Chapter 76 Disorders of Peripheral Nerves
Clinical Approach to Disorders of Peripheral Nerves
Pathological Processes Involving Peripheral Nerves
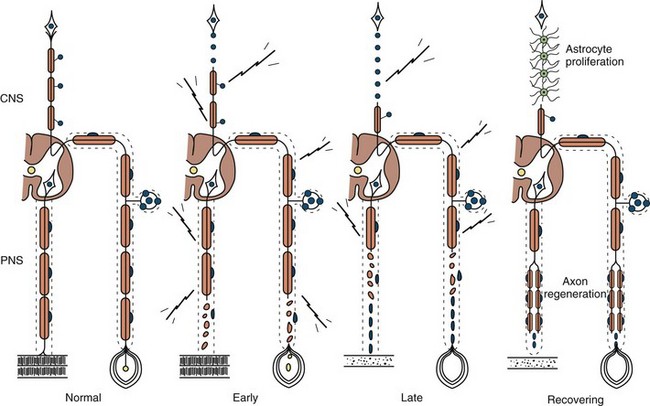
Any type of mechanical injury that causes interruption of axons leads to wallerian degeneration—that is, distal degeneration of axons and their myelin sheaths. Immediately following injury, motor weakness and sensory loss occur in the distribution of the damaged nerve, as well as complete loss of voluntary activity (with a complete lesion) or a decrease in motor unit action potential (MUAP) recruitment (with a partial lesion) on needle electromyogram (EMG). However, the axons remain excitable distally, since distal conduction failure is not completed until 10 to 11 days later as the distal nerve trunk becomes progressively unexcitable. On nerve conduction studies, the amplitude of the compound muscle action potential (CMAP) begins to decline by the second day after injury and reaches its nadir by the fifth to sixth. For sensory axons, the loss of sensory nerve action potential (SNAP) is delayed by another 2 to 3 days; distal SNAP remains normal for 5 to 6 days and then decreases rapidly to reach its nadir by 10 to 11 days after injury (see Fig. 32B.9 in Chapter 32B). The temporal sequence of wallerian degeneration is length dependent, occurring earlier in shorter than in longer distal nerve stumps. Denervation potentials (fibrillation potentials) are typically seen on needle EMG in some affected muscles 10 to 14 days after injury and become full after 3 weeks from acute nerve injury. Axonal interruption initiates secondary morphological changes of the nerve cell body, termed chromatolysis, and the proximal axonal caliber becomes smaller. Regeneration from the proximal stump begins as early as 24 hours following transection but proceeds slowly at a maximal rate of 2 to 3 mm/day and is often incomplete. The quality of recovery depends on the degree of preservation of the Schwann cell/basal lamina tube and the nerve sheath and surrounding tissue, the distance of the site of injury from the cell body, and the patient’s age.
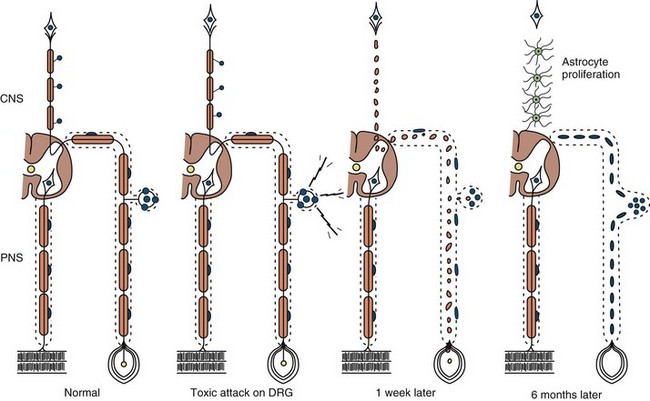
Axonal degeneration (or axonopathy), the most common pathological reaction of peripheral nerve, signifies distal axonal breakdown and is presumably caused by metabolic derangement within neurons or ischemia. Systemic metabolic disorders, toxin exposure, vasculitis, and some inherited neuropathies are the usual causes of axonal degeneration. The myelin sheath breaks down concomitantly with the axon in a process that starts at the most distal part of the nerve fiber and progresses toward the nerve cell body, hence the term dying-back or length-dependent polyneuropathy (Fig. 76.1). A similar sequence of events may occur simultaneously in centrally directed sensory axons, resulting in distal degeneration of rostral dorsal column fibers. The selective length-dependent vulnerability of distal axons could result from failure of the perikaryon to synthesize enzymes or structural proteins, from alterations in axonal transport, or from regional disturbances of energy metabolism. In some axonopathies, alterations in axon caliber, either axonal atrophy or axonal swelling, may precede distal axonal degeneration. Clinically, dying-back polyneuropathy presents with symmetrical distal loss of sensory and motor function in the lower extremities that extends proximally in a graded manner. The result is sensory loss in a stocking-like pattern, distal muscle weakness and atrophy, and loss of distal limb myotatic reflexes. Axonopathies result in low-amplitude SNAPs and CMAPs, but they affect distal latencies and conduction velocities only slightly. Needle EMG of distal muscles shows acute and/or chronic of denervation changes (see Chapter 32B). Because axonal regeneration proceeds at a maximal rate of 2 to 3 mm/day, recovery may be delayed and is often incomplete.
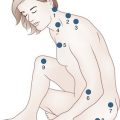
Neuronopathy designates loss of nerve cell bodies with resultant degeneration of their entire peripheral and central axons. Either anterior horn or dorsal root ganglion cells may be affected. When anterior horn cells are affected, as in anterior poliomyelitis or motor neuron disease, focal weakness without sensory loss results. Sensory neuronopathy, or dorsal polyganglionopathy, means damage to dorsal root ganglion neurons that results in sensory ataxia, sensory loss, and diffuse areflexia (Fig. 76.2). A number of toxins such as organic mercury compounds, doxorubicin, and high-dose pyridoxine produce primary sensory neuronal degeneration. Immune-mediated inflammatory damage of dorsal root ganglion neurons occurs in paraneoplastic sensory neuronopathy and Sjögren syndrome. It is often difficult to distinguish between neuronopathies and axonopathies on clinical grounds alone. Once the pathological processes are no longer active, sensory deficits become fixed, and little or no recovery takes place.
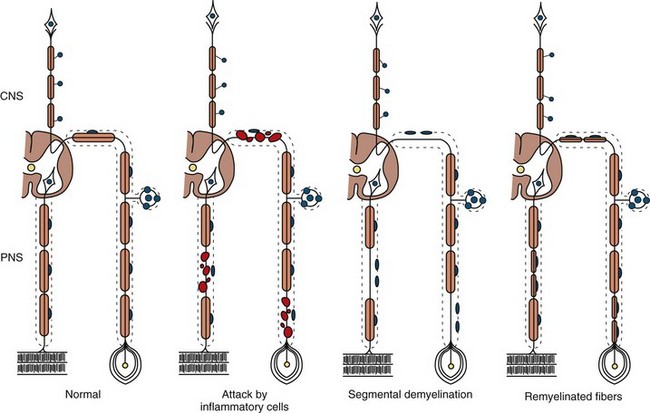
The term segmental demyelination (or myelinopathy) implies injury of either myelin sheaths or Schwann cells, resulting in breakdown of myelin with sparing of axons (Fig. 76.3). This occurs mechanically by acute nerve compression or chronic nerve entrapment and in immune-mediated demyelinating neuropathies and hereditary disorders of Schwann cell/myelin metabolism. Primary myelin damage may be produced experimentally by myelinotoxic agents such as diphtheria toxin or by acute nerve compression. Remyelination of demyelinated segments usually occurs within weeks. The newly formed remyelinated segments have thinner-than-normal myelin sheaths and internodes of shortened length. Repeated episodes of demyelination and remyelination produce proliferation of multiple layers of Schwann cells around the axon, termed an onion bulb. The physiological consequence of acquired demyelination, such as in inflammatory or compressive demyelination but not hereditary myelinopathies, is conduction block, which results in loss of the ability of the nerve action potential to reach the muscle, thereby producing weakness. Because the axon remains intact, there is little muscle atrophy. Relative sparing of temperature and pinprick sensation in many demyelinating polyneuropathies reflects preserved function of unmyelinated and small-diameter myelinated fibers. Early generalized loss of reflexes, disproportionately mild muscle atrophy in the presence of proximal and distal weakness, neuropathic tremor, and palpably enlarged nerves are all clinical clues that suggest demyelinating polyneuropathy. Nerve conduction studies or analysis of single teased nerve fiber preparations stained with osmium can confirm demyelination. Demyelination is present if motor and sensory nerve conduction velocities (NCVs) are reduced to less than 70% of the lower limits of normal, with relative preservation of response amplitudes. The presence of partial motor conduction block, temporal dispersion of CMAPs, and marked prolongation of distal motor and F-wave latencies are all features consistent with acquired demyelination (see Chapter 32B). Recovery depends on the extent of remyelination, and therefore clinical improvement may occur within days to weeks. It should be noted that in many demyelinating neuropathies, axonal degeneration may also coexist.
Classification of Peripheral Nerve Disorders
There are several patterns of peripheral nerve disease (Box 76.1). Brachial, lumbar, and sacral plexopathy are discussed in Chapter 75, and radiculopathies are discussed in Chapter 74.
Diagnostic Clues from the History
Symptoms of autonomic dysfunction can be helpful in directing attention toward specific neuropathies that have prominent autonomic symptoms. It is important to ask about orthostatic intolerance (lightheadedness, presyncopal symptoms or syncope), reduced or excessive sweating, heat intolerance, and bladder, bowel, and sexual dysfunctions. Anorexia, early satiety, nausea, and vomiting are symptoms suggestive of gastroparesis. The degree of autonomic involvement can be documented by noninvasive autonomic function studies (see Chapter 77).
Diagnostic Clues from the Examination
In multiple mononeuropathies (mononeuropathy multiplex), the neurological findings should point to simultaneous or sequential damage to two or more noncontiguous peripheral nerves. Confluent multiple mononeuropathies, such as with involvement of the fibular and tibial nerves or median and ulnar nerves, may give rise to motor weakness with sensory loss that can simulate a length-dependent peripheral polyneuropathy. EDX studies ascertain whether the primary pathological process is axonal degeneration or segmental demyelination (Box 76.2). Approximately two-thirds of patients with multiple mononeuropathies display a picture of axonal damage. Ischemia caused by systemic or nonsystemic vasculitis or microangiopathy in diabetes mellitus should be considered. Other less common causes are disorders affecting interstitial structures of nerve, namely infectious, granulomatous, leukemic, or neoplastic infiltration, including Hansen disease (leprosy) and sarcoidosis. In the event focal demyelination or motor conduction block leads to multiple mononeuropathies, multifocal acquired demyelinating sensory and motor neuropathy (Lewis-Sumner syndrome), multifocal motor neuropathy, or HNPP should be considered.
Box 76.2 Causes of Multiple Mononeuropathies
Motor deficits tend to dominate the clinical picture in acute and chronic inflammatory demyelinating polyneuropathies, hereditary motor and sensory neuropathies, and in neuropathies associated with osteosclerotic myeloma, porphyria, lead toxicity, organophosphate intoxication, and hypoglycemia (Box 76.3). The distribution of weakness provides important information. Asymmetrical weakness without sensory loss suggests a motor neuronopathy such as motor neuron disease or multifocal motor neuropathy. The facial nerve can be affected in several peripheral nerve disorders (Box 76.4). In most polyneuropathies, the legs are more severely affected than the arms, with several notable exceptions (Box 76.5). Polyradiculoneuropathies cause both proximal and distal muscle weakness. For example, proximal and distal weakness is encountered in acute and chronic inflammatory demyelinating polyradiculoneuropathies, osteosclerotic myeloma, porphyria, and diabetic lumbar radiculoplexopathy. Nerve root involvement is confirmed by denervation in paraspinal muscles on needle EMG.
Box 76.3 Polyneuropathies with Predominantly Motor Manifestations
Box 76.5 Polyneuropathies with Predominantly Upper-Limb Motor Involvement
Autonomic dysfunction of clinical importance is seen in association with specific acute (e.g., GBS) or chronic (e.g., amyloid and diabetic) sensorimotor polyneuropathies. Rarely, an autonomic neuropathy can be the exclusive manifestation of a peripheral nerve disorder, without somatic nerve involvement (Box 76.6).
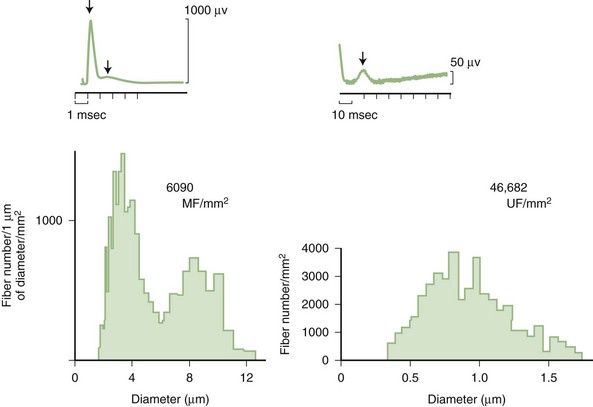
Predominant sensory involvement may be a feature of polyneuropathies caused by diabetes, carcinoma, Sjögren syndrome, dysproteinemia, acquired immunodeficiency syndrome (AIDS), vitamin B12 deficiency, celiac disease, inherited and idiopathic sensory neuropathies, and intoxications with cisplatin, thalidomide, or pyridoxine. Loss of sensation in peripheral neuropathies often involves all sensory modalities. However, the impairment may be restricted to selective sensory modalities in many situations, which makes it possible to correlate the type of sensory loss with the diameter size of affected afferent fibers (Fig. 76.4). Pain and temperature sensation are mediated by unmyelinated and small myelinated Aδ fibers, whereas vibratory sense, proprioception, and the afferent limb of the tendon reflex are subserved by large myelinated Aα and Aβ fibers. Light touch is mediated by both large and small myelinated fibers. In polyneuropathies preferentially affecting small fibers, diminished pain and temperature sensation predominate, along with spontaneous burning pain, painful dysesthesias, and autonomic dysfunction. There is preservation of tendon reflexes, balance, and motor strength, and hence few abnormal objective neurological signs are found on examination. A pattern of sensory loss that is very characteristic is distal loss of pinprick sensation, above which is a band of hyperalgesia (exaggerated pain from noxious stimuli), with normal sensation above this level. Relatively few disorders cause selective small-fiber neuropathies (Mendell and Sahenk, 2003) (Box 76.7). Selective large-fiber sensory loss is characterized by areflexia, sensory ataxia, and loss of joint position and vibration sense. Loss of joint position may also manifest as pseudoathetosis (involuntary sinuous movements of fingers and hands when the arms are outstretched and the eyes are closed) and/or a Romberg sign (disproportionate loss of balance with eyes closed compared with eyes open). Striking sensory ataxia, together with pseudoathetosis or asymmetrical truncal or facial sensory loss, directs attention to a primary disorder of sensory neurons or polyganglionopathies. The differential diagnosis of ataxic sensory neuropathies is limited (Box 76.8).
Box 76.8 Sensory Ataxic Neuropathies
Certain telltale signs of the skin and its appendages may direct the experienced examiner to a specific diagnosis (Table 76.1): alopecia is seen in thallium poisoning, tightly curled hair in giant axonal neuropathy; white transverse nail bands termed Mees lines in arsenic or thallium intoxications; purpuric skin eruptions of the legs in cryoglobulinemia and some vasculitides; skin hyperpigmentation or hypertrichosis in POEMS syndrome (polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes); telangiectasias over the abdomen and buttocks in Fabry disease; enlarged yellow-orange tonsils in Tangier disease; pes cavus and hammer toes in CMT disease; and overriding toes and ichthyosis in Refsum disease.
Table 76.1 Neuropathies with Skin, Nail, or Hair Manifestations
| Disease | Skin, Nail, or Hair Manifestations |
|---|---|
| Vasculitis | Purpura, livedo reticularis |
| Cryoglobulinemia | Purpura |
| Fabry disease | Angiokeratomas |
| Leprosy | Skin hypopigmentation |
| Osteosclerotic myeloma (POEMS syndrome) | Skin hyperpigmentation |
| Variegate porphyria | Bullous lesions |
| Refsum disease | Ichthyosis |
| Arsenic or thallium intoxication | Mees lines |
| Thallium poisoning | Alopecia |
| Giant axonal neuropathy | Curled hair |
POEMS, Polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes.
