these patients have been described to have a characteristic T2 hyperintensity in the middle cerebellar peduncles in addition to cerebral white-matter changes. Occasional female permutation carriers with FXTAS have been described and have less severe disease. Pathologically, there are characteristic eosinophilic intranuclear inclusions in neurons and astrocytes. It has been proposed that alleles with the permutation produce elevated levels of the corresponding mRNA, and this may be another example of RNA gain of function.
Sporadic Ataxias
The occurrence of progressive cerebellar ataxia clinically resembling inherited ataxias but with no definite genetic etiology has been recognized for over 100 years. The term sporadic ataxia has been used for such a process when other well-established causes of cerebellar ataxia have been excluded, though some use the term sporadic ataxia to mean ataxia with no genetic etiology. Some of the common causes of ataxia such as multiple sclerosis, strokes, and tumors can be easily excluded by imaging studies. Other causes of ataxia such as alcohol and hypothyroidism have nonspecific imaging findings and can only be diagnosed by appropriate history and laboratory studies. There is little understanding of the etiopathogenesis of truly sporadic cases of ataxia. Sporadic ataxia with childhood or young-adult onset may still have hitherto undefined single gene mutations as the underlying cause. Sporadic ataxia with onset in older adults (idiopathic late-onset ataxia) may be the result of a complex interplay of genetic and environmental factors. It should be noted that among patients with a diagnosis of sporadic ataxia, a very small percentage will test positive for one of the known gene mutations or may have one of the disorders noted in Table 72.3. It is difficult to make recommendations regarding the specific gene tests that should be obtained in a patient with sporadic ataxia. In a recent study of 112 cases of sporadic ataxia, the FA mutation was found in 4%, SCA2 in 1%, SCA3 in 2%, and SCA6 in 6% (Abele et al., 2002). The fragile X permutation may also be associated with such cases (Brussino et al., 2005). Other mutations often seen in this group of patients include the SCA8 CTG expansion and the SCA5 β-spectrin mutations. One should consider some of the mutation analyses in patients with sporadic ataxia if the family history is not very clear or the clinical picture is more typical for one of the genetically determined ataxias. Among patients with idiopathic late-onset ataxia, some have added noncerebellar signs, some do not.
Sporadic Ataxia with Added Non-Cerebellar Deficits
Patients with sporadic ataxia with added non-cerebellar deficits initially have slowly progressive ataxia but soon develop upper motor neuron signs, ophthalmoplegia, parkinsonian features, and autonomic failure. Both MRI and pathological examination show cerebellar and brainstem degeneration. Autonomic structures may be affected as well, so the clinical picture of this disorder merges with that of the olivopontocerebellar atrophy form of multiple system atrophy (MSA). Gilman et al. (2000) have shown that among patients with idiopathic progressive ataxia, 25% to 35% will transition to probable MSA over 5 to 10 years, and a similar rate was observed by Abele et al. as well (2002). Clinical evidence for this in most patients will be added parkinsonian and autonomic deficits, but in a few, only autonomic failure develops. Signs of autonomic failure include orthostatic hypotension, incontinence, and erectile dysfunction. Parkinsonian signs include rigidity and facial hypomimia. Older age at onset and a more rapid progression to a disabled state confer a higher risk of such a transition; median survival after such transition was only 3.5 years. Sphincter electromyography (EMG) and cardiovascular autonomic testing may be useful to confirm the diagnosis of MSA. Signs of autonomic failure such as orthostatic hypotension and cardiac denervation are uncommon in the inherited ataxias, though bladder symptoms are often present in late stages.
Clinical Approach to Patients with Progressive Cerebellar Ataxia
A careful clinical approach to patients presenting with progressive ataxia will allow accurate diagnosis and appropriate management (Fig. 72.13). The age at onset, the tempo of progression, associated neurological and systemic signs, and the availability of family history can all be considered in making a diagnosis. Imaging studies, especially MRI of the brain, allow the diagnosis of those disorders in which characteristic morphological abnormalities underlie the ataxia (e.g., infarcts, mass lesions, demyelinating plaques). Among the degenerative ataxias, MRI can be helpful by documenting isolated cerebellar versus pontocerebellar atrophy and the presence of additional abnormalities such as cortical atrophy or hyperintensity in the middle cerebellar peduncle. Other laboratory studies of value in a patient with progressive ataxia include thyroid function tests, vitamin E and B12 measurements, serology for syphilis, gliadin and GAD antibodies, paraneoplastic antibodies, and possibly CSF examination for pleocytosis, such as can be seen with meningeal carcinomatosis, and for oligoclonal bands. PCR analysis for the WD organism may be useful. In young adults and children, when appropriate clinical circumstances exist, specialized laboratory studies including those in Table 72.3 are indicated. Careful elicitation of family history and the details of the illness in other family members are often of value.
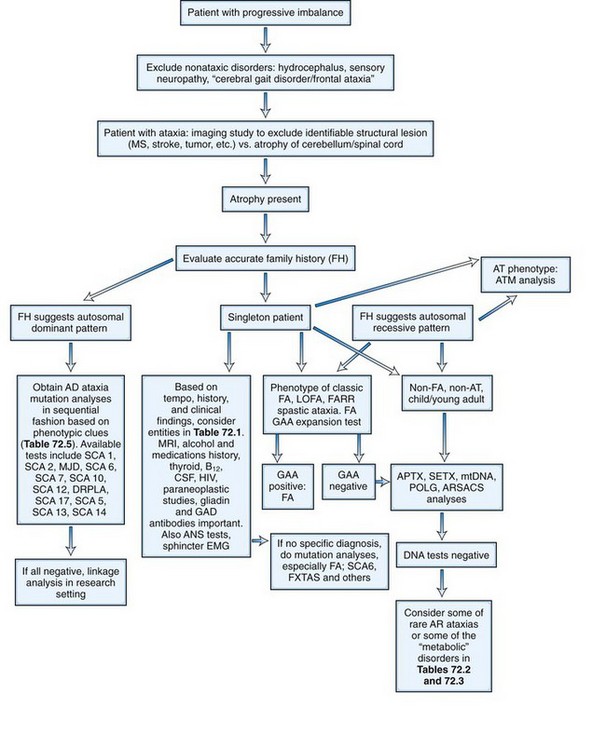
The art and science of molecular testing in inherited ataxias is an evolving one. As of now, DNA tests are available for the CAG expansions involved in SCA1, 2, 3 (MJD), 6, 7, 12, 17, and DRPLA. The CTG expansion associated with SCA8 and the ATTCT expansion in SCA10 are other existing tests. In addition, tests that detect conventional mutations in SCA5, SCA13, SCA14, and EA2 are available. Among the AR ataxias, testing can be done for the FA GAA expansion and mutations in the POLG, aprataxin, and senataxin genes. Tests for the common mutations in mtDNA that may be associated with ataxias such as MERRF, MELAS, and NARP are offered. Gene tests are also possible for AT, ARSACS, AVED, and EA syndromes and many other recently defined gene mutations can be tested for by selected labs or research facilities (see www.geneclinics.org). Patients with features compatible with an inherited ataxia as described in this chapter are possible candidates for gene testing. Such testing is very accurate and usually less expensive than many tests usually employed in neurological diagnosis. Often the choice of tests can be made based on the inheritance pattern and some of the clinical clues alluded to earlier. When family history suggests a dominant mode of inheritance, mutation analysis for the SCAs may be the only test needed. In addition, the prevalence of different genotypes can vary in different areas, probably due to genetic bias caused by strong founder effects. For instance, the FA mutation is confined to Indo-Caucasian populations. The dominant ataxias have different prevalence rates in different areas (Schols et al., 2004) (Fig. 72.14). With regard to the nucleotide repeat expansion disorders, interpretive caution may be required for many reasons. The alleles may be classified into those with normal numbers of repeats (nonpathogenic) and clearly pathogenic ( fully penetrant) alleles. In addition, alleles that have repeat numbers in the gray zone between normal and pathogenic can be found in many diseases. These have been classified as mutable normal (i.e., an allele size that is not yet known to be associated with disease signs but may expand into pathogenic range when transmitted to the next generation) and low penetrance alleles that can cause disease at a late age. The website www.geneclinics.org is an excellent resource for information regarding the evolving information about such tests, and Tables 72.9 and 72.10 summarize some of this information. The FA mutation analysis is indicated in anyone who has a phenotype compatible with classical FA, LOFA, or FARR. Since the phenotypical variability of FA has been found to be extensive, one should consider doing this test in almost all childhood or young adult–onset sporadic ataxia that cannot be readily diagnosed. In the dominant ataxias, some of the phenotypical features noted in Table 72.5 may be of use, but these serve only as very rough guides. Sequence analysis for point mutations is becoming increasingly available; a particular sequence alteration reported may be difficult to interpret, but it may be one already known to be pathogenic. If not, whether a given alteration is the real cause of the disease can only be judged based on more research—for example, the alteration occurs in a highly conserved site and can be predicted to alter protein function, or the alteration segregates with many other affected individuals in the family and cannot be found in control chromosomes from the same ethnic group. There are also no firm guidelines for the use of ataxia tests among patients with sporadic ataxia. If the disorder cannot be adequately diagnosed, at least FA, SCA6, SCA8, and fragile X analysis should be done (see also Sporadic Ataxias). If the clinical features are more typical for an inherited ataxia despite a negative family history or the family history is sketchy, the entire dominant ataxia panel should be performed.
