[level-membership-for-neurology-category]
Chapter 78 Disorders of Neuromuscular Transmission
Myasthenia Gravis
Epidemiology of Myasthenia Gravis
MG may begin at any age from infancy to very old age. Epidemiological studies report considerable variability in incidence and prevalence around the world (Meriggioli and Sanders, 2009). While methodological differences may explain some of this variability, biological and genetic factors may also play a role. Recent estimates indicate that the U.S. prevalence is approximately 20/100,000, 60,000 patients total (Phillips, 2004). Epidemiological studies have shown an increasing prevalence over the past 50 years, related to an increase in the frequency of diagnosis in elderly patients but also likely due to improved ascertainment, reduced mortality rates, and increased longevity of the population. Gender and age influence the incidence of MG; women are affected nearly three times more often than men before age 40, but the incidence is higher in males after age 50 and roughly equal during puberty. As the population ages, the average age at onset has increased correspondingly. More men than women are now affected, and the majority of MG patients in the United States are older than 50. Detailed population-based data on clinical and serological subtypes of MG (see Myasthenia Gravis Subtypes) are largely lacking.
Clinical Presentation of Myasthenia Gravis
Patients with MG seek medical attention for specific muscle weakness or dysfunction that typically worsens with activity and improves with rest. Although they may also have generalized fatigue or malaise, it is not usually the major or presenting complaint. Ptosis or diplopia is the initial symptom in approximately two-thirds of patients; nearly all will develop both within 2 years (Sanders and Massey, 2008). Difficulty chewing, swallowing, or talking is the initial symptom in one-sixth of patients, and limb weakness in 10%. Rarely, the initial weakness is limited to single muscle groups such as neck or finger extensors, hip flexors, or ankle dorsiflexors.
The course of disease is variable but usually progressive. Weakness remains restricted to the ocular muscles in approximately 10% to 15% of cases (see Ocular Myasthenia Gravis, later in this chapter), although a higher proportion has been reported in Asian populations (Meriggioli and Sanders, 2009). In the rest, weakness progresses to involve nonocular muscles during the first 3 years and ultimately involves facial, oropharyngeal, and limb muscles (generalized MG). Maximum weakness occurs during the first year in two-thirds of patients. Before the introduction of corticosteroids for treatment, approximately one-third of patients improved spontaneously, one-third became worse, and one-third died of the disease. Improvement, even remission, may occur early on but is rarely permanent (i.e., there is a subsequent relapse). Symptoms typically fluctuate over a relatively short period and then become more severe (active stage). Left untreated, an inactive stage follows the active stage, in which fluctuations in strength still occur but are attributable to fatigue, intercurrent illness, or other identifiable factors. After many years, untreated weakness becomes fixed, and the most severely involved muscles are frequently atrophic (burnt-out stage). Factors that worsen myasthenic symptoms are emotional upset, systemic illness (especially viral respiratory infections), hypothyroidism or hyperthyroidism, pregnancy, the menstrual cycle, drugs affecting NMT (see Treatment of Associated Diseases and Medications to Avoid, later in this chapter), and fever.
Physical Findings in Myasthenia Gravis
Ocular Muscles
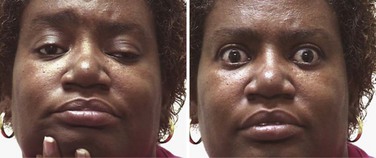
 Most MG patients have weakness of ocular muscles (Box 78.1). (Videos of MG-related ocular phenomena [Videos 78.1 and 78.2] can be found at www.expertconsult.com.) Asymmetrical weakness of several muscles in both eyes is typical, the medial rectus being more frequently and usually more severely involved. The pattern of weakness is not localizable to lesions of one or more nerves, and the pupillary responses are normal. Ptosis is usually asymmetrical (Fig. 78.1) and varies during sustained activity. To compensate for ptosis, chronic contraction of the frontalis muscle produces a worried or surprised look. Unilateral frontalis contraction is a clue that the lid elevators are weak on that side (see Fig. 78.1). When mild, ocular weakness may not be obvious on routine examination and appear only upon provocative testing (i.e., sustained upward gaze). Eyelid closure is usually weak, even when strength is normal in all other facial muscles, and may be the only residual weakness in otherwise complete remission. This is usually asymptomatic unless it is severe enough to allow soap or water in the eyes during bathing. With moderate weakness of these muscles, the eyelashes are not “buried” during forced eye closure (Fig. 78.2). Fatigue in these muscles may result in slight involuntary opening of the eyes as the patient tries to keep the eyes closed; this is called the peek sign (see Fig. 78.2).
Most MG patients have weakness of ocular muscles (Box 78.1). (Videos of MG-related ocular phenomena [Videos 78.1 and 78.2] can be found at www.expertconsult.com.) Asymmetrical weakness of several muscles in both eyes is typical, the medial rectus being more frequently and usually more severely involved. The pattern of weakness is not localizable to lesions of one or more nerves, and the pupillary responses are normal. Ptosis is usually asymmetrical (Fig. 78.1) and varies during sustained activity. To compensate for ptosis, chronic contraction of the frontalis muscle produces a worried or surprised look. Unilateral frontalis contraction is a clue that the lid elevators are weak on that side (see Fig. 78.1). When mild, ocular weakness may not be obvious on routine examination and appear only upon provocative testing (i.e., sustained upward gaze). Eyelid closure is usually weak, even when strength is normal in all other facial muscles, and may be the only residual weakness in otherwise complete remission. This is usually asymptomatic unless it is severe enough to allow soap or water in the eyes during bathing. With moderate weakness of these muscles, the eyelashes are not “buried” during forced eye closure (Fig. 78.2). Fatigue in these muscles may result in slight involuntary opening of the eyes as the patient tries to keep the eyes closed; this is called the peek sign (see Fig. 78.2).
Box 78.1 Ocular Findings in Myasthenia Gravis
• Weakness usually involves one or more ocular muscles, without overt pupillary abnormality
• Weakness is typically variable, fluctuating, and fatigable
• Ptosis that shifts from one eye to the other is virtually pathognomonic of myasthenia gravis
• With limited ocular excursion, saccades are superfast, producing ocular “quiver”
• After downgaze, upgaze produces lid overshoot (“lid twitch”)
• Pseudo-internuclear ophthalmoplegia—limited adduction, with nystagmoid jerks in abducting eye
• In asymmetrical ptosis, covering the eye that has lid ptosis may relieve contraction of the opposite frontalis
•  Passively lifting a ptotic lid may cause the opposite lid to fall (“enhanced ptosis”) (see Video 78.2)
Passively lifting a ptotic lid may cause the opposite lid to fall (“enhanced ptosis”) (see Video 78.2)
• Edrophonium may improve only some of several weak ocular muscles; others may actually become weaker
• Edrophonium may relieve asymmetrical ptosis and produce retraction of the opposite lid from frontalis contraction
• The opposite lid may droop further as the more involved lid improves after edrophonium


Oropharyngeal Muscles
Myasthenic patients may have a characteristic facial appearance. At rest, the corners of the mouth often droop downward, giving a depressed appearance. Attempts to smile often produce contraction of the medial portion of the upper lip and a horizontal contraction of the corners of the mouth without the natural upward curling, which gives the appearance of a sneer (see Fig. 78.1).
Limb Muscles
Weakness begins in limb or axial muscles in about 20% of MG patients (Kuks and Oosterhuis, 2004). Any trunk or limb muscle may be weak, but some are more often affected than others. Neck flexors are usually weaker than neck extensors, and the deltoids, triceps, and extensors of the wrist and fingers and ankle dorsiflexors are frequently weaker than other limb muscles. Rarely, MG presents initially with focal weakness in single muscle groups, such as a “dropped head syndrome” due to severe neck extensor weakness or isolated vocal cord or respiratory muscle weakness. In untreated patients with long-standing disease, weakness may be fixed, and severely involved muscles may be atrophic, giving the appearance of a chronic myopathy; this is particularly likely in muscle-specific tyrosine kinase (MuSK) antibody–positive MG (see MuSK Antibody Myasthenia Gravis, later in this chapter).
Immunopathology of Myasthenia Gravis
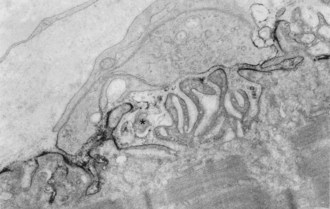
In about 80% to 85% of MG patients, weakness results from the effects of circulating anti-AChR antibodies. These antibodies bind to AChR on the terminal expansions of the junctional folds (Fig. 78.3) (Engel et al., 1977a) and cause complement-mediated destruction of the folds, accelerated internalization and degradation of AChR, and in some cases, they block ACh-AChR binding. Destruction of the junctional folds results in distortion and simplification of the postsynaptic region (see Fig. 78.4) and loss of functional AChR (Engel et al., 1977b). This leads to NMT failure and muscle weakness. MG is a paradigm for an antibody-mediated disease: the physiological abnormality is passively transferable by injection of MG immunoglobulin (Ig)G into mice, and clinical improvement follows removal of circulating antibodies by plasma exchange (see Treatment of Myasthenia Gravis, later in this chapter).
Patients with MG have increased numbers of CD4+ T cells, which regulate the production of AChR antibody (AChR-Ab). The α subunit of AChR contains the majority of T-cell recognition sites. These recognition sites may be different from those of the main immunogenic region that binding antibodies recognize. Sensitization to CD4+ T-cells spreads across the AChR complex as the disease progresses and most MG patients have T cells that recognize multiple epitopes on the AChR α-subunit (Conti-Fine et al., 1997). This epitope spread drives the synthesis of anti-AChR antibodies and accounts for the large and varied antibody repertoire of the myasthenic patient.
Recently, low-affinity IgG antibodies have been found in about two-thirds of MG patients who were seronegative using conventional anti-AChR and anti-MuSK antibody assays (Leite et al., 2008). These antibodies bind to AChRs that have been clustered into high-density arrays, suggesting that they have relatively low affinity and cannot bind strongly to AChR in solution but do bind to immobilized AChRs in a native conformation.
The Thymus in Myasthenia Gravis
The thymus is abnormal in most MG patients; 70% have lymphoid follicular hyperplasia, and more than 10% have a thymoma. Hyperplastic thymus glands from MG patients contain all the components necessary for the development of an immune response to the AChR: T cells, B cells, and plasma cells, as well as muscle-like myoid cells that express AChR. It is unlikely that the cellular alterations in the thymus are secondary to an ongoing peripheral immune response because they are absent in experimental autoimmune MG (Hohlfeld and Wekerle, 2008). In addition, thymocytes in culture spontaneously generate anti-AChR antibodies. These findings support the concept of an intrathymic pathogenesis and argue that the hyperplastic thymus is involved in the initiation of the anti-AChR immune response in patients with thymic hyperplasia. Thymic-derived AChR subunits may serve as an antigen for the autosensitization against the AChR.
Neoplastic epithelial cells in thymomas express numerous self-like antigens, including AChR, titin, and ryanodine receptor-like epitopes. MG-associated thymomas are also rich in autoreactive T cells. The regulation of potentially autoreactive T cells may be impaired in thymoma due to a deficiency in the expression of the autoimmune regulator gene (AIRE) and selective loss of T regulatory cells in human thymomas (Meriggioli and Sanders, 2009).
Myasthenia Gravis Subtypes
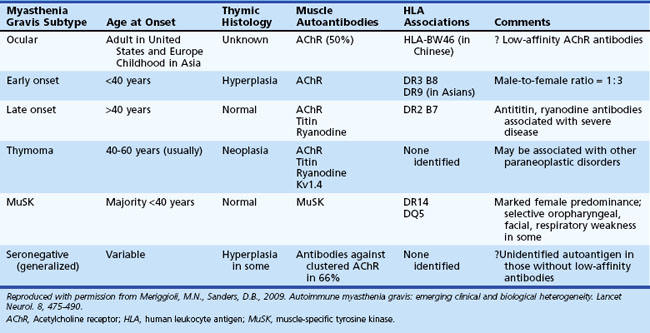
A number of MG subtypes (Table 78.1) may be identified based on the clinical presentation, age of onset, autoantibody profile, and thymic pathology (Meriggioli and Sanders, 2009). Interestingly, these subtypes appear to have unique genetic associations, strengthening the concept of distinct clinical entities and disease mechanisms.
Ocular Myasthenia Gravis
Ptosis and/or diplopia are the initial symptoms of MG in up to 85% of patients (Grob et al., 2008), and almost all patients have both symptoms within 2 years of disease onset. Myasthenic weakness that remains limited to the ocular muscles is termed ocular myasthenia gravis (OMG) and accounts for approximately 10% to 15% of all MG in Caucasian populations. If weakness remains limited to the ocular muscles after 2 years, there is a 90% likelihood that the disease will not generalize. OMG is more common in Asian populations (up to 58% of all MG patients) (Zang et al., 2007).
