[level-membership-for-neurology-category]
Chapter 77 Disorders of the Autonomic Nervous System
Classification of Autonomic Disorders
Clinical Features of Autonomic Impairment
Assessment of Autonomic Function
Functional Autonomic Disorders
Autonomic Disorders Characterized by Excessive Autonomic Outflow
Predominantly Peripheral Afferent Structural Autonomic Disorders Characterized by Impaired Autonomic Outflow
Predominantly Peripheral Efferent Structural Autonomic Disorders Characterized by Impaired Autonomic Outflow
Predominantly Central Structural Autonomic Disorders Characterized by Impaired Autonomic Outflow
Nonautonomic Disorders Causing Hypotension or Syncope to Consider in the Differential Diagnosis
At the higher control levels in the brain, autonomic integrators and signals are integrated and expressed subconsciously through the central autonomic network (Benarroch, 1997). This overlies a strong circadian rhythm of autonomic function. The autonomic nervous system consists of two large divisions, the sympathetic (thoracolumbar) outflow, and the parasympathetic (craniosacral) outflow. The two divisions are defined by their anatomical origin rather than by their physiological characteristics. The circadian rhythm of autonomic function originates in the suprachiasmatic nucleus and is conveyed to the hypothalamus and brainstem. Light falling on retinal ganglion cell dendrites (not rods or cones) in the eye and transmitted via the retinohypothalamic tract entrains this rhythm. Other key inputs to autonomic outflow originate in the insular cortex and the amygdala. The principal integration of autonomic outflow to the cardiovascular system lies in the medulla. Stretch-sensitive baroreflex mechanoreceptors in the blood vessels of the thorax and neck relay information about blood pressure and blood volume through the glossopharyngeal (from carotid arteries) and vagus (from aorta) nerves to the nucleus tractus solitarius (NTS) in the posterior medulla. Excitatory neurons from the NTS innervate the dorsal motor nucleus of the vagus, which regulates parasympathetic outflow. Inhibitory neurons project to areas in the ventrolateral medulla from which sympathetic outflow is regulated. The most important such site is the rostral ventrolateral medulla.
The autonomic nervous system exerts widespread control over homeostasis (Goldstein, 2001; Mathias and Bannister, 2002). Almost every organ system receives regulatory information from the central nervous system (CNS) through the autonomic efferents (Fig. 77.1), and increasingly we recognize that afferent input into the central autonomic network regulates not only the output of the autonomic system but also much CNS function not generally considered to be autonomic in nature. The emerging concept is pervasive integration of autonomic activities with brain and body.
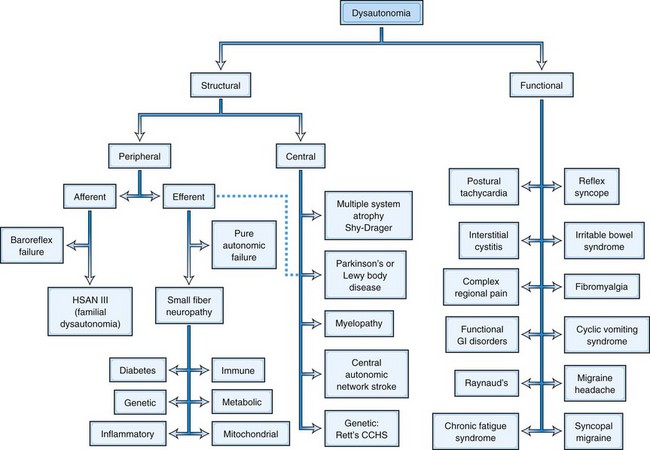
Classification of Autonomic Disorders
Since by definition, structural disorders have a known pathological substrate, they can be divided into two camps, those that involve the peripheral nervous system and those that involve central pathways. Such a division would be more challenging in functional disorders, where a specific etiopathology is generally not defined, with certain rare exceptions (see later discussion and Fig. 77.1). Peripheral disorders can be further subdivided by whether the dominant pathological process involves afferent or efferent fibers. As can be discerned from Fig. 77.1, most peripheral disorders involve efferent nerves. It should be kept in mind that few if any of the structural disorders are “pure” either in their central versus peripheral localization or in afferent versus efferent functional classification. For example, diabetes has been shown to involve central pathways, and PD involves peripheral ganglia, symbolized by the dashed line in the figure connecting that box to the efferent peripheral category.
These two basic classes of dysautonomias, structural and functional, contrast with one another in other ways as well. Patients with structural disorders, in spite of their extensive pathological changes in the nervous system, tend to harbor proportionally few symptoms; for example, the majority of patients do not realize when their systolic blood pressure drops by nearly 90 mm Hg (Arbogast et al., 2009). In contrast, patients with functional disorders harbor enormous numbers of complaints, just as disproportionate with the demonstrable pathological involvement of the autonomic nervous system but in the opposite sense (Ojha, 2011). These disorders are now being considered as multisystem rather than involving a single end-organ. Another difference relates to prognosis. Structural disorders in general carry a very poor prognosis. For example, diabetics with autonomic neuropathy have a 5-year mortality between 25% and 50% (Ewing et al., 1976). Patients with MSA survive only 7 to 9 years from onset (Schrag et al., 2008). In contrast, while functional disorders can be extremely disabling (Benrud-Larson et al., 2002; O’Leary and Sant, 1997; Spiegel et al., 2008), their prognosis for longevity is generally good.
Clinical Features of Autonomic Impairment
Cardiovascular
Normally when one stands, systolic blood pressure falls about 10 mm Hg, and diastolic pressure increases 5 mm Hg. Heart rate rises 5 to 20 beats per minute (bpm). A fall in blood pressure of 20/10 mm Hg or more in the first 3 minutes of standing defines orthostatic hypotension (Consensus Committee of the American Autonomic Society and the American Academy of Neurology, 1996). If there is not a fall in blood pressure of 20/10 on standing, but the patient is symptomatic and experiencing tachycardia, orthostatic intolerance is present, qualifying as POTS if the heart rate rise is greater than 30 bpm in the first 10 minutes in the upright position (Schondorf and Low, 1993). The most common symptoms of orthostatic hypotension are dizziness, dimming of vision, and discomfort in the neck and head. Orthostatic hypotension is greatest, and hence most easily detected, in the hour after ingesting a large breakfast. Carbohydrate acts as a depressor more than protein or fat. It is important to note that a majority of patients with orthostatic hypotension will not have the usual expected symptoms and may present with falls only (Arbogast et al., 2009), so it is vital to not rely solely on symptom reports in the elderly population.
Pulmonary
Lung function is generally preserved in autonomic failure. Patients with MSA frequently experience sleep apnea, and in consequence of these apneic spells, major perturbations in blood pressure may arise acutely. This is in part dependent on the role of carbon dioxide in determining blood pressure level in autonomic failure. Hypoventilation increases blood pressure in autonomic failure, whereas hyperventilation may lower blood pressure significantly. Patients have occasionally noted improved orthostatic tolerance while breathing through a dead space, although this has never been a recommendation in practice. Because of impaired swallowing in patients with MSA (Seppi et al., 2005), aspiration pneumonia occurs frequently and can be missed because the patient may not manifest the expected fever. With early diagnosis and treatment of aspiration pneumonia, patients often do well, returning to their prior state of health.
Gastrointestinal
Constipation occurs in many patients with autonomic failure, but patients with diabetic dysautonomia may also have frequent and often severe diarrhea as well as significant gastroparesis. The diarrhea itself can prevent adequate blood pressure control because of the associated volatility of blood volume. Special problems sometimes occur in specific dysautonomias. For example, patients with Sjögren syndrome commonly have gastroesophageal reflux and may therefore have an increased risk of esophageal carcinoma. Patients with Chagas disease may have achalasia and enlargement of the esophagus, resulting in vomiting. Achalasia is also present in the 4 “A” syndrome (Allgrove disease) characterized by alacrima, achalasia, ACTH insensitivity, and autonomic neuropathy. This syndrome has been mapped to chromosome 12q13 and produced by mutations in the AAAS gene (Handschug et al., 2001). Patients with some forms of genetic autonomic failure may have strikingly severe gastrointestinal fluid loss and bowel movements 10 or more times every day, which sometimes responds to low doses of clonidine. Postprandial angina may occur with food ingestion, usually without associated ST-T wave changes. Most patients with postprandial angina in practice probably have some degree of dysautonomia, and the depressor effect of food is most prominent in the setting of impaired autonomic reflexes. Postprandial angina tends to occur with upright posture following food (especially carbohydrates). Water intake in association with eating will usually help prevent this symptom.
The gastrointestinal dysfunction in two disorders with significant autonomic involvement, namely PD and diabetes, have been better evaluated and understood. For example, the role of diabetes in gastrointestinal dysmotility has been extensively studied. Some changes in the function of the enteric nervous system in diabetes appear to result from apoptosis of enteric neurons. Oxidative stress may play a role in the cell death process. An imbalance also exists in inhibitory and excitatory neuropeptides. These factors then result in altered gut mucosa (Chandrasekharan and Srinivasan, 2007). PD is also associated with gastrointestinal problems, affecting swallowing disorders that may lead to aspiration. Salivation is not increased or may even be decreased in these patients. From early on, individuals with PD may develop delayed gastric emptying, which later in the disease may affect jejunal absorption of levodopa. Gastric emptying of liquids is usually not delayed (Goetze et al., 2006) in PD, so alternatives when giving levodopa are a liquid solution or administering it directly into the jejunum (Jost, 2010). Constipation is also very common in this disorder and represents the most common “autonomic” manifestation of PD, affecting 70% to 80% of patients. Often, severe constipation develops before the more typical symptoms of PD are noticed (Korczyn, 1990; Jost, 2010); the cause is multifactorial. Many of the medications prescribed to treat PD, mainly anticholinergics and (perhaps more controversial) levodopa, may worsen constipation, but they are not the cause, since constipation is usually present before the diagnosis of PD is made and therefore before the onset of medications. Constipation is thought to be due to degeneration of central and peripheral parasympathetic nuclei (Jost, 2010).
Other less well-characterized dysautonomias also have gastrointestinal symptoms. Individuals with POTS complain of bloating, early satiety, nausea, pain, and alternating diarrhea and constipation (Sandroni et al., 1999). In fact, more than half of the children and adults with POTS have gastrointestinal complaints, usually reporting epigastric or lower-abdominal discomfort. Children with POTS seem to suffer nausea and vomiting more often than adults, but these findings were present in both groups (Ojha et al., 2011). Interestingly, coming from the other direction, many with functional gastrointestinal problems also have cardiovascular autonomic dysfunction, primarily sympathetic. In adults, three-eighths also had parasympathetic involvement, which was not present in the pediatric group. Neuropathy was common in both groups (Camilleri and Fealey, 1990; Chelimsky and Chelimsky, 2001). Importantly, when both a functional gastrointestinal disorder and POTS are present, the gastrointestinal symptoms may resolve with treatment of the orthostatic intolerance (Sullivan et al., 2005). The cause of the gastrointestinal symptoms in POTS in unclear and may be related to blood pooling in either the lower extremities or abdomen. Electrical activity of the stomach in POTS changes from supine to the upright position (Safder et al., 2009), suggesting either lack of accommodation or gastroparesis while upright (Buchwald et al., 1987). Cyclic vomiting syndrome (CVS) has also been associated with autonomic dysfunction, and both pediatric and adult sufferers usually have POTS associated with an autonomic neuropathy (Chelimsky and Chelimsky, 2007; Venkatesan et al., 2010).
Urinary Tract
In structural disorders, a reversal of the usual pattern of urine output occurs. Nocturia is brought on by recumbency and the attendant increase in blood pressure (Mathias et al., 2002). The weight loss during the night is often 2 to 4 pounds, and the reduction in blood volume that results partially accounts for the reduction in orthostatic tolerance seen on arising each morning. The bladder is often directly involved in dysautonomias. This autonomic involvement presents as urgency, retention, incontinence, and frequency. Urological evaluation often suggests prostatic hypertrophy in men, and surgery may be a consideration. Such surgery rarely helps patients with autonomic dysfunction and should only be considered after careful consultation between the urologist and neurologist to ascertain whether a physical obstruction is truly playing a major role. Unfortunately, the α1-antagonist class of drugs commonly used to treat prostatic hypertrophy can worsen orthostatic hypotension; conversely, the α1-agonist, midodrine, used to increase blood pressure, may occasionally increase bladder symptoms. With urine retention, urinary tract infections occur commonly. With autonomic failure, plasma renin levels are often quite low, probably because sympathetic regulation of the kidney is impaired. However, renal function is usually well preserved in most forms of autonomic failure, although not in dopamine β-hydroxylase deficiency, in which significant renal failure occurs in adulthood.
Sweating Abnormalities
Increases or decreases in sweating occur with disturbances of autonomic thermoregulatory function (Fealey, 2008; Low, 1997). Hyperhidrosis refers to conditions in which sweating is excessive for a given stimulus. It can be general or localized. General hyperhidrosis can be primary (episodic hypothermia with hyperhidrosis, or Shapiro syndrome) or secondary to other disorders. Typically, hyperhidrosis is episodic. Dramatic hyperhidrosis may occur in pheochromocytoma. Tumors may produce cytokines, which provoke fever and consequently sweating. Hyperhidrosis also occurs in powerful sympathetic excitation, such as delirium tremens or in the pressor surges of baroreflex failure.
Referral for localized hyperhidrosis (Table 77.1) is often to the neurologist. Evidence exists of enhanced sweat gland innervation coupled with increased activity of sympathetic fibers passing through T2-T4. This is especially prominent in young people. Perhaps 25% of such individuals have a positive family history of hyperhidrosis. Axillary or palmar hyperhidrosis may be so severe as to interfere with normal activities and social interactions. Several therapeutic modalities may help (Table 77.2). A major limitation in therapy of hyperhidrosis is achieving sufficient muscarinic antagonism on sweat glands without incurring unacceptable levels of muscarinic blockade elsewhere—for example, in the heart. Local application of botulinum toxin to affected skin may also be quite effective but requires repeat injections at 3- to 12-month intervals (Saadia et al., 2001). In addition, blockade of the sympathetic ganglia using pharmacological injections, radiofrequency ablations, or endoscopic gangliotomy can be very effective (Atkinson and Fealey, 2003).
Table 77.1 Pathological Hyperhidrosis Differential Diagnosis and Some Causes of Localized Hyperhidrosis
| Condition | Pathophysiological Mechanism of Sweating |
|---|---|
| Essential hyperhidrosis |
Table 77.2 Treatment Measures for Primary Hyperhidrosis
| Topical Rx | 20% Aluminum chloride hexahydrate in anhydrous ethyl alcohol (Drysol). Apply half-strength to dry skin daily or every other day, mornings, and wash off. | Irritation of skin; less effective on palms and soles, which may require occlusive (plastic wrap) technique |
| Tanning Rx, iontophoresis | Glutaraldehyde (2%-10%) solution; apply 2-4 times/week as needed. | Stains skin brown; for soles of feet only |
| For palms/soles; 15-30 mA current, 20 min. at start. Drionic battery-run unit or galvanic generator needed; 3-6 treatments/week for total of 10-15 treatments initially; 1-2 treatments/week maintenance. | Shocks, tingling may occur Difficult to use in axilla Drionic unit not effective when batteries low |
|
| Anticholinergic | Glycopyrrolate (Robinul/Robinul Forte) at 1-2 mg PO tid as needed; for intermittent/adjunctive treatment. | Dry mouth, blurred vision Contraindicated: glaucoma, GI tract obstruction, GU tract obstruction |
| Clonidine | Useful for paroxysmal localized (e.g., hemibody) hyperhidrosis; 0.1-0.3 mg PO tid or as TTS patch (0.1-0.3 mg/day) weekly. | Somnolence, hypotension, constipation, nausea, rash, impotence, agitation |
| Excision | Second and third thoracic ganglionic sympathectomy (palmar hyperhidrosis), sweat glands (axillary liposuction); recent preference is for T2 sympathectomy to limit compensatory hyperhidrosis. | Homer syndrome, dry skin, transient dysesthetic pain Postoperative scar or infection Compensatory hyperhidrosis of trunk, pelvis, legs, and feet |
| Botox | 50-100 mU of botulinum toxin A into each axilla or body area treated; high doses (200 mU) prolong effect; can be repeated. | Injection discomfort, variable Duration of effect 3-12 months Expensive Mild grip weakness when palm is treated Contraindicated in pregnancy, NMJ disease |
GI, Gastrointestinal; GU, genitourinary; mU, mouse units; NMJ, neuromuscular junction; PO, orally; Rx, prescription; tid, three times daily.
Adapted from Fealey, R.D., 2004. Disorders of sweating, in: Robertson, D., Biaggioni, I., Burnstock, G., et al. (Eds.), Primer on the Autonomic Nervous System, second ed. Elsevier, New York, pp. 354-357.
Idiopathic hyperhidrosis must be distinguished from compensatory hyperhidrosis due to lower body hypohidrosis, common in dysautonomias (Klein et al., 2003), which may paradoxically be described by patients as excessive sweating in the upper body. Patients who lose their ability to perspire over most of their body may preserve it in the neck and facial area and perspire disproportionately in these areas, which captures attention more than the loss of sweating elsewhere. In this setting, the hyperhidrosis actually mandates an evaluation for the reason for the anhidrosis, usually a structural dysautonomia of some type. Loss of sweating does not require specific medical therapy, but rather practical advice such as staying well hydrated, avoiding alcohol, and avoiding hot conditions. One of the most effective home remedies is a “wet shirt”: A tee shirt soaked in warm water and wrung out thoroughly before putting on provides some artificial perspiration that lasts 30 to 90 minutes in a hot environment. The associated surface cooling is striking and greatly increases the functional capacity of patients who must be in a hot environment.