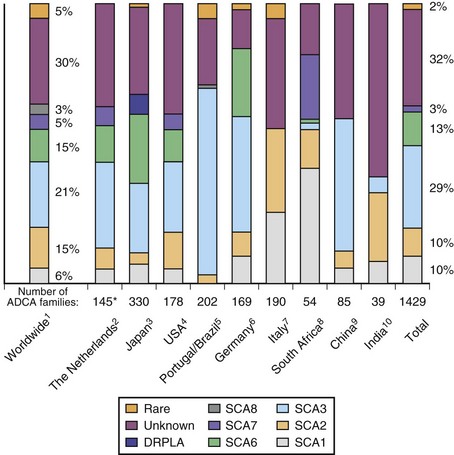
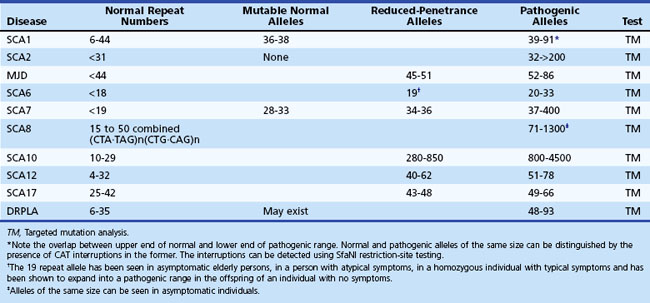
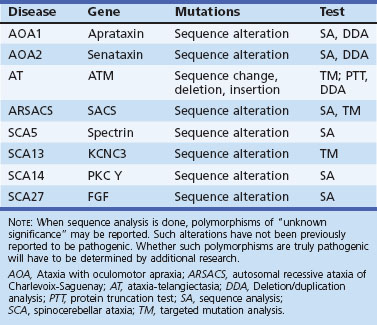
Abele M., Burk K., et al. The aetiology of sporadic adult-onset ataxia. Brain. 2002;125(Pt 5):961-968.
Ammar H., Brown S.H., et al. A case of paraneoplastic cerebellar ataxia secondary to renal cell carcinoma. South Med J. 2008;101(5):556-557.
Anheim M., Fleury M., et al. Epidemiological, clinical, paraclinical and molecular study of a cohort of 102 patients affected with autosomal recessive progressive cerebellar ataxia from Alsace, Eastern France: implications for clinical management. Neurogenetics. 2010;11(1):1-12.
Anheim M., Monga B., et al. Ataxia with oculomotor apraxia type 2: clinical, biological and genotype/phenotype correlation study of a cohort of 90 patients. Brain. 2009;132(Pt 10):2688-2698.
Anttonen A.K., Mahjneh I., et al. The gene disrupted in Marinesco-Sjogren syndrome encodes SIL1, an HSPA5 co-chaperone. Nat Genet. 2005;37(12):1309-1311.
Armstrong M.B., Robertson P.L., et al. Delayed, recurrent opsoclonus-myoclonus syndrome responding to plasmapheresis. Pediatr Neurol. 2005;33(5):365-367.
Babcock M., de Silva D., et al. Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science. 1997;276(5319):1709-1712.
Barnard R.O., Campbell M.J., et al. Pathological findings in a case of hypothyroidism with ataxia. J Neurol Neurosurg Psychiatry. 1971;34(6):755-760.
Bataller L., Wade D.F., et al. Antibodies to Zic4 in paraneoplastic neurologic disorders and small-cell lung cancer. Neurology. 2004;62(5):778-782.
Bayreuther C., Hieronimus S., et al. Auto-immune cerebellar ataxia with anti-GAD antibodies accompanied by de novo late-onset type 1 diabetes mellitus. Diabetes Metab. 2008;34(4 Pt 1):386-388.
Bernal F., Shams’ili S., et al. Anti-Tr antibodies as markers of paraneoplastic cerebellar degeneration and Hodgkin’s disease. Neurology. 2003;60(2):230-234.
Bertholon P., Chabrier S., et al. Episodic ataxia type 2: unusual aspects in clinical and genetic presentation. Special emphasis in childhood. J Neurol Neurosurg Psychiatry. 2009;80(11):1289-1292.
Bidichandani S.I., Purandare S.M., et al. Somatic sequence variation at the Friedreich ataxia locus includes complete contraction of the expanded GAA triplet repeat, significant length variation in serially passaged lymphoblasts and enhanced mutagenesis in the flanking sequence. Hum Mol Genet. 1999;8(13):2425-2436.
Bier S.A., Hile D.C. Emergency department presentation of a rare neurological disorder. J Emerg Med. 2010;38(4):452-455.
Boesch S., Sturm B., et al. Neurological effects of recombinant human erythropoietin in Friedreich’s ataxia: a clinical pilot trial. Mov Disord. 2008;23(13):1940-1944.
Bomar J.M., Benke P.J., et al. Mutations in a novel gene encoding a CRAL-TRIO domain cause human Cayman ataxia and ataxia/dystonia in the jittery mouse. Nat Genet. 2003;35(3):264-269.
Brkanac Z., Fernandez M., et al. Autosomal dominant sensory/motor neuropathy with Ataxia (SMNA), Linkage to chromosome 7q22-q32. Am J Med Genet. 2002;114(4):450-457.
Brussino A., Gellera C., et al. FMR1 gene premutation is a frequent genetic cause of late-onset sporadic cerebellar ataxia. Neurology. 2005;64(1):145-147.
Burk K., Bosch S., et al. Sporadic cerebellar ataxia associated with gluten sensitivity. Brain. 2001;124(Pt 5):1013-1019.
Burk K., Globas C., et al. Cognitive deficits in spinocerebellar ataxia 2. Brain. 1999;122(Pt 4):769-777.
Burke J.R., Wingfield M.S., et al. The Haw River syndrome: dentatorubropallidoluysian atrophy (DRPLA) in an African-American family. Nat Genet. 1994;7(4):521-524.
Bushara K.O., Goebel S.U., et al. Gluten sensitivity in sporadic and hereditary cerebellar ataxia. Ann Neurol. 2001;49(4):540-543.
Campuzano V., Montermini L., et al. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271(5254):1423-1427.
Cancel G., Durr A., et al. Molecular and clinical correlations in spinocerebellar ataxia 2: a study of 32 families. Hum Mol Genet. 1997;6(5):709-715.
Carlson K.M., Andresen J.M., et al. Emerging pathogenic pathways in the spinocerebellar ataxias. Curr Opin Genet Dev. 2009;19(3):247-253.
Cavalier L., Ouahchi K., et al. Ataxia with isolated vitamin E deficiency: heterogeneity of mutations and phenotypic variability in a large number of families. Am J Hum Genet. 1998;62(2):301-310.
Chen Y.Z., Bennett C.L., et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am J Hum Genet. 2004;74(6):1128-1135.
Chu G., Mayne L. Xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy: do the genes explain the diseases? Trends Genet. 1996;12(5):187-192.
Chung M.Y., Lu Y.C., et al. A novel autosomal dominant spinocerebellar ataxia (SCA22) linked to chromosome 1p21-q23. Brain. 2003;126(Pt 6):1293-1299.
Chung M.Y., Ranum L.P., et al. Evidence for a mechanism predisposing to intergenerational CAG repeat instability in spinocerebellar ataxia type I. Nat Genet. 1993;5(3):254-258.
Combarros O., Infante J., et al. Celiac disease and idiopathic cerebellar ataxia. Neurology. 2000;54(12):2346.
Connolly A.M., Dodson W.E., et al. Course and outcome of acute cerebellar ataxia. Ann Neurol. 1994;35(6):673-679.
Cooke W.T., Smith W.T. Neurological disorders associated with adult coeliac disease. Brain. 1966;89(4):683-722.
Cossee M., Durr A., et al. Friedreich’s ataxia: point mutations and clinical presentation of compound heterozygotes. Ann Neurol. 1999;45(2):200-206.
Costanzi-Porrini S., Tessarolo D., et al. An interrupted 34-CAG repeat SCA-2 allele in patients with sporadic spinocerebellar ataxia. Neurology. 2000;54(2):491-493.
Date H., Onodera O., et al. Early-onset ataxia with ocular motor apraxia and hypoalbuminemia is caused by mutations in a new HIT superfamily gene. Nat Genet. 2001;29(2):184-188.
Demos M.K., Macri V., et al. A novel KCNA1 mutation associated with global delay and persistent cerebellar dysfunction. Mov Disord. 2009;24(5):778-782.
Diener H.C., Dichgans J., et al. Improvement of ataxia in alcoholic cerebellar atrophy through alcohol abstinence. J Neurol. 1984;231(5):258-262.
Dohlinger S., Hauser T.K., et al. Magnetic resonance imaging in spinocerebellar ataxias. Cerebellum. 2008;7(2):204-214.
Dupre N., Gros-Louis F., et al. Clinical and genetic study of autosomal recessive cerebellar ataxia type 1. Ann Neurol. 2007;62(1):93-98.
Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol. 2010;9(9):885-894.
Durr A., Cossee M., et al. Clinical and genetic abnormalities in patients with Friedreich’s ataxia. N Engl J Med. 1996;335(16):1169-1175.
Elsheikh B.H., Maher W.E., et al. Cerebellar atrophy associated with human immunodeficiency virus infection. Arch Neurol. 2010;67(5):634-635.
Engert J.C., Berube P., et al. ARSACS, a spastic ataxia common in northeastern Quebec, is caused by mutations in a new gene encoding an 11.5-kb ORF. Nat Genet. 2000;24(2):120-125.
Fearnley J.M., Stevens J.M., et al. Superficial siderosis of the central nervous system. Brain. 1995;118(Pt 4):1051-1066.
Filla A., De Michele G., et al. The relationship between trinucleotide (GAA) repeat length and clinical features in Friedreich ataxia. Am J Hum Genet. 1996;59(3):554-560.
