[level-membership-for-neurology-category]
Chapter 79 Disorders of Skeletal Muscle
Striated muscle is the tissue that converts chemical energy into mechanical energy. The component processes include (1) excitation and contraction occurring in the muscle membranes, (2) the contractile mechanism itself, (3) various structural supporting elements that allow the muscle to withstand the mechanical stresses, and (4) the energy system that supports the activity and integrity of the other three systems. The logical categorization of myopathies is according to the part of the system involved. Until recently, this was impossible because the molecular basis of muscle activity was unknown. Scientific advances since the 1980s made this classification possible. Abnormalities in the membrane ion channels (channelopathies) involved in muscle excitation cause various forms of myotonia and periodic paralysis (see Chapter 64). The complex of proteins that include dystrophin, the sarcoglycans, and α-laminin constitute a vital structural mechanism linking the contractile proteins with the extracellular supporting structures. Defects in these proteins are the basis of many forms of muscular dystrophy. Although knowledge remains incomplete, it seems reasonable to modify the classic description of the myopathies to incorporate the new information. For this reason, in the sections that follow, disease descriptions are under the heading of their known molecular defect where possible; the classic appellation appears parenthetically. Before describing the illnesses themselves, we first review the techniques used in the clinical evaluation of patients.
Muscle Histology
The technique of muscle biopsy is not difficult. Under local anesthesia, a small incision made over the muscle allows, with careful dissection, removal of a small strip of muscle. Needle biopsies are useful in some situations. Histochemical studies of frozen sections are essential for proper interpretation. A transverse section of normal muscle shows fibers that are roughly of equal size and average approximately 60 mm in transverse diameter (Fig. 79.1). The muscle fibers of infants and young children are proportionately smaller. Each fiber consists of hundreds of myofibrils separated by an intermyofibrillar network containing aqueous sarcoplasm, mitochondria, and the sarcoplasmic reticulum with the associated transverse tubular system. Surrounding each muscle fiber is a thin layer of connective tissue (the endomysium). Strands of connective tissue group fibers into a fascicle, separated from each other by the perimysium. Groups of fascicles are collected into muscle bellies surrounded by epimysium.
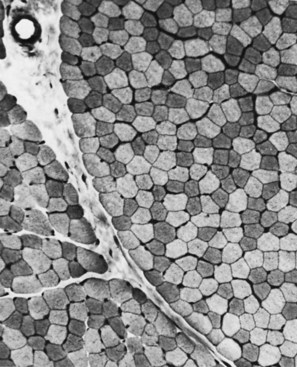
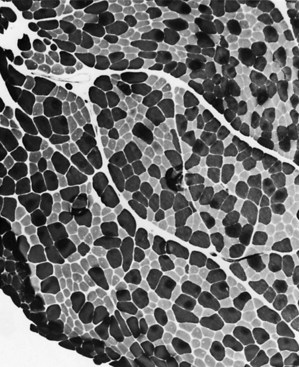
Situated at the periphery of the fibers are the sarcolemmal nuclei. The fibers are of different types. The simplest division is into type 1 and type 2 fibers, best demonstrated with the histochemical reaction for myosin adenosine triphosphatase (ATPase) (Fig. 79.2). The type 1 and type 2 fibers are roughly equivalent to slow and fast fibers or to oxidative and glycolytic fibers in human muscle. The best demonstration of the intermyofibrillar network pattern is with the histochemical reactions for oxidative enzymes, such as reduced nicotinamide adenine dinucleotide dehydrogenase. A regular network extends across the whole fiber. In addition to the routine stains with hematoxylin and eosin, modified Gomori-trichrome, myosin ATPase, and nicotinamide adenine dinucleotide dehydrogenase, the use of other special stains demonstrates fat (Sudan black or oil red 0), complex carbohydrates (periodic acid–Schiff), amyloid (Congo red), or specific enzymes (e.g., phosphorylase, succinic dehydrogenase, cytochrome oxidase). Immunocytochemical techniques demonstrate the location and integrity of structural proteins such as dystrophin. They also characterize cell types in biopsy samples with inflammatory changes.
Changes of Denervation
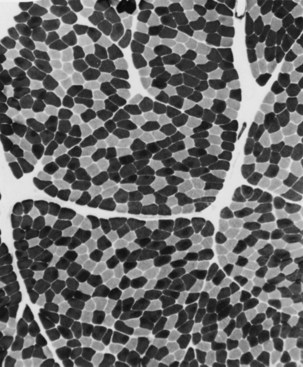
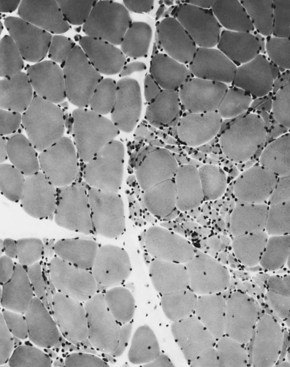
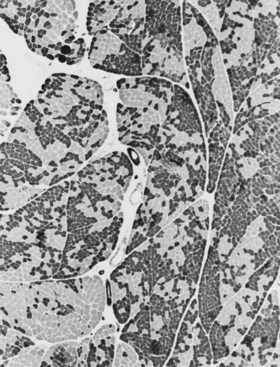
When muscle loses its nerve supply, muscle fibers atrophy, often resulting in fiber squeezing into the spaces between normal fibers and assuming an angulated appearance (Fig. 79.3). Scattered angulated fibers appear early in denervation. Sometimes, picturesque changes in the intermyofibrillar network occur, as in the target fiber, which characterizes denervation and reinnervation. This is a three-zone fiber on which the intermediate zone stains more darkly, and the central “bull’s eye” stains much lighter than normal tissue (Fig. 79.4). Often a neighboring nerve twig reinnervates a denervated fiber. This results in the same anterior horn cell supplying two or more contiguous fibers. If that nerve twig then undergoes degeneration, instead of only one small angulated fiber being produced, a small group of atrophic fibers develops. Group atrophy suggests denervation (Fig. 79.5). As the process continues, large groups of geographical atrophy occur in which entire fascicles are atrophic. In addition to the change in size, a redistribution of the fiber types occurs as well. Normally a random distribution of type 1 and 2 muscle fiber types exists, sometimes incorrectly called a checkerboard or mosaic pattern. The same process of denervation and reinnervation results in larger and larger groups of contiguous fibers supplied by the same nerve. Because all fibers supplied by the same nerve are of the same fiber type, groups of type 1 fibers next to groups of type 2 fibers replace the normal random pattern. This fiber type grouping is pathognomonic of reinnervation (Fig. 79.6). When long-standing denervation is present, the atrophic muscle fibers almost disappear, leaving small clumps of pyknotic nuclei in their place.
Myopathic Changes
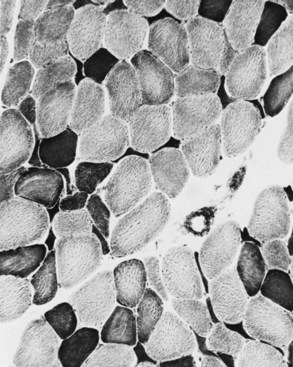
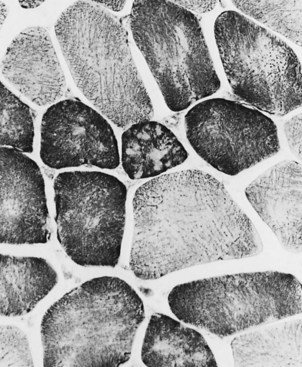
Changes in the intermyofibrillar network pattern are common in myopathic disorders. There is often a moth-eaten, whorled change to the intermyofibrillar network in LGMD and facioscapulohumeral dystrophy (FSHD) (Fig. 79.7); the intermyofibrillar network loses its orderly arrangement and swirls, resembling the current in an eddying stream. These changes may be seen in several diseases but tend to be much more common in the myopathies.
Other Changes
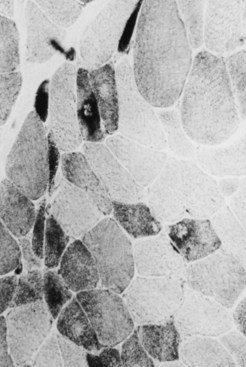
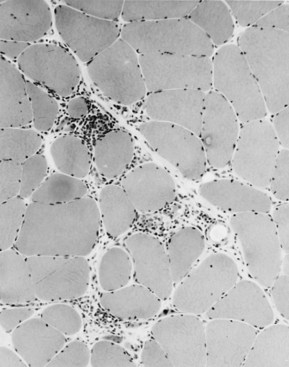
Selective changes in fiber types occur. Type 2 fiber atrophy is one of the most common abnormalities seen in muscle (Fig. 79.8). Type 2 atrophy, particularly if limited to type 2B fibers, is nonspecific and indicates muscle disuse. If a limb is casted and the muscle examined some weeks later, selective atrophy of type 2 fibers is noted. Any chronic systemic illness tends to produce type 2 atrophy. It occurs in rheumatoid arthritis, nonspecific collagen vascular diseases, cancer (hence the name cachectic atrophy), mental retardation in children, and pyramidal tract disease. Type 2B fiber atrophy can also result from chronic corticosteroid administration. Therefore, type 2 fiber atrophy should probably be regarded as a nonspecific result of anything less than robust good health.

Specific Disorders
Muscular Dystrophies
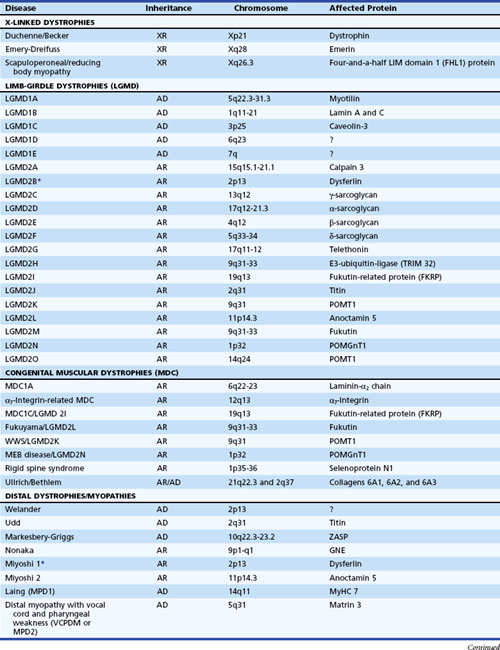
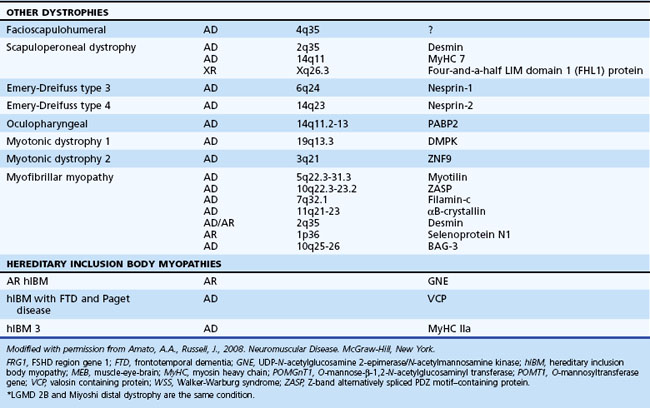
The muscular dystrophies are a group of hereditary muscle disorders that occur at all ages and with varying degrees of severity. The traditional classification is on clinical grounds. Increasing information about the molecular basis of these disorders provides both reassurance and puzzlement to clinicians (Table 79.1). Different dystrophies are due to distinct molecular abnormalities; however, patients with similar molecular defects may show a wide variability in phenotype not always easily explained.