Electrodiagnostic Studies
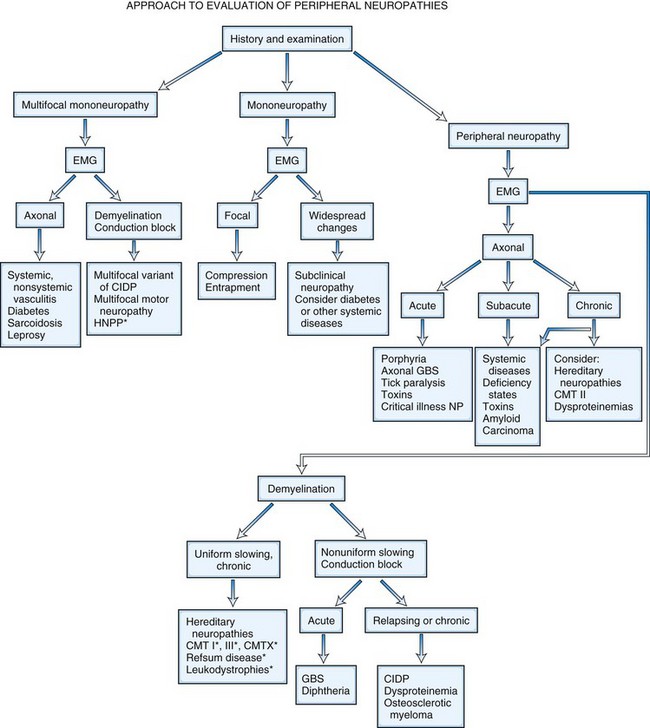
It is helpful to follow a decision-making pathway based initially on the overall pattern of distribution of deficits, followed by the electrophysiological findings, and finally the clinical course (Fig. 76.5). EDX studies, carefully performed and directed to the particular clinical situation, play a key role in the evaluation by (1) confirming the presence of neuropathy, (2) precisely locating focal nerve lesions, and (3) giving information as to the nature of the underlying nerve pathology (Gooch and Weimer, 2007; Wilbourn, A.J., 2002) (see Chapter 32B).
Because routine sensory nerve conduction studies assess only large myelinated fibers, such studies may be entirely normal in selective small fiber neuropathies. Quantitative sensory testing assessing cold and heat-pain thresholds, tests of sudomotor function, and skin biopsy with analysis of intraepidermal nerve fiber density may be helpful in confirming the unmyelinated nerve fiber abnormalities (Devigili et al., 2008). Since sweating mediated by unmyelinated sympathetic cholinergic fibers is often impaired, the quantitative sudomotor axon reflex (QSART) that evaluates sweating is a highly specific and sensitive method (sensitivity of 80%) to confirm small nerve fiber damage. Quantitative sensory testing assessing both vibratory and thermal detection thresholds has become a useful addition to the bedside sensory examination in controlled clinical trials. Its use in routine clinical practice remains limited because the test is still subjective in that it requires patient cooperation and is time consuming.
Nerve and Skin Biopsy
Skin punch or blister biopsies that demonstrate loss of intraepidermal nerve fibers is an alternative method for documenting small fiber neuropathy (Panoutsopoulou, 2009). Only unmyelinated intraepidermal networks of nerve fibers can be demonstrated by immunostaining with the panaxonal marker protein gene product 9.5, studied best with the use of confocal microscopy. Age, gender, and site of skin biopsy have a profound effect on epidermal nerve fiber density. The density of intraepidermal nerve fibers is reduced in skin biopsies obtained from patients with idiopathic, HIV-associated, diabetic, and other sensory neuropathies (Kennedy, 2004). Skin punch biopsy is most useful in patients with suspected small fiber neuropathy, when nerve conduction studies, are normal. The diagnosis of small-fiber neuropathy is best accomplished when at least two abnormal results are met, including positive clinical findings, quantitative sensory testing, QSART, and skin biopsy examinations (Devigili et al., 2008). Skin punch biopsy only detects the presence of skin nerve abnormalities and rarely leads to a specific etiological diagnosis. The skin biopsy also does not permit the study of myelinated fibers unless a thicker biopsy including dermis is obtained. Finally, unlike sural nerve biopsy, the interstitial pathological processes of the nerves cannot be studied.
Nerve biopsy (other than for the diagnosis of vasculitis and neoplasia) should be performed only in centers with established experience with the surgical procedure, handling of nerve specimens, and pathological technique; otherwise little useful information is likely to be obtained. The sural nerve is selected most commonly for biopsy, because the resultant sensory deficit is restricted to a small area over the heel and dorsolateral aspect of the foot, and because its morphology has been well characterized in health and disease. The superficial fibular nerve is an alternative lower-extremity cutaneous nerve suitable for biopsy and has the advantage of allowing simultaneous biopsy of the peroneus brevis muscle through the same incision. This combined distal nerve and muscle biopsy procedure increases the yield of identifying suspected vasculitis (Collins et al., 2000; Vital et al., 2006). In contrast, adding a proximal muscle (e.g., quadriceps) to a cutaneous nerve biopsy (e.g., sural) does not significantly increase the diagnostic yield compared to nerve biopsy alone (Bennett et al., 2008). In patients with proximal involvement of the lower limbs, the intermediate cutaneous nerve of the thigh combined with a muscle biopsy can be performed. When the risk of complication is increased from a biopsy of the lower limbs (e.g., in significant distal leg ischemia, edema) or the neuropathy is preferentially more pronounced in the upper limbs, a cutaneous nerve biopsy of superficial radial or antebrachial nerves may be performed. When the imaging studies indicate a plexus or nerve root pathological process (e.g., inflammatory, infiltrative), a fascicular biopsy of the affected nerve by an expert surgeon may provide invaluable information. Nerve biopsy has proved to be particularly informative when techniques such as single teased fiber preparations, semithin sections, ultrastructural studies, and morphometry are applied to quantitate the nerve fiber pathology. Nowadays, relatively few disorders remain in which a nerve biopsy is essential for diagnosis (Pleasure, 2007; Said, 2002) (Box 76.9). In general, nerve biopsy is most frequently diagnostic in suspected vasculitis, amyloid neuropathy, and leprosy. It is helpful in the recognition of CIDP, inherited disorders of myelin, and some rare axonopathies in which distinctive axonal changes occur, such as in giant axonal neuropathy and polyglucosan body disease. The availability of molecular genetic tests for several CMT neuropathies, HNPP, and familial transthyretin amyloidosis has decreased the necessity for nerve biopsy in these conditions.
Box 76.9 Indications for Nerve Biopsy
Nerve biopsy is an invasive procedure and is associated with as high as 15% complication rate—particularly minor wound infections, wound dehiscence, and stump neuromas. Approximately one-third of patients (particularly those without much sensory loss initially) report unpleasant sensory symptoms at the sural nerve biopsy site that are still present 1 year after the biopsy (Gabriel et al., 2000). The area of the original sensory deficit declines by 90% after 18 months because of collateral reinnervation (Theriault et al., 1998). The complications may be greater if substantial foot ischemia is present or if the patient smokes cigarettes.
Other Laboratory Tests
Several serum autoantibodies with reactivity to various components of peripheral nerve have been associated with peripheral neuropathy syndromes, and reference laboratories offer panels of nerve antibodies for sensory, sensorimotor, and motor neuropathies. It must be emphasized that the clinical relevance of most autoantibodies has not been established for patient treatment, and their use is not cost-effective (Vernino and Wolfe, 2007). Those of greatest clinical utility are listed in Table 76.2 (Kissel, 1998). An ever-increasing number of molecular genetic tests for inherited neuropathies is available at reference laboratories (see Hereditary Neuropathies, later).
Table 76.2 Neuropathies Associated with Serum Autoantibodies
| Autoantibody | Disease (% Positive) |
|---|---|
| ANTIBODIES AGAINST GANGLIOSIDES | |
| GM1 (polyclonal IgM) | Multifocal motor neuropathy (70%) |
| GM1, GD1a (polyclonal IgG) | Guillain-Barré syndrome (30%) |
| GQ1b (polyclonal IgG) | Miller Fisher variant of Guillain-Barré syndrome (>95%) |
| ANTIBODIES AGAINST GLYCOPROTEINS | |
| Myelin-associated glycoprotein (MAG; monoclonal IgM) | IgM monoclonal gammopathy of undetermined significance neuropathy (50%) |
| ANTIBODIES AGAINST RNA-BINDING PROTEINS | |
| Anti-Hu, antineuronal nuclear antibody 1 (ANNA1) | Malignant inflammatory polyganglionopathy (>95%) |
Ig, Immunoglobulin.
Mononeuropathies
Definition and Classification of Mononeuropathies
Peripheral nerve injuries are classified based on functional status of the nerve and histological findings. Seddon divided peripheral nerve injury into three classes: neurapraxia, axonotmesis, and neurotmesis. This classification remains popular, particularly among surgeons, because of its correlation to outcome (Seddon, 1975). Later, Sunderland (1991) revised the classification into five degrees that have better prognostic implications.
Neurotmesis (Fifth-Degree Nerve Injury)
Entrapment neuropathy is defined as a mononeuropathy caused by focal compression or mechanical distortion of a nerve within a fibrous or fibro-osseous tunnel or less commonly by other structures such as bone, ligament, other connective tissues, blood vessels, or mass lesions. Compression, constriction, angulation, and stretching are important mechanisms that produce nerve injury at certain vulnerable anatomical sites (Tables 76.3 and 76.4). The term entrapment is a useful one in that it implies that compression occurs at particular sites where surgical intervention is often required to release the entrapped nerve, such as in the case of the median nerve at the wrist in moderate to severe carpal tunnel syndrome. Overuse has been implicated as the cause of entrapment neuropathies in certain occupations, including the playing of musical instruments by professional musicians.
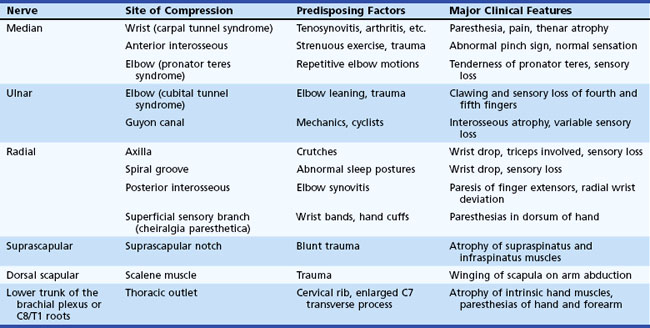
The characteristic electrophysiological feature of entrapment neuropathy is either short-segment conduction delay (i.e., focal slowing) or conduction block across the site of entrapment (see Chapter 32B). In severe cases, wallerian degeneration gives rise to denervation and reinnervation in affected muscles. Nerve conduction studies together with needle EMG are essential for diagnosis and reliable documentation of the site and severity of nerve entrapment. Although plain radiography, computed tomography (CT), and magnetic resonance imaging (MRI) may be of occasional value in identifying rare structural abnormalities, these imaging procedures are not necessary for routine diagnosis.
Mononeuropathies of the Upper Extremities
Entrapment neuropathies of the upper extremities are shown in Table 76.3.
Median Nerve
Median Nerve Entrapment at the Wrist (Carpal Tunnel Syndrome)
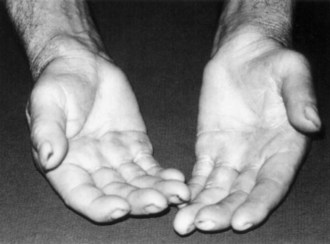
Symptoms consist of nocturnal pain and paresthesias, most often confined to the thumb, index, and middle fingers, but may be reported to involve the entire hand. Patients complain of tingling numbness and burning sensations, often awakening them from sleep. Referred pain may radiate to the forearm and even as high as the shoulder (Stevens et al., 1999). Symptoms are often provoked after excessive use of the hand or wrist or during ordinary activities such as driving or holding a phone, book, or newspaper, in which the wrist is assumed in either a flexed or extended posture. Objective sensory changes may be found in the distribution of the median nerve, most often impaired two-point discrimination, pinprick and light touch sensation, or occasionally hyperesthesia in the thumb, index, and middle fingers, with sparing of the skin over the thenar eminence. Thenar (abductor pollicis brevis muscle) weakness and atrophy may be present in advanced cases of CTS (Fig. 76.6). Flexing the patient’s hand at the wrist for 1 minute (Phalen maneuver) or hyperextension of the wrist (reversed Phalen maneuver) can reproduce the symptoms, is present in about 80% of patients, and is rarely false positive. A positive Tinel sign, in which percussion of the nerve at the carpal tunnel causes paresthesias in the distribution of the distal distribution of the median nerve, is present in approximately 60% of affected patients but is not specific for CTS and may be false positive.
Work-related wrist and hand symptoms (repetitive motion injury) from cumulative trauma in the workplace have received increasing attention by the general public in recent years (Thomsen et al., 2002). Although a proportion of these cases have bona fide CTS, longitudinal natural history data suggest that the majority of industrial workers do not develop symptoms of CTS (Nathan et al., 1998). Symptoms consistent with hand and wrist arthritis in a variety of occupational settings are now recognized as being much more common than CTS (Dillon et al., 2002). CTS appears to occur in work settings that include repetitive forceful grasping or pinching, awkward positions of the hand and wrist, direct pressure over the carpal tunnel, and the use of handheld vibrating tools. Increased risk for the syndrome has been found in meat packers, garment workers, butchers, grocery checkers, electronic assembly workers, musicians, dental hygienists, and housekeepers. The highest reported incidence of work-related CTS, based on the number of carpal tunnel surgeries performed, was 15% among a group of meat packers. Although computer keyboard use has long been thought to be related to developing carpal tunnel symptoms, recent data provide no convincing correlation between intensive keyboard use and the subsequent development of CTS (Papanicolaou et al., 2001; Stevens et al., 2001).
Diseases and conditions that have been found to predispose to the development of CTS include pregnancy, diabetes, obesity, age, rheumatoid arthritis, hypothyroidism, amyloidosis, gout, acromegaly, certain mucopolysaccharidoses, arteriovenous shunts for hemodialysis, old fractures at the wrist, and inflammatory diseases involving tendons or connective tissues at the wrist level (Becker et al., 2002). On rare occasions, CTS may be familial, and some patients with CTS have carpal canals that are significantly narrower than average.
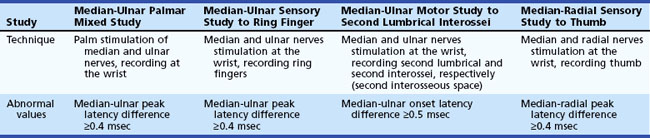
The most commonly performed EDX test for CTS are the median nerve sensory and motor conduction studies, which exhibit delayed sensory or motor latencies across the wrist in about 70% of patients. However, these studies are not sensitive enough in the diagnosis of CTS and fail to detect up to a third of patients with CTS, particularly those with mild and early symptoms. Recording the median latency at short distances over the course of the median nerve from palm to wrist and/or comparing this latency with the latency for the ulnar or radial nerve at the same distance (internal comparison nerve conduction studies) increase the sensitivity of these nerve conduction studies (Stevens, 1997) (Table 76.5).
In cases with only mild sensory symptoms, treatment with splints in neutral position, nonsteroidal antiinflammatory drugs (NSAIDs), and local corticosteroid injection often suffice. Withdrawal of provoking factors is also important. Although nonoperative treatments have been advocated (Osterman et al., 2002), a comparison of splinting versus surgery suggested that the latter may have a better long-term outcome than the former (Gerritsen et al., 2002). Use of a range of devices and appliances to protect the hand against CTS, including gel-padded gloves, has shown little if any improvement in objective measures of nerve function. Severe sensory loss and thenar atrophy suggest the need for surgical carpal tunnel release. Open surgical sectioning of the volar carpal ligament or fiberoptic techniques are often successful, with more than 90% of patients having prompt resolution of pain and paresthesias (Mirza and King, 1996). Improvement in distal latencies may lag behind the relief of symptoms. Comparing with preoperative values, nerve conduction studies demonstrate improvement in those with moderate abnormalities preoperatively, whereas patients with severe or no abnormalities on baseline nerve conduction studies had poorer results (Bland, 2001). A correlation between patients seeking workers’ compensation who hire attorneys and poorer operative outcomes has also been reported (Katz et al., 2001a). Older individuals may not improve as much as younger patients (Porter et al., 2002), and factors such as poor mental health, significant alcohol consumption, longer disease duration, and male gender also portend a poorer outcome. Rarely, symptoms persist after operation. Poor surgical results usually are associated with incomplete sectioning of the transverse ligament, surgical damage of the palmar cutaneous branch of the median nerve by an improperly placed skin incision, scarring within the carpal tunnel, or an incorrect preoperative diagnosis. Surgical reexploration may be required in diagnostically certain cases with poor response to the initial operation (Steyers, 2002).