Flanigan K., Gardner K., et al. Autosomal dominant spinocerebellar ataxia with sensory axonal neuropathy (SCA4), clinical description and genetic localization to chromosome 16q22.1. Am J Hum Genet. 1996;59(2):392-399.
Fleming J., Spinoulas A., et al. Partial correction of sensitivity to oxidant stress in Friedreich ataxia patient fibroblasts by frataxin-encoding adeno-associated virus and lentivirus vectors. Hum Gene Ther. 2005;16(8):947-956.
Fogel B.L., Perlman S. Clinical features and molecular genetics of autosomal recessive cerebellar ataxias. Lancet Neurol. 2007;6(3):245-257.
Friedman J.H., Fernandez H.H., et al. REM behavior disorder and excessive daytime somnolence in Machado-Joseph disease (SCA-3). Mov Disord. 2003;18(12):1520-1522.
Gerard A., Sarrot-Reynauld F., et al. Neurologic presentation of Whipple disease: report of 12 cases and review of the literature. Medicine (Baltimore). 2002;81(6):443-457.
Giacchetti M., Monticelli A., et al. Mitochondrial DNA haplogroups influence the Friedreich’s ataxia phenotype. J Med Genet. 2004;41(4):293-295.
Gilman S., Junck L., et al. Cerebral glucose hypermetabolism in Friedreich’s ataxia detected with positron emission tomography. Ann Neurol. 1990;28(6):750-757.
Gilman S., Little R., et al. Evolution of sporadic olivopontocerebellar atrophy into multiple system atrophy. Neurology. 2000;55(4):527-532.
Gordon M.F., Abrams R.I., et al. Bismuth subsalicylate toxicity as a cause of prolonged encephalopathy with myoclonus. Mov Disord. 1995;10(2):220-222.
Gottlieb J.A., Luce J.K. Cerebellar ataxia with weekly 5-fluorouracil administration. Lancet. 1971;1(7690):138-139.
Hadjivassiliou M., Grunewald R., et al. Gluten ataxia in perspective: epidemiology, genetic susceptibility and clinical characteristics. Brain. 2003;126(Pt 3):685-691.
Hadjivassiliou M., Sanders D.S., et al. Gluten sensitivity: from gut to brain. Lancet Neurol. 2010;9(3):318-330.
Hagenah J.M., Zuhlke C., et al. Focal dystonia as a presenting sign of spinocerebellar ataxia 17. Mov Disord. 2004;19(2):217-220.
Hakonen A.H., Heiskanen S., et al. Mitochondrial DNA polymerase W748S mutation: a common cause of autosomal recessive ataxia with ancient European origin. Am J Hum Genet. 2005;77(3):430-441.
Halazonetis T.D., Shiloh Y. Many faces of ATM: eighth international workshop on ataxia-telangiectasia. Biochim Biophys Acta. 1999;1424(2-3):R45-R55.
Harding A.E. Friedreich’s ataxia: a clinical and genetic study of 90 families with an analysis of early diagnostic criteria and intrafamilial clustering of clinical features. Brain. 1981;104(3):589-620.
Harding A.E. The clinical features and classification of the late-onset autosomal dominant cerebellar ataxias. A study of 11 families, including descendants of the “the Drew family of Walworth”. Brain. 1982;105(Pt 1):1-28.
Harding A.E. Classification of the hereditary ataxias and paraplegias. Lancet. 1983;1(8334):1151-1155.
Hebert M.D. Targeting the gene in Friedreich ataxia. Biochimie. 2008;90(8):1131-1139.
Herman-Bert A., Stevanin G., et al. Mapping of spinocerebellar ataxia 13 to chromosome 19q13.3-q13.4 in a family with autosomal dominant cerebellar ataxia and mental retardation. Am J Hum Genet. 2000;67(1):229-235.
Hillbom M., Muuronen A., et al. The clinical versus radiological diagnosis of alcoholic cerebellar degeneration. J Neurol Sci. 1986;73(1):45-53.
Hoffmann L.A., Jarius S., et al. Anti-Ma and anti-Ta associated paraneoplastic neurological syndromes: 22 newly diagnosed patients and review of previous cases. J Neurol Neurosurg Psychiatry. 2008;79(7):767-773.
Holmes S.E., Hearn E.O, et al. Why is SCA12 different from other SCAs? Cytogenet Genome Res. 2003;100(1-4):189-197.
Honnorat J., Trouillas P., et al. Autoantibodies to glutamate decarboxylase in a patient with cerebellar cortical atrophy, peripheral neuropathy, and slow eye movements. Arch Neurol. 1995;52(5):462-468.
Hou J.G., Jankovic J. Movement disorders in Friedreich’s ataxia. J Neurol Sci. 2003;206(1):59-64.
Houlden H., Johnson J., et al. Mutations in TTBK2, encoding a kinase implicated in tau phosphorylation, segregate with spinocerebellar ataxia type 11. Nat Genet. 2007;39(12):1434-1436.
Ikeda Y., Dick K.A., et al. Spectrin mutations cause spinocerebellar ataxia type 5. Nat Genet. 2006;38(2):184-190.
Iwaki A., Kawano Y., et al. Heterozygous deletion of ITPR1, but not SUMF1, in spinocerebellar ataxia type 16. J Med Genet. 2008;45(1):32-35.
Jackson J.F., Currier R.D., et al. Spinocerebellar ataxia and HLA linkage: risk prediction by HLA typing. N Engl J Med. 1977;296(20):1138-1141.
Jellinek E.H., Kelly R.E. Cerebellar syndrome in myxoedema. Lancet. 1960;2(7144):225-227.
Jellinger K., Heiss W.D., et al. The ataxic (cerebellar) form of Creutzfeldt-Jakob disease. J Neurol. 1974;207(4):289-305.
Jen J.C., Graves T.D., et al. Primary episodic ataxias: diagnosis, pathogenesis and treatment. Brain. 2007;130(Pt 10):2484-2493.
Johansson J., Forsgren L., et al. Expanded CAG repeats in Swedish spinocerebellar ataxia type 7 (SCA7) patients: effect of CAG repeat length on the clinical manifestation. Hum Mol Genet. 1998;7(2):171-176.
Juvonen V., Hietala M., et al. Clinical and genetic findings in Finnish ataxia patients with the spinocerebellar ataxia 8 repeat expansion. Ann Neurol. 2000;48(3):354-361.
Karthikeyan G., Lewis L.K., et al. The mitochondrial protein frataxin prevents nuclear damage. Hum Mol Genet. 2002;11(11):1351-1362.
Karthikeyan G., Santos J.H., et al. Reduction in frataxin causes progressive accumulation of mitochondrial damage. Hum Mol Genet. 2003;12(24):3331-3342.
Kawai Y., Takeda A., et al. Cognitive impairments in Machado-Joseph disease. Arch Neurol. 2004;61(11):1757-1760.
Klebe S., Durr A., et al. New mutations in protein kinase C[gamma] associated with spinocerebellar ataxia type 14. Ann Neurol. 2005;58(5):720-729.
Klockgether T. Sporadic ataxia with adult onset: classification and diagnostic criteria. Lancet Neurol. 2010;9(1):94-104.
Knight M.A., Gardner R.J., et al. Dominantly inherited ataxia and dysphonia with dentate calcification: spinocerebellar ataxia type 20. Brain. 2004;127(Pt 5):1172-1181.
Koob M.D., Moseley M.L., et al. An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8). Nat Genet. 1999;21(4):379-384.
Kwakwa H.A., Ghobrial M.W. Primary cerebellar degeneration and HIV. Arch Intern Med. 2001;161(12):1555-1556.
Land R., Carter J., et al. Gynaecology meets neurology: paraneoplastic cerebellar degeneration. Aust N Z J Obstet Gynaecol. 2005;45(1):79-81.
Lavin M.F., Birrell G., et al. ATM signaling and genomic stability in response to DNA damage. Mutat Res. 2005;569(1-2):123-132.
Le Ber I., Bouslam N., et al. Frequency and phenotypic spectrum of ataxia with oculomotor apraxia 2: a clinical and genetic study in 18 patients. Brain. 2004;127(Pt 4):759-767.
Le Ber I., Moreira M.C., et al. Cerebellar ataxia with oculomotor apraxia type 1: clinical and genetic studies. Brain. 2003;126(Pt 12):2761-2772.
Leehey M.A. Fragile X-associated tremor/ataxia syndrome: clinical phenotype, diagnosis, and treatment. J Investig Med. 2009;57(8):830-836.
Lin X., Ashizawa T. Recent progress in spinocerebellar ataxia type-10 (SCA10). Cerebellum. 2005;4(1):37-42.
Lock R.J., Pengiran Tengah D.S., et al. Ataxia, peripheral neuropathy, and anti-gliadin antibody. Guilt by association? J Neurol Neurosurg Psychiatry. 2005;76(11):1601-1603.
Lodi R., Taylor D.J., et al. Mitochondrial dysfunction in Friedreich’s ataxia. Biol Signals Recept. 2001;10(3-4):263-270.
Luque F.A., Furneaux H.M., et al. Anti-Ri: an antibody associated with paraneoplastic opsoclonus and breast cancer. Ann Neurol. 1991;29(3):241-251.
Mani J., Chaudhary N., et al. Cerebellar ataxia due to lead encephalopathy in an adult. J Neurol Neurosurg Psychiatry. 1998;65(5):797.
Manto M., Marmolino D. Cerebellar ataxias. Curr Opin Neurol. 2009;22(4):419-429.
Mariotti C., Gellera C., et al. Ataxia with isolated vitamin E deficiency: neurological phenotype, clinical follow-up and novel mutations in TTPA gene in Italian families. Neurol Sci. 2004;25(3):130-137.
