Chapter 8 Disorders of Intracellular Transport and Intercellular Conduction
Disorders of Intercellular Trafficking
Formation of Protein Subunits
As membrane proteins are formed in the ER, they undergo folding to their correct functional conformation. Occasionally, this process does not work correctly. In order to ensure that nonfunctional proteins are not targeted to the myocyte membrane, a process called unfolded protein response occurs. ER-associated degradation (ERAD) is responsible for the elimination and control of the buildup of aberrant proteins, preventing the aggregation of toxic nonfunctional proteins.1 These misfolded proteins are recognized by chaperones such as Hsp70/Hsp40, which interact directly with the misfolded protein and aid in its translocation into the cytoplasm for degradation by the proteasome. Evidence is now beginning to accumulate that the ERAD pathway participates not only in the removal of proteins that are not folded correctly but also in the rapid degradation of excess proteins. Thus, a protein is made from RNA in larger quantities than needed under normal conditions but is degraded via the ERAD pathway prior to insertion into cell membranes. When a cell needs a rapid increase of a particular protein, the ERAD system is turned off and more of the protein is trafficked to the cell membrane for use. An example of this is the adenosine-5′-triphosphate (ATP)–sensitive potassium (K[ATP]) channel, whose biogenesis and surface expression are controlled by ERAD. This is thought to be a mechanism for rapid increase in the availability of K(ATP) channels during cellular stress.2
Genetic Disorders of Trafficking
Normal trafficking of membrane proteins is exquisitely dependent on having particular amino acid sequences within the protein sequence. These sequences allow for the interaction of the channel protein with molecular motors, cytoskeletal elements, and scaffolding proteins, which are all important for protein movements throughout the cell. Changes in the nucleotide sequence of DNA will translate into RNA missense errors, which, in turn, lead to the production of incorrect amino acid sequences within the proteins. Thus, errors within the genome may produce proteins that are missing part or all of the appropriate trafficking sequences, thus causing genetic trafficking disorders. Since the normal cardiac rhythm is dependent on ion channels and gap junctions, abnormal ion channel and gap junction function underlie a number of cardiac rhythm disturbances. Alteration in individual channel gating function was thought to cause much of the loss of function of mutated channels, but analysis of many of the proarrhythmic mutations indicates that the true cause is the loss of trafficking to the membrane surface. For example, an alteration in the trafficking of ion channels to the cell’s surface has been shown to occur in some forms of atrial fibrillation (AF), long QT syndrome (LQTS) types 1 and 2, Brugada syndrome, and Anderson syndrome (Table 8-1).
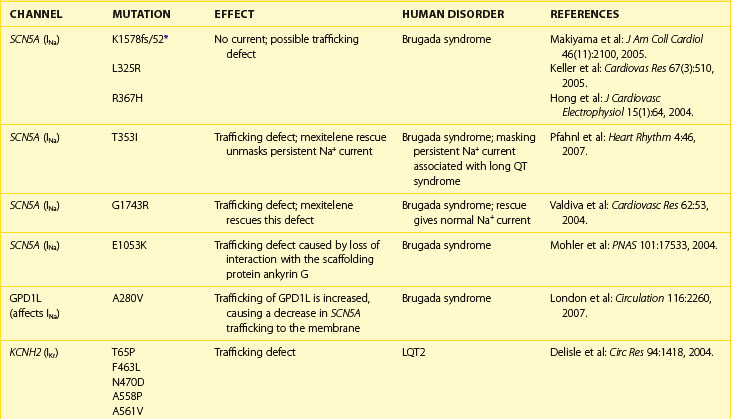
One of the more common forms of changes within protein sequences occurs when a coding change produces a protein with alternative amino acids at a given position within the protein. This shift, known as polymorphism, has two possible amino acids at particular loci within a protein but leaves the remaining amino acids in the correct location within the protein sequence. In many cases, no phenotypic change associated with polymorphisms occurs, but in rare cases, the changes at a single site can lead to the production of a protein that is nonfunctional. For example, in SCN5A, a mutation at amino acid 1053 from glutamic acid (E) to lysine (K) leads to a trafficking disorder of the sodium (Na+) channel. Written as I1053K, this mutation prevents the SCN5A protein from localizing to cell membranes by interrupting the ability of the SCN5A protein to interact with the scaffolding protein ankyrin G, which normally maintains the protein subunit at the cell membranes (see Chapter 2 and Table 8-1). This simple substitution of a single amino acid thus causes the Brugada syndrome phenotype.
Frame shift mutations may also occur. In these mutations, a coding error causes the initial RNA triplet to have either an extra nucleotide or a lost nucleotide, shifting the reading frame in a manner that causes a novel amino acid to be produced at that site. The produced protein will then have a scrambled or “nonsense” sequence of amino acids. These errors are very severe and often lead to proteins that are trafficking deficient; in addition, if the trafficking deficiency is rescued pharmacologically, these proteins are unable to form a channel with normal function. One example of this is the LQTS type 1 (LQT1) mutation that results from a frame shift at position 178 in the α-subunit of the channel that underlies the slow component of the delayed rectifier current, IK. In this case, the alanine normally found at position 178 is lost, which leads to the formation of an abnormal amino acid sequence; this sequence ends 105 amino acids later when, by chance, the combination of nucleotides forms a stop codon and the protein translation is terminated. This type of mutation, written as A178fs/105 in this case, causes the formation of a truncated form of the protein that is trafficking deficient and leads to the LQT1 phenotype (see Table 8-1).
Sodium Channels
The primary phenotype found in patients with mutations that cause loss of trafficking of sodium (Na+) channels to the cell membranes is Brugada syndrome. This phenotype is associated with high risk for sudden cardiac death. Voltage-gated Na+ channels are responsible for the rapid upstroke of the cardiac action potential and persistence of some Na+ current after rapid depolarization participates in the early phase of repolarization. Conduction velocity is, in part, dependent on both the amplitude and the rate of activation of these channels, and repolarization is normally slowed by the persistence of Na+ inward current during the early plateau phase. Therefore, decreased Na+ current leads to a shortened action potential duration. This exaggerates the normal heterogeneity of the outward current found in the heart. This heterogeneity is usually masked by the inward Na+ current leading to a voltage gradient across the ventricular wall, which is evident on the ECG as the classic Brugada’s ST-segment elevations in leads V1 through V3.3 Brugada syndrome can also result from mutations in the channel proteins that cause changes in channel function rather than a trafficking defect. Thus, loss of properly functioning Na+ channels at the cell membranes—either because of direct Na+ channel mutations or through loss of interaction of the Na+ channel with trafficking or scaffolding partners—leads to reduced Na+ current, increased heterogeneity in repolarization, and the subsequent production of re-entrant arrhythmias leading to sudden cardiac death.