Assessment of Autonomic Function
More tests of autonomic function exist than for any other neurological system. Many of these tests are readily applied at the bedside, and though easy to perform, they may be difficult to interpret in an individual patient. Most physicians who routinely follow patients with autonomic disorders develop a small armamentarium of tests that answer questions relating to the focus of their specialty. The neurologist assessing autonomic function requires tests that localize the lesion within the neuraxis and provide information about the types of fibers involved. Therefore, tests of peripheral and central sudomotor function and tests that differentiate sympathetic from parasympathetic cardiovascular function are essential. The cardiologist requires tests of blood pressure and heart rate that evaluate the mechanism of any cardiovascular dysregulation. The endocrinologist measures circulating catecholamines, corticoids, sex hormones, and rennin. These tests are centered on appreciating the hormonal impact and consequences of autonomic dysfunction (Raj et al., 2005). The ophthalmologist tests pupillary function, and the pharmacologist uses drug tests that assess normal or hypersensitive autonomic function response (Robertson et al., 2004). Despite such dramatically divergent diagnostic approaches, it is remarkable how much consensus is often achieved in terms of the actual diagnosis and therapy of an individual patient. Indeed, an interdisciplinary approach that involves multiple specialists from different disciplines may provide both broader perspective on organ-system involvement and more accurate diagnosis and may be the reason more centers are taking this direction clinically.
Regardless of the clinical evaluation setting, a careful history is obviously the critical diagnostic resource. A brief listing of important items in questioning patients is shown in Box 77.1. More detailed discussions of some of these may be found elsewhere. Key autonomic features in the physical examination are shown in Box 77.2. In this section, attention is given to highly informative autonomic tests. Table 77.3 displays a listing of widely employed tests of baroreflex function.
Box 77.1 Key Features in the Autonomic History
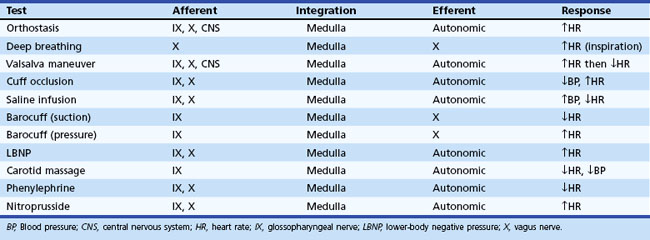
Orthostatic Test
Orthostatic symptoms are usually the most debilitating aspect of autonomic dysfunction readily amenable to therapy. For this reason, the blood pressure and heart rate response to upright posture is the starting point of any autonomic laboratory evaluation. In healthy human subjects, the cardiovascular effect of upright posture has been well defined (Low, 1997). Upon active assumption of the upright posture by standing, the vigorous contraction of large muscles leads to a transitory muscle vasodilation and a minor fall in arterial pressure for which the reflexes do not immediately compensate. However, this short-lived depressor phase is not usually seen with passive (tilt-table) upright posture. Immediately after 70- or 90-degree head-up tilt, approximately 500 mL of blood move into the veins of the legs and approximately 250 mL into the buttocks and pelvic area. A rapid vagally mediated increase in heart rate occurs, followed by a sympathetically mediated further increase. As right ventricular stroke volume declines, a depletion of blood from the pulmonary reservoir occurs, and central blood volume falls. Stroke volume falls, and cardiac output decreases about 20%. With this decline in cardiac output, blood pressure is maintained by vasoconstriction that reduces splanchnic, renal, and skeletal muscle blood flow especially, but other circulations as well.
An important aspect of evaluating responses to orthostasis is the rapid reduction in total blood volume that occurs physiologically. It is not unusual for a 12% fall in plasma volume to occur within 10 minutes of assumption of the upright posture as fluid moves from the vascular compartment into the extravascular space (Jacob et al., 2005). This accounts for the delay in appearance of symptoms in patients with mild autonomic impairment for some minutes after the actual assumption of upright posture. Therefore, the long-stand (30 minutes) test, or Schellong test, is a much more severe orthostatic stress than the short-stand (5 minutes) test commonly employed.
Sweat Testing
Although hypohidrosis rarely dominates a patient’s dysautonomia, assessment of sudomotor function is often helpful in testing for autonomic impairment (Low, 2004). Widely used tests include the thermoregulatory sweat test (TST), quantitative sudomotor axon reflex test (QSART), and sympathetic skin response (SSR).
Thermoregulatory Sweat Test
The TST is a sensitive semiquantitative test of sweating (Fealey et al., 1989). After a color indicator (quinizarin powder or povidone-iodine) is applied to the skin, the environmental temperature is increased until an adequate core temperature rise is attained (usually a 2°C rise in core temperature or a core temperature of 38.5°C, whichever is less) and the presence of sweating causes a change in the indicator. Thermal stimulation using infrared radiant heat lamps to directly heat the skin are also employed to provide more effective sweat stimulus. Estimating the percent of anterior surface anhidrosis quantitates the results, and the sweat rates may be measured as well. This test has also been helpful in assessing the status of dysautonomias over time. Some characteristic patterns of anhidrosis include (1) the peripheral pattern of distal anhidrosis, seen in distal small-fiber neuropathy and length-dependent axonal neuropathy; (2) the central patterns of distal sparing or segmental involvement, generally seen in MSA or PD; and (3) a sudotomal pattern suggesting involvement at the root or ganglion level, seen in disorders involving nerve roots or specific ganglia, such as diabetes, Sjögren disease, and pure autonomic failure. The TST pattern is therefore helpful in distinguishing between postganglionic, preganglionic, and central lesions.
Quantitative Sudomotor Axon Reflex Test
The physiological basis of the QSART is elicitation of an axon reflex mediated by the postganglionic sympathetic sudomotor axon (Low, 2004). Acetylcholine (ACh) activates the axon terminal. The impulse travels antidromically, reaches a branch-point, then travels orthodromically to release ACh from the nerve terminal. ACh traverses the neuroglandular junction and binds to M3 muscarinic receptors on eccrine sweat glands to evoke the sweat response. The QSART specifically evaluates the functional status of postganglionic sympathetic axons.
Pharmacological Tests
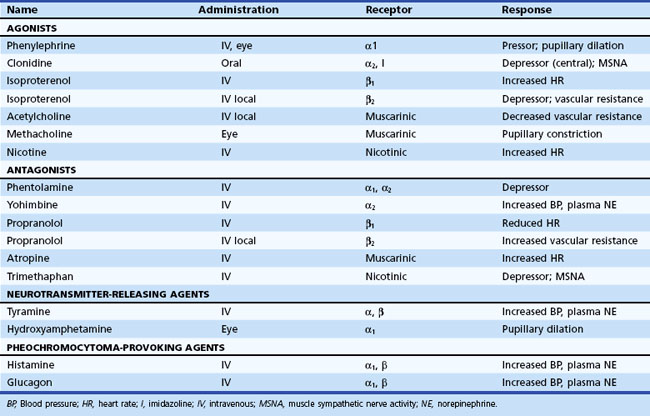
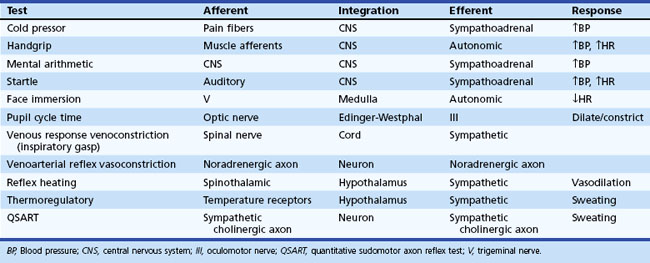
Information about prevailing sympathetic and parasympathetic activation as well as denervation hypersensitivity can be achieved by use of muscarinic and adrenergic agonists and antagonists (Robertson et al., 2004). Tables 77.4 and 77.5 provide instructive examples of how biochemical and physiological tests may be combined to make novel diagnostic discoveries.
Functional Autonomic Disorders
Functional autonomic disorders are a heterogeneous group of disorders where autonomic nervous system involvement exists, but the role of autonomic dysregulation in the pathogenesis of symptoms is unclear. Included in this group are functional gastrointestinal disorders, interstitial cystitis, migraine, cyclic vomiting syndrome (CVS), fibromyalgia, and chronic fatigue syndrome. Many of these disorders tend to coexist in the same person. For example, about 40% of individuals with migraines have symptoms of IBS and chronic aches and pains that could represent fibromyalgia (Chelimsky et al., 2009). In our experience, children with functional gastrointestinal problems have severe fatigue, headaches, and sleep problems (Ojha et al., 2011). Whitehead et al. (2002) found a high association of functional gastrointestinal disorders with fibromyalgia, chronic fatigue syndrome, temporomandibular joint disorder, and chronic pelvic pain. Often the common denominator is POTS. By report, about 50% of persons with CVS and 40% with migraine report symptoms of orthostatic intolerance (Chelimsky et al., 2009). Sullivan et al. (2005) reported 24 pediatric subjects with functional gastrointestinal syndrome and either POTS, syncope, or both. Interestingly, the gastrointestinal symptoms improved or resolved with treatment aimed at the orthostasis (fludrocortisone, sertraline, and midodrine). Chronic fatigue has been described in association with POTS (Hoad et al., 2008).
Reflex Syncope
Syncope of any type is defined as sudden transitory loss of consciousness with spontaneous recovery that is associated with a loss of postural tone (Mathias et al., 2001). Syncope accounts for more than 1% of hospital admissions. The causes of syncope range from benign to life threatening. The common underlying mechanism of syncope is a transitory decrease in cerebral perfusion. Reflex syncope (fainting) is the most common type of syncope, especially in patients without evidence of structural heart disease (Benditt, 2006; Strickberger et al., 2006). Reflex syncope most commonly occurs while the patient is standing but also occurs while seated and occasionally even while lying during sleep (Jardine et al., 2006). Finally, it can occur with exercise (at initiation or at peak exercise) or with emotional/psychological triggers (e.g., phlebotomy).
Many patients can simply be reassured about the usual benign course of reflex syncope and instructed to avoid those situations that precipitate fainting. The use of support stockings or increased salt intake may help. In young non-hypertensive patients, the most frequently affected, we utilize 2 grams of salt in the morning and 2 grams in the early afternoon. Most salt is excreted by a normal kidney within 3 to 4 hours. Patients should be taught to recognize an impending faint and urged to lie down (or sit down if that is not possible) quickly. This will not be enough for some patients, and other treatment options such as physical countermaneuvers (Wieling et al., 2004) and tilt training (Ector et al., 1998) may be necessary. These are covered in detail in the treatment section of the chapter, as are pharmacological and other interventions.
Syncopal Migraine
A subgroup of patients with syncope respond poorly to the usual management of salt supplementation and tilt training. On further questioning, these patients may consistently experience a headache with migrainous features immediately prior to or after the syncopal spell. A recent review suggests that this entity, sometimes also termed basilar migraine, may be far more common than previously suspected, accounting for about one-third of patients with syncope referred to an autonomic specialist (presumably a more complex group of patients based on the referral bias) (Curfman et al., 2011). The importance of identifying this diagnosis is its prompt response to anti-migrainous medications such as verapamil and topiramate, in this author’s experience. Further identifying features include an increased duration of loss of consciousness (up to 15 minutes in this series), and longer time to full recovery, as might be expected in a migrainous mechanism.
Carotid Sinus Hypersensitivity
Carotid sinus hypersensitivity is defined as an asystole of 3 seconds, a fall in systolic pressure of 50 mm Hg, or both in response to carotid artery massage in a patient with otherwise unexplained dizziness or syncope (Fenton et al., 2000; Mathias et al., 2001). Estimates are that 35 to 100 patients per million per year present with this condition. Although the condition has been ensconced in the medical literature since the era of Soma Weiss (1898-1942), its definition remains controversial, in part because diagnosis is by manual massage of the carotid sinus, with its inherent variability. The test should be performed with the patient supine during continuous ECG and blood pressure monitoring and recording. Longitudinal massage should be performed for 5 seconds over the site of maximal pulsation of the right carotid sinus, located between the superior border of the thyroid cartilage and the angle of the mandible. If no response is elicited, the massage is sometimes repeated on the left side supine and ultimately also on the right and then left sides with upright tilt. Unfortunately, improved practical methods for diagnosis have not emerged. Clinically, a history may exist of syncopal symptoms associated with neck pressure, a tight collar, turning the head, shaving, or swallowing; syncope may also occur spontaneously. Hypotension, bradycardia, or both may dominate the clinical picture. The form in which bradycardia predominates may be improved by demand pacing. Some patients with carotid sinus syncope ultimately require surgical denervation. Occasionally, additional symptoms of headache, dizziness, vertigo, paresthesias, homonymous hemianopsia, and hemiplegia occur in the absence of measured blood pressure or heart rate change, but this may reflect another mechanism such as a migrainous or ischemic process; the older literature terms this phenomenon Weiss-Baker syndrome.
Postural Tachycardia Syndrome
POTS is defined as an increase of at least 30 bpm on standing, associated with symptoms of sympathetic activation (Freeman et al., 2002; Jacob et al., 2000; Low et al., 1997). Orthostatic symptoms include light-headedness, palpitations, tremulousness, visual changes, discomfort or throbbing of the head, poor concentration, tiredness, weakness, and occasionally fainting. Usually, little or no fall in blood pressure occurs on standing (Shibao et al., 2005), and this characteristic should probably be incorporated into the definition. Patients may also have an elevated plasma norepinephrine concentration of 600 pg/mL or more on standing. Standing plasma norepinephrine levels greater than 2000 pg/mL occur, and such patients require careful study to exclude pheochromocytoma. Many POTS patients also have a bluish-red discoloration of skin in the lower extremities on standing. A reduced plasma volume of about 500 mL is often present.
POTS is estimated to affect 250,000 to 500,000 Americans and causes a wide range of disabilities (Benrud-Larson et al., 2002). A 4 : 1 female preponderance exists, typically in the 15- to 45-year age group. Symptom severity is sometimes catamenial. Possible reasons for these cyclical changes include an estrogen-dependent change in plasma volume or a direct estrogen receptor–mediated modulation of vascular reactivity. Other than essential hypertension, POTS is the most common chronic disorder of cardiovascular homeostasis. It is commonly encountered and accounts for frequent referrals to centers specializing in autonomic disorders.
The etiology of POTS is unknown; indeed, the condition has many different names (Box 77.3) and probably many causes. For many years, such patients were considered deconditioned and encouraged to pursue a vigorous exercise regimen. Although such regimens can be quite effective (Fu et al., 2010), it is nonetheless clear that the disorder did not arise from mere deconditioning. The onset of POTS may be abrupt, suddenly disabling a prior marathon runner or Olympic athlete, and often occurs in the wake of a viral infection, pregnancy, or major surgical procedure, encouraging consideration of an autoimmune etiology.
Box 77.3 Postural Tachycardia Syndrome
Alternative Names
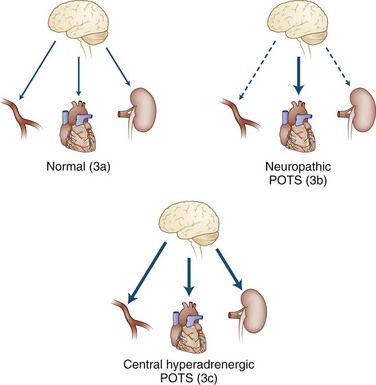
It is important to view POTS not as a disease but as a syndrome, a final common pathway by which many pathophysiological processes may present. The fundamental deficit appears to be anything that reduces the effectiveness of venous return to the heart. Some of these are simple mechanical problems that do not involve the autonomic nervous system at all, such as congenital absence of venous valves or hypermobility disorders (e.g., Ehlers-Danlos syndrome) (Gazit et al., 2003; Rowe et al., 1999) where presumably an inadequate recoil of venous elastic tissue exists to propel adequate volume back to the great veins. Other observations in patients with POTS have included the presence of autonomic neuropathy, termed neuropathic postural tachycardia syndrome (Al-Shekhlee et al., 2005), and the presence of very high adrenergic responsiveness, termed hyperadrenergic postural tachycardia syndrome, as shown in Fig. 77.2. It is not clear that these two processes are mutually exclusive. Other investigators have divided POTS into low, normal, and high blood-flow rates (Fouad et al., 1986; Stewart et al., 2007) and have found different levels of venous compliance in the three types.
Other potential pathophysiological causes include excessive venous pooling, a gravity-dependent fluid shift, diminished plasma volume or red cell mass, and dysfunction of the norepinephrine transporter. In rare specific cases, a genetic disorder has been identified. The clearest example is the illustrative but quite rare form of POTS due to norepinephrine transporter deficiency. This derives from a unique A453P mutation yielding loss of gene function. The norepinephrine transporter clears norepinephrine from the synaptic cleft. Its absence leads to a rise in synaptic and plasma norepinephrine levels with sympathetic hyperactivity (Shannon et al., 2000a).