Ulnar Nerve
Ulnar Nerve Entrapment at the Elbow
Ulnar mononeuropathy is the second most common entrapment or compression mononeuropathy, although it is considerably less common than CTS. Compression of the ulnar nerve by a thickened, fibrotic flexor carpi ulnaris aponeurosis (humeral-ulnar aponeurosis) at the entrance of the elbow’s cubital tunnel is a common cause of ulnar neuropathy (cubital tunnel syndrome). Patients with a subluxed ulnar nerve are at high risk for compression at the elbow. Also, prolonged and frequent resting of the flexed elbow on a hard surface such as a desk or armchair may result in external pressure to the nerve (ulnar groove syndrome). Occupations involving repeated flexion of the elbow may on occasion cause symptoms of ulnar neuropathy. A flexed elbow position increases both the intraneural and extraneural pressure on the nerve. The nerve at the site of repeated compression is associated with fibrous thickening, when a spindle-shaped swelling can often be felt. Other possible sources of injury of the ulnar nerve at the elbow include direct compression when the patient uses the arms to raise up in bed following surgical operations (Stewart and Shantz, 2003) or after periods of prolonged unconsciousness. The ulnar nerve at the elbow may be acutely injured as a result of fracture or dislocation involving the lower end of the humerus and the elbow joint. Occasionally, however, the nerve becomes chronically compressed years after such an injury, which often has led to cubitus valgus deformity (“tardy ulnar palsy”). The nerve may be damaged by osteophyte outgrowths resulting from arthritis of the elbow joint, by a ganglion or lipoma, by a Charcot elbow, and by the epitrochleoanconeus muscle and/or its dense fibrotendinous band. The ulnar nerve may also be involved in conditions that are known to increase the susceptibility of nerves to compression, such as diabetes mellitus or HNPP. Ulnar neuropathy at the elbow segment may also occur without any apparent cause.
Compared to evaluating entrapment neuropathies such as CTS, the EDX studies used to confirm and localize ulnar nerve entrapment at the elbow are more challenging. Localizing ulnar neuropathy at the elbow relies upon the demonstration of focal demyelination across the elbow, namely slowed motor conduction velocity (>10 meters per second) or conduction block (localized reduction in CMAP amplitude and area of >20%-30%) or both. Focal slowing or conduction block may be found in the elbow segment in more than 75% of cases (Azrieli et al., 2003). Performing additional ulnar motor conduction study, recording the first dorsal interosseous muscle in addition to recording the abductor digiti minimi muscle, increases the yield of finding focal slowing or conduction block. In the remaining patients, localization becomes less precise because of predominant axonal loss. To provide the extra nerve length needed during elbow flexion, the ulnar nerve is anatomically redundant in the ulnar groove when the elbow is extended, and this can cause measurement errors. A flexed position of the elbow (70 to 90 degrees) is preferred to the extended position when doing ulnar motor conduction studies to localize an ulnar lesion at the elbow. Short-segment incremental studies (“inching”) by stimulating the ulnar nerve in successive 1-cm increments across the elbow, looking for either an abrupt drop in amplitude or increase in latency, is a useful technique that helps to precisely localize the ulnar nerve lesion. Electrophysiological tests are helpful in differentiating between an ulnar neuropathy and a C8 nerve root or brachial plexus lesion. Ulnar sparing in ulnar sensory studies points to C8 radiculopathy, and needle EMG of C8 muscles innervated by the median nerve (e.g., abductor pollicis brevis, flexor pollicis longus) and radial nerve (e.g., extensor indicis proprius) helps exclude a C8 root lesion or a lower brachial plexopathy. MRI of the elbow may reveal a space-occupying lesion or anomalous structures impinging on the nerve or demonstrate nerve enlargement and increased signal intensity, even in the absence of localizing electrophysiological abnormalities (Vucic et al., 2006). High-resolution sonography at the elbow is also reported to accurately detect thickening of the ulnar nerve at the elbow (Beekman et al., 2004).
Conservative treatment should be attempted in patients with mild or intermittent sensory symptoms or in those with symptoms brought on by occupational causes. Avoidance of repetitive elbow flexion and extension or direct pressure on the elbow may alleviate the symptoms. Elbow protectors are helpful in patients with a history of excessive elbow leaning. Conservative treatment should be continued for at least 3 months before surgery is considered. Several surgical approaches to an ulnar nerve lesion at the elbow are possible, each with its proponents and critics. Techniques include simple release of the flexor carpi ulnaris aponeurosis, anterior transposition of the nerve trunk, and resection of the medial epicondyle. The choice of procedure should be tailored to the specific lesion found at surgery and may be assisted by short-segment incremental electrophysiological studies (“inching”). Transposition of the nerve trunk carries a higher rate of complications than ulnar neurolysis (Biggs and Curtis, 2006). Depending on the type of surgery and the severity and duration of neuropathy, response to these procedures will vary. Only about 60% of patients, especially those with symptoms of less than 1 year’s duration, benefit from surgery; some experience worsening of symptoms. It appears that those with more thickening of the nerve at the time of diagnosis (as determined by sonography) have a more unfavorable outcome, and those with electrophysiological signs of demyelination across the elbow have a more favorable course (Beekman et al., 2004).
Radial Nerve
Radial Nerve Compression in the Arm
Radial nerve compression in the arm often occurs at the spiral groove of the humerus during drunken sleep wherein the arm is draped over a chair (Saturday-night palsy) (Spinner et al., 2002). The radial nerve may be also injured following fractures of the humerus. Radial nerve lesions at the axilla are much less common and may result from crutches or from the weight of a sleeping partner’s head (honeymoon palsy). The radial nerve is also often involved in isolation or in combination with other single nerves in multifocal motor neuropathy with conduction block.
Posterior Interosseous Neuropathy
Lesions of the posterior interosseous nerve (PIN) are uncommon and usually occur in association with trauma, fracture, soft-tissue masses (e.g., lipomas, gangliomas), exuberant synovium motor neuropathy (i.e., rheumatoid arthritis). On rare occasions, a PIN lesion is a manifestation of neuralgic amyotrophy, with acute arm pain followed within few days by weakness (Hashizume et al., 1996). The clinical manifestations of a PIN lesion are dropped fingers and inability to extend them at the metacarpophalangeal joints. Radial deviation of the wrist on wrist extension is often pathognomic and is due to weakness of the extensor carpi ulnaris muscle with sparing of the extensor carpi radialis muscle, the latter innervated by the main trunk of the radial nerve. EMG study confirms the diagnosis by demonstrating normal radial SNAP and denervation of the muscles supplied by the posterior interosseous nerve, with sparing of more proximal radial-innervated muscles including the brachioradialis, extensor carpi radialis, and triceps muscles.
In rheumatoid arthritis, local injection of corticosteroids may be helpful. If the syndrome is progressive, surgical exploration, including synovectomy or decompression of the posterior interosseous nerve, may become necessary (Shergill et al., 2001).
Radial Tunnel Syndrome
Patients with persistent tennis elbow (lateral epicondylitis) are sometimes given a diagnosis of radial tunnel syndrome, an extremely rare and highly controversial entrapment of the radial nerve or its posterior interosseous branch within the radial tunnel (Rosenbaum, 1999). The nerve appears most vulnerable to entrapment at the level of the supinator muscle. These patients present with forearm pain and tenderness at the lateral epicondyle and slightly distally into the forearm, with no associated muscle weakness or sensory loss in the radial or PIN distribution. Pain is induced by extension of the middle finger or supination with the elbow extended. The EMG study, including radial nerve conduction studies recording the extensor digitorum communis and extensor indicis proprius, is almost always normal. Local steroid injection may temporarily relieve symptoms. In patients with persistent pain, surgical division of the supinator muscle has been advocated, with variable results.
Superficial Radial Sensory Neuropathy (Cheiralgia Paresthetica)
Cheiralgia paresthetica is a mononeuropathy of the superficial dorsal sensory branch of the radial nerve. It occurs as a result of trauma from tight wristbands or handcuffs, or may result from intravenous cannulation, fracture of the wrist, or wrist surgery (e.g., plating of forearm bones after fracture). It is also associated with de Quervain tenosynovitis, an inflammatory condition of thumb extensor muscles, predominantly extensor pollicis brevis (Lanzetta and Foucher, 1995). In de Quervain tenosynovitis, there is tenderness of the anatomical snuffbox and thumb extensor tendons with forced ulnar deviation while holding the thumb wrapped in the palm. Paresthesias and pain in the distribution of the superficial sensory branch of the radial nerve characterize this benign self-limiting condition. A small area of hypoesthesia in the dorsoradial aspect of the hand is frequently identified. Nerve conduction study results can show a low-amplitude or absent dorsal radial SNAP.
Musculocutaneous Nerve
The musculocutaneous nerve arises from the lateral cord of the brachial plexus, with fibers originating from the C5-C6 roots via the upper trunk. The nerve innervates and penetrates the coracobrachialis muscle and courses down the anterior aspect of the upper arm between the two muscles it innervates—the biceps brachii and brachialis. It then terminates as a sensory nerve, the lateral antebrachial cutaneous nerve, which innervates the lateral forearm to the base of thumb. This nerve may be damaged with shoulder dislocations, following general anesthesia, or with vigorous exercise such as weight lifting or repetitive movements such as occurs in carpet carriers, where the nerve is repeatedly compressed by carrying carpets on the shoulder while held in place by the arm (Sander et al., 1997). The musculocutaneous nerve may be also involved in neuralgic amyotrophy (idiopathic brachial plexus neuropathy). The differential diagnosis includes C5 or C6 radiculopathy, upper trunk or lateral cord brachial plexopathy, and rupture of the biceps tendon.
Suprascapular Nerve
The suprascapular nerve is a pure motor branch of the upper trunk of the brachial plexus with innervation from the C5 and C6 roots. It then passes through the suprascapular notch, covered by the transverse scapular ligament, to innervate the supraspinatus muscle. It wraps around the spinoglenoid notch of the scapular spine and innervates the infraspinatus muscle. Entrapment at the suprascapular notch occurs after repetitive forward traction of the shoulders—a condition seen in certain athletes, especially volleyball players. This nerve also may be involved in a restricted form of neuralgic amyotrophy (idiopathic brachial plexus neuropathy). Diffuse aching pain in the posterior aspect of the shoulder exacerbated by overhead activities is a cardinal symptom. The pain has an articular characteristic because the acromioclavicular joint and surrounding structures are innervated by the suprascapular nerve. Atrophy and weakness are confined to the infraspinatus and supraspinatus muscles. Slow and steady abduction of the arm starting from a vertical position alongside the chest is not possible with a severe lesion of the suprascapular nerve. Tendon ruptures of the rotator cuff have to be considered in the differential diagnosis. EMG shows denervation restricted to the supraspinatus and infraspinatus muscles. Local corticosteroid injection may give temporary relief of pain, although surgery is sometimes required (Antoniou et al., 2001). In entrapment at the spinoglenoid notch, pain is usually absent, and there is atrophy, weakness, and denervation of the infraspinatus muscle only.
Intercostobrachial Nerve
The intercostobrachial nerve is a cutaneous sensory nerve derived from the second and third thoracic nerve roots. It supplies the skin on the medial surface of the upper arm and axilla, as well as the adjacent chest wall. It may be injured in a modified radical mastectomy and other surgical procedures involving the axilla and lateral pectoral region (Wallace et al., 1996).
Double Crush Syndrome
When a sizable cohort of patients with EDX evidence of distal upper-limb entrapment neuropathies was found to have either electrophysiological or radiological and clinical evidence of cervical radiculopathy, Upton and McComas proposed that focal compression of single nerve fibers proximally might so alter axoplasmic transport as to render the distal nerve more susceptible to symptomatic entrapment neuropathy. They termed this the double crush syndrome. Although the concept of double crush syndrome has since been invoked in a wide variety of entrapment neuropathies, often as an explanation for failure of decompressive surgeries of the neck or limb or as a rationale to decompress a nerve in multiple proximal-to-distal sites along its course, this phenomenon is of uncertain validity (Wilbourn and Gilliatt, 1997).
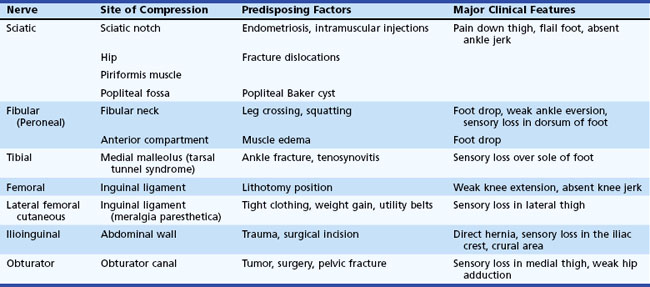
Mononeuropathies of the Lower Extremities
Entrapment neuropathies of lower limbs are shown in Table 76.4.
Sciatic Nerve
Sciatic Nerve Lesions at the Sciatic Notch
The sciatic nerve is occasionally vulnerable to entrapment as it crosses over the sciatic notch leaving the pelvis. Most sciatic nerve lesions result from trauma such as bullet and stab wounds, fractures, dislocations, hematomas in the posterior thigh compartment, misplaced intramuscular injections, and complications of hip replacement surgery (Plewnia et al., 1999). Recurrent sciatic mononeuropathy may be caused by endometriosis involving the nerve at the sciatic notch. Direct compression of the sciatic nerve is rare but occasionally occurs during coma, anesthesia, or prolonged sitting on a hard surface (toilet seat palsy). Either or both divisions of the nerve may be compressed by a Baker cyst in the popliteal fossa.
A complete sciatic nerve lesion results in weakness of knee flexors and all muscles below the knee, as well as sensory loss of the entire foot and leg below the knee except for a region supplied by the saphenous nerve over the medial leg. The fibular division is more commonly involved than the tibial in proximal lesions of the sciatic nerve. Partial sciatic nerve lesions often affect the fibular nerve more than the tibial nerve and may mimic a common fibular neuropathy. This is explained by the fewer fascicles with limited supportive tissue within the fibular nerve, which is also taut and secured at the sciatic notch and fibular neck. In such patients, evidence of denervation in the short head of the biceps femoris and tibialis posterior muscles and abnormal sural or medial plantar SNAPs help localize partial proximal sciatic nerve lesions (Yuen and So, 1999). Occasionally the common fibular nerve gets injured selectively (Katirji and Wilbourn, 1994).
Piriformis Syndrome
On rare occasions, the piriformis muscle may entrap the sciatic nerve trunk as it passes through or over the piriformis muscle. Since the term was coined in 1947 by Robinson, the piriformis syndrome has been subject to controversy (Fishman and Schaeffer, 2003; Stewart, 2003). The syndrome fell out of fashion with the advancement of radiological techniques (myelography, CT, MRI) that often demonstrate that most patients with sciatica have nerve root compression and occasionally lesions of the sacral plexus or sciatic nerve at other locations. The typical patient with piriformis syndrome has a history of buttock trauma and experiences maximal buttock pain during prolonged sitting (e.g., driving, biking), bending at the waist, or activity that requires hip adduction and internal rotation (e.g., cross-country skiing) (Kirschner et al., 2009). The neurological and EDX examinations are usually normal. A bedside test maneuver in which the hip is placed passively in adduction, internal rotation, and flexion of the hip may reproduce the pain and is considered diagnostic. Imaging is usually normal but occasionally shows hypertrophy of the piriformis muscle or abnormal vessels or bands in the region of the piriformis muscle. MR neurography may show sciatic nerve hyperintensity at the sciatic notch, a more specific sign of nerve entrapment (Filler et al., 2005). Treatment consists of exercises that include prolonged stretching of the piriformis muscle by flexion, adduction, and internal rotation of the hip. CT- or MRI-guided corticosteroid injection into the piriformis muscle may alleviate the symptoms; a positive response is used as a confirmatory test. Surgical sectioning of the piriformis muscle is indicated in cases resistant to conservative therapy, with good outcome in carefully selected patients (Filler et al., 2005).