Generalized Myasthenia Gravis
Patients with generalized myasthenia gravis (GMG) may have either early-onset (EOMG) or late-onset (LOMG) disease, with the cutoff age usually defined as age 40. EOMG patients are more often female and typically have anti-AChR antibodies and enlarged hyperplastic thymus glands. LOMG patients are more often male and may have antibodies to striated muscle proteins such as titin and the ryanodine receptor in addition to anti-AChR antibodies. The presence of these anti-muscle antibodies, particularly anti–ryanodine receptor antibodies, has been associated with more severe generalized or predominantly oropharyngeal weakness and frequent myasthenic crises (Romi et al., 2005). LOMG patients without thymoma usually have a normal or atrophic thymus, but relatively few histological studies are available in this age group, as thymectomy after age 50 is unusual.
MuSK-Antibody Myasthenia Gravis
Antibodies to MuSK have been reported in up to 50% of patients with GMG who lack AChR antibodies (Guptill and Sanders, 2010) and have recently been reported in OMG as well (Bau et al., 2006; Caress et al., 2005). The reported incidence of MuSK-antibody myasthenia gravis (MMG) varies among geographic regions, the highest being closer to the equator and the lowest closer to the poles (Vincent and Lang, 2006). Genetic or environmental factors (or both) presumably play a role in these differences. MMG predominantly affects females and begins from childhood through middle age. In some patients, the clinical findings are indistinguishable from anti-AChR-positive MG, with fluctuating ocular, bulbar, and limb weakness. However, many MMG patients have predominant weakness in cranial and bulbar muscles, frequently with marked atrophy of these muscles (Fig. 78.5). Others have prominent neck, shoulder, and respiratory weakness, with little or no involvement of ocular or bulbar muscles. Electrodiagnostic abnormalities may not be as widespread as in other forms of MG, and it may be necessary to examine different muscles to demonstrate abnormal NMT (Stickler et al., 2005). The potentially more limited distribution of physiological abnormalities also may limit the interpretation of microphysiological and histological studies in MMG, inasmuch as the muscles usually biopsied for these studies may be normal.
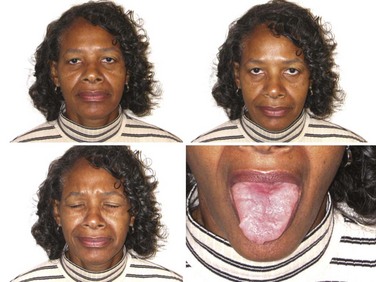
Many MMG patients do not improve with cholinesterase inhibitors (ChEIs); some actually become worse, and many have profuse fasciculations with these medications (Hatanaka et al., 2005). Disease severity tends to be worse, but most improve dramatically with PLEX or corticosteroids (Sanders et al., 2003). More immunosuppression is typically necessary, though long-term outcome is generally good (Guptill and Sanders, 2010). Thymic changes are absent or minimal (Lauriola et al., 2005; Leite et al., 2005), and the role of thymectomy in MMG is not yet clear (Guptill and Sanders, 2010; Sanders et al., 2003). The diagnosis of MMG may be elusive when the clinical features, electrodiagnostic findings, and response to ChEIs differ from typical MG.
Seronegative Myasthenia Gravis
MG patients who lack both anti-AChR and anti-MuSK antibodies (“double-seronegative MG”) are clinically heterogeneous. The true frequency of “seronegative MG” may be quite low, as certain patients may have low-affinity anti-AChR antibodies that can only be detected using specialized assays (see Immunopathology of Myasthenia Gravis, earlier).
Genetics of Myasthenia Gravis
Several correlations exist between MG and the human leukocyte antigen (HLA) genes. Certain HLA types (-DR2, -DR3, -B8, -DR1) predispose to MG (see Table 78.1), whereas others may offer resistance to disease. HLA-B8, -DR2, and -DR3 types occur more commonly in patients with EOMG; HLA-B7 and -DR2 in LOMG; and HLA-DR1 in OMG (see Table 78.1). MMG is associated with HLA-DR14-DQ5 (Niks et al., 2006). Different HLA associations have been reported in Asian MG patients, including an association of OMG with HLA-BW46 in Chinese patients (Meriggioli and Sanders, 2009). Non-HLA genes (PTPN22, FCGR2, CHRNA1) have also been found to be associated with MG; some are also associated with other autoimmune diseases and thus may represent a nonspecific susceptibility to autoimmunity. An exception to this is the CHRNA1 gene, which encodes the α subunit of the AChR and may provide pathogenetic clues specific for MG (Meriggioli and Sanders, 2009).
Diagnostic Procedures in Myasthenia Gravis
Edrophonium Chloride Test
The most important consideration in performance of the edrophonium test is the choice of endpoint. Only unequivocal improvement in strength of an affected muscle is acceptable as a positive result. For this reason, resolution of eyelid ptosis, improvement in strength of a single paretic extraocular muscle, or clear improvement of dysarthria have been proposed as the only truly valid endpoints because observed function in these muscles is largely independent of fluctuating effort (Fig. 78.6). Interpret changes in strength of other muscles cautiously, especially in a suggestible patient.
The edrophonium test is reported to be positive in 60% to 95% of patients with OMG and in 72% to 95% with GMG (Pascuzzi, 2003). Improvement after edrophonium is not unique to MG; it is also seen in congenital myasthenic syndromes, the Lambert-Eaton syndrome, intracranial aneurysms, brainstem lesions, cavernous sinus tumors, end-stage renal disease, and in muscle diseases affecting the ocular muscles.
 The optimal dose of edrophonium varies among patients and cannot be predetermined. In a study of OMG, the mean dose of edrophonium that gave a positive response was 3.3 mg for ptosis and 2.6 mg for ocular muscle dysfunction (Kupersmith et al., 2003). The lowest effective dose can be determined by injecting small incremental doses up to a maximum total of 10 mg. Inject an initial test dose of 2 mg, and monitor the response for 60 seconds. Subsequent injections of 3 and 5 mg may then be given, but if clear improvement is seen within 60 seconds after any dose, the test is positive, and no further injections are necessary (see Video 78.1). Weakness that develops or worsens after injection of 10 mg or less also indicates an NMT defect, as this dose will not weaken normal muscle.
The optimal dose of edrophonium varies among patients and cannot be predetermined. In a study of OMG, the mean dose of edrophonium that gave a positive response was 3.3 mg for ptosis and 2.6 mg for ocular muscle dysfunction (Kupersmith et al., 2003). The lowest effective dose can be determined by injecting small incremental doses up to a maximum total of 10 mg. Inject an initial test dose of 2 mg, and monitor the response for 60 seconds. Subsequent injections of 3 and 5 mg may then be given, but if clear improvement is seen within 60 seconds after any dose, the test is positive, and no further injections are necessary (see Video 78.1). Weakness that develops or worsens after injection of 10 mg or less also indicates an NMT defect, as this dose will not weaken normal muscle.
Common side effects of edrophonium are increased salivation and sweating, nausea, stomach cramps, and fasciculations. Serious complications (bradyarrhythmia or syncope) have been reported in only 0.16% of edrophonium tests (Ing et al., 2005). These symptoms generally resolve with rest in the supine position. Atropine (0.4-2 mg) should be available for intravenous (IV) injection if bradycardia is severe.
Autoantibodies in Myasthenia Gravis
Acetylcholine Receptor Antibodies
Assays measuring antibodies that react with AChR proteins are generally regarded as specific serological markers for MG. The most widely utilized test for MG is the AChR-Ab binding assay. This assay tests serum for binding to purified AChR from human skeletal muscle labeled with radioiodinated α-bungarotoxin. The reported sensitivity of this assay is approximately 85% for GMG and 50% for OMG (Stålberg et al., 2010). Nearly all thymomatous MG patients have elevated AChR antibodies.
AChR antibodies cross-link the AChRs in the membrane and increase their rate of degradation. The AChR-modulating antibody assay measures the rate of loss of labeled AChR from cultured human myotubes. About 10% of MG patients who do not have elevated binding antibodies have AChR-modulating antibodies. Many patients with thymomatous MG have high levels of AChR-modulating antibodies, often exceeding 90% loss of AChR (Vernino and Lennon, 2004).
Antistriational Muscle Antibodies
StrAbs are not pathogenic and are also found in one-third of patients with thymoma who do not have MG and in one-third of MG patients without thymoma. They are more frequent in older MG patients and in those with more severe disease, suggesting that disease severity is related to a more vigorous humoral response against multiple muscle antigens (Romi et al., 2005).
Electrodiagnostic Testing in Myasthenia Gravis
Repetitive nerve stimulation (RNS) is the most commonly used electrophysiological test of NMT. At low rates of stimulation (2-5 Hz), RNS depletes the store of readily releasable ACh at diseased motor end-plates, causing failure of NMT. Characteristically in MG, there is a decrementing response of at least 10% to trains of 2- to 3-Hz stimulation (see Chapter 32B). This may be present at baseline or after a period of exercise (postactivation exhaustion). Although a seemingly simple test, careful attention to proper technique is important to avoid technical errors. The sensitivity of RNS for diagnosing MG reportedly ranges from 53% to 100% in GMG and 10% to 48% in OMG (Meriggioli and Sanders, 2004; Stålberg et al., 2010). RNS is more likely to be abnormal in a proximal or facial muscle and in clinically weak muscles. For maximal diagnostic yield, test several muscles, particularly those that are weak. If RNS is normal and there exists a high suspicion for an NMJ disorder, perform SFEMG of at least one symptomatic muscle.
SFEMG (see Chapter 32B) is the most sensitive clinical test of NMT and shows increased jitter in some muscles in almost all patients with MG (Stålberg et al., 2010). Jitter is greatest in weak muscles but is usually abnormal even in muscles with normal strength. Sixty percent of patients with OMG show increased jitter in a limb muscle, but this does not predict the subsequent development of generalized myasthenia.
In the rare patient who has weakness restricted to a few limb muscles, only a weak muscle may show abnormal jitter. This is particularly true in some patients with MMG (Stickler et al., 2005) (see MuSK-Antibody Myasthenia Gravis, earlier).
Recently, measuring jitter with concentric needle electrodes (CNE) has been proposed as an alternative to the specially designed (reusable) single-fiber electrode (Stålberg and Sanders, 2009). Interpret the results with caution, particularly in borderline cases, as signals recorded with the CNE may represent the summation of more than one single-fiber action potential, which will decrease the apparent jitter.
Ocular Cooling
Myasthenic weakness typically improves with muscle cooling. This is the basis of the “ice-pack” test, in which cooling of a ptotic lid improves lid elevation. Assess improvement in ptosis after placing an ice pack over the ptotic eyelid, usually for 2 minutes. Positive responses can occur even when edrophonium tests are negative. A meta-analysis of six studies showed this test to have high sensitivity and specificity in MG, suggesting that it may be useful in patients with lid ptosis, particularly if the edrophonium test is negative or contraindicated (Larner, 2004).
Comparison of Diagnostic Techniques in Myasthenia Gravis
Plan diagnostic testing based on the clinical presentation and distribution of weakness (Table 78.2). The edrophonium test is often diagnostic in patients with ptosis or ophthalmoparesis but is less useful in assessing other muscles. The presence of serum AChR or anti-MuSK antibodies virtually ensures the diagnosis of MG, but their absence does not exclude it. RNS confirms impaired NMT but is frequently normal in mild or purely ocular disease. Almost all patients with MG have increased jitter, and normal jitter in a weak muscle excludes MG as the cause of the weakness. Neither electrodiagnostic test is specific for MG, because increased jitter, even abnormal RNS, occurs in other motor unit disorders that impair NMT.