Mastocytosis may underlie POTS in some individuals (Shibao et al., 2005). The cause may be an increased number or an increased responsiveness of mast cells. Release of histamine and prostaglandin D2 into the circulation dominate the clinical picture. Characteristic chronic skin changes (erythematous acneiform papular lesions) occur in a minority of patients, but red flushing and urticaria are common during attacks. Palpitations, with or without chest pain, headaches, nausea, vomiting, diarrhea, and dyspnea may occur. Perhaps 25% of cases are familial (autosomal dominant). Many patients respond well to treatment with H1 and H2 antagonists, but some have severe attacks with hypotension, requiring treatment by epinephrine infusion. Increases in blood pressure also occur. During severe attacks, disturbances in mental status may seem out of proportion to the hypotension. Patients may also seem to be unconscious for 5 to 20 minutes after syncope, but they may indicate after recovery that they heard what was said to them in the minutes after syncope but were unable to speak or reply to questions. Along with histamine and prostaglandin D2, substantial quantities of heparin are present in mast cells; during attacks, sometimes enough heparin is released to increase the partial thromboplastin time.
POTS is not a stand-alone disorder. Although specific data are still forthcoming, POTS has several comorbidities in both children and adults, such as chronic fatigue syndrome (Freeman and Komaroff, 1997; Schondorf and Freeman, 1999) and migraine. Most patients with POTS have multisystem involvement including sleep problems, upper and lower gastrointestinal complaints, headaches with migrainous features, chronic pain in various locations, Raynaud-like symptoms, and severe fatigue (Ojha et al., 2011). POTS highlights the striking contrast between structural and functional autonomic disorders. Patients with this disorder generally voice many complaints involving several systems, whereas those with severe orthostatic hypotension (mean fall of 90 mm Hg in systolic blood pressure) frequently have minimal or no symptoms at all (Arbogast et al., 2009).
The interactions of deconditioning, sleep disturbance, chronic fatigue, and POTS itself are difficult to assess. Perhaps more challenging is the fact that many patients with POTS respond to their illness by reducing physical activity, and upon presentation to physicians, they therefore have POTS and are deconditioned. In individual patients, several therapies may prove helpful (Shannon et al., 2002), including propranolol 10 to 20 mg three times daily; increased dietary salt; fludrocortisone 0.1 mg orally daily; clonidine 0.05 mg once or twice daily; and midodrine 5 mg orally twice daily. Finally, some patients benefit from an exercise program. This should be approached cautiously, beginning in the most severe patients with no more than “orthostatic exercise,” standing against a wall for incrementally increased periods each day. Therapy is discussed in more detail in the treatment section.
Functional Gastrointestinal Disorders
Despite highly varied presenting symptom complexes, current understanding suggests a biopsychosocial model as the underlying process for most functional gastrointestinal disorders (FGID). This model contains three main components, a genetic/environmental predisposition, pathophysiological changes, and a psychological contribution. It is important to understand that the psychological contribution is not considered to be the cause of these disorders, but rather a comorbid process either primary or secondary to the functional pain disorder. Twin and other genetic studies have demonstrated both a genetic and an environmental predisposition to the development of these disorders (Kalantar et al., 2003; Locke et al., 2000). Genetic factors have included the genotype for reduced interleukin (IL)-10 (antiinflammatory cytokine) production, certain SERT polymorphisms (Yeo et al., 2004), increased messenger ribonucleic acid (mRNA) expression of p11 (a protein critical to 5-HT1B receptor function) in the sigmoid biopsies of IBS-C (Camilleri et al., 2007), and α2-adrenoceptor polymorphism that affects motility (Kim et al., 2004). Environment plays a role through learned behavior from parents who seek more frequent medical attention. For example, children of patients with IBS utilize more healthcare resources than children of healthy control parents (Levy et al., 2000).
Altered motility in the context of stress may play a role in IBS, though these mild abnormalities do not usually explain the disorder. We have also found that gastric electrical activity becomes abnormal in the upright position in children with POTS (a disorder with overlapping symptoms with FGID) (Safder et al., 2010). This may explain the frequent symptom of nausea. Hyperalgesia is very common, from increased rectal sensation to distention of anal balloons to increased sensitivity and discomfort with normal physiological functions (Munakata et al., 1997; Naliboff et al., 1997). The role of either increased intestinal bacterial in the foregut (small-bowel bacterial overgrowth) or altered bacteria is well documented. Some IBS sufferers have small-bowel bacterial overgrowth; others do not fulfill the classic definition for this disease but have greater numbers of bacteria than healthy controls. Furthermore, the use of nonabsorbable antibiotics like rifaximin often improves gastrointestinal symptoms (Pimentel et al., 2000; Posserud et al., 2007). The role of bacteria in the pathogenesis of IBS is further supported by (1) the fact that a third of patients with IBS or dyspepsia describe their symptoms as having begun after an acute enteric infection (Mearin et al., 2005), and (2) that about a quarter of patients who suffer an acute gastrointestinal illness will then develop an FGID (Gwee et al., 1999). More recently, these disorders that were originally defined by the absence of anatomical or inflammatory markers are now found to have increased inflammatory cells (Chadwick et al., 2002).
It is not the goal of this chapter to enumerate every functional disorder, but to list those most commonly encountered in a general neurologist’s practice. IBS is defined as 3 months of abdominal discomfort relieved by a bowel movement or associated with a change in bowel movement frequency or consistency and occurs in 10% to 20% of the general population. Chronic idiopathic nausea also occurs with some frequency. Finally, CVS is characterized by episodes of vomiting alternating with normal baseline health. Though less frequent than other disorders, it sometimes falls in the province of the neurologist because it is highly responsive to antimigrainous strategies, in particular triptans, dihydroergotamine infusion, and prevention using verapamil or tricyclic agents. This disorder has been associated with a mitochondrial polymorphism, and CoQ10 and l-carnitine supplementation may be highly beneficial in some patients (Boles et al., 2005; Ropper, 1994).
Autonomic Disorders Characterized by Excessive Autonomic Outflow
Autonomic Storm and Takotsubo Cardiomyopathy
Emotional stress can cause cardiac injury and sometimes death. Until recently, neurologists have emphasized this connection more than cardiologists. Dr. Martin Samuels has been an especially forceful advocate for the study of this phenomenon (Samuels, 2007). Autonomic storms following cerebral catastrophe may have dramatic physical findings and sometimes ECG abnormalities. Indeed, some of the most dramatic T-wave inversions in the intensive care unit are observed in such patients (Rabinstein et al., 2004). The terminology for these autonomic storms is somewhat chaotic because of lack of systematic study and a profusion of names for the condition. These hypersympathetic states have been termed paroxysmal sympathetic storms, autonomic storms, diencephalic seizures, diencephalic epilepsy, autonomic dysreflexia, or simply dysautonomia. In their extreme form, such autonomic storms result in acute alterations in body temperature, blood pressure, heart rate, respiratory rate, sweating, and muscle tone. They occur with severe head trauma, in post-resuscitation encephalopathy, in intracerebral hemorrhage, with brain tumors, and sometimes in hydrocephalus. Immediately after such a catastrophe, a massive catecholamine surge occurs that can induce seizures, neurogenic pulmonary edema, and myocardial injury. Excessive sympathetic outflow declines or goes away within a few days in most patients, but some patients develop recurrent paroxysms of sympathetic overactivity for weeks or months (Rabinstein et al., 2004).
The pathophysiology of these autonomic storms remains uncertain, but heightened activity of diencephalic or brainstem sympathoexcitatory pathways appears to be the major substrate of these episodes. Autonomic storms may be much more common than suggested by the surprisingly sparse reports in the literature. Clinical features of autonomic storms include tachycardia, hypertension (often with widening of the pulse pressure), fever, tachypnea, sweating, flushing, and pupillary dilation. Intracranial pressure increases during the episodes, with the autonomic dysfunction usually preceding the rise in intracranial pressure. The basis for the diagnosis of autonomic storm is the characteristic dysautonomic spells in a patient with acute intracranial disease. It must be distinguished from other conditions such as neuroleptic malignant syndrome, serotonergic syndrome, malignant hyperthermia, and lethal catatonia. Additional disorders that may manifest sudden unexplained autonomic hyperactivity include Guillain-Barré syndrome (Ropper, 1994) and myelopathy. Severe hypertension, but bradycardia rather than tachycardia, are characteristic of the Cushing reflex syndrome, which occurs in patients with compression of the brainstem.
Takotsubo “Broken Heart” Syndrome
A less dramatic but analogous condition has been recognized in postmenopausal women (Gold et al., 2005; Kurisu et al., 2002; Wittstein et al., 2005). It mimics myocardial infarction and is characterized by chest pain and shortness of breath. It was described initially in Japan as tako tsubo (octopus trap) syndrome and in the United States as apical ballooning syndrome or broken heart syndrome (Wittstein et al., 2005). The first two names describe the appearance of the left ventricle on imaging; it seems to stretch, balloon out, and weaken (Fig. 77.3). The third name derives from the severe emotional stress, such as loss of a family member, that may trigger the condition. In approximately 20% of patients, the inciting factor seems to be severe physical stress such as trauma, surgery, or severe pain. Other names for takotsubo cardiomyopathy are neurogenic myocardial stunning, catecholaminergic myocardial stunning, and stress-induced cardiomyopathy.
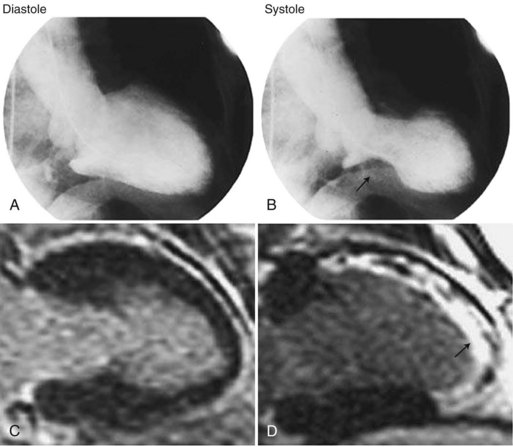
Patients with takotsubo cardiomyopathy do not have coronary artery occlusion or coronary artery spasm during their presentation, but two-thirds have abnormal myocardial blood flow, apparently due to dysfunction in the microvasculature of the heart. Diagnosis is from nonspecific ST-T abnormalities, ST elevation, or QT prolongation with large negative T waves, often occurring over days in succession (Fig. 77.4). Common markers of myocardial infarction (troponin, creatine kinase) are only slightly raised, confirming that there is more stunning of the heart than permanent heart muscle damage. Plasma and urinary catecholamines are typically elevated (Wittstein et al., 2005). The reason this syndrome occurs in some individuals but not others seeming to suffer comparable levels of stress remains unknown. Treatment is supportive, and while 95% of patients experience complete recovery, approximately 10% will have recurrence over a 4-year period.
The pathophysiology of myocardial stunning probably derives from exaggerated calcium influx into the myocardial cells due to excessive norepinephrine release into the myocardium. Norepinephrine activates release of cyclic adenosine monophosphate (c-AMP), which in turn activates a calcium channel allowing calcium influx and potassium outflow. The potassium outflow may account for the frequently encountered peaked T waves seen on ECG. Pathological examination of the heart muscle reveals coagulative myocytolysis (also known as myofibrillar degeneration or contraction band necrosis) characterized by cell death in a hypercontracted state and abnormal cross-band formations. Similar lesions can be induced in animal models through stimulation of the lateral hypothalamus, limbic cortex, mesencephalic reticular formation, and stellate ganglia (Samuels, 2007).
Predominantly Peripheral Afferent Structural Autonomic Disorders Characterized by Impaired Autonomic Outflow
Familial Dysautonomia
Familial dysautonomia (FD, also referred to as Riley-Day syndrome in the older literature) occurs predominantly in individuals of Ashkenazi Jewish extraction carrying mutations in the IB kinase–associated protein gene (IKBKAP). The mutations cause impaired expression of the normal protein product IB kinase–associated protein (IKAP). IKAP deficiency may impair normal expression of neurotransmitters (Axelrod et al., 2006; Axelrod and Hilz, 2003). FD is part of a group of disorders termed hereditary sensory and autonomic neuropathies (HSANs) and is classified as HSAN III in this scheme. HSAN I and V are the other members of the group, with predominant involvement of small sensory and autonomic fibers, where loss of pain sensation is a major clinical issue. Other HSANs involve larger and generally nonautonomic fibers.
FD patients have inadequate development and limited survival of sensory and autonomic neurons. Although the literature has generally emphasized efferent sympathetic involvement, more recent studies suggest that the predominant pathophysiology originates from loss of afferent nerve function, particularly baroreceptor information (Norcliffe-Kaufmann et al., 2010) but also likely including visceral and chemoceptor information (Bernardi et al., 2003). Peripheral nerves demonstrate a reduction in the unmyelinated and small myelinated neuronal populations. In addition, there is evidence of impaired CNS myelination, especially in the optic radiation and middle cerebellar peduncle, which probably accounts for patients’ frequent visual complaints and ataxia (Axelrod et al., 2010).
Identification of the gene defect has not yet led to curative therapy (Tutaj et al., 2006). Preventive and supportive strategies include measures to maintain eye moisture, fundoplication with gastrostomy to provide nutrition and avoid risk of aspiration, use of central agents such as benzodiazepines and clonidine to control vomiting and the dysautonomic crisis, and fludrocortisone and midodrine to combat cardiovascular lability (Freeman, 2003). The result of these improved supportive measures is that approximately half of FD patients now reach adulthood.
Baroreflex Failure
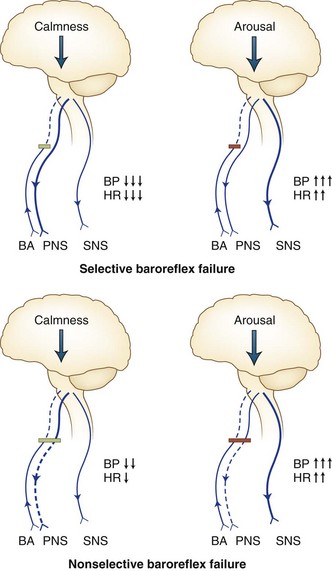
Acute baroreflex failure, perhaps the most dramatic neurological disorder of blood pressure regulation, may display stress-induced systolic blood pressure surges of more than 300 mm Hg. It provides evidence of the great capability of the human CNS to generate unbuffered cardiovascular excitation (Ketch et al., 2002; Timmers et al., 2003) (Fig. 77.5). The clinical syndrome resembles pheochromocytoma more than a typical dysautonomia. Human baroreflexes defend against excessive peaks or dips in blood pressure. Baroreflex failure occurs when afferent baroreceptive input via the vagus or glossopharyngeal nerves or their central connections becomes impaired. Wide excursions of blood pressure and heart rate result. Such excursions may derive from endogenous factors such as anger or drowsiness, which result in high and low pressures, respectively. They may also derive from exogenous factors such as environmental stressors like excessive cold or bright light.
Jordan Syndrome
Abnormalities in the vascular baroreceptors, the glossopharyngeal or vagal nerves, or their brainstem connections can all potentially lead to baroreflex failure. Trauma from injury, tumor, radiation, surgical intervention, or brainstem stroke may also cause baroreflex failure. It occurs in familial paraganglioma syndrome owing to bilateral local tumor growth invading structures at or near the glossopharyngeal and vagal nerves. Radiation therapy for throat carcinoma may incur collateral damage to cranial nerves (Seppi et al., 2005). This damage tends to occur after an interval of months and, in some cases, years after the irradiation, perhaps reflecting local fibrosis as the pathophysiology of nerve damage. In addition, patients who have received radiotherapy for head and neck cancer may present with lightheadedness or syncope due to baroreceptor damage. A baroreflex failure patient with impaired function of the nucleus tractus solitarii but no history of radiation, tumor, or trauma was ultimately diagnosed with Leigh syndrome. Two genetic disorders that appear to entail baroreflex dysfunction have been described: the Groll-Hirschowitz syndrome, in which carotid sinus nerve dysfunction, progressive sensory neuropathy, and duodenal diverticula occur; and the syndrome of autosomal dominant hypertension and brachydactyly with loss of baroreflex buffering. Progressive nerve deafness begins at about age 6 and is complete at age 12 in the face of cochleosaccular degeneration but normal vestibular function. Multiple diverticula with jejunoileal ulceration associated with malabsorption and intestinal loss of serum protein may be associated. Peripheral nerve biopsy shows demyelination. Death occurs in early adult life.
The typical pattern in volatile hypertension is baseline pressures in the high normal or hypertensive range (Fig. 77.6), but with pressor surges accompanied by tachycardia lasting minutes to hours. These pressor surges are elicited by mental or physical stress, during which sympathetic outflow is increased, and are characterized by palpitations and often severe headaches. Profuse sweating occurs during many attacks. Tremulousness, anxiety, and irritability are typical of these episodes, sometimes acting as the triggering event for the surge. Mild and transitory elevations in plasma glucose occasionally occur, as well as a positive correlation between blood pressure and intraocular pressure. During such pressor surges, plasma norepinephrine levels reach values not much less than those seen in pheochromocytoma.