Common Fibular (Peroneal) Nerve
Common Fibular (peroneal) Neuropathy at the Fibular Neck
Compression of the common fibular nerve is the most frequent compressive neuropathy in the lower extremity. This nerve is particularly vulnerable to direct pressure in the region of the fibular neck as it passes through the origin of the peroneus longus muscle. Intraoperative compression due to improper positioning or padding during anesthesia is the leading cause of acute common fibular neuropathy at the fibular neck. Weight loss, habitual leg crossing, or unrecognized pressure on the nerve in hospitalized critically ill, debilitated, or unconscious patients may also be responsible for this nerve injury (Katirji, 1999). Devices that may compress the fibular nerve include casts, orthoses, pneumatic compression, antithrombotic stockings, bandages and straps. Fibular nerve stretch injury may result from an acute forceful foot inversion or prolonged squatting (strawberry pickers palsy). Blunt trauma (e.g., post fibular fracture, knee dislocation) and open injury (e.g., lacerations) account for a significant number of cases. Fibular nerve injury is also a known complication of knee surgery, including arthroscopic surgery and lateral meniscus repair. Up to half of patients without a clear cause of fibular mononeuropathy across the fibular head have intraneural ganglia (Young et al., 2009). These are formed when disruption of the capsule of the superior tibiofibular joint results in dissection of synovial fluid along the articular branch of the fibular nerve. Other mass lesions such as osteochondromas or schwannomas are much less common.
EDX studies are useful for localizing lesions and may provide clues to the underlying cause and a guide to prognosis. Although it is often possible by nerve conduction studies to demonstrate focal conduction block across the fibular head, contrary to common belief, the most frequent pathophysiological process is axonal loss, regardless of the cause (Katirji and Wilbourn, 1988). Axon-loss lesions reveal diffusely low or absent fibular motor and sensory amplitudes. In contrast to ulnar lesions across the elbow and CTS, localized slowing in the region of the fibular head is not common. Needle EMG demonstrates denervation in common fibular–innervated muscles but not in the short head of the biceps femoris (innervated by the common fibular division of the sciatic nerve in the thigh), in the L5 nerve root–innervated muscles, such as the tibialis posterior, flexor digitorum longus, tensor fascia lata and gluteus medius, or the low lumbar paraspinal muscles. MRI is effective in visualizing intraneural ganglia and other soft-tissue masses or tumors.
The prognosis is uniformly good in cases of acute demyelinating lesions, whereas recovery is delayed in those with axonal lesions and stretch injuries. The distal fibular motor amplitude recording (tibialis anterior) serves as an accurate estimate of the extent of axonal loss and a good prognostic indicator of foot drop. Hence, fibular nerve studies should be performed bilaterally and compared. Bracing with a custom-made plastic ankle-foot orthosis is necessary to improve the gait in the presence of severe foot drop. The few patients who do not improve spontaneously after 3 months, or those who have pain or a slowly progressive fibular nerve lesion, may require MRI studies and surgical exploration (Kim and Kline, 1996).
Sural Nerve Lesions
Although the vast majority of sural nerve lesions are iatrogenic as the result of diagnostic sural nerve biopsy or sural nerve harvesting for nerve grafts, mononeuropathy of the sural nerve has been reported with a number of other conditions including lower-limb vein-stripping surgery, Baker cyst or ankle joint surgery, local trauma, tightly laced high-topped footwear such as ski boots or ice skates, and rarely as the initial presentation of vasculitic mononeuritis multiplex (Stickler et al., 2006).
Femoral Nerve
Femoral Nerve Lesions
The majority of femoral nerve lesions are iatrogenic (Al-Hakim and Katirji, 1993). Pelvic lesions follow a variety of gastrointestinal, vascular, urological, or gynecological operations such as abdominal hysterectomy, radical prostatectomy, renal transplantation, and abdominal aortic repair. During these procedures, the femoral nerve becomes compressed between the lateral blade of the retractor and the pelvic wall; the incidence of these lesions is significantly reduced when self-retractors are avoided. Acute retroperitoneal hematoma is often iatrogenic following anticoagulant therapy, pelvic operations, or femoral vessel catheterization such as for cardiac catheterization. At the inguinal ligament, the femoral nerve may become kinked during lithotomy positioning, particularly when the leg is held in extreme hip flexion and external rotation, used during vaginal delivery, vaginal hysterectomy, prostatectomy, and laparoscopy. Total hip replacement, particularly surgical revisions and complicated reconstructions, may result in femoral nerve injury.
Needle EMG reveals denervation of the quadriceps muscle. The iliacus muscle is often normal in inguinal lesions but shows denervation in femoral nerve lesions in the pelvis. Needle EMG of the thigh adductor muscles, innervated by the L2, L3, L4 roots via the obturator nerve, helps distinguish femoral nerve lesions from upper lumbar radiculopathy or plexopathy. Nerve conduction studies have prognostic value, since the amplitude and area of the femoral CMAP is a very good quantitative measure of motor axonal loss (Kuntzer et al., 1997). CT or MRI of the pelvis are urgently indicated in patients with suspected retroperitoneal hematoma or pelvic mass lesion.
Other Lower-Extremity Mononeuropathies
Lateral Femoral Cutaneous Nerve Entrapment (Meralgia Paresthetica)
Lateral femoral cutaneous nerve SNAP is technically difficult to measure and may be absent in healthy subjects, particularly women and obese individuals. Asymmetrical low-amplitude or absent potential on the symptomatic side is a confirmatory finding. Electrophysiological studies of the femoral nerve and quadriceps femoris and iliacus muscles are normal, which helps exclude lumbar radiculopathy and plexopathy. A local anesthetic nerve block may have diagnostic value (Haim et al., 2006). Treatment consists of symptomatic measures such as rest, analgesics, and weight loss. Postural abnormalities should be corrected. Neurolysis is rarely beneficial.
Obturator Neuropathy
Entrapment produces radiating pain from the groin down the inner aspect of the thigh, often difficult to distinguish from the pain of a recent procedure or trauma. There is weakness of hip adduction and sensory impairment in the upper medial thigh. Many patients appear to have hip-flexor weakness as a false localizing sign. Although this phenomenon may be explained by pain, it is more likely due to mechanical disadvantage of the hip flexors in the presence of weak thigh adductors. CT or MRI scanning of the pelvis is helpful in finding primary or metastatic pelvic tumors. EMG testing is essential for diagnosis by detecting selective denervation of the thigh adductor muscles, with normal quadriceps and iliacus muscles, thus excluding other causes of hip weakness including femoral nerve lesions, upper lumbar (L2, L3 or L4) radiculopathy or plexopathy, and diabetic amyotrophy (diabetic proximal neuropathy) (Sorenson et al., 2002).
Migrant Sensory Neuritis of Wartenberg
In this rarely reported but not uncommon condition, a pure and relapsing-remitting sensory mononeuritis multiplex is associated with loss of sensation and pain in the distribution of the affected nerves. The onset is usually sudden, and pain is precipitated by movements and (especially) stretching of the affected limbs. Many different cutaneous nerves can be involved. Motor nerve fibers are not affected. Laboratory tests fail to detect any underlying cause, but on occasion a sural nerve biopsy demonstrates inflammatory changes or a vasculitis, with patchy loss of nerve fibers and evidence of axonal degeneration suggestive of an ischemic process. Rarely, immunoglobulin (Ig)G deposits are also observed around blood vessels. The pain and areas of sensory loss often recover over weeks to months, but the improvement may be partial. Symptoms may recur at the same or other sites. The condition is often misdiagnosed as other diseases such as multiple sclerosis, but the discrete areas of sensory deficit and nerve irritation in several cutaneous nerves should indicate the proper diagnosis. The differential diagnosis should always include conditions like diabetes mellitus, leprosy, vasculitis, sarcoidosis, sensory perineuritis, and rarely HNPP (Nicolle et al., 2001; Zifko and Hahn, 1997).
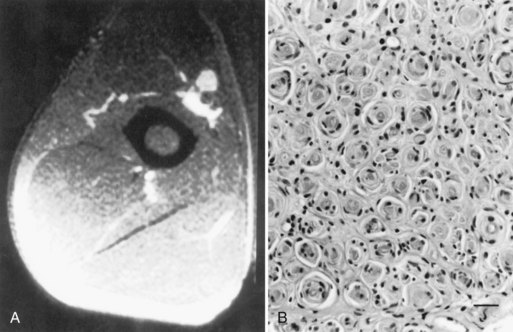
Localized Perineurial Hypertrophic Mononeuropathy
A slowly progressive painless mononeuropathy that cannot be localized to entrapment sites and is caused by a focal fusiform enlargement of the affected nerve, termed localized hypertrophic neuropathy or perineurioma, is an uncommon condition affecting young adults (Simmons et al., 1999). Although any nerve may be involved, it often occurs in the radial, posterior interosseous, tibial, and sciatic nerves. The fusiform enlargement is mainly composed of “onion bulblike whorls” formed by layers of perineurial cells (Fig. 76.7, B). The lamellae of the whorls stain for epithelial membrane antigen. The cause of the perineurial cell proliferation is unknown. It typically results in painless, slowly progressive weakness and atrophy in the distribution of the affected nerve. Sensory symptoms are minor, although sensory nerve fibers are obviously involved. EDX study results show an axonal mononeuropathy and help in the precise localization of the focal nerve lesion. MRI shows a focal enlargement of the affected nerve, increased signal on T2-weighted images, and enhancement with gadolinium (see Fig. 76.7, A).
Hereditary Neuropathies
The hereditary neuropathies constitute a complex heterogeneous group of diseases that usually share the clinical features of insidious onset and indolent course over years to decades. The number of hereditary disorders for which a metabolic or molecular defect is known is rapidly increasing, allowing a more accurate classification. For those inherited neuropathies for which the underlying genetic abnormality has yet to be identified, the classification still depends on the clinical phenotype, mode of inheritance, and class of neurons predominantly affected. Major advances in understanding the molecular basis of inherited neuropathies have come from identifying chromosomal loci or causative genes for a given disease phenotype, leading to identification of an ever-increasing number of genes coding for a specific gene product essential to myelin or axonal function (Berger et al., 2002; Kamholz et al., 2000; Lupski, 1998; Scherer, 2006).
Although hereditary neuropathies are common disorders, their inherited nature may go unrecognized in a surprisingly large percentage of patients (Klein, 2007). Eliciting historical evidence of long-standing neuromuscular symptoms; obtaining detailed family histories; looking for skeletal abnormalities such as hammer toes, high arches, or scoliosis; and more importantly, performing neurological evaluations in relatives of patients are essential in identifying a previously unsuspected inherited neuropathy. Because of the paucity of positive symptoms, patients may not volunteer information about their own or relatives’ conditions. For example, paresthesias are spontaneously reported three times more commonly in acquired than in inherited neuropathies. Even in the face of a truly negative family history, the possibility of an inherited neuropathy cannot be dismissed. Such a situation may arise in cases of early death of one or both parents, few blood relatives, or autosomal recessive disease. Also, available diagnostic deoxyribonucleic acid (DNA) testing has shown that about a third of isolated cases of inherited neuropathies may arise from de novo gene mutations (Boerkoel et al., 2002). It is advisable to consider the possibility of an inherited neuropathy in any patient with a chronic “acquired” neuropathy that remains cryptogenic or refractory to treatment.
Charcot-Marie-Tooth Disease (Hereditary Motor and Sensory Neuropathy)
The syndrome of peroneal muscular atrophy, or CMT disease, was first described in 1886 by Charcot and Marie in Paris and Tooth in London (Charcot and Marie, 1886; Tooth, 1886). CMT disease is the most common inherited neuropathy, with an estimated prevalence of 10 to 41 per 100,000 (Martyn and Hughes, 1997). Clinical studies combined with electrophysiological and sural nerve biopsy findings of a large number of families with peroneal muscular atrophy have allowed a separation into two main groups: (1) the demyelinating form, or CMT1 (hereditary motor and sensory neuropathy [HMSN-I]), in which there are marked reductions in motor NCVs and nerve biopsy findings of demyelination and onion bulb formation; and (2) the axonal form of CMT disease, or CMT2 (HMSN-II), in which motor NCVs are normal or near normal, and nerve biopsy reveals axonal loss without prominent demyelination (Harding, 1995). The peroneal muscular atrophy phenotype without sensory involvement on either clinical or electrophysiological examination has been classified as hereditary distal spinal muscular atrophy. A more severe form of demyelinating neuropathy with onset occurring in early childhood is referred to as Dejerine-Sottas disease.
Charcot-Marie-Tooth Disease Type 1
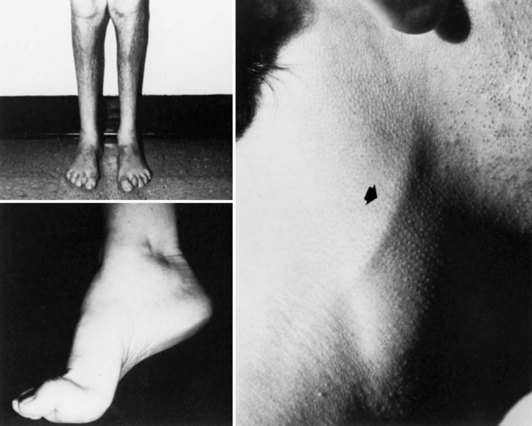
In CMT1, symptoms often begin during the first or second decade of life. It is characterized by slowly progressive weakness, muscular wasting, and sensory impairment predominantly involving the distal legs. Foot deformities and difficulties in running or walking resulting from symmetrical weakness and wasting in the intrinsic foot, peroneal, and anterior tibial muscles are often present. In two-thirds of patients, the upper limbs are involved later in life. Inspection reveals pes cavus and hammer toes in nearly three-quarters of adult patients, mild kyphosis in approximately a tenth, and palpably enlarged hypertrophic peripheral nerves in a quarter (Fig. 76.8). The foot deformities occur because of long-term muscular weakness and imbalance between the intrinsic extensor and long extensor muscles of the feet and toes (a similar process causes clawing of the fingers in more advanced cases). Absent ankle reflexes are universal and frequently associated with absent or reduced knee and upper limb reflexes. Some degree of distal sensory impairment (diminished vibration sense and light touch in the feet and hands) is usually discovered by examination but rarely gives rise to symptoms. Occasionally, patients have an essential or postural upper-limb tremor. Such cases have been referred to as Roussy-Lévy syndrome, but current evidence suggests that this is not a separate clinical or genetic entity.
Severity of neuropathy in affected family members varies considerably. Approximately 10% of patients with slowed NCVs may remain asymptomatic. In women with CMT1, the disease may exacerbate during pregnancy. Such worsening is temporary in about a third of patients but becomes progressive in the remainder. Slow deterioration in strength and decline in axonal function continues throughout adulthood, although much of this deterioration likely represents the effects of aging superimposed on decreased reserves (Verhamme et al., 2009).
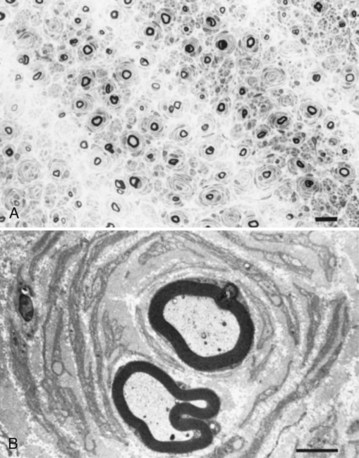
Motor nerve conduction studies show uniform slowing by more than 25% of the lower limits of normal in all nerves. Motor conduction of upper-limb nerves proves more useful than studies of lower-extremity nerves because distal denervation in the feet is often severe and virtually complete. A conduction velocity below 38 m/sec in the forearm segment of the median nerve is proposed as a cutoff value to distinguish between CMT1 and CMT2. Although this cutoff is useful, it can be misleading if applied too rigidly. SNAPs are usually absent with surface recordings. Routine hematological and biochemical studies are normal. CSF is also normal, which helps differentiate the condition from chronic inflammatory demyelinating polyneuropathy, in which the CSF protein is usually elevated. Sural nerve biopsy typically shows the changes of a hypertrophic neuropathy, characterized by onion bulb formation, increased frequency of fibers with demyelinated and remyelinated segments, an increase in endoneurial area, and loss of large myelinated fibers (Fig. 76.9).
Gene mutations, predominantly affecting genes for myelin and Schwann cell proteins, have been recognized that account for about three-quarters of families with CMT1 (see Chapter 40).