Treatment of Myasthenia Gravis
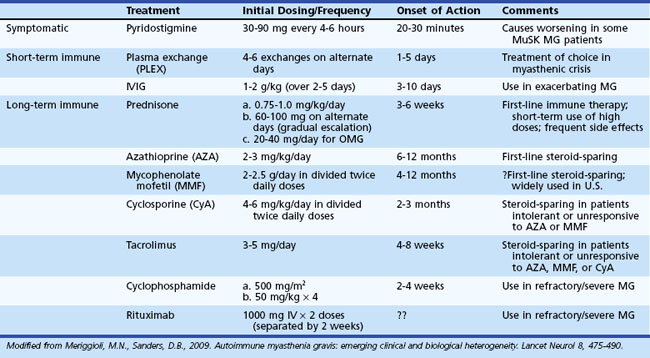
The outlook for patients with MG has improved considerably in recent years, largely due to advances in intensive care medicine and the use of immunomodulating agents. The therapeutic goal is to return the patient to normal function as rapidly as possible while minimizing the side effects of therapy. A number of therapeutic options are available (Table 78.3), but treatment must be individualized according to the extent (ocular versus generalized) and severity (mild to severe) of disease, and the presence or absence of concomitant disease (including but not limited to other autoimmune diseases and thymoma). The basis of treatment decisions for individual patients is the predicted course of disease and the predicted response to specific treatments. Successful treatment of MG requires close medical supervision and long-term follow-up. Consider the return of weakness after a period of improvement as a herald of further progression requiring reassessment of current treatment and evaluation for underlying systemic disease or thymoma.
Symptom Management: Cholinesterase Inhibitors
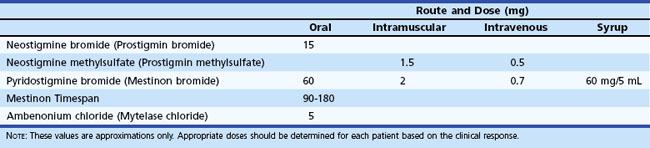
Pyridostigmine bromide and neostigmine bromide are the most commonly used ChEIs. Pyridostigmine is generally preferred because it has a lower frequency of gastrointestinal side effects and longer duration of action. The initial oral dose in adults is 30 to 60 mg every 4 to 8 hours. The equivalent dose of neostigmine is 7.5 to 15 mg. In infants and children, the initial oral dose of pyridostigmine is 1 mg/kg, and of neostigmine is 0.3 mg/kg (Table 78.4). Pyridostigmine is available as syrup (60 mg/5 mL) for children or for nasogastric tube administration in patients with impaired swallowing. A timed-release tablet of pyridostigmine (180 mg) is useful as a bedtime dose for patients who are too weak to swallow in the morning. Its absorption is erratic, however, leading to possible overdosage and underdosage, and it is not useful during waking hours.
Preliminary studies of an antisense oligonucleotide (EN101) that blocks expression of a splice isoform of acetylcholinesterase have been published (Sussman et al., 2008). The drug appears to be safe and the beneficial effects long lasting—many hours compared to 3 to 5 hours for pyridostigmine. Further studies are ongoing.
Short-Term (Rapid-Onset) Immune Therapies
Intravenous Immunoglobulin
Improvement in MG occurs in 50% to 100% of MG patients after infusion of high-dose intravenous immunoglobulin (IVIG), typically given at a dose of 2 g/kg given over 2 to 5 days. Improvement usually begins within 1 week and lasts for several weeks or months. Class I evidence supports the use of IVIG to treat patients with refractory exacerbations of MG (Donofrio et al., 2009), but there is little evidence to advise the clinician on the proper dosing of IVIG and duration of therapy. A recent double-blind placebo-controlled trial in MG patients with worsening weakness showed that IVIG induced rapid improvement in muscle strength, but this effect was more pronounced and likely more clinically significant in patients with moderate to severe MG (Zinman et al., 2007).
IVIG induces rapid improvement in patients with severe disease or crisis, reduces perioperative morbidity prior to surgery, and may be used chronically in selected refractory patients. IVIG may be particularly useful as an alternative to PLEX in children with limited vascular access. Although IVIG has demonstrated similar efficacy to PLEX in the treatment of MG exacerbations, it is unclear whether it is as effective in true MG crisis, since the published comparison studies generally used suboptimal PLEX regimens and did not directly compare onset of improvement. Common side effects of IVIG include headaches, chills, and fever, which usually improve if infusion rates are slowed. Serious side effects are rare but include renal toxicity, stroke, leukopenia, and aseptic meningitis. Lyophilized forms of IVIG may be associated with greater prevalence of adverse events in patients with neuromuscular diseases (Nadeau et al., 2010).
Long-Term Immune Therapies
Corticosteroids
Corticosteroids were the first immunosuppressants to be widely used in MG and remain the most commonly used immune therapy today. Prednisone produces marked improvement or complete relief of symptoms in more than 75% of MG patients (Pascuzzi et al., 1984) and some improvement in most of the rest. Much of the improvement occurs in the first 6 to 8 weeks, but strength may increase to total remission in the following months, even while tapering the dose. Patients with recent onset of symptoms have the best responses, but those with chronic disease also may respond. The severity of disease does not predict the ultimate improvement. Patients with thymoma usually respond well to prednisone, before or after removal of the tumor.
Transitory worsening of weakness occurs in approximately one-third to half of patients treated with high-dose daily prednisone (Pascuzzi et al., 1984). This usually begins within the first 7 to 10 days with high prednisone doses and lasts for several days. In mild cases, ChEIs usually manage this worsening. However, hospitalization or administration of PLEX or IVIG during steroid initiation is advisable in patients with significant oropharyngeal or respiratory symptoms.
An alternative approach favored by some is to begin prednisone with 20 mg/day and increase the dose by 10 mg every 1 to 2 weeks until improvement begins. The dose is maintained until improvement is maximum, and then tapered as above. Exacerbations still may occur with this protocol, but the onset of such worsening and the therapeutic response are less predictable. A similar dose schedule is common in OMG (see Ocular Myasthenia Gravis, later in this chapter).
Prednisone is inexpensive, has a quick onset of response, and an established track record in MG, but its use is limited by the numerous and frequent side effects (Table 78.5), the severity and frequency of which increase when high doses are given for more than 1 month. Most side effects improve with dose reduction and become minimal at less than 20 mg every other day. A low fat, low-sodium diet, and exercise will minimize the weight gain associated with prednisone use. Supplemental calcium and vitamin D with bisphosphonate therapy are useful to counter osteopenia, particularly in postmenopausal women. Treat patients with peptic ulcer disease or symptoms of gastritis accordingly. Prednisone is contraindicated in patients with untreated tuberculosis.
| Side Effect | Treatment/Prevention |
|---|---|
| Weight gain/fluid retention | Low calorie, low-fat, sodium-restricted diet; exercise |
| Glucose intolerance | Monitor blood glucose/treat |
| Osteopenia/osteoporosis/avascular necrosis | Calcium and vitamin D supplementation, bisphosphonates |
| Hypertension | Monitor/treat |
| Cataracts/glaucoma | At least yearly ophthalmological evaluation |
| Steroid myopathy | Exercise/high-protein diet |
| Peptic ulcer disease | Proton pump inhibitors, H2 blockers |
Prednisone given with azathioprine, cyclosporine, mycophenolate mofetil (MMF), or other immunosuppressant drugs may produce more benefit than either drug alone (see next section, Immunosuppressant Drugs).
Immunosuppressant Drugs
Several immunosuppressant drugs are reportedly effective in MG (see Table 78.3). Azathioprine (AZA) is a purine antimetabolite that interferes with T- and B-cell proliferation and is the nonsteroidal immunosuppressant with the longest track record in MG. It improves weakness in most patients, but benefit may not be apparent for 6 to 12 months. The initial dose is 50 mg/day, which increases by 50 mg/day every 7 days to a total of 150 to 200 mg/day (2-3 mg/kg/day). After achieving maximum benefit, slowly taper the dose to the minimal effective dose, which may be as low as 50 mg/day. Symptom recurrence follows discontinuation or reduction below the minimal effective dose. Patients may respond better and more rapidly when starting prednisone concurrently. An idiosyncratic reaction with flulike symptoms occurs within 10 to 14 days after starting AZA in 15% to 20% of patients; this reaction requires stopping the drug. The use of divided doses after meals or by dose reduction minimizes gastrointestinal irritation. Leukopenia and even pancytopenia can occur at any time during treatment but are not common. Liver toxicity is also possible and heralded by elevations in the serum transaminases. To guard against this, monitor complete blood cell counts and liver enzymes every week during the first month, every 1 to 3 months for a year, and every 3 to 6 months thereafter. Reduce the dose if the peripheral white blood cell (WBC) count falls below 3500 cells/mm3, and then gradually increase after the WBC count rises. Stop the drug immediately if counts fall below 1000 WBC/mm3. Also discontinue treatment if the serum transaminase concentration exceeds twice the upper limit of normal, and restart at lower doses after values become normal. There are rare reports of AZA-induced pancreatitis, but the cost-effectiveness of monitoring serum amylase concentrations is not established.
MMF selectively blocks purine synthesis, thereby suppressing both T- and B-cell proliferation. Pilot studies and retrospective series indicate efficacy in MG (Hehir et al., 2010; Meriggioli et al., 2003). However, data from two randomized controlled trials failed to show additional benefit of MMF over 20 mg daily prednisone as initial immunotherapy of MG (The Muscle Study Group, 2008) and did not show a significant steroid-sparing effect of MMF in patients on prednisone (Sanders et al., 2008). Several factors have been cited as possible explanations for these negative results, including the generally mild disease of the patients, the better-than-expected response to relatively low-dose daily prednisone, and the short duration of the studies (Sanders and Siddiqi, 2008). The clinical efficacy of MMF in MG remains an open question, but it continues to be widely used in the treatment of MG as monotherapy or as a steroid-sparing agent, mainly because many experts are convinced that it is effective, and it has a favorable side-effect profile.
Cyclosporine (CYA) is a potent immunosuppressant that binds to the cytosolic protein, cyclophilin (immunophilin), of immunocompetent lymphocytes, especially T lymphocytes. This complex of CYA and cyclophilin inhibits calcineurin, which activates transcription of interleukin 2 (IL-2). It also inhibits lymphokine production and interleukin release and leads to reduced function of effector T cells. Retrospective analyses have reported improvement in most MG patients taking CYA, with or without corticosteroids (Ciafaloni et al., 2000). Many medications interact with CYA and must be avoided or used with caution. Hypertension and cumulative renal toxicity are common reactions of CYA, and we use this agent in MG only when other immunosuppressants are contraindicated or ineffective.
Recent reports indicate that tacrolimus (FK506) may be effective in the treatment of MG (Evoli et al., 2002; Ponseti et al., 2005), including a randomized (though unblinded) study in 36 de novo MG patients (Nagane et al., 2005). Use of doses from 3 to 5 mg/day have a favorable side effect profile. Tacrolimus is in the same class of immunosuppressants as CYA, with a similar mechanism of action. It appears to be less nephrotoxic than CYA at doses used in published MG reports, but hyperglycemia due to transcriptional inhibition of insulin is relatively common in transplant patients receiving tacrolimus. Pending further study, it should be considered as adjunctive therapy in refractory MG and as a steroid-sparing agent in patients intolerant or unresponsive to AZA, MMF, and CYA.
Cyclophosphamide (CP) given IV in monthly pulsed doses has been used in severe refractory GMG (de Feo et al., 2002; Drachman et al., 2002). In a randomized controlled trial, patients with refractory MG had improved muscle strength and reduced steroid requirement after pulsed doses of IV CP (500 mg/m2). There are reports of therapeutic responses in refractory MG after a one-time, high-dose (50 mg/kg) IV course of CP for 4 days, followed by rescue therapy. Side effects of CP are common and potentially serious and include myelosuppression, hemorrhagic cystitis, and an increased risk for infection and malignancy. For this reason, CP should be reserved for patients with truly refractory severe disease.
In a recent review of 1000 MG patients who received immunosuppressants for at least 1 year, all forms of MG benefited from immunosuppression: the rate of remission or minimal manifestations (Jaretzki et al., 2000) ranged from 85% in OMG to 47% in thymoma-associated disease (Sanders and Evoli, 2010). Prednisone was used in the great majority of these patients, and AZA was the first-choice nonsteroidal immunosuppressant; MMF and CYA were used as second-choice agents. Treatment was ultimately discontinued in nearly 20% of anti-AChR-Ab-positive EOMG patients, but in only 7% of thymoma cases. The risk of complications was related to drug dosage, treatment duration, and patient characteristics, the highest rate of serious side effects (20%) occurring in LOMG and the lowest (4%) in early-onset disease.
Thymectomy
Thymectomy is widely used as treatment for nonthymomatous MG, although no prospective controlled study of efficacy exists. Based on review of existing studies, the Quality Standards Subcommittee of the American Academy of Neurology concluded that MG patients undergoing thymectomy are twice as likely to attain medication-free remission, 1.6 times as likely to become asymptomatic, and 1.7 times as likely to improve (Gronseth and Barohn, 2000). However, the authors expressed uncertainty as to whether the observed improvement was due to thymectomy or explicable by differences in baseline characteristics. Their practice recommendations were that for patients with nonthymomatous autoimmune MG, thymectomy is recommended as an option to increase the probability of remission or improvement. An international prospective single-blinded randomized trial of thymectomy in nonthymomatous MG is currently ongoing, and will hopefully clarify this issue (Newsom-Davis et al., 2008).