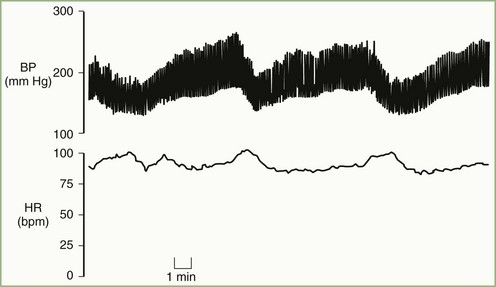
Malignant vagotonia from selective baroreflex failure (Jordan syndrome; see Fig. 77.5) presents as severe bradycardia and asystole due to surges in parasympathetic tone. Along with the hypertensive episodes encountered in the other forms of baroreflex failure, patients with this form may have episodes of hypotension with a systolic pressure below 50 mm Hg. Accompanying symptoms include fatigue and dizziness with possible progression to frank syncope. The most severe episodes tend to occur during early-morning sleep, and periods of asystole longer than 20 seconds may occur. Episodes have also occurred after administration of intravenous nitroprusside and sublingual nitroglycerin.
Treatment of baroreflex failure aims to reduce the frequency and magnitude of life-threatening surges in blood pressure and heart rate (Ketch et al., 2002). A secondary goal of therapy is to attenuate symptomatic hypotensive episodes. The pharmacological treatment of choice for blood pressure surges is clonidine (see Box 77.5). This is a physiological approach to treatment, because this agent acts centrally and peripherally to attenuate sympathetic activation and limit the extent to which pressor surges can occur. The α-adrenoreceptor blocker, phenoxybenzamine, has been relatively unsuccessful in reducing the frequency of pressor surges, although the magnitude of surges (but not tachycardia) is controlled. The sedative effects of α2-adrenoreceptor agonists such as clonidine may assist patients in preventing hypertensive episodes. In the case of clonidine, the inconvenience of frequent oral dosing can be avoided by using a transdermal preparation. Most patients with baroreflex failure will require large doses, whether oral or transdermal. To reduce the possibility of loss of a patch with consequent provocation of clonidine withdrawal, we sometimes use two #1 patches, one placed on Sunday and a second placed on Wednesday of each week, staggered this way to lessen the likelihood of inadvertent complete discontinuation of clonidine.
Predominantly Peripheral Efferent Structural Autonomic Disorders Characterized by Impaired Autonomic Outflow
Pure Autonomic Failure
The term pure autonomic failure (PAF) once encompassed many causes of autonomic failure and orthostatic hypotension (Box 77.4). This term is now restricted to a synucleinopathy with synuclein found within Lewy bodies confined to autonomic ganglia, presenting in mid- to late life (Low, 1997; Mathias et al., 2002; Robertson et al., 2004). The adrenal medulla is relatively spared. The initial feature in men is impotence, but the symptom that usually brings patients to the physician is orthostatic hypotension manifesting as unsteadiness or faintness on standing. It is worst in the morning and improves as the day progresses. Supine hypertension may occur during the night while supine. Meals, exercise, fever, or environmental heat make orthostatic hypotension more severe. Other complaints include orthostatic pain in the neck, shoulders, or occiput, relieved by lying down. Although orthostatic hypotension is defined as a decline in systolic blood pressure of 20 mm Hg and diastolic blood pressure of 10 mm Hg after at least 1 minute of standing, most PAF patients have a decrease in systolic blood pressure of 50 mm Hg or greater. This hypotension may be so severe that convulsive near-syncope occurs in a small portion of affected patients. A single measurement of upright blood pressure that does not meet the above criteria does not exclude the diagnosis. Several measurements of orthostatic blood pressure are required. About 5% of PAF patients have what appears to be angina pectoris, usually in the absence of significant angiographically demonstrable coronary atherosclerosis. PAF patients tolerate high altitude very poorly, perhaps because they hyperventilate in this situation. Even with severe supine hypertension, cardiac function can be preserved and contractility may even rise (Mathias et al., 2002; Robertson et al., 2004).
Box 77.4 Differential Diagnosis of Orthostatic Hypotension
PAF patients have greatly reduced levels of catecholamines. Plasma and urinary norepinephrine levels are usually markedly reduced, sometimes to 10% of normal. Plasma norepinephrine concentrations are always less than 200 pg/mL and often under 100 pg/mL. Plasma levels of epinephrine are also reduced but usually to a lesser extent than norepinephrine. Dopamine levels in urine are usually about 50% of normal values. Marked hypersensitivity to all pressor and depressor stimuli exists (Table 77.6).
| Depressor | Pressor |
|---|---|
| Standing | Lying |
| Food | Water |
| Hyperventilation | Hypoventilation |
| Exercise | Water immersion |
| Straining | Abdominal binding |
| Fever, environmental heat |
Autoimmune Autonomic Ganglionopathy
Also known as autoimmune autonomic neuropathy (AAN) and acute pandysautonomia (Vernino et al., 2000), autoimmune autonomic ganglionopathy typically strikes a previously healthy individual. Severe generalized sympathetic and parasympathetic autonomic failure unfolds over a few days to a few weeks. Orthostatic hypotension, fixed heart rate, anhidrosis, dry mouth, dry eyes, sexual dysfunction, constipation, and impaired pupillary function are present (Vernino et al., 2000). Anorexia, early satiety, postprandial abdominal pain and vomiting, constipation, or diarrhea may also be present. The spectrum and severity of dysautonomia is quite variable, however. Motor and sensory nerve abnormalities are typically absent. The most convincing evidence of an autoimmune pathogenesis is the demonstration of ganglionic nicotinic acetylcholine receptor (AChR) antibodies in high titers in a large proportion of these patients, the correlation of antibody level with dysautonomia severity, and the response of this disorder to intravenous globulin and plasma exchange (Vernino, 2005). Animal studies have demonstrated passive transfer of the disorder with infusion of patient serum. Recent work has confirmed antibody-mediated impairment of synaptic transmission in autonomic ganglia (Vernino et al., 2000).
Autonomic Neuropathy
Causes of autonomic and small-fiber neuropathy include diabetes or metabolic syndrome in about half of cases. Although traditional teaching might suggest that diabetic autonomic neuropathy would only occur in patients with long-standing diabetes, this is clearly no longer the case. Involvement of autonomic nerves and autonomic Schwann cells is probably an immune-mediated process that occurs at the time diabetes develops, or may antedate glucose intolerance by several months or years. In one study (Hoffman-Snyder et al., 2006), more than half of patients presenting with a small-fiber neuropathy of unknown cause had occult glucose intolerance, with over a quarter being diagnosed with frank diabetes for the first time as a result of their neuropathic presentation. Neuropathy associated with diabetes treatment is increasingly recognized as well (Freeman et al., 2010) and may result from too rapid a drop in sugar levels. The triad of rapid-onset neuropathic symptoms, unexplained weight loss (probably due to diabetic gastroparesis), and new treatment initiation for diabetes should prompt strong consideration for this diagnosis, which may respond to steroid treatment early in its course (Said et al., 2003).
Sjögren syndrome (keratoconjunctivitis sicca) is probably the second most frequent cause of small-fiber neuropathy after diabetes and may present with dryness of the eyes, ears, nose, mouth, and vagina, with associated renal tubular acidosis, mononeuritis multiplex, achlorhydria, and often associated collagen disorders. Dysautonomia is relatively common from ganglionitis or cumulative mononeuritis multiplex lesions involving autonomic areas. Both primary and secondary Sjögren syndrome patients (60%) may have autoantibodies against the M3 muscarinic receptor and impaired parasympathetic stimulation of bladder (Wang et al., 2004).
Drug-Induced Dysautonomia
Approximately 10% of drugs used in clinical practice derive their usefulness from their effect on the autonomic nervous system (Benowitz, 2004; Low et al., 2004). Moreover, at least 25% of drugs may, with overdose or toxicity, exert unwanted effects on the autonomic nervous system. These observations account for the importance physicians must attach to recognition of drug-induced autonomic impairment.
Oligohidrosis has been reported in a small number of patients receiving zonisamide, and in a proportion of these patients, hyperthermia has occurred (Low et al., 2004). Production of ciguatera toxin is by dinoflagellates consumed by reef fish. The presenting features of intoxication (most commonly after ingestion of barracuda, red snapper, or grouper) include vomiting, abdominal pain, myalgias, weakness, pruritus, and paresthesias of the mouth, face, and extremities. A peculiar “hot-and-cold reversal” occurs in which cold objects feel hot and vice versa. Cardiovascular features include bradycardia, hypotension, and in some cases, severe orthostatic hypotension. Increased vagal tone characterizes the nature of the autonomic disturbance. The bradycardia and, in part, the orthostatic hypotension reverse with atropine. The orthostatic hypotension is reversible in most cases, resolving within 4 to 6 weeks. Vacor is a toxin associated with severe autonomic neuropathy.
Dopamine β-Hydroxylase Deficiency
Dopamine β-hydroxylase (DBH) deficiency is due to selective absence of norepinephrine and all its metabolites (Robertson et al., 2005). It is described in only a few patients but has a disproportionate importance because it illuminates human noradrenergic function. Affected patients have absent sympathetic noradrenergic function but normal parasympathetic and sympathetic cholinergic function. DBH-deficient patients exhibit profound orthostatic hypotension. Although present from birth, the disorder is often unrecognized until adulthood. Symptoms in the perinatal period include vomiting, dehydration, hypotension, hypothermia, and profound hypoglycemia requiring repeated hospitalization. Exercise capacity is poor. By early adulthood, individuals have profound orthostatic hypotension, greatly reduced exercise tolerance, ptosis of the eyelids, and supine nasal stuffiness. Presyncopal symptoms include dizziness, blurred vision, dyspnea, nuchal discomfort, and chest pain. Neuropsychiatric symptoms are surprisingly mild. CNS abnormalities are not a consideration before diagnosis driven by the orthostatic hypotension. During adult life, some DBH-deficient patients develop renal function abnormalities, including raised blood urea nitrogen and creatinine levels. Life expectancy is uncertain but appears near normal.
The diagnosis is clinical, based on the findings of orthostatic hypotension, intact sweating, ptosis of the eyelids, and arched palate. Biochemical features include minimal or undetectable plasma, CSF, and urinary norepinephrine and epinephrine and a fivefold to tenfold elevation of plasma dopamine, a finding pathognomonic of DBH deficiency. Patients lack urinary normetanephrine, metanephrine, and vanillylmandelic acid. The molecular basis of DBH deficiency has been elucidated (Kim et al., 2002).
Menkes Kinky Hair Syndrome (Trichopolydystrophy, X-linked Copper Deficiency)
DBH is a copper-containing enzyme, and congenital disorders of impaired copper metabolism may present with certain features similar to DBH deficiency. Male infants with Menkes kinky hair syndrome (see Chapter 65) present with stubby, tangled, sparse hair (often white or gray in color), pudgy cheeks, spasticity, seizures, hypothermia, retarded growth, and decreased visual function. Subdural hematoma, jaundice, and osteoporosis also occur. The abnormality in copper handling leads to defective DBH functional efficiency. The incidence is between 1 in 50,000 and 1 in 100,000.
DBH deficiency is the first neurotransmitter defect with a uniquely efficacious replacement strategy. Administration of droxidopa (l-threo-3,4-dihydroxyphenylserine), or LDOPS (Freeman et al., 1999; Kaufmann et al., 2003), alleviates the orthostatic hypotension and other symptoms. Individuals do not respond well to standard therapeutic approaches for autonomic failure. Surgery can correct ptosis. Renal function should be assessed every 3 years or more often if function decreases.
Predominantly Central Structural Autonomic Disorders Characterized by Impaired Autonomic Outflow
Multiple System Atrophy
MSA, also known as Shy-Drager syndrome, is a progressive neurodegenerative disorder encompassing autonomic, extrapyramidal, cerebellar, and pyramidal features (Parikh et al., 2002; Seppi et al., 2005; Wenning et al., 2004). Extrapyramidal involvement (termed MSA-P, for parkinsonian) is about threefold more common than cerebellar involvement (termed MSA-C for cerebellar). Occasional patients feature both types. The pathological hallmark of MSA is neuronal loss and gliosis within multiple sites in the brain, intermediolateral columns, and the Onuf nucleus, with characteristic glial cytoplasmic inclusions (GCIs) containing α-synuclein and ubiquitin. These inclusions are quite distinct from Lewy bodies, which also contain ubiquitin in several respects:
1. Shape: GCIs tend to be irregular in outline, in contrast to the target-shaped concentric circular Lewy bodies of PD.
2. Cellular location: GCIs are obviously in glia, whereas Lewy bodies are in neurons.
3. Neuraxis location: GCIs dominate in the basal ganglia and pons, whereas Lewy bodies occur in midbrain, cortex, and autonomic ganglia.
The average age of onset is 53 years; no confirmed cases exist under age 30, and onset beyond age 70 is rare. No gender predilection exists. MSA has a prevalence of 0.4 per 100,000 individuals. In some patients, the disease presents as orthostatic hypotension or urinary tract symptoms, but in other cases, extrapyramidal or cerebellar symptoms predominate in the early stages. When orthostatic hypotension antedates other neurological involvement, differentiation from autonomic neuropathy, PD, diffuse Lewy body disease, or PAF may be difficult (Geser et al., 2005; Kaufmann et al., 2004; Seppi et al., 2005). Even when there is full-blown parkinsonism and severe dysautonomia, MSA and PD are difficult to separate and cannot be separated on the basis of autonomic testing alone (Riley and Chelimsky, 2003). Other clinical features are better predictors of a diagnosis of MSA, such as poor response to dopamine agonists or l-dopa, absence of significant dementia, absence of tremor, and falls early in the disease course.
Orthostatic hypotension is usually severe, and treatment is frequently complicated by supine hypertension. Supine plasma norepinephrine levels are often near normal in patients with MSA but do not rise appropriately on standing, as expected in a disorder involving central pathways. This contrasts with the low plasma norepinephrine found in the ganglionic disorder, PAF. The reduction in the lying-to-standing norepinephrine level change in MSA results from both lower than normal secretion with standing, and higher than normal secretion when supine. This accounts for the sometimes severe supine hypertension seen in MSA (Shannon et al., 2000b). Thus MSA does not reflect only a reduction of required autonomic outflow but an inversion of central autonomic regulation.
Involvement of the bladder occurs in most patients. The pathophysiology is complex, resulting from pathological changes in bladder-control elements at several levels in the neuraxis. Early complaints of urgency, frequency, and nocturia reflect dysfunction in upper motor neuron circuits, including loss of neuromelatonin-containing neurons in the striatum and loss of neurons in the cerebellum, raphe nuclei, and frontal cortex. The later picture is dominated by overflow incontinence, more consistent with a “lower motor neuron bladder,” most likely reflecting the destruction of cells in the Onuf parasympathetic nucleus of the sacral spinal cord. Post-void residuals steadily increase from year 1 to year 5 of the disease (Ito et al., 2006). In fact, a large post-void residual in a patient with a Parkinson-like disorder suggests MSA rather than PD (Hahn and Ebersbach, 2005). Failure of bladder emptying is further compromised by loss of intermediolateral horn preganglionic sympathetic innervation to the bladder, which results in loss of control of both the sphincter and collapse of the bladder neck (Kirby et al., 1986), which remains open in 53% of patients (Sakakibara et al., 2001). Detailed reviews of the subject have been published (Fowler et al., 2008, 2010).
The pathophysiology of MSA is unknown, and no cure exists, so management continues to be symptomatic (Freeman et al., 1999; Hussain et al., 2001; Jordan and Biaggioni, 2002; Wenning et al., 2005). This includes treatment of the depression, tremor, gait disturbances, orthostatic hypotension, and possible self-catheterization when urine retention is severe. However, in the last 5 years, several promising developments have occurred. A transgenic mouse model that closely mimics the disorder was developed by splicing the α-synuclein gene in front of a myelin basic protein promoter (Shults et al., 2005). Further, rifampicin was shown to inhibit aggregation of α-synuclein in vitro and in this mouse model, leading to a National Institutes of Health–sponsored trial of rifampicin in MSA begun in 2011. A trial of intravenous globulin has now been completed, and the results are pending. Finally, an international trial of the antiparkinsonian drug, rasagiline, an inhibitor of monoamine oxidase type B, has just enrolled its last patient and will be following the cohort for 1 year.
Parkinson Disease
From a clinical perspective, the autonomic manifestations of PD are virtually indistinguishable from those of MSA. Although some authors report a difference between the results of autonomic testing in MSA and PD (Chelimsky, 2008; Lipp et al., 2009), these studies compared patients with nonequivalent burdens of autonomic dysfunction, being much worse in MSA than in PD. Clearly, more severe abnormalities occur in patients with more severe clinical dysautonomia, causing patients with MSA to appear more severe in their test results than patients with PD. When patients in the PD group are matched with those of the MSA group for severity of autonomic involvement, autonomic testing cannot distinguish MSA from PD (Riley and Chelimsky, 2003). The autonomic dysfunction of PD can be astoundingly severe. Although autonomic dysfunction is clearly more prevalent in MSA than in PD, when a patient presents to the office with severe orthostatic hypotension and a PD-like syndrome, PD is more likely, simply because PD is so much more common than MSA (1000 per 100,000 versus 0.4 per 100,000) (Bonuccelli et al., 2003).
The pathophysiology of autonomic dysfunction in PD is different than in MSA. Whereas MSA exclusively involves central networks and nuclei, the Lewy bodies of PD directly involve ganglia, and hence postganglionic neurons, with degeneration of peripheral autonomic fibers. One may therefore see reduction in axon reflex sweating more frequently in PD than in MSA, though this is not always reliable. In addition, increased response to sympathetic agonists such as midodrine due to denervation supersensitivity may occur, but again this is inconsistent. Finally, this peripheral predilection in PD is the basis for the denervation seen on metaiodobenzylguanidine (MIBG) scanning of the heart, which has been touted as a diagnostic test to distinguish PD from MSA (Goldstein et al., 2009), though others do not find the same results (Geser et al., 2011). The rationale is that uptake of this compound, a congener of norepinephrine, into noradrenergic-rich tissues such as the heart will be impaired in conditions such as PAF and PD, but is comparatively normal in MSA. Finally, some authors have advocated sphincter EMG as a method of distinction, since the lower motor neuron is involved in MSA, resulting in polyphasic and bizarre motor units in this disorder but normal motor units in PD (Fowler, 2001).