Dystrophin Deficiency (Duchenne Muscular Dystrophy, Becker Muscular Dystrophy, and Atypical Forms)
An absence or deficiency of dystrophin is responsible for two disorders that cause progressive destruction of muscle. The responsible gene is located on the short arm of the X chromosome at locus Xp21. The gene is extremely large, comprising more than 2.5 million base pairs and 79 exons or coding regions. Approximately two-thirds of cases are associated with a detectable deletion or duplication of segments within the gene. The others are presumably due to point mutations too small to be detected using standard techniques. “Hot spots” for these gene deletions exist, notably between exons 43 and 52 and particularly 44 and 49 (Nobile et al., 1997). Whether the deletion is in frame or out of frame (see Chapter 40) determines whether dystrophin is absent from the muscle or present in a reduced altered form. This has clinical significance because the former is usually associated with the severe Duchenne variety of the disease (DMD), whereas the latter may cause the milder Becker variant (BMD). In BMD, the abnormal dystrophin preserves enough function to slow down the progress of the illness. Reading of the DNA code is triplet by triplet. Maintenance of this reading frame throughout the length of the gene is required for dystrophin production. If a deletion removes a multiple of three base pairs, the reading frame may be intact upstream and downstream and may make limited sense, as if the sentence “You cannot eat the cat” were changed to “You not eat the cat,” and some modified dystrophin may be formed. This is often the situation in the mild form of dystrophin deficiency. In the severe form, the reading frame is destroyed, as if a deletion resulted in the sentence “Yoc ann ote att hec at.” Exceptions to this rule exist, and frameshift deletions have been associated with the milder form of the disease, particularly at the 5′ end of the gene in exons 3 to 7.
Duchenne Muscular Dystrophy
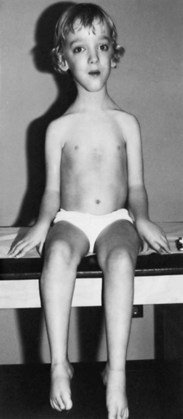
Even in the severe variety of DMD, affected children are normal at birth. During the second year when the boys begin walking, the clumsiness seen in all toddlers persists. Soon the children have to place one hand on the knee to assume an upright position when rising from the floor (Gower maneuver). Often at this stage, the calf muscles are rather firm and rubbery (pseudohypertrophy) (Fig. 79.9). Within 2 to 3 years, parents notice that the child runs properly and is never able to jump clear of the floor with both feet. In the absence of therapy, tightness across several joints in the legs is noted. The iliotibial bands and the heel cords are usually the first to become tight. This is particularly noticeable in boys who habitually walk on their toes.

Fig. 79.9 Duchenne muscular dystrophy. Calf and thigh hypertrophy in an ambulatory 8-year-old patient.
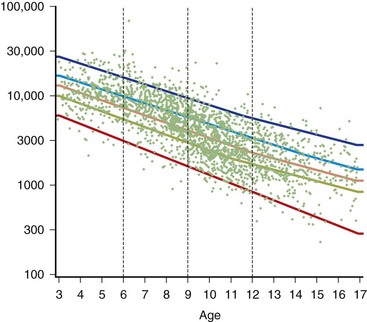
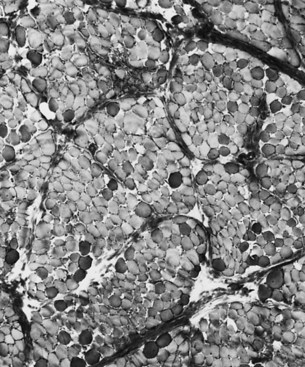
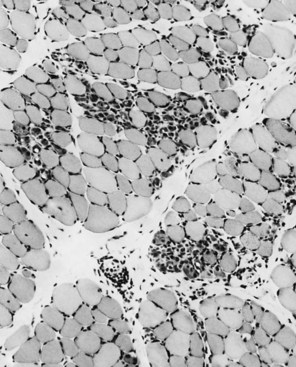
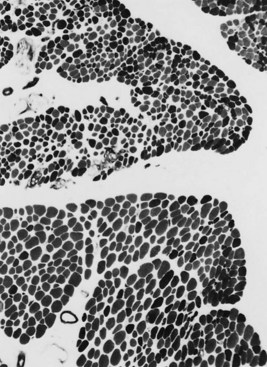
The serum concentration of creatine kinase (CK) is markedly elevated in this illness; levels greater than 10,000 mU/mL are common. Electromyography (EMG) shows myopathic changes (see Chapter 32B), and muscle biopsy demonstrates variation in the size of fibers, fibrosis, groups of basophilic fibers, and opaque or hypercontracted fibers (hyaline fibers) (Figs. 79.10 to 79.12).
Genetic Counseling
Genetic analysis of all potential carriers is advisable. In a family in which the disease is associated with a deletion, there is little problem in determining whether the woman is carrying the affected X chromosome, using techniques that are presently available. Genetics laboratories now have the ability to identify the presence of a mutant gene over the background contributed by the normal allele. This involves an analysis of the gene “dosage,” comparing two normal alleles that have a double dose against a deleted allele and a normal allele that have a single dose (Voskova-Goldman et al., 1997). The basis for current diagnosis of carrier status after identification of a deletion in a proband is by analysis of a gene dosage. Our diagnostic strategy uses fluorescence in situ hybridization (FISH) to detect female carriers with major deletions in the dystrophin gene. Unfortunately, situations exist in which a mutation is identified in a boy with “sporadic” DMD but not in the mother, and yet the mother is still a carrier. This occurrence is secondary to germline mosaicism in which the mutation in the mother lies only in a percentage of her oocytes. The estimated recurrence rate of DMD is as high as 14% even in such cases. Prenatal diagnosis using amniotic cells or chorionic villus biopsies can identify the affected fetus and, more importantly, those who are unaffected.
Other Limb-Girdle Dystrophies
The most important recent development in muscle disease relates to a group of disorders that were clearly dystrophic but defied proper classification. The traditional diagnosis of LGMDs cloaked the clinician’s uncertainty. In some patients, weakness was generally proximal, but other characteristics were disparate. Some cases were dominantly inherited, others recessively. Some had more hip than shoulder weakness, others the reverse. The illness could be mild in late life, others severe and early. There was general recognition that the rubric LGMD encompassed a group of different illnesses. Beginning with the discovery that a defect in one of the sarcoglycans caused a severe form of dystrophy occurring in North Africa, the delineation of several other entities characterized by defects in structural proteins or enzymes followed. These include the sarcoglycans, the α2 chain of laminin (merosin), calcium-activated protease, calpain-3, and others. The designation of autosomal dominant LGMD is type 1, and the designation of autosomal recessive LGMD is type 2. Subclassification with an alphabetical letter characterizes distinct genetic forms of LGMD1 and LGMD2 (see Table 79.1). The following sections outline the known protein abnormalities and then comment on the more amorphous forms of LGMD. The prevalence of these diseases as a group probably approaches 1 per 100,000 (van der Kooi et al., 1996). The autosomal recessive LGMDs are more common than the autosomal dominant LGMDs.
Autosomal Dominant Limb-Girdle Muscular Dystrophies
LGMD1A (Myotilin Deficiency)
LGMD1A is allelic to one subtype of myofibrillary myopathy and is caused by mutations in the myotilin gene located on chromosome 5q22.3-31.3 (Selcen and Engel, 2004). Myotilin is a sarcomeric protein that is present at the Z-disc. The protein is likely important in myofibrillogenesis and stabilization of the Z-disc and sarcomere.
LGMD1B (Lamin A/C Deficiency)
This myopathy is allelic with the disorder previously reported as autosomal dominant Emery-Dreifuss muscular dystrophy (EDMD) (Bonne et al., 2000; van der Kooi et al., 1996). Some patients manifest with a limb-girdle pattern of weakness, while others present with a humeral-peroneal weakness and early contractions. Cardiomyopathy with severe conduction defects and arrhythmias may also occur, with or without skeletal muscle involvement. Sudden death secondary to fatal arrhythmias is common, and pacemaker insertion is often necessary. Serum CK levels may be normal or elevated up to 25-fold. Muscle biopsies demonstrate dystrophic features with the rare occurrence of rimmed vacuoles.
LGMD1B is localized to mutations in the lamin A/C gene located on chromosome 1q11-21 (Bonne et al., 1999; van der Kooi et al., 1997). Alternative splicing of the lamin A/C messenger (m)RNA transcript produces lamins A and C. Lamin A/C is an intermediate-size filament located on the nucleoplasmic surface of the inner nuclear membrane, where it interacts with various lamin-associated proteins including emerin, the abnormal protein associated with X-linked EDMD. Lamin A/C may also bind to heterochromatin. Immunostaining of the nuclear membrane with anti-emerin antibodies is normal, helping distinguish this myopathy from X-linked EDMD. Electron microscopy reveals alterations in myonuclei, including the loss of peripheral heterochromatin, altered interchromatin texture, and fewer than normal nuclear pores (Sabatelli et al., 2001).
LGMD1C (Caveolin-3 Deficiency)
This rare myopathy usually presents in childhood with proximal leg weakness greater than arm weakness and exertional myalgias (Carbone et al., 2000; Minetti et al., 1998). Progression of weakness is variable. The clinical phenotype associated with caveolin-3 mutations is quite heterogeneous. Some patients manifest with mainly distal weakness and others with autosomal dominant rippling muscle disease (Betz et al., 2001; Vogerd et al., 2001). Serum CK levels are increased 3 to 25 times normal. In fact, some patients manifest with asymptomatic hyper-CK-emia.