We consider repeat thymectomy when relapse follows a good response to the initial surgery or if there is concern that thymic tissue removal had been incomplete. MRI with appropriate cardiac gating may be useful in identifying residual thymus tissue, although many authors believe that the clinical suspicion should be the basis upon which repeat surgery is considered (Jaretzki, 2003).
Evolving Treatments
Rituximab is a chimeric monoclonal antibody directed against the B-cell surface marker, CD20. Case reports and small case series suggest that MG patients, particularly those with anti-MuSK antibodies, may improve after treatment with rituximab (Guptill and Sanders, 2010; Meriggioli and Sanders, 2009). Controlled trials are needed in both anti-AChR and anti-MuSK-positive disease.
A single case series reports improvement in MG following treatment with etanercept (recombinant human tumor necrosis factor (TNF) receptor:Fc) (Tuzun et al., 2005). Patients with lower pretreatment plasma IL-6 and interferon (IFN)-γ levels responded better. The mechanism of action in MG is unknown, and its role in treatment is yet unproven.
Complement inhibition has been shown to be effective in experimental MG (Soltys et al., 2009), and clinical trials in human MG are underway. Autologous stem cell transplantation has been performed in refractory MG patients (Pringle and Atkins, 2005), but the role of this procedure for MG and other autoimmune disorders is unclear at this time.
Association of Myasthenia Gravis with Other Diseases
MG is often associated with other immune-mediated diseases, especially hyperthyroidism and rheumatoid arthritis. Seizures occur with increased frequency in children with MG. One-fifth of our MG patients have another disease: 7% had diabetes mellitus before corticosteroid treatment, 6% have thyroid disease, 3% have an extrathymic neoplasm, and less than 2% have rheumatoid arthritis. Reports of MG cases related to human immunodeficiency virus and after allogeneic bone marrow transplantation suggest a more than coincidental relationship. Extrathymic malignancies have been reported to be common in MG patients, especially in the older age group, possibly owing to a common background of immune dysregulation (Levin et al., 2005).
Treatment of Associated Diseases and Medications to Avoid
Use drugs that adversely affect NMT (Box 78.2) with caution. Many antibiotics fall into this category, particularly aminoglycosides, fluoroquinolones, and macrolides. Ophthalmic preparations of beta-blockers and aminoglycoside antibiotics may cause worsening of ocular symptoms. Never use d-penicillamine, because it can induce MG. When using corticosteroids to treat concomitant illness, anticipate and explain the potential adverse and beneficial effects to the patient.
Box 78.2 Medications That Adversely Affect Neuromuscular Transmission
1. Alpha-interferon, botulinum toxin, d-penicillamine, and telithromycin (Ketek) should never be used in myasthenic patients.
2. The following drugs produce worsening of myasthenic weakness in most patients who receive them. Use with caution, and monitor patient for exacerbation of myasthenic symptoms:
3. Many other drugs are reported to exacerbate the weakness in some patients with myasthenia gravis (MG). All MG and Lambert-Eaton syndrome patients should be observed for increased weakness whenever any new medication is started. An up-to-date reference document for such adverse interactions is maintained on the web site of the Myasthenia Gravis Foundation of America (http://www.myasthenia.org/drugs/reference.htm).
Special Situations
Myasthenic Crisis
In MG patients with progressive respiratory symptoms, no single factor determines the need for ventilatory support. The safest approach is to admit the patient to an intensive care unit and observe closely for impending respiratory insufficiency. Serial measurements of negative inspiratory force (NIF) provide the best measure of deteriorating respiratory function in MG. Respiratory assistance is needed when the NIF is less than −20 cm H2O, when tidal volume is less than 4 to 5 mL/kg body weight and maximum breathing capacity is less than three times the tidal volume, or when the forced vital capacity is less than 15 mL/kg body weight. Use a mask and breathing bag acutely. Noninvasive mechanical ventilation using bilevel positive-pressure ventilation (BiPAP) may avoid the need for intubation in patients in crisis without hypercapnia (Rabinstein and Wijdicks, 2002). In patients with Pco2 greater than 50 mm Hg, tracheal intubation should be done with a low-pressure, high-compliance cuffed endotracheal tube. A volume-controlled respirator set to provide tidal volumes of 400 to 500 mL and automatic sighing every 10 to 15 minutes is preferred. Check the pressure of the tube cuff frequently, and verify the tube position daily by chest radiographs. Use assisted respiration whenever possible so that the patient’s own respiratory efforts trigger the respirator. Use an oxygen-enriched atmosphere only when arterial blood oxygen values fall below 70 mm Hg. Humidify the inspired gas to at least 80% at 37°C to prevent drying of the tracheobronchial tree. Remove tracheal secretions periodically using aseptic aspiration techniques. Low-pressure, high-compliance endotracheal tubes may be tolerable for long periods and usually obviate the need for tracheostomy.
Many case series report short-term benefit from PLEX and IVIG in myasthenic crisis. Retrospective studies suggest that both are equally effective in disease stabilization (Murthy et al., 2005). Others suggest that PLEX is superior, producing more rapid respiratory improvement (Qureshi et al., 1999). We recommend PLEX in the treatment of crisis except when there is hemodynamic instability, sepsis, coagulopathy, or during the first trimester of pregnancy.
Ocular Myasthenia Gravis
Treatment of OMG requires an accurate assessment of the patient’s functional impairment and its effects on daily life. ChEIs may control symptoms adequately in some patients, but the benefit is often partial and not long lived, whereas prednisone is often quite effective. The decision to initiate steroid therapy will depend upon the risk/benefit assessment, which is significantly different in patients considering treatment for purely cosmetic reasons versus those in whom diplopia has a profound bearing on their livelihood (pilots, surgeons, etc.). Existing evidence suggests that prednisone treatment may delay or reduce the frequency of progression of OMG to generalized disease (Kupersmith et al., 2003). Start prednisone at an initial dose of 10 to 20 mg/day, with gradual increases every 3 to 5 days until achieving a clinical response. Alternatively, begin treatment at a dose of 30 to 60 mg, since the risk of steroid-associated exacerbation is less in OMG. In OMG, use a maintenance dosage of corticosteroids that does not significantly suppress the immune system and causes few major systemic adverse effects. Consider a steroid-sparing agent if this is not the case. In general, OMG is not an indication for thymectomy, but this may be effective in some patients.
Childhood Myasthenia Gravis
The onset of immune-mediated MG before age 18 is referred to as juvenile myasthenia gravis (JMG) (Andrews and Sanders, 2002). Some 20% of JMG and almost 50% of those with onset before puberty are seronegative; the distinction from a congenital myasthenic syndrome is most challenging in the latter group (see Congenital Myasthenic Syndromes, later in this chapter). Electrodiagnostic studies identify a defect in NMT but have distinguishing features in only a few forms of congenital MG. A beneficial response to PLEX exchange or IVIG may help to establish the diagnosis of autoimmune MG. Many children who are initially seronegative later develop AChR antibodies. Thymomas are rare in this age group.
Pregnancy
Therapeutic abortion is rarely if ever needed because of MG, and the frequency of spontaneous abortion is not increased. Oral ChEIs are the first-line treatment during pregnancy. Intravenous ChEIs may produce uterine contractions and are contraindicated. Prednisone is the immunosuppressive agent of choice. We do not use immunosuppressive drugs during pregnancy because of theoretical potential mutagenic effects, although others feel that AZA and even CYA can be used safely during pregnancy (Ferrero et al., 2005). Increased risk of fetal malformation has been reported when men used AZA prior to conception (Norgard et al., 2004). MMF can cause birth defects and is contraindicated during pregnancy. PLEX or IVIG are useful when requiring an immediate (albeit temporary) improvement during pregnancy, but avoid PLEX during the first trimester.
Transient Neonatal Myasthenia Gravis
A temporary form of MG affects 10% to 20% of newborns whose mothers have immune-mediated MG. The severity of symptoms in the newborn does not correlate with the severity of symptoms in the mother. The maternal antibody level correlates with the frequency and severity of transient neonatal myasthenia gravis (TNMG), and TNMG occurs only rarely in infants of seronegative mothers. Weakness may manifest in utero, particularly when maternal antibodies are directed against the fetal AChR, and may lead to arthrogryposis multiplex congenita (Barnes et al., 1995). Consider decreased fetal movement as a possible indication for PLEX or IVIG. Birth of a child with arthrogryposis should also prompt a search for MG in the mother. An affected mother who delivers an infant with TNMG is likely to have similarly affected subsequent infants. Consider prophylactic treatment with PLEX and/or steroids in a woman with a previously affected child, as the risk of recurrent TNMG is high.
Congenital Myasthenic Syndromes
The congenital myasthenic syndromes (CMS) are a group of neuromuscular junction diseases caused by genetic defects of muscle end-plate molecules involved in NMT (Engel, 2008). They are individually and collectively rare but are of interest because they produce novel insights into the understanding of NMJ physiology. Symptoms are present at birth in most forms but may go unrecognized until adolescence or adulthood, particularly when progression is gradual and clinical expression is mild. Appropriate diagnosis is important, since many of the syndromes are treatable with drugs that increase the availability of ACh at the muscle end-plate or alter the kinetics of the AChR.
ChEIs, sometimes in very high doses, improve limb muscle weakness in some forms of CMS but may worsen it in others. The weakness in some children responds to 3,4-diaminopyridine (3,4-DAP) (Harper and Engel, 2000). Thymectomy and immunosuppression are not effective.
Mutations Resulting in AChR Deficiency
This is a genetically heterogeneous group with patients having various AChR subunit or rapsyn mutations. The age of symptom onset ranges from infancy to adulthood. Clinical manifestations include hypotonia, respiratory insufficiency, weakness of ocular and bulbar muscles, and skeletal deformities. The findings on electrodiagnostic studies are variable and depend on the severity and distribution of weakness. RNS studies usually demonstrate a decrement, but the decrement may be absent or restricted to facial muscles in mild cases. Jitter is increased in all reported cases (Stålberg et al., 2010).
Choline Acetyl Transferase Deficiency
A decremental response to RNS is usually present in weak muscles. The decrement may repair with brief exercise but becomes more marked with prolonged exercise or continuous repetitive stimulation at 3 Hz for 3 to 5 minutes. Jitter also becomes progressively worse during continuous nerve stimulation (Stålberg et al., 2010). ChEIs improve strength in most children with ChAT deficiency. Symptoms tend to lessen in adolescence and adulthood, when the disease resembles mild autoimmune myasthenia gravis or a congenital myopathy. We have seen sustained symptomatic improvement in children from several families with this syndrome when 3,4-DAP is given with pyridostigmine.
Slow-Channel Congenital Myasthenic Syndrome
Variable expression results in a wide spectrum of clinical manifestations and severity in slow-channel congenital myasthenic syndrome (SCCMS), which is the only form of CMS with autosomal dominant inheritance. Severe cases present in infancy or early childhood, but mild cases may present in adulthood, as late as the seventh decade. Characteristically, the weakness in SCCMS involves muscles of the neck and distal regions of the upper limbs, and the intrinsic hand muscles and digit extensors are particularly weak and atrophic. Ptosis, ophthalmoparesis, dysarthria, dysphagia, proximal limb weakness, and respiratory insufficiency also occur in some cases. Worsening of weakness occurs with ChEI administration. RNS shows a decremental response. Repetitive discharges appear after single nerve stimulations, similar to those seen in ChEI toxicity or congenital deficiency of end-plate acetylcholinesterase (see later discussion). The underlying defect is a prolonged open time of the ACh channel. Quinidine sulfate and fluoxetine, which reduce AChR channel open time, may improve strength in this condition (Harper et al., 2003).
DOK-7 Mutations
DOK-7 is a muscle protein that activates MuSK and is critical in end-plate development and AChR aggregation. Some CMS patients with this mutation were previously characterized as having “limb-girdle myasthenia,” but the clinical manifestations of CMS associated with DOK-7 mutations may also be indistinguishable from patients with AChR deficiency (Selcen et al., 2008), including reduced fetal movements in utero and static and fatigable weakness of cranial, respiratory, and limb muscles. The electrodiagnostic findings are also indistinguishable from patients with congenital AChR deficiency. Response to ChEIs is variable, with some patients improving but many demonstrating no response. Ephedrine and 3,4-DAP may produce modest benefit.