Myelopathy
Because sympathetic outflow (T1-L2/3) and sacral parasympathetic outflow descend in the spinal cord, cord lesions commonly result in substantial autonomic impairment. This may occur in both spinal cord injury and other disorders such as multiple sclerosis (Fowler et al., 2010), neuromyelitis optica, or spinal cord lesions of any other cause. In cervical and high thoracic levels, most sympathetic and sacral parasympathetic outflow is lost (Mathias, 2004, 2006). Following spinal cord injury, the initial response is hypoexcitability (spinal shock) with flaccid paralysis, impaired tendon reflexes, and spinal autonomic dysfunction presenting as atonic bladder and bowel, vasodilation, and absent spinal autonomic reflexes. This stage lasts days to weeks, and then activity below the transected cord returns. Chronically, a quite distinct autonomic dysfunction emerges (Critchley et al., 2003).
Environmental temperature therefore needs to be carefully controlled. Hyperthermia responds to sponge bathing, the efficacy of which is enhanced by fanning. In severe cases, ice-cooled saline by intravenous infusion or urinary bladder irrigation is occasionally required. The sympathetic skin response can be useful in assessing spinal cord injury (Cariga et al., 2002). Activation of supraspinal centers and descending sudomotor neural pathways in the spinal cord are necessary for the SSR. It is absent in the feet in low spinal injuries and absent in the hand region in high spinal injuries. The presence or absence of the SSR, in addition to motor and sensory evaluation, can be a marker of spinal cord autonomic involvement and may improve classification of the extent of spinal functional deficits. The SSR can also test spinal cord sudomotor centers isolated from the brainstem that are capable of generating an SSR.
In early cord injury, vagal hyperactivity may cause excessive gastric acid secretion, with ulceration and hemorrhage (Mathias, 2002). H2 antagonists or proton pump inhibitors may be useful in such patients. In high lesions, paralytic ileus may occur, especially after ingestion of solid food. Colon dysfunction occurs commonly but may be amenable to bowel training, modification of diet, mild laxatives, and stool softeners. Urine retention, distension, and overflow occur early after cord injury. Intermittent catheterization should be used in the early stages. Persistent infection in various sites may cause secondary amyloidosis with renal infiltration and damage.
In men, both erectile and ejaculatory failure occur in early cord injury. Conversely, in the chronic phase, priapism may occur during autonomic dysreflexia. Ejaculation is often retrograde. Various approaches that include electrical stimulation and collection of seminal fluid have been used for artificial insemination. Sildenafil (Viagra) is effective for erection in spinal injuries and autonomic failure (Hussain et al., 2001). In women, menstrual cycle disruption occurs in early cord injury. Recovery is usually within a year, and successful pregnancy has occurred in both tetraplegic and paraplegic women. In high cord lesions, uterine contractions may evoke severe autonomic dysreflexia. Such patients are particularly prone to seizures and sometimes to cerebral hemorrhage in response to very severe hypertension. These individuals require antihypertensive therapy as well as medication for seizure control.
Nonautonomic Disorders Causing Hypotension or Syncope to Consider in the Differential Diagnosis
Cortisol Deficiency (Addison Disease)
Hypotension and postural hypotension occur in the late stages of cortisol deficiency, as both aldosterone and cortisol itself play important roles in the maintenance of blood pressure. Aldosterone increases available intravascular volume through renal retention of sodium, while cortisol directly influences both the endothelium and vascular smooth muscle. In endothelium, cortisol inhibits the generation of vasodilators such as nitric oxide and prostacyclin, while it enables pharmacomechanical coupling of agonists such as norepinephrine in vascular smooth muscle. When cortisol is absent, hypotension and vascular collapse due to unresponsiveness of the smooth muscle to any pharmacological agonists ensue, including those given exogenously for resuscitation. In this setting, administration of intravenous glucocorticoids is the only strategy and constitutes a medical emergency (Yang and Zhang, 2004).
Therapy of Dysautonomias
Patients with autonomic disorders have both autonomic and nonautonomic features to their illness. Some features, such as extrapyramidal and cerebellar complications in MSA and PD, are discussed elsewhere in this book (see Chapter 71). This chapter addresses treatment of the specifically autonomic aspects of these diseases, such as orthostatic hypotension and bladder and bowel issues. For most disorders, at the time of this writing, treatment is still primarily symptomatic, even where a metabolic or genetic defect has been identified.
Nonpharmacological Interventions for Orthostatic Hypotension
A range of simple physical maneuvers have been identified that can improve the patient’s ability to stand upright (Krediet et al., 2005; van Dijk et al., 2006); implementation is accomplished with a little education (Fig. 77.7). Physical counter-maneuvers possesses the additional advantage of being entirely under the patient’s control, with all the convenience and self-reliance that derive from this. Such maneuvers use muscle pump action or gravity to improve circulation to the brain and forestall a faint when the person is standing; they include leg crossing, performed by crossing the legs in a scissors pattern. The patient actively stands on both legs. It is likely that this maneuver squeezes venous vessels in the legs and abdomen so that less blood pools there. Leaning forward slightly enhances the benefit of this maneuver. Crossing the legs in the seated posture may have a similar physiological basis and is quite effective, especially for individuals with the most severe orthostatic hypotension, when even the seated posture represents a challenge. One limitation of leg crossing in the upright posture is that the patient may have reduced postural stability and be more likely to fall. In particular, few patients with MSA, for example, can benefit from upright leg crossing because of their balance problems independent of their blood pressure. However, in individuals without extrapyramidal or cerebellar abnormalities, this maneuver is so strikingly useful that many patients will have discovered its value on their own and may have been using it for a number of years before they are even diagnosed with orthostatic hypotension (Wieling et al., 2004).
Orthostatic (standing) training has been suggested as effective nonpharmacological therapy for patients with recurrent reflex syncope. This treatment involves leaning patients upright with the upper back against a wall and the feet away from the wall about 1 foot (mimicking a 70-degree angle) for 10 to 20 minutes once or twice a day. The maneuver should be performed on a carpeted floor with no sharp angular furniture nearby, in case of a faint. The initial results from this therapy are quite promising, and it has been subjected to an open-label trial with good results (Fowler et al., 2010). Although not proven to work in POTS or orthostatic hypotension, orthostatic training merits being tried for both of these disorders.
Dietary Measures
Water
In recent years, water has been recognized as a powerful pressor agent in autonomic disorders (Jordan et al., 1999). Increases in blood pressures of 40 mm Hg after ingestion of 500 mL (≈16 ounces) of tap water are quite common; occasional patients may have increases in pressure of 80 mm Hg or more. The effect of water is greatest in the hour after ingestion and is almost gone 90 minutes afterwards. Some patients will have discovered the benefit of water on their own, but for many others it is important to educate them about this very useful approach to raising blood pressure (Jordan et al., 2004; Shannon et al., 2002). Once the possible effect of water is raised with patients, it usually takes only one trial for them to recognize its value. Pure water works best because the sugar or salt content of other beverages attenuate pressor action. The ideal use of water is to drink it immediately upon awakening at the time medications are taken, to cover the hour window before medications begin to take effect.
Drugs
Fludrocortisone
For 50 years, fludrocortisone has been the mainstay of therapy for orthostatic hypotension (Freeman, 2003). However, the limited success of therapy has led to many alternative agents that are sometimes helpful (Box 77.5). Fludrocortisone has two main effects: a well-known high-dose volume effect and a less well-recognized low-dose pressor effect. As a mineralocorticoid like aldosterone, fludrocortisone initially increases blood volume secondary to sodium retention. This effect requires several days to weeks to reach its peak. Although the plasma volume may return to baseline subsequently, in many patients a residual beneficial pressor effect continues, in part due to increased peripheral vascular resistance, enhanced pressor response to norepinephrine, and extravascular fluid accumulation (edema) in the legs, which limits blood pooling. This lesser-known effect, sensitization of α-adrenergic receptors (Davies et al., 1978), allows extremely low doses to have an impact on orthostatic homeostasis in children and young adults with POTS or syncope. We often start with a half tablet (0.05 mg) every other day, with benefit beginning at about 10 days after treatment onset. Fludrocortisone is rapidly absorbed after oral ingestion and has a plasma half-life of 2 to 3 hours, yet its biological half-life is much longer (several days) because of nuclear changes in sodium handling. An additional effect of fludrocortisone is that it increases sympathetic tone and pressor response (Chobanian et al., 1979; Distler et al., 1985).
Midodrine
The most commonly used sympathomimetic agent (Low et al., 1997), midodrine is a prodrug that is hydrolyzed in the liver to its active form, desglymidodrine. The peak effect of oral midodrine occurs 1 hour after administration, and its duration of action is usually 4 to 6 hours. Side effects are generally mild and dose related and include piloerection (goose flesh) and urine retention (due to the α-agonist effect on the urinary sphincter). Because midodrine does not cross the blood-brain barrier, some central side effects such as those that might be seen with ephedrine, pseudoephedrine, or other sympathomimetic amines do not occur. The symptoms of goose flesh are transitory and not very problematic in most individuals, but occasional patients will find this a significant problem, especially in the scalp, and may even need to withdraw from the drug because of the side effect. Usually, however, the most severely affected patients with orthostatic hypotension will recognize the goose flesh sensation as a welcome signal that the blood pressure–raising effect of midodrine is present and that they will now have increased capacity to be up and around. Most patients receive 2.5 to 10 mg every 3 to 4 hours, up to a maximum of 60 mg/day. When first administering this agent to a patient who has never received it, particularly one in whom plasma norepinephrine levels are quite low, it may be worthwhile to give a 1.25-mg test dose. Midodrine should not be given after 6 pm or if the subject will be supine, owing to the risk of supine hypertension. For older patients who nap after lunch, midodrine can be prescribed as a twice-daily drug to be taken in the morning and then after the nap around 3 or 4 pm.
Droxidopa
Droxidopa is beginning to be used more in treatment of orthostatic hypotension (Freeman et al., 1999; Kaufmann et al., 2003). This agent is also a kind of prodrug. It has little effect on its own but is converted in the body directly to norepinephrine by l-aromatic amino acid decarboxylase. Droxidopa is particularly beneficial in patients with DBH deficiency because the drug directly overcomes the enzymatic defect in this genetic disorder. One DBH-deficient patient recently completed a competitive 26-mile marathon (Garland et al., 2005). However, droxidopa is also finding wider use in the treatment of other autonomic disorders including PAF and MSA. The maximum effect of the drug occurs 5 hours after ingestion. The ultimate role of LDOPS vis-à-vis direct-acting agents such as midodrine will depend on future studies comparing these two approaches. Excellent progress has been made, and the drug is likely to be released for widespread use in the next 1 or 2 years.
Pyridostigmine
The anticholinesterase inhibitor, pyridostigmine, commonly used in myasthenia gravis to increase neuromuscular junction neural traffic at the nicotinic receptor on the muscle, has been advocated for use in orthostatic hypotension (Singer et al., 2003). It increases signal strength at the other major nicotinic receptor type, located at the autonomic ganglia, resulting in greater autonomic outflow when the system is activated. As a result, the increase in blood pressure is greater in the standing position than in the lying position, and exacerbation of supine hypertension is less of an issue than it is with drugs that work independently of central autonomic drive. Treatment usually begins with 30 mg (half a 60-mg tablet) 3 times daily, and the dose is gradually increased until blood pressure standing becomes acceptable to a maximum of 90 mg tid.
Erythropoietin
A mild degree of anemia is common in autonomic failure. When comparing levels of hematocrit and hemoglobin to erythropoietin in such individuals, it appears that the erythropoietin level is lower than expected for the level of anemia encountered. For this reason, erythropoietin is an indicated therapy to improve functional capacity in patients with orthostatic hypotension and anemia. This mode of therapy is quite expensive, and third-party payers often find it difficult to understand its use for a neurological condition. The testimonials of patients about erythropoietin are quite dramatic, but long-term controlled trials are lacking. It is possible that with resolution of anemia and normalization of hematocrit, patients may look healthier to family and colleagues and may receive positive reinforcement about their condition based on this. Caution must be used in applying this mode of therapy because in other contexts (e.g., chronic renal failure) normalization of hematocrit with erythropoietin seems to increase the mortality rate (Lau et al., 2010).
Octreotide
Octreotide is a somatostatin analog that has significant effects on the gastrointestinal tract. At low doses in patients with autonomic impairment, it often elicits an increase in blood pressure, sometimes with minimal side effects. In our practice, we usually begin with 12.5 µg. Unfortunately, octreotide must be administered subcutaneously. Its effect appears within a few minutes and lasts a few hours. Almost all patients experience some increase in blood pressure if they have baseline autonomic impairment, but occasional patients experience increases in blood pressure of 40 mm Hg or more and find the drug useful enough to justify subcutaneous injection. We have not had to go beyond 25 µg in managing blood pressures in our patients. However, excessively high doses may cause nausea or altered gastrointestinal motility. Long-term use (years) may increase the risk of gallstones (Veysey et al., 1999), but we have not yet encountered that complication at this low dose.
Pacemaker Placement
Permanent dual-chamber pacemakers may sometimes be required for treatment—if, for example, severe bradycardias occur as part of the dysautonomia—but their value in reflex syncope remains controversial. The are currently not recommended, as several trials failed to show significant benefit (Connolly et al., 1999; Mosqueda-Garcia et al., 2000).
Treatment of Supine Hypertension
Supine hypertension may sometimes be a serious problem. The physiology is now better understood. Ganglionic blockade significantly reduces supine pressure, implying that it is actually in part due to inappropriate sympathetic outflow to the vasculature in the supine position (Shannon et al., 2000a, 2000b). Supine hypertension often requires specific treatment with short-acting antihypertensives at bedtime, such as captopril or propranolol. In our practice, we usually treat pressures over 170/95 mm Hg or so, though no data are available as to when to treat this problem. Patients with autonomic failure do not appear to have the usually associated higher risk of stroke because high pressure is episodic rather than continuous, and the main reason to treat is to reduce the risk associated with very high pressures (e.g., cerebral bleed).
Bowel Issues
Upper-bowel dysfunction usually manifests as early satiety. It occurs commonly in diabetes and probably accounts for the profound weight loss (may be 30–80 pounds over 1–2 months) associated with acute diabetic autonomic syndromes that may be treatment associated. Two aspects of gastric function may come as a surprise: (1) both sympathetic and parasympathetic nervous systems play key roles in gastric emptying, and pure loss of sympathetic function may lead to severe gastroparesis (Chelimsky et al., 2004); and (2) early satiety may NOT represent poor gastric emptying but rather loss of gastric accommodation (ability of the stomach to expand rapidly in compliance with a meal). To distinguish between these two possibilities, one may use a water-load test (How much water can the patient drink in 5 minutes? [normal > 300 mL] (Sood et al., 2002) and a gastric-emptying test with solid and liquid diet. Upper-bowel dysmotility may benefit from promotility agents such as metoclopramide, 10 mg 2 or 3 times daily. However, metoclopramide is contraindicated in PD, owing to brain barrier passage and central effect. It should also be used with extreme caution because of the risk of tardive dyskinesia. Other motility agents are also available, including erythromycin at an adult dose of 250 mg 2 to 3 times daily. Erythromycin is a motilin agonist that improves gastric emptying and foregut motility. Octreotide is also useful in improving foregut motility, although it may produce delayed gastric emptying. It has some effect in increasing blood pressure and has been used in postprandial hypotension.
Al-Shekhlee A., Lindenberg J.R., Hachwi R.N., et al. The value of autonomic testing in postural tachycardia syndrome. Clin Auton Res. 2005;15:219-222.
Arbogast S.D., Alshekhlee A., et al. Hypotension unawareness in profound orthostatic hypotension. Am J Med. 2009;122(6):574-580.
Atkinson J.L., Fealey R.D. Sympathotomy instead of sympathectomy for palmar hyperhidrosis: minimizing postoperative compensatory hyperhidrosis. Mayo Clin Proc. 2003;78(2):167-172.
Axelrod F.B., Chelimsky G.G., Weese-Mayer D.E. Pediatric autonomic disorders. Pediatrics. 2006;118:309-321.
Axelrod F.B., Hilz M.J. Inherited autonomic neuropathies. Semin Neurol. 2003;23(4):381-390.
Axelrod F.B., Hilz M.J., et al. Neuroimaging supports central pathology in familial dysautonomia. J Neurol. 2010;257(2):198-206.
Benarroch E.E. Central Autonomic Network: Functional Organization and Clinical Correlations. NY: Armonk; 1997.
Benditt D.G. The ACCF/AHA scientific statement on syncope: a document in need of thoughtful revision. Clin Auton Res. 2006;16(6):363-368. discussion 369-370
Benowitz N.L. Drug-induced autonomic disorders. In: Robertson I.B.D., Burnstock G., Low P.A. Primer on the Autonomic Nervous System. New York: Elsevier; 2004:336-338.