Markakis I., Alexiou E., et al. Opsoclonus-myoclonus-ataxia syndrome with autoantibodies to glutamic acid decarboxylase. Clin Neurol Neurosurg. 2008;110(6):619-621.
Mascalchi M., Salvi F., et al. Friedreich’s ataxia: MR findings involving the cervical portion of the spinal cord. AJR Am J Roentgenol. 1994;163(1):187-191.
Matilla-Duenas A., Sanchez I., et al. Cellular and molecular pathways triggering neurodegeneration in the spinocerebellar ataxias. Cerebellum. 2010;9(2):148-166.
Matsuura T., Yamagata T., et al. Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat Genet. 2000;26(2):191-194.
Matthews B.R., Jones L.K., et al. Cerebellar ataxia and central nervous system Whipple disease. Arch Neurol. 2005;62(4):618-620.
Meier T., Buyse G. Idebenone: an emerging therapy for Friedreich ataxia. J Neurol. 2009;256(Suppl 1):25-30.
Montermini L., Richter A., et al. Phenotypic variability in Friedreich ataxia: role of the associated GAA triplet repeat expansion. Ann Neurol. 1997;41(5):675-682.
Moreira M.C., Barbot C., et al. The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nat Genet. 2001;29(2):189-193.
Moreira M.C., Klur S., et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat Genet. 2004;36(3):225-227.
Musunuru K., Kesari S. Paraneoplastic opsoclonus-myoclonus ataxia associated with non-small-cell lung carcinoma. J Neurooncol. 2008;90(2):213-216.
Nanri K., Okita M., et al. Intravenous immunoglobulin therapy for autoantibody-positive cerebellar ataxia. Intern Med. 2009;48(10):783-790.
Nikali K., Suomalainen A., et al. Infantile onset spinocerebellar ataxia is caused by recessive mutations in mitochondrial proteins Twinkle and Twinky. Hum Mol Genet. 2005;14(20):2981-2990.
Nociti V., Frisullo G., et al. Refractory generalized seizures and cerebellar ataxia associated with anti-GAD antibodies responsive to immunosuppressive treatment. Eur J Neurol. 2010;17(1):e5.
Odaka M., Yuki N., et al. Bickerstaff’s brainstem encephalitis: clinical features of 62 cases and a subgroup associated with Guillain-Barre syndrome. Brain. 2003;126(Pt 10):2279-2290.
Orozco Diaz G., Nodarse Fleites A., et al. Autosomal dominant cerebellar ataxia: clinical analysis of 263 patients from a homogeneous population in Holguin, Cuba. Neurology. 1990;40(9):1369-1375.
Ouahchi K., Arita M., et al. Ataxia with isolated vitamin E deficiency is caused by mutations in the alpha-tocopherol transfer protein. Nat Genet. 1995;9(2):141-145.
Pandolfo M. Molecular basis of Friedreich ataxia. Mov Disord. 2001;16(5):815-821.
Pandolfo M. Friedreich ataxia. Semin Pediatr Neurol. 2003;10(3):163-172.
Pandolfo M., Pastore A. The pathogenesis of Friedreich ataxia and the structure and function of frataxin. J Neurol. 2009;256(Suppl 1):9-17.
Parchi P., Giese A., et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46(2):224-233.
Paulson H.L. The spinocerebellar ataxias. J Neuroophthalmol. 2009;29(3):227-237.
Pellecchia M.T., Scala R., et al. Cerebellar ataxia associated with subclinical celiac disease responding to gluten-free diet. Neurology. 1999;53(7):1606-1608.
Pennington C., Chohan G., et al. The role of cerebrospinal fluid proteins as early diagnostic markers for sporadic Creutzfeldt-Jakob disease. Neurosci Lett. 2009;455(1):56-59.
Posner J.B., Dalmau J.O. Paraneoplastic syndromes of the nervous system. Clin Chem Lab Med. 2000;38(2):117-122.
Puccio H., Koenig M. Recent advances in the molecular pathogenesis of Friedreich ataxia. Hum Mol Genet. 2000;9(6):887-892.
Rakocevic G., Raju R., et al. Stiff person syndrome with cerebellar disease and high-titer anti-GAD antibodies. Neurology. 2006;67(6):1068-1070.
Rashtak S., Snyder M.R., et al. Serology of celiac disease in gluten-sensitive ataxia or neuropathy: Role of deamidated gliadin antibody. J Neuroimmunol. 2010;230:130-134.
Rewers M. Epidemiology of celiac disease: what are the prevalence, incidence, and progression of celiac disease? Gastroenterology. 2005;128(4 Suppl 1):S47-S51.
Riess O., Rub U., et al. SCA3: neurological features, pathogenesis and animal models. Cerebellum. 2008;7(2):125-137.
Rolfs A., Koeppen A.H., et al. Clinical features and neuropathology of autosomal dominant spinocerebellar ataxia (SCA17). Ann Neurol. 2003;54(3):367-375.
Rotig A., de Lonlay P., et al. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat Genet. 1997;17(2):215-217.
Rub U., Schultz C., et al. Anatomically based guidelines for systematic investigation of the central somatosensory system and their application to a spinocerebellar ataxia type 2 (SCA2) patient. Neuropathol Appl Neurobiol. 2003;29(5):418-433.
Sabater L., Bataller L., et al. Protein kinase C[gamma] autoimmunity in paraneoplastic cerebellar degeneration and non-small-cell lung cancer. J Neurol Neurosurg Psychiatry. 2006;77(12):1359-1362.
Saiz A., Arpa J., et al. Autoantibodies to glutamic acid decarboxylase in three patients with cerebellar ataxia, late-onset insulin-dependent diabetes mellitus, and polyendocrine autoimmunity. Neurology. 1997;49(4):1026-1030.
Sakamoto N., Chastain P.D., et al. Sticky DNA: self-association properties of long GAA.TTC repeats in R.R.Y triplex structures from Friedreich’s ataxia. Mol Cell. 1999;3(4):465-475.
Salinsky M.C., Levine R.L., et al. Acute cerebellar dysfunction with high-dose ARA-C therapy. Cancer. 1983;51(3):426-429.
Sasaki H., Fukazawa T., et al. Central phenotype and related varieties of spinocerebellar ataxia 2 (SCA2): a clinical and genetic study with a pedigree in the Japanese. J Neurol Sci. 1996;144(1-2):176-181.
Sasaki H., Fukazawa T., et al. Clinical features and natural history of spinocerebellar ataxia type 1. Acta Neurol Scand. 1996;93(1):64-71.
Savitsky K., Bar-Shira A., et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268(5218):1749-1753.
Schols L., Amoiridis G., et al. Relations between genotype and phenotype in German patients with the Machado-Joseph disease mutation. J Neurol Neurosurg Psychiatry. 1996;61(5):466-470.
Schols L., Bauer P., et al. Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol. 2004;3(5):291-304.
Schorge S., van de Leemput J., et al. Human ataxias: a genetic dissection of inositol triphosphate receptor (ITPR1)-dependent signaling. Trends Neurosci. 2010;33(5):211-219.
Schulz J.B., Di Prospero N.A., et al. Clinical experience with high-dose idebenone in Friedreich ataxia. J Neurol. 2009;256(Suppl 1):42-45.
Senderek J., Krieger M., et al. Mutations in SIL1 cause Marinesco-Sjogren syndrome, a cerebellar ataxia with cataract and myopathy. Nat Genet. 2005;37(12):1312-1314.
Shakkottai V.G., Paulson H.L. Physiologic alterations in ataxia: channeling changes into novel therapies. Arch Neurol. 2009;66(10):1196-1201.
Sillevis Smitt P., Kinoshita A., et al. Paraneoplastic cerebellar ataxia due to autoantibodies against a glutamate receptor. N Engl J Med. 2000;342(1):21-27.
Sirven J.I., Fife T.D., et al. Second-generation antiepileptic drugs’ impact on balance: a meta-analysis. Mayo Clin Proc. 2007;82(1):40-47.
Stevanin G., Bouslam N., et al. Spinocerebellar ataxia with sensory neuropathy (SCA25) maps to chromosome 2p. Ann Neurol. 2004;55(1):97-104.
Stevanin G., Herman A., et al. Are (CTG)n expansions at the SCA8 locus rare polymorphisms? Nat Genet. 2000;24(3):213. author reply 215
Subramony S.H., editor. Presumed Machado-Joseph disease: four kindreds from Mississippi. Handbook of Cerebellar Diseases. New York: Marcel-Dekker, 1993.
Subramony S.H. Genetics of inherited ataxias. Continuum. 2005;11:8.
Subramony S.H., Hernandez D., et al. Ethnic differences in the expression of neurodegenerative disease: Machado-Joseph disease in Africans and Caucasians. Mov Disord. 2002;17(5):1068-1071.
Swift M., Heim R.A., et al. Genetic aspects of ataxia telangiectasia. Adv Neurol. 1993;61:115-125.
Tagliati M., Simpson D., et al. Cerebellar degeneration associated with human immunodeficiency virus infection. Neurology. 1998;50(1):244-251.
Takashima H., Boerkoel C.F., et al. Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat Genet. 2002;32(2):267-272.
Takiyama Y. Sacsinopathies: sacsin-related ataxia. Cerebellum. 2007:1-7.
Taniguchi Y., Tanji C., et al. A case report of plasmapheresis in paraneoplastic cerebellar ataxia associated with anti-Tr antibody. Ther Apher Dial. 2006;10(1):90-93.
van de Leemput J., Chandran J., et al. Deletion at ITPR1 underlies ataxia in mice and spinocerebellar ataxia 15 in humans. PLoS Genet. 2007;3(6):e108.