Congenital Acetylcholinesterase Deficiency
End-plate acetylcholinesterase (AChE) deficiency results from a recessive mutation of COLQ, the gene coding for the collagenous tail of the heteromeric AChE molecule at the muscle end-plate (Ohno et al., 1999). Presentation is usually in infancy or early childhood. The symptoms are generalized weakness, ptosis, ophthalmoparesis, bulbar and limb weakness, underdevelopment of muscles, slowed pupillary responses to light, and either no response or clinical worsening with ChEIs. Skeletal deformities include lordosis or scoliosis that worsens with prolonged standing. Nerve conduction studies reveal one or more repetitive compound muscle action potentials with single stimuli. No effective treatment has been described. ChEIs do not help and may make symptoms worse.
Lambert-Eaton Syndrome
LES results from an immune-mediated attack against the P/Q-type voltage-gated calcium channels (VGCC) on presynaptic cholinergic nerve terminals at the neuromuscular junction and in autonomic ganglia (Fig. 78.6). LES, first described in patients with lung cancer (CA-LES), also occurs as an organ-specific autoimmune disorder in the absence of cancer (NCA-LES).
LES is usually clinically quite distinct from MG. Most patients report gradual onset of lower-extremity weakness, sometimes with muscle tenderness. Dry mouth is a common symptom of autonomic dysfunction; other features are erectile dysfunction, postural hypotension, constipation, and dry eyes. Ocular and bulbar symptoms are generally not prominent (O’Neill et al., 1988; Tim et al., 2000; Wirtz et al., 2002), but are reported in some patients in a pattern suggesting MG (Burns et al., 2003; Titulaer et al., 2008). Prolonged apnea and ventilator dependence may follow use of neuromuscular blocking agents for surgery (Anderson et al., 1953), but respiratory failure is otherwise uncommon in the absence of primary pulmonary disease (Barr et al, 1993; Smith and Wald, 1996).
Diagnostic Procedures in Lambert-Eaton Syndrome
Confirmation of the diagnosis is by the following characteristic electrodiagnostic findings (see Chapter 32B): CMAPs with low amplitude, which increases during 20- to 50-Hz stimulation and after brief maximum voluntary muscle activation. Low-frequency repetitive nerve stimulation produces a decrementing response in a hand or foot muscle in almost all patients, and almost all have small CMAPs in some distal muscle (Tim et al., 2000). The characteristic increase in CMAP size after activation is more prominent in distal muscles, but it may be necessary to examine several muscles to demonstrate this important finding.
Immunoprecipitation assays demonstrate VGCC antibodies in almost all patients with CA-LES and in more than 90% with NCA-LES (Harper and Lennon, 2002). Low titers of VGCC antibodies have also been reported in SLE and rheumatoid arthritis (Lang et al., 1993). Early in the disease, these antibodies may not be detectable. Repeat antibody testing may be useful.
In a recent report, antibodies to SOX1, a transcription factor involved in neural development, were found in 64% of CA-LES patients with SCLC and in none with NCA-LES (Sabater et al., 2008). Confirmation of the presence of SOX1 antibodies are a marker for an underlying cancer in LES patients.
Immunopathology of Lambert-Eaton Syndrome
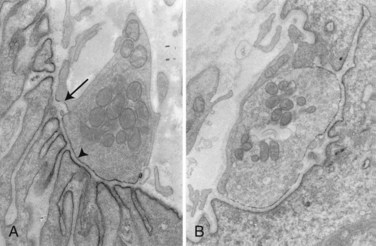
Studies indicate that P/Q VGCC are the target of disease-causing antibodies in LES. The number of motor nerve terminal active zone particles representing the VGCC is reduced (Fig. 78.7) (Fukunaga et al., 1982). Similar changes occur in mice injected with IgG from LES patients. The mechanism is probably from cross-linking of the VGCC by antibodies that down-regulate VGCC expression by antigenic modulation.

P/Q VGCC antibodies are found in up to 90% of non–immune suppressed LES patients (Lennon et al., 1995). SCLC cells are of neuroectodermal origin and contain high concentrations of VGCC. In CA-LES, the neuroectodermal antigens expressed by SCLC cells mimic VGCC and induce production of VGCC antibodies as a paraneoplastic syndrome. In NCA-LES, as in other primary autoimmune disorders, the presumption is that altered self-tolerance induces production of VGCC antibodies as part of a more general immune-mediated state. VGCC antibody titers do not correlate with disease severity among individuals, but the antibody levels may fall as the disease improves in patients receiving immunosuppression.
Treatment of Lambert-Eaton Syndrome
Tailor therapy to the individual, based on the severity of weakness, underlying disease, life expectancy, and response to previous treatment. Randomized controlled trials have shown that 3,4-DAP and IVIG improve muscle strength scores and CMAP amplitudes in patients with LES (Maddison and Newsom-Davis, 2005; McEvoy et al., 1989; Oh et al., 2009; Sanders et al., 2000; Wirtz et al., 2009). Other treatments such as PLEX, corticosteroids, and immunosuppressive agents, including rituximab (Maddison et al., 2010), may be of benefit in some patients but have not been tested in controlled trials.
Administering 3,4-DAP facilitates release of ACh from motor nerve terminals and produces clinically significant improvement of strength and autonomic symptoms in most LES patients (Tim et al., 2000). Therapeutic responses occur with doses of 5 to 25 mg 3 to 4 times a day; seizures may occur at doses higher than 100 mg/day. Concomitant use of pyridostigmine, 30 to 60 mg, 3 or 4 times a day enhances the response to 3,4-DAP. Side effects usually are negligible. Transitory perioral and digital paresthesias occur with doses greater than 10 to 15 mg. Cramps and diarrhea may occur when 3,4-DAP is given with pyridostigmine and can be minimized by reducing the dose of pyridostigmine; 3,4-DAP is a safe and effective treatment for LES but is not available for general clinical use in the United States. It is available for individual patients upon submission of a Treatment-Use Investigational New Drug application by the administering physician. Information on the application process can be obtained from Jacobus Pharmaceutical Co., Inc., Princeton, New Jersey, Fax No. 609-799-1176.
Both PLEX and IVIG provide short-term improvement in some patients with LES (Tim et al., 2000), but the results are usually not as good as in MG. If these treatments are not effective, it must be determined if weakness is sufficiently severe to warrant immunotherapy with prednisone, AZA, CYA, or rituximab. In patients with severe weakness, use PLEX or IVIG first, and add prednisone and AZA after improvement begins. Maintaining improvement may require repeated courses of treatment.
In LES patients with cancer, the response to cancer therapy determines the prognosis. In patients without cancer, treatment with immunosuppression produces improvement in many patients, but most require substantial and continuing doses of immunosuppressive medications (Maddison et al., 2001).
Myasthenia Gravis/Lambert-Eaton Syndrome Overlap Syndrome
The clinical presentations of MG and LES are usually quite distinct (Wirtz et al., 2002), but in some patients the clinical and electrodiagnostic findings may be similar, and the correct diagnosis may not obvious. Features that favor MG include prominent ocular muscle weakness, limb weakness that predominates in the arms, and normal muscle stretch reflexes (Wirtz et al., 2002). Features that favor LES include weakness that predominates in the hip girdle muscles, hypoactive or absent reflexes, and autonomic symptoms, especially dry mouth.
Patients with various overlapping features of MG and LES appear in the literature. Two examples are (1) clinical features of MG but facilitation on manual muscle testing or EMG, typical of LES; and (2) clinical and EMG patterns typical of one condition initially that change to the other later, or EMG patterns typical of MG in one muscle and of LES in another. A few patients have been reported who appear to have a true MG/LES overlap syndrome, with antibodies to both the AChR and VGCC (Kanzato et al., 1999; Katz et al., 1998; Newsom-Davis et al., 1991; Oh and Sher, 2005), and we have seen one such case among 1200 patients with acquired MG and 102 with LEMS. The ultimate diagnosis in patients with mixed features of MG and LES may be moot because most treatments are the same for both conditions. Exceptions are that we do not search for cancer other than thymoma in MG, and thymectomy is never a treatment for LES.
Botulism
Botulism is caused by a toxin produced by the anaerobic bacterium, Clostridium botulinum, that blocks the release of ACh from the motor nerve terminal (Cherington, 2007). The result is a long-lasting severe muscle paralysis. Botulism usually follows ingestion of inadequately sterilized contaminated foods. Of eight types of botulinum toxins (A, B, Cα, Cβ, D, E, F, and G), types A and B are the cause of most cases of botulism in the United States. Transmission of type E is in seafood. All forms of the toxin block ACh release from the presynaptic motor nerve terminal and the parasympathetic and sympathetic nerve ganglia. The intracellular target is the SNARE proteins of the presynaptic membrane. Neuromuscular symptoms usually begin 12 to 36 hours after ingestion of contaminated food and are preceded by nausea and vomiting. Not all people who ingest the contaminated food become symptomatic.
Clinical Features of Botulism
Infantile botulism results from the growth of C. botulinum in the immature gastrointestinal tract and the elaboration of small quantities of toxin over a prolonged period (Jones, 2002). Honey is a vehicle commonly incriminated as carrying the spores of C. botulinum that produce infantile botulism. Symptoms of constipation, lethargy, poor suck, and weak cry usually begin at approximately 4 months of age. Examination reveals weakness of the limb and oropharyngeal muscles, poorly reactive pupils, and hypoactive tendon reflexes. Most patients require ventilatory support. Demonstrating botulinum toxin in the stool or isolation of C. botulinum from stool culture confirms the diagnosis of infant botulism.
Electromyographic Findings in Botulism
Electrophysiological abnormalities in botulism tend to evolve with time and may not be present early in the disease (Padua et al., 1999). The EMG findings in botulism include:
• Reduced CMAP amplitude in at least two muscles.
• At least 20% facilitation of CMAP amplitude during tetanic stimulation.
• Persistence of facilitation for at least 2 minutes after activation.
• No postactivation exhaustion.
• Short-duration motor unit potentials resembling myopathic motor units in affected muscles on EMG.
High-frequency RNS may show pseudofacilitation without true posttetanic facilitation, but SFEMG demonstrates markedly increased jitter and blocking in virtually every case (Padua et al., 1999). Jitter and blocking may decrease as the firing rate increases, but this is not a consistent finding.
Treatment of Botulism
Treatment consists of administration of bivalent (type A and B) or trivalent (A, B, and E) antitoxin. Antibiotic therapy is not effective, since the cause of symptoms (in all but infantile botulism) is the ingestion of toxin rather than organisms. In infantile botulism, IV human botulism immune globulin (BIG-IV) neutralizes the toxin for several days after illness onset, shortens the length and cost of the hospital stay, and reduces the severity of illness (Arnon et al., 2006). Otherwise, treatment is supportive. ChEIs are usually not beneficial; 3,4-DAP may improve strength but not respiratory function. With improvements in intensive care, the mortality rate has declined to about 20%. Depending on initial severity of illness, recovery may be quite prolonged, with many patients continuing to have symptoms a year or longer after the onset of illness.
Other Causes of Abnormal Neuromuscular Transmission
A defect in NMT may be a cause of weakness in critically ill patients and is often due to administration of drugs such as antibiotics, antiarrhythmics, and nondepolarizing neuromuscular blocking agents (Gorson, 2005). Prolonged use of these agents may result in weakness due to persistent neuromuscular blockade even hours or days after discontinuation.
Anderson H.J., Churchill-Davidson H.C., et al. Bronchial neoplasm with myasthenia. Prolonged apnea after administration of succinylcholine. Lancet. 1953;ii:1291-1293.
Andrews P.I., Sanders D.B. Juvenile myasthenia gravis. In: Jones H.R., DeVivo D.C., Darras B.T. Neuromuscular Disorders of Infancy, Childhood, and Adolescence. Boston: Butterworth Heinemann; 2002:575-597.
Arnon S.S., Schechter R., Maslanka S.E., et al. Human botulism immune globulin for the treatment of infant botulism. N Engl J Med. 2006;354:462-471.
Barnes P.R.J., Kanabar D.J., Brueton L., et al. Recurrent congenital arthrogryposis leading to a diagnosis of myasthenia gravis in an initially asymptomatic mother. Neuromuscul Disord. 1995;5:59-65.
Barr C.W., Claussen G., Thomas D., et al. Primary respiratory failure as the presenting symptom in Lambert- Eaton myasthenic syndrome. Muscle Nerve. 1993;16:712-715.
Bau V., Hanisch F., Hain B., et al. Ocular involvement in MuSK antibody-positive myasthenia gravis. Klin Monbl Augenheilkd. 2006;223:81-83.
Burns T.M., Russell J.A., LaChance D.H., et al. Oculobulbar involvement is typical with Lambert-Eaton myasthenic syndrome. Ann Neurol. 2003;53:270-273.