Potassium Channels
Potassium (K+) channel mutations that decrease total channel expression at cell membranes are an important cause of LQTS types 1 and 2 in addition to other mutations that cause loss of function of channels that are trafficked to the membranes. The primary current affected in LQTS is the delayed outward rectifier K+ current, IK. Depending on the channel in which a mutation resides, the subtypes of K+ current affected (IKr or IKs) may vary, but the overall effect that manifests as LQTS is a loss of outward K+ current. This loss leads to a delay in ventricular cell repolarization and the duration of the QT interval. As with Brugada syndrome, this leads to an increase in transmural repolarization gradients. The mechanism for ventricular tachycardia (torsades de pointes, TdP) associated with LQTS has been postulated to be the occurrence of early after-depolarizations (EADs) and triggered activity caused by the action potential prolongation, which leads to re-entry facilitated by the heterogeneities of repolarization.3
An additional K+ channel trafficking mutation associated with cardiac arrhythmias and sudden cardiac death is Andersen-Tawil syndrome (see Table 8-1). This pleiotropic disorder is caused by a primary mutation in the gene KCNJ2. This gene codes for the Kir2.1 channel, which underlies the inwardly rectifying K+ channel. As with other K+ channel trafficking mutations, the primary ECG manifestation is a longer than normal QT interval, a dispersion of repolarization, and an increase in the propensity for ventricular arrhythmias likely triggered by EADs. While Andersen-Tawil syndrome has electrocardiographic similarities to LQTS, the pleiotropic nature of the disorder (periodic paralysis and dysmorphic features) helps distinguish it from other LQTSs.
Gap Junction Channels
Although deficiencies in gap junction function are associated with many different cardiac arrhythmias (see below), to date, the only arrhythmia associated with trafficking deficient mutations of gap junction proteins is AF. In some patients with AF, a subset of trafficking deficient mutations is found in connexin40 (Cx40), a primary gap junction protein in the atria.4 Interestingly, these mutations also cause a dominant negative effect on the trafficking of connexin43 (Cx43), the other connexin isoform found in the atria, leading to very low levels of cell–cell coupling between the atrial myocytes. The mechanism for this transdominant effect is not clear, but the overall effect is loss of cell–cell coupling, which leads to slowed conduction and a propensity for the formation of re-entrant arrhythmias. Trafficking mutations in Cx43, which is the major gap junction protein in the ventricular myocardium, have not been found to be associated with ventricular arrhythmias. This is primarily because Cx43 is vital for normal cardiac development and function; thus, trafficking mutations would likely cause embryonic lethality.
Intercellular Communication
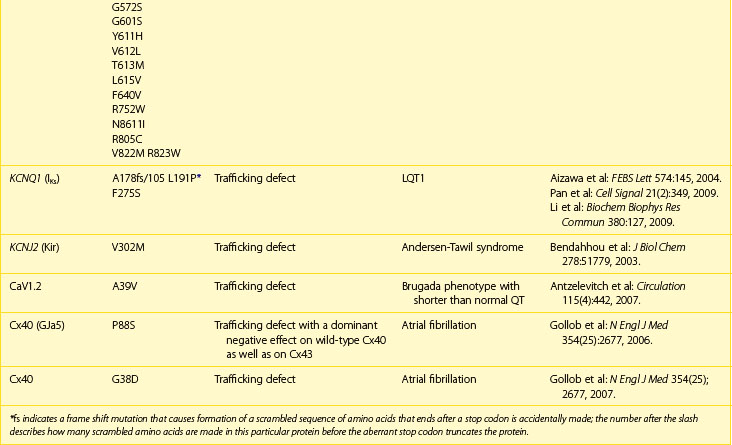
The ability for the heart to pump efficiently is dependent on the syncytial nature of the myocardium. Coordination of the contraction is maintained by passage of the electrical current through gap junctions, which are specialized membrane channels.5 Gap junctions are formed from half, or hemi-, channels inserted into the plasma membranes of individual myocytes. These hemi-channels (connexons) localize to the intercalated disc and meet head-to-head across the extracellular space with a connexon from an adjacent cell. This forms a longitudinally oriented conduit, which rapidly spreads excitation throughout the heart (Figure 8-1). The importance of gap junctions in maintaining conduction is underscored by the fact that their loss has been associated with slowed conduction and the formation of re-entrant arrhythmias.
Connexons are formed from the oligomerization of proteins from the connexin family in the Golgi apparatus. These proteins form four transmembrane domain-spanning units (see Figure 8-1). Overall, there are 21 isoforms of connexin in the human genome, five of which are found in the heart (Table 8-2).5 The most abundant connexin in the heart is Cx43, which localizes to both the atrial and the ventricular myocytes. The second most abundant connexin is Cx40, which is found in the atrial myocytes, largely co-localizing with Cx43 but is also a predominant isoform within the nodes of the heart. It is found more sparsely within the specialized conduction system in conjunction with connexin45 (Cx45). Low levels of this connexin have been reported in both the atria and the ventricles, although the physiological relevance Cx45 in these regions is unclear. The nodes of the human heart also contain connexin31.9 (Cx31.9), a low-conductance gap junction channel. Connexin37 (Cx37) occurs in the endothelial lining of the cardiac vasculature in conjunction with Cx43.5
| Cx31.9 |
Connexins follow the general pattern of membrane protein trafficking outlined above, with some interesting additions. The threading of the connexin protein into the membrane occurs, as with all membrane proteins, as it is being formed in the ER, but then the six connexins that form the connexon oligomerize, most likely beginning this process in the ER but finalizing the connexon formation within the Golgi apparatus.6 Thus, the protein subunits form the channel prior to insertion into the membrane of the cell and must be regulated to stay closed to ensure that the intracellular compartments do not exchange components.