Van Lierde A., Righini A., et al. Acute cerebellitis with tonsillar herniation and hydrocephalus in Epstein-Barr virus infection. Eur J Pediatr. 2004;163(11):689-691.
van Swieten J.C., Brusse E., et al. A mutation in the fibroblast growth factor 14 gene is associated with autosomal dominant cerebellar ataxia [corrected]. Am J Hum Genet. 2003;72(1):191-199.
Verbeek D.S. Spinocerebellar ataxia type 23: a genetic update. Cerebellum. 2009;8(2):104-107.
Verbeek D.S., Schelhaas J.H., et al. Identification of a novel SCA locus (SCA19) in a Dutch autosomal dominant cerebellar ataxia family on chromosome region 1p21-q21. Hum Genet. 2002;111(4-5):388-393.
Vuillaume I., Devos D., et al. A new locus for spinocerebellar ataxia (SCA21) maps to chromosome 7p21.3-p15.1. Ann Neurol. 2002;52(5):666-670.
Vulliemoz S., Vanini G., et al. Epilepsy and cerebellar ataxia associated with anti-glutamic acid decarboxylase antibodies. J Neurol Neurosurg Psychiatry. 2007;78(2):187-189.
Wadia N., Pang J., et al. A clinicogenetic analysis of six Indian spinocerebellar ataxia (SCA2) pedigrees. The significance of slow saccades in diagnosis. Brain. 1998;121(Pt 12):2341-2355.
Wadsworth J.D., Collinge J. Update on human prion disease. Biochim Biophys Acta. 2007;1772(6):598-609.
Waters M.F., Fee D., et al. An autosomal dominant ataxia maps to 19q13: Allelic heterogeneity of SCA13 or novel locus? Neurology. 2005;65(7):1111-1113.
Waters M.F., Minassian N.A., et al. Mutations in voltage-gated potassium channel KCNC3 cause degenerative and developmental central nervous system phenotypes. Nat Genet. 2006;38(4):447-451.
Wong D., Dwinnel M., et al. Ataxia and the role of antigliadin antibodies. Can J Neurol Sci. 2007;34(2):193-196.
Worth P.F., Houlden H., et al. Large, expanded repeats in SCA8 are not confined to patients with cerebellar ataxia. Nat Genet. 2000;24(3):214-215.
Xia H., Mao Q., et al. RNAi suppresses polyglutamine-induced neurodegeneration in a model of spinocerebellar ataxia. Nat Med. 2004;10(8):816-820.
Yabe I., Sasaki H., et al. Spinocerebellar ataxia type 14 caused by a mutation in protein kinase C gamma. Arch Neurol. 2003;60(12):1749-1751.
Yeh T.H., Lu C.S., et al. Autonomic dysfunction in Machado-Joseph disease. Arch Neurol. 2005;62(4):630-636.
Yu G.Y., Howell M.J., et al. Spinocerebellar ataxia type 26 maps to chromosome 19p13.3 adjacent to SCA6. Ann Neurol. 2005;57(3):349-354.
Zeviani M., Di Donato S. Mitochondrial disorders. Brain. 2004;127(Pt 10):2153-2172.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 72 Disorders of the Cerebellum, Including the Degenerative Ataxias
Cerebellar ataxia can result from a variety of insults to the cerebellum and its connecting pathways. The cerebellum can be the seat of pathology in many well-recognized diseases of the central nervous system (CNS) and may be involved in such processes in isolation or in combination with other structures. Ataxia can either be the major feature of the disease or one of its various clinical signs. Progressive degeneration of the cerebellum and its connections resulting in ataxia can be due to a number of genetic abnormalities, classified as inherited ataxia. Lastly, the term sporadic ataxia or idiopathic ataxia is used for those diseases in which ataxia related to cerebellar degeneration occurs in the absence of a definite genetic or acquired etiology (Klockgether, 2010). In this chapter, we discuss some of the well-defined acquired causes of ataxia, summarize the current understanding of inherited ataxias, and address the issue of sporadic ataxia.
Acquired Ataxias
In many patients, progressive ataxia results from environmental insults and other well-recognized disorders involving the nervous system. In any patient presenting with cerebellar ataxia, such disorders as ischemic or hemorrhagic stroke involving the cerebellum, previous episodes of cerebral hypoxia, primary or metastatic tumors, and CNS demyelinating diseases such as multiple sclerosis can be diagnosed by appropriate history, imaging studies, and other investigations. Many of these diseases tend to have an acute or subacute evolution rather than the chronic course associated with degenerative ataxias. There are, however, diseases that can present with ataxia in which the major imaging abnormality may be an atrophic cerebellum akin to the finding in degenerative ataxias (Table 72.1). Some of these will be briefly discussed next.
| Entity | Diagnostic Process |
|---|---|
Hypothyroidism
Occasionally, patients with hypothyroidism develop a mild dysequilibrium and gait ataxia in conjunction with their systemic symptoms. This is correspondent with pathological changes in midline cerebellar structure (Barnard et al., 1971). Thyroid functions should be tested in patients with progressive ataxia, since replacement treatment may dramatically improve the symptoms (Jellinek and Kelly, 1960).
Toxic Causes
Alcohol
Alcohol remains the major exogenous agent that causes ataxia. A significant proportion of alcoholics have midline cerebellar degeneration at autopsy. Clinically this disease is characterized by a progressive trunk ataxia with gait disturbance of a cerebellar type; upper-limb ataxia, speech difficulties, or eye movement abnormalities occur only in a small proportion of patients. This may reflect the relative sparing of the cerebellar hemispheres. Imaging studies typically reveal vermian atrophy. Chronic alcoholism can also produce significant cerebellar atrophy in the absence of major clinical deficits (Hillbom et al., 1986). Gait ataxia can dramatically improve with abstinence for over a year (Diener et al., 1984).
Chemotherapeutic Agents
Some cancer chemotherapeutic agents are recognized to produce ataxia as a side effect. 5-Fluorouracil (5FU), a fluorinated pyrimidine that acts by incorporating into ribonucleic acid (RNA) and interfering with RNA function, has been used to treat breast and gastrointestinal cancer. Conventional doses of 5FU may cause cerebellar ataxia if there is an abnormality of pyrimidine metabolism in the form of dihydropyrimidine dehydrogenase deficiency. Higher doses of 5FU can also cause a pancerebebellar syndrome that has an acute to subacute evolution (Gottlieb and Luce, 1971).
When cytosine arabinoside (ara-C) is given in high doses (>3 g/m2 for 8-12 doses, as opposed to the conventional 100-200 mg/m2 for 5-7 days), a significant number of patients develop a cerebellar syndrome. Pathologically, this is characterized by loss of Purkinje cells, gliosis, loss of dentate neurons, and spongiform changes (Salinsky et al., 1983). Signs of cerebellar dysfunction mandate immediate cessation of high-dose cytosine arabinoside.
Heavy Metals
Organic mercury poisoning has occurred in epidemic form as a result of contamination from mercury-containing fungicides. Mercury appears to be particularly toxic to cerebellar granule cells and visual cortex and causes a syndrome that includes paresthesia, ataxia, and restricted visual fields. Lead toxicity causes encephalopathy that is especially common in children. It is usually associated with abdominal colic. However, cerebellar signs can be the prominent presentation. Successful chelation therapy may achieve complete neurological recovery (Mani and Chaudhary, 1998). Gait ataxia associated with other signs such as confusion and myoclonus has also been described with bismuth toxicity due to excessive intake of bismuth subsalicylate (Pepto-Bismol) (Gordon et al., 1995). This is reversible over several weeks or months when bismuth intake is stopped.
Anticonvulsants
Transient cerebellar signs associated with supratherapeutic levels of drugs have been seen with many anticonvulsants, especially phenytoin. More persistent ataxia and documented Purkinje cell loss has been primarily seen in epileptics treated with phenytoin for prolonged periods of time. Cerebellar atrophy occurs in phenytoin-treated patients but may not always be associated with overt ataxia. The pathogenesis of this syndrome remains unclear. The various hypotheses include a direct toxic effect of phenytoin, the result of repeated hypoxia related to seizures, the effect of the seizure-related electrical discharge on cerebellar Purkinje cells, and the possibility that both the seizures and the cerebellar pathology are secondary to a unifying underlying pathology such as prenatal injuries. Another intriguing possibility is that both the seizures and the progressive cerebellar syndrome could result from a single underlying gene mutation such as can be seen in SCA10 (Matsuura et al., 2000). It is probably best to avoid phenytoin in an epileptic patient if either ataxia or cerebellar atrophy is present. In a meta-analysis of second-generation anticonvulsants (gabapentin, lamotrigine, levetiracetam, oxcarbazepine, pregabalin, tiagabine, topiramate, zonisamide) used as adjunct therapy, all of them were found to increase the risk of imbalance, with the exception of gabapentin and levetiracetam. A dose-response relation was seen with almost all the drugs (Sirven et al., 2007).
Infectious and Transmissible Diseases
Ataxia can be one of the features of postinfectious encephalomyelitis but usually accompanies a more diffuse cerebral process. A more restricted cerebellar syndrome has been seen in children after a viral syndrome. Connolly et al. (1994) have proposed that this diagnosis be considered when children develop an acute ataxic disorder not associated with a more diffuse process reflected by seizures, meningismus, or obtundation. In most children, this was preceded by a nonspecific viral syndrome or varicella, with a peak incidence at the age of 5 to 6 years. A similar syndrome following Epstein-Barr virus (EBV) or vaccinations occurred in the teenage years. Cerebrospinal fluid (CSF) may show some elevation of protein and a modest mononuclear pleocytosis, and PCR may allow identification of a specific etiology such as EBV. Magnetic resonance imaging (MRI) often reveals signal density changes in the cerebellum (Fig. 72.1). Prognosis for recovery is usually excellent, with residual dysfunction in a distinct minority. However, swelling, hydrocephalus, and herniation have been described in some and may require decompressive surgery (Van Lierde et al., 2004).