Benrud-Larson L.M., Dewar M.S., et al. Quality of life in patients with postural tachycardia syndrome. Mayo Clin Proc. 2002;77(6):531-537.
Bernardi L., Hilz M., et al. Respiratory and cerebrovascular responses to hypoxia and hypercapnia in familial dysautonomia. Am J Respir Crit Care Med. 2003;167(2):141-149.
Boles R.G., Adams K., et al. Maternal inheritance in cyclic vomiting syndrome. Am J Med Genet A. 2005;133(1):71-77.
Bonuccelli U., Lucetti C., et al. Orthostatic hypotension in de novo Parkinson disease. Arch Neurol. 2003;60(10):1400-1404.
Buchwald D., Goldenberg D.L., et al. The chronic, active Epstein-Barr virus infection syndrome and primary fibromyalgia. Arthritis Rheum. 1987;30(10):1132-1136.
Camilleri M., Andrews C.N., et al. Alterations in expression of p11 and SERT in mucosal biopsy specimens of patients with irritable bowel syndrome. Gastroenterology. 2007;132(1):17-25.
Camilleri M., Fealey R. Idiopathic autonomic denervation in eight patients presenting with functional gastrointestinal disease. Dig Dis Sci. 1990;35:609-616.
Cariga P., Catley M., et al. Organisation of the sympathetic skin response in spinal cord injury. J Neurol Neurosurg Psychiatry. 2002;72(3):356-360.
Chadwick V.S., Chen W., et al. Activation of the mucosal immune system in irritable bowel syndrome. Gastroenterology. 2002;122(7):1778-1783.
Chandrasekharan B., Srinivasan S. Diabetes and the enteric nervous system. Neurogastroenterol Motil. 2007;19(12):951-960.
Chelimsky G., Chelimsky T. Familial association of autonomic and gastrointestinal symptoms. Clin Auton Res. 2001;11(6):383-386.
Chelimsky G., Madan S., et al. A comparison of dysautonomias comorbid with cyclic vomiting syndrome and with migraine. Gastroenterol Res Pract. 2009;2009:701-719.
Chelimsky G., Wiznitzer M., et al. Vomiting and gastroparesis in thoracic myelopathy with pure sympathetic dysfunction. J Pediatr Gastroenterol Nutr. 2004;39(4):426-430.
Chelimsky T. Autonomic dysfunction. Factor S.A., Weiner W.J. Parkinson’s Disease: Diagnosis and Clinical Management, second ed, New York: Demos, 2008.
Chelimsky T., Chelimsky G. Autonomic abnormalities in cyclic vomiting syndrome. J Pediatr Gastroenterol Nutr. 2007;44:326-330.
Chobanian A.V., Volicer L., et al. Mineralocorticoid-induced hypertension in patients with orthostatic hypotension. N Engl J Med. 1979;301(2):68-73.
Consensus Committee of the American Autonomic Society and the American Academy of Neurology. Consensus statement on the definition of orthostatic hypotension, pure autonomic failure, and multiple system atrophy. Neurology. 1996;46(5):1470.
Connolly S.J., Sheldon R., et al. The North American Vasovagal Pacemaker Study (VPS). A randomized trial of permanent cardiac pacing for the prevention of vasovagal syncope. J Am Coll Cardiol. 1999;33(1):16-20.
Critchley H.D., Mathias C.J., et al. Human cingulate cortex and autonomic control: converging neuroimaging and clinical evidence. Brain. 2003;126(Pt 10):2139-2152.
Curfman D., Chilungu M., Daroff R.B., et al. Syncopal migraine. Clin Auton Res. 2011. [Epub ahead of print]
Davies I.B., Bannister R.G., et al. Fludrocortisone in the treatment of postural hypotension: altered sensitivity to pressor agents [proceedings]. Br J Clin Pharmacol. 1978;6(5):444P-445P.
Distler A., Haller H., et al. Sympathetic tone and pressor response to noradrenaline during mineralocorticoid-induced blood pressure rise in man. J Hypertens Suppl. 1985;3(4):S27-S32.
Ector H., Reybrouck T., et al. Tilt training: a new treatment for recurrent neurocardiogenic syncope and severe orthostatic intolerance. Pacing Clin Electrophysiol. 1998;21(1 Pt 2):193-196.
Ewing D.J., Campbell I.W., et al. Mortality in diabetic autonomic neuropathy. Lancet. 1976;1(7960):601-603.
Fealey R., Low P., et al. Thermoregulatory sweating abnormalities in diabetes mellitus. Mayo Clin Proc. 1989;64:617-628.
Fealey R.D. Thermoregulatory sweat test. In: Low P.A., Benarroch E.E. Clinical Autonomic Disorders. Philadelphia: Lippincott Williams & Wilkins; 2008:244-263.
Fenton A.M., Hammill S.C., et al. Vasovagal syncope. Ann Intern Med. 2000;133(9):714-725.
Fouad F.M., Tadena-Thorne L., Bravo E.L., et al. Idiopathic hypovolemia. Ann Intern Med. 1986;104(3):298-303.
Fowler C.J. Videodynamic and sphincter motor unit potential analyses in Parkinson’s disease and multiple system atrophy. J Neurol Neurosurg Psychiatry. 2001;71(5):575.
Fowler C.J., Dalton C., et al. Review of neurologic diseases for the urologist. Urol Clin North Am. 2010;37(4):517-526.
Fowler C.J., Griffiths D., et al. The neural control of micturition. Nat Rev Neurosci. 2008;9(6):453-466.
Freeman R. Treatment of orthostatic hypotension. Semin Neurol. 2003;23(4):435-442.
Freeman R., Komaroff A.L. Does the chronic fatigue syndrome involve the autonomic nervous system? Am J Med. 1997;102(4):357-364.
Freeman R., Landsberg L., et al. The treatment of neurogenic orthostatic hypotension with 3,4-DL-threo-dihydroxyphenylserine: a randomized, placebo-controlled, crossover trial. Neurology. 1999;53(9):2151-2157.
Freeman R., Lirofonis V., et al. Limb venous compliance in patients with idiopathic orthostatic intolerance and postural tachycardia. J Appl Physiol. 2002;93(2):636-644.
Freeman R., Pollack R., et al. Assessing impaired glucose tolerance and insulin resistance in polycystic ovarian syndrome with a muffin test: alternative to glucose tolerance test. Endocr Pract. 2010;16:810-817.
Fu Q., Vangundy T.B., et al. Cardiac origins of the postural orthostatic tachycardia syndrome. J Am Coll Cardiol. 2010;55(25):2858-2868.
Garland E.M., Raj S.R., et al. Case report: marathon runner with severe autonomic failure. Lancet. 2005;366(Suppl 1):S13.
Gazit Y., Nahir A.M., et al. Dysautonomia in the joint hypermobility syndrome. Am J Med. 2003;115:33-40.
Geser F., Malunda J.A., Hurtig H.I., et al. Subcortical TDP-43 pathology occurs infrequently in multiple system atrophy. Neuropathol Appl Neurobiol. 2011;37:358-365.
Geser F., Wenning G.K., et al. How to diagnose dementia with Lewy bodies: state of the art. Mov Disord. 2005;20(Suppl 12):S11-S20.
Goetze O., Nikodem A.B., et al. Predictors of gastric emptying in Parkinson’s disease. Neurogastroenterol Motil. 2006;18(5):369-375.
Gold P.W., Wong M.L., et al. Cardiac implications of increased arterial entry and reversible 24-h central and peripheral norepinephrine levels in melancholia. Proc Natl Acad Sci U S A. 2005;102(23):8303-8308.
Goldstein D.S. The Autonomic Nervous System in Health and Disease. New York: Marcel Dekker; 2001.
Goldstein D.S., Sharabi Y., et al. Cardiac sympathetic denervation preceding motor signs in Parkinson disease. Cleve Clin J Med. 2009;76(Suppl 2):S47-S50.
Gwee K.A., Leong Y.L., et al. The role of psychological and biological factors in postinfective gut dysfunction. Gut. 1999;44(3):400-406.
Hahn K., Ebersbach G. Sonographic assessment of urinary retention in multiple system atrophy and idiopathic Parkinson’s disease. Mov Disord. 2005;20(11):1499-1502.
Handschug K., Sperling S., et al. Triple A syndrome is caused by mutations in AAAS, a new WD-repeat protein gene. Hum Mol Genet. 2001;10(3):283-290.
Hoad A., Spickett G., et al. Postural orthostatic tachycardia syndrome is an under-recognized condition in chronic fatigue syndrome. QJM. 2008;101(12):961-965.
Hoffman-Snyder C., Smith B.E., et al. Value of the oral glucose tolerance test in the evaluation of chronic idiopathic axonal polyneuropathy. Arch Neurol. 2006;63(8):1075-1079.
Hussain I.F., Brady C.M., et al. Treatment of erectile dysfunction with sildenafil citrate (Viagra) in parkinsonism due to Parkinson’s disease or multiple system atrophy with observations on orthostatic hypotension. J Neurol Neurosurg Psychiatry. 2001;71(3):371-374.
Ito T., Sakakibara R., et al. Incomplete emptying and urinary retention in multiple-system atrophy: when does it occur and how do we manage it? Mov Disord. 2006;21(6):816-823.
Jacob G., Costa F., et al. The neuropathic postural tachycardia syndrome. N Engl J Med. 2000;343(14):1008-1014.
Jacob G., Raj S.R., et al. Postural pseudoanemia: posture-dependent change in hematocrit. Mayo Clin Proc. 2005;80(5):611-614.
Jardine D.L., Krediet C.T., et al. Fainting in your sleep? Clin Auton Res. 2006;16(1):76-78.
Jordan J., Biaggioni I. Diagnosis and treatment of supine hypertension in autonomic failure patients with orthostatic hypotension. J Clin Hypertens (Greenwich). 2002;4(2):139-145.
Jordan J., Shannon J.R., et al. A potent pressor response elicited by drinking water. Lancet. 1999;353(9154):723.
Jordan J., Shannon J.R., et al. Water potentiates the pressor effect of ephedra alkaloids. Circulation. 2004;109(15):1823-1825.
Jost W.H. Gastrointestinal dysfunction in Parkinson’s Disease. J Neurol Sci. 2010;289(1-2):69-73.
Kalantar J.S., Locke G.R.3rd, et al. Familial aggregation of irritable bowel syndrome: a prospective study. Gut. 2003;52(12):1703-1707.
Kaufmann H., Nahm K., et al. Autonomic failure as the initial presentation of Parkinson disease and dementia with Lewy bodies. Neurology. 2004;63(6):1093-1095.
Kaufmann H., Saadia D., et al. Norepinephrine precursor therapy in neurogenic orthostatic hypotension. Circulation. 2003;108(6):724-728.
Ketch T., Biaggioni I., et al. Four faces of baroreflex failure: hypertensive crisis, volatile hypertension, orthostatic tachycardia, and malignant vagotonia. Circulation. 2002;105(21):2518-2523.
Kim C.H., Zabetian C.P., et al. Mutations in the dopamine beta-hydroxylase gene are associated with human norepinephrine deficiency. Am J Med Genet. 2002;108(2):140-147.
Kim H.J., Camilleri M., et al. Association of distinct alpha(2) adrenoceptor and serotonin transporter polymorphisms with constipation and somatic symptoms in functional gastrointestinal disorders. Gut. 2004;53(6):829-837.
Kirby R., Fowler C., et al. Urethro-vesical dysfunction in progressive autonomic failure with multiple system atrophy. J Neurol Neurosurg Psychiatry. 1986;49(5):554-562.
Klein C.M., Vernino S., et al. The spectrum of autoimmune autonomic neuropathies. Ann Neurol. 2003;53(6):752-758.
Korczyn A.D. Autonomic nervous system disturbances in Parkinson’s disease. Adv Neurol. 1990;53:463-468.
Krediet C.T., de Bruin I.G., et al. Leg crossing, muscle tensing, squatting, and the crash position are effective against vasovagal reactions solely through increases in cardiac output. J Appl Physiol. 2005;99(5):1697-1703.
Kurisu S., Sato H., et al. Tako-tsubo-like left ventricular dysfunction with ST-segment elevation: a novel cardiac syndrome mimicking acute myocardial infarction. Am Heart J. 2002;143(3):448-455.
Lau J.H., Gangji A.S., et al. Impact of haemoglobin and erythropoietin dose changes on mortality: a secondary analysis of results from a randomized anaemia management trial. Nephrol Dial Transplant. 2010;25(12):4002-4009.
Levy R.L., Whitehead W.E., et al. Intergenerational transmission of gastrointestinal illness behavior. Am J Gastroenterol. 2000;95(2):451-456.
Lipp A., Sandroni P., et al. Prospective differentiation of multiple system atrophy from Parkinson disease, with and without autonomic failure. Arch Neurol. 2009;66(6):742-750.
Locke G.R., Zinsmeister A.R.3rd, et al. Familial association in adults with functional gastrointestinal disorders. Mayo Clin Proc. 2000;75(9):907-912.
Low P. Clinical Autonomic Disorders. Philadelphia: Lippincott-Raven; 1997.
Low P. Laboratory evaluation of autonomic failure. In: Low P., editor. Clinical Autonomic Disorders. Boston, MA: Little Brown & Co; 1997:169-196.
Low P., Schondorf R., et al. Postural tachycardia syndrome. Clinical autonomic disorders, in: Low, P. (Ed.). Philadelphia: Lippincott-Raven; 1997. pp. 681-697
Low P.A. Laboratory evaluation of autonomic function. Suppl Clin Neurophysiol. 2004;57:358-368.
Low P.A., Gilden J.L., et al. Efficacy of midodrine vs placebo in neurogenic orthostatic hypotension. A randomized, double-blind multicenter study. Midodrine Study Group. JAMA. 1997;277(13):1046-1051.
Low P.A., James S., et al. Zonisamide and associated oligohidrosis and hyperthermia. Epilepsy Res. 2004;62(1):27-34.
Mathias C.J. Autonomic Disturbances. In: Robertson I.B.D., Burnstock G., Low P.A. Spinal Cord Injuries. Primer on the Autonomic Nervous System. New York: Elsevier; 2004:298-301.
Mathias C.J. Orthostatic hypotension and paroxysmal hypertension in humans with high spinal cord injury. Prog Brain Res. 2006;152:231-243.
Mathias C.J., Bannister R. Autonomic Failure: A Textbook of Clinical Disorders of the Autonomic Nervous System. Oxford: University Press; 2002.
Mathias C.J., Deguchi K., et al. Observations on recurrent syncope and presyncope in 641 patients. Lancet. 2001;357(9253):348-353.
Mearin F., Perez-Oliveras M., et al. Dyspepsia and irritable bowel syndrome after a Salmonella gastroenteritis outbreak: one-year follow-up cohort study. Gastroenterology. 2005;129(1):98-104.
Mosqueda-Garcia R., Furlan R., et al. The elusive pathophysiology of neurally mediated syncope. Circulation. 2000;102(23):2898-2906.
Munakata J., Naliboff B., et al. Repetitive sigmoid stimulation induces rectal hyperalgesia in patients with irritable bowel syndrome. Gastroenterology. 1997;112:55-63.
Naliboff B.D., Munakata J., et al. Evidence for two distinct perceptual alterations in irritable bowel syndrome. Gut. 1997;41(4):505-512.
Norcliffe-Kaufmann L., Axelrod F., et al. Afferent baroreflex failure in familial dysautonomia. Neurology. 2010;75(21):1904-1911.
O’Leary M.P., Sant G.R. The interstitial cystitis symptom and problem indices: rationale, development, and application. In: Sant G.R., editor. Interstitial Cystitis. Philadelphia: Lippincott-Raven; 1997:271-276.
Ojha A., Chelimsky T.C., Chelimsky G. Comorbidities in pediatric patients with postural orthostatic tachycardia syndrome. J Pediatr. 2011;158:20-23.
Parikh S.M., Diedrich A., et al. The nature of the autonomic dysfunction in multiple system atrophy. J Neurol Sci. 2002;200(1-2):1-10.
Pimentel M., Chow E.J., et al. Eradication of small intestinal bacterial overgrowth reduces symptoms of irritable bowel syndrome. Am J Gastroenterol. 2000;95(12):3503-3506.
Posserud I., Stotzer P.O., et al. Small intestinal bacterial overgrowth in patients with irritable bowel syndrome. Gut. 2007;56(6):802-808.
Rabinstein A.A., Wijdicks E.F.M. The autonomic storm. In: Robertson I.B.D., Burnstock G., Low P.A. Primer on the Autonomic Nervous System. New York: Elsevier, 2004.
Raj S.R., Biaggioni I., et al. Renin-aldosterone paradox and perturbed blood volume regulation underlying postural tachycardia syndrome. Circulation. 2005;111(13):1574-1582.
Riley D.E., Chelimsky T.C. Autonomic nervous system testing may not distinguish multiple system atrophy from Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2003;74(1):56-60.
Robertson D., Biaggioni I., Burnstock G., et al. Primer on the Autonomic Nervous System. New York: Elsevier; 2004.
Robertson D., Garland E.M. Dopamine β-hydroxylase deficiency (GeneClinics). Gene Clinics. from http://www.geneclinics.org/servlet/access?db=geneclinics&id=8888891&key=YFQfVXIfQFoXz&gry=&fcn=y&fw=1017&filename=/profiles/dbh/index.html, 2005.
Ropper A.H. Intensive care of acute Guillain-Barre syndrome. Can J Neurol Sci. 1994;21(2):S23-S27.