LGMD2A (Calpain-3 Deficiency)
A careful series of studies conducted over a decade by Fardeau’s group documented the existence of a form of autosomal recessive LGMD in an inbred population on Reunion Island in the Indian Ocean (Fardeau et al., 1996). The disease, LGMD2A, was localized to chromosome 15 and was associated with a mutation in the gene for muscle-specific calcium-activated neutral protease (CANP-3 or calpain-3) (Spencer et al., 1997). Since this description, the disorder has shown a worldwide distribution and may account for as many as 20% to 26% of dystrophies with normal dystrophin and sarcoglycan (Fanin et al., 2001). It probably accounts for the majority of LGMD cases in patients of Spanish, Italian, and Brazilian ancestry. The underlying pathophysiology of the illness is uncertain. CANP-3 is not a structural protein but an enzyme. It has been suggested that the enzyme has a regulatory role in the modulation and control of transcription factors and thus of gene expression. CANP-3 also binds to titin and can cleave filamin-C; thus it may have a role in stabilizing the sarcomere.
The disease begins in childhood or in early adult life and is progressive. Most cases have been mild to moderately progressive, with loss of ambulation in adult life. Severe forms occur. Weakness occurs in the hips first and then in the shoulders. Facial strength is good, and the neck flexors and extensors are strong. Scapular winging occurs, different from that seen in FSHD, with the whole of the medial scapular border jutting backward. The posterior thigh muscles are more severely affected than the knee extensors, and the rectus abdominis muscles seem affected early. The serum concentration of CK is markedly elevated early in the course and then decreases to normal concentrations later in the illness. Muscle biopsies may demonstrate endomysial inflammatory cell infiltrate with prominent eosinophils that may lead to misdiagnosis as eosinophilic myositis (Brown and Amato 2006; Krahn et al., 2006).
LGMD2B (Dysferlin Deficiency)
Mutations in the gene encoding for dysferlin, located on chromosome 2p13, lead to clinically heterogeneous myopathy (Illarioshkin et al., 1996; Mahjneh et al., 1996). Some patients show a limb-girdle pattern of weakness (LGMD2B), while others present with weakness and atrophy of the calf muscles (Miyoshi myopathy). Furthermore, some patients have earlier involvement of the anterior tibial muscles. Dysferlinopathies account for only about 1% of LGMDs but about 60% of distal myopathies (Fanin et al., 2001). Looking at this another way, 80% of patients with dysferlinopathy manifest a distal myopathy, 8% have an LGMD pattern of weakness, and 6% have asymptomatic elevation of serum CK. Less common presentations include progressive foot drop (Illa et al., 2001; Seror et al., 2008; Vilchez et al., 2005) or axial muscle weakness with bent spine syndrome/camptocormia (Illa et al., 2001; Vilchez et al., 2005), and rigid spine syndrome (Nagashima et al., 2004).
Serum CK concentrations are markedly elevated, usually 35 to 200 times normal. Muscle biopsies demonstrate dystrophic features in severely affected muscles but nonspecific myopathic features in less affected muscles. Occasionally a striking endomysial or perivascular inflammatory process is appreciated, leading to an incorrect diagnosis of polymyositis. However, unlike polymyositis, the inflammatory cells usually do not invade non-necrotic muscle fibers. Amyloid deposition may also be evident in some cases (Spuler et al., 2008). A helpful immunohistological feature seen in some biopsies is the demonstration of membrane attack complex on the sarcolemma of non-necrotic muscle fibers. This feature is demonstrable in other dystrophies with secondary inflammatory changes (e.g., FSHD, dysferlinopathies) but not in primary inflammatory myopathies (i.e., polymyositis, dermatomyositis, inclusion body myositis) (Spuler and Engel, 1998). Immunostaining and immunoblot confirm the diagnosis. Dysferlin localizes to the sarcolemmal membrane but does not directly interact with dystrophin or the sarcoglycans. Dysferlin is thought important in membrane repair (Bansal et al., 2003; Cenacchi et al., 2005; Glover and Brown, 2007).
LGMD2C, 2D, 2E, and 2F (Sarcoglycan Deficiencies)
Large population studies are not yet available, but surveys of muscle biopsies suggest that the sarcoglycanopathies may account for more than 10% of patients with a limb-girdle pattern and positive dystrophin (Duggan et al., 1997). Of these, α-sarcoglycanopathies are most common, accounting for approximately 6% of cases, β-sarcoglycanopathies 3%, γ-sarcoglycanopathies 1%, and δ-sarcoglycanopathies less than 1%. However, sarcoglycan deficiencies may account for up to 50% of patients with muscular dystrophy in North Africa.
LGMD2G (Telethonin Deficiency)
Patients with this dystrophy may have either proximal or distal weakness (Moreira et al., 1997). Mean age of onset is approximately 12.5 years. Legs are affected more than arms, and significant weakness develops in the quadriceps and anterior tibial muscle groups. Serum CK levels are 3 to 17 times normal. Muscle pathology shows dystrophic features in addition to the frequent occurrence of rimmed vacuoles within muscle fibers.
LGMD2G links to mutations in the gene that encodes for telethonin on chromosome 17q11-12 (Moreira et al., 2000). Telethonin is one of the most abundant muscle proteins, where it localizes to the sarcomere. Telethonin may interact with the large sarcomeric proteins, titin and myosin. Abnormal telethonin may disrupt normal myofibrillogenesis.
LGMD2H
This LGMD occurs in several families of Manitoba Hutterite origin (Weiler et al., 1998) and is now known to be allelic with sarcotubular myopathy. The age of onset ranged from 8 to 27 years. Progression was slow, with most patients being ambulatory through the fourth decade of life. Serum CKs ranged from 250 to over 3000 IU/L, and EMGs revealed myopathic features. This myopathy is caused by mutation in the gene that encodes for E3-ubiquitine ligase (also known as TRIM 32) (Frosk et al., 2002). This ligase may be important for ubiquinating proteins targeted for destruction by the proteasomes (Kramerova et al., 2007). TRIM32 mutations may lead to dysregulation of myofibrillar protein turnover.
LGMD2I
This dystrophy was initially reported in a large consanguineous Tunisian family (Driss et al., 2000). Subsequently the dystrophy has shown a worldwide distribution and is the most common type of LGMD in patients of English ancestry. The clinical phenotype is variable (Brockington et al., 2001, 2002). The age of onset ranges from infancy (e.g., congenital) to the fourth decade of life. The clinical course can be similar to that of a congenital muscular dystrophy or a mild BMD. A severe cardiomyopathy can develop. Serum CKs are elevated 10 to 30 times normal in some affected younger patients but were normal in some older individuals.
LGMD2I linked to chromosome 19q13.3, where mutations in the gene that encodes for fukutin-related protein (FKRP) were identified (Brockington et al., 2002). Mutations in this gene were also seen in some patients with congenital muscular dystrophy with normal merosin (MDC1C). FKRP is a glycosyltransferase and its deficiency is associated with abnormal glycosylation of α-dystroglycan.
LGMD2J
This rare childhood-onset LGMD is caused by homozygous mutations in the titin gene located on chromosome 2q31 (Udd et al., 2005). Mutations in this gene usually cause an autosomal dominant adult onset tibial muscular dystrophy (Udd myopathy), which is discussed in the Distal Myopathy section.
LGMD2L
This disorder was initially described in 14 French Canadian patients from 8 different families (Jarry et al., 2007). Affected individuals aged 11 to 50 years presented with limb-girdle weakness associated with quadriceps atrophy and myalgias. Recently, two patients from a Finnish family manifested a Miyoshi myopathy–like phenotype (see Distal Myopathy) with involvement of the calves in the second decade of life (Bolduc et al., 2010). Serum CK concentrations can be normal or markedly increased. EMG studies show myopathic changes. Muscle biopsies demonstrate nonspecific dystrophic features with increased endomysial connective tissue associated with basal lamina duplication and collagen disorganization infiltration. The disorder is caused by mutations in a gene that maps to 11p13-p12. The candidate gene is ANO5, which encodes for anoctamin 5. This is a member of the anoctamin family of transmembrane proteins and may be a calcium-activated chloride channel.
LGMD2K, LGMD2M, LGMD2N, and LGMD2O
LGMD2M is caused by mutations in the gene that encodes for fukutin, which usually causes Fukuyama congenital muscular dystrophy (FCMD). However, mutations in the fukutin gene have also been associated with a milder myopathy (LGMD2M), particularly outside of Japan (Puckett et al., 2009; Vuillaumier-Barrot et al., 2009). Affected individuals can have normal intelligence and brain structure, have a mild limb-girdle weakness, present with only a cardiomyopathy, or have asymptomatic elevation of serum CK concentrations Some patients have remarkable steroid responsiveness (Godfrey et al., 2006).
LGMD2N is caused by mutations in POMGnT1, which encodes protein O-mannose-β-1,2-N-acetylglucosaminyl transferase. Mutations in this enzyme also cause muscle-eye-brain (MEB) disease. Mutations in POMGnT1 can be associated with milder allelic variants of muscular dystrophy and normal intelligence (LGMD2N) (Clement et al., 2008; Godfrey et al., 2007).
LGMD2O is caused by mutations in the gene encoding protein O-mannosyltransferase 2 (POMT2). Mutations in this gene can also cause Walker-Warburg syndrome and rarely is associated with a milder limb-girdle syndrome without brain involvement (Godfrey et al., 2007).
Myofibrillar Myopathy
The characteristic pathological finding in myofibrillar myopathy (MFM) is myofibrillary disruption on EM and excessive desmin accumulation in muscle fibers on immunostains (Amato et al., 1998; Dalakas et al., 2000; DeBleecker et al., 1996; Nakano et al., 1996, 1997). Desmin is not the only protein accumulating, however, so myofibrillar myopathy is the preferred term. This myopathy has been reported as desmin storage myopathy, desmin myopathy, familial desminopathy, spheroid body myopathy, cytoplasmic body myopathy, Mallory body myopathy, reducing body myopathy, familial cardiomyopathy with subsarcolemmal vermiform deposits, myopathy with intrasarcoplasmic accumulation of dense granulofilamentous material, Markesbery-Griggs myopathy, and hereditary inclusion body myopathy (hIBM) with early respiratory failure (Amato et al., 1998). The original classification of many such disorders was as forms of congenital myopathy, but it is now clear that MFM is a muscular dystrophy.
A spectrum of clinical phenotypes is associated with MFM (Amato et al., 1998). Most patients develop weakness between 25 and 45 years of age, but onset can occur from infancy to late adulthood. Either cardiac or skeletal muscles can be involved and dominate the clinical picture. Limb weakness can be predominantly distal and affect either the arms or the legs, but in others, proximal muscles are involved more than distal muscles. Facial and pharyngeal muscles are also affected. Some patients have a facioscapulohumeral or scapuloperoneal distribution of weakness.