Charcot-Marie-Tooth Disease Type 2
CMT2 constitutes one-third of all autosomal dominant CMT disease. It is associated with mutations in genes affecting intracellular processes such as axonal transport, membrane trafficking, and translation (see Chapter 40). Clinical symptoms begin later than in CMT1, most commonly in the second decade, but may be delayed until middle age or beyond. Foot and spinal deformities tend to be less prominent than in CMT1. The clinical features closely resemble those of CMT1 but differ in that peripheral nerves are not enlarged, and upper limb involvement, tremor, and general areflexia occur less frequently. However, in individual cases, it is often impossible to determine the type of CMT disease on the basis of clinical manifestation alone. Approximately 20% of affected individuals are asymptomatic. CMT2A1, linked to chromosome 1p35, is caused by a mutation in kinesin protein involved in axonal transport of synaptic vesicles (Saito et al., 1997; Zhao et al., 2001). CMT2A2, which is responsible for most CMT2 families, shares clinical features of weakness and atrophy with CMT2A1 but has an earlier onset and is more severe. It may also be associated with optic atrophy. It is caused by mutations in the mitofusin 2 (MFN2) gene, with a locus on chromosome 1p36-p35. MFN2 protein is a mitochondrial fusion protein ubiquitously expressed in many tissues including peripheral nerves. In CMT2B, which is linked to chromosome 3q13-22, there is prominent sensory loss with foot ulcerations (De Jonghe et al., 1997). A mutation in the RAB7 gene, which encodes a small guanosine triphosphatase (GTPase) late endosomal protein, has been found to be causative (Verhoeven et al., 2003). This form of CMT is clinically very similar to hereditary sensory neuropathy type 1 (HSN1) but lacks spontaneous lancinating pain. Another distinct subgroup of severely affected patients, designated CMT2C (mapped to chromosome 12q24), develop vocal cord, intercostal, and diaphragmatic muscle weakness (Klein et al., 2003). Because of respiratory failure, the life expectancy of these patients is shortened. CMT2D, mapped to chromosome 7p14, is characterized by weakness and atrophy that is more severe in the hands than in the feet (Ionasescu et al., 1996b). In CMT2E, some patients within the same kindred and with an otherwise typical CMT2 phenotype may exhibit slowed motor nerve conduction that is much below the cutoff value of 38 m/sec and a more severe clinical phenotype. This form of CMT is caused by mutations in genes that encode neurofilament light (NEFL) subunit, and patients may have axonal swelling (giant axons) and significant secondary demyelination on sural nerve biopsies (Fabrizi et al., 2006; Jordanova et al., 2003). CMT2F, caused by mutations in small heat shock protein 27 (Hsp27), is characterized by later onset (35-60 years), mild sensory impairment, and moderate to severely slowed NCVs of lower limbs but normal or mildly reduced velocities in the upper limbs. Mutation in Hsp27 may impair formation of the stable neurofilament network that is essential for the maintenance of peripheral nerves. CMT2G, reported in a Spanish family, has the same gene locus as CMT4H (see later discussion on type 4 disease) on chromosome 12q12-q13.3, with the age onset from 9 to 76 years. CMT2J, also designated as CMT2 with MPZ (myelin protein zero) gene mutation, is associated with pupillary abnormalities (Adie pupil) and hearing loss. CMT2L is caused by mutation in the HSPB8 gene and is associated with otherwise typical features of the CMT2 phenotype.
X-Linked Charcot-Marie-Tooth Disease
X-linked Charcot-Marie-Tooth disease (CMTX) is phenotypically similar to CMT1. Affected male subjects tend to be more severely affected, and females with the gene mutation may have a mild neuropathy or be asymptomatic. No male-to-male transmission occurs. CMTX accounts for 7% to 16% of all forms of CMT, making it the second most common form of CMT. It is caused by many mutations in GJB1, the gene that encodes connexin 32 (Cx32). The connexins are a family of highly related genes encoding a group of channel-forming proteins. Cx32 is expressed in Schwann cells and oligodendrocytes, regions of noncompact myelin (incisures and paranodes), as well as other non-neural cells. Some mutations of Cx32 have been reported to be associated with central nervous system (CNS) involvement with white-matter MRI and MR spectroscopy abnormalities, abnormal brainstem auditory evoked potentials, and deafness (Murru et al., 2006). An interesting phenomenon of transient and acute ataxia, dysarthria, and weakness occurring after visiting high altitudes and associated with CNS white-matter MRI abnormalities has been described in patients with two mutations: R142W and C168Y (Paulson et al., 2002). This suggests that CMTX patients should be cautioned about travel to high-altitude locations. It has been proposed that Cx32 mutations may cause these abnormalities by reducing the number of functional gap junctions between oligodendrocytes and astrocytes, making them more susceptible to changes in intercellular ions and small-molecule exchange that occur in situations of metabolic stress (e.g., high altitude). Men with CMTX show significant slowing in NCV, whereas in heterozygous women, the slowing parallels the loss of CMAP amplitude. Brainstem auditory evoked responses are often abnormal. A picture of both axonal loss and demyelination is revealed on nerve biopsy.
Charcot-Marie-Tooth Disease Type 3, or Dejerine-Sottas Disease
Motor conduction velocities are severely slowed, often to less than 10 m/sec. The CSF protein is frequently increased. Pathologically, pronounced onion bulb changes are associated with hypomyelination and loss of myelinated fibers. Defective myelination is confirmed by an increased axon-to-fiber diameter ratio. Cases of congenital hypomyelination neuropathy probably represent a variant of CMT3 at the far end of a spectrum of defective myelination. DSS is genetically heterogeneous and is caused by different structural myelin protein and transcription factor gene mutations (see Chapter 40).
Charcot-Marie-Tooth Disease Type 4
Charcot-Marie-Tooth disease type 4 (CMT4) consists of several subgroups: CMT4A, CMT4B1 and 2, CMT4C, CMT4D, CMT4E, CMT4F, and CMT4H. All have autosomal recessive inheritance and are characterized by onset in early childhood and progressive weakness leading to inability to walk in adolescence. Each subgroup is rare and tends to be more common in certain inbred populations. Common to all these subgroups is a disturbance in normal myelination of the axons. Clinical and electrophysiological features are similar in several of these subtypes with CMT3. NCV studies are slowed (20-30 m/sec). CSF protein is normal. Nerve biopsy shows loss of myelinated fibers, hypomyelination, and onion bulbs. In CMT4B1, irregular folding and redundancy of loops of myelin are evident on nerve biopsies. Children affected with CMT4B2 also exhibit congenital glaucoma leading to loss of vision. CMT4C, characterized by frequent and severe scoliosis, is linked to chromosome 5q31-q33 and is caused by SH3TC2 gene mutation (Azzedine et al., 2006). CMT4D has onset in childhood but may progress into the fifth decade of life. It is associated with dysmorphic features and hearing loss. CMT4E is a form of congenital hypomyelinating neuropathy associated with mutations in PMP22 and ERG2 (early growth response) genes. These cases are often classified as CMT3. The phenotypic presentation of CMT4F is similar to that for CMT3, but the mutations occur in the periaxin gene, which produces a membrane-associated protein solely expressed in myelinating Schwann cells. CMT4H is similar to CMT2G in terms of genetic locus but is more severe clinically, with an onset in early childhood and prominent nerve hypomyelination.
Complex Forms of Charcot-Marie-Tooth Disease
Some dominant forms of CMT have displayed features intermediate between CMT1 and CMT2, with conduction velocities between 25 m/sec and normal. Although CMT2E and CMT1X meet this criterion, more recently these forms have been classified separately as dominant intermediate CMT (DI-CMT) and include types A, B, and C. The gene defect for DI-CMTA, found in a large Italian family, has not been discovered. DI-CMTB is caused by mutations in the dynamin 2 gene. This typically presents as a classic mild to moderately severe CMT phenotype. Some families with this variety have developed neutropenia and early cataracts (Claeys et al., 2009). A mutation in tyrosyl-tRNA (transfer ribonucleic acid) synthetase has been found to be the cause of DI-CMTC, which typically displays a mild, very slowly progressive course (Jordanova et al., 2006).
A number of families with peroneal muscular atrophy exhibit additional features such as optic atrophy, pigmentary retinal degeneration, deafness, and spastic paraparesis. Cardiac involvement is encountered in occasional patients, but prospective family studies find no association between cardiomyopathy and CMT disease. A syndrome of CIDP responding to prednisone and immunosuppression has been reported in patients with inherited CMT disease due to MPZ mutation (Watanabe et al., 2002), providing evidence that nongenetic factors may play a role in clinical expression of the mutant gene. It has been suggested that any patient with a hereditary neuropathy who suffers a recent rapid deterioration should be considered as having a secondary CIDP and be treated with immunosuppressants such as corticosteroids or high-dose intravenous immunoglobulin (IVIG).
Molecular Advances of Charcot-Marie-Tooth Disease and Related Disorders
Major advances have been made in recent years in the molecular genetics of CMT or hereditary motor and sensory neuropathies (Bennett and Chance, 2001; Berger et al., 2002; Kamholz et al., 2000) (Fig. 76.10; also see Chapter 40). Most CMT1 patients have DNA rearrangements on chromosome 17p11.2. A 1.5-megabase tandem duplication accounts for 70% of CMT1A cases. As the gene for peripheral myelin protein 22 (PMP22) lies within the CMT1A duplication, it results in three copies of the dosage-sensitive PMP22 gene. PMP22 is a membrane glycoprotein found in the compact portion of the peripheral myelin sheath. The precise function of PMP22 in normal nerve remains unknown. A minority of CMT1A patients have PMP22 missense mutations similar to the natural mouse mutants, trembler and trembler-J, which feature a severe hypomyelinating neuropathy. Deletion of the same 1.5-megabase region on chromosome 17p11.2 results in a single copy of the normal PMP22 gene, a finding observed in 85% of patients with HNPP. The CMT1A duplication or HNPP deletion is caused by reciprocal recombination events that occur in male germ cell meiosis. The PMP22 duplication or deletion can be detected in blood samples using pulse-field electrophoresis followed by hybridization with specific CMT1A duplication junction fragments or cytogenetic testing with a PMP22 probe by fluorescence in situ hybridization.
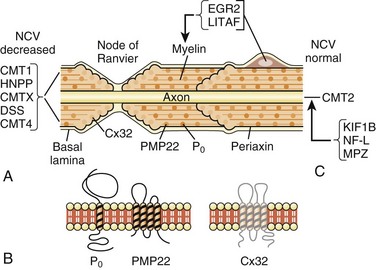
Myelin protein zero (P0; gene symbol, MPZ) is the major peripheral myelin glycoprotein and is thought to function as an adhesion molecule in the formation and compaction of peripheral myelin. It is a member of the immunoglobulin superfamily, with distinct extracellular transmembrane and intracellular domains. Mutations in the gene encoding for MPZ, located on chromosome 1q22-23, have been associated with CMT1B, DSS, and congenital hypomyelination neuropathy. Different MPZ mutations result in divergent morphological effects on myelin sheaths, consisting of uncompacting of myelin or focal myelin foldings (Gabreëls-Festen et al., 1996). Specific MPZ missense mutations have also been reported with a CMT2 phenotype, showing only mild slowing of NCVs (Marrosu et al., 1998). The Thr124 Met mutations in the MPZ gene have been detected in several families with a distinct CMT2 phenotype (CMT2J) characterized by late onset, marked sensory loss, and sometimes deafness, chronic cough, and pupillary abnormalities (De Jonghe et al., 1999).
The early growth response 2 gene (EGR2), located on chromosome 10q21-q22, encodes a zinc-finger transcription factor expressed in myelinating Schwann cells that regulates the expression of myelin proteins including PMP22, P0, Cx32, and periaxin (Kamholz et al., 2000). EGR2 gene missense mutations have been reported in patients with CMT1D, DSS, or congenital hypomyelination neuropathy (Timmerman et al., 1999; Warner et al., 1998). Respiratory compromise and cranial nerve dysfunction are commonly associated with EGR2 mutations (Szigeti, 2007).
Connexin 32 (Cx32) is a gap junction protein found in noncompacted paranodal loops and Schmidt-Lanterman incisures of Schwann cell cytoplasm, which is encoded by a four-exon gene located on chromosome Xq. As a gap junction protein, Cx32 forms small channels that facilitate transfer of ions and small molecules between Schwann cells and axons. Cx32 is also expressed in oligodendroglial cells in the CNS. This may explain the common abnormal brainstem auditory evoked potentials and other CNS abnormalities found in affected males. More than 200 different mutations in Cx32 have been identified in CMTX families. Genotype-phenotype correlations among patients with Cx32 mutations suggest that most missense mutations result in a mild clinical phenotype, whereas nonsense and frameshift mutations produce more severe phenotypes (Ionasescu et al., 1996a). Mutation analysis of Cx32 should be considered in any patient whose family history does not suggest a male-to-male transmission.
The designation CMT1C is reserved for autosomal dominant CMT1 families not linked to either CMT1A or CMT1B. Three such families showed linkage to chromosome 16p13. Missense mutations were found in the LITAF (lipopolysaccharide-induced tumor necrosis factor alpha) gene, which encodes a lysosomal protein that may play a role in protein degradation pathways (Street et al., 2003). Affected individuals in these families manifest characteristic CMT1 symptoms.
Periaxin is a cytoskeleton-associated protein that links the cytoskeleton of the Schwann cell with the basal lamina, a necessary function to stabilize the mature myelin sheath. Periaxin frameshift and nonsense mutations have been found in families with DSS and also those with autosomal recessive CMT4F (Takashima et al., 2002).
Point mutations of signal transduction proteins such as myotubularin-related protein 2 (MTMR2), N-myc downstream regulated gene 1 (NDRG1), and ganglioside-induced differentiation-associated protein 1 (GDAP1) are found in severe childhood-onset, autosomal-recessive demyelinating neuropathies or CMT4. Mutations of MTMR2 give rise to a severe demyelinating neuropathy characterized by redundant loops of focally folded myelin (Houlden et al., 2001b). GDAP1 mutations may be the most common cause of autosomal recessive CMT and can result in demyelinating as well as axonal phenotypes (Nelis et al., 2002).
In general, DNA rearrangements such as PMP22 duplication or deletion result in milder clinical phenotypes than point mutations for structural myelin proteins or the transcription factor gene, EGR2. Most known mutations cause disease manifestations in the heterozygous state, behaving as either gain-of-function or loss-of-function dominant mutations. Rare recessive mutations in PMP22, EGR2, MTMR2, NDRG1, and periaxin have been reported (Lupski, 2000). DNA sequencing tests are available for PMP22, Cx32, MPZ, EGR2, and periaxin in commercial laboratories.
CMT can also be classified by mode of inheritance (autosomal dominant, X-linked, and rarely autosomal recessive), chromosomal locus, or causative genes (Table 76.6). The classification remains fluid and may change as experts alter and revise these designations based on new molecular findings. An up-to-date listing of loci and genes can be found at the CMT mutation database (www.molgen.ua.ac.be/cmtmutations). DSS, formerly CMT3, may no longer be a useful designation because it is genetically heterogeneous, caused by different structural myelin protein and transcription factor gene mutations. One proposal is to reserve CMT3 for rare axonal types of autosomal recessive neuropathy (Vance, 2000).
Reappraisal of Electrophysiological Studies of Inherited Demyelinating Neuropathies
Lewis and colleagues revisited the topic of electrophysiological studies in inherited demyelinating neuropathies and correlated conduction changes with molecular diagnoses (Lewis et al., 2000). Uniform conduction slowing is found in CMT1A with PMP22 duplication or point mutations; CMT1B with MPZ point mutations; DSS, including PMP22, MPZ, and EGR2 gene mutations; metachromatic leukodystrophy (MLD); Cockayne disease; and globoid cell leukodystrophy. In DSS, severe nerve conduction slowing below 10 m/sec is consistently found. Temporal dispersion and amplitude reduction on proximal stimulation may be found in such cases, owing to high electrical stimulation thresholds in hypertrophic nerves.
Motor conduction block can be found in rare CMT1B patients with specific MPZ mutations (Street et al., 2002). Multifocal conduction slowing is typically seen in HNPP, CMTX, adrenomyeloneuropathy, Pelizaeus-Merzbacher disease with proteolipid protein-null mutation, and Refsum disease.
Nerve conduction velocities in CMTX with Cx32 mutations range from near normal to intermediate slowing in the 30- to 40-m/sec range. There is debate as to whether CMTX should be classified as a primary axonal or demyelinating disorder (Birouk et al., 1998). However, careful studies of individual patients suggest nonuniform conduction slowing consistent with demyelination (Gutierrez et al., 2000; Lewis, 2000). Conduction changes in affected women can be subtle and frequently are in the range found in patients with CMT2. Before considering a diagnosis of CMT2 in such cases, it is important to review the family history. If there is no male-to-male transmission, the presence of intermediate conduction velocities (>42 m/sec) in female carriers and delayed brainstem auditory evoked response latencies in affected men is highly suggestive of Cx32 mutations (Nicholson et al., 1998). In certain mutations of MPZ, NEFL, and GDAP1 genes, either a demyelinating or an axonal phenotype may be seen.