Caress J.B., Hunt C.H., Batish S.D. Anti-MuSK myasthenia gravis presenting with purely ocular findings. Arch Neurol. 2005;62:1002-1003.
Cherington M. Botulism, Elsevier, Engel A.G., editor. Neuromuscular Junction Disorders. 2007.
Ciafaloni E., Nikhar N.K., Massey J.M., et al. Retrospective analysis of the use of cyclosporine in myasthenia gravis. Neurology. 2000;55:448-450.
Conti-Fine B.M., Protti M.P., Belone M., et al. Myasthenia Gravis and Its Experimental Model: the Immunobiology of an Autoimmune Disease. Georgetown, TX: Landes Bioscience Publishers; 1997.
de Feo L.G., Schottlender J., Martelli N.A., et al. Use of intravenous pulsed cyclophosphamide in severe, generalized myasthenia gravis. Muscle Nerve. 2002;26:31-36.
Donofrio P.D., Berger A., Brannagan T.H., et al. Consensus statement: the use of intravenous immunoglobulin in the treatment of neuromuscular conditions report of the AANEM ad hoc committee. Muscle Nerve. 2009;40:890-900.
Drachman D.B., Jones R.J., Brodsky R.A. Treatment of refractory myasthenia: “rebooting” the immune system with high-dose cyclophosphamide. Neurology. 2002;58(Suppl 3):A328-A329.
Engel A.G. Congenital myasthenic syndromes, Elsevier, Engel A.G., editor. Neuromuscular Junction Disorders. 2008:285-331.
Engel A.G., Lambert E.H., Howard F.M. Immune complexes (IgG and C3) at the motor endplate in myasthenia gravis: ultrastructural and light microscopic localization and electrophysiologic correlation. Mayo Clin Proc. 1977;52:267-280.
Engel A.G., Lindstrom J.M., Lambert E.H., et al. Ultrastructural localization of the acetylcholine receptor in myasthenia gravis and its experimental autoimmune model. Neurology. 1977;27:307-315.
Evoli A., Di Schino C., Marsili F., et al. Successful treatment of myasthenia gravis with tacrolimus. Muscle Nerve. 2002;25:111-114.
Ferrero S., Pretta S., Nicoletti A., et al. Myasthenia gravis: management issues during pregnancy. Eur J Obstet Gynecol Reprod Biol. 2005;121:129-138.
Fukunaga H., Engel A.G., Osame M., et al. Paucity and disorganization of presynaptic membrane active zones in the Lambert-Eaton myasthenic syndrome. Muscle Nerve. 1982;5:686-697.
Gorson K.C. Approach to neuromuscular disorders in the intensive care unit. Neurocrit Care. 2005;3:195-212.
Grob D., Brunner N.G., Namba T., et al. Lifetime course of myasthenia gravis. Muscle Nerve. 2008;37:141-149.
Gronseth G.S., Barohn R.J. Practice parameter: thymectomy for autoimmune myasthenia gravis (an evidence-based review). Neurology. 2000;55:5-15.
Guptill J.T., Sanders D.B. Update on MuSK antibody positive myasthenia gravis. Curr Opin Neurol. 2010;23:530-535.
Harper C.M., Engel A.G. Treatment of 31 congenital myasthenic syndrome patients with 3,4-diaminopyridine. Neurology. 2000;54(Suppl 3):A395.
Harper C.M., Fukudome T., Engel A.G. Treatment of slow-channel congenital myasthenic syndrome with fluoxetine. Neurology. 2003;60:1710-1713.
Harper C.M., Lennon V.A. The Lambert-Eaton myasthenic syndrome. In: Kaminski H.J., editor. Current Clinical Neurology: Myasthenia Gravis and Related Disorders. Totowa, NJ: Humana Press; 2002:269-291.
Hatanaka Y., Claussen G.C., Oh S.J. Anticholinesterase hypersensitivity or intolerance is common in MuSK antibody positive myasthenia gravis. Neurology. 2005;64:A79.
Hehir M.K., Burns T.M., Alpers J.P., et al. Mycophenolate mofetil in AChR-antibody positive myasthenia gravis: outcomes in 102 patients. Muscle Nerve. 2010;41:593-598.
Hohlfeld R., Wekerle H. Reflections on the “intrathymic pathogenesis” of myasthenia gravis. J Neuroimmunol. 2008;201:21-27.
Ing E.B., Ing S.Y., Ing T., et al. The complication rate of edrophonium testing for suspected myasthenia gravis. Can J Ophthalmol. 2005;35:141-144.
Jaretzki A. Thymectomy for myasthenia gravis: analysis of controversies–patient management. Neurologist. 2003;9:77-92.
Jaretzki A., Barohn R.J., Ernstoff R.M., et al. Myasthenia gravis. Recommendations for clinical research standards. Neurology. 2000;55:16-23.
Jones H.R. Infantile botulism and other acquired neuromuscular junction disorders of infancy and childhood. In: Brown W.F., Bolton C.F., Aminoff M.J. Neuromuscular Function and Disease. Philadelphia: W.B. Saunders; 2002:1697-1702.
Kanzato N., Motomura M., Suehara M., et al. Lambert-Eaton myasthenic syndrome with ophthalmoparesis and pseudoblepharospasm. Muscle Nerve. 1999;22:1727-1730.
Katz J.S., Wolfe G.I., Bryan W.W., et al. Acetylcholine receptor antibodies in the Lambert-Eaton myasthenic syndrome. Neurology. 1998;50:470-475.
Kuks J.B.M., Oosterhuis H.J.G.H. Clinical presentation and epidemiology of myasthenia gravis. In: Kaminski H.J., editor. Current Clinical Neurology: Myasthenia Gravis and Related disorders. Totowa, NJ: Humana Press; 2004:93-113.
Kupersmith M.J., Latkany R., Homel P. Development of generalized disease at 2 years in patients with ocular myasthenia gravis. Arch Neurol. 2003;60:243-248.
Lang B., Johnston I., Leys K., et al. Autoantibody specificities in Lambert-Eaton myasthenic syndrome. Ann NY Acad Sci. 1993;681:382-393.
Larner A.J. The place of the ice pack test in the diagnosis of myasthenia gravis. Int J Clin Pract. 2004;58:887-888.
Lauriola L., Ranelletti F., Maggiano N., et al. Thymus changes in anti-MuSK-positive and -negative myasthenia gravis. Neurology. 2005;64:536-538.
Leite M.I., Jacob S., Viegas S., et al. IgG1 antibodies to acetylcholine receptors in “seronegative” myasthenia gravis. Brain. 2008;131:1940-1952.
Leite M.I., Strobel P., Jones M., et al. Fewer thymic changes in MuSK antibody-positive than in MuSK antibody-negative MG. Ann Neurol. 2005;57:444-448.
Lennon V.A., Kryzer T.J., Griesmann G.E., et al. Calcium-channel antibodies in the Lambert-Eaton syndrome and other paraneoplastic syndromes. N Engl J Med. 1995;332:1467-1474.
Levin N., Abramsky O., Lossos A., et al. Extrathymic malignancies in patients with myasthenia gravis. J Neurol Sci. 2005;237:39-43.
Maddison P., Lang B., Mills K., et al. Long-term outcome in Lambert-Eaton myasthenic syndrome without lung cancer. J Neurol Neurosurg Psychiatry. 2001;70:212-217.
Maddison P., McConville J., Farrugia M.E., et al. The use of rituximab in myasthenia gravis and Lambert-Eaton myasthenic syndrome. J Neurol Neurosurg Psychiatry. 2010;82:671-673.
Maddison, P., Newsom-Davis, J., 2005. Treatment for Lambert-Eaton myasthenic syndrome. Cochrane Database Syst Rev CD003279.
McEvoy K.M., Windebank A.J., Daube J.R., et al. 3,4-Diaminopyridine in the treatment of Lambert-Eaton myasthenic syndrome. N Engl J Med. 1989;321:1567-1571.
Meriggioli M.N., Rowin J., Richman J.G., et al. Mycophenolate mofetil for myasthenia gravis: a double-blind, placebo-controlled pilot study. Ann NY Acad Sci. 2003;998:494-499.
Meriggioli M.N., Sanders D.B. Myasthenia gravis: diagnosis. Semin Neurol. 2004;24:31-39.
Meriggioli M.N., Sanders D.B. Autoimmune myasthenia gravis: emerging clinical and biological heterogeneity. Lancet Neurology. 2009;8:475-490.
Murthy J.M.K., Meena A.K., Chowdary C.V.S., et al. Myasthenic crisis: clinical features, complications and mortality. Neurol India. 2005;53:37-40.
Nadeau J.A., Bhibhatbhan A., McDougall D., et al. Identification and comparison of adverse events for preparations of IVIG in patients with neuromuscular disorders. Clin Neurol Neurosurg. 2010;112:467-469.
Nagane Y., Utsugisawa K., Obara D., et al. Efficacy of low-dose FK506 in the treatment of myasthenia gravis–a randomized pilot study. Eur Neurol. 2005;53:146-150.
Newsom-Davis J., Cutter G., Wolfe G.I., et al. Status of the thymectomy trial for nonthymomatous myasthenia gravis patients receiving prednisone. Ann N Y Acad Sci. 2008;1132:344-347.
Newsom-Davis J., Leys K., Vincent A., et al. Immunological evidence for the co-existence of the Lambert-Eaton myasthenic syndrome and myasthenia gravis in two patients. J Neurol Neurosurg Psychiatry. 1991;54:452-453.
Niks E.H., Kuks J.B.M., Roep B.O., et al. Strong association of MuSK antibody-positive myasthenia gravis and HLA-DR14-DQ5. Neurology. 2006;66:1772-1774.
Norgard B., Pedersen L., Jacobsen J., et al. The risk of congenital abnormalities in children fathered by men treated with azathioprine or mercaptopurine before conception. Aliment Pharmacol Ther. 2004;19:679-685.
O’Neill J.H., Murray N.M., Newsom-Davis J. The Lambert-Eaton myasthenic syndrome. A review of 50 cases. Brain. 1988;111:577-596.
Oh S.J., Claussen G.G., Hatanaka Y., et al. 3,4-Diaminopyridine is more effective than placebo in a randomized, double-blind, cross-over drug study in LEMS. Muscle Nerve. 2009;40:795-800.
Oh S.J., Sher E. MG and LEMS overlap syndrome: case report with electrophysiological and immunological evidence. Clin Neurophysiol. 2005;116:1167-1171.
Ohno K., Brengman J.M., Felice K.J., et al. Congenital end-plate acetylcholinesterase deficiency caused by a nonsense mutation and an A–>G splice-donor-site mutation at position +3 of the collagenlike-tail-subunit gene (COLQ): how does G at position +3 result in aberrant splicing? Am J Hum Genet. 1999;65:635-644.
Padua L., Aprile I., Lo Monaco M., et al. Neurophysiological assessment in the diagnosis of botulism: usefulness of single-fiber EMG. Muscle Nerve. 1999;22:1388-1392.
Pascuzzi R.M. The edrophonium test. Semin Neurol. 2003;23:83-88.
Pascuzzi R.M., Coslett B., Johns T.R. Long-term corticosteroid treatment of myasthenia gravis: report of 116 patients. Ann Neurol. 1984;15:291-298.
Phillips L.H. The epidemiology of myasthenia gravis. Semin Neurol. 2004;24:17-20.
Ponseti J.M., Azem Z., Fort J.M., et al. Long-term results of tacrolimus in cyclosporine- and prednisone-dependent myasthenia gravis. Neurology. 2005;64:1641-1643.
Pringle C.E., Atkins H.L. Stem cell transplant results in sustained remission of generalized myasthenia gravis. J Neurol Sci. 2005;238:S95-S96.
Qureshi A.I., Choudhry M.A., Akbar M.S., et al. Plasma exchange versus intravenous immunoglobulin treatment in myasthenic crisis. Neurology. 1999;52:629-632.
Rabinstein A., Wijdicks E.F.M. BiPAP in acute respiratory failure due to myasthenic crisis may prevent intubation. Neurology. 2002;59:1647-1649.
Romi F., Skeie G.O., Gilhus N.E., et al. Striational antibodies in myasthenia gravis–reactivity and possible clinical significance. Arch Neurol. 2005;62:442-446.
Sabater L., Titulaer M., Saiz A., et al. SOX1 antibodies are markers of paraneoplastic Lambert-Eaton myasthenic syndrome. Neurology. 2008;70:924-928.
Sanders D.B., El Salem K., Massey J.M., et al. Clinical aspects of MuSK antibody positive seronegative MG. Neurology. 2003;60:1978-1980.
Sanders D.B., Evoli A. Immunosuppressive therapies in myasthenia gravis. Autoimmunity. 2010;43:428-435.