Muscle histology demonstrates variability in fiber size, increased central nuclei, and occasionally type 1 fiber predominance (Amato et al., 1998; DeBleecker et al., 1996; Nakano et al., 1996, 1997). Muscle fibers with rimmed vacuoles may also be evident. Two major types of lesions are evident on light and EM: hyaline structures and nonhyaline lesions. The hyaline structures are cytoplasmic granular inclusions that are typically eosinophilic on hematoxylin and eosin, and dark blue-green or occasionally red on modified Gomori trichrome stains. They appear as cytoplasmic bodies, spheroid bodies, or Mallory bodies on EM. The nonhyaline lesions appear as dark green areas of amorphous material on Gomori trichrome stains. On EM, these nonhyaline lesions correspond to foci of myofibrillar destruction and consist of disrupted myofilaments, Z-disc-derived bodies, dappled dense structures of Z-disc origin, and streaming of the Z-disc. Immunohistochemistry reveals that both the hyaline and nonhyaline lesions contain desmin, myotilin, and numerous other proteins (Amato et al., 1998; DeBleecker et al., 1996; Nakano et al., 1996, 1997). Interestingly, abnormal muscle fibers also abnormally express several cyclin-dependent kinases in the cytoplasm, including CDC2, CDK2, CDK4, and CDK7 (Amato et al., 1998).
The pathogenesis of MFM is multifactorial (Selcen et al., 2004). Mutations in the myotilin gene on chromosome 5q22-31 cause many autosomal dominant cases. They are thus allelic to LGMD1A (Selcen and Engel, 2004). Mutations in the ZASP gene (chromosome 10 q22.3-10q23.2) cause other autosomal dominant cases (also known as Markesbery-Griggs distal myopathy) (Selcen and Engel, 2005). Less common mutations have been identified in the genes that encode for desmin, αB-crystallin, filamin C, selenoprotein N, Bag-3, and four-and-a-half LIM domain 1 (FHL-1). Of note, almost all of these proteins are important in the formation and stabilization of the Z-disc.
Congenital Muscular Dystrophies
Laminin-α2 (Merosin) Deficiency
In milder forms of the disease, delayed onset of symptoms and mild weakness occur (Tan et al., 1997). Complete absence of the laminin-α2 protein usually causes marked muscle destruction, but partial deficiencies cause less severe symptoms. This situation is akin to that seen in dystrophin deficiency. Diagnosis depends on the demonstration of altered merosin in muscle (Sewry et al., 1997). Commercial antibodies are available for this purpose, but not all antibodies demonstrate the abnormality, particularly if part of the laminin-α2 chain is present. As in dystrophin deficiency, it may be advantageous to use at least two different antibodies. Skin or muscle biopsy reveals merosin deficiency. The ultimate diagnosis rests on showing an abnormality in the merosin gene on chromosome 6 because laminin-α2 reduction also occurs as a secondary phenomenon in some of the other myopathies with membrane instability.
Other Forms of Classical MDC Type 1
There are patients with identical symptoms who do not have a mutation in the laminin α2 gene (Fig. 79.13). Frequently the muscle symptoms are milder, and children gain function as they grow older and may walk independently. These myopathies progress more slowly than merosin-negative MDCD. Despite a shortened lifespan, many survive to adulthood.
MDC type 1B links to mutations in the gene that encodes for the α7 subunit of α7β1D integrin (Hayashi et al., 1998). Integrin is a sarcolemmal protein that binds to merosin and is probably important for structural integrity of the muscle membrane. MDC1C with normal merosin occurs in patients with mutations in FKRP (Brockington et al., 2002). As noted in the LGMD section, mutations involving this protein have also been described in LGMD2I. Therefore, as with the dystrophinopathies, sarcoglycanopathies, and merosinopathies, a wide range of clinical phenotypes is possible. FKRP plays a role in glycosylation of other proteins. α-Dystroglycan, which binds merosin, is normally heavily glycosylated. The normal glycosylation of α-dystroglycan is impaired with FKRP mutations, and this most likely also results in loss of stability of the muscle membrane.
Fukuyama-Type Muscular Dystrophy
Inheritance of Fukuyama-type muscular dystrophy, like the other congenital dystrophies, is autosomal recessive. The myopathy links to mutations in the gene that encodes for fukutin, located on chromosome 9q31-33 (Kobayashi et al., 1998). Secondary loss of laminin α2 and α-dystroglycan occur. Fukutin is an enzyme associated with the Golgi complex that may participate in the glycosylation of other proteins. As already noted, α-dystroglycan is heavily glycosylated. Fukutin mutation likely results in abnormal glycosylation of α-dystroglycan and perhaps other important proteins, altering the normal three-dimensional protein structure and the integrity of the glycoprotein-dystrophin complex. This complex is expressed in the central nervous system (CNS) and probably accounts for the severe CNS manifestations associated with Fukuyama-type muscular dystrophy.
Affected children are usually normal at birth. Some are floppy, and joint contractures are present in 70% by the age of 3 months, with the hip, knee, and ankle commonly involved. These children are often severely mentally retarded, sometimes to the extent that speech is never developed, and either major motor or absence seizures are common (Fig. 79.14). Another curious finding is asymmetry of the skull, often noted on clinical examination. Muscle-strength testing is almost impossible, but weakness is diffuse and often disabling so that the child never learns to walk. Weakness occurs in the face and neck. Usually these children are completely dependent on their parents and never attain any degree of unsupervised activity. The muscle disease is moderately or slowly progressive, and survival into early adult life is common.

The Fukuyama variant does not resemble DMD, and the only real difficulty is in differentiating it from other forms of congenital dystrophy. Serum CK concentration is usually markedly elevated, and muscle biopsy shows dystrophic changes with variability in fiber size and fibrosis. Internal nuclei are common, and the biopsy specimen easily distinguishes this disorder from other congenital nonprogressive myopathies and acquired myopathies such as polymyositis. As in the other congenital dystrophies, muscle biopsy may show changes that indicate an additional neurogenic component. The changes in fiber size may not be random, and some fascicles contain much smaller fibers than others. This nonrandom change differs from denervation atrophy, in which there is a wide random variability in fiber size within individual fascicles (Fig. 79.15).
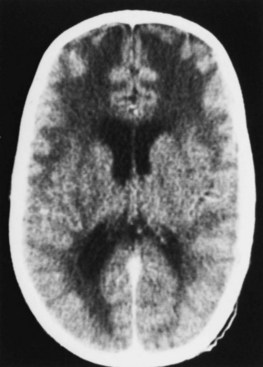
MRI and CT scans show a variety of abnormalities, but the most striking is the presence of lucencies, particularly in the frontal area (Fig. 79.16). These changes seldom extend to the genu of the corpus callosum and spare the medial subependymal regions along the trigones and occipital horns. As the children grow older, these lucencies disappear in sequence from the occipital to the frontal region in a fashion resembling the progression of normal myelination. Occasionally, marked pallor of the myelin in the centrum semiovale, together with mild gliosis or edema, is noted. Postmortem examination reveals numerous brain malformations including agyria, pachygyria, and microgyria. The cortex may have a cobblestone appearance, absence of gray-matter lamination, and other abnormal cytoarchitectural features. Heterotopias are present in the brainstem and basal meninges, as is micropolygyria of the cerebellum. Ventricular dilatation, enlarged sulci, and aqueductal stenosis are other associated features. In short, there are marked abnormalities in the architecture of the brain.
Walker-Warburg Syndrome and Muscle-Eye-Brain Disease
The combination of muscular dystrophy, lissencephaly, cerebellar malformations, and severe retinal and eye malformations characterize Walker-Warburg syndrome and the related muscle-eye-brain disease (Haltia et al., 1997). Both are associated with more severe eye abnormalities than Fukuyama-type muscular dystrophy. Walker-Warburg syndrome is the more catastrophic disease, with death often occurring within the first 2 years; eye changes include microphthalmia, colobomas, congenital cataracts and glaucoma, corneal opacities, retinal dysplasia and nonattachment, hypoplastic vitreous, and optic atrophy. Muscle-eye-brain disease is milder and characterized by high myopia and possibly a preretinal membrane or gliosis, but severe structural abnormalities of the eye are not present.
The CNS findings are also different. MRI may be a useful technique to separate the entities (van der Knaap et al., 1997). The changes in Walker-Warburg syndrome are more severe, with various combinations of hydrocephalus, aqueductal stenosis, cerebellar and pontine hypoplasia with a small posterior vermis, Dandy-Walker malformations, and an agyric or pachygyric cobblestone cortex. T1-weighted images show diffuse decreased white-matter signal; T2-weighted images show an increased signal compatible with a defect in myelination. In muscle-eye-brain disease, white-matter changes are focal and the cortical changes milder.
In Walker-Warburg syndrome, mutations have been identified in genes that encode for four proteins: POMT1 and POMT2, fukutin, and FKRP. However, these account for a minority of cases (Beltran-Valero de Bernabe et al., 2002; Cormand et al., 2001; Diesen et al., 2004; van Reeuwijk et al., 2005). Mutations in the POMT1 gene on chromosome 9q31-33 account for 20% of Walker-Warburg syndrome cases (van Reeuwijk et al., 2005). Some cases of muscle-eye-brain disease are caused by mutations in the gene that encodes for O-mannose β-1,2-N-acetylglucosaminyl transferase (POMGnT1) located on chromosome 1p3 (Yoshida et al., 2001). POMT1 forms a complex with POMT2 to catalyze the first step in O-mannosyl glycylation (Muntoni and Voit, 2004, 2005), and subsequently the transfer of N-acetylglucosamine to O-mannose of glycoproteins is catalyzed by POMGnT1.
Other Congenital Muscular Dystrophies
Ullrich Congenital Muscular Dystrophy
Ullrich congenital muscular dystrophy, also known as atonic-sclerotic dystrophy, is associated with neonatal weakness, multiple contractures, and distal hyperlaxity. Affected children often have marked protrusion of the calcanei in their feet. The clinical course is static or slowly progressive. Serum CKs are normal or only slightly elevated. Mutations in subunits of collagen type VI cause the disorder. Thus, it is allelic to the more benign Bethlem myopathy (Camacho-Vanegas et al., 2001).
Congenital Muscular Dystrophies with Rigid Spine Syndrome
This disorder presents in infancy with hypotonia, weakness, and delayed motor milestones. Reduced mobility of the spine is marked, and many children also develop scoliosis and contractures at the knees and elbows. Serum CK concentrations are normal or moderately elevated. Muscle biopsies may reveal type 1 predominance, increased internal nuclei, and other nonspecific myopathic features. In some kinships, the myopathy links to mutations in the selenoprotein N1 gene located on chromosome 1p3 (Moghadaszadeh et al., 2001). Other myopathic disorders (i.e., Emery-Dreifuss muscular dystrophy) can also be associated with rigid spine syndrome.