Practical Molecular Diagnostic Testing for Patients with Charcot-Marie-Tooth Disease and Related Disorders
Molecular diagnostic testing should be considered in CMT and related peripheral neuropathies. Commercial reference laboratories can detect point mutations or PMP22 duplication/deletion by DNA sequencing of PMP22, Cx32, MPZ, EGR2, periaxin, GDAP1 and NEFL, among others, in samples of peripheral blood (Table 76.7). It is, however, advisable to avoid the temptation to obtain an all-inclusive “battery” of available genetic tests. In families with at least two generations with the disease, known male-to-male transmission, and uniform conduction slowing, the PMP22 duplication test should be obtained and, if normal, followed by PMP22 and MPZ DNA sequencing. Patients who have neither the PMP22 duplication nor male-to-male transmission should be screened for Cx32 mutations. For those patients displaying an axonal pattern, the MFN2 mutation should be investigated first, as this accounts for 33% of cases of CMT2 (England et al., 2009). Given the high spontaneous mutation rate, the diagnoses of CMT1A and HNPP should be considered even in the absence of a positive family history. Because of the severe reactions to vincristine and other chemotherapeutic neurotoxic drugs in CMT patients, before initiating cancer chemotherapy, it is best to rule out CMT1A in any patient with either unexplained chronic neuropathy or a family history of neuropathy (Graf et al., 1996).
DNA sequencing tests for Cx32, MPZ, and NEFL mutations are available for selected patients with the axonal CMT2 phenotype. The PMP22 duplication test followed by DNA sequencing of PMP22, MPZ, EGR2, and periaxin should be considered in childhood cases with severe demyelinating neuropathy suggestive of DSS or congenital hypomyelination neuropathy. Information about available clinical gene tests can be found at www.genetests.org.
Treatment and Management
The rates of progression of CMT1 and CMT2 are slow, disability occurs relatively late, and lifespan may be normal. Management is mainly symptomatic. Patients should be instructed in proper foot care and advised to wear broad, well-fitting shoes. Insoles may be used to distribute body weight more evenly in patients with a foot deformity. Ankle-foot braces or orthopedic procedures are indicated to correct severe footdrop. Patients should be warned to avoid neurotoxic drugs because of greater susceptibility to agents such as vincristine. Issues like genetic counseling, family planning, prenatal diagnosis, and psychological concerns must be carefully approached, preferably by a multidisciplinary team including a genetic counselor. Recent studies suggest that CMT is associated with higher risk for complication during delivery (Hoff et al., 2005). Also, during pregnancy the symptoms of CMT may worsen.
Despite astonishing advances in the molecular genetics of CMT, there is still no effective treatment available for any form of the disease. Rats overexpressing PMP22 worsen with progesterone administration. This has led to proposed use of progesterone antagonists for CMT1A, though potentially unacceptable side effects have prevented such trials. The same animal model showed a convincing clinical and pathological improvement following administration of ascorbic acid, a known promoter of myelination. This prompted a multicenter double-blind placebo-controlled study on the use of ascorbic acid at 1 or 3 g/day in CMT1A. Ascorbic acid was found to be well tolerated, but no significant benefit was demonstrated in the CMT neuropathy score at 12 months (Micallef et al., 2009). The possible beneficial role of neurotrophins, particularly neurotrophin 3 (NT3), in CMT1A and nerve regeneration has recently been demonstrated in a small pilot study (Sahenk et al., 2005). Some CMT2 patients with MPZ mutations may respond to corticosteroids (Donaghy et al., 2000).
Hereditary Neuropathy with Liability to Pressure Palsies
HNPP is an autosomal dominant disorder of peripheral nerves leading to increased susceptibility to mechanical traction or compression. It occurs with an estimated prevalence of 16 per 100,000 population. Patients have recurrent episodes of isolated mononeuropathies, typically affecting, in order of decreasing frequency, the fibular nerve, ulnar nerve, brachial plexus, radial nerve, and median nerves. Painless brachial plexopathy is seen in up to a third of patients. Recurrent plexopathy can also be caused by another condition, hereditary neuralgic amyotrophy, which is often preceded by severe ipsilateral limb pain. Most HNPP patients experience the initial episode in the second or third decade of life. Attacks usually are provoked by compression, slight traction, or other minor trauma. Most episodes are of sudden onset, painless, and usually followed by complete recovery within days or weeks. Less-common presentations include transient positionally induced sensory symptoms, progressive mononeuropathy, chronic sensory polyneuropathy, CMT phenotype with pes cavus, and a diffuse chronic sensorimotor neuropathy resembling chronic inflammatory demyelinating polyneuropathy (Mouton et al., 1999). About 15% of mutation carriers remain asymptomatic.
Nerve conduction studies in patients with HNPP associated with PMP22 deletion typically demonstrate a characteristic pattern of prolonged distal motor latencies with only mild slowing in forearm segments of median and ulnar nerves, focal slowing of median, ulnar, and fibular nerves at compression sites, and diffuse reduction of SNAP amplitudes (Dubourg et al., 2000; Mouton et al., 1999). The median motor nerve conduction is typically above 38 m/sec, but sensory studies demonstrate velocities in the demyelinating range and reduced or absent SNAPs. Prolonged median distal motor latencies and abnormal sensory conduction studies are frequently found in asymptomatic carriers (Infante et al., 2001).
Sural nerve biopsy specimens demonstrate focal sausagelike thickenings of myelin termed tomacula (Fig. 76.11), segmental demyelination, and axonal loss.

Linkage studies show a 1.5-megabase deletion of chromosome 17p11.2-12 that includes the PMP22 gene and corresponds to the duplicated region in CMT1A in 85% of affected patients with HNPP. The remaining patients have a variety of mutations in PMP22 that lead to frameshift or nonsense mutations causing functional changes in the protein (Lenssen et al., 1998; van de Wetering et al., 2002). Exactly how the deletion of PMP22 causes HNPP remains unclear. One small study of PMP22 expression in dermal nerve myelin from patients with CMT1A or HNPP as well as healthy controls found that levels of PMP22 were highly variable in patients with CMT1A but not in patients with HNPP or in healthy controls, although most patients with HNPP did exhibit PMP22 overexpression. The investigators’ hypothesis was that the variability in PMP22 levels plays an important role in the differences in pathology and disability seen in CMT1A versus HNPP (Katona et al., 2009). Molecular diagnosis of the 17p11.2 deletion is available in reference laboratories and has replaced nerve biopsy for the diagnosis of HNPP. Testing should be considered regardless of family history in any patient presenting with painless multiple mononeuropathies, brachial plexopathy, or recurrent demyelinating neuropathy (Tyson et al., 1996). The primary treatment strategy is to prevent nerve injury by avoiding pressure damage.
Giant Axonal Neuropathy
Giant axonal neuropathy (GAN) is a rare autosomal recessive multisystemic neurodegenerative disorder of intermediate filaments affecting the peripheral and CNS. GAN presents as a slowly progressive axonal sensorimotor neuropathy in early childhood and leads to death by late adolescence. Most affected children have tightly curled hair and distal leg weakness. Some develop a peculiar gait disturbance with a tendency to walk on the inner edges of the feet (Fig. 76.12). With disease progression, evidence of CNS involvement occurs, including optic atrophy, nystagmus, cerebellar ataxia, upper motor neuron signs and intellectual decline, and abnormal visual, auditory, and somatosensory evoked potentials. MRI of the brain demonstrates cerebellar and cerebral white-matter abnormalities. Nerve conduction studies show reduced CMAP and SNAP amplitudes with normal to only slightly reduced conduction velocities. Sural nerve biopsy demonstrates the pathognomonic changes of large focal axonal swellings that contain densely packed disorganized neurofilaments (Fig. 76.13). Axonal function and axoplasmic transport are impaired. The disease locus was mapped to chromosome 16q24. GAN is caused by mutations of the GAN gene, which encodes a novel protein called gigaxonin (Bomont et al., 2000; Kuhlenbäumer et al., 2002). Gigaxonin is a ubiquitously expressed cytoskeletal protein and a member of the BTB/kelsch superfamily, which is involved in diverse cellular functions and may be important in the cross-linking of intermediate filaments (Herrmann and Griffin, 2002).
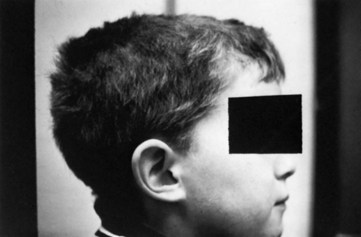
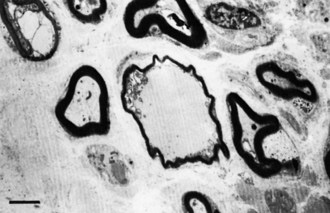
Hereditary Sensory and Autonomic Neuropathy
Hereditary sensory and autonomic neuropathies (HSANs) are a clinically and genetically heterogenous group of neuropathies characterized by prominent sensory loss and variable autonomic features but without significant motor involvement (Verhoeven et al., 2006). Currently these neuropathies are divided into five main groups based on the inheritance, clinical features, and type of sensory neurons involved (Table 76.8). Compared with CMT, HSANs are distinctly rare. Molecular genetic studies have identified seven disease-causing gene mutations in these disorders. More genetic discoveries are likely forthcoming; a study of 100 patients with familial HSAN and isolated patients diagnosed with HSAN found that only 14% of the isolated cases and 31% of the familial cases were found to have mutations in the known causative genes (Rotthier et al., 2009). The pronounced sensory loss in HSAN predisposes these patients to unnoticed recurrent trauma, leading to neuropathic (Charcot) joints, nonhealing ulcers, infections, and osteomyelitis resulting in acral mutilations (acrodystrophic neuropathy). These complications are preventable by careful avoidance of trauma to the insensitive extremities.
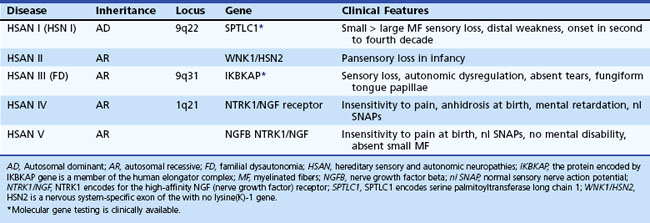
Hereditary Sensory and Autonomic Neuropathy Type I
HSAN type I is an autosomal dominant disorder and the most common hereditary sensory neuropathy (Houlden et al., 2006). Symptoms begin in the second to fourth decade with sensory loss and subsequent tissue injury mainly affecting the feet and legs. Sensory loss initially affects pain and temperature perception more than touch-pressure sensation, but it involves all modalities as the disease progresses. Autonomic involvements are limited to hypohidrosis. Patients present with calluses on the soles, painless stress fractures of the feet, neuropathic foot and ankle joints, and recurrent plantar ulcers. If ulcers are neglected and become infected, severe acromutilation may result (acrodystrophic neuropathy) (Fig. 76.14). Lancinating or shooting pains are often prominent and are considered the hallmark feature of HSAN-I. Distal muscle weakness and wasting are present in advanced cases. Such motor involvement, albeit without the lancinating pain, may create diagnostic confusion with CMT2B with dense sensory loss (linked to chromosome 3q13) or with certain axonal MPZ gene mutations. Variable neural hearing loss or rarely spastic paraparesis may be seen in HSAN-I. Some families present with burning feet or neurogenic arthropathy, suggesting clinical as well as genetic heterogeneity both within and between families with HSAN-I (Dyck et al., 2000a; Houlden et al., 2006).
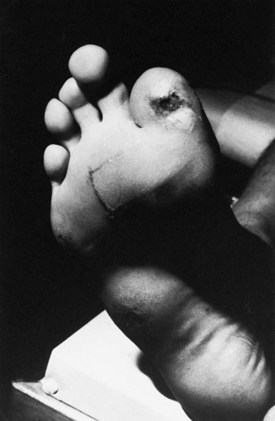
SNAP amplitudes are reduced late in the disease. Motor conduction velocities remain normal, but CMAP amplitudes are reduced in advanced cases. Sural nerve biopsy confirms a severe loss of small myelinated axons and, to a lesser degree, of unmyelinated and large myelinated fibers (Fig. 76.15). Pathological evidence of regenerative activity is usually minimal. HSAN-I has been mapped to chromosome 9q22.1-22.3. Mutations in the SPTLC1 gene encoding a subunit of serine palmitoyltransferase are identified in 90% of patients (Dawkins et al., 2001). These mutations result in increased de novo ceramide synthesis. Since ceramide plays a role in the regulation of programmed cell death, the neuronal degeneration in HSAN-I may be caused by ceramide-induced apoptosis. Molecular genetic testing for HSAN-I is clinically available. Families without linkage to chromosome 9q22 have been described, suggesting genetic heterogeneity (Auer-Grumbach et al., 2000). A subtype, HSAN-IB, is associated with paroxysmal cough, cough syncope, and gastroesophageal reflux (Spring et al., 2005). Additional features include hoarseness of voice and hearing deficit; motor involvement, acral mutilations and ulceration are usually absent. The association of cough and gastroesophageal reflux is not unique to HSAN-IB, as it has also been associated with CMT2 with MPZ mutation.
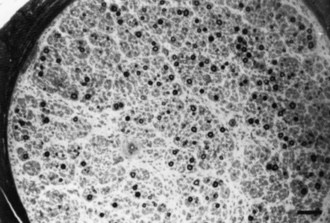
Hereditary Sensory and Autonomic Neuropathy Type II
HSAN-II is recessively inherited and rarely begins later than infancy. All sensory modalities of distal upper and lower limbs and, to a lesser extent, of trunk and face are affected. The hands, feet, lips, and tongue are at risk for mutilation because of generalized sensory loss and insensitivity to pain. Autonomic symptoms are minimal, and mental development is normal. There is loss of tendon reflexes. Rarely, associations with spastic paraplegia, retinitis pigmentosa, mild motor weakness, or neurotrophic keratitis have been described. The clinical course is slowly progressive, with progressive axonal loss. SNAPs are absent. Sural nerve biopsy specimens show almost complete absence of myelinated fibers and reduced unmyelinated fiber populations (Fig. 76.16). Mutations in the HSN2 nervous-system-specific exon of the with-no-lysine(K)-1 (WNK1) gene on chromosome 12q13.33 causes HSAN-II (Shekarabi et al., 2008). All mutations result in a truncation of the HSN2 protein, with the protein loss or inactivation (or both) causing the peripheral neuropathy. The exact function of HSN2 protein remains unknown, but it may play a role in the development or maintenance of sensory neurons or accompanying Schwann cells.
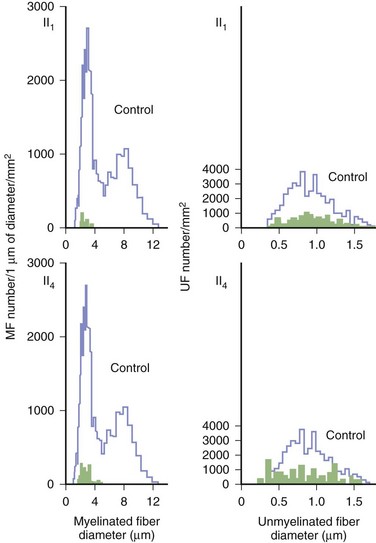
Hereditary Sensory and Autonomic Neuropathy Type III (Riley-Day Syndrome)
Motor NCVs are generally normal, whereas SNAP amplitudes are frequently reduced. A marked reduction in the density of unmyelinated axons and small myelinated fibers is seen in sural nerve biopsy specimens, even in the youngest patients. The number of neurons in the sympathetic, parasympathetic, and spinal ganglia is reduced. Peripheral blood vessels also demonstrate lack of autonomic nerve terminals. Linkage studies have mapped the gene locus to chromosome 9q31-q33. The diagnosis is established by molecular genetic testing of the IKBKAP gene, which encodes an essential protein named IKAP of the human elongation complex. The major FD mutation is a splice mutation that results in aberrant tissue-specific messenger (m)RNA splicing (Slaugenhaupt and Gusella, 2002). The elongator complex is thought to be involved in the regulation of cell-surface transport of exocytosis, and its impairment results in the dysregulation of neural endocytosis.