Once the connexons are formed, they traffic out to the plasma membrane within vesicles and in some cases, such as Cx43, within caveolae with the aid of microtubules.7,8 For nonpolarized cells such as epithelial cells, a nondirected movement of connexons occurs, when connexons are trafficked outward to any region of the cell membrane. Polarized cells such as cardiomyocytes show directionality in the trafficking, with connexons being shuttled to the membrane domains in which they will reside and function. In the cardiac myocyte, this is the intercalated disc. From here, connexons move through the lipid bilayer and accumulate at the edges of gap junctional plaques, increasing the size of the plaque. As the protein is added, older protein that is designated to be removed from the plaque coalesces in the center of the plaque, and removal occurs from there.9 This turnover is rapid, occurring within a few hours, giving an exquisite regulatory control to the level of junctional coupling between cells.
Abnormalities in Intercellular Communication Causing Cardiac Arrhythmias
Changes in Gap Junction Conductance Without Changes in Connexin Amount or Location
The gap-junctional membrane provides low-resistance pathways for current flow between myocardial cells as well as for the passage of small molecules (up to 1 kDa). The permeability of a gap junction to the flow of ions that carry current (i.e., junctional conductance) is determined by the number of junctional channels, the proportion of the channels that are in the open state, and the permeability (conductance) of the open channel. The permeability of the gap junction channel in the open state, or unitary conductance, is determined by the particular connexin isoform or combination of isoforms that form the channel.5
The conductance of gap junctional channels may change under pathophysiological conditions without any change in the quantity or location of the connexin protein, through a change in the average conductance of gap junction channels. An important cause of reduction in Cx43 gap junction coupling that can occur in the ventricular ischemic arrhythmogenic substrate is low intracellular pH (below 6.5), an effect that can be facilitated by high intracellular Ca2+. Effects of Ca2+ and pH are caused by a change in the open probability of the gap junction channel rather than by single-channel conductance. pH becomes an important direct influence at the low pH (<6.5) associated with hypoxia and ischemia by directly closing the channels, but ischemia and hypoxia may also lead to changes in connexon configuration that contribute to decreased gap junction conductance.6
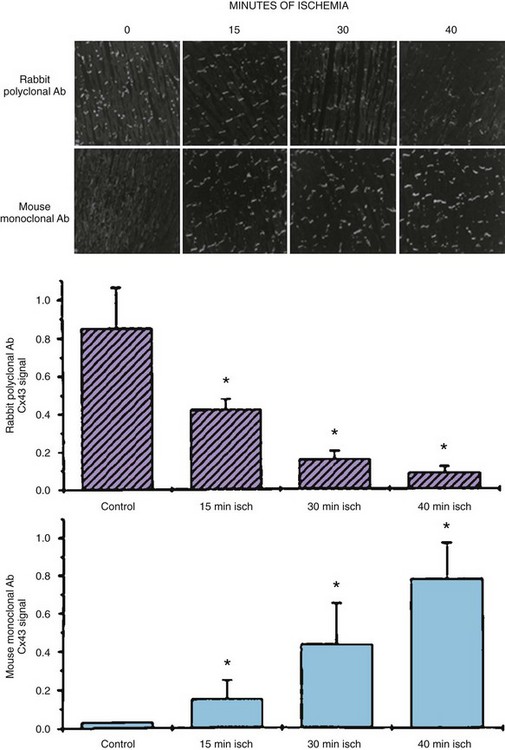
Changes in the phosphorylation state of connexins during remodeling can change gap junction channel function.6 For example, Cx43 is normally phosphorylated at multiple serine residues. Phosphorylation plays a number of roles. It might be necessary for maintaining hemi-channels in their closed state until docking occurs in the membrane of the intercalated disc. It also appears to be necessary for the normal opening and closing of gap junction channels. Changes in the phosphorylation state during acute ischemia can decrease the open probability of gap junction channels, probably prior to the change in Cx43 quantity that occurs following approximately 15 minutes of ischemia.
Changes in Quantity of Connexin Protein
Atrial Remodeling
The atria, including the sleeves of the thoracic veins, contain Cx40, Cx43, and Cx45.10 Cx40 is expressed two- to threefold higher in the right atrium than in the left.11 Distribution is heterogeneous, with areas containing large amounts of Cx40 adjacent to regions with minimal or no Cx40. Cx43 is more homogeneously distributed in the atria, although some reports have suggested an increased level of Cx43 in the right atrial free wall, as compared with the left atrial free wall. The functional significance of this difference in expression is unknown. The presence of multiple connexin isoforms in the atria may result in the formation of gap junction channels that have multiple isoform configurations with different regulatory and physiological characteristics. Gap junctions may comprise Cx40 alone, Cx43 alone, or Cx45 alone (homomeric and homotypic). The channels may also be homomeric but heterotypic; for example, homomeric Cx40 connexons may be coupled with homomeric Cx43 or Cx45 connexons. Channels may also be heteromeric but heterotypic; that is, a single connexon may be composed of different connexins such as Cx40 and Cx43, and coupled with another connexon comprising different connexins.6
Remodeling of gap junctions in the atrial myocardium has been documented in atrial fibrillation, an arrhythmia often associated with aging.10 Fibrosis associated with aging and fibrillation occurs in the form of fine longitudinally oriented collagenous microsepta and diminishes intercellular connections.12 It might decrease connexin quantity, although the relationships of connexin quantity to this kind of gap junction remodeling have not been determined. More extensive fibrosis that occurs in other atrial pathologies is often associated with decreased connexin as well (see below) and may play a larger role in the formation of the atrial arrhythmogenic substrate.
Studies on quantification of Cx43 and Cx40 have been done mostly in right atrial tissue from human and animal models of atrial fibrillation.10 Connexin quantification in the left atrium has rarely been done. This represents a significant problem in relating connexin remodeling to fibrillation, which often originates in the left atrium or pulmonary veins. Increase, decrease Cx40, and no change in the quantity of Cx40 have been reported.10 A common finding that is unrelated to quantitative changes in humans or animal models has been an increased heterogeneity in Cx40 distribution manifested as increased regions, in which Cx40 containing myocytes are located adjacent to myocytes mostly lacking Cx40. Cx43 may increase, decrease, or not change in quantity.10 Cx45, which has only been measured in postoperative AF, does not change. No quantitative data indicates how heteromeric or heterotypic gap junction channels might be affected. Together, these disparate data suggest that changes in connexins are variable. Their relationship to the occurrence of AF is uncertain but has been supported by some studies in transgenic murine models (see below).