Sanders D.B., Massey J.M. Clinical features of myasthenia gravis, Elsevier, Engel A.G., editor. Neuromuscular Junction Disorders. 2008:229-252.
Sanders D.B., Massey J.M., Sanders L.L., et al. A randomized trial of 3,4-diaminopyridine in Lambert-Eaton myasthenic syndrome. Neurology. 2000;54:603-607.
Sanders D.B., McDermott M., Thornton C., et al. A trial of mycophenolate mofetil with prednisone as initial immunotherapy in myasthenia gravis. Neurology. 2008;71:394-399.
Sanders D.B., Siddiqi Z.A. Lessons from two trials of mycophenolate mofetil in myasthenia gravis. Ann N Y Acad Sci. 2008;1132:249-253.
Selcen D., Milone M., Shen X.M., et al. Dok-7 myasthenia: phenotypic and molecular genetic studies in 16 patients. Ann Neurol. 2008;64:71-87.
Smith A.G., Wald J. Acute ventilatory failure in Lambert-Eaton myasthenic syndrome and its response to 3,4-diaminopyridine. Neurology. 1996;46:1143-1145.
Soltys J., Kusner L.L., Young A., et al. Novel complement inhibitor limits severity of experimentally myasthenia gravis. Ann Neurol. 2009;65:67-75.
Stålberg E.V., Sanders D.B. Jitter recordings with concentric needle electrodes. Muscle Nerve. 2009;40:331-339.
Stålberg E.V., Trontelj J.V., Sanders D.B. Myasthenia gravis and other disorders of neuromuscular transmission. In: Single Fiber EMG. Fiskebåckskil, Sweden: Edshagen Publishing House; 2010:218-266.
Stickler D.E., Massey J.M., Sanders D.B. MuSK-antibody positive myasthenia gravis: clinical and electrodiagnostic patterns. Clin Neurophysiol. 2005;116:2065-2068.
Sussman J.D., Argov Z., McKee D., et al. Antisense treatment for myasthenia gravis. Experience with Monarsen. Ann N Y Acad Sci. 2008;1132:283-290.
The Muscle Study Group. A trial of mycophenolate mofetil with prednisone as initial immunotherapy in myasthenia gravis. Neurology. 2008;71:394-399.
Tim R.W., Massey J.M., Sanders D.B. Lambert-Eaton myasthenic syndrome. Electrodiagnostic findings and response to treatment. Neurology. 2000;54:2176-2178.
Titulaer M.J., Wirtz P.W., Wintzen A.R., et al. Lambert-Eaton myasthenic syndrome with pure ocular weakness. Neurology. 2008;70:86-87.
Tuzun E., Meriggioli M.N., Rowin J., et al. Myasthenia gravis patients with low plasma IL-6 and IFN-[gamma] benefit from etanercept treatment. J Autoimmun. 2005;24:261-268.
Vernino S., Lennon V.A. Autoantibody profiles and neurological correlations of thymoma. Clin Cancer Res. 2004;10:7270-7275.
Vincent A., Lang B. The prevalence of MuSK antibody positive myasthenia gravis worldwide. J Neuroimmunol. 2006;178:233.
Wirtz P.W., Sotodeh M., Nijnuis M., et al. Difference in distribution of muscle weakness between myasthenia gravis and the Lambert-Eaton myasthenic syndrome. J Neurol Neurosurg Psychiatry. 2002;73:766-768.
Wirtz P.W., Verschuuren J.J., van Kijk J.G., et al. Efficacy of 3,4-diaminopyridine and pyridostigmine in the treatment of Lambert-Eaton myasthenic syndrome: a randomized, double-blind, placebo-controlled, crossover study. Clin Pharmacol Ther. 2009;86:44-48.
Zang X., Yang M., Xu J., et al. Clinical and serological study of myasthenia gravis in HuBei Province, China. J Neurol Neurosurg Psychiatry. 2007;78:386-390.
Zinman L., Ng E., Bril V. IV immunoglobulin in patients with myasthenia gravis: a randomized controlled trial. Neurology. 2007;68:837-841.
[/level-membership-for-neurology-category][not-level-membership-for-neurology-category]
Chapter 78 Disorders of Neuromuscular Transmission
Myasthenia Gravis
Epidemiology of Myasthenia Gravis
MG may begin at any age from infancy to very old age. Epidemiological studies report considerable variability in incidence and prevalence around the world (Meriggioli and Sanders, 2009). While methodological differences may explain some of this variability, biological and genetic factors may also play a role. Recent estimates indicate that the U.S. prevalence is approximately 20/100,000, 60,000 patients total (Phillips, 2004). Epidemiological studies have shown an increasing prevalence over the past 50 years, related to an increase in the frequency of diagnosis in elderly patients but also likely due to improved ascertainment, reduced mortality rates, and increased longevity of the population. Gender and age influence the incidence of MG; women are affected nearly three times more often than men before age 40, but the incidence is higher in males after age 50 and roughly equal during puberty. As the population ages, the average age at onset has increased correspondingly. More men than women are now affected, and the majority of MG patients in the United States are older than 50. Detailed population-based data on clinical and serological subtypes of MG (see Myasthenia Gravis Subtypes) are largely lacking.
Clinical Presentation of Myasthenia Gravis
Patients with MG seek medical attention for specific muscle weakness or dysfunction that typically worsens with activity and improves with rest. Although they may also have generalized fatigue or malaise, it is not usually the major or presenting complaint. Ptosis or diplopia is the initial symptom in approximately two-thirds of patients; nearly all will develop both within 2 years (Sanders and Massey, 2008). Difficulty chewing, swallowing, or talking is the initial symptom in one-sixth of patients, and limb weakness in 10%. Rarely, the initial weakness is limited to single muscle groups such as neck or finger extensors, hip flexors, or ankle dorsiflexors.
The course of disease is variable but usually progressive. Weakness remains restricted to the ocular muscles in approximately 10% to 15% of cases (see Ocular Myasthenia Gravis, later in this chapter), although a higher proportion has been reported in Asian populations (Meriggioli and Sanders, 2009). In the rest, weakness progresses to involve nonocular muscles during the first 3 years and ultimately involves facial, oropharyngeal, and limb muscles (generalized MG). Maximum weakness occurs during the first year in two-thirds of patients. Before the introduction of corticosteroids for treatment, approximately one-third of patients improved spontaneously, one-third became worse, and one-third died of the disease. Improvement, even remission, may occur early on but is rarely permanent (i.e., there is a subsequent relapse). Symptoms typically fluctuate over a relatively short period and then become more severe (active stage). Left untreated, an inactive stage follows the active stage, in which fluctuations in strength still occur but are attributable to fatigue, intercurrent illness, or other identifiable factors. After many years, untreated weakness becomes fixed, and the most severely involved muscles are frequently atrophic (burnt-out stage). Factors that worsen myasthenic symptoms are emotional upset, systemic illness (especially viral respiratory infections), hypothyroidism or hyperthyroidism, pregnancy, the menstrual cycle, drugs affecting NMT (see Treatment of Associated Diseases and Medications to Avoid, later in this chapter), and fever.
Physical Findings in Myasthenia Gravis
Ocular Muscles
Most MG patients have weakness of ocular muscles (Box 78.1). (Videos of MG-related ocular phenomena [Videos 78.1 and 78.2] can be found at www.expertconsult.com.) Asymmetrical weakness of several muscles in both eyes is typical, the medial rectus being more frequently and usually more severely involved. The pattern of weakness is not localizable to lesions of one or more nerves, and the pupillary responses are normal. Ptosis is usually asymmetrical (Fig. 78.1) and varies during sustained activity. To compensate for ptosis, chronic contraction of the frontalis muscle produces a worried or surprised look. Unilateral frontalis contraction is a clue that the lid elevators are weak on that side (see Fig. 78.1). When mild, ocular weakness may not be obvious on routine examination and appear only upon provocative testing (i.e., sustained upward gaze). Eyelid closure is usually weak, even when strength is normal in all other facial muscles, and may be the only residual weakness in otherwise complete remission. This is usually asymptomatic unless it is severe enough to allow soap or water in the eyes during bathing. With moderate weakness of these muscles, the eyelashes are not “buried” during forced eye closure (Fig. 78.2). Fatigue in these muscles may result in slight involuntary opening of the eyes as the patient tries to keep the eyes closed; this is called the peek sign (see Fig. 78.2).
Box 78.1 Ocular Findings in Myasthenia Gravis
• Weakness usually involves one or more ocular muscles, without overt pupillary abnormality
• Weakness is typically variable, fluctuating, and fatigable
• Ptosis that shifts from one eye to the other is virtually pathognomonic of myasthenia gravis
• With limited ocular excursion, saccades are superfast, producing ocular “quiver”
• After downgaze, upgaze produces lid overshoot (“lid twitch”)
• Pseudo-internuclear ophthalmoplegia—limited adduction, with nystagmoid jerks in abducting eye
• In asymmetrical ptosis, covering the eye that has lid ptosis may relieve contraction of the opposite frontalis
• Passively lifting a ptotic lid may cause the opposite lid to fall (“enhanced ptosis”) (see Video 78.2)
• Edrophonium may improve only some of several weak ocular muscles; others may actually become weaker
• Edrophonium may relieve asymmetrical ptosis and produce retraction of the opposite lid from frontalis contraction
• The opposite lid may droop further as the more involved lid improves after edrophonium
Oropharyngeal Muscles
Myasthenic patients may have a characteristic facial appearance. At rest, the corners of the mouth often droop downward, giving a depressed appearance. Attempts to smile often produce contraction of the medial portion of the upper lip and a horizontal contraction of the corners of the mouth without the natural upward curling, which gives the appearance of a sneer (see Fig. 78.1).
Limb Muscles
Weakness begins in limb or axial muscles in about 20% of MG patients (Kuks and Oosterhuis, 2004). Any trunk or limb muscle may be weak, but some are more often affected than others. Neck flexors are usually weaker than neck extensors, and the deltoids, triceps, and extensors of the wrist and fingers and ankle dorsiflexors are frequently weaker than other limb muscles. Rarely, MG presents initially with focal weakness in single muscle groups, such as a “dropped head syndrome” due to severe neck extensor weakness or isolated vocal cord or respiratory muscle weakness. In untreated patients with long-standing disease, weakness may be fixed, and severely involved muscles may be atrophic, giving the appearance of a chronic myopathy; this is particularly likely in muscle-specific tyrosine kinase (MuSK) antibody–positive MG (see MuSK Antibody Myasthenia Gravis, later in this chapter).
Immunopathology of Myasthenia Gravis
In about 80% to 85% of MG patients, weakness results from the effects of circulating anti-AChR antibodies. These antibodies bind to AChR on the terminal expansions of the junctional folds (Fig. 78.3) (Engel et al., 1977a) and cause complement-mediated destruction of the folds, accelerated internalization and degradation of AChR, and in some cases, they block ACh-AChR binding. Destruction of the junctional folds results in distortion and simplification of the postsynaptic region (see Fig. 78.4) and loss of functional AChR (Engel et al., 1977b). This leads to NMT failure and muscle weakness. MG is a paradigm for an antibody-mediated disease: the physiological abnormality is passively transferable by injection of MG immunoglobulin (Ig)G into mice, and clinical improvement follows removal of circulating antibodies by plasma exchange (see Treatment of Myasthenia Gravis, later in this chapter).
Patients with MG have increased numbers of CD4+ T cells, which regulate the production of AChR antibody (AChR-Ab). The α subunit of AChR contains the majority of T-cell recognition sites. These recognition sites may be different from those of the main immunogenic region that binding antibodies recognize. Sensitization to CD4+ T-cells spreads across the AChR complex as the disease progresses and most MG patients have T cells that recognize multiple epitopes on the AChR α-subunit (Conti-Fine et al., 1997). This epitope spread drives the synthesis of anti-AChR antibodies and accounts for the large and varied antibody repertoire of the myasthenic patient.
Recently, low-affinity IgG antibodies have been found in about two-thirds of MG patients who were seronegative using conventional anti-AChR and anti-MuSK antibody assays (Leite et al., 2008). These antibodies bind to AChRs that have been clustered into high-density arrays, suggesting that they have relatively low affinity and cannot bind strongly to AChR in solution but do bind to immobilized AChRs in a native conformation.