Other Regional Forms of Muscular Dystrophies
X-Linked Emery-Dreifuss Dystrophy (Emerin Deficiency)
Inheritance of the most common form of EDMD is as an X-linked recessive trait. The gene for EDMD (STA) is located at Xq28 and encodes for the nuclear membrane protein, emerin. Emerin localizes to the inner nuclear membrane, from which it projects into the nucleoplasm (Manilal et al., 1996). Emerin belongs to a family of lamina-associated structural proteins and is important in nuclear membrane organization and its attachment to heterochromatin.
Facioscapulohumeral Dystrophy
FSHD is inherited as an autosomal dominant trait. Its prevalence is around 1 to 2 per 100,000 population. Most cases (FSHD type 1) are caused by a deletion in a 3.3-kb repeating sequence termed D4Z4 at chromosome 4q35 (Tawil and Van Der Maarel, 2006). Digestion by the endonuclease, EcoRI, produces a fragment shorter than 34 kb in most families who have the illness, compared to more than 40 kb in normal individuals. Recent studies have demonstrated that this D4Z4 region is hypomethylated compared to normal (de Greef et al., 2009).
Some 5% of patients with FSHD do not have a deletion (so-called FSHD type 2), but similar to FSHD1, the D4Z4 region is hypomethylated, and this hypomethylation causes the myopathy. The DUX4 gene within the D4Z4 region appears to be responsible. The DUX4 protein may control expression of other genes. Hypomethylation of the gene may lead to overexpression of DUX4 and dysregulation of transcription of other genes (de Greef et al., 2009).
With FSHD1, the severity of the illness bears a relationship to the size of the deletion: the smallest fragments tend to be associated with severe illness. Another phenomenon exhibited by these families is anticipation of the illness, where cases occur with more severity and at a younger age with successive generations (Tawil and Van Der Maarel, 2006). This suggests the mutation is a dynamic one that may become increasingly severe with each generation, as is the case with myotonic dystrophy. That said, FSHD varies in severity even within the same family. Some patients may have mild facial weakness that can go unnoticed throughout life. Others may have total facial paralysis and severe weakness of most other muscles in the body, causing wheelchair confinement by the age of 9 or 10 years. In a prototypical case, the onset is during adolescence. Facial weakness expresses itself as difficulty blowing up balloons or drinking through a straw. The child sleeps with the sclera of the eyes showing through partially opened lids. Facial expression is relatively preserved, but the smile is often flattened and transverse as opposed to the upward curve of the usual smile. When the patient attempts to whistle, the lips move awkwardly and have a peculiar pucker (Fig. 79.17). The mouth also may have a pouting quality called bouche de tapir.

DNA studies reliably establish the diagnosis for FSHD1, but routine testing for FSHD2 by assessment of the methylation status of the D4Z4 region is not commercially available. The serum CK concentration usually is elevated several-fold above normal. Muscle biopsy may show general dystrophic features or tiny fibers scattered throughout the biopsy sample that suggest neuropathic change, or scattered inflammatory cellular foci associated with muscle fibers and in the interstitial tissue in patients with more severe disease (Fig. 79.18). EMG shows myopathic potentials (see Chapter 32B).
Treatment of FSHD is supportive. There is no known way of reversing the illness. If lack of scapular fixation renders the patient unable to raise the arms above the head, surgical stabilization of the scapula may be beneficial (Twyman et al., 1996). This is particularly true in the majority of patients with preserved deltoid function. It is more usual for ambulatory patients to undertake this procedure. Wheelchair-confined patients, because of the severity of the general weakness, do not find the problems in shoulder movements to be out of proportion to the rest of the muscular weakness. Such patients find a forearm orthosis or ball-bearing feeder device to be more useful than undertaking rather lengthy surgery.
A trial of prednisone in a small group of patients did not have dramatic results.
Scapuloperoneal Syndromes
Weakness of the muscles of the shoulder and the anterior compartment of the lower leg are the early symptoms of scapuloperoneal syndromes. Some forms of scapuloperoneal dystrophy may relate to FSHD, but most cases show no linkage to the FSH site on 4q35 (Tawil and Van Der Maarel, 2006). Mutations in the desmin gene on chromosome 2q35 and FHL1 on Xq26.3 have been demonstrated to cause some cases of scapuloperoneal dystrophy; these are also categorized as myofibrillar myopathies. In FSHD, a discrepancy exists between the strength of the ankle dorsiflexors, which are weak, and the plantar flexors, which are strong. The same is true of the scapuloperoneal syndrome, but facial muscles are spared. Often the patient presents with a foot drop, and examination reveals the shoulder weakness. Biopsy, EMG, and other laboratory tests reveal nonspecific myopathic features. Because other hereditary scapuloperoneal syndromes due to neuropathy and anterior horn cell disease exist, it is important to confirm the diagnosis with appropriate tests in any patient with a scapuloperoneal syndrome. Ankle-foot orthoses are the only useful treatment and may improve function by correcting foot drop.
Oculopharyngeal Muscular Dystrophy
Oculopharyngeal muscular dystrophy (OPMD) is another illness with an uneven geographical distribution, although now recognized worldwide. It is an inherited autosomal dominant disorder with almost complete penetrance. Foci of the illness occur in Quebec, Germany, Uruguay (Montevideo), and the Spanish-American populations of Colorado, New Mexico, and Arizona. Isolated families appear throughout the rest of the world. The disease is caused by mutations (expansion of GCG repeats) in the gene that encodes for polyadenylate-binding protein nuclear 1 (PABPN1, formerly PABP2) located on chromosome 14q11.2-13 (Brais et al., 1995; Stajich et al., 1996). This nuclear protein is involved in mRNA polyadenylation, but the mechanisms by which mutations cause OPMD are not yet clear.
OPMD usually begins in the fifth or sixth decade of life, but onset can be in the fourth decade. Patients present with eye-muscle weakness and mild ptosis. Initially the ptosis may be quite asymmetrical, but as the muscles weaken, both lids eventually become severely ptotic, and eye movements diminish in all directions (Fig. 79.19). Considerable variation in the severity of the extraocular palsies exists, but ptosis is constant. Concomitant with or shortly after the development of ocular symptoms, patients notice difficulty swallowing. Saliva pools in the pharynx, and at the extreme stages of the illness, dysphagia may be complete. Facial weakness occurs in a number of patients, and hip and shoulder weakness is common in the late stages. Death may occur from emaciation and starvation. The terminal event is often pneumonia initiated by aspiration of secretions. Although the symptoms associated with this disease may be severe, patients’ lifespans may be unaffected, making management of their nutritional status all the more important.
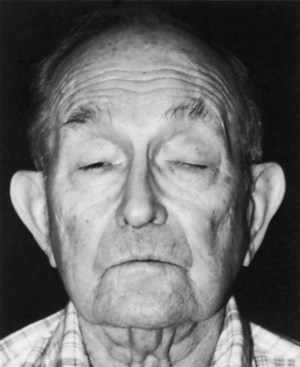
Fig. 79.19 Oculopharyngeal dystrophy. Facial appearance of patient who has ptosis and no eye movements.
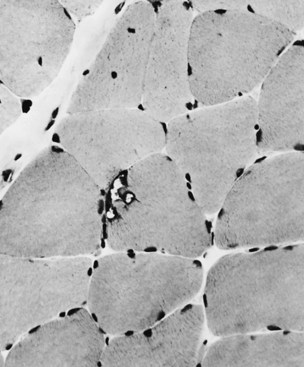
Muscle biopsy can usually be avoided, since genetic testing reveals the diagnosis. However, biopsy of weak muscles may show dystrophic findings, random variation in fiber size, necrotic fibers, some fibrosis, and occasional internal nuclei. In addition, fibers may contain autophagic vacuoles (rimmed vacuoles) (Fig. 79.20), a feature common to this illness as well as to inclusion body myositis and various hereditary distal myopathies/dystrophies.
Distal Muscular Dystrophies/Distal Myopathies
Several muscle diseases show a predominantly distal pattern of weakness (Udd, B., 2009). Initial diagnosis of the distal myopathies is often a hereditary or acquired neuropathy or motor neuron disease, because distal dysfunction is more characteristic of neuropathic disorders. Mild elevations in serum CK occur in neuropathic disorders, but CK concentrations over 500 IU/L should raise suspicion of a myopathic process. Serum CK concentrations can be normal in some distal myopathies, therefore CK alone does not exclude a myopathy. EMG distinguishes a distal myopathy from a neuropathic disorder. Furthermore, myasthenia gravis can rarely present with predominantly distal weakness, so repetitive nerve stimulation can also be helpful. Other myopathies associated with distal weakness include myotonic dystrophy type 1, inclusion body myositis, and certain forms of congenital myopathy (e.g., myofibrillar myopathy). Age of onset, inheritance pattern, histopathology, and pattern of weakness are most useful in distinguishing the distal myopathies from one another.
Miyoshi Myopathy
Inherited as an autosomal recessive disease, Miyoshi myopathy begins early, often in adolescence (Bejaoui et al., 1995). Although originally described in Japanese individuals, this common distal myopathy occurs worldwide. The weakness is characteristically in the foot plantar flexors, with severe gastrocnemius atrophy. This causes a thin, tapering leg. Patients are unable to stand on their toes, and they walk up stairs in a clumsy, jerky fashion. The illness is progressive, although it remains confined to the legs. Ultimately, hip weakness develops, and ambulation may become difficult in midlife. The serum CK concentration is extremely elevated and reaches levels of several thousand international units even before the patient becomes symptomatic. Mutations in the gene that encodes the sarcolemmal protein, dysferlin, located on chromosome 2p12-14, are responsible for Miyoshi myopathy. The same gene is responsible for LGMD2B (discussed earlier). Families exist in which some members exhibit a distal myopathy and others the more traditional proximal variety. The muscle biopsy shows dystrophic changes but no autophagic vacuoles.
Udd Myopathy
Serum CK concentration is normal or only slightly elevated. Muscle biopsy specimens demonstrate myopathic features along with rimmed vacuoles. The genetic basis is mutation in the gene that encodes for titin on chromosome 2q31-33 (Hackman et al., 2002). Titin is a giant sarcomeric protein that serves as a ligand for calpain-3.
Markesbery-Griggs Myopathy
Because some of the clinical and laboratory features are similar to Udd distal myopathy, they were thought to be allelic disorders. However, the identification of mutations in the gene encoding ZASP separates it from Udd myopathy with titin mutations (Griggs et al., 2007).
Serum CK concentration is normal or usually only slightly increased. The ECG may demonstrate conduction defects or arrhythmia. An echocardiogram may reveal a dilated or hypertrophic cardiomyopathy. EMG demonstrates markedly increased insertional and spontaneous activity with fibrillation potentials, positive sharp waves, and myotonic discharges. Motor units are myopathic in morphology and recruit early. Muscle biopsies demonstrate fibers with rimmed vacuoles and other features seen in the myofibrillar myopathies (Selcen and Carpén, 2008). Therefore, Markesbery-Griggs myopathy can be classified as one of the myofibrillar myopathies, discussed in greater detail later.