Ventricular Remodeling
A decrease in total Cx43 occurs in the ischemic region as early as 15 minutes after experimental coronary occlusion (Figure 8-2). A change in the phosphorylation state of Cx43 that accompanies acute ischemia also occurs.13,14 Total phosphorylation is markedly decreased such that the ratio of dephosphorylated Cx43 to phosphorylated Cx43 increases. Dephosphorylation occurs at sites at S325, S328, S330, or in all three, which may contribute to decreased cell coupling by decreasing the number of functional gap junctions in intercalated discs.13,14 After more than 6 hours of ischemia resulting from coronary occlusion, Cx43 disappears from the necrotic infarct core but remains in reduced amounts in surviving myocytes that form border zones and then remains decreased (5% to 30% of normal) for days to months.15
In the border zones of healed infarcts, significant interstitial fibrosis is present. Collagen fibrils separate myofibers and distort the interconnections between cells. Fewer gap junctions per unit of intercalated disc length are present, and the long transversely oriented gap junctions in the interplicate regions are essentially absent.16 As a result, the number of cells connected to an individual ventricular myocyte is reduced with side-to-side contacts selectively affected. Normal organization of gap junctions as a prominent ring enclosing small spots is no longer discernible in some border zone myocytes adjacent to the necrotic core.15 Comparatively fewer labeled gap junctions are organized in discrete intercalated discs, and many are spread laterally over the cell (see section on lateralization).15 A reduction of Cx43 expression by more than 50% has been measured in experimental and clinical heart failure and hypertrophy, although quantitative data differ, depending on the cause, the duration, and the regions of the ventricle sampled.17,18
Upregulation of Cx45 in conjunction with downregulation of Cx43 at the end stage of human failing ventricles has been reported.19 An increase or no change in the quantity of Cx45 in the face of a decrease in Cx43 levels would increase the Cx45/Cx43 ratio, potentially promoting the formation of more heterotypic gap junctions with lower conductance than in normal hearts. As in ischemia, a decrease in the phosphorylated form and an increase in the dephosphorylated form of Cx43 are evident in experimental as well as clinical heart failure, with a substantial decrease in both pS365 as well as in pS325/328/330, which would also be expected to decrease conductance.20
Cx43 gap junctions within the intercalated discs of the hypertrophied myocardium are reduced.21 In hypertrophic cardiomyopathy, intercalated discs in regions of myocyte disarray do not show the standard stepwise morphology but are abnormally enlarged and show abundant Cx43 immunostaining. In areas of myofiber disarray, Cx43 gap junctions are no longer confined to intercalated discs but are dispersed over the surfaces of myocytes (see next section).
Normal gap junction amount and localization in ventricular myocytes likely depend on normal mechanical coupling via cell–cell adhesion junctions.22 Carvajal syndrome is caused by a recessive mutation in desmoplakin, a protein that links desmosomal adhesion molecules in intercalated discs, to the myocyte cytoskeleton. Naxos disease is caused by a recessive mutation in plakoglobin (γ-catenin), that links N-cadherins in the discs to actin and desmosomal cadherins to desmin. In Naxos disease, Cx43 is expressed abundantly but fails to localize to gap junctions. Both diseases are associated with a reduction in Cx43.22
Changes in Location of Connexin Protein: Lateralization
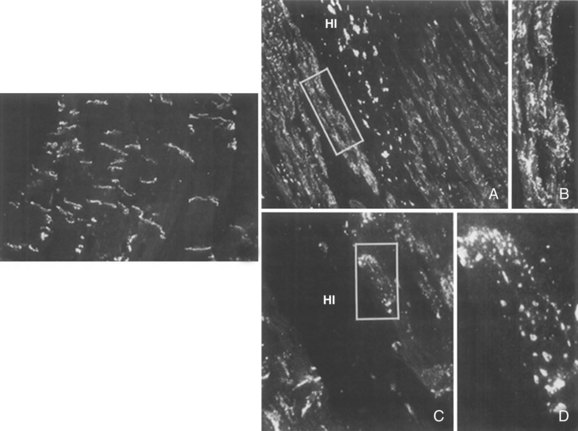
Connexin remodeling in ventricles is often associated with Cx43 dispersion over the cell surface outside the intercalated disc, a process called lateralization. Lateralization is not as prevalent in the atria as in the ventricles, although it has been described in AF.10 Lateralization appears to be intimately related to remodeling that leads to a decrease in connexin protein at intercalated discs. After acute ischemia, some of the Cx43 rapidly moves to lateral nonintercalated disc membranes within 30 minutes.13,23 In chronic ischemia and infarction, lateralization of Cx43 occurs in infarct border zones mainly in the surviving myocytes that are adjacent to the necrotic regions (Figure 8-3).15 The amount of lateralized Cx43 is not commensurate with lateralized gap junctions. In a canine model, at 5 days after occlusion, very few lateral gap junctions were detected by ultrastructure analysis despite abundant lateral immunofluorescent antibody to Cx43 (Heather S. Duffy, unpublished observation). In chronic infarcts, lateral gap junctions have been detected, but they are not as numerous as would be expected if most of the lateralized Cx43 formed gap junctions.15 Gap junctions are still present in intercalated discs after lateralization. However, the normal organization as a prominent ring around the myocyte enclosing small regions of gap junctional plaques within the intercalated disc is no longer discernible.15 The long ribbon-shaped junctions in the interplicate disc appear to be reduced in conjunction with the widening of the intercellular space.16 Lateralization of Cx43 also occurs in heart failure and hypertrophy.17,18 The amount of lateralized Cx43 as detected by immunofluorescence is much greater than any increase in identifiable gap junctions. Decreases in transverse conduction velocity following coronary occlusion argue against lateralized gap junctions forming functional channels between adjacent myocytes.