Rowe P.C., Barron D.F., et al. Orthostatic intolerance and chronic fatigue syndrome associated with Ehlers-Danlos syndrome. J Pediatr. 1999;135(4):494-499.
Saadia D., Voustianiouk A., et al. Botulinum toxin type A in primary palmar hyperhidrosis: randomized, single-blind, two-dose study. Neurology. 2001;57(11):2095-2099.
Safder S., Chelimsky T.C., O’Riordan M.A., et al. Autonomic testing in functional gastrointestinal disorders: implications of reproducible gastrointestinal complaints during tilt table testing. Gastroenterol Res Pract. 2009;2009:868-896.
Safder S., Chelimsky T.C., O’Riordan M.A., et al. Gastric electrical activity becomes abnormal in the upright position in patients with postural tachycardia syndrome. J Pediatr Gastroenterol Nutr. 2010;51:314-318.
Said G., Lacroix C., et al. Inflammatory vasculopathy in multifocal diabetic neuropathy. Brain. 2003;126(Pt 2):376-385.
Sakakibara R., Hattori T., et al. Videourodynamic and sphincter motor unit potential analyses in Parkinson’s disease and multiple system atrophy. J Neurol Neurosurg Psychiatry. 2001;71(5):600-606.
Samuels M.A. The brain-heart connection. Circulation. 2007;116(1):77-84.
Sandroni P., Opfer-Gehrking T.L., et al. Postural tachycardia syndrome: clinical features and follow-up study. Mayo Clin Proc. 1999;74(11):1106-1110.
Schondorf R., Freeman R. The importance of orthostatic intolerance in the chronic fatigue syndrome. Am J Med Sci. 1999;317(2):117-123.
Schondorf R., Low P. Idiopathic postural orthostatic tachycardia syndrome: an attenuated form of acute pandysautonomia? Neurology. 1993;43(1):132-136.
Schrag A., Wenning G.K., et al. Survival in multiple system atrophy. Mov Disord. 2008;23(2):294-296.
Seppi K., Schocke M.F., et al. How to diagnose MSA early: the role of magnetic resonance imaging. J Neural Transm. 2005;112(12):1625-1634.
Shannon J.R., Diedrich A., Biaggioni I., et al. Water drinking as a treatment for orthostatic syndromes. Am J Med. 2002;112(5):355-360.
Shannon J.R., Flattem N.L., Jordan J., et al. Orthostatic intolerance and tachycardia associated with norepinephrine-transporter deficiency. N Engl J Med. 2000;342(8):541-549.
Shannon J.R., Jordan J., Dietrich A., et al. Sympathetically mediated hypertension in autonomic failure. Circulation. 2000;101(23):2710-2715.
Shibao C., Arzubiaga C., et al. Hyperadrenergic postural tachycardia syndrome in mast cell activation disorders. Hypertension. 2005;45(3):385-390.
Shults C.W., Rockenstein E., et al. Neurological and neurodegenerative alterations in a transgenic mouse model expressing human alpha-synuclein under oligodendrocyte promoter: implications for multiple system atrophy. J Neurosci. 2005;25(46):10689-10699.
Singer W., Opfer-Gehrking T.L., et al. Acetylcholinesterase inhibition: a novel approach in the treatment of neurogenic orthostatic hypotension. J Neurol Neurosurg Psychiatry. 2003;74(9):1294-1298.
Sood M.R., Schwankovsky L.M., et al. Water load test in children. J Pediatr Gastroenterol Nutr. 2002;35(2):199-201.
Spiegel B., Strickland A., et al. Predictors of patient-assessed illness severity in irritable bowel syndrome. Am J Gastroenterol. 2008;103(10):2536-2543.
Stewart J.M., Medow M.S., et al. Cutaneous neuronal nitric oxide is specifically decreased in postural tachycardia syndrome. Am J Physiol Heart Circ Physiol. 2007;293(4):H2161-H2167.
Strickberger S.A., Benson D.W., et al. AHA/ACCF Scientific Statement on the evaluation of syncope: from the American Heart Association Councils on Clinical Cardiology, Cardiovascular Nursing, Cardiovascular Disease in the Young, and Stroke, and the Quality of Care and Outcomes Research Interdisciplinary Working Group; and the American College of Cardiology Foundation: in collaboration with the Heart Rhythm Society: endorsed by the American Autonomic Society. Circulation. 2006;113(2):316-327.
Sullivan S., Hanauer J., et al. Gastrointestinal symptoms associated with orthostatic intolerance. J Pediatr Gastroenterol Nutrition. 2005;40:425-428.
Timmers H.J., Wieling W., et al. Denervation of carotid baro- and chemoreceptors in humans. J Physiol. 2003;553(Pt 1):3-11.
Tutaj M., Marthol H., et al. Effect of physical countermaneuvers on orthostatic hypotension in familial dysautonomia. J Neurol. 2006;253(1):65-72.
van Dijk N., Quartieri F., et al. Effectiveness of physical counterpressure maneuvers in preventing vasovagal syncope: the Physical Counterpressure Manoeuvres Trial (PC-Trial). J Am Coll Cardiol. 2006;48(8):1652-1657.
Venkatesan T., Prieto T., Barbol A., et al. Autonomic nerve function in adults with cyclic vomiting syndrome: a prospective study. Neurogastroenterol Motil. 2010;22:1303-1307.
Vernino S. Neurologic autoantibodies: cautious enthusiasm. Neurology. 2005;65(11):1688-1689.
Vernino S., Low P., et al. Autoantibodies to ganglionic acetylcholine receptors in autoimmune autonomic neuropathies. N Engl J Med. 2000;343:847-855.
Veysey M.J., Thomas L.A., et al. Prolonged large bowel transit increases serum deoxycholic acid: a risk factor for octreotide induced gallstones. Gut. 1999;44(5):675-681.
Wang F., Jackson M.W., et al. Passive transfer of Sjogren’s syndrome IgG produces the pathophysiology of overactive bladder. Arthritis Rheum. 2004;50(11):3637-3645.
Wenning G.K., Colosimo C., et al. Multiple system atrophy. Lancet Neurol. 2004;3(2):93-103.
Wenning G.K., Geser F., et al. Therapeutic strategies in multiple system atrophy. Mov Disord. 2005;20(Suppl 12):S67-S76.
Whitehead W.E., Palsson O., et al. Systematic review of the comorbidity of irritable bowel syndrome with other disorders: what are the causes and implications? Gastroenterology. 2002;122(4):1140-1156.
Wieling W., Colman N., et al. Nonpharmacological treatment of reflex syncope. Clin Auton Res. 2004;14(Suppl 1):62-70.
Wittstein I.S., Thiemann D.R., et al. Neurohumoral features of myocardial stunning due to sudden emotional stress. N Engl J Med. 2005;352(6):539-548.
Yang S., Zhang L. Glucocorticoids and vascular reactivity. Curr Vasc Pharmacol. 2004;2(1):1-12.
Yeo A., Boyd P., et al. Association between a functional polymorphism in the serotonin transporter gene and diarrhoea predominant irritable bowel syndrome in women. Gut. 2004;53(10):1452-1458.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 77 Disorders of the Autonomic Nervous System
Classification of Autonomic Disorders
Clinical Features of Autonomic Impairment
Assessment of Autonomic Function
Functional Autonomic Disorders
Autonomic Disorders Characterized by Excessive Autonomic Outflow
Predominantly Peripheral Afferent Structural Autonomic Disorders Characterized by Impaired Autonomic Outflow
Predominantly Peripheral Efferent Structural Autonomic Disorders Characterized by Impaired Autonomic Outflow
Predominantly Central Structural Autonomic Disorders Characterized by Impaired Autonomic Outflow
Nonautonomic Disorders Causing Hypotension or Syncope to Consider in the Differential Diagnosis
At the higher control levels in the brain, autonomic integrators and signals are integrated and expressed subconsciously through the central autonomic network (Benarroch, 1997). This overlies a strong circadian rhythm of autonomic function. The autonomic nervous system consists of two large divisions, the sympathetic (thoracolumbar) outflow, and the parasympathetic (craniosacral) outflow. The two divisions are defined by their anatomical origin rather than by their physiological characteristics. The circadian rhythm of autonomic function originates in the suprachiasmatic nucleus and is conveyed to the hypothalamus and brainstem. Light falling on retinal ganglion cell dendrites (not rods or cones) in the eye and transmitted via the retinohypothalamic tract entrains this rhythm. Other key inputs to autonomic outflow originate in the insular cortex and the amygdala. The principal integration of autonomic outflow to the cardiovascular system lies in the medulla. Stretch-sensitive baroreflex mechanoreceptors in the blood vessels of the thorax and neck relay information about blood pressure and blood volume through the glossopharyngeal (from carotid arteries) and vagus (from aorta) nerves to the nucleus tractus solitarius (NTS) in the posterior medulla. Excitatory neurons from the NTS innervate the dorsal motor nucleus of the vagus, which regulates parasympathetic outflow. Inhibitory neurons project to areas in the ventrolateral medulla from which sympathetic outflow is regulated. The most important such site is the rostral ventrolateral medulla.
The autonomic nervous system exerts widespread control over homeostasis (Goldstein, 2001; Mathias and Bannister, 2002). Almost every organ system receives regulatory information from the central nervous system (CNS) through the autonomic efferents (Fig. 77.1), and increasingly we recognize that afferent input into the central autonomic network regulates not only the output of the autonomic system but also much CNS function not generally considered to be autonomic in nature. The emerging concept is pervasive integration of autonomic activities with brain and body.
Classification of Autonomic Disorders
Since by definition, structural disorders have a known pathological substrate, they can be divided into two camps, those that involve the peripheral nervous system and those that involve central pathways. Such a division would be more challenging in functional disorders, where a specific etiopathology is generally not defined, with certain rare exceptions (see later discussion and Fig. 77.1). Peripheral disorders can be further subdivided by whether the dominant pathological process involves afferent or efferent fibers. As can be discerned from Fig. 77.1, most peripheral disorders involve efferent nerves. It should be kept in mind that few if any of the structural disorders are “pure” either in their central versus peripheral localization or in afferent versus efferent functional classification. For example, diabetes has been shown to involve central pathways, and PD involves peripheral ganglia, symbolized by the dashed line in the figure connecting that box to the efferent peripheral category.
These two basic classes of dysautonomias, structural and functional, contrast with one another in other ways as well. Patients with structural disorders, in spite of their extensive pathological changes in the nervous system, tend to harbor proportionally few symptoms; for example, the majority of patients do not realize when their systolic blood pressure drops by nearly 90 mm Hg (Arbogast et al., 2009). In contrast, patients with functional disorders harbor enormous numbers of complaints, just as disproportionate with the demonstrable pathological involvement of the autonomic nervous system but in the opposite sense (Ojha, 2011). These disorders are now being considered as multisystem rather than involving a single end-organ. Another difference relates to prognosis. Structural disorders in general carry a very poor prognosis. For example, diabetics with autonomic neuropathy have a 5-year mortality between 25% and 50% (Ewing et al., 1976). Patients with MSA survive only 7 to 9 years from onset (Schrag et al., 2008). In contrast, while functional disorders can be extremely disabling (Benrud-Larson et al., 2002; O’Leary and Sant, 1997; Spiegel et al., 2008), their prognosis for longevity is generally good.
Clinical Features of Autonomic Impairment
Cardiovascular
Normally when one stands, systolic blood pressure falls about 10 mm Hg, and diastolic pressure increases 5 mm Hg. Heart rate rises 5 to 20 beats per minute (bpm). A fall in blood pressure of 20/10 mm Hg or more in the first 3 minutes of standing defines orthostatic hypotension (Consensus Committee of the American Autonomic Society and the American Academy of Neurology, 1996). If there is not a fall in blood pressure of 20/10 on standing, but the patient is symptomatic and experiencing tachycardia, orthostatic intolerance is present, qualifying as POTS if the heart rate rise is greater than 30 bpm in the first 10 minutes in the upright position (Schondorf and Low, 1993). The most common symptoms of orthostatic hypotension are dizziness, dimming of vision, and discomfort in the neck and head. Orthostatic hypotension is greatest, and hence most easily detected, in the hour after ingesting a large breakfast. Carbohydrate acts as a depressor more than protein or fat. It is important to note that a majority of patients with orthostatic hypotension will not have the usual expected symptoms and may present with falls only (Arbogast et al., 2009), so it is vital to not rely solely on symptom reports in the elderly population.
Pulmonary
Lung function is generally preserved in autonomic failure. Patients with MSA frequently experience sleep apnea, and in consequence of these apneic spells, major perturbations in blood pressure may arise acutely. This is in part dependent on the role of carbon dioxide in determining blood pressure level in autonomic failure. Hypoventilation increases blood pressure in autonomic failure, whereas hyperventilation may lower blood pressure significantly. Patients have occasionally noted improved orthostatic tolerance while breathing through a dead space, although this has never been a recommendation in practice. Because of impaired swallowing in patients with MSA (Seppi et al., 2005), aspiration pneumonia occurs frequently and can be missed because the patient may not manifest the expected fever. With early diagnosis and treatment of aspiration pneumonia, patients often do well, returning to their prior state of health.
Gastrointestinal
Constipation occurs in many patients with autonomic failure, but patients with diabetic dysautonomia may also have frequent and often severe diarrhea as well as significant gastroparesis. The diarrhea itself can prevent adequate blood pressure control because of the associated volatility of blood volume. Special problems sometimes occur in specific dysautonomias. For example, patients with Sjögren syndrome commonly have gastroesophageal reflux and may therefore have an increased risk of esophageal carcinoma. Patients with Chagas disease may have achalasia and enlargement of the esophagus, resulting in vomiting. Achalasia is also present in the 4 “A” syndrome (Allgrove disease) characterized by alacrima, achalasia, ACTH insensitivity, and autonomic neuropathy. This syndrome has been mapped to chromosome 12q13 and produced by mutations in the AAAS gene (Handschug et al., 2001). Patients with some forms of genetic autonomic failure may have strikingly severe gastrointestinal fluid loss and bowel movements 10 or more times every day, which sometimes responds to low doses of clonidine. Postprandial angina may occur with food ingestion, usually without associated ST-T wave changes. Most patients with postprandial angina in practice probably have some degree of dysautonomia, and the depressor effect of food is most prominent in the setting of impaired autonomic reflexes. Postprandial angina tends to occur with upright posture following food (especially carbohydrates). Water intake in association with eating will usually help prevent this symptom.
The gastrointestinal dysfunction in two disorders with significant autonomic involvement, namely PD and diabetes, have been better evaluated and understood. For example, the role of diabetes in gastrointestinal dysmotility has been extensively studied. Some changes in the function of the enteric nervous system in diabetes appear to result from apoptosis of enteric neurons. Oxidative stress may play a role in the cell death process. An imbalance also exists in inhibitory and excitatory neuropeptides. These factors then result in altered gut mucosa (Chandrasekharan and Srinivasan, 2007). PD is also associated with gastrointestinal problems, affecting swallowing disorders that may lead to aspiration. Salivation is not increased or may even be decreased in these patients. From early on, individuals with PD may develop delayed gastric emptying, which later in the disease may affect jejunal absorption of levodopa. Gastric emptying of liquids is usually not delayed (Goetze et al., 2006) in PD, so alternatives when giving levodopa are a liquid solution or administering it directly into the jejunum (Jost, 2010). Constipation is also very common in this disorder and represents the most common “autonomic” manifestation of PD, affecting 70% to 80% of patients. Often, severe constipation develops before the more typical symptoms of PD are noticed (Korczyn, 1990; Jost, 2010); the cause is multifactorial. Many of the medications prescribed to treat PD, mainly anticholinergics and (perhaps more controversial) levodopa, may worsen constipation, but they are not the cause, since constipation is usually present before the diagnosis of PD is made and therefore before the onset of medications. Constipation is thought to be due to degeneration of central and peripheral parasympathetic nuclei (Jost, 2010).
Other less well-characterized dysautonomias also have gastrointestinal symptoms. Individuals with POTS complain of bloating, early satiety, nausea, pain, and alternating diarrhea and constipation (Sandroni et al., 1999). In fact, more than half of the children and adults with POTS have gastrointestinal complaints, usually reporting epigastric or lower-abdominal discomfort. Children with POTS seem to suffer nausea and vomiting more often than adults, but these findings were present in both groups (Ojha et al., 2011). Interestingly, coming from the other direction, many with functional gastrointestinal problems also have cardiovascular autonomic dysfunction, primarily sympathetic. In adults, three-eighths also had parasympathetic involvement, which was not present in the pediatric group. Neuropathy was common in both groups (Camilleri and Fealey, 1990; Chelimsky and Chelimsky, 2001). Importantly, when both a functional gastrointestinal disorder and POTS are present, the gastrointestinal symptoms may resolve with treatment of the orthostatic intolerance (Sullivan et al., 2005). The cause of the gastrointestinal symptoms in POTS in unclear and may be related to blood pooling in either the lower extremities or abdomen. Electrical activity of the stomach in POTS changes from supine to the upright position (Safder et al., 2009), suggesting either lack of accommodation or gastroparesis while upright (Buchwald et al., 1987). Cyclic vomiting syndrome (CVS) has also been associated with autonomic dysfunction, and both pediatric and adult sufferers usually have POTS associated with an autonomic neuropathy (Chelimsky and Chelimsky, 2007; Venkatesan et al., 2010).