Nonaka Myopathy/Autosomal Recessive Hereditary Inclusion Body Myopathy
Nonaka and colleagues initially described this early adult-onset distal myopathy in Japan. Other groups reported similar patients with so-called autosomal recessive hereditary inclusion body myopathy (hIBM) (Udd, B., 2009). The clinical phenotype is similar to that of Markesbery-Griggs and Udd myopathies, with weakness initially involving the anterior tibial muscles in the legs and extensor forearm muscles. However, inheritance is in an autosomal recessive fashion and onset occurs at less that 30 years of age. The muscle biopsy results are also quite similar to those of Markesbery-Griggs, Udd, and Welander myopathies in that rimmed vacuoles are observed. Further EM reveals 15- to 18-nm tubular filaments typical of inclusion body myositis. However, unlike inclusion body myositis, no significant inflammatory process occurs in this hIBM.
Nonaka myopathy/autosomal recessive inclusion body myopathy is caused by mutations in the gene encoding for UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase, or GNE, on chromosome 9p1-q1 (Eisenberg et al., 2001). The mechanisms by which mutations in this gene lead to the myopathy are unknown but may relate to a secondary reduction in sialic acid production.
Laing Distal Myopathy
This autosomal dominant distal myopathy is characterized by weakness of the anterior tibial muscle groups and the neck flexors (Laing et al., 1995). Onset is in childhood or early adult life. Serum CK concentrations are normal or only slightly elevated, and EMG is myopathic. Cardiomyopathy is sometimes the initial symptom. Unlike Markesbery-Griggs myopathy, Udd myopathy, and Nonaka myopathy/hIBM, rimmed vacuoles are not a feature of the muscle specimen in Laing distal myopathy. Mutations in the slow/beta cardiac myosin heavy chain 1 (MyHC1) gene or MYH7 located on chromosome 14q11 are causative (Lamont et al., 2006). MyHC is the major myosin isoform expressed in type 1 muscle fibers. Of note, MYH7 mutations have also been identified in hyaline body myopathy (discussed later in Congenital Myopathies) (Tajsharghi et al., 2003).
Myotonic Dystrophies
Myotonic Dystrophy Type 1
The mutation is not in the coding region of the gene, and other experiments with knockout mice or mice that overexpress the protein show no marked abnormality in the muscle or other systems. Studies have demonstrated that the transcribed mRNA is directly toxic, in part by sequestering RNA binding proteins such as muscleblind protein, thereby leading to abnormal splicing of various mRNA transcripts, including those of muscle chloride ion channel (Mankodi et al., 2002; Mulders et al., 2009; Osborne et al., 2009; Wheeler and Thornton, 2007).
The more typical picture is of an illness beginning in early teenage life, starting with noticeable weakness of the hands and often foot drop. DM1 is one of the rare forms of dystrophy that seems to affect the distal muscles more severely. A predilection for neck muscle involvement exists, and the sternocleidomastoid muscles are often atrophic and poorly defined. A rather long face with a mournful expression is accentuated by hollowing of the temples associated with masseter and temporalis atrophy. In the fully developed disease, the eyes are hooded and the mouth slack and often tented (Fig. 79.21). The muscular weakness is not limited to the distal muscles; shoulder, hip, and leg weakness may be quite prominent.
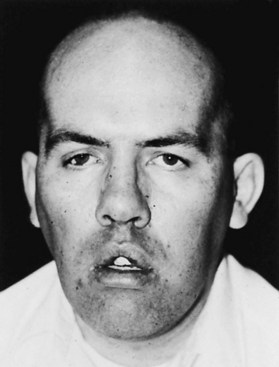
Cardiac disease is a common complication of DM1. Understanding of the heart component has increased by recent advances in both molecular techniques and cardiological investigations. Conduction disturbances and tachyarrhythmias occur commonly in DM1. The cardiomyopathy correlates in severity with the neuromuscular disease and the extent of the molecular defect in some but not all studies; the rate of progression differs widely among individuals. The cause of sudden death may be ventricular arrhythmias or complete heart block, and this can occur at an early stage of disease. Some studies show a familial tendency toward cardiac complications. The histopathology is of fibrosis (primarily in the conducting system and sinoatrial node), myocyte hypertrophy, and fatty infiltration (Phillips and Harper, 1997). EM shows prominent I-bands and myofibrillar degeneration. Suggestions for clinical management include a careful cardiac history and a 12-lead ECG at least every year, with a low threshold for use of 24-hour Holter monitoring. General anesthetic carries some risks, and the occurrence of complications, usually respiratory, was almost 10% in one series (Mathieu et al., 1997).
Excessive daytime somnolence is a common problem in DM1 (Damian et al., 2001; Giubilei et al., 1999). The uncontrollable urge to sleep may be mistaken for narcolepsy. It is accompanied by a disturbance of the nighttime sleep pattern. Patients have an abnormal central ventilatory response, without the usual hyperpnea produced by an increasing carbon dioxide concentration. This is associated with an abnormal sensitivity to barbiturates, morphine, and other drugs that depress the ventilatory drive. Anesthesiologists must be aware of the possibility of complications with these drugs. Several other organs are commonly involved in the disease. Cataracts are almost universal. Detection may only be by slit-lamp examination. Commonly, multihued specks appear in the anterior and posterior subcapsular zones. Endocrine abnormalities include disturbances of the thyroid, pancreas, hypothalamus, and gonads. Testicular atrophy, with disappearance of the seminiferous tubules, leads to male infertility. In the female, habitual abortion and menstrual irregularities are common. Although diabetes mellitus is no more common in the myotonic population than in the general population, a glucose tolerance test is often associated with abnormally high glucose levels, particularly late in the test. An associated overproduction of insulin seems to be due to abnormal resistance of the insulin receptor. Smooth muscle involvement accounts for several problems. Cholecystitis and symptoms referable to gallbladder function are frequent. Mild dysphagia and decreased peristalsis in the hypopharynx and proximal esophagus are present. Patients often complain of constipation and urinary tract symptoms.
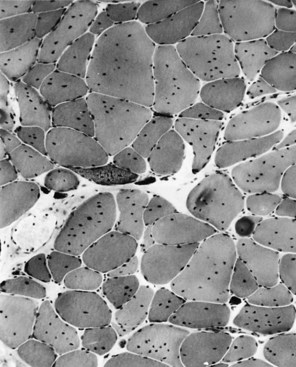
DNA analysis is the definitive diagnostic test for myotonic dystrophy. Patients with clinical evidence of disease and individuals at risk require testing. Prenatal diagnosis is reliable and uses chorionic villus biopsies or cultured amniotic cells. EMG is the most helpful laboratory study. In addition to the presence of myopathic features, the characteristic myotonic discharges are seen (see Chapter 32B). On insertion of the needle, bursts of repetitive potentials are noted. These potentials wax and wane in both amplitude and frequency; when played over a loudspeaker, they resemble the sound of a diving propeller airplane and are called dive bomber or motorcycle potentials. Muscle biopsy in the fully developed illness is markedly abnormal (Figs. 79.22 and 79.23), demonstrating random variability in the size of fibers and fibrosis. In addition, multiple nuclei pepper the interior of the fibers. Ring fibers are numerous, in which small bundles of myofibrils are oriented at 90 degrees to the majority, rather like a thread wrapped around a stick. Other laboratory studies are less helpful. Serum muscle enzymes are often abnormal. Some patients demonstrate low levels of immunoglobulin G. Abnormalities seen on head MRI include cerebral atrophy, increased white-matter signals on T2-weighted images, and thickening of the cranial vault (Miaux et al., 1997). These abnormalities have little significance.
Supportive treatment of DM1 includes the use of ankle-foot orthoses to treat foot drop. Wrist splints are less useful. In theory, they should give added function to the hand, but most myotonic patients prefer not to use them. Breathing exercises and postural drainage can be helpful in severe myotonia to ward off frequent respiratory infections. Drugs to treat myotonia include quinine, phenytoin, procainamide, mexiletine, and acetazolamide. Their lack of efficacy in clinical situations is probably because myotonic patients are bothered more by weakness than myotonia. Modafinil, 200 to 400 mg/day, improves hypersomnolence in DM1 patients (Damian et al., 2001).
Congenital Myotonic Dystrophy
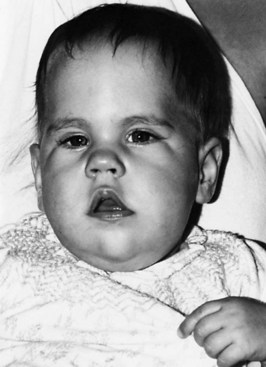
Congenital myotonic dystrophy occurs in infants of affected mothers. Cardinal features are hypotonia, facial paralysis, failure to thrive, and feeding difficulties. Affected infants are prone to frequent respiratory infections, which may develop into pneumonia. The upper lip forms an inverted V or “shark mouth” (Fig. 79.24). Clubfeet are common, and the children are severely mentally retarded. The cause of the neonatal illness is a marked increase in the trinucleotide repeat region that seems to occur with maternal transmission, particularly if the mother has a sizable expansion herself.
Myotonic Dystrophy Type 2 or Proximal Myotonic Myopathy
Whereas DM1 has a predilection for distal muscles, patients with proximal myotonic myopathy (PROMM) experience proximal stiffness, pain, and weakness (Moxley, 1996; Udd et al., 1997). Like DM1, PROMM is an autosomal dominant trait, and cataracts are characteristic. Gonadal atrophy and cardiac abnormalities occur, but less frequently than in DM1.
The genetic defect has been localized to mutations in the gene that encodes for zinc finger 9 (ZNF9) on chromosome 3q21 (Liquori et al., 2001). The mutations are expanded CCTG repeats in intron 1. As with DM1, this expanded repeat leads to expression of a toxic pre-mRNA, which as in DM1, sequesters RNA binding proteins, leading to aberrant splicing of other mRNA species including those of ion channels (Wheeler and Thornton, 2007; Mulders et al., 2009; Osborne et al., 2009).
Channelopathies
A group of illnesses that range from myotonic syndromes to the periodic paralyses results from abnormalities in ion channels (Cannon, 2010; Matthews et al., 2010). The molecular basis for these illnesses reorients classification. The ion channels are fundamentally important in controlling the passage of ions across the cell membrane and in the shift of ions from one cell compartment to another. These proteins are associated with the cell membrane and are responsible for such phenomena as the muscle action potential. Electrical potential across the cell membrane (voltage-gated) or by ligands such as glutamate influence the proteins. Segments of these proteins have amino acid sequences that are remarkably similar across a wide range of species (conserved segments). It is logical that any change in the conserved segment of a protein might produce trouble for the organism. Some of these mutations are presumably lethal, affecting as they do such an important functional component of the cell. Other mutations may produce intermittent symptoms (e.g., the periodic paralyses).
