Chapter 8 Disorders of Iron Homeostasis
Iron Deficiency and Overload
Plasma Ferritin Concentrations
Plasma ferritin concentrations are helpful in the detection of both iron deficiency and iron overload.1 Plasma ferritin concentrations decline with storage iron depletion; a plasma ferritin concentration less than 12 mg/L is virtually diagnostic of absence of iron stores. The only known conditions that may lower the plasma ferritin concentration independently of a decrease in iron stores are hypothyroidism and ascorbate deficiency, but these conditions only rarely cause problems in clinical interpretation. Increased plasma ferritin concentrations may indicate increased storage iron, but a number of disorders may increase the plasma ferritin level independently of the body iron store. Plasma ferritin is an acute-phase reactant with increased ferritin synthesis, a nonspecific response that is part of the general pattern of the systemic effects of inflammation.1,2 Thus, fever, acute infections, rheumatoid arthritis, and other chronic inflammatory disorders elevate the plasma ferritin concentration. Both acute and chronic damage to the liver, as well as to other ferritin-rich tissues, may increase plasma ferritin concentration through an inflammatory process or by releasing tissue ferritins from damaged parenchymal cells.1
| Increased Iron Requirements |
|---|
Iron Deficiency and Coexisting Disorders
Detection of iron deficiency in the presence of chronic infectious, inflammatory, or malignant disorders that increase plasma hepcidin is more problematic than in the absence of such disorders.2 Even if iron lack contributes to the anemia of chronic disorders, the increase in plasma hepcidin will lead to a fall in the transferrin concentration (or total iron-binding capacity) and an increase in the plasma ferritin concentration.2,3 Because the serum transferrin receptor concentration is less affected by inflammation, its measurement usually can determine whether iron stores are absent.4 If uncertainty remains, BM examination is definitive. If iron deficiency is present, iron stores are absent; if the anemia of chronic disorders alone is responsible, iron stores are present and typically increased.
Oral Iron Therapy
Oral iron therapy should begin with a ferrous iron salt5 taken separately from meals in three or four divided doses and supplying a daily total of 150 to 200 mg of elemental iron in adults or 3 mg of iron per kilogram of body weight in children. Simple ferrous preparations are the best absorbed and least expensive. Ferrous sulfate is the most widely used, either as tablets containing 60 to 70 mg of iron for adults or as a liquid preparation for children. Administration between meals maximizes absorption. In patients with a hemoglobin concentration less than 10 g/dL, this regimen initially provides approximately 40 to 60 mg of iron daily for erythropoiesis, permitting RBC production to increase to two to four times normal and the hemoglobin concentration to rise by approximately 0.2 g/dL/day. An increase in the hemoglobin concentration of at least 2 g/dL after 3 weeks of therapy generally is used as the criterion for an adequate therapeutic response. For milder anemia, a single daily dose of approximately 60 mg of iron per day may be adequate. After the anemia has been fully corrected, oral iron should be continued to replace storage iron, either empirically for an additional 4 to 6 months or until the plasma ferritin concentration exceeds approximately 50 µg/L.
Table 8-2 Differential Diagnosis of Microcytic Hypochromic Anemia
| Decreased Body Iron Stores |
|---|
Parenteral Iron Therapy
Five parenteral iron preparations are approved for use in the United States: higher and lower molecular-weight iron dextran, sodium ferric gluconate complex, iron sucrose, and ferumoxytol.6 Each has been widely used, often in hemodialysis patients receiving recombinant human erythropoietin, but neither prospective, randomized controlled comparisons among the five agents nor long-term safety studies have been done. Although infrequent, immediate life-threatening anaphylactic reactions constitute the most serious risk associated with use of either intramuscular or intravenous iron preparations, may have a fatal outcome, and can occur with all five parenteral iron preparations.7 Delayed but severe serum sickness–like reactions may also develop, with fever, urticaria, adenopathy, myalgias, and arthralgias.
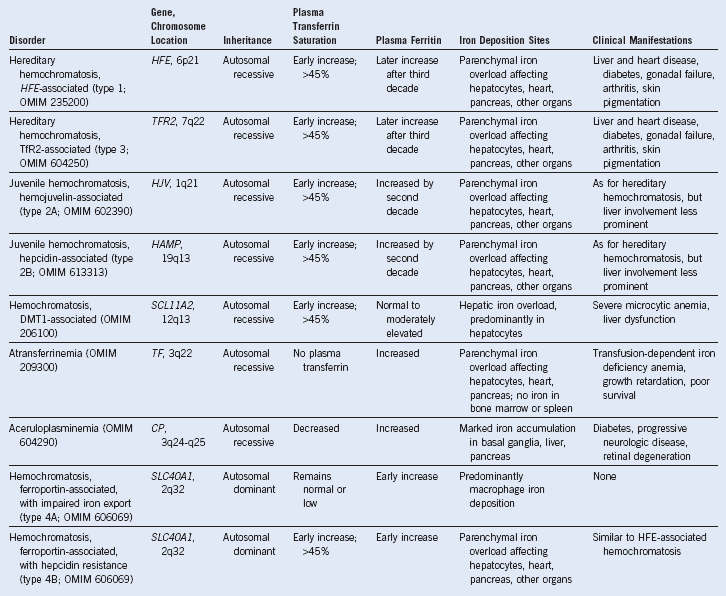
| Hereditary Iron Overload |
|---|
GRACILE, Growth retardation, aminoaciduria, cholestasis, iron overload, lactic acidosis, and early death; HFE, gene for the hemochromatoisis protein, HFE.