Fig. 72.1 Signal density change in the cerebellum in a child with acute cerebellar ataxia of childhood.
(Courtesy Dr. V. Vedanarayanan, Department of Pediatrics, University of Mississippi Medical Center.)
Similarly, a combination of ataxia, ophthalmoplegia, and other lower cranial nerve palsies can occur as a result of brainstem encephalitis (Bickerstaff encephalitis). There appears to be significant overlap between this disease and the Miller-Fisher variant of Guillain-Barré syndrome. In a report of 62 cases diagnosed as Bickerstaff encephalitis, most had some changes in consciousness level, 40% had Babinski signs, and 60% had tetraparesis; in addition, anti-GQ1 antibody was present in 66%. MRI brain abnormalities occurred only in 30% (Odaka et al., 2003).
Human immunodeficiency virus (HIV) infection can result in many neurological syndromes including ataxia. The majority of patients with ataxia in the presence of HIV infection have discrete, well-recognized lesions such as lymphomas, chronic meningeal infection, progressive multifocal leukoencephalopathy, or toxoplasmosis. Approximately 30% of patients with HIV dementia have an ataxic syndrome at the onset of their illness before cognitive decline begins. Other authors have described isolated progressive cerebellar ataxia that does not evolve into HIV dementia in a small number of patients (Elsheikh et al., 2010; Kwakwa and Ghobrial, 2001; Tagliati et al., 1998). These patients had a rapid evolution of their ataxia leading to chair-bound status in less than a year; MRI revealed cerebellar atrophy. Pathological examination has revealed marked granular cell loss.
Creutzfeldt-Jakob disease (CJD) should be considered in patients presenting with progressive ataxia. CJD is typically a rapidly progressive dementing illness related to the accumulation of mutant prion protein (PrP), which is a posttranslational modification of the normal prion protein. Among sporadic CJD patients, over 70% have ataxia (Parchi et al., 1999). Jellinger et al. (1974) noted that the ataxic variant of CJD began with minor behavior symptoms followed by ataxia. Upper motor neuron signs were frequent; myoclonus occurred only in 25%, and dementia evolved late. Survival among these patients was slightly longer than in typical CJD, with a mean time to death of 16 months and a range of 7 weeks to 8 years. Pathologically, the cerebellum shows striking granule cell loss. A recent review noted that both the iatrogenic and variant form of CJD (human bovine spongiform encephalopathy) usually have ataxia at the onset, as does the inherited form known as Gerstmann-Sträussler-Scheinker syndrome (GSS) (Wadsworth and Collinge, 2007). Prion protein gene mutation analysis is helpful in the familial cases. Iatrogenic cases are usually homozygous for codon 129, and the same codon is all methionine in the variant cases. Tonsillar biopsy for immunocytochemistry for PrP and Western blot studies for abnormal PrPSc (scrapie prion protein) can be useful in the variant cases as well. CSF protein biomarker studies involving 14-3-3, S100b, and tau are often useful in diagnosing CJD (Pennington et al., 2009).
Whipple disease (WD) is a multisystemic infectious disease caused by Tropheryma whippelii. The classic symptoms include diarrhea, weight loss, and abdominal pain. The pathognomic symptoms of CNS involvement are oculomasticatory myorhythmia (OMM) and oculo-facial-skeletal myorhythmia. Many cases of CNS WD are not diagnosed until postmortem (Gerard et al., 2002), but it is postulated that all patients have CNS involvement. Cerebellar ataxia is a common feature of CNS WD. It has been reported to be present in 45% of cases, more common than OMM (Matthews et al., 2005). Since untreated WD is fatal and therapy is available, it is paramount important to consider WD in the differential diagnosis when a patient presents with ataxia along with gastrointestinal symptoms. Those with possible CNS WD should undergo multiple duodenal biopsies if needed. Polymerase chain reaction (PCR) assay of CSF, brain MRI, and a stereotactic cerebral biopsy can also be helpful. Prolonged antibiotic therapy with drugs able to cross the blood-brain barrier is recommended. A regimen with intravenous ceftriaxone sodium, 2 g twice daily for 2 weeks, followed by oral trimethoprim-sulfamethoxazole, double strength, 1 tablet twice daily, achieved good results (Matthews et al., 2005). CSF should be analyzed repeatedly to monitor efficacy. Treatment can be discontinued only when the results of CSF PCR are negative.
Autoimmune Causes
Paraneoplastic Cerebellar Degeneration
Paraneoplastic cerebellar degeneration is a rapidly progressive pancerebellar syndrome that reaches its nadir within a few months of onset. It produces a severe ataxic disease associated with dysarthria and oscillopsia. Diplopia and vertigo may also occur. Many patients also develop other neurological signs including dementia, extrapyramidal signs, hearing loss, and dysphagia. MRI typically shows atrophy of the entire cerebellum, though some high-signal density changes may occur in the deep white matter. CSF usually shows a mononuclear pleocytosis, and oligoclonal bands may be present. It is believed that in many cases the syndrome results from an autoimmune process triggered by the cancer, but evidence for this cannot be found in every patient. Several autoantibodies have been described in these patients. The anti-Yo antibody is usually seen in ovarian cancer and primarily causes a cerebellar syndrome. The anti-Hu (type 1 antineuronal nuclear antibody [ANNA1]) antibody is seen with small-cell cancer of the lung and typically causes a multifocal disorder, the most common of which is a sensory ganglionopathy. Purkinje cell degeneration has been noted in about 25% of patients with anti-Hu antibody. The anti-Ri (ANNA2) antibody that recognizes two closely related neuronal RNA-binding proteins designated Nova-1 and Nova-2 has been seen in patients with a opsoclonus-myoclonus-ataxia syndrome in the setting of breast cancer and non–small cell lung carcinoma (Luque et al., 1991; Musunuru and Kesari, 2008). Patients who exhibit a combination of ataxia and Lambert-Eaton syndrome against a background of small-cell lung cancer may have antibodies to voltage-gated calcium channels (VGCC). Cerebellar degeneration in Hodgkin disease often occurs months to years after the lymphoma presents and is associated with the anti-Tr antibody (Bernal et al., 2003). Less common antibodies that have been related to paraneoplastic cerebellar ataxia with or without additional signs include the anti-Ta, anti-Ma, and anti-CV2 antibodies and autoantibodies against metabotropic glutamate receptor and protein kinase C gamma (Hoffmann et al., 2008; Posner and Dalmau, 2000; Sabater et al., 2006; Sillevis et al., 2000). The zic4 antibody against the neuronal zinc-finger protein is also associated with small-cell lung cancer and ataxia (Bataller et al., 2004). When routine studies are unfruitful, positron emission tomography (PET) scan may be useful to detect small cancers (Land et al., 2005). The eradication of underlying cancer usually results in improvement or resolution of ataxia (Ammar et al., 2008). Plasmapheresis has been reported effective in the reduction of autoantibodies and improvement of symptoms (Armstrong et al., 2005; Bier and Hile, 2010; Hoffmann et al., 2008; Taniguchi et al., 2006).
Ataxia with Gluten Sensitivity
Gluten sensitivity is a systemic autoimmune disease with diverse manifestations. Celiac disease is only one aspect of this entity. Celiac disease is an autoimmune disorder of the small intestine caused by an immune reaction to gliadin, a prolamin (gluten protein) found in wheat. Its prevalence is between 1 in 1750 to 1 in 105 people in the United States, varying on the definition (Rewers, 2005). Cerebellar ataxia has been reported since 1966 in this group of patients (Cooke and Smith, 1966). Vitamin deficiency from malabsorption or concomitant autoimmune diseases may account for the ataxia. A more controversial area is the association of antigliadin antibodies in the etiology of sporadic ataxia with no overt evidence for small bowel disease.
Hadjivassiliou et al. (2010) reported that 47% (101/215) of patients with idiopathic sporadic ataxia had serological evidence of gluten sensitivity. Other studies have shown a lower prevalence of gliadin antibodies in patients with ataxia or none at all (Burk et al., 2001; Combarros et al., 2000; Pellecchia et al., 1999; Wong et al., 2007). Bushara et al. (2001) and Lock et al. (2005) reported that patients with idiopathic ataxia and those with definite genetic forms of ataxia had a similar occurrence of gluten sensitivity, raising doubts about the significance of the gliadin antibodies. Contrary data have been reported by Hadjivassiliou et al. (2003), noting that while there was a 14% prevalence of gluten sensitivity among inherited cases, the figure was 41% among 132 cases of sporadic ataxia. Gluten-free diet was shown to improve measures of ataxia over a 1-year period, associated with elimination of the antigliadin antibodies, but this was not a randomized study. Patients with gluten sensitivity usually have had slowly progressive ataxia associated with brisk tendon reflexes and peripheral neuropathy. Nervous system pathology was characterized by cerebellar Purkinje cell loss, infiltration by T lymphocytes and posterior column degeneration. A variable proportion of patients had typical celiac disease on duodenal biopsy, even when there were only minor gastrointestinal symptoms; some had either normal biopsies or simply lymphocytic infiltrates, so the role of gluten sensitivity in the etiology of sporadic ataxia remains controversial. Presence of antigliadin antibodies should prompt further testing including antideaminated gliadin antibody, antitransglutaminase antibody, and small-bowel biopsy. If celiac disease is confirmed, gluten-free diet may offer a good chance for symptomatic relief (Rashtak et al., 2010).