Ion Channels
The calcium channel in muscle is made of five subunits: α1, which is the most important and forms the ion pore across the membrane; α2, β, γ, and δ. The type of α1 subunit determines the sensitivity of the calcium channel. In muscle, this is sensitive to dihydropyridines such as nifedipine. These are L-type channels because of their long-lasting effect. The subunit is formed from four similar transmembrane regions (domains D1 to D4), each made of six membrane-spanning proteins, S1 to S6, all linked in series by “loops” that extend into the cytoplasm or extracellularly; S4 is highly charged by virtue of the richness of positively charged amino acids. It may confer voltage sensitivity to the channel. Neurological conditions linked to abnormalities in the calcium channel include hypokalemic periodic paralysis (Cannon, 2010; Matthews et al., 2010).
The sodium channel is very similar in its makeup. The α subunit, a 260-kD protein, confers the sodium channel activity with four domains made of six membrane-spanning proteins connected in similar fashion. Again, the S4 segment is highly charged, which might make it suitable for responding to voltage changes. Mutations in a gene for the sodium channel are the most common cause of hyperkalemic periodic paralysis and paramyotonia congenita. Some families with hypokalemic periodic paralysis (hypoKPP type 2) have mutations in the sodium channel (Bulman et al., 1999).
Calcium Channel Abnormalities (Familial Hypokalemic Periodic Paralysis Type 1)
The inheritance of familial hypokalemic periodic paralysis is autosomal dominant. Most affected patients (hypoKPP1) have mutations in the CACL1A3 gene, which encodes for the α1 subunit of the dihydropyridine-sensitive calcium channel located on chromosome 1q31-32. In hypokPP2, mutations are found in the SCNA4A sodium channel gene, which is more commonly associated with hyperKPP, paramyotonia congenita, and the potassium-aggravated myotonias, which are discussed in a separate section. Most mutations in hypoKPP1 and hypoKPP2 involve the voltage-sensitive S4 segment of domains 2 and 4. Most of these mutations result in a substitution of histidine for arginine. The mechanisms for the episodic weakness in hypoKPP are not completely understood, but these substitutions may result in aberrant depolarization via the proton gating pore during attacks (Cannon, 2010).
Sodium Channel Abnormalities
Potassium-Sensitive Periodic Paralysis
The potassium-sensitive periodic paralyses and myotonias are associated with mutations in the SCNA4A gene that encodes the α subunit of the sodium channel; the gene is located on chromosome 17q. Even in early descriptions of these disorders, the association of decreased electrical activity, paralysis, and signs of hyperactivity (paramyotonia) were recognized. The proper activity of the sodium channel depends on a complicated series of activation and deactivation processes that open the pore to allow the passage of sodium but also protect the cell against inadvertent excess sodium flux. The channel may exist in a number of different states: closed, open, and inactivated. Physiological studies have suggested that the domain IV S3 segment has a dominant role in the recovery of inactivated channels, whereas the S4 segment is concerned with deactivation and inactivation of the open channel (Ji et al., 1996). Among the several known mutations of the sodium channel, some impair fast inactivation of the sodium channel or shift the impulse to hyperpolarization. Evidence exists that two common mutations in hyperkalemic periodic paralysis in domain II (S5) and domain IV (S6) cause defective slow inactivation, accentuating the weakness (Hayward et al., 1997). Mutations within the domain III-IV linker, which cause myotonia with or without weakness, do not impair slow inactivation.
Hyperkalemic Periodic Paralysis
Inheritance of potassium-sensitive periodic paralysis (hyperKPP) is autosomal dominant, with strong penetrance and involvement of both sexes (Matthews et al., 2010). Onset of symptoms is during infancy or early childhood. The infant’s cry may become suddenly altered or unusual, or the child may be found lying quietly in the crib. Some parents notice an unusual stare as these babies develop, particularly on exposure to cold. The first attack commonly occurs in the first few weeks of school because of the enforced sitting. By adolescence, the attacks are characteristic. Often, rest after exercise provokes an attack, and the weakness develops quite rapidly, often within a matter of minutes. The weakness is milder than in the hypokalemic variety, and the attacks last for a shorter period. Patients may be able to walk off the symptoms if they undertake exercise early in the attack. Many prefer not to do so because an attack itself is mild and followed by a period of relative freedom from symptoms. In addition to rest after exercise, other provocative factors include exposure to cold, anesthesia, and sleep. Patients often avoid fruit juices with high potassium content, having noticed their deleterious effect. Many patients have two kinds of attacks, light and heavy. During a light attack, there is a feeling of fatigue and mild weakness that usually disappears in less than an hour. A heavy attack, however, may be associated with more severe paralysis, even to the point where the patient is unable to arise from the chair or bed. The frequency varies from two or three mild attacks a day to episodes months apart. Residual weakness may occur in middle age and beyond.
Paramyotonia Congenita
Mutations in the SCNA4A gene also lead to paramyotonia congenita. This condition may or may not be associated with episodic weakness (Matthews et al., 2010). Unlike the myotonia in myotonic dystrophy, in paramyotonia, repeated exercise accentuates myotonia, a feature most easily appreciated in the eyelids. Patients may complain of this as aching or stiffness. A clinical test is to have the patient forcibly close his or her eyes in a repetitive manner. After each repetition, the difficulty with relaxation may be accentuated until eventually the patient cannot open the eyes at all. Exposure to cold not only worsens the myotonia but may provoke muscle weakness, symptoms patients may notice when swallowing ice cream or going out into winter weather to shovel snow. A useful test for paramyotonia is to soak a small towel in ice water and lay it over the patient’s eyes for 2 minutes. Eyelid myotonia is demonstrable by having the patient sustain an upward gaze for a few seconds and then look down. The eyelids remain up, baring the sclera above the iris. When muscle is sufficiently chilled, the paramyotonia disappears, and the muscle is flaccid and paralyzed. The weakness may far outlast the exposure to cold, and it is common for the muscle not to regain its full use for hours after returning to room temperature. Strong voluntary contraction also may be associated with a long-lasting decrease in strength, which is not clearly due to an increase in myotonia. Immersing the forearm in ice water may also produce obvious weakness, which may have been lacking on the initial examination.
The diagnosis of potassium-sensitive conditions relies on demonstration of the genetic defects. However, the diagnosis should be suspected when high serum potassium levels coincide with bouts of weakness. In patients with paramyotonia, EMG of the resting muscles at room temperature shows myotonic discharges that are present on percussion or with movement of the needle. The most remarkable findings are demonstration of decrementing CMAP amplitudes on the repeated short exercise test, which is worse when performed with the extremity cooled (Fournier et al., 2004, 2006). Additionally, there are often after-discharges evident following brief activity on CMAP studies.
Hypokalemic Periodic Paralysis Type 2
Although the majority of patients with hypokalemic periodic paralysis (type 1) have mutations in the calcium channel, approximately 9% of cases are due to mutation in the SCNA4A muscle sodium channel gene, as previously discussed (hypoKPP2) (Bulman et al., 1999; Cannon, 2010; Sternberg et al., 2001). The clinical and laboratory features are similar to the more common type of hypokalemic periodic paralysis, but there are some differences that may help distinguish the subtypes. Patients with hypoKPP2 may be more likely to have severe myalgias following the paralytic attacks, tubular aggregates on muscle biopsy rather than vacuoles, and worsening of symptoms on acetazolamide.
Potassium Channelopathy
Andersen-Tawil Syndrome
Although the occurrence of cardiac dysrhythmias with periodic weakness is well recognized, it has not received the attention it deserves. A series of reports emphasized the constellation of findings: periodic paralysis, cardiac dysrhythmias, and dysmorphic features. These are termed Andersen-Tawil syndrome (ATS) (Sansone et al., 1997). The disorder is a genetically heterogeneous syndrome inherited in an autosomal dominant fashion. Some cases are caused by mutations in the potassium channel gene (KCNJ2) located on chromosome 17q23.
Chloride Channelopathy
Myotonia Congenita
Two major forms of myotonia congenita exist: autosomal dominant and autosomal recessive (Matthews et al., 2010). Both are associated with abnormalities in the chloride channel, the gene for which (CLCN1) resides on chromosome 7q35. Introducing the mutant chloride channel into a cell system abolishes the chloride current and deranges the normal function of the chloride channel (Fahlke et al., 1997). The patient’s muscles show reduced chloride conductance and greater than normal membrane resistance. The action potential in a normal muscle cell is associated with an outflow of potassium, which may accumulate in the transverse tubules simply because the physical structure of the tubule does not favor easy diffusion. Ordinarily, this does not present a problem because the chloride conductance is so large that the relatively free passage of chloride ions negates the effect of any small change in potassium. With impeded chloride conductance, the increase in potassium concentration in the transverse tubules may lead to enough depolarization to activate the sodium channels again and hence lead to repetitive electrical discharge of the membrane, producing electrical and clinical myotonia.
In myotonia congenita, EMG shows well-marked myotonia with none of the associated dystrophic features. Unlike paramyotonia congenita, the short-exercise nerve conduction study test is usually normal (Fournier et al., 2004, 2006). Muscle biopsy may demonstrate an absence of type 2B fibers, an unexplained finding, and also may reveal some increase in the size of fibers and internal nuclei as well as other mild nonspecific changes.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 79 Disorders of Skeletal Muscle
Striated muscle is the tissue that converts chemical energy into mechanical energy. The component processes include (1) excitation and contraction occurring in the muscle membranes, (2) the contractile mechanism itself, (3) various structural supporting elements that allow the muscle to withstand the mechanical stresses, and (4) the energy system that supports the activity and integrity of the other three systems. The logical categorization of myopathies is according to the part of the system involved. Until recently, this was impossible because the molecular basis of muscle activity was unknown. Scientific advances since the 1980s made this classification possible. Abnormalities in the membrane ion channels (channelopathies) involved in muscle excitation cause various forms of myotonia and periodic paralysis (see Chapter 64). The complex of proteins that include dystrophin, the sarcoglycans, and α-laminin constitute a vital structural mechanism linking the contractile proteins with the extracellular supporting structures. Defects in these proteins are the basis of many forms of muscular dystrophy. Although knowledge remains incomplete, it seems reasonable to modify the classic description of the myopathies to incorporate the new information. For this reason, in the sections that follow, disease descriptions are under the heading of their known molecular defect where possible; the classic appellation appears parenthetically. Before describing the illnesses themselves, we first review the techniques used in the clinical evaluation of patients.