Electrophysiological Mechanisms of Cardiac Arrhythmias
Role of Gap Junction Remodeling in Re-entrant Excitation
Conduction velocity decreases monotonically with a reduction in intercellular coupling as would occur with gap junction remodeling.24 Conduction becomes discontinuous, and the safety factor increases, which differs from the fall in safety factor that occurs with a reduction of INa. Conduction can slow to about one fifteenth of normal at gap junction coupling, which is reduced by a factor of approximately 100, whereas minimum possible conduction velocity with reduced INa is by about a factor of 3.24 Therefore, gap junction uncoupling can cause the necessary slowed conduction that enables re-entry. Decreased gap junction coupling during remodeling also can alter the anisotropic properties of the myocardium, an important factor in arrhythmogenesis. A uniform reduction of transverse and longitudinal gap junction conductance causes a uniform longitudinal and transverse reduction of conduction velocity with an increase in the anisotropic ratio, since transverse conduction decreases more than longitudinal conduction does.25 Changes in the characteristics of anisotropic propagation from uniform to nonuniform may also be a consequence of gap junction remodeling.12
Gap junction coupling influences the refractory properties of the myocardium as well. Significant differences in refractory periods (heterogeneity) between closely adjacent regions predispose the myocardium to conduction block of premature impulses in those regions with longer refractory periods and allows conduction in those regions with shorter refractory periods, which can initiate re-entrant excitation.26 The time courses of repolarization of myocytes in different regions of the ventricles and atria are different because of intrinsic differences in repolarizing membrane currents. These intrinsic differences in the ventricles are expressed in the action potentials of isolated myocytes, in which the time course of repolarization is longest in the deep subepicardial/midmyocardial myocytes (M cells) of the left ventricular free wall and the deep endocardial layers of the septum and papillary muscles and shorter in the epicardial and subendocardial myocytes.27 However, a significant transmural gradient does not occur in situ, likely caused by current flow through gap junctions when cells are well coupled.28 Current flows from myocytes repolarizing early to myocytes repolarizing late because of the intrinsic differences in the time course of repolarization, lengthening the repolarization in cells with a shorter time course and shortening the repolarization in cells with a longer time course. Decreased gap junction coupling with gap junction remodeling decreases homogeneity (increases heterogeneity) by enabling the differences in intrinsic membrane properties to be expressed.28
Re-entry Caused by Changes in Gap Junction Channel Conductance Without Changes in Connexin Amount or Location
Ventricular arrhythmias that occur within the first minutes of coronary artery occlusion (phase 1a) are mostly caused by re-entrant excitation with slow conduction resulting from membrane depolarization, before conduction slowing caused by gap junction uncoupling. A second phase of arrhythmias that occurs later (phase 1b) may also be caused by re-entry at a time when uncoupling occurs because of the effects of hypoxia, acidification, and dephosphorylation on gap junction channel function.26 Uncoupling causes a substantial increase in internal longitudinal (axial) resistance and conduction velocity over a period of about 30 minutes before conduction block occurs.29,30 Alterations in the configuration of connexon channels in gap junctions over a slightly longer time course, up to 3 hours, may also contribute to the decreased conductance before irreversible cell damage occurs.23 Increased heterogeneity of refractoriness that contributes to conduction block during both phases may be partly related to uncoupling. Cx43 quantity begins to decrease shortly after coronary occlusion, probably contributing to more long-term slowing of conduction during phase 1b arrhythmia.14
Re-entry Caused by Gap Junction Remodeling Characterized by Changes in Connexin Quantity
The decrease in connexin protein that accompanies atrial and ventricular pathology is often associated with the occurrence of arrhythmias. Although a re-entrant mechanism is a likely cause, it has not always been directly demonstrated. A decrease in connexin protein should decrease the number of functioning gap junctions, intercellular coupling, and conduction velocity and also increase heterogeneity of refractoriness. The exact quantitative relationship between reduction of connexin protein levels and changes in conduction velocity and refractoriness is uncertain. This problem of relating connexin reduction to changes in conduction has been addressed in studies of transgenic murine models. For example, in the atria of transgenic murine models with downregulation of Cx40 in the absence of changes in Cx43 and Cx45, almost complete reduction of Cx40 was shown to result in decreased atrial conduction velocity of 30% to 36%.31 The decrease in Cx40 is heterogeneous, as is the decrease in conduction, likely causing block in some regions devoid of most Cx40. Such heterogeneities also occur in animal models as well as in patients with AF.10 Refractory periods have not been measured in the transgenic murine models but are heterogeneous in both animal models and patients, which is a possible result of heterogeneous gap junction remodeling. Some slowing of atrial conduction also occurs with decreased Cx43. The presence of Cx43, therefore, may be maintaining conduction in the absence of Cx40. In a clinical study, it was found that the ratio Cx43/[Cx43+Cx40] was directly related to propagation velocity and that the Cx40/[Cx43+Cx40] ratio was inversely related to propagation velocity.32 This suggests that changing the ratio of homomeric channels to heteromeric channels will have effects on propagation velocity, reflecting the functions of individual channels. As described above, in some clinical studies on AF, an upregulation of Cx40 has been demonstrated.10 Its effects on conduction or other electrophysiological properties are unknown, but it may contribute to the occurrence of heterogeneities in conduction and refractoriness.
The link between atrial gap junction remodeling and arrhythmias is circumstantial. An increased occurrence of spontaneous and induced atrial arrhythmias in transgenic models of reduced Cx40 has been demonstrated, although re-entrant circuits have not been mapped.33 Induction and termination by pacing is suggestive of a re-entrant mechanism. Re-entry has been shown to cause rapid atrial arrhythmias in both animal models and clinical cases.10 The relative contributions of gap junction remodeling and other properties of the arrhythmogenic substrate have not been determined.