FD is a potentially life-threatening disorder with a high mortality rate due to aspiration pneumonia or autonomic crises. Improved supportive treatment has extended the survival of patients into adulthood. There is a greater than 50% probability for infants with familial dysautonomia to reach 30 years of age (Gold-Von Simson and Axelrod, 2006).
Hereditary Sensory and Autonomic Neuropathy Type IV
HSAN-IV is a rare autosomal recessive disorder characterized by congenital insensitivity to pain and anhidrosis, leading to repeated episodes of fever, thick and calloused skin, dystrophic nails, self-mutilating behavior, and mild mental retardation associated with emotional lability and hyperactivity. Tendon reflexes, muscle strength, and SNAPs are preserved, but sympathetic skin responses are absent. Biopsy of sensory nerves shows selective total or near-total loss of unmyelinated axons and small myelinated fibers. Confirmation of a neuropathic abnormality in cases of congenital indifference to pain without apparent neurological signs therefore depends on the morphometric study of unmyelinated and myelinated fiber populations in nerve biopsy specimens, and is supported by quantitative sensory testing and lack of sweating by the quantitative sudomotor axon reflex test. Intradermal histamine injection produces a wheal but no flare response. Skin biopsy has demonstrated a lack of intradermal nerve fibers and sweat glands devoid of nerve fibers (Verzé et al., 2000). The gene locus for HSAN-IV maps to chromosome 1q21-22. Mutations in the NTRK1 (formerly trkA) gene encoding the tyrosine kinase receptor for nerve growth factor (NGF) have been described in patients with HSAN-IV (Indo, 2002). These findings indicate that the NGF-trkA system plays a crucial role in the development of unmyelinated nociceptive and sudomotor fibers.
Clinically similar cases with selective loss of only small myelinated fibers have been designated HSAN type V. These patients do not have mental retardation. Mutations in the NTRK1 gene have been found in some cases, suggesting that the two disorders may be allelic (Houlden et al., 2001a). A related disorder with loss of deep pain perception but normal sweating has been linked to chromosome 1p13.2-p11.2, encoding the NGFB gene (Minde et al., 2004).
Neuropathy Associated with Spinocerebellar Ataxias
Friedreich ataxia is an autosomal recessive neurodegenerative disease characterized by degeneration of large sensory neurons and spinocerebellar tracts, cardiomyopathy, and increased incidence of diabetes (see Chapter 72). Even in the early stages of the disease, examination reveals lower-limb areflexia and impaired joint position and vibration sense, with preserved pain and temperature sensation. Pes cavus and hammer toes occur in approximately 90% of cases.
Motor NCVs are normal or slightly reduced, and SNAPs are invariably reduced or absent. A selective loss of large myelinated fibers occurs in the sural nerve. Friedreich ataxia is the result of a large GAA triplet repeat expansion on chromosome 9q13-q21.1, leading to loss of frataxin expression (see Chapter 72).
Peripheral nerve involvement has been found in association with other spinocerebellar ataxias (SCAs), most notably SCA3 in older patients, SCA4, SCA18, and prominently in SCA25. An autosomal recessive SCA and peripheral neuropathy with raised serum α-fetoprotein unrelated to ataxia telangiectasia has been reported (Izatt et al., 2004).
Primary Erythromelalgia
Inherited or primary erythromelalgia is a rare autosomal dominant neuropathic condition characterized by recurrent attacks of erythema and intense burning pain of the hands and feet in response to mild warmth or moderate exercise. To get relief, patients often attempt to cool the affected areas. These symptoms may lead to significant disability. Patients function normally between episodes. Mutations in the voltage-gated sodium channel Na(v)1.7, encoded by the gene SCN9A, is responsible for this syndrome. Na(v)1.7 is preferentially expressed in small dorsal-root ganglia neurons. Its mutations result in the increased activity of dorsal root ganglion neurons and enhanced sensation of pain (Choi et al., 2006; Waxman, 2007). Other mutations in SCN9A that cause a loss of Na(v)1.7 function result in the opposing condition of “congenital inability to experience pain,” emphasizing the importance of sodium channels in nociception (Cox et al., 2006). A third disorder has also been linked to Na(v)1.7: paroxysmal extreme pain disorder, which was formerly known as familial rectal pain syndrome (Fertleman et al., 2007). The linkage between pain and temperature in erythromelalgia is in keeping with other diseases of sodium channels. No treatment is consistently effective. Aspirin, lidocaine (gel, patch, or infusion), anticonvulsants, tricyclic antidepressants, and vasoactive drugs have been used with varying success. Paroxysmal extreme pain disorder almost universally responds to carbamazepine, although the response is often incomplete.
Familial Amyloid Polyneuropathy
Familial amyloid polyneuropathy (FAP) is a group of autosomal dominant disorders characterized by the extracellular deposition of amyloid proteins in peripheral nerves and other organs. Amyloid is a fibrillar conformation of a protein characterized by (1) green birefringence of Congo red–stained sections viewed in polarized light, (2) the presence of nonbranched 10-nm amyloid fibrils on electron microscopy, and (3) a β-pleated sheet structure on x-ray diffraction. More than 20 different proteins are known to undergo conformational changes leading to the generation of amyloid deposits in tissues. These deposits may be widespread or restricted to certain organs (localized amyloidosis). The clinical presentation depends on the organs involved and the size of amyloid fibrils. In FAP, one of three aberrant proteins (transthyretin, apolipoprotein A1, or gelsolin [Falk et al., 1997]) can be found in the peripheral nerves. The latter two are rare and restricted to a few families. In acquired primary systemic amyloidosis, polypeptides of immunoglobulin light-chain origin are deposited in tissues as amyloid, which leads to the term AL amyloid (see Primary Systemic Amyloidosis, later).
The classification of FAP was traditionally based on clinical presentation. Progress in understanding the protein composition and molecular genetics of these disorders justifies a different approach (Table 76.9).
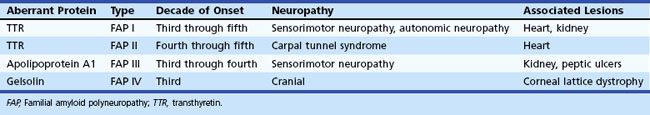
Transthyretin Amyloidosis (Familial Amyloid Polyneuropathy Types I and II)
TTR-related amyloid polyneuropathies demonstrate two disparate clinical phenotypes. The original cases described by Andrade in Portugal are referred to as FAP type I. Two other large foci of patients are found in Sweden and Japan. This is the most common form of FAP and has been observed in 30 different countries and in many ethnic groups (Ando et al., 2005). The neuropathy begins insidiously in the third and fourth decades with dissociated sensory impairment (loss of pain and thermal sense) in the lower extremities, often associated with lancinating pain and paresthesias. Because of its similarity to other neuropathies, it may be difficult to diagnose this condition (Ikeda, 2007; Plante-Bordeneuve et al., 2007). Autonomic dysfunction commonly includes impotence, postural hypotension, bladder dysfunction, distal anhidrosis, and abnormal pupils with scalloped margins. Gastrointestinal symptoms characterized by constipation alternating with diarrhea, delayed gastric emptying, and weight loss may be prominent. Eventually panmodality sensory loss, distal wasting, weakness, and areflexia develop. Systemically, amyloid is deposited in the ocular vitreous, heart, and kidneys. The disorder is relentlessly progressive. Patients usually die of cardiac or renal failure or malnutrition 10 to 15 years after the onset.
More than 90 different amino acid substitutions of the TTR protein have been identified as causing the clinical FAP-I and FAP-II phenotypes. Among these, substitution of valine by methionine at position 30 (Val30Met) is by far the most frequent and found in clusters in distinct areas of Portugal, Sweden, and Japan. Other TTR variants including isoleucine 33, alanine 60, and tyrosine 77 substitutions have similar features of generalized polyneuropathy with varying degrees of autonomic involvement. A serine substitution at position 84 and histidine at position 58 are the two TTR mutations seen most commonly in FAP-II. The age of onset varies greatly with specific TTR mutations. Even in families with the Val-30Met mutation, variation in age of onset is observed in different geographic regions (Ikeda et al. 2002). Specific TTR mutations may produce unique phenotypes with predominantly cardiac or leptomeningeal amyloidosis (Hund et al., 2001).
Apolipoprotein A1 Amyloidosis (Familial Amyloid Polyneuropathy Type III, Van Allen)
The clinical manifestations of the type III variant have much in common with those of type I, except for early renal involvement and a high incidence of duodenal ulcers. Uremia is the most common cause of death, typically occurring 12 to 15 years after the onset of neuropathy. Sixteen mutations of apolipoprotein A1 are known to cause FAP. Those involving codons 1 to 75 more commonly cause hepatic and renal amyloidosis, while those involving codons 173 to 178 cause cardiac, laryngeal, and cutaneous amyloidosis (Eriksson et al., 2009).
DNA Diagnosis of Familial Amyloid Polyneuropathy
The diagnosis of FAP requires several steps. Initially, the diagnosis is established by confirming the presence of amyloid in nerve and muscle, rectal biopsies, a sample of subcutaneous abdominal fat, or any other tissues that had been removed at the time of unrelated procedures. Sampling error, however, confounds these tests. Nerve biopsies have shown 83% sensitivity and salivary gland biopsies, 67% (Plante-Bordeneuve et al., 2007). Immunostaining using specific antibodies against one of the three aberrant proteins may identify the responsible protein. In sporadic cases, the more common AL amyloidosis should be excluded by a search for clonal plasma cell dyscrasia (see Primary Systemic Amyloidosis, later). If no evidence of plasma cell dyscrasia exists, TTR can be identified by isoelectric focusing of the serum, which separates variant and wild-type TTR. The finding of a variant TTR should prompt genetic testing. DNA isolated from peripheral leukocytes or tissue can be amplified with the polymerase chain reaction and specific oligonucleotide primers used to amplify regions of the gene of interest, thereby demonstrating specific point mutations. DNA testing for the most common TTR mutations (Met-30, lle-33, Ala-60, Tyr-77, Ser-84) is available in reference laboratories. A negative result, however, does not exclude a TTR mutation. Mutation analysis of the entire TTR gene detects more than 99% of amyloidogenic mutations. Genetic testing has become the most reliable form of testing for FAP and should be considered in appropriate patients with cryptogenic progressive axonal neuropathy affecting small-fiber nerves in a length-dependent fashion and the autonomic nervous system.
Treatment
The prognosis of TTR amyloidosis varies with the specific mutation, age of onset, and organ involvement. Because there is no specific treatment, supportive measures are essential for both the neuropathy and specific organ system involved, including cardiac pacing, hemodialysis, parenteral nutrition, and physical therapy. Because over 90% of TTR is synthesized in the liver, orthotopic liver transplantation has been proposed as definite therapy for this disorder. Transplantation results in rapid clearance of the variant TTR from serum; it may halt progression of neurological deficits in patients with mild neuropathy and stop the rate of axonal degeneration; autonomic dysfunction remains largely unchanged (Adams et al., 2000; Stangou and Hawkins, 2004). The reported 5-year survival rate after liver transplantation is approximately 80%. Overall, 60% stabilize, 20% improve, and 20% do poorly. Liver transplantation is recommended for patients who are younger than 50 years, have mild neuropathy (walking unaided), and have no significant cardiac or renal involvement. Transplantation is not effective in cardiac or leptomeningeal amyloidosis. In some cases of non-Val30Met mutation, despite liver transplantation, the amyloidogenic TTR continues to be deposited in the heart and vitreous, based on the existing “template” of amyloid (Stangou et al., 1998). Combined liver and renal transplantation has been performed in a few patients with severe renal involvement. In patients with TTR Tyr77 mutation, progressive neuropathy and cardiomyopathy may continue after liver transplantation. Combined liver and heart transplantation in one patient with the same mutation resulted in mild improvement of the peripheral neuropathy. Alternative treatment modalities with agents that stabilize the variant TTR and inhibit fibril formation are under investigation.
Porphyric Neuropathy
Acute hepatic porphyrias are a group of autosomal dominant inherited metabolic disorders that manifest as acute or subacute severe life-threatening motor neuropathy, abdominal pain, autonomic dysfunction, and neuropsychiatric manifestations (Albers and Fink, 2004). Its gene is thought to be present in 1 in 80,000 people, although only one-third of affected persons ever manifest symptoms of the disease. The main forms of porphyria include AIP, variegate porphyria, and hereditary coproporphyria. The basic defects are a 50% reduction in hydroxymethylbilane synthase (also known as porphobilinogen deaminase) in AIP, of protoporphinogen IX oxidase in variegate porphyria, and of coproporphinogen oxidase in coproporphyria, all of which result in abnormalities of heme synthesis. Each abnormality of enzyme activity provokes a compensatory overproduction of porphyrins and their precursors through the negative feedback regulation by heme of the first and rate-limiting enzyme of the heme biosynthetic pathway, δ-aminolevulinic acid synthase (ALAS). A fourth disorder referred to as plumboporphyria is inherited as an autosomal recessive trait and is caused by a deficiency of δ-aminolevulinic acid dehydratase (ALAD) (Table 76.10). More than 90 mutations in the porphobilinogen deaminase (PBGD) gene have been identified that decrease enzyme activity and cause AIP (Elder et al., 1997). These partial enzyme defects remain latent until precipitating factors trigger acute attacks. Precipitating factors include certain drugs, the menstrual cycle, alcohol, hormones, and fasting (either intentional or during an intercurrent illness). Precipitating factors induce hepatic δ-ALAS, the rate-limiting enzyme in heme biosynthesis, leading to the overproduction and overexcretion of PBG and δ-ALA.
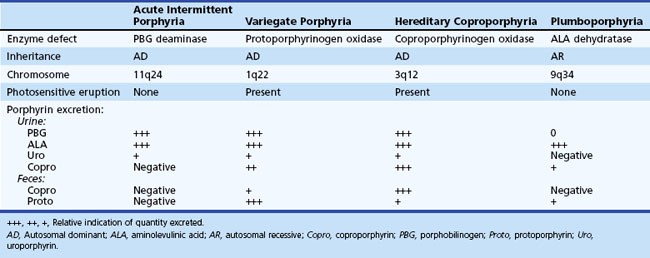
Clinical Features of the Acute Porphyric Attack
The neurological manifestations of all forms of the acute porphyrias are identical. All clinical symptoms can be explained by the dysfunction of the autonomic nervous system, peripheral nervous system (PNS), and CNS. Characteristically, porphyric attacks first occur during the third and fourth decades of life and are five times more common and severe in women than men. The most frequent presenting symptoms are abdominal pain, nausea, vomiting, and severe constipation. These symptoms may occur several days before neurological manifestations. The autonomic manifestations are prevalent and include persistent tachycardia, labile hypertension, orthostatic hypotension, and difficulty with micturition. Only a few patients progress to develop the more ominous motor neuropathy or CNS involvement including psychosis and seizures. Onset of the predominantly motor neuropathy is subacute, with generalized, proximal, or asymmetrical muscle weakness developing over days or weeks. The arms rather than the legs may be affected first, and proximal muscles may be preferentially involved. Muscular activity before the onset of symptoms may influence the pattern of weakness. Facial and bulbar weakness is common. In severe cases, flaccid quadriplegia with respiratory failure ensues. This picture may resemble GBS. Rapidly progressive muscle wasting is a striking feature. Tendon reflexes are diminished or absent, but paradoxically, ankle jerks may be retained. Sensory impairment may occur in a distal stocking-glove distribution or may affect the trunk and proximal limbs in an unusual bathing-suit pattern. In exceptional cases, a bilateral radial motor neuropathy without abdominal pain may be the only manifestation of AIP (King et al., 2002). The rate of improvement varies. Some patients rapidly recover, suggesting a reversible acute toxic-metabolic neuronal injury. Those with fixed weakness caused by axonal degeneration improve slowly (mean time to recovery, 10.6 months for proximal muscles and nearly twice as long for distal muscles). The protean CNS manifestations during severe attacks include anxiety, confusion, delirium, seizures, and coma.