Ataxia and Anti–Glutamic Acid Decarboxylase Antibodies
A number of reports have noted the presence of antibodies to glutamic acid decarboxylase (GAD) among a small number of patients with progressive ataxia (Bayreuther et al., 2008; Honnorat et al., 1995; Rakocevic et al., 2006; Saiz et al., 1997). The patients have usually been middle-aged women. Ataxia was often associated with peripheral neuropathy, rarely associated with neuropathy or slow saccades and in some cases with stiff person syndrome. Many patients had multiple organ specific antibodies including those directed against thyroid, parietal cells, and pancreatic islet cells, and insulin-dependent diabetes was an invariable accompaniment. The antibodies occurred in higher titers than those found in adult-onset diabetes, and there is evidence that they bind to presynaptic nerve terminals around cerebellar Purkinje cells. GAD is the enzyme that synthesizes γ-aminobutyric acid (GABA) from glutamate, and it is believed that the antibodies may be pathogenic because of their binding to GABA terminals. Intravenous immunoglobulin or immunosuppressants may cause partial remission of symptoms (Markakis et al., 2008; Nanri et al., 2009; Nociti et al., 2010; Vulliemoz et al., 2007).
Superficial Siderosis
Superficial siderosis occurs when iron and hemosiderin are deposited in the pial and subpial regions of the brain. Clinical features include ataxia, hearing loss, and cognitive decline (Fearnley et al., 1995). Superficial siderosis results from bleeds that are often covert, such as with cryptic vascular malformations, or more obvious, such as with tumors or prior surgery. Diagnosis can be made by the characteristic hypointensity over CNS structures in T2-weighted images on MRI (Fig. 72.2). The disease can be controlled if the source of bleeding is found and obliterated.
Inherited Ataxias
The initial recognition that progressive balance difficulties related to pathology in the cerebellum and its connecting pathways could have a genetic basis is attributed to Nicholas Friedreich, who published a series of papers describing siblings with such a disease, now known as Friedreich ataxia (FA). As early as 1893, Pierre Marie recognized the clinical and genetic heterogeneity among the inherited ataxias. During much of the 20th century, inherited ataxias that did not quite fit the Friedreich mould were often labeled as Marie ataxia. It eventually became apparent that many patients have progressive ataxia of a degenerative nature, clinically and pathologically resembling patients with inherited ataxias, and yet have no discernible genetic basis. Much of the last 100 years saw detailed clinical and pathological descriptions of patients with various forms of ataxia and attempts at understanding them on a clinicopathological basis. The classification of these diseases based on the underlying neuropathology reflects attempts at such understanding. Nevertheless, the clinical, neuropathological, and genetic heterogeneity of the disorders did not allow a universally acceptable classification. Harding (1983), based on an extensive clinical study of many families, wrote a clinical-genetic classification that served as a prelude to an increasingly gene-based classification of the inherited ataxias (Harding, 1983). This section will summarize the current information regarding inherited forms of ataxia.
Autosomal Recessive Ataxias
As the autosomal recessive ataxias have been elucidated at the molecular genetic level, a semblance of a pathogenetic classification may be emerging, albeit subject to revisions or additions. The genetic abnormalities discovered so far have either pointed to abnormalities in mitochondria, oxidative stress, abnormalities of deoxyribonucleic acid (DNA) repair mechanisms, possibly abnormal protein folding, or changes in cytoskeletal proteins. Table 72.2 lists various autosomal recessive ataxias based on specific gene loci. Most of these diseases begin in childhood or early adult life; however, onset even late in life is possible with autosomal recessive ataxias. Singleton patients may occur in many families. Typically, parents do not manifest symptoms, since they are heterozygous for the mutation. If the sibship size is large, affected siblings may be found, and the disorder will affect both males and females. Parental consanguinity may be found but is not essential.
| Entity | Gene locus/gene | Mutation |
|---|---|---|
AOA1, Ataxia with oculomotor apraxia type 1; AOA2, ataxia with oculomotor apraxia type 2; ARCA1, autosomal recessive cerebellar ataxia type 1; ARSACS, autosomal recessive ataxia of Charlevoix-Saguenay; ATLD, ataxia-telangiectasia-like disorder; IOSCA, infantile-onset spinocerebellar ataxia; MIRAS, mitochondrial recessive ataxia syndrome; MSS, Marinesco-Sjögren syndrome; SCAN1, spinocerebellar ataxia with axonal neuropathy.
Friedreich Ataxia
Clinical Features
Friedreich ataxia is the most common inherited ataxia, with a prevalence of 1 in 50,000 Caucasians. Classical descriptions of the disease include an age at onset below 25 years, typically early in adolescence (Harding, 1981). Initial symptoms include gait difficulties and clumsiness. Neurological examination reveals gait ataxia, loss of proprioceptive sense in the lower limbs, and absence of deep tendon reflexes (Pandolfo, 2003). These findings are related to early pathology in dorsal root ganglion cells, and occasional patients may be mistaken for having a hereditary sensory neuropathy. However, most will exhibit signs indicating involvement of the CNS including dysarthria, some upper motor neuron findings such as extensor plantar responses, and eye movement abnormalities such as square-wave jerks. Rarely, patients may present with cardiac disease or a spinal deformity and are later found to develop neurological disease; they tend to lose ambulation in about 9 to 15 years. At this stage, patients have increasing ataxia of both upper and lower limbs, profound proprioceptive loss, areflexia, weakness of lower-limb muscles, flexor spasms, and increasing dysarthria and dysphagia. Optic atrophy and hearing loss may occur in a minority of patients.
Systemic abnormalities include abnormal electrocardiograms, hypertrophic cardiomyopathy in about 50% of the patients, and diabetes in 10%. The hypertrophic cardiomyopathy may later become a dilated cardiomyopathy and rhythm problems such as atrial fibrillation may add to the cardiac morbidity. Skeletal abnormalities such as spinal deformities and foot deformities are common (Fig. 72.3). The mean age of death among patients with FA has been reported to be late in the fourth decade, but this information comes from clinical studies done prior to the availability of FA mutation analysis. Also, the availability of better diabetic and cardiac care as well as adaptive rehabilitation techniques can allow much longer survival. Cause of death is often cardiac, but respiratory compromise related to the motor difficulties and spinal deformity may also contribute.
Nerve conduction studies show early absence or reduction of sensory nerve potentials in a diffuse fashion, reflecting the loss of large sensory axons in peripheral nerves. Central motor conduction studies show abnormalities that evolve over the course of the disease and may reflect disease progression. MRI scans of the brain reveal no abnormalities in the cerebellum; rather, the upper cervical cord shows atrophy. Signal density changes may be seen in the posterior columns by MRI (Mascalchi et al., 1994). Functional imaging has been reported to show decreased cerebellar blood flow, but there is cerebral glucose hypermetabolism in ambulatory patients with FA (Gilman et al., 1990). Pathologically, there is loss of dorsal root ganglion cells, resultant degeneration of the dorsal columns, degeneration of spinocerebellar and corticospinal tracts, and loss of cells in the cerebellar dentate nucleus.
The Friedreich Ataxia Mutation
The mutation in FA is an unstable expansion of a repeated trinucleotide (GAA) sequence within the first intron of the FRDA gene on chromosome 9q13-21.1 (Campuzano et al., 1996) (Fig. 72.4). Expanded alleles have 66 to over 1000 repeats. In nearly 95% of affected persons, the GAA expansion occurs in both alleles (homozygous expansion), although the size of the expansion can be different in each.
Since the identification of the FA mutation, we have realized that many patients with a phenotype very atypical for classical FA carry the FA mutation (Durr et al., 1996; Filla et al., 1996; Montermini et al., 1997). Some 15% of patients with homozygous GAA expansion have neurological signs identical to those of classical FA but have an age at onset later than 25 years (late-onset FA [LOFA]). Other patients with the GAA expansion have the typical age at onset but retain their tendon reflexes or even have exaggerated reflexes (FA with retained reflexes [FARR]). There are patients who combine a later onset with retained reflexes as well; in addition, very late onset of mild gait ataxia, spastic paraparesis, and even chorea have been occasionally associated with the FA GAA expansion (Hou and Jankovic, 2003). The variation of expansion size may account for the clinical diversities. As with other trinucleotide repeat diseases, there is an inverse correlation between the size of the GAA repeat and the age at onset; this correlation is better with the smaller of the two expanded alleles. Cardiomyopathy and diabetes also tend to occur in patients with larger expansions (>700 GAA repeats). However, not all of the variation in age at onset and severity of disease can be correlated with GAA size alone. Other genetic and possibly environmental factors may contribute to this variation. In addition, variation in clinical picture can result from somatic mosaicism of the repeat size, with different tissues having significant differences in the size of the expansions (Bidichandani et al., 1999). Recently it has been reported that the U mitochondrial (mt)DNA haplogroup can delay the age at onset and have a lower rate of cardiomyopathy in FA (Giacchetti et al., 2004).
Point Mutations
Approximately 5% of patients with clinical findings compatible with FA have only one expanded allele; however, such patients harbor point mutations in the unexpanded allele. Sequencing of the unexpanded allele to document a point mutation will have to be done in this situation. Point mutations located in the carboxy terminal half of the mature frataxin appear to be associated with a typical FA phenotype (Cossee et al., 1999). Missense mutations in the amino terminal half of the protein, such as the G130V mutation, appear to result in a milder phenotype with less ataxia, greater spasticity, and no dysarthria. To date, all the patients with point mutations have been compound heterozygotes with one expanded allele. Homozygous point mutations will cause complete lack of frataxin, and this may be incompatible with life. This has been further supported by the fact that complete knockout of the gene in animal models is embryonically lethal.