In the ventricles, reduction of Cx43, either by targeting the genes that control Cx43 formation or the genes controlling other proteins related to gap junction location in intercalated discs, causes a significant decrease in gap junction coupling and ventricular arrhythmias related to abnormal conduction and refractoriness, which are likely to be re-entrant (Figure 8-4). Reduction of Cx43 in the ventricles by as much as 50% by disruption of one allele of the Cx43 gene does not significantly slow conduction.34 However, it does predispose the myocardium to the effects of “second hits” on gap junctions to slow conduction such as ischemia (see below). A very large reduction of Cx43, by more than 75%, significantly slows conduction by approximately 50% in the transverse and approximately 20% in the longitudinal direction, and increases the anisotropic ratio (see Figure 8-4).35,36 Conduction slowing does not seem to be commensurate with the marked reduction in Cx43 in transgenic murine models, and it is not certain if this quantitative relationship applies to the human heart (see below).
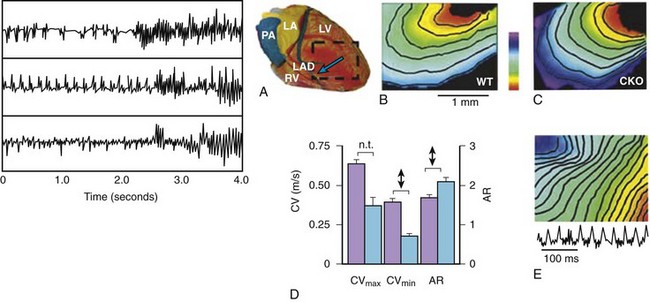
Since remodeling of Cx43 associated with a decrease in quantity is usually heterogeneous, regions of the myocardium nearly devoid of Cx43 may occur both in pathology and in transgenic murine models. These regions cause very slow conduction or conduction block and may be the critical feature leading to re-entry. Although the upregulation of other connexins must be considered as an explanation for the unexpected maintenance of conduction in the face of a marked reduction of Cx43 in transgenic models, this has not been demonstrated. Another important factor concerning gap junction control of conduction in murine ventricles is the very small size of the myocytes and the concomitant high resistance of the intracellular compartment in relation to gap junction resistance and an upregulation of Na+ channels, both rendering conduction less dependent on gap junction coupling than it might be in humans.24
Beyond the observation that arrhythmias are associated with Cx43 remodeling, direct proof that remodeling of gap junctions is the cause of re-entry caused by slow conduction in pathology is sparse. The diseased heart in which gap junction remodeling occurs usually has alterations in sarcolemmal ion channel function as well as structural changes such as fibrosis, making it difficult to define the relative role of gap junction remodeling in the arrhythmogenic substrate. In a canine model of myocardial infarction, in which there is a decrease in Cx43 and lateralization (see below), re-entrant circuits causing ventricular tachycardia have been mapped in the epicardial border zone.36 It is likely that interaction between the reduced Na+ current in border zone myocytes with reduced gap junction coupling causes the slow conduction and block necessary for re-entry. Re-entrant circuits have also been mapped in border zones of healed human infarcts which have gap junction remodeling.21,26 Although gap junction remodeling can influence the refractory period, re-entry dependent on heterogeneities in the effective refractory period has only been shown in a canine model of pacing-induced heart failure.37
Additional evidence to show the relationship between conduction slowing caused by Cx43 reduction and re-entrant arrhythmias comes from studies on transgenic murine models of gap junction remodeling, in which changes in sarcolemmal ion channel function and myocardial structure changes do not occur. Cx43 heterozygous mice are abnormally susceptible to the development of ventricular tachycardia in response to acute ischemia, despite the minimal effects of the reduction in Cx43 by itself (~50%) on conduction.38 The effects of ischemia to reduce gap junction coupling may be enhanced and thus cause more severe conduction slowing leading to re-entrant excitation, although re-entrant circuits have not been mapped. This has important implications for clinical arrhythmias, since hearts with chronic pathology causing reduction of Cx43 (e.g., failure and hypertrophy) that, in itself, may not be sufficient to cause arrhythmias may be more susceptible following an ischemic event.
Murine hearts with conditionally inactivated Cx43 genes, resulting in Cx43 expression reduced by up to 90% with extensive areas completely devoid of Cx43, exhibit spontaneously occurring ventricular tachycardia/fibrillation and sudden cardiac death as well as inducible ventricular arrhythmias indicative of a re-entrant mechanism.35 Arrhythmias do not occur if Cx43 is not reduced to less than 40%.39 Other transgenic murine models with reduction of Cx43 also have demonstrated ventricular tachyarrhythmias.40 Re-entrant circuits that have been mapped have the characteristics of anisotropic re-entry with conduction velocity more rapid in the longitudinal direction and functional lines of conduction block of transversely conducting wavefronts.39 Small rotors may also form in regions with inexcitable obstacles formed by myocytes devoid of Cx43. Abnormal impulse initiation (see below) cannot be excluded.
Although the evidence is limited, Cx45 may be upregulated in heart failure.19 Transgenic mice overexpressing Cx45, without changes in Cx43 gap junctions, exhibit enhanced spontaneous or induced ventricular arrhythmias suggesting a re-entrant mechanisms.41 Overexpression of Cx45 in cells normally expressing Cx43 significantly reduces intercellular coupling, since Cx43 and Cx45 may form low-conductance heteromeric channels.