Treatment and Management
The treatment of patients with acute hepatic porphyria involves three important steps: (1) prevention of attacks, (2) attempts to repress hepatic ALAS activity, thereby reducing porphyrin production, and (3) supportive care. Ideally, attacks should be prevented by avoiding drugs and situations that induce them. Among the inducing drugs, barbiturates are the most common precipitants, followed by sulfonamides, analgesics, nonbarbiturate hypnotics, anticonvulsants, and female sex hormones. Intentional fasting and alcohol consumption should be avoided. Gonadotropin-releasing hormone agonists may benefit women with recurrent attacks related to the menstrual cycle. The attack must be treated promptly as outlined in Fig. 76.17. First, all offending drugs are removed, and any intercurrent infection is treated. A high-carbohydrate diet orally or by nasogastric feeding (at least 400 g daily or the equivalence of glucose or levulose infusions) results in reduced porphyrin precursor production. Persistent symptoms or neurological deficits that progress for 24 hours after carbohydrate loading are indications for treatment with hematin (a hydroxide of heme), which represses the activity of hepatic ALAS and may restore cytochrome functions by replenishing an endogenous heme deficit. Hematin therapy at the recommended dose of 6 infusions of 4 mg/kg body weight at 12-hour intervals has resulted in consistent reduction of porphyrin precursors in serum and urine and clinical improvement in more than 80% of attacks. The only placebo-controlled study suggested a more modest clinical benefit of IV hematin. Early administration of hematin is advocated to correct the metabolic insult before neuronal damage becomes irreversible.
Fabry Disease
Fabry disease (angiokeratoma corporis diffusum) is an X-linked lysosomal storage disorder caused by a deficiency of the lysosomal enzyme α-galactosidase A, which results in the accumulation of the glycolipid globotriacylceramide (ceramide trihexoside) within vascular endothelial cells of the kidneys, heart, brain, and skin. Over time, progressive vascular disease leads to renal failure, cardiac disease, and strokes. Skin involvement gives rise to the typical angiokeratomas, which are dark red, punctate telangiectasias found mainly over the lower part of the trunk, buttocks, and scrotum (Fig. 76.18). A painful small-fiber neuropathy develops in childhood or adolescence. In fact, any boy or young man with severe painful sensory neuropathy should be suspected as having Fabry disease, and any skin lesions should be carefully scrutinized because they are usually sparse and may be easily overlooked. Distal paresthesias and lancinating pain are intensified by exertion, fever, or hot environments. Autonomic dysfunction includes diminished sweating, impaired tear and saliva formation, and decreased intestinal motility. Some patients have hearing loss (Barras and Maire, 2006; Vibert et al., 2006). Except for impairment of temperature sensation, overt neurological signs are absent. Female carriers often show clinical involvement but rarely develop renal failure, which is characteristically seen in affected men.
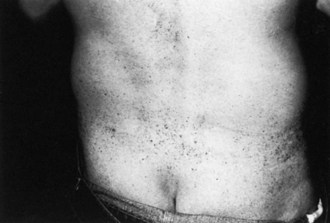
On nerve conduction studies, the conduction velocities are mildly reduced in two-thirds of patients. Deposition of glycolipid in small neurons of sensory and peripheral autonomic ganglia results in neuronal degeneration and selective loss of small myelinated and unmyelinated fibers in sural nerve biopsy specimens. Ultrastructurally, perineurial, endothelial, and perithelial cells contain characteristic lamellated glycolipid inclusions. Leukocyte preparations or skin fibroblasts are used for the diagnostic α-galactosidase assay. Screening for Fabry disease is recommended for patients with small-fiber neuropathy of unknown cause (Tanislav et al., 2011).
Analgesics, phenytoin, or carbamazepine, along with avoidance of aggravating factors, are effective for pain relief. Recombinant α-galactosidase-A replacement therapy with recombinant α-galactosidase A (agalsidase beta) has been shown to be safe and effective in the removal of microvascular endothelial deposits of globotriacylceramide from target organs (Eng et al., 2001) and in improvement of autonomic function (Ries et al., 2006). Agalsidase alfa has also been shown to be beneficial in cardiac, renal, neuropathic pain, and quality-of-life measures (Mehta et al., 2009). Renal transplantation may correct the biochemical defect, resulting in relief from pain and partial restoration of sweating.
Leukodystrophies with Neuropathy
The leukodystrophies result from inherited abnormalities of myelin metabolism that may affect both the CNS and PNS. Overall incidence of the leukodystrophies in the United States is 1 in 7663 live births (Bonkowsky et al., 2010). Peripheral nerve involvement is seen in metachromatic leukodystrophy (MLD), Krabbe disease, adrenomyeloneuropathy, and Cockayne syndrome. Recognition of an associated neuropathy may be helpful in the differential diagnosis of the underlying leukodystrophy. Missense mutations in proteolipid protein, a major protein component of CNS myelin, cause a spectrum of X-linked CNS disorders without peripheral neuropathy, including Pelizaeus-Merzbacher disease and hereditary spastic paraparesis. Proteolipid protein is also expressed in Schwann cells and compact peripheral myelin. Absence of proteolipid protein expression caused by a frameshift mutation has been reported to produce a demyelinating neuropathy with less severe CNS manifestations (Gabern et al., 1997).
Metachromatic Leukodystrophy
Three main clinical forms have been divided by age of onset: late-infantile (6 months-2 years), juvenile (3-16 years), and adult. Peripheral nerve involvement characterized by a progressive gait disorder, hypotonia, and lower-limb areflexia is an early manifestation that frequently precedes CNS involvement in late-infantile and early-juvenile MLD. In contrast, behavioral abnormalities and progressive dementia predominate over subtle neuropathic signs in adult-onset MLD. A homozygous missense mutation has been described in an adult patient presenting with an isolated polyneuropathy without CNS involvement (Felice et al., 2000). Marked uniform slowing of nerve conduction is seen in late-infantile and juvenile cases. Reduced NCVs and delayed visual and somatosensory evoked potential latencies are present in most adult cases. Extensive segmental demyelination and abnormally thin myelin sheaths are seen in nerve biopsy specimens of all MLD variants, along with metachromatic inclusions within Schwann cells and macrophages. Peripheral nerve biopsy therefore offers a means of confirming the diagnosis, although this is rarely needed now. The diagnosis of MLD is supported by MRI of the brain and confirmed by an increased urinary sulfatide excretion and abnormal ASA enzyme assays in leukocytes or fibroblasts.
Globoid Cell Leukodystrophy
Globoid cell leukodystrophy, or Krabbe disease, is an autosomal recessive disease caused by an inherited deficiency of the lysosomal enzyme galactocerebroside β-galactosidase. The gene for Krabbe disease has been localized to chromosome 14q31. The disorder is characterized by extensive CNS and peripheral nerve demyelination and the presence of multinucleated macrophages (globoid cells) in the cerebral white matter. The classic presentation in early infancy consists of rapidly progressive deterioration in intellectual and motor development, accompanied by hypertonicity, opisthotonic posture, optic atrophy, and seizures. In the late-onset form, peripheral neuropathy and spasticity may be the only manifestations. Peripheral nerve involvement is demonstrated by marked uniform slowing of motor conduction velocities. Segmental demyelination, together with ultrastructurally characteristic tubular or crystalloid inclusions within Schwann cells and macrophages, is seen in the sural nerve. Hematopoietic stem cell transplantation provides a source of the missing enzyme and can thereby prevent and reverse the CNS manifestations (Krivit et al., 1998).
Adrenomyeloneuropathy
Adrenomyeloneuropathy (AMN), the adult phenotype of adrenoleukodystrophy, is an X-linked recessive disorder of fatty acid metabolism characterized by adrenal insufficiency, progressive myelopathy, and peripheral neuropathy. A defect in beta-oxidation of saturated very-long-chain fatty acids (VLCFA) in peroxisomes leads to the accumulation of tetracosanoic (C24:0) and hexacosanoic (C26:0) acid in tissues and body fluids in affected patients. A significant increase of VLCFA levels in plasma, fibroblasts, or both allows reliable detection in patients and heterozygote female carriers. The defective gene (ABCD1) is located in the region Xq28 and codes for a peroxisomal membrane protein referred to as ALD protein that belongs to the ABC transporter protein family (Moser, 1997). AMN manifests in the second to third decades with progressive spastic paraparesis, distal muscle weakness, sensory loss, and sphincter disturbances. Neurological features frequently are preceded by clinical or laboratory evidence of hypoadrenalism. Approximately 10% of patients have primary adrenal insufficiency without evidence of nervous system involvement. At least 20% of female carriers develop spastic paraparesis similar to that in men, but less severe and later in onset.
Electrophysiological studies are helpful in identifying peripheral nerve involvement that may escape clinical detection because of prominent upper motor neuron signs. Nerve conduction studies demonstrate a distal axonopathy with low CMAP amplitudes and mildly reduced NCVs. Fewer than 10% of patients have significant nerve conduction slowing suggestive of demyelination (van Geel et al., 1996). Sural nerve biopsy shows loss of myelinated fibers, occasional small onion bulbs, and curvilinear lamellar lipid inclusions in Schwann cells. Brain MRI study results are abnormal, demonstrating white-matter changes in roughly half of patients with AMN at some time in the course of their disease.
Dietary restriction of VLCFA combined with the administration of oleic and erucic acids (Lorenzo oil) lower plasma levels of VLCFA but have no effect in arresting the rate of neurological progression (van Geel et al., 1999). Adrenal insufficiency responds readily to corticosteroid replacement. In contrast to the cerebral form, patients with AMN are not considered suitable candidates for bone marrow transplantation.
Phytanic Acid Storage Disease (Refsum Disease)
Chronic dietary treatment, by restricting the exogenous sources of phytanic acid (<10 mg/day) and its precursor, phytol, results in reduction of serum phytanic acid levels and clinical improvement. The diet should provide sufficient calories to avoid weight loss. Plasma exchange has been used to lower toxic serum phytanic acid levels more rapidly in critically ill patients. Docosahexaenoic acid has been found to be deficient in peroxisomal disorders, but supplementation does not lead to clinical improvement even though docosahexaenoic acid plasma levels do increase (Parker et al., 2010).
Abetalipoproteinemia (Bassen-Kornzweig Syndrome)
Clinical manifestations of vitamin E deficiency can also be found in familial vitamin E deficiency caused by mutations of the α-tocopherol transfer protein gene. These gene mutations result in the failure to incorporate α-tocopherol in very-low-density lipoprotein in the liver and lead to low vitamin E levels in the absence of malabsorption (Martinello et al., 1998).
Mitochondrial Cytopathies and Polyneuropathy
Inborn errors of mitochondrial metabolism encompass a heterogenous group of multisystemic disorders caused by mutations in either mitochondrial or nuclear DNA. These mutations impair cellular energy metabolism of virtually every organ, but in particular the CNS and muscles. Involvement of the PNS also occurs in several mitochondrial cytopathies (Bouillot et al., 2002; Stickler et al., 2006). However, mitochondrial cytopathy remains an uncommon cause for isolated and progressive neuropathy. A mixed demyelinating and axonal neuropathy is seen in MERRF (myoclonic epilepsy and ragged red fibers). In the syndrome of neurogenic muscle weakness, ataxia, and retinitis pigmentosa (NARP) caused by a mutation of the ATP6 gene, an axonal neuropathy predominates and may be the presenting feature. In fact, the first case of an established mitochondrial cytopathy and neuropathy was described in this syndrome. In sensory ataxic neuropathy, dysarthria, and ophthalmoplegia (SANDO), a multiple mitochondrial depletion disorder, a severe sensory axonal neuropathy and ophthalmoplegia associated with other features such as migraine, depression, and cyclooxygenase (COX)-deficient muscle fibers develops after age 30 (Fadic et al., 1997). Mutations in the nuclear DNA polymerase (POLG), crucial for mitochondrial DNA replication, results in epilepsy, ataxia, headache, myoclonus, late-onset ophthalmoplegia, and a sensorimotor neuropathy (Tzoulis et al., 2006).
A demyelinating neuropathy, at times severe, is seen in MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes). The majority of patients (≈75%) have a predominantly axonal neuropathy, half predominantly sensory; the remainder have only electrophysiological abnormalities (Kaufmann et al., 2006). Endocrine involvement, especially of thyroid and pancreas, is common, and impaired glucose metabolism and hypothyroidism may contribute to the development and severity of neuropathy in MELAS. The neuropathy of MELAS appears to be more common in men than women.
In the syndrome of myoneurogastrointestinal encephalopathy (MNGIE), an autosomal recessive condition with a loss-of-function mutation in the thymidine phosphorylase gene, a mixed axonal and demyelinating neuropathy may be present. At times, the neuropathy may follow a course mimicking CIDP (Bedlack et al., 2004). However, these patients often have no proximal weakness, suffer from gastrointestinal dysfunction due to intestinal pseudo-obstruction, may have CNS demyelination or lactic acidosis, exhibit extraocular abnormalities, and show no response to immunomodulatory treatments. Measurement of thymidine level in the serum or activity of thymidine phosphorylase in buffy coat supports the diagnosis. DNA sequencing will confirm the diagnosis by demonstrating mutations in the thymidine phosphorylase gene. A presentation similar to CMT with distal muscle atrophy and pes cavus has also been observed in patients with MNGIE (Said et al., 2005). The course of neuropathy in MNGIE, however, may become rapid and pain may be prominent. The role of nutritional factors secondary to gastrointestinal dysmotility in the development and intensity of the neuropathy in such patients should not be ignored. Nerve biopsies performed in few patients with MNGIE has shown variable abnormalities including reduction in myelinated fiber density, demyelination and remyelination, myelin sheath irregularity, and a high proportion of fibers with axonal degeneration and axonal atrophy. Schwann cells, axons, and endothelial cells may contain abnormal mitochondria, including paracrystalline inclusions. Recent reports suggest that allogenic stem cell transplantation corrects biochemical derangements in the blood of MNGIE patients, but its clinical efficacy remains unproven (Hirano et al., 2006).
In another mitochondrial disorder commonly observed in children, Leigh disease, a demyelinating-remyelinating neuropathy and hypomyelination of nerves is observed. This is particularly prominent when mutations occur in nuclear genes (e.g., SURF1 and SCO2) encoding the assembly of complex IV of the respiratory chain. Navajo neurohepatopathy, a unique autosomal recessive, multisystemic, progressive syndrome prevalent in Navajo children of the southwest United States, is caused by a mutation in the mitochondrial MPV17 gene, which is believed to be involved in mitochondrial DNA maintenance and in the regulation of oxidative phosphorylation (Karadimas et al., 2006). These children develop a severe sensorimotor neuropathy associated with corneal ulcerations, acral mutilation, poor weight gain, short stature, sexual infantilism, serious systemic infections, and liver derangement, including Reye syndrome–like episodes, leading to cirrhosis and hepatic failure. Mitochondrial cytopathies should be considered in the differential diagnoses of any patient with a peripheral neuropathy and a multisystemic condition.
Inflammatory Demyelinating Polyradiculoneuropathies
Acute Inflammatory Demyelinating Polyradiculoneuropathy (Guillain-Barré Syndrome)
In 1916, Guillain, Barré, and Strohl emphasized the main clinical features of GBS: motor weakness, areflexia, paresthesias with minor sensory loss, and increased protein in CSF without pleocytosis (albuminocytological dissociation). Our current understanding of the pathology was greatly enhanced when multifocal inflammatory demyelination of spinal roots and peripheral nerves were described. The frequent finding of motor conduction blocks and reduced conduction velocities provided further electrophysiological confirmation of the widespread demyelination. Improvement in modern critical care has dramatically changed outcomes in GBS, including a reduction in the mortality rate from 33% before the introduction of positive-pressure ventilation to the current rate of approximately 5% to 10%. The diagnosis of GBS depends on clinical criteria supported by electrophysiological studies and CSF findings (Box 76.10). These diagnostic criteria define AIDP, which is the most common form of GBS in Europe and North America. However, in recent years it has been recognized that GBS is a heterogeneous disorder, and not all GBS cases are due to demyelination. An axonal immune-mediated injury may also produce a similar clinical presentation. This axonal variant of GBS is called acute motor-sensory axonal neuropathy (AMSAN) because of involvement of both motor and sensory fibers and is known for its severity and poor recovery. A second, pure motor axonal form called acute motor axonal neuropathy (AMAN) has been described in northern China, where it occurs in summer epidemics in children and young adults. The Miller Fisher syndrome (MFS) variant accounts for 6% of total GBS cases in Western countries, whereas in Taiwan the proportion is as high as 18%, suggesting a geographical difference. Other variants of GBS such as acute pandysautonomia and the pharyngeal-cervical-brachial variant occur so infrequently that no exact figure on their prevalence and incidence can be given. The classification of GBS subtypes and variants is based on the clinical picture and electrophysiological and pathological findings (Griffin et al., 1996) (Box 76.11).