The Thymus in Myasthenia Gravis
The thymus is abnormal in most MG patients; 70% have lymphoid follicular hyperplasia, and more than 10% have a thymoma. Hyperplastic thymus glands from MG patients contain all the components necessary for the development of an immune response to the AChR: T cells, B cells, and plasma cells, as well as muscle-like myoid cells that express AChR. It is unlikely that the cellular alterations in the thymus are secondary to an ongoing peripheral immune response because they are absent in experimental autoimmune MG (Hohlfeld and Wekerle, 2008). In addition, thymocytes in culture spontaneously generate anti-AChR antibodies. These findings support the concept of an intrathymic pathogenesis and argue that the hyperplastic thymus is involved in the initiation of the anti-AChR immune response in patients with thymic hyperplasia. Thymic-derived AChR subunits may serve as an antigen for the autosensitization against the AChR.
Neoplastic epithelial cells in thymomas express numerous self-like antigens, including AChR, titin, and ryanodine receptor-like epitopes. MG-associated thymomas are also rich in autoreactive T cells. The regulation of potentially autoreactive T cells may be impaired in thymoma due to a deficiency in the expression of the autoimmune regulator gene (AIRE) and selective loss of T regulatory cells in human thymomas (Meriggioli and Sanders, 2009).
Myasthenia Gravis Subtypes
A number of MG subtypes (Table 78.1) may be identified based on the clinical presentation, age of onset, autoantibody profile, and thymic pathology (Meriggioli and Sanders, 2009). Interestingly, these subtypes appear to have unique genetic associations, strengthening the concept of distinct clinical entities and disease mechanisms.
Ocular Myasthenia Gravis
Ptosis and/or diplopia are the initial symptoms of MG in up to 85% of patients (Grob et al., 2008), and almost all patients have both symptoms within 2 years of disease onset. Myasthenic weakness that remains limited to the ocular muscles is termed ocular myasthenia gravis (OMG) and accounts for approximately 10% to 15% of all MG in Caucasian populations. If weakness remains limited to the ocular muscles after 2 years, there is a 90% likelihood that the disease will not generalize. OMG is more common in Asian populations (up to 58% of all MG patients) (Zang et al., 2007).
Generalized Myasthenia Gravis
Patients with generalized myasthenia gravis (GMG) may have either early-onset (EOMG) or late-onset (LOMG) disease, with the cutoff age usually defined as age 40. EOMG patients are more often female and typically have anti-AChR antibodies and enlarged hyperplastic thymus glands. LOMG patients are more often male and may have antibodies to striated muscle proteins such as titin and the ryanodine receptor in addition to anti-AChR antibodies. The presence of these anti-muscle antibodies, particularly anti–ryanodine receptor antibodies, has been associated with more severe generalized or predominantly oropharyngeal weakness and frequent myasthenic crises (Romi et al., 2005). LOMG patients without thymoma usually have a normal or atrophic thymus, but relatively few histological studies are available in this age group, as thymectomy after age 50 is unusual.
MuSK-Antibody Myasthenia Gravis
Antibodies to MuSK have been reported in up to 50% of patients with GMG who lack AChR antibodies (Guptill and Sanders, 2010) and have recently been reported in OMG as well (Bau et al., 2006; Caress et al., 2005). The reported incidence of MuSK-antibody myasthenia gravis (MMG) varies among geographic regions, the highest being closer to the equator and the lowest closer to the poles (Vincent and Lang, 2006). Genetic or environmental factors (or both) presumably play a role in these differences. MMG predominantly affects females and begins from childhood through middle age. In some patients, the clinical findings are indistinguishable from anti-AChR-positive MG, with fluctuating ocular, bulbar, and limb weakness. However, many MMG patients have predominant weakness in cranial and bulbar muscles, frequently with marked atrophy of these muscles (Fig. 78.5). Others have prominent neck, shoulder, and respiratory weakness, with little or no involvement of ocular or bulbar muscles. Electrodiagnostic abnormalities may not be as widespread as in other forms of MG, and it may be necessary to examine different muscles to demonstrate abnormal NMT (Stickler et al., 2005). The potentially more limited distribution of physiological abnormalities also may limit the interpretation of microphysiological and histological studies in MMG, inasmuch as the muscles usually biopsied for these studies may be normal.
Many MMG patients do not improve with cholinesterase inhibitors (ChEIs); some actually become worse, and many have profuse fasciculations with these medications (Hatanaka et al., 2005). Disease severity tends to be worse, but most improve dramatically with PLEX or corticosteroids (Sanders et al., 2003). More immunosuppression is typically necessary, though long-term outcome is generally good (Guptill and Sanders, 2010). Thymic changes are absent or minimal (Lauriola et al., 2005; Leite et al., 2005), and the role of thymectomy in MMG is not yet clear (Guptill and Sanders, 2010; Sanders et al., 2003). The diagnosis of MMG may be elusive when the clinical features, electrodiagnostic findings, and response to ChEIs differ from typical MG.
Seronegative Myasthenia Gravis
MG patients who lack both anti-AChR and anti-MuSK antibodies (“double-seronegative MG”) are clinically heterogeneous. The true frequency of “seronegative MG” may be quite low, as certain patients may have low-affinity anti-AChR antibodies that can only be detected using specialized assays (see Immunopathology of Myasthenia Gravis, earlier).
Genetics of Myasthenia Gravis
Several correlations exist between MG and the human leukocyte antigen (HLA) genes. Certain HLA types (-DR2, -DR3, -B8, -DR1) predispose to MG (see Table 78.1), whereas others may offer resistance to disease. HLA-B8, -DR2, and -DR3 types occur more commonly in patients with EOMG; HLA-B7 and -DR2 in LOMG; and HLA-DR1 in OMG (see Table 78.1). MMG is associated with HLA-DR14-DQ5 (Niks et al., 2006). Different HLA associations have been reported in Asian MG patients, including an association of OMG with HLA-BW46 in Chinese patients (Meriggioli and Sanders, 2009). Non-HLA genes (PTPN22, FCGR2, CHRNA1) have also been found to be associated with MG; some are also associated with other autoimmune diseases and thus may represent a nonspecific susceptibility to autoimmunity. An exception to this is the CHRNA1 gene, which encodes the α subunit of the AChR and may provide pathogenetic clues specific for MG (Meriggioli and Sanders, 2009).
Diagnostic Procedures in Myasthenia Gravis
Edrophonium Chloride Test
The most important consideration in performance of the edrophonium test is the choice of endpoint. Only unequivocal improvement in strength of an affected muscle is acceptable as a positive result. For this reason, resolution of eyelid ptosis, improvement in strength of a single paretic extraocular muscle, or clear improvement of dysarthria have been proposed as the only truly valid endpoints because observed function in these muscles is largely independent of fluctuating effort (Fig. 78.6). Interpret changes in strength of other muscles cautiously, especially in a suggestible patient.
The edrophonium test is reported to be positive in 60% to 95% of patients with OMG and in 72% to 95% with GMG (Pascuzzi, 2003). Improvement after edrophonium is not unique to MG; it is also seen in congenital myasthenic syndromes, the Lambert-Eaton syndrome, intracranial aneurysms, brainstem lesions, cavernous sinus tumors, end-stage renal disease, and in muscle diseases affecting the ocular muscles.
The optimal dose of edrophonium varies among patients and cannot be predetermined. In a study of OMG, the mean dose of edrophonium that gave a positive response was 3.3 mg for ptosis and 2.6 mg for ocular muscle dysfunction (Kupersmith et al., 2003). The lowest effective dose can be determined by injecting small incremental doses up to a maximum total of 10 mg. Inject an initial test dose of 2 mg, and monitor the response for 60 seconds. Subsequent injections of 3 and 5 mg may then be given, but if clear improvement is seen within 60 seconds after any dose, the test is positive, and no further injections are necessary (see Video 78.1). Weakness that develops or worsens after injection of 10 mg or less also indicates an NMT defect, as this dose will not weaken normal muscle.
Common side effects of edrophonium are increased salivation and sweating, nausea, stomach cramps, and fasciculations. Serious complications (bradyarrhythmia or syncope) have been reported in only 0.16% of edrophonium tests (Ing et al., 2005). These symptoms generally resolve with rest in the supine position. Atropine (0.4-2 mg) should be available for intravenous (IV) injection if bradycardia is severe.
Autoantibodies in Myasthenia Gravis
Acetylcholine Receptor Antibodies
Assays measuring antibodies that react with AChR proteins are generally regarded as specific serological markers for MG. The most widely utilized test for MG is the AChR-Ab binding assay. This assay tests serum for binding to purified AChR from human skeletal muscle labeled with radioiodinated α-bungarotoxin. The reported sensitivity of this assay is approximately 85% for GMG and 50% for OMG (Stålberg et al., 2010). Nearly all thymomatous MG patients have elevated AChR antibodies.
AChR antibodies cross-link the AChRs in the membrane and increase their rate of degradation. The AChR-modulating antibody assay measures the rate of loss of labeled AChR from cultured human myotubes. About 10% of MG patients who do not have elevated binding antibodies have AChR-modulating antibodies. Many patients with thymomatous MG have high levels of AChR-modulating antibodies, often exceeding 90% loss of AChR (Vernino and Lennon, 2004).
Antistriational Muscle Antibodies
StrAbs are not pathogenic and are also found in one-third of patients with thymoma who do not have MG and in one-third of MG patients without thymoma. They are more frequent in older MG patients and in those with more severe disease, suggesting that disease severity is related to a more vigorous humoral response against multiple muscle antigens (Romi et al., 2005).
Electrodiagnostic Testing in Myasthenia Gravis
Repetitive nerve stimulation (RNS) is the most commonly used electrophysiological test of NMT. At low rates of stimulation (2-5 Hz), RNS depletes the store of readily releasable ACh at diseased motor end-plates, causing failure of NMT. Characteristically in MG, there is a decrementing response of at least 10% to trains of 2- to 3-Hz stimulation (see Chapter 32B). This may be present at baseline or after a period of exercise (postactivation exhaustion). Although a seemingly simple test, careful attention to proper technique is important to avoid technical errors. The sensitivity of RNS for diagnosing MG reportedly ranges from 53% to 100% in GMG and 10% to 48% in OMG (Meriggioli and Sanders, 2004; Stålberg et al., 2010). RNS is more likely to be abnormal in a proximal or facial muscle and in clinically weak muscles. For maximal diagnostic yield, test several muscles, particularly those that are weak. If RNS is normal and there exists a high suspicion for an NMJ disorder, perform SFEMG of at least one symptomatic muscle.
SFEMG (see Chapter 32B) is the most sensitive clinical test of NMT and shows increased jitter in some muscles in almost all patients with MG (Stålberg et al., 2010). Jitter is greatest in weak muscles but is usually abnormal even in muscles with normal strength. Sixty percent of patients with OMG show increased jitter in a limb muscle, but this does not predict the subsequent development of generalized myasthenia.
In the rare patient who has weakness restricted to a few limb muscles, only a weak muscle may show abnormal jitter. This is particularly true in some patients with MMG (Stickler et al., 2005) (see MuSK-Antibody Myasthenia Gravis, earlier).
Recently, measuring jitter with concentric needle electrodes (CNE) has been proposed as an alternative to the specially designed (reusable) single-fiber electrode (Stålberg and Sanders, 2009). Interpret the results with caution, particularly in borderline cases, as signals recorded with the CNE may represent the summation of more than one single-fiber action potential, which will decrease the apparent jitter.
Ocular Cooling
Myasthenic weakness typically improves with muscle cooling. This is the basis of the “ice-pack” test, in which cooling of a ptotic lid improves lid elevation. Assess improvement in ptosis after placing an ice pack over the ptotic eyelid, usually for 2 minutes. Positive responses can occur even when edrophonium tests are negative. A meta-analysis of six studies showed this test to have high sensitivity and specificity in MG, suggesting that it may be useful in patients with lid ptosis, particularly if the edrophonium test is negative or contraindicated (Larner, 2004).
Comparison of Diagnostic Techniques in Myasthenia Gravis
Plan diagnostic testing based on the clinical presentation and distribution of weakness (Table 78.2). The edrophonium test is often diagnostic in patients with ptosis or ophthalmoparesis but is less useful in assessing other muscles. The presence of serum AChR or anti-MuSK antibodies virtually ensures the diagnosis of MG, but their absence does not exclude it. RNS confirms impaired NMT but is frequently normal in mild or purely ocular disease. Almost all patients with MG have increased jitter, and normal jitter in a weak muscle excludes MG as the cause of the weakness. Neither electrodiagnostic test is specific for MG, because increased jitter, even abnormal RNS, occurs in other motor unit disorders that impair NMT.
Treatment of Myasthenia Gravis
The outlook for patients with MG has improved considerably in recent years, largely due to advances in intensive care medicine and the use of immunomodulating agents. The therapeutic goal is to return the patient to normal function as rapidly as possible while minimizing the side effects of therapy. A number of therapeutic options are available (Table 78.3
[/not-level-membership-for-neurology-category]