Testing for Iron Overload
A direct measure of body iron avoids the uncertainties inherent in the interpretation of indirect indicators of iron status. Liver biopsy is the definitive direct test for assessing iron deposition and tissue damage in iron overload, permitting measurement of the nonheme iron concentration, histochemical determination of the cellular distribution of iron between hepatocytes and Kupffer cells, and pathologic examination of the extent of tissue injury.8,9 When available as appropriately calibrated and validated techniques, new noninvasive methods using hepatic magnetic susceptibility and magnetic resonance imaging (MRI) may replace liver biopsy when only determination of the liver iron concentration is needed. MRI studies of the heart are particularly useful in patients at risk for cardiac iron deposition.10 In patients with hereditary hemochromatosis undergoing therapeutic venesection, quantitative phlebotomy provides an accurate retrospective determination of the amount of storage iron that can be mobilized for hemoglobin formation.11 When liver biopsy is contraindicated in a patient, quantitative phlebotomy occasionally is useful in establishing the diagnosis of hereditary hemochromatosis. BM aspiration and biopsy provide no information about the extent of parenchymal iron loading and are of limited value in the evaluation of iron overload. Iron overload produces no specific abnormalities in the peripheral blood.
Timing of Chelation Therapy
In all forms of transfusional iron overload, the most effective means of avoiding complications is to prevent excessive iron accumulation with early iron-chelating therapy.12 In patients who are transfusion dependent from early infancy (i.e., those with thalassemia major or other congenital refractory anemias), chelation therapy is best started after 10 to 20 transfusions, usually at approximately 3 years of age. In older patients with acquired refractory anemias who become transfusion dependent, it seems advisable to begin chelation early after transfusion of 10 to 20 units of blood. In patients with iron-loading anemias and those with sickle cell disease who are chronically transfused for prevention of complications, early therapy also seems prudent. In each of these disorders, delay in beginning chelation therapy only exposes the patient to a greater risk of iron toxicity.
1 Knovich MA, Storey JA, Coffman LG, et al. Ferritin for the clinician. Blood Rev. 2009;23:95.
2 Wessling-Resnick M. Iron homeostasis and the inflammatory response. Annu Rev Nutr. 2010;30:105.
3 Goodnough LT, Nemeth E, Ganz T. Detection, evaluation, and management of iron-restricted erythropoiesis. Blood. 2010;116:4754.
4 Skikne BS, Punnonen K, Caldron PH, et al. Improved differential diagnosis of anemia of chronic disease and iron deficiency anemia: A prospective multicenter evaluation of soluble transferrin receptor and the sTfR/log ferritin index. Am J Hematol. 2011;86:923.
5 Goddard AF, James MW, McIntyre AS, et al. Guidelines for the management of iron deficiency anaemia. Gut. 2011;60:1309.
6 Auerbach M, Ballard H. Clinical use of intravenous iron: Administration, efficacy, and safety. Hematology Am Soc Hematol Educ Program. 2010;2010:338.
7 Wysowski DK, Swartz L, Borders-Hemphill BV, et al. Use of parenteral iron products and serious anaphylactic-type reactions. Am J Hematol. 2010;85:650.
8 Brissot P, Bardou-Jacquet E, Jouanolle AM, et al. Iron disorders of genetic origin: A changing world. Trends Mol Med. 2011;17:707.
9 Pietrangelo A. Hereditary hemochromatosis: Pathogenesis, diagnosis, and treatment. Gastroenterology. 2010;139:393. 408, e1-2
10 Wood JC, Noetzli L. Cardiovascular MRI in thalassemia major. Ann N Y Acad Sci. 2010;1202:173.
11 McLaren GD, Gordeuk VR. Hereditary hemochromatosis: Insights from the Hemochromatosis and Iron Overload Screening (HEIRS) Study. Hematology Am Soc Hematol Educ Program. 2009:195.
12 Pietrangelo A. Hereditary hemochromatosis: Pathogenesis, diagnosis, and treatment. Gastroenterology. 2010;139:393. 408, e1-2