Gap Junction Remodeling Characterized by Changes in Gap Junction Location
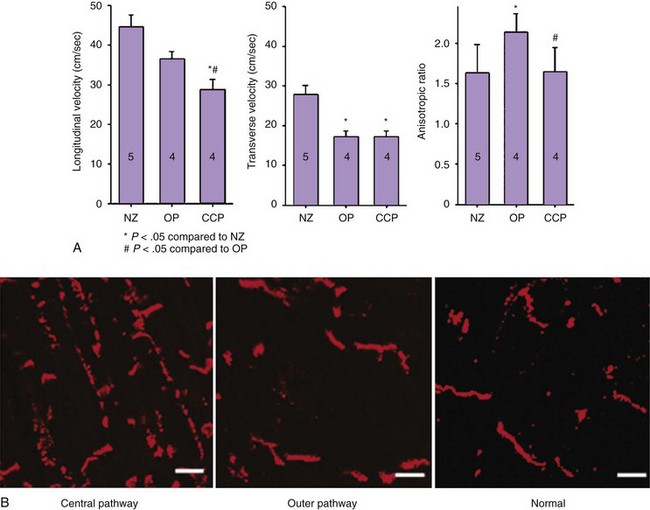
Lateralization of connexin protein may be part of the process that leads to a reduction of connexins. In most instances, a decrease in total connexins occurs despite an increase on nondisc lateral membranes. Lateralized Cx43, sometimes but not always, is dephosphorylated, which may be part of the mechanism for lateralization. The expected increase in transverse conduction velocity that would occur if new lateral gap junctions were formed has usually not been seen (Figure 8-5), and an increase in lateral gap junctions commensurate with increased lateralized connexin demonstrated by immunofluorescent studies is absent.36
Effects of Gap Junction Remodeling on Abnormal Impulse Initiation
Automaticity
The sinus node provides an example of the influence of gap junction coupling on automaticity. The sinus node is poorly coupled with the surrounding atrial myocardium through transitional cells with gap junctions formed by connexins 40, 45, 31.9, or all of them. If the two regions were well coupled, hyperpolarizing current flow from the well-polarized atrial myocardium would prevent sinus node impulse initiation. The weak coupling allows propagation from the small mass of the sinus node to the larger mass of the atrium.42 The effect of gap junction coupling on sinus node automaticity and conduction also applies to the interaction of subsidiary pacemakers with surrounding nonpacemakers. In well-coupled cells, flow of the hyperpolarizing current from nonpacemaker cells to pacemaker cells would slow spontaneous firing or even inhibit it, depending on the coupling resistance, while preventing impulses initiated in a relatively small region from propagating into the larger surrounding region. Partial uncoupling caused by gap junction remodeling would allow subsidiary pacemakers to fire and possibly drive the heart as an arrhythmia.43 Such arrhythmias might occur in any of the pathologic situations that result in atrial or ventricular gap junction remodeling, particularly in situations of heterogeneous decreases in connexin.
Conversely, the flow of current between partially depolarized regions and well-polarized regions may induce spontaneous (automatic) firing under special circumstances. In ischemic regions with decreased membrane potential adjacent to normally polarized myocytes either in nonischemic subendocardial Purkinje system or muscle, an injury current flows from the ischemic myocytes with low membrane potentials to myocytes with higher membrane potentials, which results in depolarization-induced impulse initiation (abnormal automaticity) in the latter.26 When there is normal coupling, the flow of the injury current is too large to permit spontaneous activity, since it “clamps” the cells at the low membrane potential. If uncoupling is too severe, insufficient injury current occurs to depolarize the myocytes to the level of membrane potential necessary for abnormal automaticity.44 The cause of the uncoupling is likely the low pH and elevated Ca2+, which occur during acute ischemia; however, by this time, some dephosphorylation and reduction of Cx43 have occurred (see above).
Triggered Activity
Since the occurrence of EADs is favored by the prolongation of repolarization in the presence of inciting influences such as β-adrenergic stimulation or class III antiarrhythmic drugs, gap junction remodeling leading to uncoupling and the increase in action potential duration described above, can lead to EAD formation and triggered activity, particularly in myocytes with long intrinsic action potential durations such as M cells.26 However, even in the face of moderate uncoupling, EADs may be prevented by the flow of repolarizing current from the neighboring myocardium.45 The amount of current required is dependent on the size of the EAD focus, and this, in turn, influences the degree of uncoupling that allows EAD expression. If the uncoupling is sufficient to allow the expression of EADs and triggered activity, the coupling must still be sufficient to allow the propagation of the triggered activity to the surrounding myocardium.46 In the case of acute ischemia and partial uncoupling, the flow of injury current, particularly at junctions of partially depolarized ventricular muscle with more normally polarized Purkinje fibers, can prolong repolarization time course and lead to the formation of EADs. The effects of coupling on DAD-induced triggered activity has not been specifically addressed, but the same influences of well-polarized cells on automaticity and EADs described above would be expected.
1 Ahner A, Brodsky JL. Checkpoints in ER-associated degradation: Excuse me, which way to the proteasome? Trends Cell Biol. 2004;14:474-478.
2 Yan FF, Lin CW, Cartier EA, Shyng SL. Role of ubiquitin-proteasome degradation pathway in biogenesis efficiency of b-cell ATP-sensitive potassium channels. Am J Physiol Cell Physiol. 2005;289:C1351-C1359.
3 Priori SG, Rivolta I, Napoitano C. Genetics of long QT, Brugada, and other channelopathies. In Zipes DP, Jalife J, editors: Cardiac electrophysiology: From cell to bedside, ed 4, Philadelphia: Saunders, 2004.
4 Gollob MH, Jones DL, Krahn AD, et al. Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N Engl J Med. 2006;354(25):2677-2688.
5 Duffy HS, Fort AG, Spray DC. Cardiac connexins: Genes to nexus. In: Dhein S, editor. Cardiovascular gap junctions. Basel: Karger Press, 2006.
6 Harris AL. Emerging issues of connexin channels: biophysics fills the gap. Q Rev Biophys. 2001;34(3):325-472.
7 Schubert AL, Schubert W, Spray DC, Lisanti MP. Connexin family members target to lipid raft domains and interact with caveolin-1. Biochemistry. 2002;41(18):5754-5764.
8 Shaw RM, Fay AJ, Puthenveedu MA, et al. Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell. 2007;128(3):547-560.
9 Gaietta G, Deerinck TJ, Adams SR, et al. Multicolor and electron microscopic imaging of connexin trafficking. Science. 2002;296(5567):503-507.
10 Duffy HS, Wit AL. Is there a role for remodeled connexins in AF? No simple answers. J Mol Cell Cardiol. 2008;44:4-13.
11 Vozzi C, Dupont E, Coppen SR, et al. Chamber-related differences in connexin expression in the human heart. J Mol Cell Cardiol. 1999;31(5):991-1003.
12 Spach MS, Dolber PC. Relating extracellular potentials and their derivatives to anisotropic propagation at a microscopic level in human cardiac muscle. Evidence for electrical uncoupling of side-to-side fiber connections with increasing age. Circ Res. 1986;58:356-371.
13 Lampe PD, Cooper CD, King TJ, Burt JM. Analysis of connexin43 phosphorylated at S325, S328 and S330 in normoxic and ischemic heart. J Cell Sci. 2006;119(Pt 16):3435-3442.