Pathogenesis
The presence of the expanded GAA sequence in the first intron of the gene results in reduced transcriptional efficiency leading to a partial deficiency of the protein, frataxin (Lodi et al., 2001; Pandolfo, 2001; Puccio and Koenig, 2000). The reduced transcriptional efficiency of the mutated gene has been attributed to an unusual “sticky DNA” configuration of the expanded repeat (Sakamoto et al., 1999). In addition, there is evidence for chromatin condensation and remodeling, which can further suppress frataxin transcription (Pandolfo and Pastore, 2009). The exact role of frataxin in normal biology is still unclear, but many studies suggest that it is a mitochondrial protein (Fig. 72.5). Knockout of the frataxin homolog in yeast leads to accumulation of iron in the mitochondria, a finding of interest in view of the presence of iron in the cardiac muscle in human disease (Babcock et al., 1997; Pandolfo and Pastore, 2009). Activity of enzymes such as aconitase and complex I, II, and III of the respiratory chain that contain iron-sulfur clusters is reduced in cardiac muscle biopsies of patients with FA (Rotig et al., 1997). The iron-sulfur cluster synthesis defect results from the role of frataxin in the process and probably is the cause of iron accumulation. It has been hypothesized that the presence of excess iron as well as excess oxidative stress related to impaired respiratory chain function can induce oxidative stress via the Fenton reaction and cause progressive nuclear and mitochondrial damage (Karthikeyan et al., 2002, 2003; Pandolfo and Pastore, 2009).
Treatment
Based on evidence for excess oxidative stress and poor defense mechanisms of FA tissue to such stress, antioxidant therapy has been tried over the last 10 years; as of 2008, at least 11 studies using idebenone in patients with FA have been reported (Meier and Buyse, 2009). This drug has been reasonably well tolerated in doses of 900 mg/day or more by FA patients, the main adverse effect being gastrointestinal. At doses of 5 mg/kg/day, it appeared to have beneficial effect on the cardiomyopathy of FA. In a 6-month study, idebenone given in high doses appeared to improve neurological function as measured by International Cooperative Ataxia Rating Scale (ICARS) scores in ambulatory FA patients (Schulz et al., 2009). Newer approaches now in the experimental phase include gene replacement (Fleming et al., 2005) and the use of molecules that may enhance frataxin production even in the presence of the GAA expansion (Hebert, 2008). In fact, recent open-label experience suggests some beneficial effect from erythropoietin (Boesch et al., 2008).
Mitochondrial Recessive Ataxia Syndrome
An autosomal recessive ataxic syndrome caused by a mutation in the polymerase γ (POLG) gene has been found to be the most common cause of recessive ataxia in Finland (Hakonen et al., 2005). Polymerase γ is encoded by the nuclear gene, POLG, in humans and is responsible for replication and repair of mtDNA. In a recent study report, 27 patients were found among 15 Finnish families with a carrier frequency of 1 : 125. In addition, patients with recessive ataxia from Norway, Belgium, and England shared the same core Finnish haplotype, suggesting a common European founder mutation and making it likely that this disease will be found in places other than Finland. The age at onset varied from 5 to 41 years. The neurological picture was heterogeneous, but ataxia, hypo- or areflexia, axonal neuropathy, and epilepsy were common. There was often mild cognitive impairment and psychiatric manifestations such as depression, anxiety, and hallucinations.
Infantile-Onset Spinocerebellar Ataxia
Infantile-onset spinocerebellar ataxia has been described among an isolated population in Finland. The disorder begins soon after children acquire the ability to walk. Neurological signs include ataxia, peripheral neuropathy with areflexia, athetosis of hands and feet, Babinski signs, and clumsiness. Speech becomes impaired, and sensorineural hearing loss occurs before the age of 5. Ophthalmoplegia and optic atrophy develop, and some patients have seizures. Patients usually have learning deficits and skeletal deformities as well. The gene responsible has recently been found to be C10ORF2, a nuclear gene encoding for Twinkle, an mtDNA-specific helicase, and a splice variant known as Twinky (Nikali et al., 2005) located on 10q24. Dominant mutations in the same gene cause a syndrome of progressive external ophthalmoplegia. In contrast to the latter, the recessive mutations in infantile-onset spinocerebellar ataxia do not appear to alter mtDNA maintenance.
Ataxia with Isolated Vitamin E Deficiency
The occurrence of a childhood-onset recessive ataxia associated with isolated vitamin E deficiency is related to mutations in the gene encoding the α-tocopherol transfer protein (α-TTP) on chromosome 8 (Ouahchi et al., 1995). A hepatic protein, α-TTP is involved in the processing of vitamin E for transport in the chylomicrons. The low vitamin E levels are related to impaired hepatic processing, not to deficient intestinal absorption. Deficiency of vitamin E, which has antioxidant properties, may cause neuronal degeneration. Purkinje cells are more vulnerable owing to their high metabolic activity and high oxygen demand. The severity of the phenotype associated with α-TTP mutations depends on residual protein activity. The childhood-onset disease has considerable resemblance to FA. There is progressive ataxia and evidence for a polyneuropathy with loss of proprioception and tendon reflexes. Retinitis pigmentosa and visual loss can accompany this syndrome. Patients often have head titubation. Cardiomyopathy can occur but is less common than in FA (Cavalier et al., 1998). Vitamin E levels should be obtained in all persons with sporadic ataxia of childhood or young-adult onset. Patients typically have less than 1.8 mg/L of vitamin E. Treatment with large doses of vitamin E stabilizes the neurological features, especially when started early in the disease, but some features may show worsening despite therapy (Mariotti et al., 2004).
Abetalipoproteinemia
Abetalipoproteinemia results from mutations in the MTTP gene, which codes for a subunit of the microsomal triglyceride transfer protein; this results in impaired lipid absorption in the intestine, including lipid-soluble vitamins like vitamin E. The ataxia resembles that in isolated vitamin E deficiency but is associated with evidence for malabsorption (Fogel and Perlman, 2007). Vitamin E and cholesterol levels are low, and apolipoprotein B is absent. Acanthocytes are found in peripheral blood smears. Transaminases may be high and INR elevated due to vitamin K deficiency. Serum lipoprotein electrophoresis can establish the diagnosis. Treatment involves dietary modification and vitamin replacement, which may prevent neurological complications.
Cayman Ataxia
This recessive ataxia of early childhood onset has been identified in the Grand Cayman island. Affected patients have been shown to carry two homozygous mutations in the gene that encodes caytaxin (ATCAY) on chromosome 19. Caytaxin contains a region with homology to a CRAL-TRIO domain, also present in α-TTP. A ligand similar to vitamin E may bind to this protein, and its deficiency may cause ataxia by a mechanism similar to that in vitamin E deficiency (Bomar et al., 2003).
Ataxia-Telangiectasia
Clinical Features
The occurrence of ataxia-telangiectasia (AT) has been estimated to be 1 in 20,000 to 100,000 live births (Swift et al., 1993). This disorder has its onset early in the first decade with truncal instability and impaired gait. This is followed by progressive ataxia associated with polyneuropathy, hypotonia, and loss of reflexes (Fogel and Perlman, 2007). Choreiform movements are frequent. Characteristic eye movement abnormalities are seen, with the need for a rapid head thrust for fixating on targets to one side, with the eyes following later (eye-head lag). This is referred to as oculomotor apraxia. Telangiectasia tends to appear at about 5 years of age and is most frequent on the conjunctivae (Fig. 72.6) but can also occur over the earlobes, popliteal fossa, and other locations. There is evidence for an immunodeficiency, with frequent bronchopulmonary infections. These children are at risk for malignancies, especially hematological tumors like lymphomas and leukemias; other types of cancers occur if the patient reaches adulthood. Infections related to immunodeficiency are also common. The thymus gland is typically small or absent in patients with AT. Many have decreased immunoglobulin (Ig)A levels; decreased IgE and IgM levels, lymphocytopenia, and skin anergy may also occur. In addition, elevation of α-fetoprotein (AFP) in the serum is a consistent finding in AT.
Mutation and Pathogenesis
AT is caused by truncating, missense, deletion, or insertion mutations of the ATM gene on chromosome 11 (Halazonetis and Shiloh, 1999; Savitsky et al., 1995). More than 300 mutations of this gene have been associated with AT. The protein product of ATM is a serine-threonine kinase and may be involved in checkpoint responses to double-stranded DNA breaks by conveying the signal of damage from sensor proteins such as MRN (MRE11-RAD50-NBS1 complex) to the effector proteins. Increased susceptibility to radiation damage has been shown to cause chromosomal translocations and has been used in a fibroblast survival assay as a diagnostic test. The degree of DNA damage determines the response: either cell-cycle arrest and repair or initiation of apoptosis (Lavin et al., 2005).
Laboratory Diagnosis
Serum AFP is usually elevated (>10 ng/mL), and immunoglobulin levels are low. Karyotyping often shows chromosomal abnormalities, especially a 7 : 14 translocation. Increased sensitivity of cultured fibroblasts to radiation can be documented by the colony survival assay. The definitive way of establishing diagnosis is immunoblotting for the ATM protein; 90% of patients have no protein, 10% have trace amounts, and rare patients have normal ATM levels but no kinase activity (so-called kinase-dead) (www.geneclinics.org). Further molecular confirmation can be achieved by detecting the mutation, but this can be difficult because of the size of the ATM gene and the lack of mutation “hot-spots.” Sequence analysis of the coding regions of the ATM gene will detect 90% of mutations but miss intronic alterations and heterozygous deletions; benign polymorphisms and pathogenic alterations may be difficult to distinguish. Other types of DNA-based tests include protein truncation tests (examining for premature stop codons), which can detect about 70% of the mutations, and mutation scanning using denaturing high-performance liquid chromatography (DHPLC), which has a sensitivity of 85%.
[/not-level-membership-for-neurology-category]