Urinary Tract
In structural disorders, a reversal of the usual pattern of urine output occurs. Nocturia is brought on by recumbency and the attendant increase in blood pressure (Mathias et al., 2002). The weight loss during the night is often 2 to 4 pounds, and the reduction in blood volume that results partially accounts for the reduction in orthostatic tolerance seen on arising each morning. The bladder is often directly involved in dysautonomias. This autonomic involvement presents as urgency, retention, incontinence, and frequency. Urological evaluation often suggests prostatic hypertrophy in men, and surgery may be a consideration. Such surgery rarely helps patients with autonomic dysfunction and should only be considered after careful consultation between the urologist and neurologist to ascertain whether a physical obstruction is truly playing a major role. Unfortunately, the α1-antagonist class of drugs commonly used to treat prostatic hypertrophy can worsen orthostatic hypotension; conversely, the α1-agonist, midodrine, used to increase blood pressure, may occasionally increase bladder symptoms. With urine retention, urinary tract infections occur commonly. With autonomic failure, plasma renin levels are often quite low, probably because sympathetic regulation of the kidney is impaired. However, renal function is usually well preserved in most forms of autonomic failure, although not in dopamine β-hydroxylase deficiency, in which significant renal failure occurs in adulthood.
Sweating Abnormalities
Increases or decreases in sweating occur with disturbances of autonomic thermoregulatory function (Fealey, 2008; Low, 1997). Hyperhidrosis refers to conditions in which sweating is excessive for a given stimulus. It can be general or localized. General hyperhidrosis can be primary (episodic hypothermia with hyperhidrosis, or Shapiro syndrome) or secondary to other disorders. Typically, hyperhidrosis is episodic. Dramatic hyperhidrosis may occur in pheochromocytoma. Tumors may produce cytokines, which provoke fever and consequently sweating. Hyperhidrosis also occurs in powerful sympathetic excitation, such as delirium tremens or in the pressor surges of baroreflex failure.
Referral for localized hyperhidrosis (Table 77.1) is often to the neurologist. Evidence exists of enhanced sweat gland innervation coupled with increased activity of sympathetic fibers passing through T2-T4. This is especially prominent in young people. Perhaps 25% of such individuals have a positive family history of hyperhidrosis. Axillary or palmar hyperhidrosis may be so severe as to interfere with normal activities and social interactions. Several therapeutic modalities may help (Table 77.2). A major limitation in therapy of hyperhidrosis is achieving sufficient muscarinic antagonism on sweat glands without incurring unacceptable levels of muscarinic blockade elsewhere—for example, in the heart. Local application of botulinum toxin to affected skin may also be quite effective but requires repeat injections at 3- to 12-month intervals (Saadia et al., 2001). In addition, blockade of the sympathetic ganglia using pharmacological injections, radiofrequency ablations, or endoscopic gangliotomy can be very effective (Atkinson and Fealey, 2003).
Table 77.1 Pathological Hyperhidrosis Differential Diagnosis and Some Causes of Localized Hyperhidrosis
| Condition | Pathophysiological Mechanism of Sweating |
|---|---|
| Essential hyperhidrosis |
Table 77.2 Treatment Measures for Primary Hyperhidrosis
| Topical Rx | 20% Aluminum chloride hexahydrate in anhydrous ethyl alcohol (Drysol). Apply half-strength to dry skin daily or every other day, mornings, and wash off. | Irritation of skin; less effective on palms and soles, which may require occlusive (plastic wrap) technique |
| Tanning Rx, iontophoresis | Glutaraldehyde (2%-10%) solution; apply 2-4 times/week as needed. | Stains skin brown; for soles of feet only |
| For palms/soles; 15-30 mA current, 20 min. at start. Drionic battery-run unit or galvanic generator needed; 3-6 treatments/week for total of 10-15 treatments initially; 1-2 treatments/week maintenance. | Shocks, tingling may occur Difficult to use in axilla Drionic unit not effective when batteries low |
|
| Anticholinergic | Glycopyrrolate (Robinul/Robinul Forte) at 1-2 mg PO tid as needed; for intermittent/adjunctive treatment. | Dry mouth, blurred vision Contraindicated: glaucoma, GI tract obstruction, GU tract obstruction |
| Clonidine | Useful for paroxysmal localized (e.g., hemibody) hyperhidrosis; 0.1-0.3 mg PO tid or as TTS patch (0.1-0.3 mg/day) weekly. | Somnolence, hypotension, constipation, nausea, rash, impotence, agitation |
| Excision | Second and third thoracic ganglionic sympathectomy (palmar hyperhidrosis), sweat glands (axillary liposuction); recent preference is for T2 sympathectomy to limit compensatory hyperhidrosis. | Homer syndrome, dry skin, transient dysesthetic pain Postoperative scar or infection Compensatory hyperhidrosis of trunk, pelvis, legs, and feet |
| Botox | 50-100 mU of botulinum toxin A into each axilla or body area treated; high doses (200 mU) prolong effect; can be repeated. | Injection discomfort, variable Duration of effect 3-12 months Expensive Mild grip weakness when palm is treated Contraindicated in pregnancy, NMJ disease |
GI, Gastrointestinal; GU, genitourinary; mU, mouse units; NMJ, neuromuscular junction; PO, orally; Rx, prescription; tid, three times daily.
Adapted from Fealey, R.D., 2004. Disorders of sweating, in: Robertson, D., Biaggioni, I., Burnstock, G., et al. (Eds.), Primer on the Autonomic Nervous System, second ed. Elsevier, New York, pp. 354-357.
Idiopathic hyperhidrosis must be distinguished from compensatory hyperhidrosis due to lower body hypohidrosis, common in dysautonomias (Klein et al., 2003), which may paradoxically be described by patients as excessive sweating in the upper body. Patients who lose their ability to perspire over most of their body may preserve it in the neck and facial area and perspire disproportionately in these areas, which captures attention more than the loss of sweating elsewhere. In this setting, the hyperhidrosis actually mandates an evaluation for the reason for the anhidrosis, usually a structural dysautonomia of some type. Loss of sweating does not require specific medical therapy, but rather practical advice such as staying well hydrated, avoiding alcohol, and avoiding hot conditions. One of the most effective home remedies is a “wet shirt”: A tee shirt soaked in warm water and wrung out thoroughly before putting on provides some artificial perspiration that lasts 30 to 90 minutes in a hot environment. The associated surface cooling is striking and greatly increases the functional capacity of patients who must be in a hot environment.
Assessment of Autonomic Function
More tests of autonomic function exist than for any other neurological system. Many of these tests are readily applied at the bedside, and though easy to perform, they may be difficult to interpret in an individual patient. Most physicians who routinely follow patients with autonomic disorders develop a small armamentarium of tests that answer questions relating to the focus of their specialty. The neurologist assessing autonomic function requires tests that localize the lesion within the neuraxis and provide information about the types of fibers involved. Therefore, tests of peripheral and central sudomotor function and tests that differentiate sympathetic from parasympathetic cardiovascular function are essential. The cardiologist requires tests of blood pressure and heart rate that evaluate the mechanism of any cardiovascular dysregulation. The endocrinologist measures circulating catecholamines, corticoids, sex hormones, and rennin. These tests are centered on appreciating the hormonal impact and consequences of autonomic dysfunction (Raj et al., 2005). The ophthalmologist tests pupillary function, and the pharmacologist uses drug tests that assess normal or hypersensitive autonomic function response (Robertson et al., 2004). Despite such dramatically divergent diagnostic approaches, it is remarkable how much consensus is often achieved in terms of the actual diagnosis and therapy of an individual patient. Indeed, an interdisciplinary approach that involves multiple specialists from different disciplines may provide both broader perspective on organ-system involvement and more accurate diagnosis and may be the reason more centers are taking this direction clinically.
Regardless of the clinical evaluation setting, a careful history is obviously the critical diagnostic resource. A brief listing of important items in questioning patients is shown in Box 77.1. More detailed discussions of some of these may be found elsewhere. Key autonomic features in the physical examination are shown in Box 77.2. In this section, attention is given to highly informative autonomic tests. Table 77.3 displays a listing of widely employed tests of baroreflex function.
Box 77.1 Key Features in the Autonomic History
Orthostatic Test
Orthostatic symptoms are usually the most debilitating aspect of autonomic dysfunction readily amenable to therapy. For this reason, the blood pressure and heart rate response to upright posture is the starting point of any autonomic laboratory evaluation. In healthy human subjects, the cardiovascular effect of upright posture has been well defined (Low, 1997). Upon active assumption of the upright posture by standing, the vigorous contraction of large muscles leads to a transitory muscle vasodilation and a minor fall in arterial pressure for which the reflexes do not immediately compensate. However, this short-lived depressor phase is not usually seen with passive (tilt-table) upright posture. Immediately after 70- or 90-degree head-up tilt, approximately 500 mL of blood move into the veins of the legs and approximately 250 mL into the buttocks and pelvic area. A rapid vagally mediated increase in heart rate occurs, followed by a sympathetically mediated further increase. As right ventricular stroke volume declines, a depletion of blood from the pulmonary reservoir occurs, and central blood volume falls. Stroke volume falls, and cardiac output decreases about 20%. With this decline in cardiac output, blood pressure is maintained by vasoconstriction that reduces splanchnic, renal, and skeletal muscle blood flow especially, but other circulations as well.
An important aspect of evaluating responses to orthostasis is the rapid reduction in total blood volume that occurs physiologically. It is not unusual for a 12% fall in plasma volume to occur within 10 minutes of assumption of the upright posture as fluid moves from the vascular compartment into the extravascular space (Jacob et al., 2005). This accounts for the delay in appearance of symptoms in patients with mild autonomic impairment for some minutes after the actual assumption of upright posture. Therefore, the long-stand (30 minutes) test, or Schellong test, is a much more severe orthostatic stress than the short-stand (5 minutes) test commonly employed.
Sweat Testing
Although hypohidrosis rarely dominates a patient’s dysautonomia, assessment of sudomotor function is often helpful in testing for autonomic impairment (Low, 2004). Widely used tests include the thermoregulatory sweat test (TST), quantitative sudomotor axon reflex test (QSART), and sympathetic skin response (SSR).
Thermoregulatory Sweat Test
The TST is a sensitive semiquantitative test of sweating (Fealey et al., 1989). After a color indicator (quinizarin powder or povidone-iodine) is applied to the skin, the environmental temperature is increased until an adequate core temperature rise is attained (usually a 2°C rise in core temperature or a core temperature of 38.5°C, whichever is less) and the presence of sweating causes a change in the indicator. Thermal stimulation using infrared radiant heat lamps to directly heat the skin are also employed to provide more effective sweat stimulus. Estimating the percent of anterior surface anhidrosis quantitates the results, and the sweat rates may be measured as well. This test has also been helpful in assessing the status of dysautonomias over time. Some characteristic patterns of anhidrosis include (1) the peripheral pattern of distal anhidrosis, seen in distal small-fiber neuropathy and length-dependent axonal neuropathy; (2) the central patterns of distal sparing or segmental involvement, generally seen in MSA or PD; and (3) a sudotomal pattern suggesting involvement at the root or ganglion level, seen in disorders involving nerve roots or specific ganglia, such as diabetes, Sjögren disease, and pure autonomic failure. The TST pattern is therefore helpful in distinguishing between postganglionic, preganglionic, and central lesions.
Quantitative Sudomotor Axon Reflex Test
The physiological basis of the QSART is elicitation of an axon reflex mediated by the postganglionic sympathetic sudomotor axon (Low, 2004). Acetylcholine (ACh) activates the axon terminal. The impulse travels antidromically, reaches a branch-point, then travels orthodromically to release ACh from the nerve terminal. ACh traverses the neuroglandular junction and binds to M3 muscarinic receptors on eccrine sweat glands to evoke the sweat response. The QSART specifically evaluates the functional status of postganglionic sympathetic axons.
Pharmacological Tests
Information about prevailing sympathetic and parasympathetic activation as well as denervation hypersensitivity can be achieved by use of muscarinic and adrenergic agonists and antagonists (Robertson et al., 2004). Tables 77.4 and 77.5 provide instructive examples of how biochemical and physiological tests may be combined to make novel diagnostic discoveries.
Functional Autonomic Disorders
Functional autonomic disorders are a heterogeneous group of disorders where autonomic nervous system involvement exists, but the role of autonomic dysregulation in the pathogenesis of symptoms is unclear. Included in this group are functional gastrointestinal disorders, interstitial cystitis, migraine, cyclic vomiting syndrome (CVS), fibromyalgia, and chronic fatigue syndrome. Many of these disorders tend to coexist in the same person. For example, about 40% of individuals with migraines have symptoms of IBS and chronic aches and pains that could represent fibromyalgia (Chelimsky et al., 2009). In our experience, children with functional gastrointestinal problems have severe fatigue, headaches, and sleep problems (Ojha et al., 2011). Whitehead et al. (2002) found a high association of functional gastrointestinal disorders with fibromyalgia, chronic fatigue syndrome, temporomandibular joint disorder, and chronic pelvic pain. Often the common denominator is POTS. By report, about 50% of persons with CVS and 40% with migraine report symptoms of orthostatic intolerance (Chelimsky et al., 2009). Sullivan et al. (2005) reported 24 pediatric subjects with functional gastrointestinal syndrome and either POTS, syncope, or both. Interestingly, the gastrointestinal symptoms improved or resolved with treatment aimed at the orthostasis (fludrocortisone, sertraline, and midodrine). Chronic fatigue has been described in association with POTS (Hoad et al., 2008).
Reflex Syncope
Syncope of any type is defined as sudden transitory loss of consciousness with spontaneous recovery that is associated with a loss of postural tone (Mathias et al., 2001). Syncope accounts for more than 1% of hospital admissions. The causes of syncope range from benign to life threatening. The common underlying mechanism of syncope is a transitory decrease in cerebral perfusion. Reflex syncope (fainting) is the most common type of syncope, especially in patients without evidence of structural heart disease (Benditt, 2006; Strickberger et al., 2006). Reflex syncope most commonly occurs while the patient is standing but also occurs while seated and occasionally even while lying during sleep (Jardine et al., 2006). Finally, it can occur with exercise (at initiation or at peak exercise) or with emotional/psychological triggers (e.g., phlebotomy).
Many patients can simply be reassured about the usual benign course of reflex syncope and instructed to avoid those situations that precipitate fainting. The use of support stockings or increased salt intake may help. In young non-hypertensive patients, the most frequently affected, we utilize 2 grams of salt in the morning and 2 grams in the early afternoon. Most salt is excreted by a normal kidney within 3 to 4 hours. Patients should be taught to recognize an impending faint and urged to lie down (or sit down if that is not possible) quickly. This will not be enough for some patients, and other treatment options such as physical countermaneuvers (Wieling et al., 2004) and tilt training (Ector et al., 1998) may be necessary. These are covered in detail in the treatment section of the chapter, as are pharmacological and other interventions.
Syncopal Migraine
A subgroup of patients with syncope respond poorly to the usual management of salt supplementation and tilt training. On further questioning, these patients may consistently experience a headache with migrainous features immediately prior to or after the syncopal spell. A recent review suggests that this entity, sometimes also termed basilar migraine, may be far more common than previously suspected, accounting for about one-third of patients with syncope referred to an autonomic specialist (presumably a more complex group of patients based on the referral bias) (Curfman et al., 2011). The importance of identifying this diagnosis is its prompt response to anti-migrainous medications such as verapamil and topiramate, in this author’s experience. Further identifying features include an increased duration of loss of consciousness (up to 15 minutes in this series), and longer time to full recovery, as might be expected in a migrainous mechanism.
Carotid Sinus Hypersensitivity
Carotid sinus hypersensitivity is defined as an asystole of 3 seconds, a fall in systolic pressure of 50 mm Hg, or both in response to carotid artery massage in a patient with otherwise unexplained dizziness or syncope (Fenton et al., 2000; Mathias et al., 2001). Estimates are that 35 to 100 patients per million per year present with this condition. Although the condition has been ensconced in the medical literature since the era of Soma Weiss (1898-1942), its definition remains controversial, in part because diagnosis is by manual massage of the carotid sinus, with its inherent variability. The test should be performed with the patient supine during continuous ECG and blood pressure monitoring and recording. Longitudinal massage should be performed for 5 seconds over the site of maximal pulsation of the right carotid sinus, located between the superior border of the thyroid cartilage and the angle of the mandible. If no response is elicited, the massage is sometimes repeated on the left side supine and ultimately also on the right and then left sides with upright tilt. Unfortunately, improved practical methods for diagnosis have not emerged. Clinically, a history may exist of syncopal symptoms associated with neck pressure, a tight collar, turning the head, shaving, or swallowing; syncope may also occur spontaneously. Hypotension, bradycardia, or both may dominate the clinical picture. The form in which bradycardia predominates may be improved by demand pacing. Some patients with carotid sinus syncope ultimately require surgical denervation. Occasionally, additional symptoms of headache, dizziness, vertigo, paresthesias, homonymous hemianopsia, and hemiplegia occur in the absence of measured blood pressure or heart rate change, but this may reflect another mechanism such as a migrainous or ischemic process; the older literature terms this phenomenon Weiss-Baker syndrome.
Postural Tachycardia Syndrome
POTS is defined as an increase of at least 30 bpm on standing, associated with symptoms of sympathetic activation (Freeman et al., 2002; Jacob et al., 2000; Low et al., 1997
[/not-level-membership-for-neurology-category]