14 Beardslee MA, Lerner DL, Tadros PN, et al. Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ Res. 2000;87:656-662.
15 Smith JH, Green CR, Peters NS, et al. Altered patterns of gap junction distribution in ischemic heart disease. An immunohistochemical study of human myocardium using laser scanning confocal microscopy. Am J Pathol. 1991;139(4):801-821.
16 Luke RA, Saffitz JE. Remodeling of ventricular conduction pathways in healed canine infarct border zones. J Clin Invest. 1991;87(5):1594-1602.
17 Dupont E, Matsushita T, Kaba RA, et al. Altered connexin expression in human congestive heart failure. J Mol Cell Cardiol. 2001;33:359-371.
18 Teunissen BE, Jongsma HJ, Bierhuizen MF. Regulation of myocardial connexins during hypertrophic remodelling. Eur Heart J. 2004;25(22):1979-1989.
19 Yamada KA, Rogers JG, Sundset R, et al. Up-regulation of connexin45 in heart failure. J Cardiovasc Electrophysiol. 2003;14:1205-1212.
20 Qu J, Volpicelli FM, Garcia LI, et al. Gap junction remodeling and spironolactone-dependent reverse remodeling in the hypertrophied heart. Circ Res. 2009;104:365-371.
21 Severs NJ, Bruce AF, Dupont E, Rothery S. Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc Res. 2008;80(1):9-19.
22 Duffy HS. How do myocytes tell right from left? Circ Res. 2008;99(6):563-564.
23 Kieken F, Mutsaers N, Dolmatova E, et al. Structural and molecular mechanisms of gap junction remodeling in epicardial border zone myocytes following myocardial infarction. Circ Res. 2009;104:1103-1112.
24 Kléber AG, Rudy Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol Rev. 2004;84:431-488.
25 Jongsma HJ, Wilders R. Gap junctions in cardiovascular disease. Circ Res. 2000;86:1193-1197.
26 Wit AL, Janse MJJ. The ventricular arrhythmias of ischemia and infarction electrophyiological mechanisms, Mt. Kisco. New York: Futura Publications; 1993.
27 Antzelevitch C, Dumaine R. Electrical heterogeneity in the heart: Physiological, pharmacological and clinical implications. In: Page E, Fozzard HA, Solaro RJ, editors. The handbook of physiology. New York: Oxford University Press, 2001.
28 Lesh MD, Pring M, Spear JF. Cellular uncoupling can unmask dispersion of action potential duration in ventricular myocardium: A computer modeling study. Circ Res. 1989;65:1426-1440.
29 Kléber AG, Riegger CB, Janse MJ. Electrical uncoupling and increase of extracellular resistance after induction if ischemia in isolated, arterially perfused rabbit papillary muscle. Circ Res. 1987;61:271-279.
30 de Groot JR, Wilms-Schopman FJ, Opthof T, et al. Late ventricular arrhythmias during acute regional ischemia in the isolated blood perfused pig heart. Role of electrical cellular coupling. Cardiovasc Res. 2001;50(2):362-372.
31 Leaf DE, Feig JE, Vasquez C, et al. Connexin40 imparts conduction heterogeneity to atrial tissue. Circ Res. 2008;103:1001-1008.
32 Kanagaratnam P, Cherian A, Stanbridge RDL, et al. Relationship between connexins and atrial activation during human atrial fibrillation. J Cardiovasc Electrophysiol. 2004;15:206-213.
33 Hagendorff A, Schumacher B, Kirchhoff S, et al. Conduction disturbances and increased atrial vulnerability in connexin40 deficient mice analyzed by transesophageal stimulation. Circulation. 1999;99:1508-1515.
34 Morley GE, Vaidya D, Samie FH, et al. Characterization of conduction in the ventricles of normal and heterozygous Cx43 knockout mice using optical mapping. J Cardiovasc Electrophysiol. 1999;10:1361-1375.
35 Gutstein DE, Morley GE, Vaidya TH, et al. Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ Res. 2001;88:333-339.
36 Cabo C, Yao J, Boyden PA, et al. Heterogeneous gap junction remodeling in re-entrant circuits in the epicardial border zone of the healing canine infarct. Cardiovasc Res. 2006;72(2):241-249.
37 Poelzing S, Akar FG, Baron E, Rosenbaum DS. Heterogeneous connexin43 expression produces electrophysiologic heterogeneities across the ventricular wall. Am J Physiol Heart Circ Physiol. 2004;286:H2001-H2009.
38 Lerner DL, Yamada KA, Schuessle RB, Saffitz JE. Accelerated onset and increased incidence of ventricular arrhythmias induced by ischemia in Cx43 deficient mice. Circulation. 2000;101:547-552.
39 Danik SB, Liu F, Zhang J, et al. Modulation of cardiac gap junction expression and arrhythmic susceptibility. Circ Res. 2004;95:1035-1041.
40 Van Rijen HVM, Eckardt D, Degen J, et al. Slow conduction and enhanced anisotropy increase the propensity for ventricular tachyarrhythmias in adult mice with induced deletion of connexin43. Circulation. 2004;109:1048-1055.
41 Betsuyaku B, Nnebe NS, Sundset R, et al. Overexpression of cardiac connexin45 increases susceptibility to ventricular tachyarrhythmias in vivo. Am J Physiol Heart Circ Physiol. 2006;290:H163-H171.
42 Joyner RW, van Capelle FJ. Propagation through electrically coupled cells. How a small SA node drives a large atrium. Biophys J. 1986;50:1157-1164.
43 van Capelle FJ, Durrer D. Computer simulation of arrhythmias in a network of coupled excitable elements. Circ Res. 1980;47:454-466.
44 Huelsing DJ, Spitzerb KW, Pollard AE. Spontaneous activity induced in rabbit Purkinje myocytes during coupling to a depolarized model cell. Cardiovasc Res. 2003;59:620-627.
45 Spitzer KW, Pollard AE, Yang L, et al. Cell-to-cell electrical interactions during early and late repolarization. J Cardiovasc Electrophysiol. 2006;17:S8-S14.
46 Saiz J, Ferrero JM, Monserrat M, et al. Influence of electrical coupling on early afterdepolarizations in ventricular myocytes. IEEE Transac Biomed Engineer. 2006;46:138-147.