Muscle Histology
The technique of muscle biopsy is not difficult. Under local anesthesia, a small incision made over the muscle allows, with careful dissection, removal of a small strip of muscle. Needle biopsies are useful in some situations. Histochemical studies of frozen sections are essential for proper interpretation. A transverse section of normal muscle shows fibers that are roughly of equal size and average approximately 60 mm in transverse diameter (Fig. 79.1). The muscle fibers of infants and young children are proportionately smaller. Each fiber consists of hundreds of myofibrils separated by an intermyofibrillar network containing aqueous sarcoplasm, mitochondria, and the sarcoplasmic reticulum with the associated transverse tubular system. Surrounding each muscle fiber is a thin layer of connective tissue (the endomysium). Strands of connective tissue group fibers into a fascicle, separated from each other by the perimysium. Groups of fascicles are collected into muscle bellies surrounded by epimysium.
Situated at the periphery of the fibers are the sarcolemmal nuclei. The fibers are of different types. The simplest division is into type 1 and type 2 fibers, best demonstrated with the histochemical reaction for myosin adenosine triphosphatase (ATPase) (Fig. 79.2). The type 1 and type 2 fibers are roughly equivalent to slow and fast fibers or to oxidative and glycolytic fibers in human muscle. The best demonstration of the intermyofibrillar network pattern is with the histochemical reactions for oxidative enzymes, such as reduced nicotinamide adenine dinucleotide dehydrogenase. A regular network extends across the whole fiber. In addition to the routine stains with hematoxylin and eosin, modified Gomori-trichrome, myosin ATPase, and nicotinamide adenine dinucleotide dehydrogenase, the use of other special stains demonstrates fat (Sudan black or oil red 0), complex carbohydrates (periodic acid–Schiff), amyloid (Congo red), or specific enzymes (e.g., phosphorylase, succinic dehydrogenase, cytochrome oxidase). Immunocytochemical techniques demonstrate the location and integrity of structural proteins such as dystrophin. They also characterize cell types in biopsy samples with inflammatory changes.
Changes of Denervation
When muscle loses its nerve supply, muscle fibers atrophy, often resulting in fiber squeezing into the spaces between normal fibers and assuming an angulated appearance (Fig. 79.3). Scattered angulated fibers appear early in denervation. Sometimes, picturesque changes in the intermyofibrillar network occur, as in the target fiber, which characterizes denervation and reinnervation. This is a three-zone fiber on which the intermediate zone stains more darkly, and the central “bull’s eye” stains much lighter than normal tissue (Fig. 79.4). Often a neighboring nerve twig reinnervates a denervated fiber. This results in the same anterior horn cell supplying two or more contiguous fibers. If that nerve twig then undergoes degeneration, instead of only one small angulated fiber being produced, a small group of atrophic fibers develops. Group atrophy suggests denervation (Fig. 79.5). As the process continues, large groups of geographical atrophy occur in which entire fascicles are atrophic. In addition to the change in size, a redistribution of the fiber types occurs as well. Normally a random distribution of type 1 and 2 muscle fiber types exists, sometimes incorrectly called a checkerboard or mosaic pattern. The same process of denervation and reinnervation results in larger and larger groups of contiguous fibers supplied by the same nerve. Because all fibers supplied by the same nerve are of the same fiber type, groups of type 1 fibers next to groups of type 2 fibers replace the normal random pattern. This fiber type grouping is pathognomonic of reinnervation (Fig. 79.6). When long-standing denervation is present, the atrophic muscle fibers almost disappear, leaving small clumps of pyknotic nuclei in their place.
Myopathic Changes
Changes in the intermyofibrillar network pattern are common in myopathic disorders. There is often a moth-eaten, whorled change to the intermyofibrillar network in LGMD and facioscapulohumeral dystrophy (FSHD) (Fig. 79.7); the intermyofibrillar network loses its orderly arrangement and swirls, resembling the current in an eddying stream. These changes may be seen in several diseases but tend to be much more common in the myopathies.
Other Changes
Selective changes in fiber types occur. Type 2 fiber atrophy is one of the most common abnormalities seen in muscle (Fig. 79.8). Type 2 atrophy, particularly if limited to type 2B fibers, is nonspecific and indicates muscle disuse. If a limb is casted and the muscle examined some weeks later, selective atrophy of type 2 fibers is noted. Any chronic systemic illness tends to produce type 2 atrophy. It occurs in rheumatoid arthritis, nonspecific collagen vascular diseases, cancer (hence the name cachectic atrophy), mental retardation in children, and pyramidal tract disease. Type 2B fiber atrophy can also result from chronic corticosteroid administration. Therefore, type 2 fiber atrophy should probably be regarded as a nonspecific result of anything less than robust good health.
Specific Disorders
Muscular Dystrophies
The muscular dystrophies are a group of hereditary muscle disorders that occur at all ages and with varying degrees of severity. The traditional classification is on clinical grounds. Increasing information about the molecular basis of these disorders provides both reassurance and puzzlement to clinicians (Table 79.1). Different dystrophies are due to distinct molecular abnormalities; however, patients with similar molecular defects may show a wide variability in phenotype not always easily explained.
Dystrophin Deficiency (Duchenne Muscular Dystrophy, Becker Muscular Dystrophy, and Atypical Forms)
An absence or deficiency of dystrophin is responsible for two disorders that cause progressive destruction of muscle. The responsible gene is located on the short arm of the X chromosome at locus Xp21. The gene is extremely large, comprising more than 2.5 million base pairs and 79 exons or coding regions. Approximately two-thirds of cases are associated with a detectable deletion or duplication of segments within the gene. The others are presumably due to point mutations too small to be detected using standard techniques. “Hot spots” for these gene deletions exist, notably between exons 43 and 52 and particularly 44 and 49 (Nobile et al., 1997). Whether the deletion is in frame or out of frame (see Chapter 40) determines whether dystrophin is absent from the muscle or present in a reduced altered form. This has clinical significance because the former is usually associated with the severe Duchenne variety of the disease (DMD), whereas the latter may cause the milder Becker variant (BMD). In BMD, the abnormal dystrophin preserves enough function to slow down the progress of the illness. Reading of the DNA code is triplet by triplet. Maintenance of this reading frame throughout the length of the gene is required for dystrophin production. If a deletion removes a multiple of three base pairs, the reading frame may be intact upstream and downstream and may make limited sense, as if the sentence “You cannot eat the cat” were changed to “You not eat the cat,” and some modified dystrophin may be formed. This is often the situation in the mild form of dystrophin deficiency. In the severe form, the reading frame is destroyed, as if a deletion resulted in the sentence “Yoc ann ote att hec at.” Exceptions to this rule exist, and frameshift deletions have been associated with the milder form of the disease, particularly at the 5′ end of the gene in exons 3 to 7.
Duchenne Muscular Dystrophy
Even in the severe variety of DMD, affected children are normal at birth. During the second year when the boys begin walking, the clumsiness seen in all toddlers persists. Soon the children have to place one hand on the knee to assume an upright position when rising from the floor (Gower maneuver). Often at this stage, the calf muscles are rather firm and rubbery (pseudohypertrophy) (Fig. 79.9). Within 2 to 3 years, parents notice that the child runs properly and is never able to jump clear of the floor with both feet. In the absence of therapy, tightness across several joints in the legs is noted. The iliotibial bands and the heel cords are usually the first to become tight. This is particularly noticeable in boys who habitually walk on their toes.
Fig. 79.9 Duchenne muscular dystrophy. Calf and thigh hypertrophy in an ambulatory 8-year-old patient.
The serum concentration of creatine kinase (CK) is markedly elevated in this illness; levels greater than 10,000 mU/mL are common. Electromyography (EMG) shows myopathic changes (see Chapter 32B), and muscle biopsy demonstrates variation in the size of fibers, fibrosis, groups of basophilic fibers, and opaque or hypercontracted fibers (hyaline fibers) (Figs. 79.10 to 79.12).
Genetic Counseling
Genetic analysis of all potential carriers is advisable. In a family in which the disease is associated with a deletion, there is little problem in determining whether the woman is carrying the affected X chromosome, using techniques that are presently available. Genetics laboratories now have the ability to identify the presence of a mutant gene over the background contributed by the normal allele. This involves an analysis of the gene “dosage,” comparing two normal alleles that have a double dose against a deleted allele and a normal allele that have a single dose (Voskova-Goldman et al., 1997). The basis for current diagnosis of carrier status after identification of a deletion in a proband is by analysis of a gene dosage. Our diagnostic strategy uses fluorescence in situ hybridization (FISH) to detect female carriers with major deletions in the dystrophin gene. Unfortunately, situations exist in which a mutation is identified in a boy with “sporadic” DMD but not in the mother, and yet the mother is still a carrier. This occurrence is secondary to germline mosaicism in which the mutation in the mother lies only in a percentage of her oocytes. The estimated recurrence rate of DMD is as high as 14% even in such cases. Prenatal diagnosis using amniotic cells or chorionic villus biopsies can identify the affected fetus and, more importantly, those who are unaffected.
Other Limb-Girdle Dystrophies
The most important recent development in muscle disease relates to a group of disorders that were clearly dystrophic but defied proper classification. The traditional diagnosis of LGMDs cloaked the clinician’s uncertainty. In some patients, weakness was generally proximal, but other characteristics were disparate. Some cases were dominantly inherited, others recessively. Some had more hip than shoulder weakness, others the reverse. The illness could be mild in late life, others severe and early. There was general recognition that the rubric LGMD encompassed a group of different illnesses. Beginning with the discovery that a defect in one of the sarcoglycans caused a severe form of dystrophy occurring in North Africa, the delineation of several other entities characterized by defects in structural proteins or enzymes followed. These include the sarcoglycans, the α2 chain of laminin (merosin), calcium-activated protease, calpain-3, and others. The designation of autosomal dominant LGMD is type 1, and the designation of autosomal recessive LGMD is type 2. Subclassification with an alphabetical letter characterizes distinct genetic forms of LGMD1 and LGMD2 (see Table 79.1). The following sections outline the known protein abnormalities and then comment on the more amorphous forms of LGMD. The prevalence of these diseases as a group probably approaches 1 per 100,000 (van der Kooi et al., 1996). The autosomal recessive LGMDs are more common than the autosomal dominant LGMDs.
Autosomal Dominant Limb-Girdle Muscular Dystrophies
LGMD1A (Myotilin Deficiency)
LGMD1A is allelic to one subtype of myofibrillary myopathy and is caused by mutations in the myotilin gene located on chromosome 5q22.3-31.3 (Selcen and Engel, 2004). Myotilin is a sarcomeric protein that is present at the Z-disc. The protein is likely important in myofibrillogenesis and stabilization of the Z-disc and sarcomere.
LGMD1B (Lamin A/C Deficiency)
This myopathy is allelic with the disorder previously reported as autosomal dominant Emery-Dreifuss muscular dystrophy (EDMD) (Bonne et al., 2000; van der Kooi et al., 1996
[/not-level-membership-for-neurology-category]
